

Abstract
PURPOSE
Resistance to chemotherapeutic agents such as doxorubicin is a major reason for cancer treatment failure. At present the treatment option for metastatic breast cancer is very poor. Therefore, development of an effective therapeutic strategy to circumvent MDR of metastatic breast cancer is highly anticipated. The MDR of metastatic breast cancer cells was accompanied with the overexpression of P-gp transporter. Even though the overexpression of P-gp could be minimized by silencing with siRNA, the question is how they can be selectively targeted to the cancer cells. We propose that aptamer surface labeling of the nanoparticles could enhance the selectively delivery of p-gp siRNA into the metastatic breast cancer cells. Our hypothesis is that conjugating nanoparticles with a cancer cell specific aptamer should allow selective delivery of therapeutic drugs to tumor cells leading to enhanced cellular toxicity and antitumor effect as compared to unconjugated nanoparticles. The primary objective of this study is to develop a targeted nanocarrier delivery system for siRNA into breast cancer cells.
DESIGN METHODS
For targeted delivery, Aptamer A6 has been used which can bind to Her-2 receptors on breast cancer cells. For aptamer binding to particle surface, maleimide-terminated PEG-DSPE (Mal-PEG) was incorporated into the nanoparticles. Initially, three blank hybrid nanoparticles (i.e. F21, F31, and F40) out of nine different formulations prepared by high pressure homogenization (HPH) using different amount of DOTAP, cholesterol, PLGA or PLGA-PEG and Mal-PEG were chosen. Then protamine sulphate-condensed GAPDH siRNA (TRITC conjugated; red) or P-gp siRNA was encapsulated into those nanoparticles. Finally, the particles were incubated with aptamer A6 (FITC conjugated; green) for surface labeling.
RESULTS
Aptamer labeled-nanoparticles having PLGA are smaller in size than those having PLGA-PEG. Surface charge was reduced when the particles were labeled with aptamer. Cell transfection was increased significantly in Her-2 (+) SKBR-3 and 4T1-R cells but not in Her-2 poorly expressed MDA MB-231 and MCF-7 cells. The knockdown of P-gp was increased significantly when the particles were labeled with aptamer. No significant cellular toxicity was observed for any of these formulations.
CONCLUSION
This preliminary study concludes that aptamer-functionalized hybrid nanoparticles could be used to deliver P-gp targeted siRNA into the breast cancer cells to overcome chemoresistance.
Keywords: P-glycoprotein, Nanoparticle, Her-2, Aptamer, siRNA
Graphical abstract
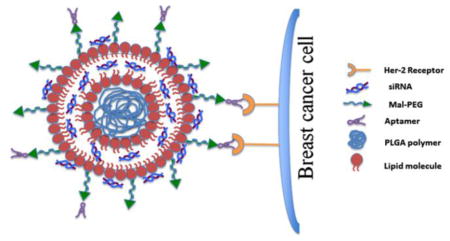
Schematic diagram showing the organization of the nanoparticles.
1. Introduction
Breast cancer is one of the most invasive and malignant diseases affecting millions, and worldwide over 508, 000 women died in 2011 due to breast cancer [1]. The primary treatment for breast cancer is surgery followed by adjuvant chemotherapy. However, there are ample evidences where recurrence of cancer metastasis is experienced by patients in spite of adjuvant chemotherapy [2]. The development of multidrug resistance (MDR) is a major obstacle to effective cancer chemotherapy [3]. Once the tumor cells develop resistance to a single class of drug, it can also show cross-resistance to other functionally and structurally unrelated drugs. This phenomenon is known as multidrug resistance (MDR). The mechanism why some tumor cells develop resistance and some don’t is unclear [4].
The actual mechanism behind MDR is yet to be revealed. Both intrinsic and acquired mechanisms have been proposed as possible causes of breast cancer drug resistance. Overexpression of adenosine-triphosphate (ATP) binding cassette (ABC) transporters, a 48-member superfamily, is one of the identified causes of MDR. Of these, the most well characterized transporter, ABCB1 (MDR1/P-glycoprotein) overexpression in breast cancer cells has been found to be strongly associated with reduction of intracellular level of anticancer drugs below their therapeutic threshold [5]. Strategies have been formulated to beat the P-gp mediated drug efflux by using chemical inhibitors though there are only a few that are clinically effective. The RNA Interference (RNAi) technology provides a potential mechanism of gene silencing and could be utilized to knockdown P-gp. Wu et al. has markedly inhibited the overexpression of MDR1 (i.e. P-gp) by siRNAs in MDR cancer cells resulting in restoration of drug sensitivity [6]. Similar findings were also observed in human MDR cells as well [7]. However, the siRNA delivery needs to be targeted specifically to cancer cells so as to avoid notorious side effects to the normal cells.
The potential of siRNA as an anticancer therapeutic depends on the availability of a carrier vehicle which will not only have higher binding affinity for siRNA but also safely administer the drugs ( i.e. siRNA) specifically and efficiently to the target cells or tissues. The carrier should protect the functional integrity of the siRNA as well as permitting their (siRNA) easy and efficient release from the vehicle within the cells. Among the numerous vehicles developed for RNAi delivery, cationic lipids and polymers are most promising because of their easy and efficient packaging with siRNAs to form nanoscale complexes (lipoplexes or polyplexes) which have shown potential in delivering siRNA [8]. Nevertheless, if the delivery vehicle is not specifically targeted to the cancer cells, problems associated with toxicity, immune or inflammatory responses, and serum instability would hinder their effective use for the treatment of cancer. To that end, several strategies have been adopted, including pegylation (i.e. coupling to PEG) of nanocomplexes and liposomal envelopment of polyplexes (to form lipopolyplexes) [9–10] to optimally protect both siRNA and nanocomplexes from the physiological barriers in vivo.
With the development of nanotechnology, nanoformulations have been widely tried for the last few years to bypass the MDR development of tumor cells [11–13]. In this study, we have explored nanotechnology to overcome chemoresistance mechanisms. It is recently reported by our collaborator that doxorubicin treatment effectively suppresses the multiorgan metastasis of doxorubicin-sensitive 4T1 cells in Balb/c mice, but not doxorubicin resistant 4T1 breast cancer cells. They also reported that down regulating nuclear expression of MDR1 P-gp (ABCB1 gene) by P-gp specific siRNA could increase the delivery of doxorubicin to doxorubicin resistant breast cancer cells [5,14]. However, unless these nanoparticles are targeted specifically to cancer cells, they will not have a significant impact in the treatment of cancer. So, we plan to overcome this problem by developing a targeted nanocarrier delivery system for siRNAs targeting P-gp into breast cancer cells. We assume that silencing P-gp will eventually help to deliver more doxorubicin into the breast cancer cells.
For targeted delivery, the aptamer technology has been used [15–17]. A single-stranded RNA or DNA oligonucleotide aptamer can fold into unique tertiary conformations [ 17] and are capable of binding with high specificity to non-nucleic acid targets like proteins [18]. Because of these unique characteristics, aptamer has been lately used for targeted delivery of drugs into cancer cells. For example, aptamer AS1411 targets nucleolin in MCF-7 cells [15], aptamer-liposome conjugates containing aptamer sgc8 has been used for drug delivery into CEM-CCRF leukemia cells [16], and PLGA-PEG nanoparticles surface functionalized with A10 2′ fluoropyrimidine RNA aptamer has been used to enhance the delivery of docetaxel in LNCaP xenograft nude mice [17]. The aptamer that has been used in this study is amino-terminal at one end (NH2-Apt-6) and it binds to epidermal growth factor receptors (ERBB2/Her-2), a master regulator of cancer progression which is overexpressed on the cell surface of breast cancer cells. In the present study, we modified our pretested nanoparticles originally prepared using DOTAP and cholesterol [19] by including PLGA or PLGA-PEG. For aptamer binding to particle surface, maleimide-terminated PEG-DSPE (Mal-PEG) was specifically incorporated into the nanoparticles [20]. Maleimide linked to PEG serves as a flexible linker that can readily associate with a variety of functional groups including thiol-and amino- group. [21]. The amino-terminal aptamer (NH2-Apt-6) presumably binds with the Mal-PEG moieties on the nanoparticle surface which in turn labels the particles for selective binding to Her-2 (+) breast cancer cells. This study is aimed to know whether labeling nanoparticles with a cancer cell specific aptamer could enhance the selective delivery of siRNA into tumor cells leading to enhanced knockdown of P-gp as compared to non-labeled nanoparticles.
2. Materials and Methods
2.1 Materials
1,2-dioleoyl-3-trimethylammonium-propane (DOTAP), Cholesterol and Maleimide-terminated PEG-DSPE (Mal-PEG) were purchased from Avanti Polar- lipids Inc. (Birmingham, AL, USA). Protamine sulfate salt Grade X, trehalose dihydrate and HPLC grade chloroform were obtained from Sigma Chemical Co. (St. Louis, MO, USA). PLGA and PLGA-PEG were purchased from Boehringer Ingelheim (Germany). Lipofectamine RNAiMAX transfection reagent was purchased from Invitrogen. Fetal bovine serum albumin (BSA), Dulbecco’s modified Eagle’s medium (DMEM) and penicillin/streptomycin antibiotics were purchased from Gibco, Invitrogen Corp. (Carlsbad, CA, USA). β-actin antibody was purchased from Sigma Chemical Co. (St. Louis, MO, USA), anti-mouse and anti-rabbit IgG HRP labeled secondary antibody were purchased from GE Healthcare (Little Chalfont, UK). GAPDH siRNA and aptamers were purchased from Life Technologies (Carlsbad, CA). Anti-P-gp antibody was purchased from Pierce, Thermo Scientific (Waltham, MA). All other reagents were of analytical grade and were supplied by Sigma Chemical Co. (St. Louis, MO, USA).
2.2 Preparation of lipid-polymer hybrid liposomes
The hybrid liposomes were prepared by an EmulsiFlex-B3 high pressure homogenizer from a mixture of two lipids cholesterol and DOTAP at the molar ratio of 1:1 [19, 22] and PLGA or PLGA-PEG (10% diblock) at different weight ratios shown in Table 1. Nine different formulations of nanoparticles were prepared into two different categories: PLGA-PEG group (i.e. F20, F21, F22, F23) and PLGA group (i.e. F30, F31, F32, F33) and F40 being the basic liposomal formulation containing only DOTAP and cholesterol. Three formulations were chosen for further experiments F21, F31 and F40; the first two representing the best combination possible from each of PLGA-PEG and PLGA group.
Table 1.
Composition of different hybrid nanoparticles (blank)
| Blank nanoparticles | DOTAP (25 mg/ml) (ml) | Cholesterol (10 mg/ml) (ml) | PLGA (50 mg/ml) (μl) | PLGA-PEG 10% diblock (50 mg/ml) (μl) | Mal-PEG (25 mg/ml) (μl) | Total volume After HPH (ml) |
|---|---|---|---|---|---|---|
| F20 | 1.8 | 2.5 | 50 | 20 | ||
| F21 | 1.8 | 2.5 | 50 | 10 | 20 | |
| F22 | 1.8 | 2.5 | 50 | 20 | 20 | |
| F23 | 1.8 | 2.5 | 100 | 20 | 20 | |
| F30 | 1.8 | 2.5 | 50 | 20 | ||
| F31 | 1.8 | 2.5 | 50 | 10 | 20 | |
| F32 | 1.8 | 2.5 | 50 | 20 | 20 | |
| F33 | 1.8 | 2.5 | 100 | 20 | 20 | |
| F40 | 1.8 | 2.5 | 20 |
The blank liposome (F40) was prepared 20 mM containing equi-molar ratio of DOTAP and cholesterol. The hybrid liposomes (F21and F31) were prepared by adding either PLGA-PEG (F21) or PLGA (F31) and Mal-PEG into the blank liposomes. For the preparation of hybrid liposomes; DOTAP, cholesterol, Mal-PEG and PLGA-PEG or PLGA were weighed and taken into a round bottom flask. They were mixed together by adding 15 ml of HPLC-grade chloroform and then dried under nitrogen gas and overnight vacuum. The resulting film of the lipid polymer mixture was hydrated in DEPC- treated water. The lipid polymer mixer was warmed and mixed at 50 C for 45 minutes by rotation and then kept at RT for 2 hours. The resultant dispersion was transferred into a scintillation vial and warmed again at 50°C for 15 minutes. The final lipid polymer dispersion was homogenized by using a high pressure homogenizer at 20,000 PSI for 5 cycles. Each time, 2.5 ml of lipid polymer dispersion was subjected to homogenization and the resultant hybrid liposomes were collected in another scintillation vial. They were kept at room temperature for 1 hour prior to overnight storage at 4°C.
2.3 Preparation of siRNA encapsulated aptamer-labeled nanoparticles
Previously, we have developed highly efficient and non-toxic siRNA-encapsulated liposomal nanoparticles for the treatment of HCV [19, 23–24]. Similar methods were employed here to encapsulate either GAPDH siRNA or P-gp targeted siRNA into the blank nanoparticles. The stepwise preparation of siRNA incorporated nanoparticles and aptamer a ssociation is outlined in Fig. 4. Briefly, freshly prepared protamine sulfate solution (2 μg at 1 μg/10 μl conc.) in DEPC-treated water was added drop wise to an aqueous solution of siRNA (1 μg at 10 pmol/μl) while vortexing the solution at a moderate speed. siRNA condensation with protamine sulfate was performed at room temperature by 40 minute incubation of the mixture. In the meantime, the blank nanoparticles were reconstituted into DEPC-treated water and kept at RT for 1 hour. Following siRNA-protamine sulfate condensation, the reconstituted blank nanoparticle was added to the mixture keeping the nanoparticle to siRNA ratio equal to 6.8: 0.66 after 1 min brief sonication in ice cold water followed by pipetting 30 times. Then the nanoparticles containing siRNA were vortexed 4 to 5 times to allow thorough mixing. Finally they were sonicated in ice cold water for 3 to 4 min to reduce the particle size. For aptamer binding to particle surface, Mal-PEG was previously incorporated into the nanoparticles. Finally, the siRNA- encapsulated particles were incubated with aptamer A6 (NH2-Apt-6) for surface labeling. 12.8 μl aptamer from a stock conc. of 58.56 μM was added so as to give a final concentration of 0.75 μM in 1 ml cell culture media. Only three (i.e. F21, F31 and F40) out of those nine formulations have been chosen for the targeted delivery of siRNA (Table 1).
Figure 4.

Schematic diagram showing the preparation and organization of the nanoparticles. (A) Illustration showing the stepwise preparation of the siRNA-encapsulated aptamer-labeled nanoparticles. (B) Schematic diagram showing the organization of the siRNA-encapsulated aptamer-labeled hybrid nanoparticles (F31) having a polymer core surrounded by the lipid bilayer. siRNA is assumed to be trapped in-between the lipid bilayer as well as on the surface of the bilayer. The surface bound aptamer (via Mal-PEG) is recognized by the Her-2 receptors on the breast cancer cells, which facilitates the entry of nanoparticles into the breast cancer cells by endocytosis.
Each formulation was then divided into three different subgroups:
Blank formulation (no siRNA, no aptamer) (F21, F31 and F40)
Formulation with siRNA (F21−Apt, F31−Apt and F40−Apt)
Formulation with siRNA & Aptamer (F21+Apt, F31+Apt and F40+Apt).
2.4 Measurement of particle size and zeta potential
The particle size of the blank particles with/without siRNA and aptamer was determined by dynamic laser light scattering method at room temperature by using a Delsa Nano C Particle Analyzer (Beckman Coulter Inc., Fullerton, CA, USA). The 20 mM nanoparticle suspension (stock) was diluted 1: 10 in water and the particle size and zeta potential were assessed using 2 ml of this diluted suspension. The particle size was reported as the mean ± standard deviation (n=4). The hydrodynamic size of the particle shown in Figure 1 corresponds to it’s diameter. Analysis of the charge density of different formulations was performed by examining their zeta potential by using a Delsa Nano C Particle Analyzer (Beckman Coulter Inc., Fullerton, CA, USA).
Figure 1.

Comparison of particle size between blank and siRNA-encapsulated aptamer-labeled nanoparticles. The size of the particles was determined by dynamic laser light scattering method at room temperature using a Delsa Nano C Particle Analyzer. The particle size was reported as the mean ± standard deviation (n=4).
2.5 Measurement of siRNA encapsulation efficiency of different hybrid nanoparticles
The amount of siRNA (%) entrapped within the particles was determined for these three different formulations (without aptamer labeling) F21, F31 and F40 by using Ribogreen Assay (Molecular Probes, Invitrogen) following the manufacturer’s protocol. The procedure in brief is as follows: the particle solution was centrifuged at 14,000 rpm (Allegra Centrifuge, Beckman Coulter Inc, Fullerton, CA) for 15 min at 4°C. The unbound siRNA present in the supernatant was separated from the pellet containing the entrapped siRNA. 500 μL of 1% sodium dodecyl sulfate (SDS) was added to the pellets and to the supernatants. These samples were then incubated at 37°C for 18 hours with gentle agitation (50 rpm). The concentration of siRNA in both the supernatants and pellets was measured by using Ribogreen Assay using Cary Eclipse Fluorescence Spectrophotometer, excitation wavelength 500 nm and emission wavelength 525 nm. The results were reported as mean ± standard deviation (n=4).
2.6 Cell culture and cell lines
Five different breast cancer cell lines (i.e. human MDA MB-231, MCF-7, SKBR-3, chemoresistant mouse 4T1-R and chemosensitive 4T1-S) were used. SKBR-3 cells were maintained in McCoy’s media supplemented with 10% FBS and all other cell lines were maintained in DMEM supplemented with 10% FBS, 1% sodium pyruvate, 1% nonessential amino acids, and 1% L-glutamine. The liver cancer cell lines Huh-7.5 and HepG2 cells were also maintained in DMEM and served as controls.
2.7 Cell viability assay
The cytotoxicity of the aptamer-labeled siRNA-encapsulated formulations prepared using F21 and F31 was assessed on 4T1-R cells by MTT assay in accordance with the manufacturer’s protocol (Sigma Chemical Co., MO, USA). The cells were transfected with blank and siRNA-entrapped aptamer-labeled nanoparticles. Twenty-four hours after transfection, the cells were incubated with MTT solution at 37°C for 2 h, and the cell viability was measured by reading the absorbance at 570 nm.
2.8 Cell uptake Study
a.)Transfection of GAPDH siRNA using nanoparticles labeled with aptamer
MDA MB-231, MCF-7, SKBR-3 and 4T1-R breast cancer cells were used to measure both qualitatively (by using a fluorescence microscope) and quantitatively (by FACS analysis) the uptake of the nanoparticles by those cells. Briefly, the cells were cultured in 12-well tissue culture plates (TCPs) for 24 hours in 10% FBS containing DMEM or McCoy’s media. The cells were then transfected with different formulations and kept at 37°C for 24 hours. For immunofluorescence, TRITC-conjugated GAPDH siRNA and FITC-conjugated aptamer were used which were monitored by a fluorescence microscope (Olympus IX-71), and photographs were taken at 10× magnification. For FACS analysis, TRITC-conjugated GAPDH siRNA and non-labeled aptamer were used. After treatment, the cells were scraped by using trypsin-EDTA, washed and resuspended in PBS and kept on ice until they were used to quantify the uptake of the nanoparticles by those cells by FACS (BD FACSCalibur). We have used Aptamer A6 (NH2-Apt-6) targeted to the HER-2 receptors on breast cancer cells the sequence of which is 5′-TGGATGGGGAGATCCGTTGAGTAAGCGGGCGTGTCTCTCTGCCGCCTTGCTATGGGG-3′ (-NH2) (Invitrogen, Life Technologies). The P-gp siRNA (5′-CGGAAGGCCUAAUGCCGAAtt-3′) was purchased from Ambion, Life Technologies [25].
b.)Transfection of P-gp specific siRNA using nanoparticles labeled with aptamer
P-gp targeted siRNA was used to transfect SKBR-3, MCF-7 (human) and 4T1-R (mouse) breast cancer cells carried by the hybrid nanoparticles (F21 and F31) labeled with/without aptamer A6. Briefly, 2×105 cells were cultured in 6-well TCPs for 24 hours. The next day, the media was replenished by 1 ml fresh media containing either 10% FBS (for nanoparticle and lipofectamine transfection) or 2% FBS (for lipofectamine transfection only). For lipofectamine transfections, typical RNAiMAX transfection procedure was followed, with 7.5 μl RNAiMAX reagent added per 25 pmol of siRNA. Here, 100 pmol siRNA in 100 μl DMEM was mixed with 30 μl lipofectamine in 100 μl DMEM and then, this 200 μl lipofectamine-siRNA complex was added to cells in 800 μl cell culture media supplemented with 10% or 2% FBS. For aptamer-labeled siRNA encapsulated nanoparticle transfection, 100 μl siRNA encapsulated aptamer-labeled nanoparticles (nanoparticle: siRNA = 6.8: 0.66) prepared following the procedure mentioned in section 2.3 was added to 900 μl cell culture media supplemented with 10% FBS giving a dilution factor of 10 and mixed by swirling. In both cases of lipofectamine and aptamer-labeled nanoparticle transfection, the cells were transfected with 100 pmol siRNA, which is equal to 100 nM siRNA based on 1 ml cell culture media. After 3 hours, an additional 1 ml cell culture media was added to the transfected cells in both nanoparticle and lipofectamine transfection. After 24 hours, all the treated cells (both lipofectamine and nanoparticle transfection) were scraped by using trypsin-EDTA, washed with PBS, pelleted and stored at −20°C for western blot analysis.
2.9 Western Blot Analysis
Approximately 30–40 μg of protein from each sample was mixed with SDS loading buffer (Lamelli sample buffer (2X) containing 2-Mercaptoethanol). The proteins were separated by Novex 4–20% Tris-Glycine gel and then transferred onto a nitrocellulose membrane (Novex, Life Technologies). The membrane was blocked with 5% non-fat dry milk in Tris-buffered saline with 0.05% Tween-20 at room temperature for an hour. The membrane was washed four times and incubated overnight at 4°C with either mouse monoclonal antibody to P-gp (C219; Pierce, Thermo Scientific) at 1:1000 dilution or a rabbit monoclonal antibody for HER-2/ErbB2 (Cell Signaling Tech., Danvers, MA) at a dilution of 1:1000. The blot was normalized by incubating with mouse monoclonal antibody for β-actin (Sigma-Aldrich) at 1:20,000 dilution. After the primary antibody incubation, the membrane was washed four times and then incubated with the secondary antibody conjugated with horse-radish peroxidase (anti-mouse or anti-rabbit) (GE HealthCare) at a dilution of 1:20,000 for 1 hour. The protein expression was detected using the ECL reagent (GE Healthcare) on a high performance chemiluminescence film (Thermo Scientific).
3. Results
3.1 Physicochemical characterization of blank and aptamer-labeled siRNA encapsulated nanoparticles - particle size and zeta potential
The particle size and zeta potential of all nine formulations are shown in Fig. 1 and Fig. 2. The particle size of the blank nanoparticles (i.e. blank formulations; F21, F31, F40 etc.) and siRNA-encapsulated aptamer-labeled nanoparticles (F21+Apt, F31+Apt, F40+Apt etc.) has been compared among different batches in the respective range of 70 percentile (Fig. 1). The incorporation of PLGA-PEG or PLGA in the formulations has shown differential impact on the size of the blank hybrid nanoparticles. Among different batches of PLGA-PEG group (i.e. F20, F21, F22, F23) with or without siRNA encapsulation and aptamer labeling, the smallest particles were generated by F21 formulation whereas, among different batches of PLGA group ( i.e. F30, F31, F32, F33) with siRNA encapsulation and aptamer labeling, the smallest particles were generated by F31 formulation. When F21 and F31 were compared with only blank liposomes (i.e. F40); it was noticed that in case of hybrid F21 formulation, the incorporation of PLGA-PEG has reduced the average particle size from ~156±20 nm (F40 blank) to ~100±12 nm (F21 blank). Whereas in case of hybrid F31, the incorporation of PLGA has decreased the particle size from ~156±20 nm (F40 blank) to ~138±12 nm (F31 blank). Once the hybrid nanoparticles were used to encapsulate siRNA and labeled with aptamer, the particle size increases dramatically irrespective of the formulation types which was measured as follows: F21 bank vs. F21+Apt (~100±12 nm vs. ~270±10 nm), F31 blank vs. F31+Apt (~138±12 nm vs. ~237±12 nm) and F40 black vs. F40+Apt (~156±20 nm vs. ~220±28 nm). However, the particle size of F31+Apt at 70% (~237±12 nm) is slightly smaller than that of F21+Apt (~270±10 nm). This increase in particle size after siRNA incorporation has also been reported previously [22].
Figure 2.

Comparison of zeta potential between blank and siRNA-encapsulated aptamer-labeled nanoparticles. The zeta potential of the particles was measured by using a Delsa Nano C Particle Analyzer. The zeta potential was reported as the mean ± standard deviation (n=4).
The surface charge of all nine different formulations was also compared and shown in Fig. 2. The surface charge of the hybrid nanoparticles (i.e. F21; 49±9 mV and F31; 45±6 mV) was shown higher than that of the blank liposomes (F40; 31±4 mV) (Fig. 2). The labeling of aptamer and the encapsulation of siRNA to F21 and F31 formulations changed their respective surface charge from 49±9 mV (F21) to 32±2 mV (F21+Apt) and 45±6 mV (F31) to 31±2 mV (F31+Apt). In general, in both PLGA-PEG group and PLGA group, the encapsulation of siRNA and aptamer labeling had decreased the surface charge of their respective blank hybrid particles (i.e. F20+Apt vs. F20 blank, F30+Apt vs. F30 blank and so on). The surface charge of the hybrid nanoparticles decreased after siRNA incorporation was due to the partial neutralization of the positive charge of the nanoparticles by the negatively charged siRNA. However, the surface charge of F40 blank particles (31±4 mV) has not changed significantly when the particles were exposed to siRNA and aptamer (i.e. F40+Apt) (34±4 mV). The inability of labeling aptamer to the F40 blank liposomes (due to the absence of Mal-PEG) as well as the lesser siRNA encapsulation (Fig. 3) are assumed to be the sole reason for those particles not having a reduced surface charge.
Figure 3.

Determination of siRNA’s encapsulation efficiency of the selected nanoparticles. The entrapped siRNA in three formulations (i.e. F21, F31 and F40) was decomplexed by exposure to 1% SDS for 18 hours and then measured by Ribogreen Assay. The results were reported as mean ± standard deviation (n=4).
3.2 siRNA encapsulation efficiency of the nanoparticles
The siRNA encapsulation efficiency of the hybrid nanoparticles (without aptamer labeling) was determined by Ribogreen assay as shown in Fig. 3. The siRNA encapsulation efficiency of F21 (without aptamer) (≈55±2) and that of F31 (without aptamer) (≈64±4) was greater than that of F40 (without aptamer) (≈49±2), which exhibits that the % entrapment of siRNA increases with the addition of the polymer. Between PLGA-PEG and PLGA group, F31 (PLGA) showed better encapsulation efficiency than that of F21 (PLGA-PEG).
Cryo-TEM images of only lipid-based nanoparticles before and after siRNA encapsulation have been shown in our previous publications [19, 22]. A plausible illustration of the lipid-polymer hybrid nanoparticles of F31 after siRNA encapsulation and aptamer labeling is shown in Fig 4.
3.3 Cytotoxicity of the formulations into breast cancer cells
The cytotoxicity of F21 and F31 formulations was determined in 4T1-R (mouse) breast cancer cells (Fig. 5).
Figure 5.

Measurement of cell toxicity of different formulations loaded with different concentration of P-gp siRNA on 4T1-R cells. The cells were transfected with different amount of siRNA-entrapped aptamer-labeled nanoparticles prepared using F21 and F31 for 24h and the cell cytotoxicity was measured by MTT assay following the manufacturer’s protocol. The results represent mean ± standard deviation (n=4).
The formulations (i.e. F21 and F31) were prepared by encapsulating varying concentrations of siRNA. The concentration of hybrid particles and protamine sulphate used for siRNA packaging also increased proportionally along with siRNA concentration. The per cent cytotoxicity of F31 formulation was observed ≈11±2, ≈11±3, ≈13±3, ≈14±2, ≈17±2 and ≈20±3 at the siRNA concentration of 25, 50, 75, 100, 125 and 150 pmol, respectively. The cell cytotoxicity of F21 formulation was ≈19±2, ≈22±3, ≈23±3, ≈24±2, ≈25±2 and ≈32±3 respectively with the corresponding siRNA concentrations ranging from 25 to 150 pmol. Nanoparticles prepared using F21 showed enhanced cytotoxicity as compared to F31.
3.4 Assessment of Her-2 and P-gp expression in different cell lines
The expression of Her-2 has been assessed in SKBR-3, MCF-7, MDA MB-231 (human) and 4T1-R and 4T1-S (mouse) breast cancer cells and in Huh-7.5 and HepG2 liver cancer cells (Fig. 6A). Among the breast cancer cells, the highest expression of Her-2 receptors was observed in SKBR-3 cells. Compared to SKBR-3 cells, the expression of Her-2 was much lower as observed in MCF-7 and MDA MB-231 breast cancer cells. Both 4T1-R (chemoresistant) and 4T1-S (chemosensitive) cells showed high level of Her-2 expression without much differences among themselves. Between two liver cancer cells, Her-2 was expressed at a lower level in Huh-7.5 cells whereas it was absent in HepG2 cells. Differential expression of P-gp is also observed in different human and mouse breast cancer cell lines (Fig. 6B). The expression of P-gp was low in MDA MB-231 and moderately low in SKBR-3 cells. On the contrary, the expression of P-gp was significantly high in both human MCF-7 and mouse 4T1-R/4T1-S cells. However, between 4T1-R and 4T1-S cells, higher expression of P-gp was observed in 4T1-R cells than 4T1-S cells. The liver cancer cells Huh-7.5 and HepG2 show a medium level of P-gp expression. Similar level of Her-2 and P-gp expression was observed from at least two separate experiments.
Figure 6.

Measurement of the expression of Her-2 (A.) and P-gp (B.) in different mouse (i.e. 4T1-R and 4T1-S) and human breast cancer cells (i.e. SKBR-3, MCF-7 and MDA MB-231) and human liver cancer cells (i.e. Huh-7.5 and HepG2) by Western blot analysis. The cells were cultured for 24h and then they were scraped by using trypsin-EDTA, washed with PBS, pelleted and then equal amount of proteins was loaded to measure the expression of Her-2 (1:1000 dilution) and P-gp (1:1000 dilution) with β-actin (1:20,000 dilution) running as the loading control.
3.5 In vitro transfection study
The transfection efficiency of the nanoparticles was determined qualitatively by fluorescence microscopy using TRITC-conjugated GAPDH siRNA with or without FITC-labeled aptamer (Fig. 7 and Fig. 8) and also quantitatively by FACS analysis using TRITC-conjugated GAPDH siRNA and unlabeled aptamer (Fig. 9). When the Her-2 highly expressed SKBR-3 cells were transfected with F31+Apt (Fig. 7A), the cells were bound with a significant amount of aptamer on the cell surface as well as fully loaded with siRNA within the cytoplasm. A similar finding was also observed when the cells were transfected with F21+Apt formulation (Fig. 7B). In contrast, when Her-2 poorly expressed MDA MB-231 (Fig. 7C) and MCF-7 cells (Fig. 7D) were transfected with F31+Apt, the cells were seen to receive TRITC-labeled siRNA (red) in the cytoplasm with a minimal level of FITC-aptamer (green) bound to the surface of the cells. Similarly, compared to non-aptamer labeled hybrid nanoparticles (Fig. 8A), the delivery of siRNA into SKBR-3 cells was significantly elevated (Fig. 8B) when highly Her-2 positive SKBR-3 cells were transfected with aptamer labeled hybrid nanoparticles (F31+Apt). This study clearly indicates that the particles are using aptamers to bind to the cells to deliver the siRNA into the cells.
Figure 7.

Testing the delivery efficiency of aptamer-labeled nanoparticles into the Her-2 highly/poorly expressed breast cancer cells. Her-2 (highly expressed) SKBR-3 cells were targeted by aptamer-labeled F31 (A.) and aptamer-labeled F21 nanoparticles (B.); whereas Her-2 (poorly expressed) MDA MB-231 (C.) and MCF-7 cells (D.) were targeted by aptamer-labeled F31 nanoparticles (Aptamer: green; siRNA: red). Fluorescence photographs of siRNA deposition into the cells were captured at 10× magnification by using a fluorescence microscope.
Figure 8.

Testing the delivery efficiency of F31 nanoparticles into the Her-2 (+) SKBR-3 cells without aptamer (A.) or with aptamer labeling (B.). SKBR-3 cells were transfected with/without aptamer-labeled F31 nanoparticles for 24h and the delivery of siRNA into the cells was imaged using a fluorescence microscope at 10× magnification.
Figure 9.

Comparison of GAPDH siRNA (TRITC conjugated; Red) delivery by different formulations with/without aptamer labeling into the breast cancer cells (A. 4T1-R, B. SKBR-3 and C. MCF-7) and D. HepG2 liver cancer cells by FACS analysis after 24h or transfection. The results are reported as mean ± standard deviation (n=3).
The results obtained from immunofluorescence are further substantiated by FACS analysis. Her-2 expressed mouse 4T1-R cells (Fig. 9A) and human SKBR-3 cells (Fig. 9B) were transfected with nanoparticles containing GAPDH siRNA (TRITC labeled). We observed that the level of transfection was higher when the particles were tagged with aptamer than when they were devoid of it. In contrast, when MCF-7 cells (Fig. 9C) with low Her-2 expression were transfected with/without aptamer labeled nanoparticles, there was not much difference in the level of transfection between aptamer and non-aptamer labeled nanoparticles. Similarly, the Her-2 (−) HepG2 liver cancer cells (Fig. 9D) showed an insignificant difference in siRNA transfection between aptamer-labeled and non-aptamer labeled nanoparticles. This is because none or low level of Her-2 receptors on the cell surface could enhance the specific binding or engulfment of those particles into the cells. The presence of Her-2 receptors facilitates aptamer binding and therefore there was a marked increase in siRNA delivery with aptamer-labeled nanoparticles compared to non-aptamer-labeled nanoparticles. For example, FACS analysis revealed that the delivery of siRNA has increased 2.5 fold into the Her-2 (+) 4T1-R cells and 1.7 fold into the SKBR-3 cells when the particles were labeled with aptamer (Fig. 10). These results confirm that the aptamer is enhancing the delivery of nanoparticles into the breast cancer cells through the Her-2 receptors.
Figure 10.

Fold increase in siRNA delivery into different breast (i.e. MCF-7, SKBR-3 and 4T1-R) and liver cancer cells (HepG2) by aptamer-labeled nanoparticles compared to non-aptamer-labeled nanoparticles measured by FACS analysis. The results are reported as mean ± standard deviation (n=3).
3.6 Determination of the knockdown efficiency of P-gp-targeted siRNA encapsulated aptamer-labeled nanoparticles
The results shown in Fig. 7, 8, 9 and 10 indicate that when the particles are labeled with aptamer, it increases the delivery of siRNA into the breast cancer cells significantly than non-labeled nanoparticles. The functional activity of siRNA (to knock-down P-gp) delivered by aptamer/non-aptamer labeled nanoparticles has been also assessed. The knockdown of P-gp by P-gp specific siRNA has been examined in three breast cancer cell lines that were transfected with or without aptamer labeled nanoparticles (i.e. SKBR-3, 4T1-R and MCF-7 cells) and compared to lipofectamine transfection which served as a positive control. In Fig. 11, it is quite evident that the knockdown of P-gp has increased significantly when the cells were transfected with aptamer-labeled nanoparticles. A semiquantitative analysis of the knock down efficiency of the nanoparticles in different cell lines was performed using Image J. In 4T1-R cells (Fig. 11 top panel), P-gp knockdown was ~65% (with aptamer) compared to ~29% (without aptamer) and ~26% with lipofectamine (lipofectamine with 10% FBS). Whereas in SKBR-3 cells (Fig. 11 middle panel), the knockdown was ~82% (with aptamer) than ~40% (without aptamer). In MCF-7 cells (Fig. 11 bottom panel), aptamer-labeled nanoparticles showed ~96% knockdown of P-gp compared to ~62% with non-aptamer-labeled nanoparticles. Hence, the silencing of P-gp has been improved significantly when the particles were labeled with aptamer.
Figure 11.

Knockdown of P-gp in 4T1-R (top panel), SKBR-3 (middle panel) and MCF-7 (bottom panel) breast cancer cells with/without aptamer-labeled F31 nanoparticles compared to lipofectamine transfection. In both cases of lipofectamine and aptamer-labeled nanoparticle transfection, the cells were transfected with 100 pmol siRNA for 24h, and then the cells were scraped by using trypsin-EDTA, washed with PBS, pelleted and the expression of P-gp was measured by Western blot analysis.
4. Discussion
The multi-drug resistance in breast cancer cells has been associated with the expression of a membrane protein called Permeability glycoprotein (P-glycoprotein or P-gp) that acts as an efflux pump and selectively transports chemotherapeutic agents out of the cell. We have used siRNA technology to knockdown P-gp in human and mouse breast cancer cells by using a new class of lipid-polymer hybrid nanoparticles that can efficiently and selectively deliver siRNAs to the target cells. For targeted delivery of siRNA into the breast cancer cells, we have used an aptamer that binds specifically to the Her-2 receptors overexpressed on the surface of the breast cancer cells.
Previously, we have developed a nanosomal formulation capable of inhibiting 85% of Hepatitis C virus (HCV) replication in an in vitro cell culture model [19]. Lipid nanoparticles incorporating siRNA targeted the 5′-UTR region of HCV were delivered to almost 100% of cells with minimal cytotoxicity and a significant knockdown efficiency of HCV. Also, a systemic administration of combinatorial siRNA nanosomes was noted to considerably reduce HCV replication in a liver tumor-xenotransplant mouse model of HCV without any recognized noticeable liver injury [23]. In another study, we have shown that a unique combination of lipid based nanoparticles containing DOTAP, cholesterol and high mobility group protein facilitated the delivery of both circular and linear DNA into the poorly transfected Plasmodium falciparum-infected red blood cells [26].
In the present study, we have used liposome- based nanoparticles partially substituted by polymer (PLGA or PLGA-PEG) for transfection of siRNA. PLGA is a polymer of two monomers; lactic acid and glycolic acid, these constituents can be combined in different proportions. The ratio of lactic to glycolic acid in PLGA was 65 : 35 which we have used in this study. These organic polymers have controlled biodegradability; excellent biocompatibility and they offer enhanced transfection efficiency combined with reduced cytotoxicity as compared to liposomes only [27]. Also, liposomes generally undergo interparticle fusion due to their mobile nature leading to self-decomposition. It has been reported that addition of polymer to the liposome-based nanoparticles results in improved RNAi efficiency, for example, lipid-polymer hybrid nanoparticles have been used to co-deliver siRNA and Gemcitabine for effective treatment of pancreatic cancer [28]. On the other hand, introduction of PEG moiety into PLGA has also been attempted so as to further reduce the hydrophobicity and negative charge on the surface and enhance interaction with the negatively charged siRNA. It has been reported that core-shell structured nanoparticles containing block co-polymers like PEG [9] forms a protective outer coating around the polyplex core containing cationic polymers complexed with siRNA and shields it by steric-stabilization. In our formulation, we have predicted that inclusion of PLGA or PLGA-PEG into the particles should not only prevent intra-fusion among liposomal particles resulting in the formation of larger particles but also they help to sustain aptamer stability and biding specificity. Recently, we have optimized another type of lipid-polymer hybrid nanoparticles to effectively knockdown mutant p53 in mouse osteosarcoma cells for the treatment of osteosarcoma (unpublished data). We have seen that the substitution of lipids by polymer in the nanoparticle formulation decreased the particle size and overall cytotoxicity but helped to increase siRNAs encapsulation efficiency within the particles (unpublished data). Similar pattern of the physiochemical properties of the hybrid nanoparticles has also been observed in the current study. The particle size decreases with the addition of polymer (PLGA) due to enhanced interfacial stabilization [29]. PEG chains are hydrophilic and orient themselves towards external aqueous phase improving overall solubility and decreasing aggregation, which results in further decrease of the particle size.
In this study, excess amount of PLGA or PLGA-PEG compared to Mal-PEG has been used because it is anticipated that excess PLGA or PLGA-PEG compared to Mal-PEG will sequester Mal-PEG-bound aptamer on the surface so that aptamer sequence self-entanglement due to electronic interaction would be minimized. It is assumed that the siRNA has been encapsulated into the particles because of its condensing nature with protamine sulphate. The particles were also vortexed vigorously to remove loosely bound siRNAs from the particle surface so that only aptamer on the particle surface can bind to the target cells.
The positively charged hybrid nanoparticles self-assemble with the negatively charged siRNAs through multiple electrostatic interactions forming stacks of lipid bilayers with siRNAs trapped in-between. Nanoparticles have high surface area to volume ratios [30] and multiple aptamers could be labeled on the particle surface. Mal-PEG present in the formulation attaches to the surface of the lipid bilayer and helps in aptamer functionalization (Fig. 4). Aptamer has amino-functional group through which they associate with Mal-PEG on the surface of the nanoparticles and facilitates target binding. Aptamer A6 is specific and it binds to Her-2 receptors overexpressed on the surface of breast cancer cells and helps in efficient cellular internalization of the nanoparticles.
Of the nine formulations developed, the best formulation based on the particle size and surface charge from the PLGA and PLGA-PEG group was measured F31 and F21, respectively. Again, F31 was found to be superior to F21 in terms of its particle size (after siRNA encapsulation and aptamer association) and siRNA encapsulation efficiency. Also, the cell cytotoxicity caused by the F21 formulation was higher than that of F31 formulation. As such, between these two groups (i.e. PLGA vs. PLGA-PEG), F31 (PLGA group) was chosen over F21 (PLGA-PEG group) to continue the rest of the study that included the measurement of the transfection efficiency and the knockdown of P-gp.
In this study, the siRNA used to knockdown P-gp was chosen from a published report [25]. Meng et al. has selected the siRNA from a panel of siRNAs undergone a high throughput screening of their efficacy to knock down P-gp on a multi-drug resistant breast cancer cell line [25]. They have showed a significant knockdown of P-gp at the heterogeneous tumor sites in the multi-drug resistant human breast cancer xenograft model where doxorubicin and the P-gp targeted siRNA were co-delivered by silica nanoparticles. We have used this custom siRNA to knock down P-gp in human and mouse breast cancer cells. The efficacy of P-gp targeted siRNA culminated with our delivery system was tested in both Her-2 (+) and Her-2 (−) breast cancer cells by using both aptamer-labeled and non-aptamer-labeled hybrid nanoparticles. The aptamer-labeled hybrid nanoparticles developed in our lab showcased a better knockdown of P-gp compared to lipofectamine as shown in Fig. 11. Another encouraging factor is that the hybrid nanoparticles can accomplish knocking down P-gp at a high serum concentration (i.e. 10% FBS) (as compared to lipofectamine transfection) which not only assures the protection of siRNA from the serum nucleases but also it brings hopes that the aptamer-labeled hybrid nanoparticles can be successfully administered in vivo to treat breast cancer in patients. Though P-gp is usually detected as a major band around 140–170 kDa in Western blotting, in our study, the major band of P-gp was detected between 55 and 65 kDa. It has been reported by Yoshimura et al. that a discrete band at 95 kDa and a sharp band at 55 kDa representing P-gp was observed with the KB-C2 cells [31]. However, when the cells were treated with trypsin, the intensity of the major140 kDa band declined whereas the intensity of the 95 kDa and 55 kDa band improved. They observed that even with the mild exposure of trypsin, the P-gp within the cells is separated into two small parts P1 and P2 whose combined molecular mass (150 kDa) is equal to the molecular mass of P-gp. Experiments on drug-resistant KB cells [32] also revealed that the 170 kDa protein after digestion with proteolytic enzyme trypsin got cleaved into three different fragments 70, 55 and 40 kDa. The separation of P-gp into 95 and 54–56 kDa fragments was also reported by Greenberger et al. [33]. In our study, the most significant changes of P-gp expression was always displayed by the 55–65 kDa band. Though trypsinization was known to induce fragmentation of P-gp, the reason behind the superior expression of 55–65 kDa band among other fragments is poorly understood which deems a thorough further investigation.
Knocking down P-gp by P-gp specific siRNA could increase the delivery of cancer drug to the breast cancer cells. However, since P-gp is just a single member of the vast ABC superfamily, it is quite likely that knocking down P-gp can indirectly induce the selection of other clones that express a different ABC member with overlapping drug selectivity. To resolve this, we are planning to assess the gene silencing of MRP (i.e. multidrug resistance protein) and BCRP (i.e. breast cancer resistance protein) proteins by different aptamer-labeled hybrid nanoparticles. If knocking down a single MDR gene is not enough for a long term inhibition of drug resistance, then two or three different siRNAs-targeted to MDR gene will be encapsulated into this aptamer-labeled hybrid nanoparticles. We have previously shown that multiple siRNAs targeted to the highly conserved 5′-untranslated region (UTR) of the HCV genome could be encapsulated into lipid nanoparticles for the treatment of HCV. Similar approaches will be considered to encapsulate multiple siRNAs should there be any requirements of taking all out actions to eradicate MDR.
In this study, we anticipate that development of a targeted delivery of siRNA specific to MDR gene using a nanocarrier system has the potential to overcome chemoresistance of breast cancer cells to therapeutic drugs such as doxorubicin. By enhancing the knockdown of MDR gene (i.e. P-gp), the aptamer-labeled P-gp siRNA encapsulated nanoparticles would produce a greater cellular internalization and direct accumulation of drugs (doxorubicin) in the nuclear compartment of breast cancer cells. If the P-gp specific siRNA is not selectively targeted to the breast cancer cells, it will not have a significant impact in the treatment of cancer. As such, by enhancing the knockdown of multidrug resistant genes, this aptamer-labeled targeted nanoparticle will open the door for the enhanced delivery of doxorubicin for the treatment of chemoresistance breast cancer.
Acknowledgments
This work is funded in part by the Louisiana Cancer Research Consortium, NIMHD grant number 5G12MD007595, NIGMS grant number 8UL1GM118967, CUR from Xavier University of Louisiana, BoR SURE Grant and NSF.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide : Sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–E386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 2.Peto R, Davies C, Godwin J, Gray R, Pan HC, Clarke M, Cutter D, Darby S, McGale P, Taylor C, et al. Comparisons between different polychemotherapy regimens for early breast cancer: meta-analyses of long-term outcome among 100,000 women in 123 randomized trials. Lancet. 2012;379:432–444. doi: 10.1016/S0140-6736(11)61625-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eastham JA, Grafton W, Martin CM, Williams BJ. Suppression of primary tumor growth and the progression to metastasis with p53 adenovirus in human prostate cancer. J Urol. 2000;164:814–819. doi: 10.1097/00005392-200009010-00055. [DOI] [PubMed] [Google Scholar]
- 4.Wehland M, Bauer J, Magnusson NE, Infanger M, Grimm D. Biomarkers for anti-angiogenic therapy in cancer. Int J Mol Sci. 2013;14:9338–9364. doi: 10.3390/ijms14059338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bao L, Haque A, Jackson K, Hazari S, Moroz K, Jetly R, Dash S. Increased expression of P-glycoprotein is associated with doxorubicin chemoresistance in the metastatic 4T1 breast cancer model. Am J Pathol. 2011;178:838–852. doi: 10.1016/j.ajpath.2010.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hao W, William H, Jin-ming Y. Small interfering RNA-induced suppression of MDR1 (P-glycoprotein) restores sensitivity to multidrug-resistant cancer cells. Cancer Res. 2003;63:1515–1519. [PubMed] [Google Scholar]
- 7.Stierlé V, Laigle A, Jollès B. The Reduction of P-Glycoprotein Expression by Small Interfering RNAs Is Improved in Exponentially Growing Cells. Oligonucleotides. 2004;14:191–198. doi: 10.1089/oli.2004.14.191. [DOI] [PubMed] [Google Scholar]
- 8.Howard KA. Delivery of RNA interference therapeutics using polycation-based nanoparticles. Adv Drug Deliv Rev. 2009;61:710–720. doi: 10.1016/j.addr.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 9.Davis ME. The first targeted delivery of siRNA in humans via a self-assembling, cyclodextrin polymer-based nanoparticle: from concept to clinic. Mol Pharm. 2009;6:659–668. doi: 10.1021/mp900015y. [DOI] [PubMed] [Google Scholar]
- 10.Peer D, Eun JP, Yoshiyuki M, Christopher VC, Motomu S. Systemic Leukocyte-Directed siRNA Delivery Revealing Cyclin D1 as an Anti-Inflammatory Target. Science. 2008;319:627–630. doi: 10.1126/science.1149859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Verdiere AC, Dubernet C, Nemati F, Soma E, Appel M, Ferte J, Bernard S, Puisieux F, Couvreur P. Reversion of multidrug resistance with polyalkylcyanoacrylate nanoparticles: towards a mechanism of action. Br J Cancer. 1997;76:198–205. doi: 10.1038/bjc.1997.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moghimi SM, Hunter AC. Poloxamers and poloxamines in nanoparticle engineering and experimental medicine. Trend Biotechnol. 2000;18:412–420. doi: 10.1016/s0167-7799(00)01485-2. [DOI] [PubMed] [Google Scholar]
- 13.Wong HL, Bendayan R, Rauth AM, Wu XY. Development of solid lipid nanoparticles containing ionically-complexed chemotherapeutic drugs and chemosensitizers. J Pharm Sci. 2004;93:1993–2004. doi: 10.1002/jps.20100. [DOI] [PubMed] [Google Scholar]
- 14.Bao L, Hazari S, Mehra S, Kaushal D, Moroz K, Dash S. Increased expression of P-glycoprotein and doxorubicin chemoresistance of metastatic breast cancer is regulated by miR-298. Am J Pathol. 2012;180:2490–2503. doi: 10.1016/j.ajpath.2012.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Soundararajan S, Chen W, Spicer EK, Courtenay-Luck N, Fernandes DJ. The nucleolin targeting aptamer AS1411 destabilizes Bcl-2 messenger RNA in human breast cancer cells. Cancer Res. 2008;68:2358–2365. doi: 10.1158/0008-5472.CAN-07-5723. [DOI] [PubMed] [Google Scholar]
- 16.Kang H, O’Donoghue MB, Liu H, Tan W. A liposome-based nanostructure for aptamer directed delivery. Chem Commun (Camb) 2010;46:249–251. doi: 10.1039/b916911c. [DOI] [PubMed] [Google Scholar]
- 17.Farokhzad OC, Cheng J, Teply BA, Sherifi I, Jon S, Kantoff PW, Richie JP, Langer R. Targeted nanoparticle-aptamer bioconjugates for cancer chemotherapy in vivo. PNAS. 2006;103:6315–6320. doi: 10.1073/pnas.0601755103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Edwards KA, Baeumner AJ. Synthesis of a liposome incorporated 1-carboxyalkylxanthine-phospholipid conjugate and its recognition by an RNA aptamer. Talanta. 2006;71:365–372. doi: 10.1016/j.talanta.2006.04.031. [DOI] [PubMed] [Google Scholar]
- 19.Kundu AK, Chandra PK, Hazari S, Pramar YV, Dash S, Mandal TK. Development and optimization of nanosomal formulations for siRNA delivery to the liver. Eur J Pharm Biopharm. 2012;80:257–267. doi: 10.1016/j.ejpb.2011.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Woodrow KA, CUY, Booth CJ, Saucier-Sawyer JK, Wood MJ, Saltzman WM. Intravaginal gene silencing using biodegradable polymer nanoparticles densely loaded with small-interfering RNA. Nat Mater. 2009;8:526–533. doi: 10.1038/nmat2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou J, Rossi JJ. Cell-type-specific, Aptamer-functionalized Agents for Targeted Disease Therapy. Mol Ther Nucleic Acids. 2014;3:E169. doi: 10.1038/mtna.2014.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kundu AK, Chandra PK, Hazari S, Ledet G, Pramar YV, Dash S, Mandal TK. Stability of lyophilized siRNA nanosome formulations. Int J Pharm. 2012;423:525–534. doi: 10.1016/j.ijpharm.2011.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chandra PK, Kundu AK, Hazari S, Chandra S, Bao L, Ooms T, Morris GF, Wu T, Mandal TK, Dash S. Inhibition of hepatitis C virus replication by intracellular delivery of multiple siRNAs by nanosomes. Mol Ther. 2012;20:1724–1736. doi: 10.1038/mt.2012.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kundu AK, Hazari S, Chinta DD, Pramar YV, Dash S, Mandal TK. Development of nanosomes using high- pressure homogenization for gene therapy. J Pharm Pharmacol. 2010;62:1103–1111. doi: 10.1111/j.2042-7158.2010.01140.x. [DOI] [PubMed] [Google Scholar]
- 25.Meng H, Mai WX, Zhang H, Xue M, Xia T, Lin S, Wang X, Zhao Y, Ji Z, Zink J, Nel AE. Codelivery of an Optimal Drug/siRNA Combination Using Mesoporous Silica Nanoparticles To Overcome Drug Resistance in Breast Cancer in Vitro and in Vivo. ACS Nano. 2013;7:994–1005. doi: 10.1021/nn3044066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gopalakrishnan AM, Kundu AK, Mandal TK, Kumar N. Novel nanosomes for gene delivery to Plasmodium falciparum-infected red blood cells. Sci Rep. 2013;3:1534. doi: 10.1038/srep01534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aigner A. Gene silencing through RNA interference (RNAi) in vivo: strategies based on the direct application of siRNAs. J Biotechnol. 2006;124:12–25. doi: 10.1016/j.jbiotec.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 28.Zhao X, Li F, Li Y, Wang H, Ren H, Chen J, Nie G, Hao J. Co-delivery of HIF1α siRNA and gemcitabine via biocompatible lipid-polymer hybrid nanoparticles for effective treatment of pancreatic cancer. Biomaterials. 2015;46:13–25. doi: 10.1016/j.biomaterials.2014.12.028. [DOI] [PubMed] [Google Scholar]
- 29.Budhian A, Siegel SJ, Winey KI. Haloperidol-loaded PLGA nanoparticles: systematic study of particle size and drug content. Int J Pharm. 2007;336:367–75. doi: 10.1016/j.ijpharm.2006.11.061. [DOI] [PubMed] [Google Scholar]
- 30.Liao J, Liu B, Jliu, Zhang J, Chen K, Liu H. Cell-specific aptamers and their conjugation with nanomaterials for targeted drug delivery. Expert Opin Drug Deliv. 2015;12:493–506. doi: 10.1517/17425247.2015.966681. [DOI] [PubMed] [Google Scholar]
- 31.Yoshimure A, Kuwazuru Y, Sumizawa T, Ichikawa M, Ikeda S, Uda T, Akiyama S. Cytoplasmic orientation and two-domain structure of the multidrug transporter, P-glycoprotein, demonstrated with sequence specific antibodies. J Biol Chem. 1989;264:16282–16291. [PubMed] [Google Scholar]
- 32.Bruggemann EP, Germann UA, Gottesman MM, Pastan I. Two different regions of P-glycoprotein [corrected] are photoaffinity-labeled by azidopine. J Biol Chem. 1989;264:15483–15488. [PubMed] [Google Scholar]
- 33.Greenberger LM, Lisanti CJ, Silva JT, Horwitz SB. Domain mapping of the photoaffinity drug-binding sites in P- glycoprotein encoded by mouse mdr1b. J Biol Chem. 1991;266:20744–20751. [PubMed] [Google Scholar]