

Abstract
MicroRNAs (miRNAs) are 19–25 nucleotide RNAs that regulate messenger RNA translation and stability. Recently, we performed a conditional knockout (CKO) of the miRNA-processing enzyme Dicer during mouse retinal development and showed an essential role for miRNAs in the transition of retinal progenitors from an early to a late competence state (Georgi and Reh [2010]: J Neurosci 30:4048–4061). Notably, Dicer CKO progenitors failed to express Ascl1 and generated ganglion cells beyond their normal competence window. Because Ascl1 regulates multiple Notch signaling components, we hypothesized that Notch signaling is downregulated in Dicer CKO retinas. We show here that Notch signaling is severely reduced in Dicer CKO retinas, but that retinal progenitors still retain a low level of Notch signaling. By increasing Notch signaling in Dicer CKO progenitors through constitutive expression of the Notch intra-cellular domain (NICD), we show that transgenic rescue of Notch signaling has little effect on the competence of retinal progenitors or the enhanced generation of ganglion cells, suggesting that loss of Notch signaling is not a major determinant of these phenotypes. Nevertheless, transgenic NICD expression restored horizontal cells, suggesting an interaction between miRNAs and Notch signaling in the development of this cell type. Furthermore, while NICD overexpression leads to robust glial induction in control retinas, NICD overexpression was insufficient to drive Dicer-null retinal progenitors to a glial fate. Surprisingly, the presence of transgenic NICD expression did not prevent the differentiation of some types of retinal neurons, suggesting that Notch inactivation is not an absolute requirement for the initial stages of neuronal differentiation.
Keywords: dicer, notch, microRNA, retina, development
INTRODUCTION
During mouse retinal development multipotent progenitors produce the various retinal cells types in a conserved manner; ganglion cells, horizontal cells, and cone photoreceptors are generated early in embryogenesis, most amacrine cells are generated in the latter part of embryonic development, and the majority of rod photoreceptors, bipolar cells, and Müller glia are generated after birth. This progressive change in the cell types generated over development is thought to be due in part to intrinsic changes in progenitor cell competence. Although numerous transcription factors have been identified that are important for differentiation of specific retinal cell types (reviewed in Ohsawa and Kageyama, 2008), the specific molecular changes that determine the intrinsic competence state of a progenitor cell are not fully understood.
With their ability to regulate broad transcriptional programs through post-transcriptional regulation of hundreds of genes simultaneously, microRNAs (miRNAs) are attractive candidates for regulators of intrinsic retinal progenitor cell (RPC) competence, and as key effectors of cell-type specification and differentiation. miRNAs are processed from longer primary transcripts through several cleavage events, eventually being incorporated into the RNA-induced silencing complex (RISC) in a mature, 19–25 nucleotide form. Through partial sequence complimentarity of the miRNA, the RISC complex binds to target messenger RNAs (mRNAs), leading to their translational repression and/or degradation (reviewed in Fabian et al., 2010). With few known exceptions, processing by the RNase III enzyme Dicer is an obligate step in miRNA maturation, and Dicer conditional knockout (CKO) is a common technique to assay for general miRNA function in a tissue.
Recently, through conditional deletion of Dicer, we showed an essential role for miRNAs in controlling the changing competence of the retinal progenitor cells (Georgi and Reh, 2010). In the absence of Dicer, RPCs were unable to progress from an early to a late progenitor state and markers of late progenitors, such as Ascl1 and Sox9, were absent. In addition, the Dicer-deficient progenitors continued to generate ganglion cells well beyond their normal competence window, and these progenitors failed to generate late cell types, including rods, bipolar cells, and Müller glia.
Several of the defects in the Dicer CKO retina were consistent with changes in the Notch signaling pathway. First, among the late progenitor markers that are absent from Dicer CKO retinal progenitors is the proneural basic helix-loop-helix (bHLH) transcription factor Acheatescute 1 (Ascl1). Previously we showed that Ascl1 is required for normal Notch signaling during retinal development, and that in the Ascl1 knockout retina, numerous Notch signaling components are downregulated (Nelson et al., 2009). Based on these data, we hypothesized that the absence of Ascl1 in Dicer CKO retinas would lead to a reduction of Notch signaling. Second, studies have shown that a reduction of Notch signaling can lead to enhanced ganglion cell generation (Austin et al., 1995; Ahmad et al., 1997; Dorsky et al., 1997; Henrique et al., 1997; Waid and McLoon, 1998; Silva et al., 2003; Kubo et al., 2005; Nelson et al., 2006, 2007). Because the increased and extended generation of ganglion cells is one of the hallmarks of the Dicer CKO phenotype, we hypothesized that this defect might also be due to a reduction in Notch signaling during retinal development. Third, previous studies have indicated that Notch signaling is required for the differentiation of mature Müller glia (Furukawa et al., 2000; Hojo et al., 2000; Bernardos et al., 2005), and that the NICD transgene is a potent gliogenic signal (Jadhav et al., 2006a). Since no Muller glia develop in the Dicer CKO retinas, reduced Notch signaling might be responsible.
To test the possibility that changes in Notch signaling underlie some or all of the phenotype caused by the loss of Dicer, we assayed Notch signaling components in Dicer CKO retinas. In addition, we combined the Dicer deletion with both Notch signaling loss of function and gain of function experiments. We found that loss of Dicer leads to decreases in several Notch pathway components, consistent with our hypothesis and confirming Davis et al. (2011) who recently showed a decrease in Hes5 and Notch1 mRNA expression in Dicer CKO retina. However, rescue of Notch signaling using a transgenic Notch intracellular domain (NICD) approach does not restore the normal developmental program to the Dicer-deficient progenitors, as ganglion cells continue to be generated in the postnatal retina. Similarly, NICD transgene expression does not rescue expression of Ascl1, suggesting that the defects in Notch signaling observed in Dicer CKO retinas are downstream of the loss of Ascl1. Lastly, we find that rescue of the reduced Notch signaling with NICD overexpression does not drive gliogenesis in the Dicer CKO retina. These data indicate that the observed reduction in Notch signaling is not sufficient to explain the absence of Müller glia in Dicer CKO retinas, and suggest a role for Dicer in regulating the gliogenic competence of retinal progenitors.
METHODS
Generation of Animals
Generation and genotyping of αPax6cre; Dicerfl/+ and αPax6cre; R26EYFP; Dicerfl/fl mice was described previously (Georgi and Reh, 2010). ROSA-NICD mice were obtained from Jackson Laboratories (Bar Harbor, ME), and genotyped according to supplier’s protocols. These were crossed to Dicerfl/fl mice to generate Dicerfl/+; ROSA-NICD mice, of which siblings were crossed to generate Dicerfl/fl; ROSA-NICD mice carrying two copies of the ROSA-NICD allele. These were then crossed to αPax6cre; Dicerfl/+ mice to generate αPax6cre; Dicerfl/fl; ROSA-NICD and αPax6cre; Dicerfl/+; ROSA-NICD mice. αPax6cre mice were also crossed to ROSA-NICD mice to generate αPax6cre; ROSA-NICD mice. Animal housing and care was carried out by the Department of Comparative Medicine at the University of Washington. All procedures were done in compliance with the standards and protocols set forth by the University of Washington Institutional Animal Care and Use Committee.
Histological Analysis
Tissue was processed for frozen and paraffin sections as described previously (Georgi and Reh, 2010). Primary antibodies used included: Bhlhb5 (Santa Cruz Biotechnology, Santa Cruz, CA), DNER (1:200, R&D, Minneapolis), Id1 (1:200, Biocheck, Foster City, CA), and Tbr2 (Abcam, Cambridge, MA), Other antibodies used are listed in Georgi and Reh (2010). Some early postnatal retinas (P0 + 1) were cultured for 24 h before fixation to increase the nuclear GFP immunofluorescence signal-to-noise ratio for imaging. In all cases, described phenotypes were identical to those observed by immunofluorescence on tissue fixed immediately after dissection. In situ hybridization and probe synthesis was performed as described previously using antisense digoxigenin-labeled RNA probes (Nelson et al., 2009). Fluorescence immunohistochemistry was performed as previous described (Georgi and Reh, 2010). Slides were visualized by confocal microscopy using a Nikon A1 confocal microscope (Melville, NY). Images were acquired as 1024 × 1024 Tiff files, and analyzed using Nikon NIS-Elements and Photoshop CS4 (Adobe, San Jose, CA). All figure panels are oriented in the same direction, with peripheral retina to the right, ganglion cell layer at the bottom.
Statistical Analysis
At P0, cells were counted and normalized per 100 μm of YFP+ retina, as measured along the outer edge of the retina. αPax6cre Dicerfl/+ retinas were used as controls for comparisons to αPax6cre Dicerfl/+ ROSA-NICD (NICD+) retinas. At P5, C57Bl/6 (WT) mice were used as controls for comparison with the same spatial normalization, and similar central regions of retina were used for quantification. For Supporting Information figures, counts were normalized to the average of one of the conditions as indicated to show relative changes in cell counts. In all conditions, retinas from three animals were used for counts. Statistical analysis of cell counts was performed using an unpaired two-tailed t test with Prism 5 (Graph Pad Software, La Jolla, CA). Data are presented at mean ± SEM.
Retinal Culture and DAPT Treatment
Postnatal day 0 (P0) retinal explants were dissected, cultured, and treated with the γ-secretase inhibitor DAPT as previously described (Nelson et al., 2007).
EdU Birthdating
Postnatal day 3 (P3) pups were administered 45 μg 5-ethynyl-2′-deoxyuridine (Invitrogen, Carlsbad, CA) through subcutaneous injection. EdU visualization was performed as described by the manufacturer using the Click-iT Edu Alexa Fluor 647 Imaging Kit.
RESULTS
Notch Signaling is Decreased in Dicer CKO Retinas
We have previously shown that retinas lacking the transcription factor Ascl1 show a significant decrease in the levels of numerous components of the Notch pathway (Nelson et al., 2009). Because Dicer conditional knockout (CKO) progenitors also lack Ascl1 expression (Georgi and Reh, 2010), we hypothesized that many of these genes may also be downregulated in Dicer CKO areas. To test this hypothesis, we analyzed Notch pathway gene expression in αPax6cre; R26EYFP; Dicerfl/fl mice. In these mice, the αPax6cre transgene is expressed transiently beginning at approximately E10.5 in peripheral retinal progenitors, followed by expression in a subset of amacrine cells beginning at E14.5, and continuing into adulthood (Marquardt et al., 2001; Yaron et al., 2006; Lefebvre et al., 2008). By using a R26EYFP reporter, we tracked those cells in which the αPax6cre transgene mediated recombination, indicating regions of Dicer deficiency. Since the transgene is expressed in the peripheral one third to one half of the retina, the central retina serves as an internal control.
Conditional deletion of Dicer results in a substantial decrease in expression of several components of the Notch pathway, as assessed by in situ hybridization and immunofluorescence on retinas from postnatal day 0 (P0) mouse pups (see Fig. 1). In each panel, an arrowhead indicates the Dicer CKO domain, while an arrow indicates a normal region of the retina. The Notch ligand Dll3 [Fig. 1(A)], as well as the Notch effector Hes5 [Fig. 1(B)], and Notch receptor Notch1 [Fig. 1(C)] show decreases in expression levels in Dicer CKO areas (green, arrowheads) when compared to wild type mRNA levels (arrows). Immunohistochemistry for the Notch ligand DNER [Fig. 1(D), red] shows reduced staining in Dicer CKO areas (green, arrowheads). However, although two Notch ligands are down-regulated in the Dicer CKO cells, in situ hybridization and immunohistochemistry for the Notch ligand Dll1 does not show an observable difference in Dicer CKO areas (data not shown). These data suggest that in the absence of Dicer, several components of the Notch signaling pathway are downregulated at P0, leading to a generalized decrease in Notch signaling, as shown by decreased levels of the Notch effector Hes5. Furthermore, Dll3, Hes5, and Notch1 were previously shown to be substantially reduced in Ascl1 knockout retinas (Nelson et al., 2009), suggesting that loss of Ascl1 may underlie the observed decrease in Notch signaling.
Figure 1.
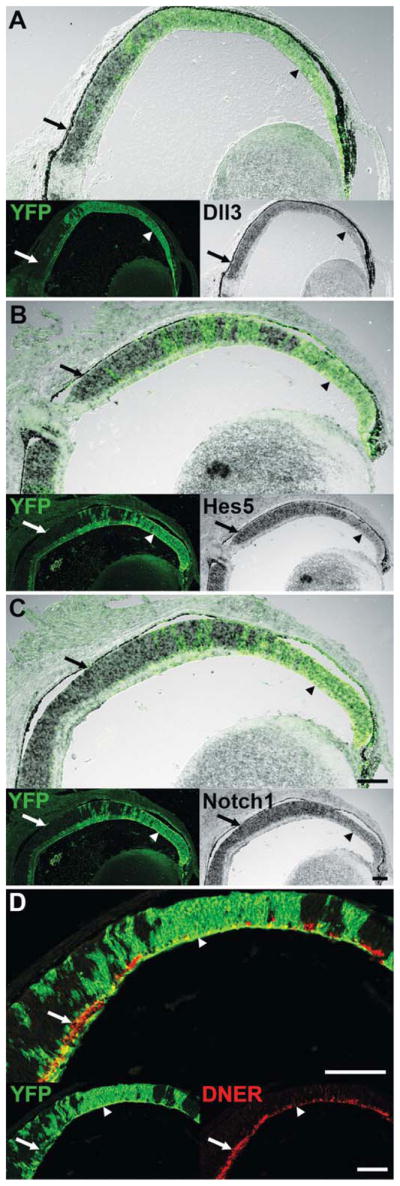
Characterization of Notch signaling components at P0. A–D, YFP staining (green) indicates areas of Dicer CKO. A–C, In situ hybridization for Dll3, Hes5, and Notch1 shows reduced transcript levels in Dicer CKO areas (green, arrowheads) compared to wild type areas (arrows). D, Immunofluorescence staining for DNER (red) shows reduced expression in Dicer CKO areas (green, arrowhead) compared to wild type areas (arrow). Scale bars: 100 μm. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Dicer CKO Retinal Progenitors are Responsive to DAPT-Mediated Notch Inhibition
Because our in situ data indicated a substantial reduction in the Notch receptor Notch1 and other components in this pathway at P0 in Dicer CKO areas, we wanted to determine if Dicer-null retinal progenitors still demonstrate any active Notch signaling at this age. We have previously shown that pharmacological inhibition of Notch signaling using the γ-secretase inhibitor DAPT leads to coordinate downregulation of progenitor genes with concomitant neuronal differentiation (Nelson et al., 2007). To test if Dicer-null retinal progenitors still display active Notch signaling, we cultured P0 αPax6cre; R26EYFP; Dicerfl/fl (CKO) retinal explants in the presence of DMSO or DAPT for 48 h (see Fig. 2).
Figure 2.

Immunofluorescence staining of P0 Dicer CKO retinas after 48 h in culture. A–D, YFP staining (green, arrowheads) indicates areas of Dicer CKO. A,C, Immunofluorescence staining for the progenitor markers Sox2 (red) and PH3 (blue), as well as the apoptotic marker AC3 (red) and the photoreceptor marker Otx2 (blue) shows no change after 48h in DMSO from that described previously in vivo (Georgi and Reh, 2010). B,D, After 48 h in the Notch inhibitor DAPT, Dicer CKO cells (green, arrowheads) show a downregulation of Sox2 (red) and PH3 (blue), with a concomitant upregulation of Otx2 (blue), similar to that observed in wild type cells. Unlike wild type cells, Dicer CKO cells also show an induction of apoptosis, as indicated by increased AC3 staining (red). Scale bars: 100 μm. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Incubation of the explants in DMSO [Fig. 2(A,C)] did not cause any apparent change in the expression of the markers in either the wild type or Dicer CKO regions of retina from what we have previously reported from our in vivo analysis (Georgi and Reh, 2010). Dicer CKO areas (green, arrowheads) show an elevated expression of the progenitor marker Sox2 [Fig. 2(A), red], and a small decline in the mitotic marker phosphohistone 3 (PH3) [Fig. 2(A), blue]. In addition, these cells show a low level of apoptosis [Fig. 2(C), red], and a reduced number of Otx2+ photoreceptor precursors [Fig. 2(C), blue]. After 48 h of DAPT treatment, however, both wild type regions of retina, as well as the Dicer CKO progenitors (green, arrowheads), show a substantial reduction in Sox2 immunoreactivity [Fig. 2(B), red], and a complete absence of PH3 staining [Fig. 2(B), blue]. Furthermore, Dicer CKO progenitors show a marked upregulation of Otx2 staining [Fig. 2(D), blue], indicative of photoreceptor differentiation. DAPT treatment also leads to a substantial induction of the apoptotic marker activated caspase 3 (AC3) in Dicer CKO cells, suggesting a requirement for Dicer in the survival of neurons generated from Dicer CKO progenitors.
These data suggest that Dicer CKO progenitors are actively receiving a Notch receptor-mediated signal, and that this low level of Notch signaling is sufficient to maintain the progenitor population. Similar to wild type progenitors, Dicer CKO progenitors are able to respond to a decrease in Notch signaling by downregulating progenitor genes, exiting the cell cycle, and upregulating neuronal genes. Furthermore, Dicer CKO cells show a large increase in apoptosis upon DAPT-mediated differentiation when compared to similarly-treated wild type cells, suggesting a failure of neuronal differentiation and survival in the absence of miRNAs. Indeed, Dicer CKO retinal areas contain apoptotic cells in vivo and these areas are progressively lost during development (Georgi and Reh, 2010).
Transgenic Overexpression of NICD is Insufficient to Rescue the Dicer CKO Phenotype
Previous studies have indicated that under certain conditions a reduction in Notch signaling can lead to increased ganglion cell differentiation. Because one of the most striking phenotypes observed in Dicer CKO retinas is increased and prolonged ganglion cell production, we sought to rescue the observed reduction in Notch signaling using the ROSA-NICD trans-gene. In these mice, cre-mediated recombination leads to constitutive Notch activity through transcription of the intracellular domain of the mouse Notch1 gene (Murtaugh et al., 2003). Areas that have undergone recombination can also be visualized using an IRES nuclear GFP reporter, which is co-transcribed with the NICD [Fig. 3(A), arrowhead]. In addition, a subset of amacrine cells express αPax6cre postnatally, driving IRES-GFP expression [Fig. 3(A), arrow].
Figure 3.

Immunofluorescence staining of P0 Dicer CKO + NICD retinas. A–F, Nuclear GFP staining (green) indicates areas of Dicer CKO and NICD expression (CKO+NICD). A–D,F are P0 + 1 div. A, Recombination takes place in two domains: embryonically in progenitor cells (arrowhead), and in a subset of amacrine cells starting in late embryonic development, and continuing through adulthood (arrow). The late progenitor marker Ascl1 (red) is absent from CKO+NICD areas, similar to the Dicer CKO retina (Georgi and Reh, 2010). B, The progenitor marker Sox2 (red) is upregulated, and the mitotic marker PH3 (blue) is reduced in CKO+NICD areas, similar to Dicer CKO retina [Fig. 2(A)]. C, Otx2+ (blue) and recoverin+ (red) photoreceptors are absent from CKO+NICD areas, though Dicer CKO retinas contain Otx2+ cells [Fig. 2(C)]. D, Brn3+ ganglion cells (D, red), are present throughout CKO+NICD areas, similar to Dicer CKO retinas. E, Colabeling for Prox1 (red) and neurofilament (NF, blue), indicates the presence of horizontal cells in CKO+NICD areas, unlike Dicer CKO retinas, where they are lost by P0. F, Bright Pax6+ (red) and Bhlhb5+ (white) amacrine cells are absent from NICD+CKO areas, similar to that observed in Dicer CKO retinas. Scale bar: 100 μm. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
To test whether restoring Notch signaling can rescue the over-production in ganglion cells observed after Dicer deletion, we generated αPax6cre; Dicerfl/fl; ROSA-NICD (CKO+NICD) mice for analysis. In these animals the ROSA-NICD transgene is activated at the same time and in the same domain in which Dicer is deleted, allowing for an examination of the effects of constitutive Notch activity on the αPax6cre; Dicerfl/fl (Dicer CKO) background phenotype. The results of this analysis are shown in Figure 3. We previously reported that at P0, Dicer CKO progenitors show an absence of the late progenitor markers Sox9 and Ascl1, but express elevated levels of Sox2 and decreased levels of PH3 (Georgi and Reh, 2010). The Dicer CKO+NICD retina looks similar to Dicer CKO alone, and lacks both Ascl1 [Fig. 3(A), red] and Sox9 (data not shown) immunoreactivity in CKO+NICD areas [Fig. 3(A), green]. In addition, both the progenitor marker Sox2 [Fig. 3(B), red] and the mitotic marker PH3 [Fig. 3(B), blue] are present in Dicer CKO+NICD areas, with elevated Sox2 immunoreactivity similar to that observed in Dicer CKO retinas. These data indicate that NICD trans-gene expression does not rescue the loss of late progenitor markers seen in Dicer CKO retinas.
In addition to changes in the progenitor gene expression, we previously reported that the loss of Dicer also leads to an absence of mature horizontal and amacrine cells, a reduction in photoreceptors, and an increase in ectopically-located ganglion cells at P0 (Georgi and Reh, 2010). We therefore analyzed the Dicer CKO+NICD retinas for the expression of markers for these cell types. We found that expression of the NICD in Dicer CKO progenitors fails to rescue the effects of Dicer deletion on the production of most retinal cell types. We found that the photoreceptor markers Otx2 [Fig. 3(C), blue] and recoverin [Fig. 3(C), red] are even more dramatically affected in the Dicer CKO+NICD areas (green) than in the Dicer CKO, now showing a near complete absence from these areas (Supporting Information Fig. 1; 0.83 ± 0.64 vs. 22 ± 2 Otx2+ cells per 100 μm of retina; Dicer CKO+NICD vs. Dicer CKO; p = 0.0006). The amacrine markers, Pax6 and Bhlhb5 are mostly missing from the Dicer CKO+NICD regions of retina [Fig. 3(F)], similar to their absence from Dicer CKO retinas (Georgi and Reh, 2010; Supporting Information Fig. 1). Brn3+ ganglion cells are still present in Dicer CKO+NICD areas, as they were in the Dicer CKO [Fig. 3(D), red; Supporting Information Fig. 1; 11 ± 1.4 vs. 11 ± 2.1 Brn3+ cells per 100 μm of retina; Dicer CKO+NICD vs. Dicer CKO, p = 0.9], and often show a similar ectopic localization. Thus, although Notch signaling is substantially reduced following deletion of Dicer in retinal progenitors, increasing Notch signaling does not rescue many of the observed changes in retinal development, and in some cases makes them more severe. Furthermore, these data suggest that contrary to prior models, Notch inactivation is not an absolute requirement for neuronal differentiation.
Despite the fact that most aspects of the Dicer CKO phenotype are not improved with Notch ICD over-expression, horizontal cells appear to be rescued. We previously reported that in the Dicer CKO retina, Prox1/Neurofilament+ horizontal cells were significantly reduced at P0, despite an overproduction during embryogenesis. However, in Dicer CKO+ NICD retinas, we found significantly more horizontal cells, as indicated by costaining for Prox1 [Fig. 3(E), red] and Neurofilament [Fig. 3(E), blue; Supporting Information Fig. 1; 1.3 ± 0.16 vs. 0.48 ± 0.06 colabeled cells per 100 μm of retina; Dicer CKO+NICD vs. Dicer CKO, p = 0.0094]. These data suggest a role for Notch signaling in the survival and/or genesis of Dicer CKO horizontal cells.
Constitutively Active Notch Does Not Prevent Neuronal Differentiation
Previous studies have reported that expression of the NICD transgene in retinal progenitors inhibits their terminal differentiation and the production of neurons (Jadhav et al., 2006a). Therefore, it was surprising that in the Dicer CKO+NICD retinal regions we found that both Brn3+ ganglion cells and Prox1/NF+ horizontal cells were generated [Fig. 3(D,E)]. We were uncertain if this was simply an effect of an abnormal developmental program in the absence of miRNAs. To further investigate the effect of constitutively active Notch signaling during retinal development, we looked at littermate αPax6cre; Dicerfl/+; ROSA-NICD (NICD+) mice at P0. Although the analysis detailed in Figures 4 and 5 is from Dicer heterozygous mice, the phenotype observed in αPax6cre; Dicerfl/+; ROSA-NICD mice matches that observed in αPax6cre; ROSA-NICD mice (data not shown), indicating that loss of a single copy of Dicer does not affect the observed phenotype.
Figure 4.

Immunofluorescence staining of P0 NICD+ retinas. A–F, Nuclear GFP staining (green) indicates areas of NICD expression (NICD+). Note that scattered GFP+ cells are present in wild type areas of retina due to αPax6 expression in a subset of amacrine cells. A,C–F are P0 + 1 div. A, NICD+ areas (green) lack a distinct ganglion cell layer (GCL, arrowhead) and inner nuclear layer (INL, arrow). Ascl1+ progenitor cells (red) are present in NICD+ retinal areas, with a domain of expression expanded into the GCL and INL. B, NICD+ progenitors show normal expression of the mitotic marker PH3 (red). C, NICD+ progenitors express elevated levels of Sox2 (red). D, The early photoreceptor marker Otx2 (blue) is expressed in NICD+ areas, but the late photoreceptor marker recoverin (red) is not. E, Brn3+ ganglion cell precursors (red) and Tbr2+ amacrine cell precursors (blue) are present in NICD+ areas (arrow), but Brn3+/Tbr2+ mature ganglion cells are absent from the vitreal surface (arrowhead). F, Bright Pax6+ (red) and Bhlhb5+ (white) mature amacrine cells are absent from NICD+ areas. Scale bar: 100 μm. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Figure 5.

Immunofluorescence staining of P5 NICD+ retinas. A–G, Nuclear GFP staining (green) indicates areas of NICD expression (NICD+). Note that scattered GFP+ cells are present in wild type areas of retina due to αPax6 expression in a subset of amacrine cells. A, NICD+ areas show a distinct outer nuclear layer (ONL), but disorganized inner nuclear layer (INL), and lack a ganglion cell layer (GCL). Faint Otx2+ photoreceptors and bright Otx2+ bipolar cells (red) are present in NICD+ areas of retina, but the mature photoreceptor marker recoverin (blue) is severely reduced. B, Bright Pax6+ amacrine cells (red) are present in NICD+ areas of retina, but show disrupted lamination. C, Bright Bhlhb5+ mature amacrine cells (red) are absent from NICD+ areas of retina, but some cells express low levels of Bhlhb5 (arrowheads). D, NICD+ Müller glia (arrowheads) express elevated levels of Sox2 (red), compared to wild type Müller glia (arrows), and show an expanded domain of expression. Tbr2+ mature ganglion cells and amacrine precursors (blue) are absent from NICD+ areas. E, NICD+ Müller glia (arrowheads) express elevated levels of Id1 (red), compared to wild type Müller glia (arrows). F, NICD+ Müller glia (arrowheads) express elevated levels of Cralbp (red), compared to wild type Müller glia (arrows). G, EdU was injected at P3. Centrally-located NICD+ glia lack EdU (red, arrowhead), indicating they were postmitotic at P3. Scale bar: 100 μm. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
The activation of Notch signaling in retinal progenitors leads to a striking phenotype at P0 (see Fig. 4). Areas in which NICD is expressed (Fig. 4, green) show abnormal retinal lamination, with an absence of a distinct ganglion cell layer [Fig. 4(A), arrowhead] and inner nuclear layer [Fig. 4(A), arrow]. Instead, the progenitor domain is expanded into the ganglion cell layer, as shown by labeling for Ascl1 [Fig. 4(A), red] and Sox9 (data not shown). Progenitor cells in the NICD expressing domain also label for Sox2 [Fig. 4(C), red] and there is no statistically significant difference in the number of PH3 cells [Fig 4(B), red] when compared with wild type retina (Supporting Information Fig. 2; 4.1 ± 0.53 vs. 7.1 ± 0.95 cells per 100 μm of retina; NICD+ vs. Control, p = 0.052).
The absence of a ganglion cell layer in the NICD+ regions of retina is consistent with earlier reports showing an inhibition of neuronal differentiation from Notch activation. However, staining for the photoreceptor markers Otx2 [Fig. 4(D), blue] and Prdm1 (data not shown) indicates that substantial numbers of photoreceptors continue to be produced from Notch ICD expressing progenitors. Quantification of Otx2+ cells at this age shows no significant difference from control (Supporting Information Fig. 2; 33 ± 3.7 vs. 45 ± 3.5 cells per 100 μm of retina; NICD+ vs. Control, p = 0.088). Co-staining with Otx2 and Ascl1 shows minimal overlap, suggesting normal photoreceptor production with concomitant down-regulation of progenitor gene expression in the presence of the NICD transgene (data not shown). Nevertheless, although the initial stages of photoreceptor differentiation appear to occur at near normal levels in the presence of active Notch signaling, the later stages of photoreceptor differentiation seem to be inhibited, as recoverin, a marker of maturing photoreceptors [Fig. 4(D), red], is significantly reduced in the NICD+ regions of retina compared to control (Supporting Information Fig. 2; 0.6 ± 0.42 vs. 5.1 ± 0.31 cells per 100 μm of retina; NICD+ vs. Control, p = 0.001).
Because these results indicated that retinal progenitors can undergo initial stages of differentiation to a photoreceptor fate in the presence of constitutive Notch signaling, we sought to assess differentiation of other cell types. Costaining for the ganglion cell markers Brn3 [Fig. 4(E), red] and Tbr2/Eomes [Fig. 4(E), blue] shows a severe reduction in ganglion cells at the vitreal surface of NICD+ areas of retina [Fig. 4(E), arrowhead; Supporting Information Fig. 2; 3.4 ± 0.49 vs. 16 ± 0.44 Brn3+ cells at the vitreal surface per 100 μm of retina; NICD+ vs. Control, p < 0.0001]. Surprisingly, however, Brn3+ and Tbr2+ cells are present in the neuroblastic layer of NICD+ areas of retina [Fig. 4(E), arrow], suggesting a continued genesis of these neurons, or a failure of migration. Quantification of ectopic Brn3+ cells indicates a significant increase compared to control (2.6 ± 0.71 vs. 0.05 ± 0.05 cells per 100 μm of retina; NICD+ vs. Control, p = 0.023). Tbr2 and Brn3 do not colocalize in this region, suggesting that they mark different populations of newly postmitotic cells. Nearly all Tbr2+ cells co-stain with Pax6 (data not shown), however, supporting the idea that these cells represent amacrine precursors (Mao et al., 2008). To determine if mature amacrine cells are generated in the presence of a constitutively active Notch signal, we stained for Pax6 [Fig. 4(F), red] and Bhlhb5 [Fig. 4(F), blue]. NICD+ areas [Fig. 4(F), green] are mostly devoid of mature amacrine cells, suggesting a failure or delay of amacrine cell differentiation in the presence of the NICD transgene (Supporting Information Fig. 2; 2.34 ± 0.57 vs. 36 ± 3.1 bright Pax6+ cells per 100 μm of retina; NICD+ vs. Control, p = 0.0004). Thus, in contrast to a previous report, retinal progenitors can undergo initial differentiation toward photoreceptors, and several types of inner retinal neurons are generated in the presence of a constitutive Notch signal.
Constitutively-Active Notch Is Sufficient to Block DAPT-Mediated Differentiation
Because our data indicated that several aspects of the Dicer CKO phenotype were not rescued by NICD overexpression, and because numerous retinal neurons were generated in the presence of the NICD, we sought to determine if the NICD transgene was truly activating the Notch pathway in these cells. To test this, we cultured retinas from αPax6cre; Dicerfl/fl; ROSA-NICD (Dicer CKO+NICD) and αPax6cre; Dicerfl/+; ROSA-NICD (NICD+) mice with DAPT to determine if NICD expression can rescue Notch signaling downstream of the Notch receptor. As expected, treatment with DAPT led to a downregulation of the progenitor markers Sox2 and PH3 in adjacent wild type cells (Supporting Information Fig. 3), but this effect was rescued by expression of the NICD transgene in both Dicer CKO+NICD and NICD+ mice. Similarly, DAPT treatment induced strong Otx2 staining in adjacent wild type retinal cells (Supporting Information Fig. 3), but this effect was rescued in both Dicer CKO+NICD and NICD+ mice. Furthermore, we saw no induction of apoptosis in Dicer CKO+NICD cells, suggesting a failure of these cells to differentiate in response to DAPT. These data strongly suggest that the NICD transgene is activating the Notch pathway downstream of the Notch receptor, and that Notch signaling is rescued in Dicer CKO+NICD cells.
Constitutively-Active Notch is Gliogenic at P5
Previous studies have indicated that NICD overexpression is strongly gliogenic, producing large numbers of Müller glia in late retinal development (Jadhav et al., 2006a). We have confirmed this finding, and find that at P0, NICD-expressing progenitors begin to express low levels of the glial markers Cralbp and S-100, but have not yet exited the cell cycle or acquired a mature glial morphology (data not shown). To assess the ability of the NICD to drive glial differentiation later in development, we looked at postnatal day 5 (P5). Areas of retina expressing the NICD transgene (Fig. 5, green, arrowheads) show an expansion of glial cells towards the vitreal surface, as assessed with staining for numerous glial markers, including Sox2 [Fig. 5(D), red], Cralbp [Fig. 5(F), red], and S-100 (data not shown). In addition, each of these markers is expressed at substantially higher levels than neighboring wild type glia [Fig. 5(D–F), arrows], suggesting an extremely strong induction of the glial phenotype by NICD expression. Id1, which also marks Müller glia (Ueki and Reh, manuscript in preparation), shows a similar expression pattern as well [Fig. 5(E), red], with a significant increase in Id1+ cells when compared with wild type retina (Supporting Information Fig. 4; 61 ± 4.4 vs. 29 ± 0.65 cells per 100 μm of retina; NICD+ vs. Control; p = 0.002). Each of these markers shows highest expression in the most central NICD+ areas, with lower levels of glial induction at the far periphery (data not shown).
Although these cells resembled Müller glia by their marker expression, we tested whether they had exited the cell cycle by labeling with the thymidine analog EdU at postnatal day 3 (P3), followed by sacrifice two days later. While central regions of NICD-expressing cells show an absence of EdU staining at P5 [Fig. 5(G), red], more peripheral regions show substantial EdU staining (data not shown). Staining for the mitotic marker PH3 at P5 shows only a few cells undergoing active division in NICD+ areas of retina, always located at the far peripheral edge (data not shown). Quantification of these peripheral PH3+ cells indicates a significant decrease when compared with wild type retinas (Supporting Information Fig. 4; 0.67 ± 0.07 vs. 2.5 ± 0.64 cells per 100 μm of retina; NICD+ vs. Control; p = 0.048). These data suggest that at P3 central NICD+ progenitors had already exited the cell cycle and begun differentiating into glial cells, while more peripheral progenitors had not yet exited the cell cycle.
While the glial population is expanded in NICD+ areas of retina, glial cells are not the only cell type present in these areas. In particular we noted a well-defined Outer Nuclear Layer (ONL) devoid of nuclear glial markers [Fig. 5(A)]. To determine if photoreceptors are present in these areas, we stained for Otx2 and recoverin. Similar to our results at P0, we see robust expression of Otx2 in the ONL [Fig. 5(A), red], but reduced recoverin staining [Fig. 5(A), blue]. Quantification of these stains indicates a moderate decrease in the average thickness of the Otx2+ photoreceptor layer when compared with wild type retinas (Supporting Information Fig. 4; 57 ± 3.7 μm vs. 93 ± 3.6 μm; NICD+ vs. Control; p = 0.002), and a large decrease in the number of recoverin+ photoreceptors (Supporting Information Fig. 4; 1.4 ± 0.23 vs. 14 ± 1.3 cells per 100 μm of retina; NICD+ vs. Control; p = 0.0006). These results support our conclusion that the initial stages of photoreceptor development are normal in the presence of the NICD trans-gene, but that photoreceptor differentiation is inhibited or delayed.
Staining for Otx2 also indicates the presence of bright Otx2+ presumptive bipolar cells in NICD+ regions of retina, though these cells are no longer locates in the Inner Nuclear Layer (INL), but often shifted towards the vitreal surface of the retina [Fig. 5(A), red]. Quantification of these bright Otx2+ cells indicates no significant decrease when compared with wild type retinas (Supporting Information Fig. 4). Staining with Pax6 shows the presence of bright Pax6+ cells, suggestive of amacrine cells, though these cells show an inverted lamination with respect to the bright Otx2+ presumptive bipolar cells, with the bright Pax6+ cells now being located proximal to the ONL [Fig. 5(B), red]. This inverted lamination is most striking in central regions of retina, but partially resolves towards the periphery. Quantification of these bright Pax6+ cells indicates no significant decrease when compared with wild type retinas (Supporting Information Fig. 4). While these cells stain brightly for Pax6 and Prox1 (data not shown), they express only faint levels of at the mature amacrine and bipolar marker Bhlhb5 [Fig. 5(C), red, arrowhead], suggesting delayed or inhibited differentiation. Indeed, quantification of Bhlhb5+ cells indicates a significant decrease when compared to wild type retinas (Supporting Information Fig. 4; 0.98 ± 0.46 vs. 11 ± 0.5 cells per 100 μm of retina; NICD+ vs. Control; p = 0.0001) In addition, staining for the markers Tbr2 [Fig. 5(A), blue] and Brn3 (data not shown) indicates an absence of amacrine and ganglion cell precursors in NICD-expressing areas of retina, and a reduced number of ganglion cells at the vitreal surface, when compared to wild type (Supporting Information Fig. 4; 0.93 ± 0.56 vs. 4.7 ± 0.38 Brn3+ cells per 100 μm of retina; NICD+ vs. Control; p = 0.0053).
Our data support a gliogenic role for Notch signaling during late retinal development, but indicate that the initial differentiation of progenitors is not inhibited by constitutive Notch signaling. Many retinal neurons are generated by NICD-expressing progenitors, but these cells show failed or delayed full differentiation, as well as abnormal lamination at P5. Our data suggest that Notch signaling must be downregulated early in neuronal differentiation to allow for full progression of the differentiation program, and that in the presence of a constitutive Notch signal, neurons fail to express mature markers.
Dicer-Null Progenitors are Refractory to Notch-Driven Gliogenesis
Because NICD expression is strongly gliogenic at P5 in control retinas, but Müller glia were not generated in the Dicer CKO retina (Georgi and Reh, 2010), we sought to assess the phenotype of Dicer CKO+NICD retinal progenitors at this same age. Previously we have shown that Dicer-null progenitors persist to P5, inappropriately continue to generate ganglion cells, and fail to differentiate into late cell types (Georgi and Reh, 2010). As was seen at P0 in Dicer CKO+ NICD retinas, photoreceptor and amacrine cell differentiation is also inhibited at P5 (data not shown), but nascent ganglion cells are still present in NICD-expressing areas [Fig. 6(A′), green], as assessed by staining for Brn3 [Fig. 6(A′), red]. To show that Dicer CKO+NICD progenitors are generating ganglion cells beyond their normal competence window, we injected mouse pups with EdU at P3, and stained at P5. Many Brn3+ ganglion cell precursors colabel with EdU [Fig. 6(A′), blue, arrowheads], indicating that they were generated from progenitors at P3, well beyond the normal window of ganglion cell genesis. Staining for Sox2 and Id1 [Fig. 6(B,C), red], which are present in both progenitors and Müller glia (Ueki and Reh, manuscript in publication), shows no strong induction of expression, as was seen in NICD+ retinas at this same age. Similarly, Dicer CKO+NICD areas of retina show no expression of the glial marker Cralbp [Fig. 6(D′), red]. Lastly, similar to our results at P0, we see a rescue of Prox1/NF+ horizontal cells at P5 in Dicer CKO+NICD areas of retina [Fig. 6(E′), arrowheads].
Figure 6.

Immunofluorescence staining of P5 Dicer CKO + NICD retinas. A–F, nuclear GFP staining (green) indicates areas of Dicer CKO and NICD expression (CKO+ NICD). Note that scattered GFP+ cells are present in wild type areas of retina due to αPax6 expression in a subset of amacrine cells. A–E, left panels focus on wild type expression of indicated markers. A′–E′, right panels focus on CKO+NICD expression of indicated markers. A–A′, EdU was injected at P3. CKO+NICD areas contain cells colabeled with the ganglion cell marker Brn3 (red) and EdU (blue), indicating birth at P3. B–B′, CKO+NICD areas contain cells that express normal levels of Sox2 (red), and show progenitor morphology. C–C′, CKO+NICD areas contain progenitor cells that express Id1 (red) at lower levels that nearby wild type Müller glia. D–D′, CKO+NICD areas lack expression of the Müller glial marker Cralbp (red). E–E′, Costaining with Prox1 (red) and neurofilament (NF, blue) reveals the presence of horizontal cells in CKO+NICD areas, though these cells are absent from Dicer CKO areas at P5 (Georgi and Reh, 2010). F, Occasional groups of CKO+NICD cells show induction of gliogenesis (arrow) as indicated by Cralbp staining (red). These cells are almost always located in close proximity to wild type cells. Scale bars: 100 μm. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Although the majority of Dicer CKO+NICD progenitors are refractory to the Notch gliogenic signal, occasional groups of GFP+ glial cells are observed in Dicer CKO+NICD retinas [Fig. 6(F), arrow]. These groups are almost always located centrally, and are often in direct contact with nearby wild type cells. It is not clear what is responsible for the rescue of Notch-promoted gliogenesis in these cells, as they represent a minority of Dicer CKO+NICD progenitors. However, similar phenotypic variation was observed in Dicer CKO progenitors, and we have previously posited that this may be due to incomplete recombination of Dicer in these cells, or rescue by a cell non-autonomous signal from nearby wild type cells (Georgi and Reh, 2010).
Our data show that although Dicer CKO progenitors exhibit reduced Notch signaling, this reduction is not responsible for the observed absence of gliogenesis, as transgenic rescue of Notch signaling is not sufficient to confer gliogenic competence. Our data indicate that Dicer is required for Notch-driven gliogenesis during retinal development, and support our conclusions that Dicer-null progenitors fail to progress in their intrinsic competence state. Indeed, our data and that of Jadhav et al. (2006a) suggest that under normal conditions early retinal progenitors are refractory to the Notch gliogenic signal. Thus, because Dicer CKO progenitors are arrested as early progenitors, they lack the intrinsic competence to respond to a Notch gliogenic signal.
DISCUSSION
Recently we showed that conditional knockout of the microRNA-processing enzyme Dicer during retinal development prevents progenitor cells from progressing from an early to a late competence state, leading to an absence of late-generated cell types, and an extended period of ganglion cell genesis (Georgi and Reh, 2010). In addition, we showed that Sox9 and Ascl1, two markers of late progenitors, are absent from Dicer-null progenitors. Because Ascl1-null retinas show decreased levels of several Notch-signaling components (Nelson et al., 2009), we sought to assess Notch signaling in Dicer CKO retinas. We show here that Notch signaling is decreased in P0 Dicer CKO retinas at the ligand (Dll3 and DNER), receptor (Notch1), and effector (Hes5) levels. These data parallel similar data showing a reduction in Notch1 and Hes5 levels at E17.5 by Davis et al. (2011).
Although Notch signaling is severely reduced in Dicer CKO progenitors at P0, we show that this progenitor pool is maintained by an active Notch signal that can be pharmacologically inhibited by DAPT, leading to neuronal differentiation. These data suggest that a low residual level of Notch1 receptor is sufficient to mediate this Notch signal, or that the progenitor pool is maintained by signaling through Notch3, which is also present in the developing retina (Lindsell et al., 1996). Surprisingly, we showed a large increase in apoptotic cells upon DAPT-induced differentiation. These data are consistent with our previous observations that Dicer CKO regions show increased apoptosis and are progressively lost during retinal development. Our results suggest that Dicer is likely required at several stages in the process of neuronal differentiation, and that in the absence of miR-NAs, newly generated neurons eventually undergo apoptosis.
Previous studies have implicated a decrease in Notch signaling with increased ganglion cell genesis (Austin et al., 1995; Ahmad et al., 1997; Dorsky et al., 1997; Henrique et al., 1997; Waid and McLoon, 1998; Silva et al., 2003; Kubo et al., 2005; Nelson et al., 2006, 2007), leading us to hypothesize that decreased Notch signaling may underlie the increased and extended ganglion cell genesis observed in Dicer CKO retinas. To test this hypothesis directly we transgenically overexpressed the Notch Intracellular Domain (NICD) specifically in retinal progenitor cells that had lost Dicer. Transgenic expression of the NICD leads to a constitutively-active Notch signal, and has been previously reported to prevent cellular differentiation (Jadhav et al., 2006a). Strikingly, NICD overexpression had no effect on ganglion cell genesis from Dicer-null progenitor cells, suggesting that decreased Notch signaling does not underlie this phenotype [Fig. 7(D)]. Davis et al. (2011) came to a similar conclusion after observing normal levels of Notch1 and Hes5 transcript at E15.5, a time at which ganglion cell genesis is already increased.
Figure 7.

Summary of observed phenotypes. A, During normal retinal development, early progenitor cells give rise to ganglion, cone, horizontal, and amacrine cells, and transition to late progenitor cells which give rise to amacrine, rod, and bipolar cells. Remaining progenitor cells develop into Müller glial cells. B, In Dicer CKO retinas, late progenitor cells are lost, and no rod, bipolar, or Müller glial cells are generated. Instead, early progenitors persist, and generate an excess of ganglion cells, fewer cone photoreceptors, as well as horizontal and amacrine cells. However, none of these cell types fully mature or express mature markers, as indicated by shorter bars in the neuronal maturation axis. In addition, horizontal cells are completely lost by P0 (Georgi and Reh, 2010; Davis et al., 2011). C, In NICD overexpressing retinas, all neuronal types are formed, but they do not fully differentiate or express mature markers. In addition, an excess of Müller glia are generated. D, In Dicer CKO + NICD retinas, late progenitors and their postmitotic derivitives are still lost. In addition, ganglion cells are still generated in excess. Horizontal cells are now rescued postnatally, but no photoreceptors are generated. These cell types do not seem to fully differentiate or express mature markers. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
If Notch signaling is not responsible for the increased and extended production of retinal ganglion cells in Dicer CKO retinas, what underlies this phenotype? It is possible that retention of progenitor cells in an early competence state is sufficient to explain these results. Ascl1 and Sox9 expression mark a “late” stage in retinal progenitors (Jasoni and Reh, 1996; Poche et al., 2008), when they are no longer competent to generate ganglion cells (Brzezinski and Reh, unpublished observations), and thus the absence of Ascl1 expression caused by the Dicer deletion correlates with the apparent inability of the cells to progress to the late progenitor competence state. Although restoring Notch to the progenitor cells was not sufficient to drive their progression in competence, expression of Ascl1 might be. Alternatively, it has been shown that Sonic Hedgehog (Shh) signaling regulates retinal ganglion cell production (Neumann and Nuesslein-Volhard, 2000; Zhang and Yang, 2001; Wang et al., 2005; Sakagami et al., 2009), and Davis et al. (2011) recently showed that Shh signaling is decreased in Dicer CKO retinas as well. Further experiments will be needed to determine if this decrease in Shh signaling underlies the observed overproduction of ganglion cells.
While NICD overexpression in Dicer CKO retinas had no effect on ganglion cell genesis or markers of late progenitor character [Fig. 7(D)], it did modify the Dicer CKO phenotype in two ways. First, while horizontal cells are initially generated in excess in Dicer CKO retinas, followed by complete loss by P0, NICD overexpression rescued horizontal cell survival at P0 and P5 [Fig. 7(D)]. These data suggest an interaction between miRNAs and Notch signaling in the survival of horizontal cells. Second, while Dicer CKO progenitors normally produce some photoreceptor precursors as well as ganglion cells, NICD expression abolished photoreceptor precursor generation [Fig. 7(D)]. This result is particularly surprising because photoreceptors were specified normally when NICD was expressed in control retinas [Fig. 7(C), see below]. It is not clear why a constitutively active Notch signal is able to prevent photoreceptor genesis only in the absence of functional miRNAs, but it is possible that miRNAs normally dampen Notch signaling or modify its output by regulating downstream effectors. Thus, in the absence of miRNA-mediated inhibition of Notch signaling, transgenic NICD overexpression may lead to a greater-than-normal inhibition of photoreceptor production. While our results seem to contradict this model, indicating that several components of Notch signaling are decreased after Dicer CKO, this decrease likely does not reflect a direct influence of miRNAs on Notch signaling. Rather, this decrease seems to be a result of Ascl1 loss, and this may therefore occlude any direct effects of miRNAs on Notch signaling. Indeed, numerous Notch signaling components are targets of miRNAs in many other systems and organisms (Kwon et al., 2005; Lai et al., 2005; Ivey et al., 2008; Garzia et al., 2009; Kefas et al., 2009; Li et al., 2009; Song et al., 2009; Pang et al., 2010; Wang et al., 2010; Vallejo et al., 2011).
A surprising result of our experiments is our observation that neuronal production is not inhibited by NICD overexpression in retinal progenitors. Instead, we see the generation of numerous retinal cell types, as assessed using markers of immature ganglion cells, amacrine cells, photoreceptors, and bipolar cells [Fig. 7(C)]. Our results instead suggest that completion of neuronal differentiation is inhibited or delayed in the presence of a constitutive Notch signal, and that neuronal precursors are generated normally. Although Jadhav et al. (2006a) concluded that neurons were not generated when the NICD transgene was expressed in the retina using Chx10-cre, their data do not contradict our observations. To assess for the presence of neurons at postnatal day 10 (P10), they stained for β III tubulin (Tuj1), which marks differentiating and mature retinal neurons (Sharma and Netland, 2007). In our experiments we do not see evidence of terminally differentiated neurons, which is consistent with their observed lack of Tuj1 staining. In addition, Jadhav et al. (2006a) described several neuronal genes that were downregulated by microarray or in situ hybridization at P10, including blue cone opsin, neurofilament, calbindin, NRL, and m-cone opsin. Most of these genes are only expressed in mature or differentiating neurons, and their absence from P10 retinas further supports our conclusions that neuronal maturation is arrested by NICD overexpression. Furthermore, because Jadhav et al. (2006a) assessed the NICD overexpression phenotype only at a late age (P10), it is possible that any neurons generated during retinogenesis would have already been converted into Müller glia by this time, similar to their observations that NICD transgene expression in Shh-cre expressing retinal neurons leads to their conversion into Müller glia by P11.
Although our data suggest that neuronal specification can happen in the presence of a constitutive Notch signal, experimental evidence nevertheless supports a role for Notch signaling in maintaining a pool of undifferentiated progenitors, and numerous studies have indicated that Notch inhibition is sufficient to induce premature cell-cycle exit and differentiation (Jadhav et al., 2006b; Yaron et al., 2006; Nelson et al., 2007). Based on our data, it is unclear if Notch inhibition is necessary for cell-cycle exit, however. It is nevertheless possible that neurons generated under these conditions were able to escape from active Notch signaling through other means, such as downstream inhibition of Notch effectors. Further investigation will be needed to determine the levels of Notch signaling activity present in NICD-overexpressing cells.
Our data support the conclusion that Notch activity drives gliogenesis late in retinal development, as we observed strong induction of the glial phenotype at P5 in control animals [Fig. 7(C)]. However, this induction was strongest in central retina, and EdU labeling indicated that glial cell cycle exit takes place in a central-to-peripheral gradient. These data suggest that progenitor cells must reach a particular developmental stage before they are competent to respond to Notch-promoted gliogenesis and cell cycle exit. In support of this conclusion, we observed that Dicer CKO retinal progenitors, which do not express markers of late progenitors, are unable to differentiate into Müller glia in the presence of the NICD trans-gene. From these observations we conclude that the decreased Notch signaling in Dicer CKO retinas is not responsible for the observed lack of glial differentiation, but that Dicer CKO progenitors lack intrinsic competence to undergo gliogenesis.
Though our data indicate that Dicer is required for retinal progenitors to differentiate into Müller glia, it is likely that this phenotype is indirect, due to a failure of Dicer-null retinal progenitor cells to progress beyond an early competence state. One late marker that is absent from Dicer CKO progenitors is the transcription factor Sox9, which plays an important role in gliogenesis (Poche et al., 2008; Muto et al., 2009; Yokoi et al., 2009). While it is possible that Dicer CKO progenitors are unable to respond to the Notch gliogenic signal because they lack Sox9, our data suggest that Sox9 is not in itself sufficient to confer gliogenic competence, as wild type progenitors are not yet competent to differentiate as Müller glia at P0, even though they express Sox9. Interestingly, while it has been reported that Notch signaling drives Sox9 expression in retinal progenitors (Muto et al., 2009), NICD overexpression was not sufficient to rescue Sox9 expression in Dicer-deficient progenitors (data not shown), suggesting a requirement for additional upstream factors.
It is possible that early progenitors are not competent to respond to a Notch gliogenic signal because their chromatin lacks a permissive epigenetic landscape. In support of this hypothesis, previous studies have shown a requirement for chromatin remodeling in the development of astrocytes both in vivo and in vitro (Takizawa et al., 2001; Shimozaki et al., 2005; Setoguchi et al., 2006; Hatada et al., 2008). Further studies will be needed to determine the epigenetic landscapes of Dicer CKO and early retinal progenitors, and how these changes relate to gliogenic potential.
It is therefore still unclear if Dicer is directly required for Müller glial differentiation, as any role for Dicer in this process may be occluded by a secondary lack of gliogenic competence. Experiments using conditional knockout of Dicer in late retinal progenitors will be needed to elucidate the role that miRNAs play in retinal gliogenesis. Nevertheless, our data establish an important role for Dicer in the maintenance of Notch signaling during retinal development, and in promoting the progression of progenitor cell competence.
Supplementary Material
Acknowledgments
Contract grant sponsor: National Science Foundation Fellowship; contract grant numbers: RO1 EY013475, PO1 GM081619-01.
Contract grant sponsor: Graduate Neuroscience Training Grant; contract grant number: 5 T32 GM07108.
The authors thank Dr. Ruth Ashery-Padan and Dr. Rachel Wong for their generous gift of mice used in this study, Dr. Byron Hartman for in situ probes used, and Dr. Jack Saari for Cralbp antibody.
Footnotes
Additional Supporting Information may be found in the online version of this article.
References
- Ahmad I, Dooley CM, Polk DL. Delta-1 is a regulator of neurogenesis in the vertebrate retina. Dev Biol. 1997;185:92–103. doi: 10.1006/dbio.1997.8546. [DOI] [PubMed] [Google Scholar]
- Austin CP, Feldman DE, Ida JA, Jr, Cepko CL. Vertebrate retinal ganglion cells are selected from competent progenitors by the action of Notch. Development. 1995;121:3637–3650. doi: 10.1242/dev.121.11.3637. [DOI] [PubMed] [Google Scholar]
- Bernardos RL, Lentz SI, Wolfe MS, Raymond PA. Notch-Delta signaling is required for spatial patterning and Muller glia differentiation in the zebrafish retina. Dev Biol. 2005;278:381–395. doi: 10.1016/j.ydbio.2004.11.018. [DOI] [PubMed] [Google Scholar]
- Davis N, Mor E, Ashery-Padan R. Roles for Dicer1 in the patterning and differentiation of the optic cup neuroepithelium. Development. 2011;138:127–138. doi: 10.1242/dev.053637. [DOI] [PubMed] [Google Scholar]
- Dorsky RI, Chang WS, Rapaport DH, Harris WA. Regulation of neuronal diversity in the Xenopus retina by Delta signalling. Nature. 1997;385:67–70. doi: 10.1038/385067a0. [DOI] [PubMed] [Google Scholar]
- Fabian MR, Sonenberg N, Filipowicz W. Regulation of mRNA translation and stability by microRNAs. Annu Rev Biochem. 2010;79:351–379. doi: 10.1146/annurev-biochem-060308-103103. [DOI] [PubMed] [Google Scholar]
- Furukawa T, Mukherjee S, Bao ZZ, Morrow EM, Cepko CL. rax, Hes1, and notch1 promote the formation of Muller glia by postnatal retinal progenitor cells. Neuron. 2000;26:383–394. doi: 10.1016/s0896-6273(00)81171-x. [DOI] [PubMed] [Google Scholar]
- Garzia L, Andolfo I, Cusanelli E, Marino N, Petrosino G, De Martino D, Esposito V, et al. MicroRNA-199b-5p impairs cancer stem cells through negative regulation of HES1 in medulloblastoma. PLo S One. 2009;4:e4998. doi: 10.1371/journal.pone.0004998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgi SA, Reh TA. Dicer is required for the transition from early to late progenitor state in the developing mouse retina. J Neurosci. 2010;30:4048–4061. doi: 10.1523/JNEUROSCI.4982-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatada I, Namihira M, Morita S, Kimura M, Horii T, Nakashima K. Astrocyte-specific genes are generally demethylated in neural precursor cells prior to astrocytic differentiation. PLoS One. 2008;3:e3189. doi: 10.1371/journal.pone.0003189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henrique D, Hirsinger E, Adam J, Le Roux I, Pourquie O, Ish-Horowicz D, Lewis J. Maintenance of neuroepithelial progenitor cells by Delta-Notch signalling in the embryonic chick retina. Curr Biol. 1997;7:661–670. doi: 10.1016/s0960-9822(06)00293-4. [DOI] [PubMed] [Google Scholar]
- Hojo M, Ohtsuka T, Hashimoto N, Gradwohl G, Guillemot F, Kageyama R. Glial cell fate specification modulated by the bHLH gene Hes5 in mouse retina. Development. 2000;127:2515–2522. doi: 10.1242/dev.127.12.2515. [DOI] [PubMed] [Google Scholar]
- Ivey KN, Muth A, Arnold J, King FW, Yeh RF, Fish JE, Hsiao EC, et al. MicroRNA regulation of cell lineages in mouse and human embryonic stem cells. Cell Stem Cell. 2008;2:219–229. doi: 10.1016/j.stem.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jadhav AP, Cho SH, Cepko CL. Notch activity permits retinal cells to progress through multiple progenitor states and acquire a stem cell property. Proc Natl Acad Sci USA. 2006a;103:18998–19003. doi: 10.1073/pnas.0608155103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jadhav AP, Mason HA, Cepko CL. Notch 1 inhibits photoreceptor production in the developing mammalian retina. Development. 2006b;133:913–923. doi: 10.1242/dev.02245. [DOI] [PubMed] [Google Scholar]
- Jasoni CL, Reh TA. Temporal and spatial pattern of MASH-1 expression in the developing rat retina demonstrates progenitor cell heterogeneity. J Comp Neurol. 1996;369:319–327. doi: 10.1002/(SICI)1096-9861(19960527)369:2<319::AID-CNE11>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Kefas B, Comeau L, Floyd DH, Seleverstov O, Godlewski J, Schmittgen T, Jiang J, et al. The neuronal micro-RNA miR-326 acts in a feedback loop with notch and has therapeutic potential against brain tumors. J Neurosci. 2009;29:15161–15168. doi: 10.1523/JNEUROSCI.4966-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo F, Takeichi M, Nakagawa S. Wnt2b inhibits differentiation of retinal progenitor cells in the absence of Notch activity by downregulating the expression of proneural genes. Development. 2005;132:2759–2770. doi: 10.1242/dev.01856. [DOI] [PubMed] [Google Scholar]
- Kwon C, Han Z, Olson EN, Srivastava D. Micro-RNA1 influences cardiac differentiation in Drosophila and regulates Notch signaling. Proc Natl Acad Sci USA. 2005;102:18986–18991. doi: 10.1073/pnas.0509535102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai EC, Tam B, Rubin GM. Pervasive regulation of Drosophila Notch target genes by GY-box-, Brd-box-, and K-box-class micro RNAs. Genes Dev. 2005;19:1067–1080. doi: 10.1101/gad.1291905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre JL, Zhang Y, Meister M, Wang X, Sanes JR. gamma-Protocadherins regulate neuronal survival but are dispensable for circuit formation in retina. Development. 2008;135:4141–4151. doi: 10.1242/dev.027912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Guessous F, Zhang Y, Dipierro C, Kefas B, Johnson E, Marcinkiewicz L, et al. MicroRNA-34a inhibits glioblastoma growth by targeting multiple oncogenes. Cancer Res. 2009;69:7569–7576. doi: 10.1158/0008-5472.CAN-09-0529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsell CE, Boulter J, diSibio G, Gossler A, Weinmaster G. Expression patterns of Jagged. Delta1, Notch1, Notch2, and Notch3 genes identify ligand-receptor pairs that may function in neural development. Mol Cell Neurosci. 1996;8:14–27. doi: 10.1006/mcne.1996.0040. [DOI] [PubMed] [Google Scholar]
- Mao CA, Kiyama T, Pan P, Furuta Y, Hadjantonakis AK, Klein WH. Eomesodermin, a target gene of Pou4f2, is required for retinal ganglion cell and optic nerve development in the mouse. Development. 2008;135:271–280. doi: 10.1242/dev.009688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquardt T, Ashery-Padan R, Andrejewski N, Scardigli R, Guillemot F, Gruss P. Pax6 is required for the multipotent state of retinal progenitor cells. Cell. 2001;105:43–55. doi: 10.1016/s0092-8674(01)00295-1. [DOI] [PubMed] [Google Scholar]
- Murtaugh LC, Stanger BZ, Kwan KM, Melton DA. Notch signaling controls multiple steps of pancreatic differentiation. Proc Natl Acad Sci USA. 2003;100:14920–14925. doi: 10.1073/pnas.2436557100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muto A, Iida A, Satoh S, Watanabe S. The group E Sox genes Sox8 and Sox9 are regulated by Notch signaling and are required for Muller glial cell development in mouse retina. Exp Eye Res. 2009;89:549–558. doi: 10.1016/j.exer.2009.05.006. [DOI] [PubMed] [Google Scholar]
- Nelson BR, Gumuscu B, Hartman BH, Reh TA. Notch activity is downregulated just prior to retinal ganglion cell differentiation. Dev Neurosci. 2006;28:128–141. doi: 10.1159/000090759. [DOI] [PubMed] [Google Scholar]
- Nelson BR, Hartman BH, Georgi SA, Lan MS, Reh TA. Transient inactivation of Notch signaling synchronizes differentiation of neural progenitor cells. Dev Biol. 2007;304:479–498. doi: 10.1016/j.ydbio.2007.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson BR, Hartman BH, Ray CA, Hayashi T, Bermingham-McDonogh O, Reh TA. Acheatescute like 1 (Ascl1) is required for normal delta-like (Dll) gene expression and notch signaling during retinal development. Dev Dyn. 2009;238:2163–2178. doi: 10.1002/dvdy.21848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann CJ, Nuesslein-Volhard C. Patterning of the zebrafish retina by a wave of sonic hedgehog activity. Science. 2000;289:2137–2139. doi: 10.1126/science.289.5487.2137. [DOI] [PubMed] [Google Scholar]
- Ohsawa R, Kageyama R. Regulation of retinal cell fate specification by multiple transcription factors. Brain Res. 2008;1192:90–98. doi: 10.1016/j.brainres.2007.04.014. [DOI] [PubMed] [Google Scholar]
- Pang RT, Leung CO, Ye TM, Liu W, Chiu PC, Lam KK, Lee KF, Yeung WS. MicroRNA-34a suppresses invasion through downregulation of Notch1 and Jagged1 in cervical carcinoma and choriocarcinoma cells. Carcinogenesis. 2010;31:1037–1044. doi: 10.1093/carcin/bgq066. [DOI] [PubMed] [Google Scholar]
- Poche RA, Furuta Y, Chaboissier MC, Schedl A, Behringer RR. Sox9 is expressed in mouse multipotent retinal progenitor cells and functions in Muller glial cell development. J Comp Neurol. 2008;510:237–250. doi: 10.1002/cne.21746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakagami K, Gan L, Yang XJ. Distinct effects of Hedgehog signaling on neuronal fate specification and cell cycle progression in the embryonic mouse retina. J Neurosci. 2009;29:6932–6944. doi: 10.1523/JNEUROSCI.0289-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setoguchi H, Namihira M, Kohyama J, Asano H, Sanosaka T, Nakashima K. Methyl-CpG binding proteins are involved in restricting differentiation plasticity in neurons. J Neurosci Res. 2006;84:969–979. doi: 10.1002/jnr.21001. [DOI] [PubMed] [Google Scholar]
- Sharma RK, Netland PA. Early born lineage of retinal neurons express class III beta-tubulin isotype. Brain Res. 2007;1176:11–17. doi: 10.1016/j.brainres.2007.07.090. [DOI] [PubMed] [Google Scholar]
- Shimozaki K, Namihira M, Nakashima K, Taga T. Stage- and site-specific DNA demethylation during neural cell development from embryonic stem cells. J Neurochem. 2005;93:432–439. doi: 10.1111/j.1471-4159.2005.03031.x. [DOI] [PubMed] [Google Scholar]
- Silva AO, Ercole CE, McLoon SC. Regulation of ganglion cell production by Notch signaling during retinal development. J Neurobiol. 2003;54:511–524. doi: 10.1002/neu.10156. [DOI] [PubMed] [Google Scholar]
- Song G, Zhang Y, Wang L. MicroRNA-206 targets notch3, activates apoptosis, and inhibits tumor cell migration and focus formation. J Biol Chem. 2009;284:31921–31927. doi: 10.1074/jbc.M109.046862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takizawa T, Nakashima K, Namihira M, Ochiai W, Uemura A, Yanagisawa M, Fujita N, et al. DNA methylation is a critical cell-intrinsic determinant of astrocyte differentiation in the fetal brain. Dev Cell. 2001;1:749–758. doi: 10.1016/s1534-5807(01)00101-0. [DOI] [PubMed] [Google Scholar]
- Vallejo DM, Caparros E, Dominguez M. Targeting Notch signalling by the conserved miR-8/200 microRNA family in development and cancer cells. EMBO J. 2011;30:756–769. doi: 10.1038/emboj.2010.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waid DK, McLoon SC. Ganglion cells influence the fate of dividing retinal cells in culture. Development. 1998;125:1059–1066. doi: 10.1242/dev.125.6.1059. [DOI] [PubMed] [Google Scholar]
- Wang Y, Dakubo GD, Thurig S, Mazerolle CJ, Wallace VA. Retinal ganglion cell-derived sonic hedgehog locally controls proliferation and the timing of RGC development in the embryonic mouse retina. Development. 2005;132:5103–5113. [Google Scholar]
- Wang Z, Li Y, Kong D, Ahmad A, Banerjee S, Sarkar FH. Cross-talk between miRNA and Notch signaling pathways in tumor development and progression. Cancer Lett. 2010;292:141–148. doi: 10.1016/j.canlet.2009.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaron O, Farhy C, Marquardt T, Applebury M, Ashery-Padan R. Notch1 functions to suppress cone-photoreceptor fate specification in the developing mouse retina. Development. 2006;133:1367–1378. doi: 10.1242/dev.02311. [DOI] [PubMed] [Google Scholar]
- Yokoi H, Yan YL, Miller MR, BreMiller RA, Catchen JM, Johnson EA, Postlethwait JH. Expression profiling of zebrafish sox9 mutants reveals that Sox9 is required for retinal differentiation. Dev Biol. 2009;329:1–15. doi: 10.1016/j.ydbio.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XM, Yang XJ. Regulation of retinal ganglion cell production by Sonic hedgehog. Development. 2001;128:943–957. doi: 10.1242/dev.128.6.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.