

Abstract
Objective
The prevalence of obesity varies between ethnic groups. No GWAS for BMI has been conducted in continental Africans.
Methods
We performed a GWAS for body mass index (BMI) in 1,570 West Africans (WA). Replication was conducted in independent samples of WA (n=1,411) and African Americans (AA) (n=9,020).
Results
We identified a novel genome-wide significant African-specific locus for BMI (SEMA4D, rs80068415; MAF = 0.008, p = 2.10×10−8). This finding replicated in independent samples of WA (p = 0.013) and AA (p = 0.017). Individuals with obesity had higher serum Sema4D levels compared to those without obesity (p < 0.0001), and elevated levels of serum Sema4D were associated with increased obesity risk (OR=4.2; p < 1×10−4). The prevalence of obesity was higher in individuals with the CT vs. TT genotypes (55.6% vs. 22.9%).
Conclusions
A novel variant in SEMA4D was significantly associated with BMI. Carriers of the C allele were 4.6 BMI units heavier than carriers of the T allele (p = 0.0007). This variant is monomorphic in Europeans and Asians, highlighting the importance of studying diverse populations. While there is evidence for the involvement of Sema4D in inflammatory processes, this study is the first to implicate SEMA4D in obesity pathophysiology.
Keywords: BMI, Obesity, Genetics
Introduction
In 2014, the World Health Organization reported that over 1.9 billion adults were overweight and that about 600 million of these adults were obese. Most of the world's population now lives in countries where death due to overweight and obesity is greater than from underweight (http://www.who.int/mediacentre/factsheets/fs311/en/). A significant proportion of the death and morbidity attributable to obesity results from its contribution to specific disease outcomes, including diabetes (44%), ischemic heart disease (23%), and certain cancers (7 - 41%)(1). The prevalence of obesity, however, is not uniform across ethnic groups, and African Americans, for example, display the highest age-adjusted rates of obesity (47.8%) in the United States (http://www.cdc.gov/obesity/data/adult.html).
Despite the huge influence of lifestyle and culture on the prevalence of obesity, it is well documented that obesity clusters in families, with heritability estimates as high as 60%(2). Thus understanding the genetic basis of excess weight gain is a major biomedical research goal. Over 20 GWAS and meta-analyses for BMI have been published and more than 90 susceptibility loci identified (3). However, none of these studies have focused on continental Africans. In 2005, we published the first linkage study for obesity in West Africans, which identified several obesity linkage regions(4). Here, we report our efforts to extend these findings by conducting GWAS for BMI in 1,570 unrelated West Africans. We identified a novel genome-wide significant variant in SEMA4D. To further understand the significance of this gene in obesity, we evaluated the association of serum Sema4D levels with obesity. Together, these analyses provide evidence for a novel BMI locus at SEMA4D, and highlight the importance of conducting genomic analyses in diverse populations(5).
Methods
Study design
Individuals included in the GWAS discovery samples were drawn from the Africa America Diabetes Mellitus (AADM) study, a large, ongoing genetic epidemiology study of type 2 diabetes (T2D) and related traits, including obesity(6). Demographic information was collected using standardized questionnaires across the AADM study centers in Nigeria, Ghana and Kenya. Anthropometric and other clinical parameters were measured by trained study staff during a clinic visit. Weight was measured in light clothes on an electronic scale to the nearest 0.1 kg, and height was measured with a stadiometer to the nearest 0.1 cm. BMI was computed as weight (kg) divided by the square of height in meters (m2). Obesity was defined as BMI ≥ 30 kg/m2. The diagnosis of T2D was based on the 1999 America Diabetes Association Expert Committee(7, 8) Criteria: a fasting plasma glucose concentration ≥ 126 mg/dl (7.0 mmol/l), a 2-h post load value in the oral glucose tolerance test ≥ 200 mg/dl (11.1 mmol/l) on more than one occasion, or taking medication for physician-diagnosed T2D.
Genotyping and quality control
A total of 1,822 individuals were genotyped at the Children's Hospital of Philadelphia (CHOP) core laboratory using the Affymetrix Axiom® PanAFR array (www.affymetrix.com). Given that the discovery GWAS was restricted to West Africans with planned replication in African Americans (who have predominantly West African ancestry), we excluded 332 non-West Africans. In addition, we removed duplicate (n=11) and sex-discordant (n=13) individuals for a total of 1576 participants included in the analysis. A total of 2,217,748 SNPs were subjected to QC filters as follows: 1) call rate ≤ 90% (excluded 22,509 SNPs), Hardy-Weinberg p-value < 10−6 (excluded 10,282), MAF < 0.01 (excluded 46,562), and non-autosomal SNPs (excluded 61,087); a total of 2,077,308 SNPs in 1576 individuals were retained and carried forward for discovery GWAS analyses. Human Genomic reference 19 (hg19) was used in these analyses.
Imputation
Imputation was performed using the 1000 Genomes reference data (1000G Phase 1 V3, http://www.sph.umich.edu/csg/abecasis/MACH/download/) with a total of 1092 individuals (AFR 246, AMR 181, ASN 286, and EUR 379)(9). The MaCH-Admix(10) algorithm was used for imputation. Dosage r2 and quality scores were used to measure imputation quality. Dosage r2 is the squared Pearson correlation between the estimated allele dosages and the true experimental genotype. The quality score is the average posterior probability for the most likely genotype(11). For imputed variants for which we had experimental genotypes, the imputation quality was high: the first (Q1) and third (Q3) quartiles of r2 scores were 0.9824, and 0.9964, respectively, and Q1 and Q3 of quality scores were 0.9965, and 0.9992. A total of 28,032,917 SNPs were successfully imputed (i.e. the imputation yield). After applying the filters of Minor Allele Count (MAC) < 5 and r2 < 0.3(12), a total of 14,620,866 SNPs were excluded. The Q1 and Q3 of r2 scores were 0.7840, and 0.9578, respectively, and Q1 and Q3 of quality scores were 0.9739, and 0.9939, respectively, in the remaining 13,412,051 imputed SNPs. The minimum quality score was 0.5701.
Statistical analyses
To minimize the potential effect of population structure, we adjusted all analyses by the first two principal components (PC1 and PC2) obtained from R package, SNPRelate (13), which generates genetic covariance matrix followed by the extraction of eigenvalues and eigenvectors for the calculation of PCs. Genome-wide association analyses were performed using mach2qtl (14) which accepts imputed dosages or posterior probabilities from MaCH-Admix output. BMI was regressed on age, sex, T2D status, duration of T2D, and the first two principal components of the genotypes. Six individuals missing phenotype data were excluded for a total of 1570 subjects included in the GWAS. Although genome-wide significance and suggestive evidence of association are usually set at p-value = 5 × 10−8 and 5 × 10−7 respectively (15, 16), we used the more conservative p-values of 2.5 × 10−8 and 2.5 × 10−7 as our threshold values given the greater genomic diversity and, thus, greater number of tests in West Africans. Analysis of serum Sema4D (sSema4D) concentration by obesity status was performed using regression, and discrete trait analyses were performed using logistic regression with adjustments for sex, age, T2D, and duration of T2D. For this set of analyses, two-tailed p-values < 0.05 were considered statistically significant. Metal was used to conduct fixed-effects meta-analysis (http://genome.sph.umich.edu/wiki/METAL_Documentation).
ELISA Assay for Serum soluble Sema4D
An enzyme-linked Immunoabsorbent Assay (ELISA) Kit for SEMA4D (Cloud-Clone Corp [www.cloud-clone.us]) was used to measure serum soluble SEMA4D (sSEMA4D) levels in the 1,343 unrelated individuals in the GWAS study. This test is a sandwich enzyme immunoassay in which the microplate is pre-coated with an antibody specific to sSEMA4D. Standards or samples are then added to the appropriate microplate wells with a biotin-conjugated antibody specific to SEMA4D. Next, Avidin conjugated to Horseradish Peroxidase (HRP) is added to each microplate well and incubated. After TMB substrate solution is added, only those wells that contain sSEMA4D, biotin-conjugated antibody and enzyme-conjugated Avidin will exhibit a change in color. The enzyme-substrate reaction is terminated by the addition of sulphuric acid solution and the color change is measured spectrophotometrically at a wavelength of 450 nm. The concentration of sSEMA4D in the samples is then determined by comparing the optical densities (OD) of the samples to the standard curve. The measurement unit of sSEMA4D is ng/ml. The third quartile value of sSEMA4D was used to categorize sSema4D into binary trait; log-transformed values of sSEMA4D were used for continuous trait analyses.
De novo genotyping for validation and replication
Using the Sequenom iPLEX platform, we performed de novo genotyping in 1,506 West Africans who were not included in the discovery sample to perform exact replication (1,411 were available for analysis after QC). Replication analyses were also performed in 9,020 African Americans obtained from the Atherosclerosis Risk in Communities study (ARIC, n = 3,137)(17), Cleveland Family Study (CFS, n = 653)(18), Howard University Family Study (HUFS, n = 1,976)(19), Jackson Heart Study (JHS, n = 2,187)(20), and Multi-Ethnic Study of Atherosclerosis (MESA, n = 1,611)(21). Human genomic reference (hg19) was used in the analysis. LiftOver (https://genome.ucsc.edu/cgi-bin/hgLiftOver) was used to convert genome coordinates and genome annotation between assemblies. The Generalized Linear Mixed Model Association Test (GMMAT, https://www.hsph.harvard.edu/han-chen/software) was used to perform association tests. We modeled cryptic relatedness as a random effect and included covariates for sex, age, T2D status, and the first two principal components (PC1 and PC2). Score tests (SCORE and VAR values) were performed for each variant in the GWAS. Results were transformed into Wald test values (beta = SCORE/var, SE = 1/sqrt(var)) to compare with our discovery GWAS results.
Power calculation was performed using QUANTO (http://biostats.usc.edu/software). Power estimation included the respective mean and standard deviation of BMI, the effect size, and allele frequency for discovery, and replication samples. Power was estimated for an additive genetic model and two-tail tests with α of 2.5 × 10−8 (i.e., 0.05/2000000) for discovery GWAS, and α of 0.05 for the validation and replication samples.
Results
The participants in the discovery GWAS had a relatively high mean BMI, reflecting the fact that these individuals were drawn from a T2D case control study (Table 1). Overall, the discovery GWAS identified one variant that reached genome-wide significance and 5 with suggestive evidence of association (Manhattan Plot, Figure 1; QQ Plot (λ = 1.01), Figure S1; Table 2). SNP rs80068415 (C allele, MAF 0.008) was significantly associated with BMI (β 5.87, p-value 2.10 × 10−8, power = 0.81). This variant is located in the intron of SEMA4D (Sema domain, immunoglobulin domain, transmembrane domain, and short cytoplasmic domain 4D; GeneID: 10507, 9q22.2). The regional plot of rs80068415 is displayed in Figure 2. Results of the discovery and replication analyses are presented in Table 3. To increase our confidence in the rs80068415 association, we genotyped the SNP in a separate sample of our West African participants (Table 1); we replicated the association with BMI using the genotyped data (n = 1,411, C allele, MAF = 0.0086, p-value 1.31× 10−2). In addition, this association was replicated in a large sample of African Americans (Table 1; n = 9,020, C allele: MAF = 0.006, p value = 1.69 × 10−2). We observed a consistent direction of effect across all three samples included in our meta-analysis of rs80068415 (C allele, direction +++, β(SE) = 2.889(0.508); p=1.34 × 10−8; Table 3). The mean BMI of CT heterozygotes (31.90 ± 5.99 kg/m2) is significantly higher compared to the TT homozygotes (26.62 ± 5.26 kg/m2; p= 7.0 × 10−6). Similarly, the percentage of obesity (BMI ≥ 30kg/m2) in CT heterozygotes is 55.56% comparing with 22.87% in TT homozygotes with an odds ratio of 4.22 [1.66, 10.72].
Table 1.
Basic Characteristics of Discovery and Replication studies
| Discovery | Replications | ||
|---|---|---|---|
| GWAS/validation * | West Africans | African Americans | |
| N (% in male) | 1,570 (41.75%) | 1,411 (45.09%) | 9,020 (41.03%) |
| Age (Years) | 54 (47, 61) | 52 (43, 61) | 51 (45, 59) |
| Weight (Kg) | 69.75 (61.22, 79.82) | 66.60 (56.46, 77.00) | NA |
| Height (M) | 1.64 (1.58, 1.70) | 1.64 (1.58, 1.70) | NA |
| Body Mass Index (Kg/m2) | 26.06 (22.77, 29.60) | 24.45 (21.29, 28.27) | 29.34 (25.51, 34.12) |
| Fat Mass (Kg) | 21.57 (14.54, 30.49) | 18.05 (11.57, 26.11) | NA |
| Percent Fat Mass (%) | 31.55 (22.00, 40.90) | 27.73 (19.88, 36.75) | NA |
| T2D (%) | 902 (57.93%) | 625 (41.50%) | 1,509 (16.54%) |
| Duration of T2D (Years) | 1 (0, 7) | 0 (0, 4) | NA |
| Waist (cm) | 91.53 (84.00, 99.5) | 88.00 (78.30, 96.50) | NA |
| HIP (cm) | 101.0 (94.5, 108.0) | 97.0 (90.0, 104.3) | NA |
| Ratio of Waist and Hip | 0.91 (0.86, 0.95) | 0.90 (0.85, 0.95) | NA |
| Hypertension (%) | 961 (61.84%) | 730 (49.22%) | NA |
| SBP (mmHg) | 134.5 (121, 153) | 130.5 (115.5, 149.5) | NA |
| DBP (mmHg) | 80 (72.5, 89) | 79 (70.5, 88) | NA |
Discrete variables presented as numbers (%);
for continue variables presented as median (Q1, Q3)
Figure 1.
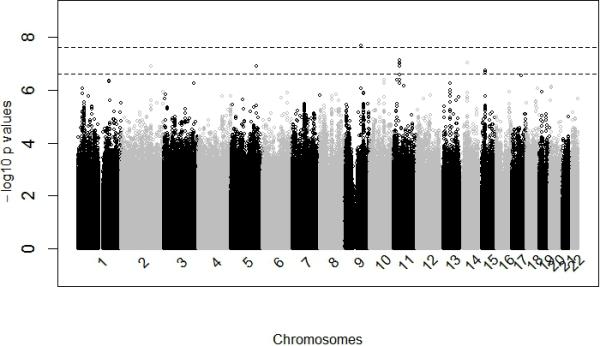
Genome-wide association results. The –log10(p-value) of associations are presented on the y-axis. The x-axis represents loci positions (bp). The dashed lines represent –log10 (2.5×10-8), and -log10(2.5×10-7), for genome-wide and suggestive statistical significance levels, respectively.
Table 2.
SNPs displaying genome-wide and suggestive evidence of association with BMI in the GWAS
| Chr | SNPs | BPs | Risk/Alt | MAF | Effect | stderr | P values | Region | Power | Genes | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Genome Wide Significant Threshold * | 9q22.2 | rs80068415 | 92093127 | C/T | 0.0076 | 5.876 | 1.049 | 2.10E-08 | intron | 0.81 | SEMA4D |
| Genome-Wide Suggestive Threshold ** | 2q31.1 | rs74349012 | 169709682 | A/G | 0.0268 | 2.848 | 0.539 | 1.30E-07 | intron | 0.41 | NOSTRIN |
| 5q32 | rs116300832 | 146333749 | A/G | 0.0326 | 3.082 | 0.583 | 1.26E-07 | intron | 0.72 | PPP2R2B | |
| 11p13 | rs139014794 | 35293132 | T/G | 0.0351 | 2.782 | 0.517 | 7.56E-08 | intron | 0.49 | SLC1A2 | |
| 14q21.1 | 14:42277454 | 42277454 | G/GGAGA | 0.022 | 3.607 | 0.675 | 9.16E-08 | intron | 0.55 | LRFN5 | |
| 15q15.1 | rs7178777 | 41634588 | A/C | 0.4647 | −1.234 | 0.237 | 1.85E-07 | missense | 0.81 | NUSAP1 |
0.05/(number of tests) = 2.5E-8
0.5/(number of tests) = 2.5E-7
Figure 2.

SEMA4D regional plot. The x-axis represents positions (Mb), and the y-axis represents –log10(p-values). Recombination rate and r2 scores were calculated from AFR samples in the 1000 Genomes Project (phase1, V3).
Table 3.
Meta Analysis Results for rs80068415
| Discovery | Replication | Meta Analysis | ||
|---|---|---|---|---|
| West Africans | West Africans | African Americans | ||
| N | 1,570 | 1,411 | 9,020 | |
| Risk/Alt. | C/T | C/T | C/T | |
| MAF (C allele) | 0.0076 | 0.0086 | 0.0058 | |
| Direction | + | + | + | +++ |
| Beta (SE) | 5.876(1.049) | 2.452(0.987) | 1.717(0.987) | 2.889(0.508) |
| P values | 2.10E-08 | 1.31E-02 | 1.69E-02 | 1.34E-08 |
| HetPVal | 0.004 | |||
We also observed suggestive evidence of association at 5 loci: 1) rs74349012 (MAF 0.03) located in the intron of NOSTRIN (Gene ID 115677, 2q31.1) with a p-value of 1.30 × 10−7 (regional plot: Figure S2); 2) rs116300832 (MAF 0.03) located in the intron of PPP2R2B (Gene ID 5521, 5q32) with p-value of 1.26 × 10−7 (regional plot: Figure S3); 3) two SNPs (rs139014794 and rs145047868) and an indel (35314600 bp) in the intron of SLC1A2 (Gene ID 6505, 11p13-p12) with p-values of 7.56 × 10−8, 9.54 × 10−8, and 9.28 × 10−8, respectively (regional plot: Figure S4); 4) an indel located at 42277454 bp in the intron of LRFN5 (Gene ID 145581, 14q21.1) with p-value 9.16 × 10−8 and 5) two exonic SNPs (rs7178634 and rs7178777) in NUSAP1 (Gene ID: 51203, 15q15.1) with p-values of 2.15 × 10−7, and 1.85 × 10−7, respectively (regional plots: Figures S5 and S6, respectively).
A total of 1,343 of the individuals in the GWAS were assayed for circulating sSEMA4D levels to evaluate the relationship between sSEMA4D, obesity and rs80068415 (Figure 3). We observed that mean sSEMA4D levels were significantly higher in individuals with obesity compared to individuals without obesity (p-value < 0.0001; Figure 3A) after adjusting for age, sex, T2D, and duration of T2D for the pooled data and for age, T2D and duration of T2D in gender-stratified analyses. A positive association was also found for BMI as a continuous trait (p= 0.0007). The partial age- and sex-adjusted correlation between obesity and sSEMA4D as binary trait was 0.21, and sSEMA4D was associated with a significant risk of obesity with an odds ratio of 4.22 (95% CI: 3.05, 5.83; p < 1×10−4) after adjustment for sex, age, and T2D. Log transformed sSEMA4D, as continuous trait was also significantly associated with BMI (p=0.00435). Carriers of the C allele were about 4.6 BMI unit heavier than carriers of the T allele (p = 0.0007; Figure 3B).
Figure 3. Relationships between sSema4D levels, obesity, and rs80068415.

(A)Mean and standard deviation of serum Sema4D levels (ng/ml) in obese and non-obese, by gender, with p-values < 0.0001 in males, females, and combined. (B) Mean and standard deviation of BMI (kg/m2) by alleles of rs80068415 (p-value = 0.0007)
We investigated the association between 2,481 SNPs within SEMA4D with sSema4D level in 1,343 individuals that were included in the discovery GWAS. Our top result was with rs184536170, which was significantly associated with sSEMA4D (beta=0.69, p-value=3.5 × 10−4; adjusted p-value= 0.01). The association between our significant BMI variant, rs80068415, with sSEMA4D was nominally significant (p=0.05367). rs80068415 is located 10kb from rs184536170 with r2 < 0.1. Three additional SNPs within SEMA4D were also associated with sSEMA4D, rs114379626 (p=0.01328, r2 with rs80068415 = 0.26), rs183701477 (p= 0.0204, r2 with rs80068415 = 0.55), and rs34488061 (p=0.004047, r2 with rs80068415 = 0.64) but these associations did not remain significant after correcting for multiple testing.
Discussion
In this first GWAS for BMI in continental Africans, we identified a novel BMI locus, SEMA4D. We replicated this finding in 1,411 West African samples and 9,020 African American samples. Meta-analysis of the discovery and replication data further supports this finding. We also showed that circulating sSEMA4D level was significantly higher in individuals with obesity compared to those without obesity. Based on ENCODE data (22), lead SNP rs80068415 displays marked transcriptional activity (flanking the active transcription start site) in a diverse population of T-cells (T- helper cells, primary T-cells, T-regulatory cells). This SNP also overlaps an enhancer in numerous tissues, including primary hematopoietic stem cells, and GATA2, a transcription factor bound by CHIP-seq. The C variant results in GATA2 motif disruption. No associated eQTL has been reported. The evidence from this study and the ENCODE data suggest that the C allele of r80068415 is probably not the causal variant but more likely tracking a regulatory process that is controlling putative obesity-related genes. rs80068415 is likely acting through either another functional SNP in LD with it or trans-regulatory elements affecting obesity-related gene function. Deep sequencing of SEMA4D in African ancestry populations along with the use of a range of functional genetic techniques including gene editing could help elucidate the mechanism(s) by which rs80068415 is associated with BMI in Africans and African Americans. Functionally, SEMA4D has been shown to play a role in cell-cell signaling, immune response, and bone formation(23). In the context of the findings of this paper, SEMA4D's role in immune response is promising given the association between inflammation and obesity. SEMA4D is a 150-kDa transmembrane glycoprotein expressed by platelets, activated B cells, dendritic cells, kidney, osteoclasts, and resting T-cells. When these cells are activated, SEMA4D is proteolytically cleaved, generating a biologically-active 120-kDa fragment with paracrine/autocrine properties (24). sSema4D is a pleiotropic protein that has also been associated with many other inflammatory/autoimmune diseases including multiple sclerosis and Non Alcoholic Steato-Hepatitis (NASH). Under these conditions SEMA4D seems to be upregulated at the protein level through the activation and differentiation of T-cells and promotion of Th1 (T helper 1) differentiation (23, 25). In fact, T-cells have been shown to have an early and critical role in obesity. Obesity is associated with an increased accumulation of T-cells which are the major source of SEMA4D(26, 27). In addition, Th1 polarization in adipose tissues during diet induced obesity in mice has been reported(28).
The prevalent Th1 pattern of secreted cytokines may be regarded as a possible mechanism contributing to obesity related-comorbidities, including low-grade inflammation and insulin resistance (IR). Indeed, IR and obesity are closely related and mechanisms underlying them have been extensively studied(25, 26). Interestingly, SEMA4D's intracellular domain has been associated with activity of a serine kinase whose activation leads to insulin signaling inhibition, hence IR(26). While there is evidence for the involvement of SEMA4D in inflammatory processes through the regulation of T-cells, this study is the first to implicate SEMA4D in obesity pathophysiology and to suggest that SEMA44D may be a key player in the triangular relationship between obesity, inflammation, and metabolic comorbidities especially IR.
In summary, we identified and replicated a novel variant in SEMA4D (rs80068415) that is significantly associated with BMI in African ancestry individuals. Furthermore, we observed that circulating Sema4D levels were considerably higher in individuals with obesity compared with those without obesity. The putative role of SEMA4D in obesity appears to involve regulation of the transcription start site. The lead SNP, rs80068415, is polymorphic in Africans but monomorphic in other 1000G populations, providing a potential explanation for why this association has not been observed in published GWAS of European and Asian populations. This work highlights the importance of conducting genomic studies in diverse populations and identifies a novel locus that may improve our understanding of BMI-related physiology.
Supplementary Material
What is already known about this subject?
Over 20 GWAS and/or meta-analysis for Body Mass Index/Obesity have been published, and more than 90 susceptibility loci identified.
None of these studies focused on continental Africans.
Heritability of obesity has been estimated to be as high as 60%.
What does this study add?
Semaphorin-4D (SEMA4D) was identified as a novel susceptibility locus for BMI in populations of African ancestry.
Individuals with obesity had significantly higher serum Sema4D levels compared to those without obesity.
Acknowledgments
The authors thank the participants in the AADM project, their families, and their physicians. The crucial role of the study coordinators, as well as field and laboratory staff is gratefully acknowledged. The study was supported in part by the Intramural Research Program of the National Institutes of Health in the Center for Research on Genomics and Global Health (CRGGH). The CRGGH is supported by the National Human Genome Research Institute, the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), the Center for Information Technology (CIT), and the Office of the Director at the National Institutes of Health (1ZIAHG200362). Support for participant recruitment and initial genetic studies of the AADM study was provided by NIH grant No. 3T37TW00041-03S2 from the National Institute of Minority Health and Health Disparities (NIMHD). The project was also supported in part by the NIDDK grant DK-54001.
The Atherosclerosis Risk in Communities Study is carried out as a collaborative study supported by National Heart, Lung, and Blood Institute contracts (HHSN268201100005C, HHSN268201100006C, HHSN268201100007C, HHSN268201100008C, HHSN268201100009C, HHSN268201100010C, HHSN268201100011C, and HHSN268201100012C). The authors thank the staff and participants of the ARIC study for their important contributions. Funding for GENEVA was provided by National Human Genome Research Institute grant U01HG004402 (E. Boerwinkle). Support for the Cleveland Family Study was provided by NHLBI grant number RO1 HL46380. Support for genotyping was provided by NHLBI Contract N01-HC-65226. The Jackson Heart Study is supported and conducted in collaboration with Jackson State University (HHSN268201300049C and HHSN268201300050C), Tougaloo College (HHSN268201300048C), and the University of Mississippi Medical Center (HHSN268201300046C and HHSN268201300047C) contracts from the National Heart, Lung, and Blood Institute (NHLBI) and the National Institute for Minority Health and Health Disparities (NIMHD). This manuscript was not prepared in collaboration with JHS investigators and does not necessarily reflect the opinions or views of JHS, or the NHLBI. Funding for CARe genotyping was provided by NHLBI Contract N01-HC-65226. MESA and the MESA SHARe project are conducted and supported by the National Heart, Lung, and Blood Institute (NHLBI) in collaboration with MESA investigators. Support for MESA is provided by contracts N01-HC- 95159, N01-HC-95160, N01-HC-95161, N01-HC-95162, N01-HC-95163, N01-HC-95164, N01-HC-95165, N01-HC-95166, N01-HC-95167, N01-HC-95168, N01-HC-95169 and CTSA UL1-RR-024156. Funding for SHARe genotyping was provided by NHLBI Contract N02-HL-64278. Genotyping was performed at Affymetrix (Santa Clara, California, USA) and the Broad Institute of Harvard and MIT (Boston, Massachusetts, USA) using the Affymetric Genome-Wide Human SNP Array 6.0.
Footnotes
DISCLOSURE: The authors declare no conflict of interest.
AUTHOR CONTRIBUITONS: RC, AA, and GC designed the study; GC and JZ analyzed the data; GC, AD, AA, and RC drafted the manuscript; OF, GO, BE, KA, AA, JA, TJ, JO, CA, and RC directed data collection; AD, and LL performed experiment; all authors participated in the critical revision of the manuscript and have seen and approved the final version.
Web Resources:
The 1000 Genomes Project, http://www.1000genomes.org/
MaCH-Admix, http://www.unc.edu/~yunmli/MaCH-Admix/
Affymetrix Axiom PanAFR array, http://www.affymetrix.com/
QUANTO, http://biostats.usc.edu/software
Human Subjects: Discovery data (AADM) was initially approved by the Institutional Review Boards (IRB) of the Howard University and then by the National Human Genome Institute (NHGRI) at NIH. All replication data except HUFS data was accessed through an approved request for controlled-access data from dbGaP (pmid 17898773) as follows: ARIC (phs000280.v2.p1, phs000090.v2.p1), CFS (phs000284.v1.p1), JHS (phs000286.v4.p1, phs000499.v2.p1), and MESA (phs000209.v13.p1, phs000420.v6.p3). Details of each of these studies has been previously described (ARIC [2646917], CFS [7881656], JHS [16320381], and MESA [12397006]). The ARIC study was approved by the Institutional Review Boards (IRB) of the University of North Carolina at Chapel Hill, Johns Hopkins University, University of Mississippi Medical Center, Wake Forest University, University of Minnesota, Brigham and Women's Hospital, and Baylor College of Medicine. CFS was approved by the University Hospitals Case Medical Center. The Jackson Heart Study was approved by the IRB of the University of Mississippi Medical Center, Jackson State University, and Tougaloo College. MESA was approved by the IRB at each of the six field centers. HUFS data was approved by the IRB of the Howard University.
References
- 1.Cefalu WT, Bray GA, Home PD, Garvey WT, Klein S, Pi-Sunyer FX, et al. Advances in the Science, Treatment, and Prevention of the Disease of Obesity: Reflections From a Diabetes Care Editors' Expert Forum. Diabetes Care. 2015;38:1567–1582. doi: 10.2337/dc15-1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maes HH, Neale MC, Eaves LJ. Genetic and environmental factors in relative body weight and human adiposity. Behav Genet. 1997;27:325–351. doi: 10.1023/a:1025635913927. [DOI] [PubMed] [Google Scholar]
- 3.Welter D, MacArthur J, Morales J, Burdett T, Hall P, Junkins H, et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014;42:D1001–1006. doi: 10.1093/nar/gkt1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen G, Adeyemo AA, Johnson T, Zhou J, Amoah A, Owusu S, et al. A genome-wide scan for quantitative trait loci linked to obesity phenotypes among West Africans. Int J Obes (Lond) 2005;29:255–259. doi: 10.1038/sj.ijo.0802873. [DOI] [PubMed] [Google Scholar]
- 5.Rosenberg NA, Huang L, Jewett EM, Szpiech ZA, Jankovic I, Boehnke M. Genome-wide association studies in diverse populations. Nat Rev Genet. 2010;11:356–366. doi: 10.1038/nrg2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rotimi CN, Dunston GM, Berg K, Akinsete O, Amoah A, Owusu S, et al. In search of susceptibility genes for type 2 diabetes in West Africa: the design and results of the first phase of the AADM study. Ann Epidemiol. 2001;11:51–58. doi: 10.1016/s1047-2797(00)00180-0. [DOI] [PubMed] [Google Scholar]
- 7.Screening for type 2 diabetes. Diabetes Care. 2000;23(Suppl 1):S20–23. [PubMed] [Google Scholar]
- 8.Engelgau MM, Narayan KM, Herman WH. Screening for type 2 diabetes. Diabetes Care. 2000;23:1563–1580. doi: 10.2337/diacare.23.10.1563. [DOI] [PubMed] [Google Scholar]
- 9.Delaneau O, Zagury JF, Marchini J. Improved whole-chromosome phasing for disease and population genetic studies. Nat Methods. 2013;10:5–6. doi: 10.1038/nmeth.2307. [DOI] [PubMed] [Google Scholar]
- 10.Liu EY, Li M, Wang W, Li Y. MaCH-admix: genotype imputation for admixed populations. Genet Epidemiol. 2013;37:25–37. doi: 10.1002/gepi.21690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genetic epidemiology. 2010;34:816–834. doi: 10.1002/gepi.20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Monda KL, Chen GK, Taylor KC, Palmer C, Edwards TL, Lange LA, et al. A meta-analysis identifies new loci associated with body mass index in individuals of African ancestry. Nat Genet. 2013 doi: 10.1038/ng.2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zheng X, Levine D, Shen J, Gogarten SM, Laurie C, Weir BS. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics. 2012;28:3326–3328. doi: 10.1093/bioinformatics/bts606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Y, Willer C, Sanna S, Abecasis G. Genotype imputation. Annu Rev Genomics Hum Genet. 2009;10:387–406. doi: 10.1146/annurev.genom.9.081307.164242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pedroso I, Breen G. Gene set analysis and network analysis for genome-wide association studies. Cold Spring Harb Protoc. 2011:2011. doi: 10.1101/pdb.top065581. [DOI] [PubMed] [Google Scholar]
- 16.Dudbridge F, Gusnanto A. Estimation of significance thresholds for genomewide association scans. Genet Epidemiol. 2008;32:227–234. doi: 10.1002/gepi.20297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu B, Zheng Y, Alexander D, Manolio TA, Alonso A, Nettleton JA, et al. Genome-wide association study of a heart failure related metabolomic profile among African Americans in the Atherosclerosis Risk in Communities (ARIC) study. Genetic epidemiology. 2013;37:840–845. doi: 10.1002/gepi.21752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dean DA, 2nd, Goldberger AL, Mueller R, Kim M, Rueschman M, Mobley D, et al. Scaling Up Scientific Discovery in Sleep Medicine: The National Sleep Research Resource. Sleep. 2016;39:1151–1164. doi: 10.5665/sleep.5774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adeyemo A, Gerry N, Chen G, Herbert A, Doumatey A, Huang H, et al. A genome-wide association study of hypertension and blood pressure in African Americans. PLoS Genet. 2009;5:e1000564. doi: 10.1371/journal.pgen.1000564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fox ER, Sarpong DF, Cook JC, Samdarshi TE, Nagarajarao HS, Liebson PR, et al. The relation of diabetes, impaired fasting blood glucose, and insulin resistance to left ventricular structure and function in African Americans: the Jackson Heart Study. Diabetes Care. 2011;34:507–509. doi: 10.2337/dc10-0838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bild DE, Bluemke DA, Burke GL, Detrano R, Diez Roux AV, Folsom AR, et al. Multi-Ethnic Study of Atherosclerosis: objectives and design. Am J Epidemiol. 2002;156:871–881. doi: 10.1093/aje/kwf113. [DOI] [PubMed] [Google Scholar]
- 22.Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012;40:D930–934. doi: 10.1093/nar/gkr917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Worzfeld T, Offermanns S. Semaphorins and plexins as therapeutic targets. Nat Rev Drug Discov. 2014;13:603–621. doi: 10.1038/nrd4337. [DOI] [PubMed] [Google Scholar]
- 24.Lu QY, Dong NZ, Wang Q, Yi WX, Wang YX, Zhang SJ, et al. Increased Levels of Plasma Soluble Sema4D in Patients with Heart Failure. PLoS One. 2013:8. doi: 10.1371/journal.pone.0064265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bertola A, Bonnafous S, Anty R, Patouraux S, Saint-Paul MC, Iannelli A, et al. Hepatic expression patterns of inflammatory and immune response genes associated with obesity and NASH in morbidly obese patients. PLoS One. 2010;5:e13577. doi: 10.1371/journal.pone.0013577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu H, Ghosh S, Perrard XD, Feng L, Garcia GE, Perrard JL, et al. T-cell accumulation and regulated on activation, normal T cell expressed and secreted upregulation in adipose tissue in obesity. Circulation. 2007;115:1029–1038. doi: 10.1161/CIRCULATIONAHA.106.638379. [DOI] [PubMed] [Google Scholar]
- 27.Mizui M, Kumanogoh A, Kikutani H. Immune semaphorins: novel features of neural guidance molecules. J Clin Immunol. 2009;29:1–11. doi: 10.1007/s10875-008-9263-7. [DOI] [PubMed] [Google Scholar]
- 28.Strissel KJ, DeFuria J, Shaul ME, Bennett G, Greenberg AS, Obin MS. T-cell recruitment and Th1 polarization in adipose tissue during diet-induced obesity in C57BL/6 mice. Obesity (Silver Spring) 2010;18:1918–1925. doi: 10.1038/oby.2010.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.