

Abstract
We report here on an approach targeting the host reactive cysteinome to identify inhibitors of host factors required for the infectious cycle of Flaviviruses and other viruses. We used two parallel cellular phenotypic screens to identify a series of covalent inhibitors, exemplified by QL-XII-47, that are active against dengue virus. We show that the compounds effectively block viral protein expression and that this inhibition is associated with repression of downstream processes of the infectious cycle, and thus significantly contributes to the potent antiviral activity of these compounds. We demonstrate that QL-XII-47’s antiviral activity requires selective, covalent modification of a host target by showing that the compound's antiviral activity is recapitulated when cells are preincubated with QL-XII-47 and then washed prior to viral infection and by showing that QL-XII-47R, a non-reactive analog, lacks antiviral activity at concentrations more than 20-fold higher than QL-XII-47's IC90. QL-XII-47’s inhibition of Zika virus, West Nile virus, hepatitis C virus, and poliovirus further suggests that it acts via a target mediating inhibition of these other medically relevant viruses. These results demonstrate the utility of screens targeting the host reactive cysteinome for rapid identification of compounds with potent antiviral activity.
1. Introduction
The rapid evolution of antiviral resistance coupled with the continued emergence of new viral pathogens together create a need for new antiviral approaches with the potential for broad-spectrum activity. Recent successful antiviral drug development efforts against human immunodeficiency virus (HIV) and hepatitis C virus (HCV) have primarily focused on inhibition of viral enzymes that are responsible for catalyzing viral genome replication, polyprotein processing during viral assembly, and virion release. Due to the selectivity of these drugs for their viral targets, their potential for broad-spectrum activity extends to other members of their viral family but not beyond. Host-targeted antivirals may provide an important alternative because they have the potential to be effective against multiple viral pathogens and because their barriers to resistance may be higher than those of drugs that act via viral targets (Bekerman and Einav, 2015). These characteristics may be especially useful in antiviral approaches directed at new and rapidly emerging pathogens.
Covalent inhibitors targeting proteins with reactive cysteines (the reactive “cysteinome”) are of increasing interest as drug targets due to growing appreciation for the biological function of such cysteines as catalytic residues in many enzyme classes and as sites of post-translational modifications in many proteins (Backus et al., 2016; Weerapana et al., 2010). Targeting functionally important cysteines has recently enabled the development of selective covalent inhibitors of a number of different human kinases (Backus et al., 2016; Cohen et al., 2007; Honigberg et al., 2010; Kwiatkowski et al., 2014; Lanning et al., 2014; Tan et al., 2015; Wu et al., 2014), as well as proteases (van der Linden et al., 2016), phosphatases, and E2 ubiquitin transferases (Singh et al., 2011).
To explore the host reactive cysteinome as a source of novel antiviral targets, we chose dengue virus (DV) as an example. An enveloped virus with a positive-strand RNA genome, DV is transmitted to humans through the bite of an infected Aedes mosquito. Recent estimates suggest approximately 50 to 100 million symptomatic DV infections per year and roughly 3.9 billion people at risk in 128 countries (Bhatt et al., 2013; Stanaway et al., 2016). While several DV vaccines are being developed (Rothman and Ennis, 2016), there is still a crucial need for effective antiviral therapies to treat or prevent disease and to limit transmission. Host-targeted inhibitors of DV and related biomedically important flaviviruses may be of particular interest in the absence of more traditional antivirals targeting the viral protease or polymerase. Since RNAi, Crispr-Cas9, and chemical biology screens have identified many host factors and functions required for DV infection (Ang et al., 2010; Chu and Yang, 2007; Heaton et al., 2010; Marceau et al., 2016; Savidis et al., 2016; Sessions et al., 2009; Zhang et al., 2016), we reasoned that members of the host reactive cysteinome should be well-represented within the set of host factors that can act as pharmacological intervention points to interfere with DV infection.
Here we report screening of a small library of acrylamide-containing heterocycles to identify compounds that inhibit DV via selective activity against members of the host reactive cysteinome. A family of related compounds, exemplified by the quinoline QL-XII-47, inhibit DV at concentrations that are non- cytotoxic with antiviral activity that is strongly correlated with inhibition of translation of the viral genome. Structure-activity relationship (SAR) data as well as time-of-addition studies suggest strongly that QL-XII-47 acts via covalent modification of a host target. QL-XII-47’s inhibition of West Nile and Zika viruses (WNV and ZIKV), as well as other medically important viruses such as poliovirus (PV) and hepatitis C virus (HCV), support its covalent targeting of a host factor and demonstrate its multispectrum antiviral activity.
2. Materials and methods
2.1 Cell lines and cytotoxicity assays
All cell lines were cultured and cytotoxicity of compounds was assessed as previously described (de Wispelaere et al., 2013). CC50 values were determined by non-linear regression analysis.
2.2 Compound synthesis and characterization
Cycloheximide and mycophenolic acid were purchased from Sigma-Aldrich and Calbiochem, respectively. The DV2419-447 peptide (Schmidt et al., 2010) was provided by Aaron Schmidt and Stephen Harrison. Synthesis of QL- XII-47 (also known as QL47), QL-XII-47R (also known as QL47R) and QL-X-138 were previously described (Wu et al., 2016; Wu et al., 2014). Detailed descriptions of synthetic methods and compound characterization are provided in Supplementary Material.
2.3 Antibodies
Monoclonal antibodies against DV E and C proteins were produced from culture supernatants of hybridomas D1-4G2-4-15 (ATCC HB-112) and 6F3.1 (Bulich and Aaskov, 1992), respectively. Other antibodies were obtained from vendors as described in Supplementary Material.
2.4 Viruses
All work with infectious virus was performed in a biosafety level 2 (BSL2) laboratory using additional safety practices as approved by the Harvard Committee on Microbiological Safety. Virus strains utilized were DV2 New Guinea C, DV1 West Pac 74, DV3 CH53489, DV4 TVP-376, WNV Kunjin, ZIKV PF-251013-18, HCV JFH1, and PV Mahoney.
Infections and titrations of infectious virus were performed as previously described for DV, WNV and ZIKV (de Wispelaere et al., 2013), HCV (Lindenbach, 2009; Rodgers et al., 2012), and PV (Rueckert and Pallansch, 1981). IC90 values were determined by non-linear regression analysis.
2.5 Reporter viral particles (RVP) production and screen
Reporter viral particles (RVP) were produced as described (de Wispelaere et al., 2013) using pCDNA6.2-D2.CprME, a plasmid that encodes DV2 structural proteins (Ansarah-Sobrinho et al., 2008) and pWIIrep-REN-IB, a plasmid that encodes a WNV reporter replicon (Pierson et al., 2006).
For screening, Huh7 cells were seeded in white 96-well plates. Medium supplemented with each compound was added to each well followed by addition of DV2 RVP. Plates were incubated at 37°C until 24 hours post-infection, when luciferase activity was measured using a Renilla-Glo luciferase assay system (Promega) and a Perkin Elmer EnVision plate reader.
2.6 Replicon screen
Stable T-REx-293-DGZ cells (Ansarah-Sobrinho et al., 2008) were seeded in 384-well clear-bottom, black plates and were pretreated with 0.5 µM of MPA for 48 hours to reduce the preexisting intracellular pool of GFP. Fresh medium supplemented with each compound was added, and the plates incubated for 48 hours at 37°C. Fluorescence signal in each well was quantified with the ImageXpress Velos Laser Scanning Cytometer using no-image flattening and regions of interest to count objects.
2.7 Reporter replicon assays
Plasmids encoding reporter replicons were as follows: pDENrep-FH, wildtype DV2 reporter replicon (Holden et al., 2006); DV2(GVD) reporter replicon with inactive NS5 polymerase (de Wispelaere et al., 2013); pSGR-JFH1/Luc-GND encoding the HCV(GND) reporter replicon (Kato et al., 2005); and pBS-EMCV-Fluci, which expresses firefly luciferase under the control of the EMCV IRES (Murakami et al., 2008).
Experiments with the wild-type DV2 replicon were performed as previously described (de Wispelaere et al., 2013). For the DV2(GVD) replicon, the HCV(GND) replicon, and the EMCV-IRES reporter, cells seeded in a 24-well plate were treated with small molecules, and immediately transfected with in vitro transcripts using the Lipofectamine MessengerMAX transfection reagent (ThermoFisher Scientific). Following collection of cells at indicated times, samples were processed according to the instructions in the luciferase assay system (Promega) and read using a Perkin-Elmer EnVision plate reader. Data are reported as a function of the DMSO-treated samples.
2.8 In situ hybridization (ISH) and immunofluorescence analysis (IFA)
ISH detection of DV RNA was performed without protease treatment of samples using the QuantiGene ViewRNA ISH Cell Assay (Affymetrix) and a DV specific probe (Affymetrix, VF1-10744) following conditions recommended by the manufacturer. IFA of DV2 C and NS5 proteins was performed as described (de Wispelaere et al., 2013). Image data were collected via wide-field imaging using the 40x objective on a Nikon Eclipse TE-2000-U microscope with a Hamamatsu Orca-ER camera and MetaMorph software.
2.9 Quantitative RT-PCR (RT-qPCR) and Western blotting
Procedures were performed essentially as in previous publications (Carocci and Yang, 2016; de Wispelaere et al., 2013).
2.10 Statistical analyses
An unpaired t-test was used to compare quantitative data. GraphPad Prism was used for all statistical analyses. Statistically significant differences between experimental samples and DMSO-treated samples are shown by asterisks in the figures (***, P < 0.001; **, 0.001 < P < 0.01; *, 0.01 < P < 0.05).
3. Results
3.1 Parallel phenotypic screens interrogating two distinct modes of antiviral activity
Due to the clinical success of covalent kinase inhibitors targeting EGFR and BTK as anti-cancer drugs (Cheng et al., 2016; Davids and Brown, 2014), much effort has recently been invested to discover and develop covalent inhibitors of the one-third of the human kinome that has a reactive cysteine residue in or near the ATP-binding site (Liu et al., 2013b) as well as non-kinase targets such as G12C KRAS (Patricelli et al., 2016) and the hepatitis C virus NS3/4A protease (Hagel et al., 2011). To explore the broader applicability of this inhibitor strategy, we built a collection of 378 heterocyclic, acrylamide-derivatized compounds for use in phenotypic and target-based screens. The pyrimidine, purine, quinoline, pyrrolopyrimidine, and pyrazolopyrimidine scaffolds in this library are mostly based on core structures that have been rich sources of ATP-competitive kinase inhibitors. To interrogate this small library for inhibitors of DV, we devised complementary screens to monitor two distinct antiviral modes of action: inhibition of productive viral entry to prevent infection and inhibition of steady-state replication of the viral RNA genome to inhibit an already-established infection.
DV entry requires attachment of the virion to the plasma membrane, followed by clathrin-mediated uptake of the virion. Fusion of the viral and endosomal membranes upon endosomal acidification allows escape of the nucleocapsid to the cytosol and subsequent translation of the incoming DV RNA genome. To screen for compounds that block infection, we utilized a single-cycle reporter virus (DV2 RVP) that expresses luciferase upon successful viral entry and translation of the incoming viral RNA (Ansarah-Sobrinho et al., 2008). The screen was performed by incubating Huh7 cells with the library followed by infection with DV2 RVP (Figure 1A) and analysis of luciferase reporter activity at 24 hours post-infection.
Figure 1. Description of the screens that led to the identification of covalent inhibitors of DV2.

A. In the reporter viral particles (RVP) screen, Huh7 cells were treated with a subset of 250 compounds (final concentration 1.9 µM), then infected with DV2 RVP. Renilla luciferase signal encoded by the WNV replicon was measured in cells at 24 hours post-infection.
B. In the replicon screen, T-REx-293-DGZ cells were pre-treated for 48 hours with 0.5 µM mycophenolic acid (MPA), then treated with a subset of 288 compounds (final concentration 3.3 µM) for 48 hours after which GFP signal encoded by the DV2 replicon was measured.
C. Data for both screens (light grey circles for RVP; dark grey triangles for replicon) are plotted as a percentage of the DMSO-treated cells with background values set as the signal from mock-infected cells for the RVP screen and signal from cells treated with 10 µM MPA for the replicon screen. Molecules causing an inhibition of signal > 50% were identified as “hits” (the 50% selection cut-off is represented as a red dotted line). The compounds are distributed into 5 groups along the x axis based on their core structure (shown below the axis): 1 – purine; 2 – pyrimidine; 3 – pyrazolopyrimidine; 4 – pyrrolopyrimidine; 5 – quinoline. The remaining 56 compounds include various structures and are grouped under “others”. The quinoline compounds further studied in the present work are identified by red symbols. QL-XII-47 is identified by a crossed-out symbol.
In our second screen, we sought compounds targeting post-entry steps of the DV infectious cycle. For this, we used the previously described T-REx-293-DGZ cell line (Ansarah-Sobrinho et al., 2008) under conditions in which the cells stably replicate a DV2 subgenomic RNA bearing the virus’s seven nonstructural (NS) genes as well as a GFP reporter gene (Figure 1B). Translation of the replicon RNA gives rise to GFP expression, with GFP fluorescence providing an indirect measure of steady-state abundance of the viral RNA (Clyde et al., 2008; Leardkamolkarn and Sirigulpanit, 2012). GFP fluorescence was quantified at 48 hours post-compound treatment. For both screens, compounds causing a 50% inhibition in luciferase signal or GFP fluorescence were chosen for further study (Figure 1C).
Primary screening hits that exhibited cytotoxicity or that lacked reproducible antiviral activity in secondary assays were removed from our analysis (data not shown). The remaining 21 compounds included molecules from multiple structural classes with different biological activities, suggesting that they target distinct host cell processes. We focused our efforts on a set of six related tricyclic quinoline inhibitors [QL-XI-13, QL-X-134, QL-X-138 (Wu et al., 2016), QL-XII-47 (Wu et al., 2014), QL-XII-54, QL-XII-115] that were originally designed to inhibit BTK, BMX, and other Tec family kinases (Liu et al., 2013a) and that had high activity in both the DV2 RVP and replicon screens (Figure 1C, red inset).
We quantified anti-DV2 IC90 values, defined as the concentration at which single-cycle viral yield was reduced by ten-fold, and CC50 values by non-linear regression analyses (Table 1 and Figure S1) to confirm that limited or no toxicity occurs at the concentrations at which potent antiviral activity is observed. Since the six compounds exhibited different antiviral potencies when added to the cells post-infection (Table 1 and Figure S1), we performed subsequent experiments at two non-cytotoxic concentrations (2 and 10 µM) to ensure detection of significant concentration-dependent inhibition in the individual assays and to allow correlation of antiviral activity against live DV2 (Table 1 and Figure S1 and S2) with the individual activities (Figure 3).
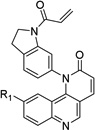
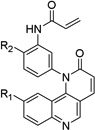
Table 1. Anti-DV2 activity of QL-X-134, QL-X-138, QL-XI- 13, QL-XII-47, QL-XII-54 and QL-XII-115.
Huh7 cells were infected with DV2 at a multiplicity of infection (MOI) of 1 then treated with a range of small molecules concentrations. Cell viability and viral yield were measured 24 hours later. CC50 and IC90 values – respectively, corresponding to the concentrations of small molecule that lead to 50% cytotoxicity and to 90% reduction in the number of infectious progeny virions produced – were determined by non-linear regression (see Figure S1). The CC50 values obtained after 48 hours treatments were of the same order as those obtained after 24 hours treatments (data not shown).
| Scaffold | Compound | R1 | R2 | IC90 (µM) |
CC50 (µM) |
|---|---|---|---|---|---|
|
QL-XII-47 |
|
0.4 | 78 | |
|
QL-XI-13 |
|
H | 2.9 | 126 |
| QL-X-138 |
|
Me | 3.5 | 77 | |
| QL-X-134 |
|
Me | 2.2 | 75 | |
| QL-XII-54 |
|
Me | 0.5 | >160 | |
| QL-XII-115 |
|
Me | 0.5 | >160 |
Figure 3. QL-XII-47 and related quinolines block translation of the DV2 RNA.

A. Huh7 cells were electroporated with a DV2 reporter replicon and treated with inhibitors at 48 hours post-electroporation. Luciferase activity was quantified at 72 h post-electroporation and is plotted as a percentage of the DMSO-treated samples. Mean values and standard deviation from n=3 independent experiments are shown.
B. Huh7 cells were transfected with a DV2(GVD) reporter replicon, and immediately treated with compounds. Luciferase activity was quantified at 6 hours post-transfection and is plotted as a percentage of the DMSO-treated samples. Mean values and standard deviation from n=3 independent experiments are shown.
C. The assay described in panel B was used to determine IC50 of each small molecule against the DV2(GVD) replicon. Cells were treated with a range of small molecules concentrations, and the concentrations that lead to 50% inhibition in firefly luciferase signal (IC50) were calculated using the nonlinear fit variable slope model (GraphPad Software). IC90 values obtained against the infectious virus in Table 1 and Figure S1 were plotted versus the IC50 values for inhibition of viral translation in the DV2(GVD) replicon assay, and the resulting curve was fit by linear regression to illustrate correlation of antiviral potency and inhibition of translation.
D. Huh7 cells were mock infected or infected with DV2 (MOI of 10) for 1 hour and then were treated with DMSO or 2 µM QL-XII-47. Cover slips were collected at 24 hours post-infection and stained for the DV2 C (in green) and NS5 (in red) proteins. Nuclei were stained with DAPI (in blue). The images were taken at 400x magnification, and representative images from n>3 independent experiments are shown.
E. Huh7 cells were mock-infected or infected with DV2 (MOI of 1) for 1 hour, and then treated with inhibitors at a final concentration of 2 µM. At 24 hours post- infection, the cell lysates were collected and analyzed for the presence of DV2 E and NS5 proteins, as well as for the presence of GAPDH by Western blotting. A representative experiment out of n=2 repeats is shown.
3.2 QL-XII-47 and related quinolines do not affect viral entry
While initial time-of-addition experiments performed with live DV2 (Figure 2A) suggested that the major effect of these compounds is on events that occur after entry of the virion into the host cell, the antiviral activity observed under these different conditions may also reflect the kinetics of covalent modification of the compounds’ antiviral target(s). Consistent with this interpretation, QL-XII-47 and QL-XII-54, the two compounds that exhibited significant antiviral activity in the co-treatment experiment, also have the highest antiviral potency (Table 1). To assess more directly whether the compounds affect DV entry, we monitored localization of the incoming viral genomic RNA over time via in situ hybridization (ISH) with a probe specific for the DV2 genome and fluorescence microscopy (Figure 2B). Internalization of the viral genome was observed in all samples at 1 and 4 hours post-infection, suggesting that QL-XII-47 does not affect attachment or clathrin-mediated uptake of DV2. For vehicle-treated control cells, the abundance of fluorescent foci representing genomic RNA was diminished between 10 and 16 hour time points followed by a burst of signal at 19 hours post-infection, presumably reflecting sequestration of the viral RNA in membrane-associated replication complexes and then synthesis of new RNA copies accessible to the ISH probe. Cells treated with QL-XII-47 exhibit levels of incoming RNA comparable to the vehicle control at 4 hours post-infection; however, the number of fluorescent foci appears to be relatively unchanged at all time points between 4 and 19 hours post-infection. This suggested that DV2 is blocked at a point prior to replication of the RNA genome.
Figure 2. QL-XII-47 and related quinolines do not affect viral entry.

A. In time-of-addition studies, Huh7 cells were infected with DV2 at MOI of 1. The cells were treated with 2 µM of small molecules concomitant with the infection (co), post-infection (post), or at both times (co+post). The infectious virus released to the supernatants at 24 hours post-infection was quantified by FFA. Representative data (mean ±standard deviation of experimental duplicates) out of n=2 independent experiments are shown.
B. To monitor the DV2 genomic RNA, Huh7 cells were mock-infected, or infected with DV2 at a MOI of 10 for 1 hour and then treated with DMSO or 2 µM of QL-XII-47. Cover slips were collected at the indicated times post-infection, and DV2 RNA was detected by in situ hybridization assays (in red). Nuclei were stained with 4’,6-diamidino-2-phenylindole (DAPI, in blue). Representative images taken at 400x magnification from one of n>3 independent experiments are shown.
C. To quantify the abundance of intracellular DV2 RNA, Huh7 cells were mock- infected, infected with DV2 at a MOI of 1 for 1 hour and then were treated with DMSO or 2 µM of QL-XII-47. Control cells were infected with DV2 that had been pretreated with DV2419-447 (5 µM, 37°C, 15 min). Total RNA was collected at the indicated times post-infection, and DV2 RNA was quantified by RT-qPCR. The results were normalized to GAPDH mRNA and are expressed as fold increase over mock-infected controls. Representative data (mean ±standard deviation of experimental duplicates) out of n=2 independent experiments are shown.
While initial time-of-addition experiments performed with live DV2 (Figure 2A) suggested that the major effect of these compounds is on events that occur after entry of the virion into the host cell, the antiviral activity observed under these different conditions may also reflect the kinetics of covalent modification of the compounds’ antiviral target(s). Consistent with this interpretation, QL-XII-47 and QL-XII-54, the two compounds that exhibited significant antiviral activity in the co-treatment experiment, also have the highest antiviral potency (Table 1). To assess more directly whether the compounds affect DV entry, we monitored localization of the incoming viral genomic RNA over time via in situ hybridization (ISH) with a probe specific for the DV2 genome and fluorescence microscopy (Figure 2B). Internalization of the viral genome was observed in all samples at 1 and 4 hours post-infection, suggesting that QL-XII-47 does not affect attachment or clathrin-mediated uptake of DV2. For vehicle-treated control cells, the abundance of fluorescent foci representing genomic RNA was diminished between 10 and 16 hour time points followed by a burst of signal at 19 hours post-infection, presumably reflecting sequestration of the viral RNA in membrane-associated replication complexes and then synthesis of new RNA copies accessible to the ISH probe. Cells treated with QL-XII-47 exhibit levels of incoming RNA comparable to the vehicle control at 4 hours post-infection; however, the number of fluorescent foci appears to be relatively unchanged at all time points between 4 and 19 hours post-infection. This suggested that DV2 is blocked at a point prior to replication of the RNA genome.
To assess whether QL-XII-47 blocks endosomal acidification or E- catalyzed membrane fusion, we quantified intracellular DV2 RNA by RT-qPCR assay at various points post-infection. Whereas vehicle-treated cells show a greater than ten-fold increase in viral RNA by 24 hours post-infection, cells treated with DV2419-447, a synthetic peptide previously shown to inhibit E- catalyzed membrane fusion (Schmidt et al., 2010), exhibit decreased DV2 RNA abundance from 9 hours post-infection onward, presumably due to degradation of virus that fails to escape the endosome. In contrast, QL-XII-47-treated cells show no change in intracellular DV2 RNA between 5 and 24 hours post-infection (Figure 2C), suggesting that membrane fusion and endosomal escape occur in the presence of QL-XII-47 and related compounds and that these inhibitors affect a process downstream of viral entry.
3.3 QL-XII-47 and related quinolines block translation of the DV2 RNA
To examine the effects of the QL compounds on viral translation and RNA replication, we utilized a subgenomic DV2 replicon RNA in which a firefly luciferase reporter gene replaces the viral structural genes (Holden et al., 2006). Transient delivery of in vitro transcribed replicon RNA by electroporation or transfection by-passes viral entry and provides a model in which luciferase activity initially reflects translation of the input RNA (≤ 6 hours) but at later time points (≥ 24 hours post-electroporation) reflects both translation and steady-state replication of the viral RNA. When inhibitor treatment was started at 48 hours post-electroporation and luciferase activity was quantified at 72 hours, we observed robust inhibition of the DV2 replicon by QL-XII-47 and related compounds as well as by cycloheximide (CHX), a general inhibitor of translation, and mycophenolic acid (MPA), a validated inhibitor of Flavivirus RNA replication (Diamond et al., 2002) (Figure 3A). To decouple effects on viral translation and RNA replication, we utilized a DV2(GVD) replicon, which cannot synthesize viral RNA due to mutation of the GDD catalytic triad of the viral NS5 RNA-dependent RNA polymerase (de Wispelaere et al., 2013). As expected, CHX caused a severe reduction in luciferase activity measured at 6 hours post-transfection due to inhibition of translation whereas MPA had no effect (Figure 3B). The effect of QL-XII-47 and related compounds more closely resembled that of CHX in these experiments, with IC50 values against the DV2(GVD) replicon closely paralleling the compounds’ antiviral potency against live DV2 (Figure 3C). Importantly, we showed that this inhibition in reporter protein expression was not due to an inhibition of the accumulation of viral RNA in cells (Figure S3).
We confirmed that QL-XII-47 severely reduces the abundance of viral structural and non-structural proteins in DV2-infected cells by performing immunofluorescence staining for the viral core and NS5 proteins (Figure 3D). Further strengthening the association between inhibition of viral translation and antiviral activity, Western blot analysis showed that QL-XII-47 causes a strong reduction in the abundance of viral proteins (E, NS5) without significantly affecting the abundance of the host housekeeping protein GAPDH (Figure 3E). The link between antiviral activity and inhibition of viral translation is also supported by the observation that the other QL compounds also inhibit viral translation (Figure 3B) and reduce steady-state viral protein expression (Figure 3E), although the correlation of these effects appeared to vary between compounds (e.g., QL-X-138 and QL-XII-115) and may reflect additional modes of antiviral activity for these compounds.
3.4 QL-XII-47 acts via a selective covalent mechanism
In time-of-addition experiments, we observed that a six-hour pretreatment of cells with QL-XII-47 was sufficient to recapitulate the inhibition of live DV2 infection and DV2 translation in the DV2(GVD) replicon assay observed when QL-XII-47 is added post-infection/transfection (Figures 4A and 4B). To test explicitly whether QL-XII-47 is an obligate covalent inhibitor, we synthesized an analog, QL-XII-47R, in which the acrylamide is replaced by a propyl amide incapable of forming a covalent bond with cysteine residues (Figure 4C). QL-XII- 47R fails to inhibit DV2 at concentrations below 10 µM, approximately twenty-fold higher than QL-XII-47’s IC90 value (Figure 4D), demonstrating that the acrylamide moiety is required for potent antiviral activity. Related quinolines that also bear the acrylamide but have varying substituents on the quinoline notably also lack the antiviral potency of QL-XII-47 (Figure 4E). Collectively, these data show that QL-XII-47’s antiviral activity is due to specific covalent inhibition of one or several host targets present in the uninfected cell.
Figure 4. The acrylamide moiety is required for the antiviral activity of QL-XII-47.

A. Huh7 cells were pre-treated with 2 µM QL-XII-47 for 6 hours and then washed prior to DV2 infection (MOI of 1) (pre-treat), or were treated with 2 µM QL-XII-47 for 24 hours post-infection (post-treat). Infectious virus released to the supernatants at 24 hours post-infection was quantified by FFA. Representative data (mean ±standard deviation of experimental duplicates) out of n=2 independent experiments are shown.
B. Huh7 cells were pre-treated with 2 μM QL-XII-47 for 6 hours and then washed prior to transfection (pre-treat) or were treated with 2 µM QL-XII-47 for 12 hours starting at the time of transfection (post-treat) with a DV2(GVD) reporter replicon. Luciferase activity was quantified at 12 hours post-transfection and is plotted as a percentage of the DMSO-treated samples. Representative data (mean ± standard deviation of experimental duplicates) out of n=2 independent experiments are shown.
C. Structures of parent compound QL-XII-47 and QL-XII-47R, a derivative in which the acrylamide moiety is replaced with a non-reactive propyl amide group.
D. The concentration-dependent effects of QL-XII-47 and QL-XII-47R were assessed by infection of Huh7 cells with DV2 (MOI of 1) followed by addition of inhibitors. Viral yield was measured 24 hours later and is plotted as a percentage of the DMSO control. Representative data (mean ± standard deviation of experimental duplicates) from n≥2 independent experiments are shown.
E. Huh7 cells were infected with DV2 (MOI of 1), then treated with the indicated compounds. Infectious virus released to the supernatants at 24 hours post- infection was quantified by FFA. Representative data (mean ± standard deviation of experimental duplicates) out of n>2 independent experiments are shown. The chemical structure of each molecule is shown on the side of the graph.
3.5 QL-XII-47 has broad-spectrum antiviral activity
Since a major hypothesized benefit of host-targeted antivirals is their potential for multispectrum activity, we examined QL-XII-47’s activity against other viruses. QL-XII-47 inhibits DV strains representative of the other three DV serotypes, West Nile virus (WNV), and Zika virus (ZIKV) with potencies comparable to that observed for DV2 (Figure 5A). We additionally observed that QL-XII-47 significantly inhibits other positive-sense RNA viruses including hepatitis C virus (HCV; Family Flaviviridae, genus Hepacivirus) and poliovirus (PV; Family Picornaviridae, genus Enterovirus) (Figure 5B) and that this antiviral activity is correlated with inhibition of viral translation as assessed using an HCV replicon and a luciferase reporter RNA under control of the Encephalomyocarditis virus IRES (EMCV; Family Picornaviridae, genus Cardiovirus) (Figure 5C). Similarly to DV2, we showed that QL-XII-47 antiviral activity is strictly dependent of the presence of the acrylamide moiety (Figure 5B). These results demonstrate the broad-spectrum activity of QL-XII-47 and provide additional support for the idea that the antiviral activity is mediated by host(s) target(s) rather than a viral target.
Figure 5. QL-XII-47 exhibits broad antiviral activity.

A. Huh7 cells were infected with DV1, DV2, DV3, DV4, WNV, or ZIKV at MOI of 1, then treated with 2 µM of QL-XII-47. Infectious virus released to the supernatants at 24 hours post-infection was quantified by FFA (DV), or plaque- formation assay (PFA) (WNV and ZIKV). PFU, plaque-forming unit. Representative data (mean ± standard deviation of experimental duplicates) out of n=2 independent experiments are shown.
B. For HCV, Huh7 cells were infected at MOI of 1, and treated with 2 µM of QL- XII-47. Infectious virus released to the supernatants at 24 hours post-infection was quantified by TCID50 assay. For PV, HeLa cells were infected at a MOI of 0.75, and treated with 2 µM of QL-XII-47 or QL-XII-47R. The infectious virus released to the supernatants at 8 hours post-infection was quantified by PFA. Representative data (mean ± standard deviation of experimental duplicates) out of n≥2 independent experiments are shown.
C. Huh7 cells were transfected with a HCV(GND) reporter replicon or with a EMCV IRES reporter RNA, and immediately treated with 2 µM of QL-XII-47. The luciferase activity was quantified at 6 hours post-transfection and is plotted as a percentage of the DMSO-treated samples. Representative data (mean ± standard deviation of experimental duplicates) out of n≥2 independent experiments are shown.
4. Discussion
4.1 Viral translation as a target for antiviral intervention
The screens were designed to identify compounds that act via two complementary modes of action: inhibition of productive viral entry and inhibition of steady-state replication of the viral genome. The QL compounds notably scored as potent antivirals in both screens, reflecting the importance of viral translation, a step in the infectious cycle that relies almost exclusively on the machinery of the host cell. While viral translation is necessary for all viruses, plus-sense RNA viruses like DV are also exquisitely dependent on efficient translation of the incoming viral genome in order to establish viral infection (Edgil et al., 2003). Accordingly, limiting viral protein expression by the QL compounds is associated with inhibition of downstream viral processes including genome replication (Figure 3A) and DV2 assembly and/or egress (data not shown). Targeting viral translation may therefore be an especially effective antiviral strategy against flaviviruses, like DV, and against other plus-sense RNA viruses. Consistent with this idea, we recently demonstrated that lactimidomycin, a natural product that inhibits translation elongation, also exhibits potent activity against DV and a number of other RNA viruses (Carocci and Yang, 2016); moreover, Wang and co-workers described a benzomorphan that inhibits DV in cellulo and in vivo by targeting translation (Wang et al., 2011). The antiviral activity of these compounds in mammalian cells of diverse origins as well as in mosquito cells (Figure S4) further supports the idea that these compounds target a broadly conserved process.
Since the antiviral activity of the QL compounds occurs in the absence of discernible cytotoxicity, a key question is how this antiviral selectivity is achieved. Although all viruses depend on the host translational machinery, the diverse mechanisms that they employ for translation initiation suggests that there are opportunities to inhibit viral translation selectively. QL-XII-47 and the other related covalent inhibitors discovered in this study appear to have the desired selectivity and thus provide useful tool compounds for investigating this question. While QL-XII-47 and related compounds have known activities against Tec, BTK, and related kinases (Wu et al., 2016; Wu et al., 2014), preliminary experiments utilizing other inhibitors of these kinases as well as RNAi indicate that these kinases are not important for antiviral activity (data not shown). Thus, we are currently employing chemoproteomic approaches that take advantage of the covalent mechanism of this compound to elucidate the full spectrum of proteins that are modified by QL-XII-47 and to ascertain which targets are the most relevant to the compound’s inhibition of viral translation and antiviral activity. We note that although the QL compounds characterized in this study all inhibit DV2 translation, additional activities may contribute to their antiviral activities, as evidenced by variation in the magnitude of their effects on steady-state protein abundance (Figure 3B, 3D, and 3E) and their apparent effects on the downstream processes of RNA replication (Figure 3A) and virion assembly/egress (data not shown). Further efforts elucidating the molecular mechanisms and targets of QL-XII-47 will be reported in due course.
4.2 Covalent inhibitor library screens targeting the reactive cysteinome as a source for host-targeted antivirals
Although concerns about off-target effects, potentially short half-lives and potential toxicities have conventionally limited enthusiasm for the deliberate development of covalent inhibitors as drugs, there are many examples of approved drugs that work via covalent mechanisms (Niphakis and Cravatt, 2014). The potential advantages of covalent inhibitors include a decreased need for extensive optimization of inhibitor potency and pharmacokinetics due to full and irreversible inactivation of the target protein even with transient exposure. This can accelerate pharmacological validation of new antiviral targets both in vitro and in vivo. In addition, the covalent mechanism of these inhibitors may provide a valuable tool for target discovery when the antiviral target is not yet known.
Targeting of reactive cysteines has emerged as a successful strategy for development of covalent inhibitors for use as selective chemical probes and as drugs against multiple enzyme classes. Through comparison of different targeting ligands and different cysteine reactive groups, it is now appreciated that there are both important non-covalent and reactivity-based factors that can confer inhibitor specificity. QL-XII-47 and a set of closely related quinolines exhibit potent antiviral activity against DV2 associated with inhibition of viral translation that was not observed with other members of the heterocyclic acrylamide library we screened (Figure 1), demonstrating that the presence of an acrylamide moiety is insufficient for antiviral activity and that the potent antiviral activity of QL-XII-47 and related compounds is due to selective covalent inhibition.
The discovery of QL-XII-47 and the related QL compounds as primary screening “hits” with potent antiviral activity (sub-micromolar IC90 values) against DV and other positive-sense RNA viruses illustrates the utility of our approach and highlights the benefits of an appropriately selective covalent inhibitor strategy in achieving potent inhibition. The work we report here provides proof of concept for targeting the host reactive cysteinome as a relatively rapid route to the discovery of novel host-targeted antivirals as tool compounds and as leads for antivirals development efforts.
Supplementary Material
Highlights.
Targeting the host cysteinome leads to the discovery of broad-spectrum antivirals
The covalent inhibitor QL-XII-47 potently inhibits dengue virus
QL-XII-47 and related compounds block viral protein expression
QL-XII-47 exhibits broad-spectrum antiviral activity
Acknowledgments
We gratefully acknowledge Ted Pierson for the plasmids used for DV2 RVP production and the DV2 replicon cell line; Eva Harris for the DV2 replicon plasmid; Takaji Wakita for the HCV and EMCV plasmids; Charles Rice for HCV plasmids; John Aaskov for the 6F3.1 hybridoma; Aaron Schmidt and Stephen Harrison for the DV2419-447 peptide; and Lee Gehrke, Aravinda de Silva, Michael Diamond, Didier Musso and Nathalie Pardigon for viruses. We thank the Harvard ICCB-Longwood Screening Facility for instrument usage and support. E.S was supported by a National Science Foundation Graduate Research Fellowship. This research was partially funded by F. Hoffmann-La Roche Ltd and NIH grant U54AI057159 NERP018 (NERCE).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ang F, Wong AP, Ng MM, Chu JJ. Small interference RNA profiling reveals the essential role of human membrane trafficking genes in mediating the infectious entry of dengue virus. Virology journal. 2010;7:24. doi: 10.1186/1743-422X-7-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansarah-Sobrinho C, Nelson S, Jost CA, Whitehead SS, Pierson TC. Temperature-dependent production of pseudoinfectious dengue reporter virus particles by complementation. Virology. 2008;381:67–74. doi: 10.1016/j.virol.2008.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backus KM, Correia BE, Lum KM, Forli S, Horning BD, Gonzalez-Paez GE, Chatterjee S, Lanning BR, Teijaro JR, Olson AJ, Wolan DW, Cravatt BF. Proteome-wide covalent ligand discovery in native biological systems. Nature. 2016;534:570–574. doi: 10.1038/nature18002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekerman E, Einav S. Infectious disease. Combating emerging viral threats. Science. 2015;348:282–283. doi: 10.1126/science.aaa3778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, Drake JM, Brownstein JS, Hoen AG, Sankoh O, Myers MF, George DB, Jaenisch T, Wint GR, Simmons CP, Scott TW, Farrar JJ, Hay SI. The global distribution and burden of dengue. Nature. 2013;496:504–507. doi: 10.1038/nature12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulich R, Aaskov JG. Nuclear localization of dengue 2 virus core protein detected with monoclonal antibodies. J Gen Virol. 1992;73(Pt 11):2999–3003. doi: 10.1099/0022-1317-73-11-2999. [DOI] [PubMed] [Google Scholar]
- Carocci M, Yang PL. Lactimidomycin is a broad-spectrum inhibitor of dengue and other RNA viruses. Antiviral Res. 2016;128:57–62. doi: 10.1016/j.antiviral.2016.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H, Nair SK, Murray BW. Recent progress on third generation covalent EGFR inhibitors. Bioorg Med Chem Lett. 2016;26:1861–1868. doi: 10.1016/j.bmcl.2016.02.067. [DOI] [PubMed] [Google Scholar]
- Chu JJ, Yang PL. c-Src protein kinase inhibitors block assembly and maturation of dengue virus. Proc Natl Acad Sci U S A. 2007;104:3520–3525. doi: 10.1073/pnas.0611681104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clyde K, Barrera J, Harris E. The capsid-coding region hairpin element (cHP) is a critical determinant of dengue virus and West Nile virus RNA synthesis. Virology. 2008;379:314–323. doi: 10.1016/j.virol.2008.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen MS, Hadjivassiliou H, Taunton J. A clickable inhibitor reveals context-dependent autoactivation of p90 RSK. Nat Chem Biol. 2007;3:156–160. doi: 10.1038/nchembio859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davids MS, Brown JR. Ibrutinib: a first in class covalent inhibitor of Bruton's tyrosine kinase. Future Oncol. 2014;10:957–967. doi: 10.2217/fon.14.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wispelaere M, LaCroix AJ, Yang PL. The small molecules AZD0530 and dasatinib inhibit dengue virus RNA replication via Fyn kinase. J Virol. 2013;87:7367–7381. doi: 10.1128/JVI.00632-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond MS, Zachariah M, Harris E. Mycophenolic Acid Inhibits Dengue Virus Infection by Preventing Replication of Viral RNA. Virology. 2002;304:211–221. doi: 10.1006/viro.2002.1685. [DOI] [PubMed] [Google Scholar]
- Edgil D, Diamond MS, Holden KL, Paranjape SM, Harris E. Translation efficiency determines differences in cellular infection among dengue virus type 2 strains. Virology. 2003;317:275–290. doi: 10.1016/j.virol.2003.08.012. [DOI] [PubMed] [Google Scholar]
- Hagel M, Niu D, St Martin T, Sheets MP, Qiao L, Bernard H, Karp RM, Zhu Z, Labenski MT, Chaturvedi P, Nacht M, Westlin WF, Petter RC, Singh J. Selective irreversible inhibition of a protease by targeting a noncatalytic cysteine. Nat Chem Biol. 2011;7:22–24. doi: 10.1038/nchembio.492. [DOI] [PubMed] [Google Scholar]
- Heaton NS, Perera R, Berger KL, Khadka S, Lacount DJ, Kuhn RJ, Randall G. Dengue virus nonstructural protein 3 redistributes fatty acid synthase to sites of viral replication and increases cellular fatty acid synthesis. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:17345–17350. doi: 10.1073/pnas.1010811107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holden KL, Stein DA, Pierson TC, Ahmed AA, Clyde K, Iversen PL, Harris E. Inhibition of dengue virus translation and RNA synthesis by a morpholino oligomer targeted to the top of the terminal 3' stem-loop structure. Virology. 2006;344:439–452. doi: 10.1016/j.virol.2005.08.034. [DOI] [PubMed] [Google Scholar]
- Honigberg LA, Smith AM, Sirisawad M, Verner E, Loury D, Chang B, Li S, Pan Z, Thamm DH, Miller RA, Buggy JJ. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:13075–13080. doi: 10.1073/pnas.1004594107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato T, Date T, Miyamoto M, Sugiyama M, Tanaka Y, Orito E, Ohno T, Sugihara K, Hasegawa I, Fujiwara K, Ito K, Ozasa A, Mizokami M, Wakita T. Detection of anti-hepatitis C virus effects of interferon and ribavirin by a sensitive replicon system. J Clin Microbiol. 2005;43:5679–5684. doi: 10.1128/JCM.43.11.5679-5684.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski N, Zhang T, Rahl PB, Abraham BJ, Reddy J, Ficarro SB, Dastur A, Amzallag A, Ramaswamy S, Tesar B, Jenkins CE, Hannett NM, McMillin D, Sanda T, Sim T, Kim ND, Look T, Mitsiades CS, Weng AP, Brown JR, Benes CH, Marto JA, Young RA, Gray NS. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature. 2014;511:616–620. doi: 10.1038/nature13393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanning BR, Whitby LR, Dix MM, Douhan J, Gilbert AM, Hett EC, Johnson TO, Joslyn C, Kath JC, Niessen S, Roberts LR, Schnute ME, Wang C, Hulce JJ, Wei B, Whiteley LO, Hayward MM, Cravatt BF. A road map to evaluate the proteome-wide selectivity of covalent kinase inhibitors. Nat Chem Biol. 2014;10:760–767. doi: 10.1038/nchembio.1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leardkamolkarn V, Sirigulpanit W. Establishment of a stable cell line coexpressing dengue virus-2 and green fluorescent protein for screening of antiviral compounds. J Biomol Screen. 2012;17:283–292. doi: 10.1177/1087057111426903. [DOI] [PubMed] [Google Scholar]
- Lindenbach BD. Measuring HCV infectivity produced in cell culture and in vivo. Methods Mol Biol. 2009;510:329–336. doi: 10.1007/978-1-59745-394-3_24. [DOI] [PubMed] [Google Scholar]
- Liu F, Zhang X, Weisberg E, Chen S, Hur W, Wu H, Zhao Z, Wang W, Mao M, Cai C, Simon NI, Sanda T, Wang J, Look AT, Griffin JD, Balk SP, Liu Q, Gray NS. Discovery of a selective irreversible BMX inhibitor for prostate cancer. ACS Chem Biol. 2013a;8:1423–1428. doi: 10.1021/cb4000629. [DOI] [PubMed] [Google Scholar]
- Liu Q, Sabnis Y, Zhao Z, Zhang T, Buhrlage SJ, Jones LH, Gray NS. Developing irreversible inhibitors of the protein kinase cysteinome. Chem Biol. 2013b;20:146–159. doi: 10.1016/j.chembiol.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marceau CD, Puschnik AS, Majzoub K, Ooi YS, Brewer SM, Fuchs G, Swaminathan K, Mata MA, Elias JE, Sarnow P, Carette JE. Genetic dissection of Flaviviridae host factors through genome-scale CRISPR screens. Nature. 2016;535:159–163. doi: 10.1038/nature18631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami K, Kimura T, Osaki M, Ishii K, Miyamura T, Suzuki T, Wakita T, Shoji I. Virological characterization of the hepatitis C virus JFH-1 strain in lymphocytic cell lines. J Gen Virol. 2008;89:1587–1592. doi: 10.1099/vir.0.83618-0. [DOI] [PubMed] [Google Scholar]
- Niphakis MJ, Cravatt BF. Enzyme inhibitor discovery by activity-based protein profiling. Annu Rev Biochem. 2014;83:341–377. doi: 10.1146/annurev-biochem-060713-035708. [DOI] [PubMed] [Google Scholar]
- Patricelli MP, Janes MR, Li LS, Hansen R, Peters U, Kessler LV, Chen Y, Kucharski JM, Feng J, Ely T, Chen JH, Firdaus SJ, Babbar A, Ren P, Liu Y. Selective Inhibition of Oncogenic KRAS Output with Small Molecules Targeting the Inactive State. Cancer Discov. 2016;6:316–329. doi: 10.1158/2159-8290.CD-15-1105. [DOI] [PubMed] [Google Scholar]
- Pierson TC, Sanchez MD, Puffer BA, Ahmed AA, Geiss BJ, Valentine LE, Altamura LA, Diamond MS, Doms RW. A rapid and quantitative assay for measuring antibody-mediated neutralization of West Nile virus infection. Virology. 2006;346:53–65. doi: 10.1016/j.virol.2005.10.030. [DOI] [PubMed] [Google Scholar]
- Rodgers MA, Villareal VA, Schaefer EA, Peng LF, Corey KE, Chung RT, Yang PL. Lipid metabolite profiling identifies desmosterol metabolism as a new antiviral target for hepatitis C virus. J Am Chem Soc. 2012;134:6896–6899. doi: 10.1021/ja207391q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman AL, Ennis FA. Dengue Vaccine: The Need, the Challenges, and Progress. The Journal of infectious diseases. 2016;214:825–827. doi: 10.1093/infdis/jiw068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueckert RR, Pallansch MA. Preparation and characterization of encephalomyocarditis (EMC) virus. Methods Enzymol. 1981;78:315–325. [PubMed] [Google Scholar]
- Savidis G, McDougall WM, Meraner P, Perreira JM, Portmann JM, Trincucci G, John SP, Aker AM, Renzette N, Robbins DR, Guo Z, Green S, Kowalik TF, Brass AL. Identification of Zika Virus and Dengue Virus Dependency Factors using Functional Genomics. Cell Rep. 2016;16:232–246. doi: 10.1016/j.celrep.2016.06.028. [DOI] [PubMed] [Google Scholar]
- Schmidt AG, Yang PL, Harrison SC. Peptide inhibitors of dengue- virus entry target a late-stage fusion intermediate. PLoS Pathog. 2010;6:e1000851. doi: 10.1371/journal.ppat.1000851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sessions OM, Barrows NJ, Souza-Neto JA, Robinson TJ, Hershey CL, Rodgers MA, Ramirez JL, Dimopoulos G, Yang PL, Pearson JL, Garcia-Blanco MA. Discovery of insect and human dengue virus host factors. Nature. 2009;458:1047–1050. doi: 10.1038/nature07967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh J, Petter RC, Baillie TA, Whitty A. The resurgence of covalent drugs. Nature reviews. Drug discovery. 2011;10:307–317. doi: 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- Stanaway JD, Shepard DS, Undurraga EA, Halasa YA, Coffeng LE, Brady OJ, Hay SI, Bedi N, Bensenor IM, Castañeda-Orjuela CA, Chuang T-W, Gibney KB, Memish ZA, Rafay A, Ukwaja KN, Yonemoto N, Murray CJL. The global burden of dengue: an analysis from the Global Burden of Disease Study 2013. The Lancet Infectious Diseases. 2016;16:712–723. doi: 10.1016/S1473-3099(16)00026-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan L, Akahane K, McNally R, Reyskens KM, Ficarro SB, Liu S, Herter-Sprie GS, Koyama S, Pattison MJ, Labella K, Johannessen L, Akbay EA, Wong KK, Frank DA, Marto JA, Look TA, Arthur JS, Eck MJ, Gray NS. Development of Selective Covalent Janus Kinase 3 Inhibitors. J Med Chem. 2015;58:6589–6606. doi: 10.1021/acs.jmedchem.5b00710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Linden WA, Schulze CJ, Herbert AS, Krause TB, Wirchnianski AA, Dye JM, Chandran K, Bogyo M. Cysteine Cathepsin Inhibitors as Anti-Ebola Agents. ACS Infect Dis. 2016;2:173–179. doi: 10.1021/acsinfecdis.5b00130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang QY, Kondreddi RR, Xie X, Rao R, Nilar S, Xu HY, Qing M, Chang D, Dong H, Yokokawa F, Lakshminarayana SB, Goh A, Schul W, Kramer L, Keller TH, Shi PY. A translation inhibitor that suppresses dengue virus in vitro and in vivo. Antimicrob Agents Chemother. 2011;55:4072–4080. doi: 10.1128/AAC.00620-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weerapana E, Wang C, Simon GM, Richter F, Khare S, Dillon MB, Bachovchin DA, Mowen K, Baker D, Cravatt BF. Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature. 2010;468:790–795. doi: 10.1038/nature09472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Hu C, Wang A, Weisberg EL, Chen Y, Yun CH, Wang W, Liu Y, Liu X, Tian B, Wang J, Zhao Z, Liang Y, Li B, Wang L, Wang B, Chen C, Buhrlage SJ, Qi Z, Zou F, Nonami A, Li Y, Fernandes SM, Adamia S, Stone RM, Galinsky IA, Wang X, Yang G, Griffin JD, Brown JR, Eck MJ, Liu J, Gray NS, Liu Q. Discovery of a BTK/MNK dual inhibitor for lymphoma and leukemia. Leukemia. 2016;30:173–181. doi: 10.1038/leu.2015.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Wang W, Liu F, Weisberg EL, Tian B, Chen Y, Li B, Wang A, Wang B, Zhao Z, McMillin DW, Hu C, Li H, Wang J, Liang Y, Buhrlage SJ, Liang J, Liu J, Yang G, Brown JR, Treon SP, Mitsiades CS, Griffin JD, Liu Q, Gray NS. Discovery of a potent, covalent BTK inhibitor for B-cell lymphoma. ACS Chem Biol. 2014;9:1086–1091. doi: 10.1021/cb4008524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Miner JJ, Gorman MJ, Rausch K, Ramage H, White JP, Zuiani A, Zhang P, Fernandez E, Zhang Q, Dowd KA, Pierson TC, Cherry S, Diamond MS. A CRISPR screen defines a signal peptide processing pathway required by flaviviruses. Nature. 2016;535:164–168. doi: 10.1038/nature18625. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.