

Abstract
Both sleep loss and pathogens can enhance brain inflammation, sleep, and sleep intensity as indicated by electroencephalogram delta (δ) power. The pro-inflammatory cytokine interleukin-1 beta (IL-1β) is increased in the cortex after sleep deprivation (SD) and in response to the Gram-negative bacterial cell-wall component lipopolysaccharide (LPS), although the exact mechanisms governing these effects are unknown. The nucleotide-binding domain and leucine-rich repeat protein-3 (NLRP3) inflammasome protein complex forms in response to changes in the local environment and, in turn, activates caspase-1 to convert IL-1β into its active form. SD enhances the cortical expression of the somnogenic cytokine IL-1β, although the underlying mechanism is, as yet, unidentified. Using NLRP3-gene knockout (KO) mice, we provide evidence that NLRP3 inflammasome activation is a crucial mechanism for the downstream pathway leading to increased IL-1β-enhanced sleep. NLRP3 KO mice exhibited reduced non-rapid eye movement (NREM) sleep during the light period. We also found that sleep amount and intensity (δ activity) were drastically attenuated in NLRP3 KO mice following SD (homeostatic sleep response), as well as after LPS administration, although they were enhanced by central administration of IL-1β. NLRP3, ASC, and IL1β mRNA, IL-1β protein, and caspase-1 activity were greater in the somatosensory cortex at the end of the wake-active period when sleep propensity was high and after SD in wild-type but not NLRP3 KO mice. Thus, our novel and converging findings suggest that the activation of the NLRP3 inflammasome can modulate sleep induced by both increased wakefulness and a bacterial component in the brain.
Keywords: Sleep deprivation, NLRP3 inflammasome, interleukin-1 beta, caspase-1, EEG delta power, inflammation, lipopolysaccharide, cortex
1 Introduction
Many brain areas—including the cortex, thalamus, ventral lateral preoptic area, median preoptic nucleus, basal forebrain, and brainstem—are known to modulate NREM sleep (Brown et al., 2012; Szymusiak, 2010; Saper et al., 2010). In particular, somatosensory cortex activity modulates sleep and the electroencephalogram (EEG) (Timofeev et al., 2013). Increased wakefulness, such as that occurring during sleep deprivation (SD), enhances the expression of pro-inflammatory molecules in the somatosensory cortex including interleukin-1 beta (IL-1β) (Zielinski et al., 2011; Imeri and Opp, 2009). IL-1β expression and protein levels in the cortex exhibit diurnal variations that coincide with sleep propensity (Krueger et al., 2010; Taishi et al., 1998; 2012). IL-1β applied centrally or to the periphery enhances NREM sleep in rabbits, rats, and mice (Zielinski et al., 2011; Obal et al., 1995; Olivadoti and Opp, 2008; Toth and Opp, 2001), although higher dosages can inhibit sleep (Opp et al., 1991). Nevertheless, the exact mechanisms for cortical IL-1β regulation of sleep are unknown.
EEG NREM sleep delta (δ) power spectra (0.5-4 Hz frequency range; also referred to as slow-wave activity) is often used as an indicator of sleep need after acute SD (Davis et al., 2011). NREM sleep δ power is, in part, regulated by a thalamo-cortico-thalamo loop (Steriade, 2006). While the exact mechanism(s) responsible for the NREM sleep δ power are unknown, pro-inflammatory molecules tend to enhance it (Zielinski et al., 2011; Imeri and Opp, 2009), for example, IL-1β enhances NREM sleep δ power in animals when applied to the brain (Tobler et al., 1994; Krueger et al., 1984), although mice exhibit reductions after IL-1β is applied to the periphery. This might be due to the activation of the anti-somnogenic properties of the hypothalamic-pituitary-axis (Krueger et al., 2010; Zielinski et al., 2012). Moreover, inhibiting IL-1β pharmacologically can attenuate NREM sleep δ power responses from SD (Takahahi et al., 1997).
The Gram-negative bacterial cell wall component lipopolysaccharide (LPS) enhances sleep, NREM sleep δ power, and cortical IL-1β expression (Zielinski et al., 2011; Imeri and Opp, 2009). These responses, however, differ depending upon the route of administration, dosage, and pathogen or related component. LPS administered to the periphery or to the brain enhances various cytokines in the brain/cortex, including IL-1β (Noh et al., 2014; Henry et al., 2008, 2009; Zielinski et al., 2012; Lawson et al., 2013). Notwithstanding, the precise mechanisms of how pathogens and their components enhance cortical IL-1β-mediated sleep are not well understood.
Inflammasomes are inducible protein complexes that form in reaction to various biological and chemical substances, as well as pathogens and their components, including LPS, which act through their respective pattern recognition receptors (PRRs) (Elliott and Sutterwala, 2015). The nucleotide-binding oligomerization domain-like receptor (NLR) family members NLRP1, NLRP3 and NLRC4 and the AIM2-like receptor (ALR) family member AIM2 have all been shown to be able to form inflammasome complexes in response to either pathogen-associated molecular patterns (PAMPs) or danger-associated molecular patterns (DAMPs). Upon activation, NLRP3 recruits the adaptor molecule ASC [apoptosis-associated speck-like protein containing a C-terminal caspase-recruitment domain (CARD)], which in turn recruits the cysteine protease caspase-1. Caspase-1 activation subsequently results in the processing of pro-IL-1β and pro-IL-18 into their mature secreted forms.
1.1 Conceptual overview of the NLRP3 inflammasome in sleep regulation
Growing evidence indicates that the NLRP3 inflammasome in the brain is involved in certain behavior, such as stress-mediated depression-like and anxiety-like behavior (Zhang et al., 2015; Wong et al., 2016). Currently, the role of the NLRP3 inflammasome in sleep regulation remains unknown. We hypothesized that the NLRP3 inflammasome functions to enhance sleep and NREM sleep δ power. Using two experimental models that have been previously shown to increase IL-1β, SD and LPS administration (Reviewed Zielinski and Krueger 2011), we determined the effects of increased wakefulness on the activation of the NLRP3 inflammasome in the somatosensory cortex. In addition, we determined the effect of increased wakefulness which enhances NREM δ during recovery sleep, and central administration of LPS on sleep and NREM sleep δ power in mice specifically lacking NLRP3 so as to investigate the effects of a well-known somnogenic substance that also activates NLRP3 inflammasomes within the brain. To our knowledge, mice lacking NLRP3 are not reported to show any gross behavioral deficits or alterations in motor activity, though they exhibit attenuated immunological responses to LPS and muramyl dipeptide (Zhang, Z-T et al., 2016; Kovarova et al., 2012). We believe these data show, for the first time, converging evidence that the NLRP3 inflammasome plays a key role in spontaneous sleep and sleep responses following SD and LPS challenge. Compared to controls, mice lacking NLRP3 showed decreased spontaneous NREM sleep, reduced NREM sleep and NREM sleep δ power responses to SD, and failed to increase caspase-1 activity and IL-1β protein in the somatosensory cortex after SD. Moreover, LPS infusion in the lateral ventricle produced differences paralleling those seen with SD.
2 Methods
2.1 Animals
Two-month-old male NLRP3 KO mice (B6.129S6-Nlrp3tm1Bhk, which were backcrossed with C57BL6J mice for 10 generations) and C57BL/6J wild-type (WT) control mice (N = 7-8 per genotype) were obtained from Jackson Laboratories (Bar Harbor, ME, USA) and bred and maintained for 2 generations at the Veterans Affairs (VA) Boston Healthcare System animal resource facilities. Mice were genotyped using primers as indicated by Jackson Laboratories. Mice were housed individually and water and food were provided ad libitum throughout the experiments. Mice were maintained on a 12:12 h light/dark cycle [light onset Zeitgeber (ZT) 0] at 22 ± 3 °C. All experimental protocols were approved by the VA Boston Healthcare system Institutional Animal Care and Use Committee and were in compliance with the National Institutes of Health guidelines.
2.2 Polysomnography surgery and analysis
Mice used for polysomnography surgery were anesthetized with isoflurane (1-2%) and surgically implanted with an EEG electrode in the somatosensory cortex (1 mm posterior to bregma and 1 mm lateral to the midline) and a ground electrode over the cerebellum (0.5 mm posterior to lambda placed centrally) as previously described (Zielinski et al., 2013). Mice were also implanted with two electromyogram (EMG) electrodes in the nuchal muscles. The electrodes were attached to a pedestal and formed into a head mount with dental cement. Mice were tethered to wireless transponders (F20-EET transponders; Data Sciences International, St. Paul, MN, USA) on a swivel mounted system (Neurotargeting Systems, Inc., Chestnut Hill, MA, USA) allowing the mice to move freely in their cage as previously described (Zielinski et al., 2013). The cages were positioned on top of receiver plates that functioned to detect potential data from FM signals from the transponders to a data exchange matrix (Data Sciences International, St. Paul, MN, USA). The signals were transferred to a computer using the Dataquest A.R.T system. The EEG and EMG signals were converted to European data files and then filtered below 0.1 Hz and above 40 Hz.
Polysomnography data were analyzed manually in 10-second epochs off-line using SleepSign Software (Kissei Comtech Co., Ltd., Japan). Vigilance states—including NREM sleep, rapid eye movement (REM) sleep, and waking—were determined in 2-h time blocks as previously described (Zielinski et al., 2013). Vigilance state episode duration and episode frequencies were determined in 12-h light and dark time blocks and during periods of experimental treatments as previously described (Zielinski et al., 2012). Fast Fourier transformations of EEG signals (μV2) using a Hanning window were made within each 10-second epoch for each sleep state. EEG power spectra were analyzed in 0.25 Hz bins in the frequency range of 0.5–20 Hz for 24-h periods. Additionally, NREM sleep δ power spectra in the EEG (0.5–4 Hz) was determined as a percentage of total EEG power spectra across a 24-h period in 2-h time bins for each individual mouse. EEG power spectra analysis between 0.5-20 Hz frequency ranges during the specified duration of the experimental treatment sleep states was normalized as a fractional percentage of total power spectra within the 0.5–20 Hz range of the respective control treatment. EEG power spectra analysis was performed for light and dark periods during for each sleep state during spontaneous sleep and sleep after experimental treatments.
2.3 Experimental treatments
Mice used for polysomnography treatments were allowed to sleep ad libitum, and sleep was recorded for 24 h beginning at dark onset (ZT 12). Mice were continuously sleep deprived by the gentle handling method for 6 h prior to dark onset (ZT 6-12). The 6 h of SD was placed at this time-of-day because IL-1β expression and NREM sleep are at their nadir which enhances the ability to detect SD-induced increases in cortical IL-1β, EEG δ power, and NREM sleep (as previously described Zielinski et al., 2012). Recovery sleep was recorded for 24 h as previously described (Zielinski et al., 2013). Mice were infused intracerebroventricularly (ICV) with LPS from E. coli (L2630, lot 122M9001V, Sigma-Aldrich, St. Louis, MO, USA; 100 ng in 5 μl of 0.9% NaCl), carrier-free IL-1β (R & D Systems, Minneapolis, MN, USA; 10 ng in 5 μl of 0.9% NaCl), or the vehicle (0.9% NaCl per 5 min) for 5 min prior to dark onset (ZT 12), and sleep was recorded for 24 h. All mice received the same LPS batch. Additional mice that were allowed ad libitum sleep were sacrificed for molecular analysis after the active-dark period at light onset (ZT 0) and after the less active light period at dark onset (ZT 12) and served as controls. Further, a group of mice were sleep deprived for 6 h prior to dark onset (post-SD ZT 12) and culled immediately thereafter for molecular analysis.
2.4 Tissue collection
Mice were anesthetized with isoflurane, decapitated, brains removed, and bilateral somatosensory cortices (between bregma at 1.98 mm to -2.46 mm and ± 2 mm in the dorsal ventral directions relative to the parietotemporal ridge) were dissected on a frozen petri dish as previously described (Zielinski et al., 2012). The brain tissue was flash frozen in liquid nitrogen and stored at -80 °C for analysis of protein and gene expression.
2.5 Gene expression analysis
Tissue was homogenized and RNA was extracted with Trizol reagent (Thermo-Fisher Scientific, Waltham, MA, USA) according to the manufacturer's instructions. cDNA was prepared from each RNA sample using a TaqMan Reverse Transcription kit (Applied Biosystems, Foster City, CA, USA) according to the manufacturer's instructions. NLRP3 (Mm00840904_m1), ASC (Mm00445747_g1), and IL-1β (Mm00434228_m1) primers were chosen using Primer Express Software (Applied Biosystems, Foster City, CA, USA). 18s ribosomal RNA (Hs99999901_s1) was used as an internal standard for normalization. Levels of mRNA of the genes of interest were analyzed using real-time polymerase chain reaction (RT-PCR) as previously described (Zielinski et al., 2014). Gene expression values of the genes of interest were used to quantify differences in light vs. dark periods and SD vs. time-of-day matched ad libitum sleeping controls using the cycle threshold (Ct) value method as previously described (Zielinski et al., 2014). Briefly, the change in values were calculated by subtracting the mean 18s Ct value from the control treatment. The gene expression was determined using the formula 2ˆ – (change in Ct for experimental treatment from the baseline control mean) – (change in Ct for the baseline experimental treatment from the baseline control mean).
2.6 Protein analysis
Tissue was homogenized using N-PER (Thermo-Fisher Scientific, Waltham, MA, USA) according to the manufacturer's instructions. Protein amounts were quantified using a bicinchoninic acid protein assay (BCA) kit (Thermoscientific, Waltham, MA, USA) according to the manufacturer's instructions. Equal aliquots containing 100 μg amounts of protein were used to determine caspase-1 activity using an enzyme-linked immunosorbent assay (ELISA) according to the manufacturer's instructions (Biovision Inc., Milpitas, CA, USA). In addition, equal aliquots containing 100 μg amounts of protein were used to determine IL-1β protein levels using an ELISA kit according to the manufacturer's instructions (R & D Systems, Minneapolis, MN, USA; sensitivity 4.8 pg/mL).
2.7 Statistical analysis
One-, two-, and three-way analysis of variance were used to determine statistical significance for sleep and molecular data analysis with IBM SPSS software (Chicago, IL, USA). Independent and paired t-tests were used for post-hoc analysis. Significance was set at p < 0.05. Bonferroni corrections were made for multiple comparisons. Data are presented as means ± standard error of the measurement (SEM).
3 Results
3.1 Spontaneous sleep
To determine the role of NLRP3 gene in spontaneous sleep, we assessed sleep architecture of NLRP3 KO mice using polysomnography. Modest yet significant differences in spontaneous sleep duration and EEG power spectra were found between NLRP3 KO mice and WT controls (Figs 1 and 2).
Fig 1.
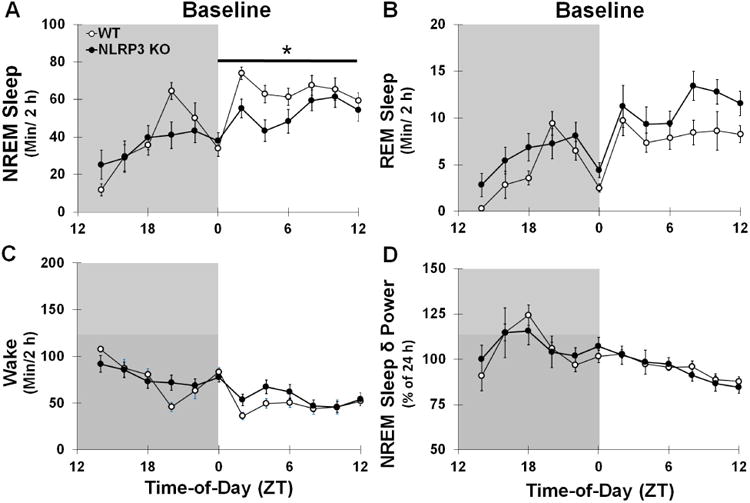
Sleep State (min/2 h time bin) and NREM Sleep δ power (0.5-4 Hz frequency range; percentage normalized to 24 h baseline) during baseline spontaneous sleep in WT and NLRP3 KO mice. Significantly greater amounts of NREM sleep (A) and REM sleep (B) were found during the light periods compared to the dark periods and vice versa for wake (C) and NREM sleep δ power (D) in both genotypes. NLRP3 KO mice exhibited significantly reduced NREM sleep during the light period (ZT 0-12) compared to KO mice, which occurred primarily in the beginning of the light period. N = 8 per genotype. Gray background indicates dark period (ZT 12-0). White background indicates light periods (ZT 0-12). Bar and star indicates significant difference over light period. The Bonferroni corrected α value was set at p < 0.004.
Fig 2.
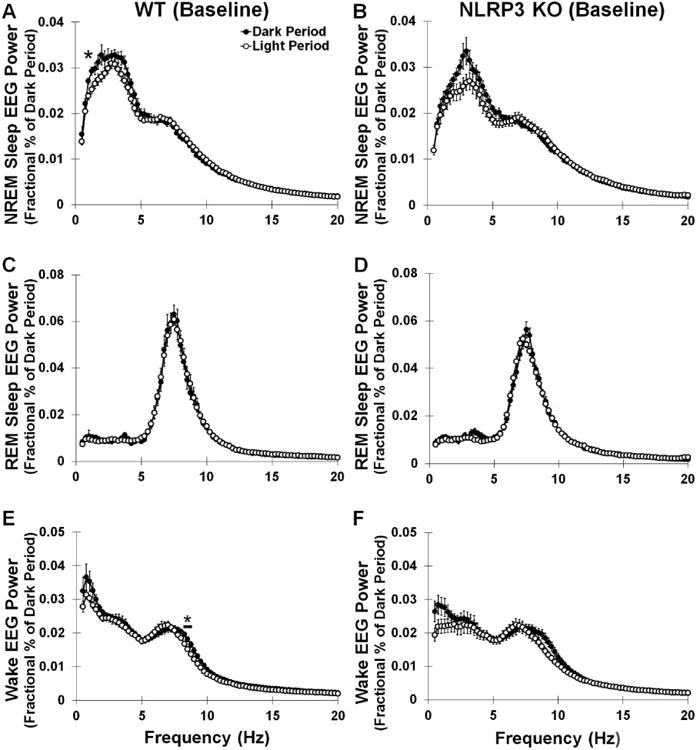
Sleep state power spectra (0.5-20 Hz frequency range) during dark and light periods of baseline spontaneous sleep in WT and NLRP3 KO mice. WT and NLRP3 KO mice demonstrated significantly greater EEG power spectra during NREM sleep and wake dark periods compared to the light periods in both genotypes (A, B, E, F), although little difference was found in REM sleep EEG power spectra (C, D) between light and dark periods for either genotype. However, NLRP3 KO mice exhibited some diurnal differences in particular power spectra bands during these sleep states compared to KO mice [light period vs. dark period: F (1, 553) = 52.007, p < 0.001; light period vs. dark period × frequency interaction: F (78, 553) = 4.750, p < 0.001]. Post-hoc analysis indicated that WT mice had significantly greater NREM sleep EEG power spectra in the 1.25 Hz frequency bands during the dark period vs. the light period. NLRP3 KO mice also exhibited increases in NREM sleep EEG power spectra. Post-hoc analysis also revealed that EEG power spectra during wakefulness was greater in the 8.25-8.75 Hz frequency bands during the dark period vs. light period in WT mice. N = 8 per genotype. Bars and (*) indicates significant difference between genotypes for individual EEG power spectra. The Bonferroni corrected α value was set at p < 0.0006.
3.1.1 NLRP3 KO mice sleep less during light period
Spontaneous NREM and REM sleep exhibited a diurnal pattern in both NLRP3 KO and WT mice with greater durations occurring during the light period compared to the dark period [NREM sleep (Fig 1A): F (1, 14) = 17.157, p = 0.001; WT: 165.3 ± 18.3 min; NLRP3 KO: 106.4 ± 30.8 min; REM sleep (Fig 1B): WT: 25.0 ± 4.5 min; NLRP3 KO: 32.9 ± 6.6 min] occurring during the light period compared to the dark period [F (1, 14) = 14.069, p = 0.002]. The amount of spontaneous wakefulness showed a diurnal rhythm in both NLRP3 KO and WT mice with larger amounts in the dark period vs. the light period [F (1, 14) = 18.347, p = 0.001; WT: 190.4 ± 20.8 min; NLRP3 KO: 139.2 ± 35.7 min] (Fig 1C). Spontaneous NREM and REM sleep values during light and dark periods for WT mice were similar to those previously reported in the literature (Zielinski et al., 2012). However, NLRP3 KO mice had reduced amounts of NREM sleep (-68.7 ± 22.8 min) during the light period compared to that of WT mice [genotype: F (1, 14) = 4.981, p = 0.042].). No significant differences between genotypes were found for sleep episode durations or episode frequencies during light or dark periods (Table 1). However, a main effect was found in NLRP3 KO mice and WT mice for greater NREM sleep, REM sleep, and waking episode frequencies and REM sleep and waking episode durations during the dark period compared to the light period [F (1, 14) = 39.626, p < 0.001; F (1, 14) = 45.990, p < 0.001; F (1, 14) = 20.591, p < 0.001; F (1, 14) = 5.331, p = 0.037; F (1, 14) = 13.852, p = 0.002, respectively]. This contributed to the diurnal variations in sleep state amounts.
Table 1.
Spontaneous sleep state episode durations and episode frequencies.
| WT | NLRP3 KO | |||
|---|---|---|---|---|
|
| ||||
| Dark (ZT 12-0) | Light (ZT 0-12) | Dark (ZT 12-0) | Light (ZT 0-12) | |
| NREM sleep | ||||
| Duration (min/12 h) | 0.93 ± 0.11 | 0.90 ± 0.04 | 0.92± 0.16 | 0.92± 0.12 |
| Frequency (number/12 h) | 261.38 ± 36.30 | 438.00* ± 24.10 | 280.88± 50.34 | 385.88*± 54.42 |
| REM sleep | ||||
| Duration (min/12 h) | 0.43 ± 0.06 | 0.39 ± 0.07 | 0.57± 0.09 | 0.53± 0.08 |
| Frequency (number/12 h) | 59.75 ± 6.66 | 133.00* ± 15.70 | 68.50 ± 12.29 | 136.13*± 15.84 |
| Wake | ||||
| Duration (min/12 h) | 2.35 ± 0.41 | 0.76* ± 0.09 | 2.56± 0.79 | 1.08± 0.12 |
| Frequency (number/12 h) | 241.25 ± 36.19 | 377.13* ± 22.34 | 261.38± 47.68 | 336.50*± 51.52 |
The table shows differences in spontaneous NREM sleep, REM sleep, and wake episode durations and episode frequencies during dark and light periods for WT and NLRP3 KO mice.
indicate significant difference between dark and light periods. N = 8 per genotype. The Bonferroni corrected α value was set at p < 0.03.
3.1.2 NLRP3 KO mice have altered EEG power spectra
Since the NLRP3 inflammasome is a major activator of mature IL-1β which increases NREM sleep EEG δ power, we investigated EEG power spectra frequencies during sleep states of spontaneous sleep and wakefulness. In NLRP3 KO mice and WT mice, a main effect was found for spontaneous NREM sleep EEG δ power spectra (0.5-4 Hz frequency range) being greater during the dark period compared to the light period [F (1, 14) = 5.885, p = 0.029] (Fig 1D). However, this diurnal variation was evident only in WT mice [WT: F (1, 7) = 6.546, p = 0.038]. A main effect was found for WT and NLRP3 KO mice exhibiting greater NREM sleep EEG power spectra (0.5-20 Hz frequency range) during the dark period compared to the light period [light period vs. dark period: F (1, 1106) = 58.635, p < 0.001] (Figs 2A and 2B). This effect varied depending upon the EEG frequency power spectra band and between genotypes [light period vs. dark period × frequency interaction: F (78, 1106) = 5.020, p < 0.001; light period vs. dark period × genotype interaction: F (78, 1106) = 3.348, p < 0.001]. In NLRP3 KO and WT mice, a main effect was observed for greater REM sleep EEG power spectra (0.5-20 Hz frequency range) during the dark period compared to the light period [light period vs. dark period: F (78, 1106) = 1.541, p = 0.002] (Figs 2C and 2D). Wake EEG power spectra values (0.5-20 Hz frequency range) were greater during the dark period compared to the light period in WT and NLRP3 KO mice [light period vs. dark period main effect: F (1, 1106) = 80.510, p < 0.001] (Figs 2E and 2F).
3.1.3 NLRP3 KO mice do not demonstrate greater values of molecular components of the NLRP3 inflammasome pathway during times of high sleep propensity
We found that WT mice exhibited greater caspase-1 activity in the somatosensory cortex at the beginning of the light period (ZT 0) compared to the start of the dark period (ZT 12) [F (1, 14) = 19.271, p = 0.001] (Fig 3A). However, somatosensory cortex caspase-1 activity levels during the beginning of the light and dark periods were similar in NLRP3 KO mice. In the somatosensory cortex, greater inflammasome-related component mRNA expression levels were found at the beginning of the light period compared to the commencement of the dark period in WT mice [NLRP3: F (1, 14) = 16.127, p = 0.001; ASC: F (1, 14) = 11.588, p = 0.004; IL-1β: F (1, 14) = 17.384, p = 0.001] (Table 2). However, NLRP3 KO mice demonstrated similar inflammasome-related component mRNA expression levels at the beginning of the light period compared to the start of the dark period. A main effect was observed for elevated IL-1β protein levels in the somatosensory cortex at the start of the light period (ZT 0) compared to beginning of the dark period (ZT 12) [F (1, 28) = 10.588, p = 0.003] (Fig 3B). Notwithstanding, an interaction was observed between the two mouse genotypes and the photoperiod for IL-1β protein levels [F (1, 28) = 6.237, p = 0.019]. IL-1β protein levels were greater during the light period than the dark period for WT but not NLRP3 mice [WT: F (1, 14) = 12.222, p = 0.004]. WT mice had greater IL-1β protein levels than NLRP3 KO mice [genotype main effect: F (1, 28) = 6.838, p = 0.014], although this effect only occurred during the beginning of the light period [F (1, 28) = 9.797, p = 0.007].
Fig 3.
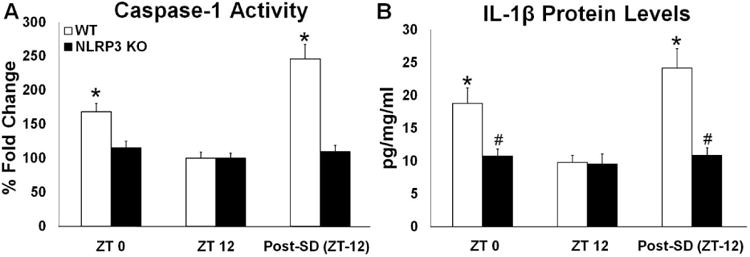
Caspase-1 activity and IL-1β protein levels in the somatosensory cortex during light and dark periods during baseline sleep and post-SD (post-SD). WT but not NLRP3 KO mice exhibited significant enhancements in caspase-1 activity during the light period (ZT 0) compared to the dark period (ZT 12)(A). Caspase-1 activity post-SD was also significantly enhanced in WT but not NLRP3 KO mice when compared to time-of-day matched controls (ZT (12)(A). Similarly, IL-1β protein levels were significantly greater during the light period (ZT 0) compared to the dark period (ZT 12) and post-SD compared to time-of-day matched controls (ZT (12) only in WT control mice (B). N = 8 per genotype. (*) indicates significant difference between baseline and treatment. (#) indicates significant difference between genotypes. The Bonferroni corrected α value was set at p < 0.02.
Table 2.
NLRP3 inflammasome-related gene expression during light and dark periods and after SD.
| WT | NLRP3 KO | |||||
|---|---|---|---|---|---|---|
|
| ||||||
| (ZT 12) | (ZT 0) | Post-SD (ZT 12) | (ZT 12) | (ZT 0) | Post-SD (ZT 12) | |
| NLRP3 | 1.05 ± 0.11 | 1.97* ± 0.10 | 2.88* ± 0.44 | Not detectable | Not detectable | Not detectable |
| ASC | 1.02 ± 0.08 | 1.84* ± 0.20 | 2.55* ± 0.44 | 1.05 ± 0.12 | 1.18# ± 0.16 | 0.96# ± 0.07 |
| IL-1β | 1.05 ± 0.12 | 2.46* ± 0.33 | 2.32* ± 0.28 | 1.05 ± 0.11 | 1.01# ± 0.06 | 1.15# ± 0.17 |
The table displays differences in NLRP3, ASC, and IL-1β gene expression (fold change) after the light period (ZT 12) and after the dark period (ZT 0) and immediately post-SD (post-SD).
indicate significant difference between ZT 12 and ZT 0. N = 8 per genotype.
indicate significant difference between SD [post-SD (ZT 12)] and baseline spontaneous sleep (ZT 12). The Bonferroni corrected α value was set at p < 0.02.
3.2 Responses to SD
Homeostatic enhanced NREM sleep responses to SD were attenuated in NLRP3 KO mice compared to WT controls (Fig 4). Furthermore, NLRP3 KO mice did not demonstrate enhancement of inflammasome-related molecular components within the somatosensory cortex after SD (Fig 3).
Fig 4.
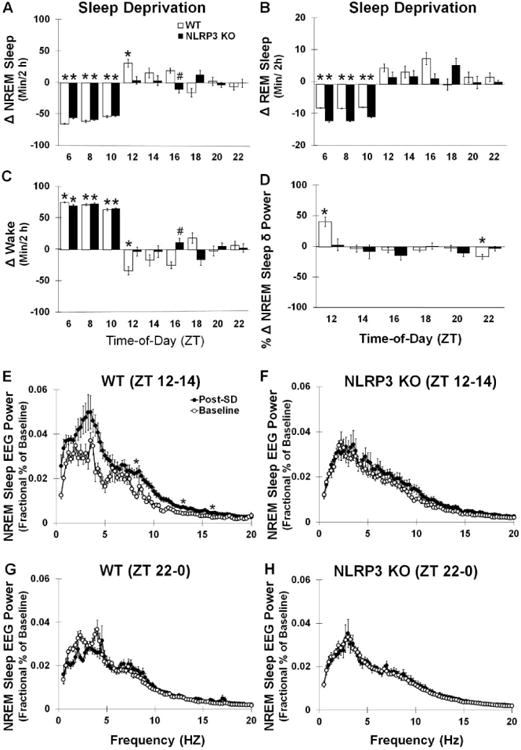
Sleep during SD and recovery sleep responses to SD. Both WT and NLRP3 mice exhibited significant reductions in (A) NREM and (B) REM sleep durations and enhanced (C) waking during the 6 hours of SD occurring prior to dark onset (ZT 6-12). WT mice but not NLRP3 KO mice had significantly enhanced recovery sleep after SD during NREM and REM sleep states. WT mice also exhibited significantly enhanced NREM sleep δ power during the first 2 hours post-SD (ZT 12-14) (D, E). WT and NLRP3 KO mice both exhibited significant enhancements in NREM sleep EEG power spectra (0.5-20 Hz frequency range) post-SD compared to baseline values, although post-hoc analysis only indicated that WT mice had enhancements in the 8.25, 13, and 16 Hz frequency bands. Significant reductions in NREM sleep δ power and NREM sleep EEG power spectra (0.5-20 Hz frequency range) 10-12 h post-SD (ZT 22-0)(i.e., negative rebound) (G) were found in WT mice, which was not found in NLRP3 KO mice (D, F, H). N = 8 per genotype. Bars and (*) indicate significant difference between baseline and treatment. (#) indicates significant difference between genotypes. The Bonferroni corrected α value was set at p < 0.03 for sleep states during sleep deprivation, p < 0.008 for sleep states during recovery sleep, and p < 0.0006 for EEG power frequency bands.
3.2.1 Recovery sleep responses after SD are attenuated in NLRP3 KO mice
SD by gentle handling was effective in both WT and NLRP3 KO mice in preventing NREM and REM sleep during the procedure when compared with baseline spontaneous sleep [NREM sleep (WT): 94.8 ± 2.1 %; (NLRP3 KO): 97.4 ± 1.1 %) (Fig 4A); NREM sleep: F (1, 28) = 377.310, p < 0.001; REM sleep (WT): 98.9 ± 0.6 %; (NLRP3 KO): 93.7 ± 2.1 %) (Fig 4B)]; REM sleep: F (1, 28) = 194.911, p < 0.001]. Conversely, wakefulness was enhanced in both genotypes during the SD procedure when compared with baseline values [F (1, 28) = 441.262, p < 0.001] (Fig 4C).
An interaction was found between the SD treatment and mouse genotypes for NREM sleep duration during the first 6 h of sleep occurring after SD (ZT 12-18, i.e., recovery sleep) [F (1, 28) = 5.898, p = 0.022] (Fig 4A). WT mice showed enhanced NREM sleep duration during recovery sleep compared to baseline values [F (1, 14) = 11.187, p = 0.005; duration change: 63.5 ± 16.0 min]. However, NREM sleep responses during recovery sleep were not significantly different from baseline values in NLRP3 KO mice [-6.2 ± 11.8 min]. The enhancement in NREM sleep duration displayed in WT mice was, in part, the result of an increased frequency of NREM sleep episodes [F (1, 14) = 4.765, p = 0.047; baseline: 96.38 ± 18.15; recovery sleep: 187.88 ± 37.78]. A main effect was found for mice having enhanced REM sleep duration during the first 6 h of recovery sleep compared to their baseline values [F (1, 28) = 5.190, p = 0.031] (Fig 4B). This effect, though, was only seen in WT mice [WT: F (1, 14) = 13.888, p = 0.002; WT: 14.3 ± 2.7 min change; NLRP3 KO: 3.8 ± 3.2 min]. An increased frequency of REM sleep episodes contributed to the enhanced REM sleep durations was found during recovery sleep of WT mice [F (1, 14) = 6.531, p = 0.023; baseline: 15.63 ± 2.58; recovery sleep: 45.88 ± 11.55]. A main effect was found for reduced wakefulness occurring during the first 6 h of recovery sleep after SD compared to baseline values [treatment main effect: F (1, 28) = 5.190, p = 0.031]. This main effect was attributed to reductions in wakefulness found in WT but not NLRP3 KO mice [treatment × genotype interaction: F (1, 28) = 5.871, p = 0.022; WT (treatment): F (1, 14) = 12.594, p = 0.003: WT: -77.8 ± 17.9 min; NLRP3 KO: 2.4 ± 14.5 min] (Fig 4C). In WT mice, the attenuation in wakefulness ensued, in part, from a reduction in the duration of waking episode bouts during sleep recovery compared to baseline [F (1, 14) = 6.034, p = 0.028; baseline: 4.06 ± 0.99 min; recovery sleep: 1.46 ± 0.37 min].
3.2.2 The homeostatic enhancement in NREM sleep delta power after SD is attenuated in NLRP3 KO mice
WT mice exhibited enhanced NREM sleep δ power (0.5-4 Hz frequency range) values during the first 2 h of recovery sleep after SD compared to spontaneous baseline values [WT: F (1, 14) = 11.125, p = 0.005]. This effect was not found in NLRP3 KO mice [treatment × genotype interaction: F (1, 28) = 4.216, p = 0.049] (Fig 4D). A main effect was found for lower NREM sleep δ power 11-12 h after SD compared to baseline values [F (1, 28) = 6.832, p = 0.014]. However, WT mice demonstrated a negative rebound in NREM sleep δ power [WT: F (1, 14) = 11.337, p = 0.005], which was not observed in NLRP3 KO mice [treatment × genotype interaction: F (1, 28) = 8.524, p = 0.007].
To confirm the homeostatic alterations in NREM sleep δ power after SD found in WT mice, we evaluated EEG power spectra (0.5-20 Hz frequency range) during the first 2 h of recovery sleep. A main effect was shown in NLRP3 KO and WT mice for enhanced NREM sleep EEG power spectra during the first 2 h of recovery sleep compared to baseline values [F (1, 1106) = 304.084, p < 0.001](Figs 4E and 4F). This effect depended upon the frequency of the power spectra and the mouse genotype that was, in part, evident in the NREM sleep δ power findings [treatment × frequency × genotype interaction: F (78, 1106) = 1.754, p < 0.001; treatment × genotype interaction: F (1, 1106) = 71.344, p < 0.001; frequency × genotype interaction: F (78, 1106) = 2.105, p < 0.001; treatment × frequency interaction: F (78, 1106) = 2.214, p < 0.001; genotype: F (1, 1106) = 50.582, p < 0.001].
We also determined the NREM sleep EEG power spectra (0.5-20 Hz frequency range) occurring 11-12 h after SD based upon the significant attenuation in NREM sleep δ power exhibited in WT mice. NREM sleep EEG power spectra was different in particular frequency bands and between genotypes that was consistent with the NREM sleep δ power findings [treatment × frequency interaction: F (78, 1106) = 1.943, p < 0.001; genotype × frequency interaction: F (78, 1106) = 1.543, p = 0.002; treatment × frequency interaction: F (78, 553) = 2.522, p < 0.001] (Figs 4G and 4H).
3.2.3 NLRP3 inflammasome-related molecular components are enhanced in the somatosensory cortex after SD
Immediately after SD (ZT 12), caspase-1 activity in the somatosensory cortex was enhanced relative to baseline time-of-day (ZT 12) controls in WT but not NLRP3 KO mice [WT: F (1, 14) = 30.029, p < 0.001] (Fig 3A). In WT mice, NLRP3, ASC, and IL-1β mRNA expression in the somatosensory cortex was increased after SD compared to baseline values [NLRP3: F (1, 14) = 16.127, p = 0.001; ASC: F (1, 14) = 11.588, p = 0.004, IL-1β: F (1, 14) = 17.384, p = 0.001] (Table 2). NLRP3 mice did not exhibit significant SD-induced differences in the expression of the measured NLRP3 inflammasome-related genes. Main effects were found for greater IL-1β protein levels in WT than NLRP3 KO mice and for SD enhancing IL-1β protein levels [genotype: F (1, 28) = 13.549, p = 0.001; treatment; F (1, 28) = 13.549, p = 0.001] (Fig 3B). NLRP3 KO mice did not show significant increased IL-1β protein levels after SD. This SD-induced effect was only found in WT mice [treatment × genotype interaction: F (1, 28) = 12.819, p = 0.001; WT (treatment): F (1, 14) = 20.537, p < 0.001].
3.3 Responses to LPS
To determine the role of NLRP3-induced sleep by the somnogenic bacterial cell wall component LPS in the brain, we determined sleep architecture responses after ICV infusions of LPS. First, we analyzed sleep responses after ICV infusions of the vehicle at dark onset (ZT 0) (Fig 5). Following ICV vehicle infusions, NLRP3 KO mice demonstrated reduced sleep over a 24 h period compared to WT mice [F (1, 26) = 9.629, p = 0.005], with lower amounts occurring during both the light (ZT 0-12) and dark periods (ZT 12-0) [light period: F (1, 26) = 6.058, p = 0.021; -58.8 ± 22.1 min difference; dark period: F (1, 26) = 4.892, p = 0.036; 39.2 ± -14.2 min difference]. NLRP3 KO mice exhibited greater REM sleep duration during the dark period compared to WT mice (ZT 12-0) [F (1, 26) = 13.786, p = 0.001; 14.1 ± 3.4 min difference]. However, REM sleep duration during the light period and during wakefulness were not different between genotypes. Second, we determined sleep and NREM sleep EEG δ power spectra responses to ICV applied LPS, which were attenuated in NLRP3 KO mice compared to WT controls.
Fig 5.
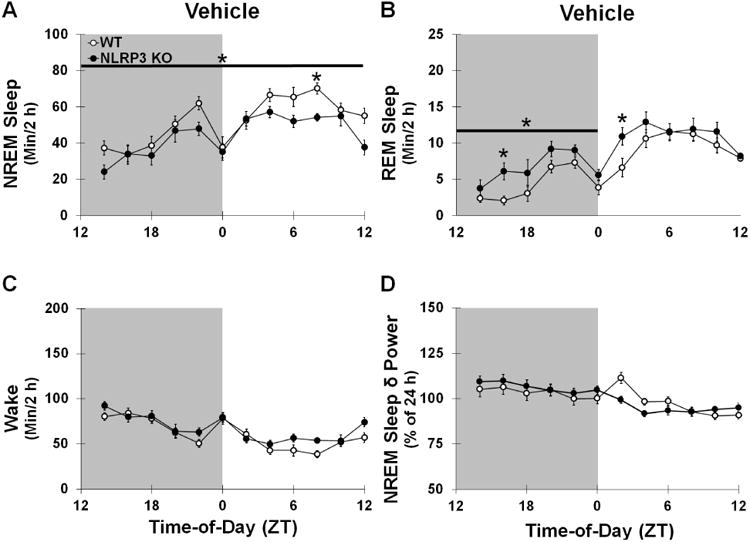
Sleep State (min/2 h time bin) and NREM Sleep δ power (0.5-4 Hz frequency range; percentage normalized to 24 h baseline) after ICV infusion of the vehicle. Significantly greater amounts of NREM sleep (A) and REM sleep (B) were found during the light periods compared to the dark periods and vice versa for wake (C) and NREM sleep δ power (D) in both genotypes. NLRP3 KO mice exhibited significantly reduced NREM sleep during light and dark periods and greater REM sleep during the dark period compared to WT control mice. Gray background indicates dark period (ZT 12-0). White background indicates light periods (ZT 0-12). N = 14 per genotype. (*) indicates significant difference between genotypes. Bar and star indicates significant difference over light and/or dark periods. The Bonferroni corrected α value was set at p < 0.004.
3.3.1 Enhanced sleep responses after ICV infusions of LPS are attenuated in NLRP3 KO mice
An interaction was observed between the NLRP3 KO and WT mouse genotypes and ICV LPS and between vehicle and LPS treatment responses for NREM sleep during the first 12 h post-LPS infusion [treatment × genotype interaction: F (1, 38) = 17.055, p < 0.001] (Fig 6A). WT mice exhibited enhanced NREM sleep during the first 12 h post-LPS infusion compared to that of the vehicle [F (1, 19) = 11.851, p = 0.003; WT: 111.6 ± 38.6 min]. Nevertheless, there were no significant differences in NREM sleep episode frequencies or episode durations after LPS infusions compared to the vehicle. NREM sleep responses to LPS were not significantly different from the vehicle for NLRP3 KO mice and displayed a slight but non-significant reduction in NREM sleep [NLRP3 KO: -45.5 ± 14.4 min]. A genotype × treatment interaction was found for the amount of REM sleep during the first 12 h post-LPS infusion compared to vehicle values for NLRP3 KO and WT mice [F (1, 38) = 8.782, p = 0.005] (Fig 6B). Yet, post-hoc analysis indicated that LPS did not significantly alter REM sleep durations in either genotype (WT: 9.2 ± 6.3 min; NLRP3 KO: -11.8 ± 2.1 min). The enhanced NREM sleep duration responses seen in WT mice contributed to a reduction in their wakefulness that was only found in WT mice [treatment × genotype main interaction: F (1, 38) = 17.803, p < 0.001; WT (treatment): F (1, 19) = 11.346, p = 0.003; WT: -120.8 ± 42.0 min; NLRP3 KO: 57.4 ± 16.1 min] (Fig 6C). The reduced wakefulness seen in WT was, in part, attributed to LPS-induced reductions in the durations of waking episode bouts [F (1, 19) = 7.598, p = 0.013; WT (vehicle): 1.31 ± 0.15; WT (post-LPS): 0.67 ± 0.11 min].
Fig 6.
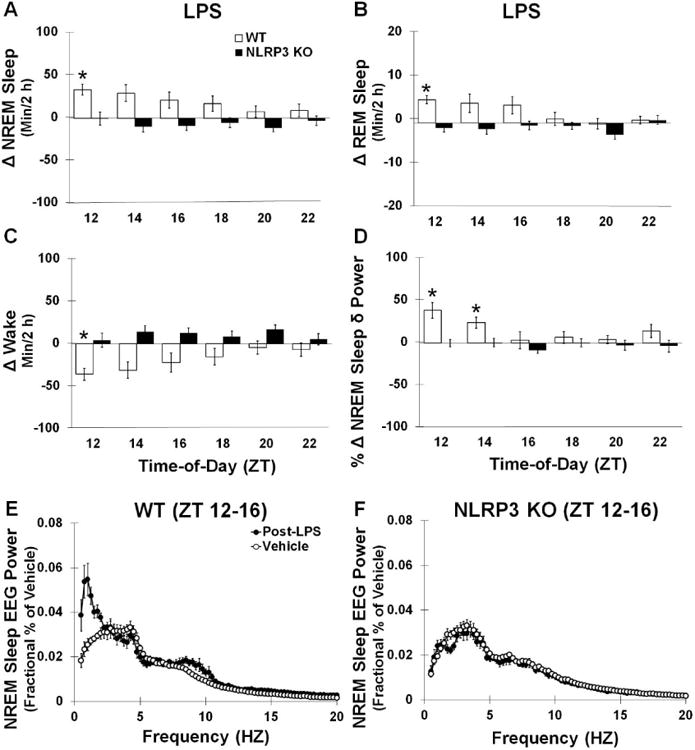
Post-LPS infusion sleep responses. WT mice but not NLRP3 KO mice had significantly enhanced NREM and reduced wakefulness (min/2 h) amounts post-LPS infusion compared to the vehicle (A). REM sleep amounts (min/2 h) were not significantly different post-LPS infusion for either genotypes (B), although reduced wakefulness post-LPS was observed in WT mice but not NLRP3 KO mice (C). NLRP3 KO mice also did not show the significant enhancements in NREM sleep δ power during the first 4 hours post-LPS infusion (ZT 12-16) as was found in WT mice (D, E, F). However, post-hoc analysis did not show significant differences at individual frequency bands. N = 7 per genotype. (*) indicates significant difference between baseline and treatment. (#) indicates significant difference between genotypes. The Bonferroni corrected α value was set at p < 0.03 for sleep states during recovery sleep and p < 0.0006 for EEG power frequency bands.
3.3.2 EEG delta power spectra enhancements after ICV infusions of LPS are attenuated in NLRP3 KO mice
Since LPS can affect NREM sleep EEG δ power, we investigated this measure after ICV infusions in NLRP3 KO and WT mice. A main effect was found for enhanced NREM sleep δ power spectra (0.5-20 Hz frequency range) during the first 4 h (ZT 12-15) post-LPS infusion compared to vehicle values and for greater NREM sleep δ power spectra in WT mice compared to NLRP3 KO mice [treatment: F (1, 24) = 7.031, p = 0.014; genotype: F (1, 24) = 4.714, p = 0.040] (Fig 6D). An interaction was observed between mouse genotypes and LPS and vehicle treatments for NREM sleep δ power spectra where WT but not NLRP3 KO mice exhibited enhancements in NREM sleep δ power spectra [genotype × treatment interaction: F (1, 24) = 11.381, p = 0.003; WT: F (1, 12) = 10.925, p = 0.006].
We further determined the NREM sleep EEG power spectra (0.5-20 Hz frequency range) during the first 4 h (ZT 12-15) post-LPS infusion compared to vehicle when NREM sleep δ power spectra was significantly enhanced in WT mice. A main effect was observed for LPS enhancing NREM sleep EEG power spectra in responses to LPS in both WT and NLRP3 KO mice [treatment: F (1, 948) = 36.384, p < 0.001] (Figs 6E and 6F). Additionally, a main effect was found for greater NREM sleep EEG power in WT mice compared to NLRP3 KO mice. However, NREM sleep EEG power responses to infusions of LPS differed in particular frequency bands depending upon the mouse genotype which was in accord with the NREM sleep δ power results [treatment × frequency × genotype: F (78, 948) = 2.995, p < 0.001; treatment × frequency: F (78, 948) = 4.797, p < 0.001; frequency × genotype; F (78, 948) = 4.953, p < 0.001; treatment × genotype: F (1, 948) = 127.491, p < 0.001].
3.4 Responses to IL-1β
As previously mentioned, IL-1β activation is located downstream of the NLRP3 inflammasome protein complex formation (Elliott and Sutterwala, 2015). Thus, we investigated the sleep and EEG power responses of NLRP3 KO mice after ICV infusions of IL-1β. Largely, NLRP3 KO mice sleep and EEG power spectra responses to IL-1β infusions were similar to WT controls (Fig 7).
Fig 7.
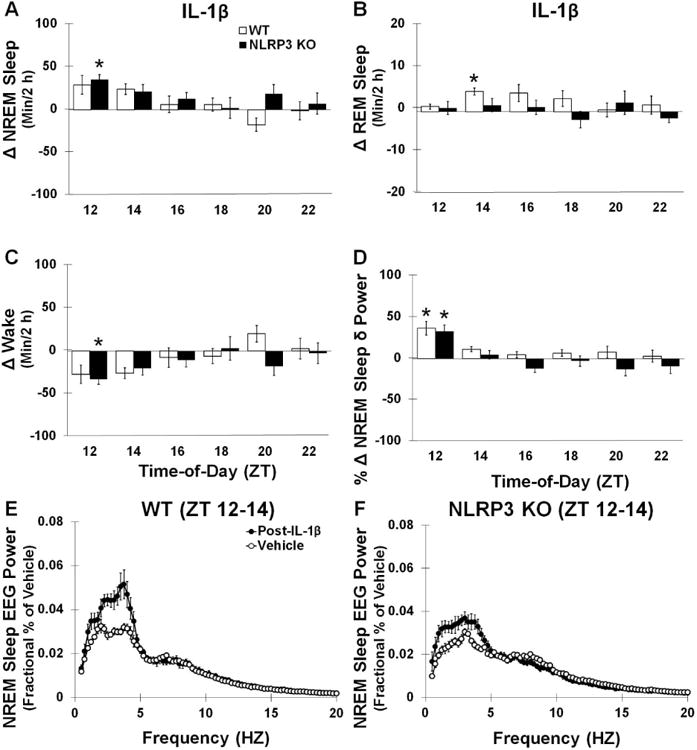
Post-IL-1β infusion sleep responses. Both WT and NLRP3 mice showed significantly enhanced NREM sleep post- IL-1β compared to the vehicle (A). However, WT mice exhibited significantly enhanced REM sleep responses 4-6 h post-infusion (ZT 14-16) which was not found in NLRP3 KO mice (B). NLRP3 KO mice exhibited significant enhancements in wakefulness during the first 2 h post-IL-1β (C). Enhancements in NREM sleep δ power spectra were also found in the first 2 h post-infusion in both WT and NLRP3 KO mice (D, E, F). (*) indicates significant difference between baseline and treatment. However, post-hoc analysis did not show significant differences at individual frequency bands. N = 7 per genotype. (#) indicates significant difference between genotypes. The Bonferroni corrected α value was set at p < 0.03 for sleep states during recovery sleep and p < 0.0006 for EEG power frequency bands.
3.4.1 Both NLRP3 KO mice and WT mice exhibit enhanced NREM sleep after ICV infusions of IL-1β
In both NLRP3 KO and WT mice, ICV infusions of IL-1β enhanced NREM sleep amounts in the first 6 h post-IL-1β compared to vehicle infusions [treatment: F (1, 38) = 11.119, p = 0.002; WT: 56.6 ± 14.4 min; NLRP3 KO: 66.3 ± 17.2 min]. There were no significant differences observed between genotypes (Fig 7A). WT mice showed reductions in the frequency of NREM sleep episodes after IL-1β infusions [F (1, 19) = 8.119, p = 0.010; vehicle: 194.79 ± 21.61; post-IL-1β: 105.29 ± 9.82] and enhancement in the duration of NREM sleep episode bouts that produced the enhanced amount of NREM sleep [F (1, 19) = 95.260, p < 0.001; vehicle: 0.60 ± 0.05 min; post-IL-1β: 1.60 ± 0.10 min]. However, NLRP3 KO mice did not show any significant difference in NREM sleep episode frequencies or episode durations after IL-1β administration compared with that after the vehicle. During the first 6 h after administration no significant main effect was found for IL-1β altering REM sleep durations in either NLRP3 KO mice or WT controls compared with the responses to the vehicle (WT: 7.5 ± 2.7 min; NLRP3 KO: 0.5 ± 4.0 min) (Fig 7B), although WT mice exhibited a significant enhancement in REM sleep during the first 2 h post-IL-1β administration. IL-1β infusions induced a reduction in wakefulness in both NLRP3 KO and WT mice compared to vehicle infusions [treatment: F (1, 38) = 9.843, p = 0.003; WT: -64.1 ± 14.3 min; NLRP3 KO: -66.9 ± 17.8 min] (Fig 7C). WT mice had reductions in the frequency of wake episodes after IL-1β treatments which, in part, contributed to the reductions in waking found after IL-1β infusions compared to the vehicle treatment [F (1, 19) = 7.862, p = 0.011; vehicle: 191.50 ± 21.61; post-IL-1β: 103.14 ± 8.58]. Nevertheless, NLRP3 KO mice did not show significant changes in waking episode frequencies or episode durations after IL-1β administration compared to with responses after the vehicle.
3.4.2 Both NLRP3 KO mice and WT mice exhibit enhanced NREM sleep EEG delta power after ICV infusions of IL-1β
IL-1β increased NREM sleep δ power spectra (0.5-4 Hz frequency range) in both NLRP3 KO and WT mice during the first 2 h after IL-1β treatment compared to the vehicle treatment [treatment: F (1,24) = 36.798, p < 0.001] (Fig 7D), although there were no significant differences in NREM sleep δ power spectra detected between genotypes.
We further determined NREM sleep EEG power spectra (0.5-20 Hz frequency range) during the first 2 h after IL-1β treatment when NREM sleep δ power was elevated in both genotypes. NREM sleep EEG power spectra was enhanced in both WT and NLRP3 KO mice in responses to IL-1β infusions compared to the vehicle [treatment: F (1, 948) = 120.133, p < 0.001] (Figs 7E and 7F). Several interactions were observed indicating that NLRP3 KO and WT genotypes responses to IL-1β differed within the power spectra analyzed yet were consistent with the NREM sleep δ power spectra findings [treatment × frequency interaction: F (78, 948) = 8.806, p < 0.001; frequency × genotype interaction; F (78, 948) = 3.738, p < 0.001; treatment × genotype interaction: F (1, 948) = 18.376, p < 0.001].
4 Discussion
Herein, we report a confluence of evidence that the NLRP3 inflammasome is a primary mechanism involved in spontaneous sleep and sleep responses after SD. NLRP3 KO mice exhibited less spontaneous NREM sleep during the light period vs. dark period and attenuated homeostatic sleep responses after SD. Greater values in the expression of the inflammasome-related components, IL-1β protein, and caspase-1 activity were found in the somatosensory cortex during times of higher sleep propensity in controls but not NLRP3 KO mice. Furthermore, NLRP3 KO mice demonstrated a lack of enhancement in NREM sleep responses to central infusions of the potent somnogenic bacterial component LPS indicating that NLRP3 inflammasomes, which are found in neurons and glia, further indicating NLRP3 inflammasomes in sleep modulation (Walsh et al., 2014; Kawana et al., 2013; Gustin et al., 2015). Moreover, sleep and EEG effects were enhanced in both NLRP3 KO and WT mice after ICV IL-1β infusions, suggesting that the NLRP3 inflammasome pathway is involved in sleep regulation, in part, through the activation of IL-1β in the brain. Collectively, these results suggest that the NLRP3 inflammasome is an important mechanism involved in sleep responses to sleep loss and components of pathogens in the brain (Fig 8).
Fig 8.
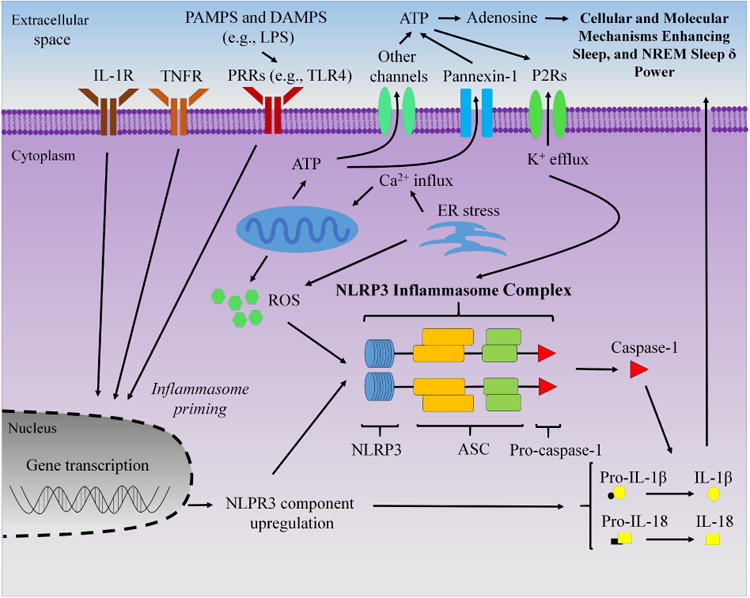
Schematic of proposed role of the non-conical NLRP3 inflammasome in neurons and glia in sleep regulation. Extracellular adenosine triphosphate (ATP) and pathogen associated molecular patterns (PAMPS) activate purine type 2 receptors (P2Rs) and pattern recognition receptors (PRRs) [e.g., Toll-like receptor 4 (TLR4) for lipopolysaccharide (LPS)], respectively, to activate the NLRP3 inflammasome protein complex. Additionally, mitochondria and endoplasmic reticulum (ER) stress likely play a role to activate the NLRP3 inflammasome, in part, through reactive oxygen species (ROS). Upon priming by LPS, interleukin-1 receptor (IL-1R) or the tumor necrosis factor receptor (TNFR), the NLRP3 inflammasome complex forms with ASC [apoptosis-associated speck-like protein containing a C-terminal caspase-recruitment domain (CARD)] to activate caspase-1, which cleaves the pro-forms of IL-1β and IL-18 into their mature active secreted forms to enhance sleep and non-rapid eye movement (NREM) sleep δ power. Arrows indicate activation or enhancement.
Mice are nocturnal and typically demonstrate a diurnal pattern of NREM and REM sleep with greater amounts happening during the more sedentary light period compared to the more active dark period (Zielinski et al 2011). The literature indicates that greater amounts of sleep during the light period vs. dark period corresponds with increased cortical expression and protein levels of IL-1β in rodents (Krueger et al., 2010; Taishi et al., 2012), but this was lacking in NLRP3 KO mice. This suggests that the NLRP3 inflammasome is involved in this cortical IL-1β and sleep pattern. Also supporting the involvement of the NLRP3 inflammasome in sleep regulation, rats given the caspase-1 inhibitor Ac-Tyr-Val-Ala-Asp chloromethyl ketone centrally at the start of the light period experience reduced NREM sleep rebound (Imeri et al., 2006). Our results showing attenuated homeostatic NREM sleep responses after SD in NLRP3 KO mice further suggest that increased wakefulness promotes sleep through the activation of the NLRP3 inflammasome.
NLRP3 KO mice lacked increases in NLRP3 inflammasome-related molecular components within the somatosensory cortex during the beginning of the light period vs. dark period and after SD. Nevertheless, enhancements in the expression of ASC and caspase-1 are common to inflammasomes and cannot be excluded for their involvement in sleep-loss-mediated changes. However, that NLRPR3 KO mice did not show enhancements in these common inflammasome-related components after SD suggests that the NLRP3 inflammasome is the primary contributing inflammasome. While the processing of IL-1β into its mature form can be accomplished by non-inflammasome-mediate mechanisms (Burm et al., 2015), NLRP3 KO mice did not exhibit increased caspase-1 activity or IL-1β protein levels in the somatosensory cortex after SD as was found in the control mice. This suggests that the NLRP3 inflammasome is a primary mechanism in IL-1β mediated sleep regulation induced by increased wakefulness. However, IL-18, which was not investigated in this study, is also activated into its mature form by caspase-1 and can promote sleep (Elliott and Sutterwala, 2015; Kubota et al., 2001). Therefore, IL-18 could also be involved in the observed attenuations in sleep responses to somnogenic stimuli we found in NLRP3 KO mice.
Our current findings that NLRP3 KO mice have attenuated NREM sleep and NREM sleep δ power spectra responses central administration of LPS suggests that these effects occur, in part, from the activation of the NLRP3 inflammasome. However, LPS can stimulate other pathways and molecules known to affect sleep, including nuclear factor-kappa B and tumor necrosis factor-alpha (TNF-α) (Zielinski et al., 2011). Interestingly, since enhanced sleep after infection can be beneficial in resolving infection (Besedovsky et al., 2012), our findings suggest that NLRP3 inflammasome activation in the cortex might have evolved as a regulatory pathway to enhance sleep to protect the host during brain infection. Differences likely exist, though, in sleep responses depending upon the pathogen component and the activation of non-canonical and canonical NLRP3 inflammasome pathways (Rivers-Auty and Brough, 2015). For example, LPS occurring from intracellular bacteria that reaches the cytosol of brain cells could potentially activate caspase 11 in mice or caspase 4 and 5 in humans providing an alternative conical inflammasome activation mechanism that enhances sleep in conditions such as meningitis.
NLRP3 inflammasome activation is complex and involves a multitude of mechanisms that are currently not well understood (Elliott and Sutterwala, 2015). The NLRP3 inflammasome appears to require a priming step for transcriptional activation such as the activation of a PRR, interleukin-1 receptor, or tumor necrosis factor receptor. In addition, a potential mechanism of activation by NLRP3 inflammasomes in the brain from sleep loss and LPS are from mitochondrial enhancement of cellular reactive oxygen species (ROS). A growing literature suggests that mitochondrial pathways, the endoplasmic reticulum, and related components are largely required for inflammasome signaling. Indeed, evidence indicates that metabolism and mitochondria are altered after sleep loss and inflammatory-related conditions associated with disturbed sleep, such as sleep apnea and cardiovascular disease (Naidoo, 2012; Gileles-Hillel et al., 2016).
Energy-related systems such as ATP and adenosine are involved in the homeostatic sleep response (Dworak et al., 2010; 2011; Krueger et al., 2010; Brown et al., 2012; Porkka-Heiskanen et al., 1997). Extracellular ATP increases in mouse basal forebrain following SD (Kalinchuk et al., 2015), whereas studies in rats showed that ATP primarily contained within the intracellular space decreases in brain areas including the frontal cortex, basal forebrain, cingulate cortex, and hippocampus following SD (Dworak et al., 2010, 2011). We note that LPS can also enhance the release of ATP into the extracellular space (Carta et al., 2015) and thus could potentially contribute to the activation of NLRP3-mediated sleep responses observed in the current study. Rats given purine type 2 analogs exhibit increased NREM sleep and NREM sleep δ power (Krueger et al. 2012), while opposing effects are found after either the P2R antagonists. Moreover, mice lacking the P2X7 receptor exhibit reduced NREM sleep and NREM sleep δ power spectra responses to SD (Krueger et al., 2010). The NLRP3 inflammasome appears to be activated by ATP-induced potassium (K+) efflux (Rivers-Auty and Brough, 2015), and high extracellular K+ pannexin 1 channel activates the inflammasome in neurons and astrocytes (Silverman et al., 2009). Animal KO studies and studies using cell slices suggest that potassium efflux modulates sleep and NREM sleep δ power (Douglas et al., 2007; Espinosa et al., 2004; Joho et al., 1999; Ding et al., 2016), which is in agreement with a role of the inflammasome in modulating sleep and NREM sleep δ power. Consequently, our findings and the literature support the idea that the NLRP3 inflammasome could be activated by energy-related mechanisms.
The cerebral cortex projects to brain regions that modulate NREM sleep including the thalamus (Jones et al., 2009; Brown et al., 2012). NLRP3 activity in additional brain areas might provide important information regarding the mechanisms of how inflammation alters sleep for particular pathologies. Since NLRP3 inflammasomes are found in neurons, astrocytes, and microglia (Walsh et al., 2014; Kawana et al., 2013; Gustin et al., 2015), the activation of particular cell types expressing the NLRP3 inflammasome likely contributes to the different sleep phenotypes that are found after SD and LPS. Indeed, a recent study demonstrated that administration of systemic LPS to selectively inhibit IL-1 receptor 1 on neurons suppresses REM sleep and NREM sleep δ power spectra while and IL-1 receptor 1 inhibition in astrocytes affects sleep fragmentation (Ingiosi and Opp, 2016). IL-1β effects on sleep, however, appear to be confined mostly to NREM sleep (Zielinski et al., 2011). The ambiguity of this effect is, in part, because of our current lack of understanding of precise mechanisms governing these two sleep states and their interaction.
Our findings indicate that NLRP3 KO mice exhibit similar diurnal differences in EEG power spectra as control mice with only small differences in particular frequency bands, but any physiological or behavioral effects of this are presently unclear. That NLRP3 KO mice had attenuated enhancements in NREM sleep δ power spectra after both SD and centrally given LPS suggest that these EEG effects are, in part, IL-1β-derived from NLRP3 inflammasome activation. SD can induce an attenuation in NREM sleep δ power spectra several hours after an initial enhancement and is referred to as a negative rebound in NREM sleep δ power (Rechtschaffen et al., 1999). The mechanism for this negative rebound is currently unknown. The lack of negative rebound in NREM sleep δ power observed in NLRP3 KO mice after SD could be compensatory due to the attenuated enhancement in NREM sleep δ power—a pattern that is similarly found in mice lacking both the IL-1 receptor 1 and TNF receptor 1 (Baracchi and Opp, 2008). However, mice lacking only the IL-1 receptor 1 exhibit enhancements in NREM sleep δ power after SD suggesting that the negative rebound in NREM sleep δ power is related to molecules other than IL-1β (Schmidt and Wisor, 2012). Nevertheless, NLRP3 is an inducible complex whereas the aforementioned transgenic mice are constitutively lacking IL-1β which could contribute to these differences.
In conclusion, our novel findings implicate the NLRP3 inflammasome in modulating sleep and NREM sleep δ power induced by spontaneous wakefulness, SD and the bacterial component LPS within the brain. Evidence indicates that IL-1β expression is enhanced in the brain with the sleep disorder breathing condition of obstructive sleep apnea and inflammatory-related pathologies (Zielinski and Krueger, 2011). Because of this, it is plausible that inflammasome activation in the brain are involved. Furthermore, it is also possible that inflammasomes are activated in the periphery in inflammatory-related conditions and/or sleep loss to enhance pro-inflammatory molecules to alter sleep and NREM sleep δ power. Thus, the NLRP3 inflammasome appears to be an important mechanism in inflammation-mediated sleep and could be a potential therapeutic target for individuals with sleep-altering pathologies.
Highlights.
Mice lacking NLRP3 have reduced sleep and EEG delta power after sleep loss
Mice lacking NLRP3 have reduced sleep and EEG delta power after LPS
NLRP3 inflammasome components are elevated in the cortex after sleep loss
Mice lacking NLRP3 have typical sleep and EEG delta power after interleukin-1 beta
Acknowledgments
This work was supported by the Department of Veterans Affairs Medical Research Service Award 2I01BX001404 (RB), Department of Veterans Affairs Medical Research Service Award I01BX002774 (RS) and Department of Veterans Affairs Career Award IBX002823A (MRZ) NIMH-MH016259 fellowship (MRZ), NINDS-NS079866 (RB), NINDS-NS064193 (DG), NIMH-MH039683 (RWM), NHLBI-HL060292 and HL095491 (RWM), NIAID-AI118719 (FSS).
Footnotes
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baracchi R, Opp MR. Sleep-wake behavior and responses to SD of mice lacking both interleukin-1 beta receptor 1 and tumor necrosis factor-alpha receptor 1. Brain Behav Immun. 2008;22:982–93. doi: 10.1016/j.bbi.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besedovsky L, Lange T, Born J. Sleep and immune function. Pflugers Arch. 2012;463:121–37. doi: 10.1007/s00424-011-1044-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown RE, Basheer R, McKenna JT, Strecker RE, McCarley RW. Control of sleep and wakefulness. Physiol Rev. 2012;92:1087–187. doi: 10.1152/physrev.00032.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burm SM, Zuiderwijk-Sick EA, 't Jong AEJ, van der Putten C, Veth J, Kondova I, Bajramovic JJ. Inflammasome-induced IL-1β secretion in microglia is characterized by delayed kinetics and is only partially dependent on inflammatory caspases. J Neuroci. 2015;35:678–87. doi: 10.1523/JNEUROSCI.2510-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carta S, Penco F, Lavieri R, Martini A, Dinarello CA, Gattorno M, Rubartelli A. Cell stress increases ATP release in NLRP3 inflammasome-mediated autoinflammatory diseases, resulting in cytokine imbalance. Proc Natl Acad Sci U S A. 2015;112:2835–40. doi: 10.1073/pnas.1424741112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis CJ, Clinton JM, Jewett KA, Zielinski MR, Krueger JM. Delta wave power: an independent sleep phenotype or epiphenomenon? J Clin Sleep Med. 2011;7:S16–8. doi: 10.5664/JCSM.1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding F, O'Donnell J, Xu Q, Kang N, Goldman N, Nedergaard M. Changes in the composition of brain interstitial ions control the sleep-wake cycle. Science. 2016;352:550–5. doi: 10.1126/science.aad4821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas CL, Vyazovskiy V, Southard T, et al. Sleep in Kcna2 knockout mice. BMC Biol. 2007;5:42. doi: 10.1186/1741-7007-5-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dworak M, Kim T, McCarley RW, Basheer R. Sleep, brain energy levels, and food intake: Relationship between hypothalamic ATP concentrations, food intake, and body weight during sleep-wake and SD in rats. Somnologie (Berl) 2011;15:111–7. doi: 10.1007/s11818-011-0524-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dworak M, McCarley RW, Kim T, Kalinchuk AV, Basheer R. Sleep and brain energy levels: ATP changes during sleep. J Neurosci. 2010;30:9007–16. doi: 10.1523/JNEUROSCI.1423-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott EL, Sutterwala FS. Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol Rev. 2015;265:35–52. doi: 10.1111/imr.12286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa F, Marks G, Heintz N, et al. Increased motor drive and sleep loss in mice lacking Kv3-type potassium channels. Genes Brain Behav. 2004;3:90–100. doi: 10.1046/j.1601-183x.2003.00054.x. [DOI] [PubMed] [Google Scholar]
- Gileles-Hillel A, Kheirandish-Gozal L, Gozal D. Biological plausibility linking sleep apnoea and metabolic dysfunction. Nat Rev Endocrinol. 2016;12:290–8. doi: 10.1038/nrendo.2016.22. [DOI] [PubMed] [Google Scholar]
- Gustin A, Kirchmeyer M, Koncina E, Felten P, Losciuto S, Heurtaux T, Tardivel A, Heuschling P, Dostert C. NLRP3 Inflammasome Is Expressed and Functional in Mouse Brain Microglia but Not in Astrocytes. PLoS One. 2015;10:e0130624. doi: 10.1371/journal.pone.0130624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry CJ, Huang Y, Wynne A, Hanke M, Himler J, Bailey MT, Sheridan JF, Godbout JP. Minocycline attenuates lipopolysaccharide (LPS)-induced neuroinflammation, sickness behavior, and anhedonia. J, Neuroinflammation. 2008;5:15. doi: 10.1186/1742-2094-5-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry CJ, Huang Y, Wynne AM, Godbout JP. Peripheral lipopolysaccharide (LPS) challenge promotes microglial hyperactivity in aged mice that is associated with exaggerated induction of both pro-inflammatory IL-1beta and anti-inflammatory IL-10 cytokines. Brain Behav Immun. 2009;23:309–17. doi: 10.1016/j.bbi.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imeri L, Bianchi S, Opp MR. Inhibition of caspase-1 in rat brain reduces spontaneous nonrapid eye movement sleep and nonrapid eye movement sleep enhancement induced by lipopolysaccharide. Am J Physiol Regul Integr Comp Physiol. 2006;291:R197–204. doi: 10.1152/ajpregu.00828.2005. [DOI] [PubMed] [Google Scholar]
- Imeri L, Opp MR. How (and why) the immune system makes us sleep. Nat Rev Neurosci. 2009;10:199–210. doi: 10.1038/nrn2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingiosi AM, Opp MR. Sleep and immunomodulatory responses to systemic lipopolysaccharide in mice selectively expressing interleukin-1 receptor 1 on neurons or astrocytes. Glia. 2016;64:780–91. doi: 10.1002/glia.22961. [DOI] [PubMed] [Google Scholar]
- Joho RH, Ho CS, Marks GA. Increased gamma- and decreased delta-oscillations in a mouse deficient for a potassium channel expressed in fast-spiking interneurons. J Neurophysiol. 1999;82:1855–64. doi: 10.1152/jn.1999.82.4.1855. [DOI] [PubMed] [Google Scholar]
- Jones EG. Synchrony in the interconnected circuitry of the thalamus and cerebral cortex. Ann N Y Acad Sci. 2009;1157:10–23. doi: 10.1111/j.1749-6632.2009.04534.x. [DOI] [PubMed] [Google Scholar]
- Kawana N, Yamamoto Y, Ishida T, et al. Reactive astrocytes and perivascular macrophages express NLRP3 inflammasome in active demyelinating lesions of multiple sclerosis necrotic lesions of neuromylelitis optica and cerebral infarction. Clinical and Experimental Neuroimmunology. 2013;4:296–304. [Google Scholar]
- Kalinchuk AV, Zant J, Haydon P, McCarley RW, Basheer R. Sleep homeostasis, adenosine and glial cells. Society for Neuroscience; Chicago: 2015. [Google Scholar]
- Krueger JM, Taishi P, De A, Davis CJ, Winters BD, Clinton J, Szentirmai E, Zielinski MR. ATP and the purine type 2 ×7 receptor affect sleep. J Appl Physiol (1985) 2010;109:1318–27. doi: 10.1152/japplphysiol.00586.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger JM, Walter J, Dinarello CA, Wolff SM, Chedid L. Sleep-promoting effects of endogenous pyrogen (interleukin-1) Am J Physiol. 1984;246:R994–9. doi: 10.1152/ajpregu.1984.246.6.R994. [DOI] [PubMed] [Google Scholar]
- Kubota T, Fang J, Brown RA, Krueger JM. Interleukin-18 promotes sleep in rabbits and rats. Am J Physiol Regul Integr Comp Physiol. 2001;281:R828–38. doi: 10.1152/ajpregu.2001.281.3.R828. [DOI] [PubMed] [Google Scholar]
- Lawson MA, McCusker RH, Kelley KW. Interleukin-1 beta converting enzyme is necessary for development of depression-like behavior following intracerebroventricular administration of lipopolysaccharide to mice. J Neuroinflammation. 2013;10:54. doi: 10.1186/1742-2094-10-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naidoo N. Roles of endoplasmic reticulum and energetic stress in disturbed sleep. Neuromolecular Med. 2012;14:213–9. doi: 10.1007/s12017-012-8179-9. [DOI] [PubMed] [Google Scholar]
- Noh H, Jeon J, Seo H. Systemic injection of LPS induces region-specific neuroinflammation and mitochondrial dysfunction in normal mouse brain. Neurochem Int. 2014;69:35–40. doi: 10.1016/j.neuint.2014.02.008. [DOI] [PubMed] [Google Scholar]
- Obal F, Jr, Fang J, Payne LC, Krueger JM. Growth-hormone-releasing hormone mediates the sleep-promoting activity of interleukin-1 in rats. Neuroendocrinology. 1995;61:559–65. doi: 10.1159/000126880. [DOI] [PubMed] [Google Scholar]
- Olivadoti MD, Opp MR. Effects of i.c.v administration of interleukin-1 on sleep and body temperature of interleukin-6-deficient mice. Neuroscience. 2008;153:338–48. doi: 10.1016/j.neuroscience.2008.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opp MR, Obal F, Jr, Krueger JM. Interleukin-1 alters rat sleep: temporal and dose-related effects. Am J Physiol. 1991;260:R52–8. doi: 10.1152/ajpregu.1991.260.1.R52. [DOI] [PubMed] [Google Scholar]
- Porkka-Heiskanen T, Strecker RE, Thakkar M, Bjorkum AA, Greene RW, McCarley RW. Adenosine: a mediator of the sleep-inducing effects of prolonged wakefulness. Science. 1997;276:1265–8. doi: 10.1126/science.276.5316.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rechtschaffen A, Bergmann BM, Gilliland MA, Bauer K. Effects of method, duration, and sleep stage on rebounds from SD in the rat. Sleep. 1999;22:11–31. doi: 10.1093/sleep/22.1.11. [DOI] [PubMed] [Google Scholar]
- Rivers-Auty J, Brough D. Potassium efflux fires the canon: Potassium efflux as a common trigger for canonical and noncanonical NLRP3 pathways. Eur J Immunol. 2015;45:27578–61. doi: 10.1002/eji.201545958. [DOI] [PubMed] [Google Scholar]
- Saper CB, Fuller PM, Pedersen NP, Lu J, Scammell TE. Sleep state switching. Neuron. 2010;68:1023–42. doi: 10.1016/j.neuron.2010.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt MA, Wisor JP. Interleukin 1 receptor contributes to methamphetamine- and SD-induced hypersomnolence. Neurosci Lett. 2012;513:209–13. doi: 10.1016/j.neulet.2012.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman WR, de Rivero Vaccari JP, Locovei S, Qiu F, Carlsson SK, Scemes E, Keane RW, Dahl G. The pannexin 1 channel activates the inflammasome in neurons and astrocytes. J Biol Chem. 2009;284:18143–51. doi: 10.1074/jbc.M109.004804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steriade M. Grouping of brain rhythms in corticothalamic systems. Neuroscience. 2006;137:1087–106. doi: 10.1016/j.neuroscience.2005.10.029. [DOI] [PubMed] [Google Scholar]
- Szymusiak R. Hypothalamic versus neocortical control of sleep. Curr Opin Pulm Med. 2010;16:530–5. doi: 10.1097/MCP.0b013e32833eec92. [DOI] [PubMed] [Google Scholar]
- Taishi P, Chen Z, Obal F, Jr, Hansen MK, Zhang J, Fang J, Krueger JM. Sleep-associated changes in interleukin-1beta mRNA in the brain. J Interferon Cytokine Res. 1998;18:793–8. doi: 10.1089/jir.1998.18.793. [DOI] [PubMed] [Google Scholar]
- Taishi P, Davis CJ, Bayomy O, Zielinski MR, Liao F, Clinton JM, Smith DE, Krueger JM. Brain-specific interleukin-1 receptor accessory protein in sleep regulation. J Appl Physiol (1985) 2012;112:1015–22. doi: 10.1152/japplphysiol.01307.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi S, Fang J, Kapás L, Wang Y, Krueger JM. Inhibition of brain interleukin-1 attenuates sleep rebound after SD in rabbits. Am J Physiol. 1997;273:R677–82. doi: 10.1152/ajpregu.1997.273.2.R677. [DOI] [PubMed] [Google Scholar]
- Timofeev I. Local origin of slow EEG waves during sleep. Zh Vyssh Nerv Deiat Im I P Pavlova. 2013;63:105–12. doi: 10.7868/s0044467713010139. [DOI] [PubMed] [Google Scholar]
- Tobler I, Borbely AA, Schwyzer M, Fontana A. Interleukin-1 derived from astrocytes enhances slow wave activity in sleep EEG of the rat. Eur J Pharmacol. 1984;104:191–2. doi: 10.1016/0014-2999(84)90391-1. [DOI] [PubMed] [Google Scholar]
- Toth LA, Opp MR. Cytokine- and microbially induced sleep responses of interleukin-10 deficient mice. Am J Physiol Regul Integr Comp Physiol. 2001;280:R1806–14. doi: 10.1152/ajpregu.2001.280.6.R1806. [DOI] [PubMed] [Google Scholar]
- Walsh JG, Muruve DA, Power C. Inflammasomes in the CNS. Nature Reviews Neuroscience. 2014;15:84–97. doi: 10.1038/nrn3638. [DOI] [PubMed] [Google Scholar]
- Wong ML, Inserra A, Lewis MD, Mastronardi CA, Leong L, Choo J, Kentish S, Xie P, Morrison M, Wesselingh SL, Rogers GB, Licinio J. Inflammasome signaling affects anxiety- and depressive-like behavior and gut microbiome composition. Molecular Psychiatry. 2016;21:797–805. doi: 10.1038/mp.2016.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu L, Liu YZ, Shen XL, Wu TY, Zhang T, Wang W, Wang YX, Jiang CL. NLRP3 inflammasome mediates chronic mild stress-induced depression in mice via neuroinflammation. Int, J, Neuropsychopharmacol. 2015;18:pyv006. doi: 10.1093/ijnp/pyv006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielinski MR, Gerashchenko L, Karpova SA, Gerashchenko D. A novel telemetric system to measure polysomnographic biopotentials in freely moving animals. J Neurosci Methods. 2013;216:79–86. doi: 10.1016/j.jneumeth.2013.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielinski MR, Kim Y, Karpova SA, McCarley RW, Strecker RE, Gerashchenko D. Chronic sleep restriction elevates brain interleukin-1 beta and tumor necrosis factor-alpha and attenuates brain-derived neurotrophic factor expression. Neurosci Lett. 2014;580:27–31. doi: 10.1016/j.neulet.2014.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielinski MR, Krueger JM. Sleep and innate immunity. In: Miller MA, editor. Encyclopedia of Bioscience, Front Biosciences Schol. Vol. 3. 2011. pp. 632–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielinski MR, Taishi P, Clinton JM, Krueger JM. 5′-Ectonucleotidase-knockout mice lack non-REM sleep responses to SD. Eur J Neurosci. 2012;35:1789–98. doi: 10.1111/j.1460-9568.2012.08112.x. [DOI] [PMC free article] [PubMed] [Google Scholar]