

Abstract
Retinoblastomas can arise from cone photoreceptor precursors in response to the loss of pRB function. Cone precursor-specific circuitry cooperates with pRB loss to initiate this process and subsequently contributes to the malignancy. Intrinsic high level MDM2 expression is a key component of the cone precursor circuitry and is thought to inactivate p53-mediated tumor surveillance that could otherwise be induced in response to pRB loss. However, the MDM2-related MDM4 has also been proposed to abrogate p53-mediated tumor surveillance in the absence of detectable MDM2 in retinoblastoma cells, bringing into question the importance of high-level MDM2 versus MDM4 expression. Here we report that high-level MDM2 but not MDM4 has a consistent critical role in retinoblastoma cell proliferation in vitro as well as in orthotopic xenografts. Reduction of either MDM2 or MDM4 weakly induced p53, yet reduction of MDM2 but not MDM4 severely impaired proliferation and survival through a p53-independent mechanism. Specifically, MDM2 up-regulated the mRNA expression and translation of another component of the cone circuitry, MYCN, in retinoblastoma cells. Moreover, MYCN was essential to retinoblastoma cell growth and tumor formation, and ectopic MYCN partially reversed the effects of MDM2 depletion, indicating that MYCN is an important MDM2 target. These findings indicate that high-level MDM2 expression is needed in order to perform a critical p53-independent function and may obviate the need for genomic alterations to the p53 pathway during retinoblastoma tumorigenesis.
Retinoblastomas initiate with exceptionally high efficiency in response to the loss of functional pRB protein.1 The tumors are thought to arise from cone photoreceptor precursors, as pRB depletion induces proliferation and development of retinoblastoma-like tumors in cone precursors but not other retinal cell types.2 Moreover, retinoblastomas have a predominant cone protein expression pattern3 and progress from a cone precursor-like state to less differentiated states with increasing genomic alterations.4, 5 The proliferation of pRB-depleted cone precursors and retinoblastoma cells depends upon cone precursor features, such as high expression of cone factors RXRγ and TRβ2, high expression of the MDM2 and MYCN oncoproteins, and SKP2-mediated down-regulation of p27.2, 3, 6, 7 These observations imply that human cone-precursor circuitry collaborates with pRB loss to initiate and sustain tumorigenesis.
The identification of the cone precursor origin of retinoblastoma provides opportunities to define cell type-specific circuitry underlying the retina’s remarkable sensitivity to pRB loss. Among other features, the cone circuitry must circumvent tumor suppressor mechanisms that are thought to effectively respond to aberrant proliferative signaling in other cell types.8 For example, in many cell types, pRB loss may deregulate E2F and enable aberrant cell cycle entry,9–11 but tumorigenesis is blocked by p53-mediated surveillance.8 This p53 response comes about because deregulated E2F can induce expression of CDKN2AARF,12–14, which allows ARF to prevent MDM2-mediated p53 degradation.15 In most cancers, this surveillance circuitry is impaired by TP53 mutation, CDKN2AARF inactivation, or MDM2 amplification.16 However, most if not all retinoblastomas lack such alterations,3, 17, 18 suggesting that p53-mediated surveillance is impaired through an alternative mechanism.
Prior studies suggested that intrinsic high-level MDM2 expression in human cone-precursors attenuates p53-mediated tumor surveillance in pRB-depleted cone precursors as well as in retinoblastoma cells.2, 3 Concordantly, peptides and small molecules such as Nutlin-3a that block the MDM2-p53 interaction impair retinoblastoma cell survival.19–21 However, it was also suggested that the p53 pathway is abrogated in retinoblastoma by chromosome 1q gains encompassing the MDM2-related gene, MDM4 (also known as MDMX),21 in the absence of detectable MDM2.22, 23 The high-level MDM4 expression in the absence of detectable MDM2 hinted at an unusual mode of p53 regulation, since MDM2 and MDM4 normally cooperate to promote p53 degradation and to impair p53-mediated transactivation.24 To reconcile the proposed lack of MDM2 with the powerful effects of Nutlin-3a it was suggested that Nutlin-3a inhibits tumorigenesis primarily by impairing the MDM4-p53 interaction.21, 25
Nevertheless, the roles of MDM4 and MDM2 in retinoblastomas remain uncertain. 1q gains encompassing MDM4 are acquired after tumor progression4 and thus could not contribute to the cone precursors’ initial proliferative response to pRB loss nor to retinoblastomas that lack 1q gains.26 Moreover, although MDM4 was found to down-regulate p53-mediated apoptosis in response to DNA damage,21 it was not shown to be essential for retinoblastoma cell proliferation in the absence of DNA damage, which may be more relevant to the escape from p53-mediated surveillance.27 Also, MDM2 and MDM4 have increasingly appreciated yet poorly defined p53-independent functions whose roles in retinoblastoma have not been explored. These issues bring into question whether intrinsically high levels of MDM2, acquired high levels of MDM4, or both inactivate the p53 pathway and/or perform other functions in retinoblastoma cells.
Here, we re-assessed the roles of high-levels of MDM2 and MDM4 in retinoblastoma cell proliferation and survival. We report that high-level expression of MDM2 but not MDM4 performs a critical p53-independent function, and we propose that the exorbitant MDM2 expression that is needed to fulfill its p53-independent role obviates the need for genomic alterations to the ARF-MDM2-p53 tumor surveillance pathway in retinoblastoma tumors.
Results
High-level MDM2 but not MDM4 is required for retinoblastoma cell proliferation and tumorigenesis
To evaluate the roles of MDM2 and MDM4, we first compared their expression in five established retinoblastoma cell lines (Y79, WERI-1, RB176, RB177, CHLAVC-RB43), in five early passage retinoblastoma cell preparations (RB212, RB214, RB216, RB217, and RB218), and in human fetal retina. Both MDM2 and MDM4 were readily detected at higher levels in all retinoblastoma cell lines and in most of the primary cultures (4/5 for MDM2 and 3/5 for MDM4) as compared to the normal retina (Fig. 1A, Supplementary Fig. S1), consistent with prior reports.3, 19–21 One primary culture expressed MDM2 at levels similar to the normal retina, possibly reflecting distinct tumor evolution.
Figure 1. MDM2 but not MDM4 maintains retinoblastoma cell proliferation.
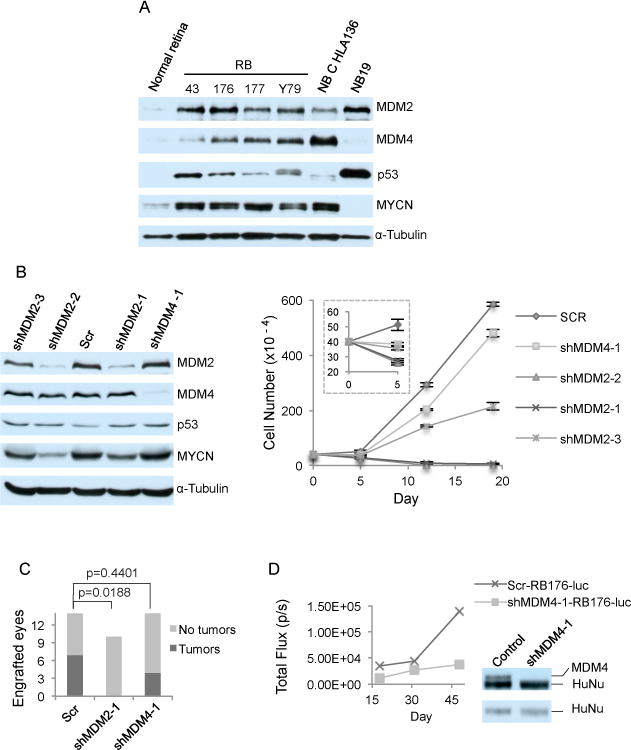
A. Western analysis of MDM2 (with SMP14), MDM4, p53, MYCN, and α-tubulin expression in post-fertilization week 19 human fetal retina and in four retinoblastoma (RB) and two neuroblastoma (NB) cell lines B. Western analysis at day 4 (left) and cell growth response (right) of RB176 cells after infection with lentivirus expressing shRNAs against MDM2 (shMDM2-1 and shMDM2-2) or against MDM4 (shMDM4-1), or expressing a scrambled shRNA control (Scr). Values and error bars denote mean and standard deviation (s.d.) of triplicate assays. C. Tumors formed up to 3.5 months after xenograft of RB176-luc cells transduced with shScr, shMDM2-1, or shMDM4-1 and engrafted into the sub-retinal space of athymic (nude) mice. P values are from two-tailed Fisher’s exact test. D. Representative tumor growth tracked over 1.5 months by bioluminescent imaging, (left) and MDM4 and HuNu expression in the tumors examined by Western blot sequentially probed with anti-HuNu (bottom) followed without stripping with anti-MDM2 (top).
Prior studies showed that high-level MDM2 expression is crucial for retinoblastoma cell proliferation and survival3 and that high-level MDM4 contributes to the retinoblastoma cell DNA damage response but has less effect in the absence of DNA damage.21 To compare their roles, we assessed the consequences of lentivirus-mediated transduction of MDM2-directed, MDM4-directed, and scrambled control shRNAs (referred to as shMDM2, shMDM4, and shScr). By four days after transduction, MDM2-directed shRNAs depleted MDM2 by ~70% and induced cell death in RB176 (Fig. 1B) as well as in WERI-1 and Y79 cells (Supplementary Fig. S2), as previously shown for Y79, RB177, and RB178.3 Cell death was associated with increased cleaved caspase 3 (CC3) in past studies of Y793 and in these analyses of RB176 (Fig. 2B), implying that MDM2 was needed to suppress apoptosis. In contrast, a similar or greater level of MDM4 knockdown only marginally diminished proliferation in RB176, WERI-1, and Y79 (Fig. 1B, Supplementary Fig. S2), in agreement with a prior analysis of MDM4 knockdown in the absence of radiation.21 MDM4 expression remained suppressed at 15 days after shMDM4 transduction in RB176 and WERI-1 (Supplementary Fig. S2A, B), demonstrating that the cells had not escaped the MDM4 knockdown but readily tolerated reduced MDM4 expression. Thus, high-level MDM2 but not MDM4 is critical to retinoblastoma cell proliferation and survival in vitro.
Figure 2. MDM2 maintains retinoblastoma cell proliferation in a p53-independent manner.
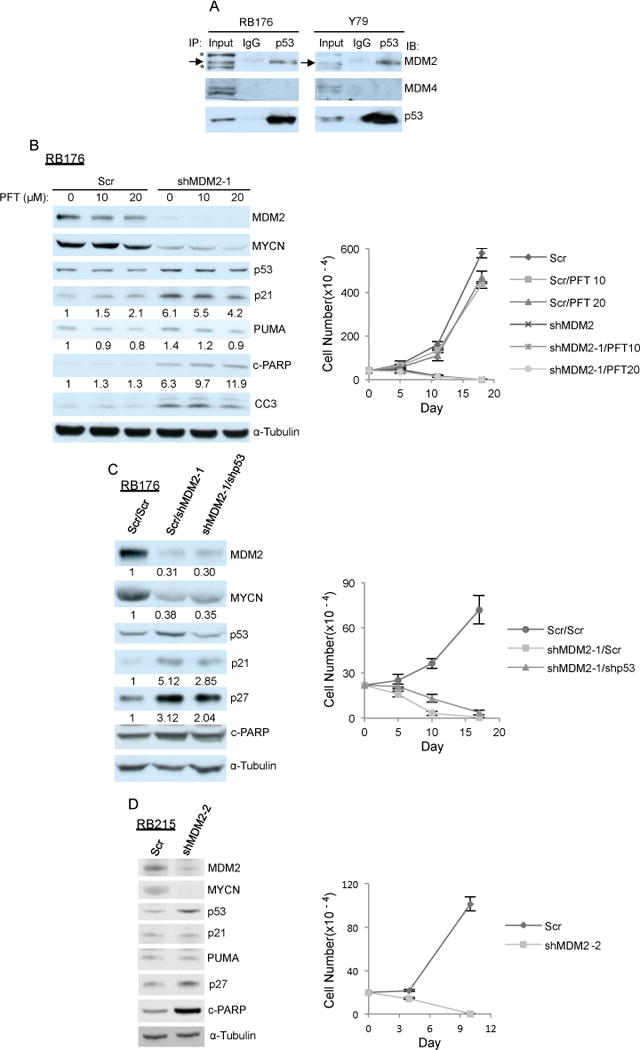
A. p53 association with MDM2 but not MDM4 in RB176 and Y79 cells detected by p53 or control IgG immunoprecipitation followed by MDM2, MDM4, and p53 western. *, a non-specific band seen with SMP-14 antibody. B. Western analysis after 6 h of treatment (left) and cell growth response (right) of RB176 cells treated with 10 μM or 20 μM PFT-α starting 4 days after infection with lentivirus expressing shRNA against MDM2 (shMDM2-1) or a scrambled control (Scr). C. Western analysis at day 4 (left) and cell growth response (right) of RB176 cells after infection with lentivirus co-expressing shMDM2 and shp53 or shMDM2 and Scr. Signal intensity was normalized to α-tubulin and expression relative to Scr control is indicated below corresponding panels. D. Western analysis at day 4 (left) and cell growth response (right) of TP53R175H/R175H CHLA- RB215 cells after infection with lentivirus expressing shMDM2-2 or a scrambled control (Scr). A similar result was obtained separately using shMDM2-1 (not shown).
To assess whether MDM2 is also required and MDM4 is also dispensable for tumorigenesis in vivo, we determined whether depletion of each prevents tumors from arising after orthotopic xenograft. Luciferase-expressing RB176-luc cells were engrafted into the sub-retinal space of nude mice two days after transduction with shScr, shMDM2, or shMDM4. Over two months, scrambled control and MDM4-depleted but not MDM2-depleted cells formed tumors (Fig. 1C). Tumors derived from cells transduced with shScr first appeared four weeks after xenograft whereas those derived from MDM4-depleted cells first appeared at seven weeks, in keeping with the slower proliferation of MDM4-depleted cells in vitro. Importantly, comparison of MDM4 levels to a human-specific protein (HuNu) revealed that the tumors remained MDM4-deficient (Fig. 1D). Thus, retinoblastomas can develop with substantially reduced MDM4 but not with reduced MDM2 expression.
High-level MDM2 enables retinoblastoma cell proliferation via a p53-independent function
We next investigated the signaling pathways through which high-level MDM2 but not MDM4 maintains retinoblastoma cell survival, initially focusing on whether high MDM2 is needed for p53 degradation and suppression of p53-mediated apoptosis. Consistent with this possibility, MDM2 but not MDM4 co-immunoprecipitated with p53 from lysates of RB176 and Y79 cells (Fig 2A). Importantly, the DO-1 p53 antibody used for this analysis co-immunoprecipitated both MDM2 and MDM4 from lysates of other cell types,28, 29 despite that DO-1 recognizes an epitope close to the p53 residues that contact MDM2.28 Thus, our findings suggest that p53 preferentially binds MDM2 in retinoblastoma cells. However, despite the preferential association of p53 with MDM2, both MDM2 knockdown and MDM4 knockdown weakly induced p53 (Fig. 1B), and co-knockdown of MDM2 and MDM4 additively but still only modestly induced p53 (Supplementary Fig. S3A). MDM2 knockdown also only modestly induced expression of p53 targets p21 and PUMA (Fig. 2B). In contrast, p53 and p21 were far more dramatically induced by 20 μM Nutlin-3a (Supplementary Fig. S3B), which is a potent antagonist of MDM2-p53 binding (Ki = 0.7 μM) and a weak antagonist of MDM4-p53 binding (Ki = 28 μM).21 The weaker p53 induction after MDM2 knockdown as compared to Nutlin-3a treatment likely relates to residual MDM2 mediating p53 degradation after MDM2 knockdown whereas the more complete inhibition of MDM2 blocks p53 degradation in response to Nutlin-3a.
The apoptotic response to MDM2 but not MDM4 knockdown despite similar induction of p53 suggested that MDM2 might promote retinoblastoma cell survival through a p53-independent mechanism. Accordingly, we used three approaches to assess whether p53 was needed for the death of MDM2-depleted retinoblastoma cells.
We first evaluated whether the death of MDM2-depleted retinoblastoma cells was suppressed by Pifithrin-α (PFT-α), which specifically blocks p53-induced transcriptional activity.30 We infected cells with shMDM2 and shScr control virus in the absence or presence of PFT-α, and analyzed expression of p53 and p21 (Fig. 2B). In cells transduced with the shScr control, PFT-α did not impede basal p21 expression, implying that there was little if any basal p53 transcriptional activity. Following MDM2-depletion, both p53 and p21 were induced, and two markers of apoptosis, cleaved PARP and cleaved caspase 3 (CC3), were induced coinciding with cell death. PFT-α decreased the induction of p21 and PUMA as previously observed,30, 31 consistent with partial p53 inhibition. However, PFT-α did not diminish the effect of MDM2 knockdown on cleaved PARP, on CC3, or on retinoblastoma cell death (Fig. 2B), suggesting that inhibition of p53-mediated transactivation did not mitigate the death of MDM2-depleted retinoblastoma cells.
Because PFT-α only partially reversed p53-induced transcriptional changes, and might not affect other p53 functions, we next evaluated the effect of shRNA-mediated p53 depletion. In RB176 cells, p53 knockdown abrogated the induction of p21 following MDM2 depletion, and slightly delayed but did not abrogate cell death associated with induction of the apoptosis marker, cleaved PARP (Fig. 2C). These findings suggested that p53 marginally contributed yet was not required for apoptosis of MDM2-depleted retinoblastoma cells.
We also examined the effect of MDM2 depletion in retinoblastoma cell line CHLA-RB215, which has a transactivation defective TP53 R175H mutation32 but no wild type TP53 allele (data not shown). In these cells, MDM2 knockdown increased p53 levels (Fig. 2D), consistent with MDM2’s ability to destabilize mutant as well as wild type p53.33 However, MDM2 knockdown did not induce p53 targets such as p21 or PUMA, in keeping with the impaired transcriptional activation of the p53 R175H mutation.32 Importantly, MDM2 depletion induced CHLA-RB215 cell death along with the apoptosis marker, cleaved PARP (Fig. 2D), conclusively showing that MDM2 has a p53-independent survival function in retinoblastoma cells.
MDM2 promotes MYCN expression through a p53-independent mechanism
Besides modestly inducing p53, MDM2 but not MDM4 knockdown markedly diminished expression of MYCN and increased expression of p27 (Fig. 1B, 2B, 2C, Supplementary Fig. S2). In RB176 cells, shRNA-mediated MDM2 knockdown by ~ 70% downregulated MYCN by ~ 65% (Fig. 2C) and increased p27 by 2- to 3-fold. Similar changes were evident in WERI-1, Y79, and CHLA-RB215 (Fig. 2D, Supplementary Fig. S2A–C), and similar effects were elicited by three different MDM2-directed shRNAs (Fig. 1B).
Interestingly, MYCN was not down-regulated following the p53 induction elicited by MDM4 knockdown or by Nutlin-3a (Fig. 1B, Supplementary Figs. S2A–C, S3A–B). Moreover, MYCN down-regulation following MDM2 knockdown was not affected by the inhibition of p53 by PFT-α or by the co-knockdown of p53 in RB176 (Fig. 2B, C) and also occurred in the p53-mutant CHLA-RB215 (Fig. 2D). Thus, MDM2 sustained MYCN expression through a p53-independent mechanism. Likewise, the increased expression of p27 following MDM2 knockdown was only modestly affected by co-knockdown of p53 in RB176 (Fig. 2C) and occurred in CHLA-RB215 (Fig. 2D), implying that it was largely p53-independent.
As decreased MYCN and increased p27 were previously found to impair retinoblastoma cell proliferation,3, 7 we examined whether these changes underlied MDM2’s p53-independent role. We first examined whether MDM2 was needed to sustain a level of MYCN that is needed for retinoblastoma survival. In RB176 (Fig. 1B, 2C) and CHLA-RB215 cells (Fig. 2D), MDM2 knockdown down-regulated MYCN by ≥ 75 % coincident with retinoblastoma cell death. Similarly, MYCN-directed shRNAs that downregulated MYCN by ≥ 75% induced cell death in RB176 and CHLA-RB215 (Fig. 3A, 3C). This implied that high-level MDM2 was required to achieve the high level of MYCN that is needed for retinoblastoma cell viability in vitro. MYCN knockdown also completely blocked production of orthotopic RB176 xenograft tumors without down-regulation of MDM2 or MDM4 (Fig. 3B), implying that MDM2 is needed to sustain the high MYCN levels that are critical to retinoblastoma tumor growth in vivo.
Figure 3. MDM2 maintains retinoblastoma cell proliferation in part by promoting MYCN expression.
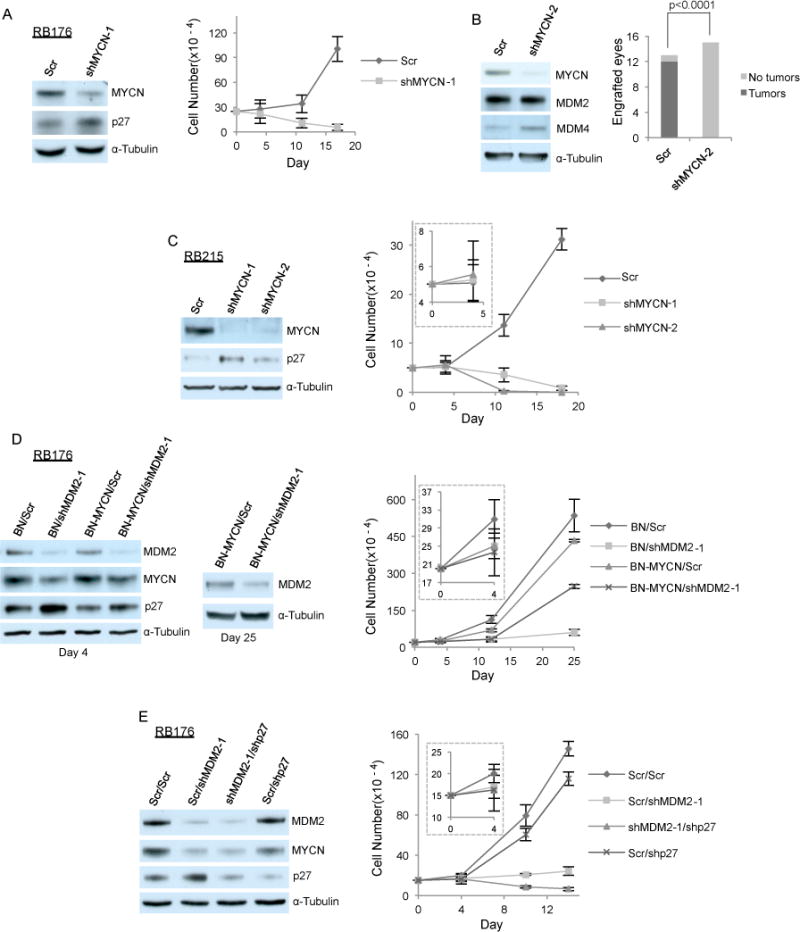
A. Western analysis at day 4 (left) and cell growth response (right) of RB176 cells after infection with lentivirus expressing shRNA against MYCN or a scrambled control (Scr). B. Tumors formed by RB176-luc cells transduced with shScr but not by those transduced with shMYCN in mouse subretinal xenografts (right, P-value from two-tailed Fisher’s exact test). Western analysis of the cells used for xenograft (left). C. Western analysis at day 4 (left) and cell growth response (right) of CHLA-RB215 cells after infection with lentivirus expressing shRNA against MYCN or Scr. D. Western analysis at days 4 and 25 (left and middle) and cell growth response (right) of RB176 cells after co-infection with lentivirus expressing shRNA against MDM2 or Scr and with lentivirus expressing MYCN under control of the EF1α promoter (BN-MYCN) or the empty vector (BN). E. Western analysis at day 4 (left) and cell growth response (right) of RB176 cells after co-infection with lentivirus expressing shMDM2 or Scr and with lentivirus expressing shp27 or Scr.
Moreover, using appropriate ratios of MDM2 knockdown and MYCN expression constructs to restore the original MYCN level partially rescued the impaired RB176 cell growth caused by MDM2 depletion (Fig. 3D). This effect of restoring the original MYCN levels, without MYCN overexpression, confirmed that MYCN is an important MDM2 target. Interestingly, this rescue was evident only when transducing limiting shMDM2 that incompletely impaired proliferation, implying that MDM2 contributes to retinoblastoma cell survival in part by maintaining MYCN and in part through additional functions. Combining p53 depletion with MYCN restoration did not further rescue the growth of MDM2-depleted RB176, and MYCN restoration failed to restore growth in MDM2-depleted CHLA-RB215 (data not shown), indicating that the additional MDM2 functions are also p53-independent.
We next examined whether MDM2 was needed to sustain sufficiently low p27 expression to enable retinoblastoma cell survival. Depending upon the experiment and cell line, MDM2 knockdown increased p27 expression by ~ 1.5- to 3-fold. However, in prior studies, a far higher-level ectopic p27 expression only modestly impaired retinoblastoma cell proliferation and survival7, 34. Using appropriate ratios of shMDM2 and shp27 to restore the original p27 level did not rescue the growth of MDM2-depleted cells (Fig. 3E), implying that the shMDM2-induced increase in p27 did not mediate retinoblastoma cell death and growth arrest. Interestingly, MYCN knockdown also induced an increase in p27 (Fig. 3A, C), as previously seen in neuroblastoma cells.35 Restoring MYCN largely prevented the increased p27 in MDM2-depleted cells (Fig. 3D), indicating that MDM2 suppresses p27 in part by sustaining high MYCN levels.
MDM2 promotes MYCN translation in retinoblastoma cells
As the p53-independent effects of MDM2 were in part mediated by MYCN, we next investigated the mechanism by which MDM2 promotes MYCN expression. Importantly, MDM2 knockdown decreased MYCN protein to a greater extent than it decreased MYCN RNA. For example, in RB176, MDM2 knockdown with two MDM2 shRNAs respectively decreased MYCN RNA by 9% and 31% at day 3 and by 17% and 52% at day 4, whereas MYCN protein expression declined by 44% and 61% at day 3 and by 64% and 90% at day 4 (Fig. 4A, B). The greater decline in MYCN protein than in MYCN RNA suggested that MYCN was regulated in part via a post-transcriptional mechanism. To test whether MDM2 regulates MYCN protein stability, we examined the MYCN half-life in RB176 cells treated with the protein synthesis inhibitor cycloheximide (CHX) starting four days after shRNA transduction. Although MYCN expression in the MDM2-knockdown cells was greatly reduced at the beginning of the treatment, the MYCN half-life was unchanged (Fig. 4C).
Figure 4. MDM2 regulates MYCN protein expression without altering its stability.
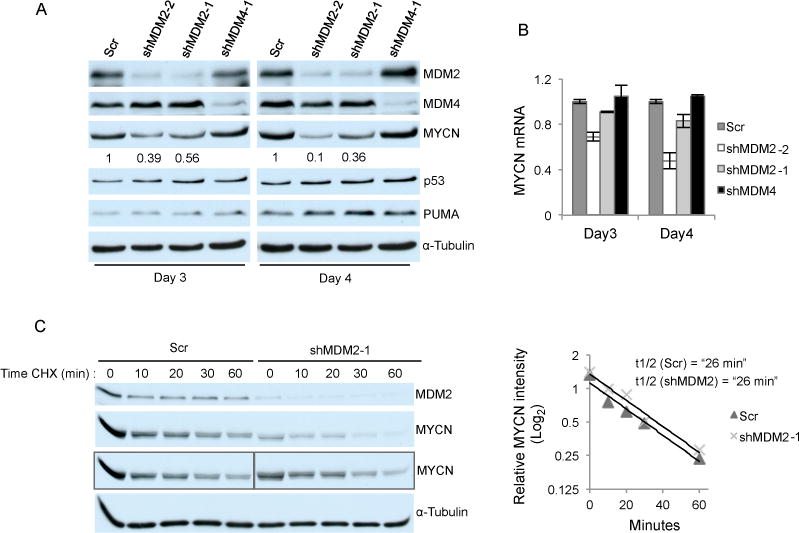
A. Western analysis of RB176 cells 3 or 4 days after infection with lentivirus expressing shRNA against MDM2 or MDM4 or with a scrambled control (Scr). Signal intensity of MYCN was normalized to α-tubulin. Numbers below the MYCN panel indicate relative MYCN expression upon MDM2 knockdown compared to Scr control. B. qRT-PCR analysis of the same samples plotted after normalization to GAPDH. Values and error bars denote mean and s.d. of duplicate assays. C. Western analysis of RB176 cells treated with 30 μM CHX 4 days after infection with lentivirus expressing shRNA against MDM2 and Scr (left). Boxed MYCN panels show either the same exposure (for Scr samples) or a longer exposure (for shMDM2 samples) of the western analysis in the above panel. The level of MYCN at each time point was quantified using the longer and more equivalent shMDM2 exposure, normalized to α-tubulin, and the Log2 values plotted (right), and MYCN half life determined. Data is representative of three independent experiments.
To evaluate whether MDM2 depletion impaired MYCN translation, we labeled nascent cellular proteins in shMDM2- and shScr-transduced RB176 cells with the alanine analog L-azidohomoalanine (AHA), then lysed and sequentially immunoprecipitated MYCN and α-tubulin (as a control), reacted the immunoprecipitated AHA-labeled protein with a TAMRA-alkyne, separated proteins by gel electrophoresis, and detected the newly synthesized immunoprecipitated MYCN and α-tubulin either by UV imaging of TAMRA fluorescence or by immunoblotting with anti-TAMRA antibody (Fig. 5A).36 When normalized to α-tubulin, newly synthesized AHA-labeled MYCN in RB176 cells transduced with MDM2 shRNAs was 20% and 13% (for shMDM-1 and shMDM-2, respectively) of that in the scrambled control (Fig. 5B). This was similar to the reductions in total MYCN protein determined by western blot and was reduced by ~ 40% and ~ 85% relative to MYCN RNA at the same time point (Fig. 5C, 5D). These results are representative of three independent experiments and MYCN translation was also reduced relative to MYCN RNA in Y79 cells (Fig. S4). Thus, we conclude that MDM2 maintains MYCN mRNA expression and promotes MYCN protein translation via a p53-independent mechanism in retinoblastoma cells.
Figure 5. MDM2 regulates MYCN translation.
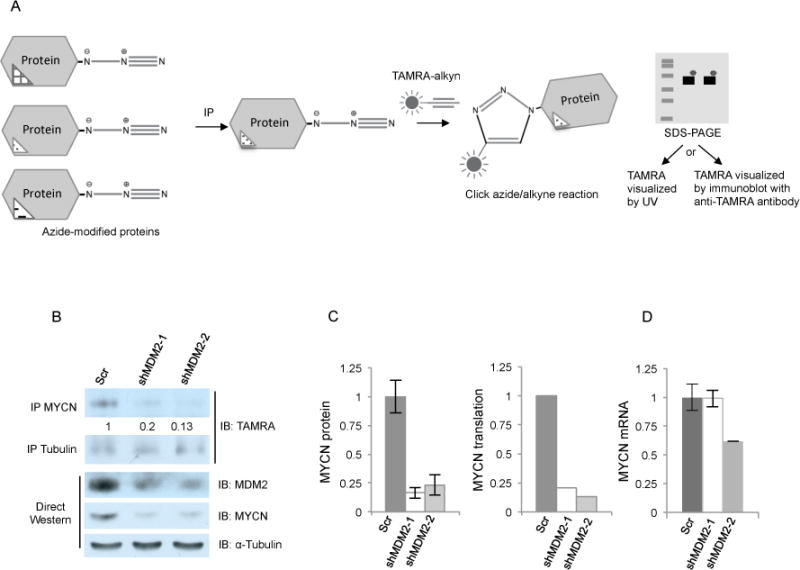
A. Diagram of protein translation assay. Nascent proteins were labeled with the azide-modified alanine analog- azidohomoalanine (AHA), immunoprecipitated, and incorporated AHA reacted with a fluorescent TAMRA labeled alkyne. TAMRA labeled proteins were separated by SDS-PAGE and detected by immunoblot using anti-TAMRA antibody (in Fig. 5b) or by UV fluorescence (in Supplementary Fig. S4). B. Protein translation assay performed on AHA-labeled RB176 cells 4 days after infection with lentivirus expressing either of two shRNAs against MDM2 or a scrambled control (Scr). AHA-labeled cell lysates either were sequentially immunoprecipitated with MYCN and then α-tubulin antibodies followed by anti-TAMRA immunoblot (top) or were used for direct Western analysis of MDM2, MYCN, and α-tubulin (bottom). C. Quantitation of total MYCN protein (the average and s.d. of two western analyses of the same lysate) and newly translated MYCN protein, both normalized to α-tubulin. D. qRT-PCR analysis of MYCN RNA from the same AHA-labeled cells as used for protein analyses (average and s.d. of triplicate technical replicates).
Discussion
Retinoblastomas are unique among human cancers in that they form with exceptionally high penetrance in response to RB1 inactivation and usually lack genomic changes in the ARF–MDM2–p53 tumor surveillance circuitry.3, 17, 18 This and other evidence suggested that retinoblastomas derive from a cell type – the cone photoreceptor precursor – in which the ARF–MDM2–p53 axis is impaired due to intrinsically high MDM2 expression, which eliminates a need for MDM2 amplification and for TP53 or CDKN2A mutation.3 Concordantly, MDM2 has a critical role in proliferating pRB-depleted cone precursors as well as in retinoblastoma cells.2, 3 Retinoblastomas have also been proposed to abrogate p53-mediated surveillance by a chromosome 1q gain-related increase in MDM4 in the absence of MDM2,21–23 at variance with a critical role for cone-related MDM2 regulation. Here, we demonstrate that high-level MDM2 but not MDM4 is needed for retinoblastoma cell proliferation and survival, yet acts predominantly via a p53-independent mechanism.
Whereas high-level MDM2 but not MDM4 was needed for proliferation and survival in each of five retinoblastoma cell lines, MDM2 knockdown and MDM4 knockdown engendered similar and only weak induction of p53 and p53 targets. Moreover, this induction of p53 was well tolerated, perhaps in part because the increase in p53 was modest and in part because of a high level of NANOS-mediated suppression of p53-activating kinases in retinoblastoma cells.37 The modest induction of p53 in response to MDM2 and/or MDM4 depletion contrasted with the dramatic induction of p53 and p53-mediated cell death in response to Nutlin-3a,20 most likely because residual MDM2 mediates p53 degradation after MDM2 knockdown whereas Nutlin-3a effectively blocks MDM2-mediated p53 degradation. Thus, the high-level MDM2 expression critical to cultured retinoblastoma cell proliferation substantially exceeds that which is needed to suppress p53-mediated apoptosis.
Our results do not support a critical role for MDM4 in the absence of MDM2 in retinoblastoma cells.21–23 In the McEvoy et al. studies,22,23 the failure to detect MDM2 protein was inconsistent with prior reports,3,18,19 including the same group’s analysis of a representative tumor.21 Furthermore, the failure to detect MDM2 RNA in microarray analyses22 could be explained by an initially incorrect MDM2 3′ untranslated sequence designation in the NCBI RefSeq database (data not shown). Regardless of the basis of the discrepancies, we detected MDM2 at or above the average level of the developing retina in each of five primary retinoblastomas and in each of five retinoblastoma cell lines. Moreover, although MDM4 depletion sensitized WERI-1 cells to DNA damaging agents it barely reduced WERI-1 cell proliferation in the absence of DNA damage,21 as confirmed here in multiple retinoblastoma cell lines. In contrast, MDM2 knockdown induced total cell death in all retinoblastoma lines examined in the absence of DNA damage. Importantly, p53-mediated tumor suppression may be more appropriately evaluated in the absence of external DNA damage since the tumor surveillance circuitry appears to respond to oncogenic rather than DNA damage signaling.27, 38, 39 Thus, our work shows that high-level MDM2 but not MDM4 is critical to retinoblastoma cell growth and survival, whereas high-level MDM4 may enhance cell proliferation and impede DNA damage-induced apoptosis.
On the other hand, MDM4 clearly promoted retinal cell survival both in Rb1/p107-mutant mouse retina and in pRB-depleted human retina.21 However, the pRB-deficient retinal cells examined in these models do not appear to represent human retinoblastoma. Tumors formed in Rb1/p107 knockout retina have a predominant amacrine or horizontal cell protein expression phenotype40 and a variant of the model (lacking Rb1 and p130 and hemizygous for p107) appeared to have a horizontal cell origin,41 whereas human retinoblastomas have a predominant cone protein expression phenotype and can derive from pRB-depleted cone precursors.2, 3 Similarly, human fetal retinal cells that proliferated in response to combined pRB loss and MDM4 overexpresion may have been retinal progenitor cells,21 which have a robust apoptotic response to pRB loss.2 In contrast, in isolated cone precursors, pRB knockdown increased survival, induced proliferation, and enabled development of retinoblastoma-like tumors. These observations suggest that MDM4 over-expression promotes survival and is synthetically oncogenic in apoptosis-prone pRB-deficient retinal progenitor cells but not in the pRB-deficient cone precursors that appear to give rise to retinoblastoma in humans.
The lack of requirement for MDM4 brings into question whether the 1q gains detected in 45 – 78% of retinoblastomas26 and more prevalent in later, less differentiated tumors4 target MDM4, concordant with evidence that 1q gains do not correlate with increased MDM4 RNA expression.42 Our data suggest that if 1q gains were to enhance MDM4 levels this might only marginally enhance retinoblastoma cell proliferation. Given MDM4’s ability to suppress p53,43 it is not surprising that high-level MDM4 impedes the p53-mediated DNA damage response and enhances proliferation. However, it is surprising and important that high-level MDM4 is not required for retinoblastoma cell proliferation and tumorigenesis. This argues against the use of drugs that selectively target MDM4 and supports the use of agents that are specific to MDM2 or that target MDM2 and MDM444 in retinoblastoma patients.
In addition to high-level expression of MDM2 and MDM4, retinoblastomas have been proposed to abrogate p53-mediated surveillance by miR-24-mediated down-regulation of p14ARF and by NANOS-mediated suppression of p53-activating kinases,37, 45 which was proposed to result from pRB loss. Thus, retinoblastoma cells may acquire several defects in the ARF–MDM2–p53 axis in addition to their intrinsic high MDM2 expression in the cell-of-origin circuitry.
The anti-proliferative effects of MDM2 depletion were not affected by pharmacologic or genetic inactivation of p53, indicating that MDM2 enables retinoblastoma cell proliferation through one or more p53-independent function. We found that a key p53-independent MDM2 function is to sustain high-level expression of MYCN. Indeed, MYCN was expressed in each of five RB1−/− retinoblastoma cell lines and in each of five RB1−/− retinoblastoma cultures at high levels similar to that of MYCN-amplified neuroblastoma cells. Although MYCN expression depended upon MDM2 in all cell lines examined, MDM2 and MYCN expression did not precisely correlate, implying that additional factors also impact MYCN levels. Importantly, MYCN was critical to retinoblastoma cell survival in vitro and for development of orthotopic xenografts, suggesting that non-MYCN amplified RB1−/− retinoblastomas depend upon high MYCN to an extent similar to their MYCN amplified RB1+/+ retinoblastoma and neuroblastoma counterparts.46, 47 We also found that MDM2 increases MYCN RNA and promotes MYCN translation, as previously seen in MYCN amplified neuroblastoma cell lines.48 As TP53, CDKN2A, and MDM2 changes are rare in primary neuroblastomas,47,49,50 MDM2 may concurrently drive MYCN expression and impede p53 expression in early neuroblastoma as well as in retinoblastoma genesis.
A detailed understanding of the mechanism by which MDM2 drives MYCN expression may provide ways to target MDM2’s p53-independent oncogenic function in retinoblastoma, neuroblastoma, and perhaps other tumors. As a first priority, it will be critical to determine whether MDM2 directly or indirectly controls MYCN expression and similarly regulates other tumorigenesis-related proteins. Indirect effects mediated by cell growth inhibition or death signaling seem unlikely because MYCN levels declined prior to overt viability changes, but cannot be ruled out. As an E3 ubiquitylation and neddylation ligase, MDM2 could regulate the stability of RNA binding proteins,51 and as an RNA binding protein itself,52 MDM2 could affect translation or microRNA function.
In summary, retinoblastoma cells consistently express high-level MDM2 that in turn sustains high expression of MYCN, a critical driver of retinoblastoma tumorigenesis. Our data suggest that the high MDM2 levels needed to enhance MYCN expression exceed those that are needed to down-regulate p53 to innocuous levels, and thus curtails the need to select for the genomic inactivation of the ARF-MDM2-p53 tumor surveillance pathway in retinoblastoma cells. The MDM2-MYCN axis provides a new therapeutic target for retinoblastoma and neuroblastoma and provokes the question whether this axis operates in additional human malignancies.
Materials and Methods
Cell culture
All retinoblastoma cells were cultured in RB medium and maintained at 37°C with 5% CO2.2 RB176 and RB177 are established cell lines.3 Y79 and WERI-RB1 were from ATCC. CHLA-VC-RB43 and CHLA-RB215 were established by culturing in IMDM with 20% fetal bovine serum (FBS) and 1x Insulin-Transferrin-Selenium (ITS) (Sigma-Aldrich), followed by over 10 passages in RB medium, will be described separately. Early passage RB212, RB214, RB216, RB217, and RB218 were explanted and cultured for less than 1 month.
shRNA and cDNA constructs
Lentiviral shRNA constructs used the pLKO.1 vector from the TRC library (Sigma-Aldrich) or were produced by inserting shRNA sequences as described (Addgene http://www.addgene.org/tools/protocols/plko/). TRC library designations and target sequences are: shMDM2-1:TRCN0000003380, 5′-CTCAGCCATCAACTTCTAGTA; shMDM2-2:TRCN0000003377, 5′-GATTCCAGAGAGTCATGTGTT; shMDM4-1:TRCN0000003858, 5′-GTTCACTGTTAAAGAGGTCAT; shMDM4-2:TRCN0000003857, 5′-CACCTAGAAGTAATGGCTCAA, shMYCN-1:TRCN0000020695, 5′-CAGCAGCAGTTGCTAAAGAAA; shMYCN-2:TRCN0000020696, 5′-CGGACGAAGATGACTTCTACT; shp53:TRCN0000003753, 5′-CGGCGCACAGAGGAAGAGAAT, shp27:TRCN0000039930, 5′-GCGCAAGTGGAATTTCGATTT. The Bidirectional Neomycin resistance (BN) lentiviral cDNA expression vector utilizes the EF1α enhancer-promoter to drive a gene of interest and a CMV minimal promoter to drive Neor as described.2 BN-MYCN was produced by inserting MYCN cDNA sequences 729–2152 (NM_001293228.1, isoform 1) into the BN PshAI site using In-Fusion (Clontech).
Lentivirus production and infections
Lentivirus was produced in 15cm dishes by reverse transfection of Lenti-X cells (Clontech) using 17.5 μg vector, 8.75 μg pVSV-G, 17.5 μg pCMV-dR8.91 and 219 μl 0.6 μg/μl polyethylenimine (PEI) (Polysciences). DNAs and PEI were separately diluted in 2.5 ml of DMEM (high glucose), incubated for 5 min, combined and incubated for 15 min, added to 3×107 Lenti-X cells suspended in 20 ml of DMEM with 10% FBS, Supernatants were collected at 48 and 68 hours post-transfection and filtered through 0.45 μm filter. shMDM2, shMDM4, and shScr infections used 5 ml of virus for 5×105 cells. For co-knockdown infections, 100 μl unconcentrated shp53 or shp27 supernatant was combined with 5 ml unconcentrated shMDM2 or shScr supernatant before infection. For MYCN experiments, BN or BN-MYCN virus was concentrated by centrifugation at 26,000 rpm (SW40Ti rotor, Beckman) at 6°C for 90 min followed by suspension in 500 μl of RB medium and infection of 5×105 cells in ~50 μl of medium, followed the next day by re-infection with unconcentrated shMDM2 or shScr. All infections were in 4 μg/ml polybrene (Sigma-Aldrich) and initiated by pipetting 20 times. After overnight culture, virus-infected cells were diluted with 1ml (for concentrated virus) or 8 ml (for unconcentrated virus) of RB medium. Infected cells were selected starting 48 h post-infection with 1–2 μg/ml puromycin (1.5 μg/ml for RB176 and WERI-RB1, 1 μg/ml for RB194, 2 μg/ml for Y79) for 48 hours for pLKO.1 constructs or with 200 μg/ml G418 for 7 days for BN constructs, and fed every 2–3 days by replacing two-thirds of the media.
Immunoprecipitation and immunoblotting
Cell lysates were prepared for Western immunoblotting by suspending 1–2×105 cells in lysis buffer (20 mM Tris pH 7.5, 200 mM NaCl, 0.5% Triton-X100, 0.5 mM EDTA, 10 mM DTT with protease (cOmplete) and phosphatase (PhosSTOP) inhibitors (Roche) on ice for 5 min, and centrifuged at 14,000 rpm for 10 min at 4°C. 20–50 ug of protein was separated by SDS-PAGE.
For co-immunoprecipitations, cells were treated with 50 μM MG13228 (Millipore) for 14 h, lysed in lysis buffer (50 mM Tris pH 7.5, 150 mM NaCl, 0.2% Triton-X100, 2 mM EDTA, 10% glycerol with inhibitors as above) for 10 min, and sonicated on ice for 10 seconds at 20% AMPL using a Qsonica Q500 sonicator. The lysate was centrifuged at 14,000 rpm as above for 10 min at 4°C. The supernatant was incubated with 2 μg of p53 DO-1 antibody (Santa Cruz) or mouse IgG with rotation for 2 hours. 30 μl Protein A/G beads (Santa Cruz) were added and incubated with rotation for 2 hours. Beads were pelleted (1,000 rpm F2402H fixed angle rotor) and washed three times with 1 ml of lysis buffer for 5 min, separated by SDS-PAGE and transferred to PVDF membranes.
Primary antibodies were MDM2: SMP14(Santa Cruz, sc-965) 1:200 and 4H26L4 (InVitrogen, 700555) 1:300 (4H26L4 was used for all MDM2 westerns unless noted), MDM4: 8C6 (Millipore, 04-1555) 1:300, MYCN: NCMII100 (Santa Cruz, sc-56729) 1:200, p53: FL393 (Santa Cruz, sc-6243) 1:300, p21: C-19 (Santa Cruz, sc-397) 1:100, p27: (Becton Dickenson, 610241) 1:1000, PUMAα/β: H-136 (Santa Cruz, sc-28226) 1:200, PARP: (Cell Signaling, #9542) 1:500, TAMRA: A-6397 (Thermo Fisher, A-6397) 1:1000, Human nuclear antigen (HuNu): 235–1 (Millipore, MAB1281)1:1000, α-tubulin: B 5-1-2 (Sigma, T5168) 1:5000; HRP-conjugated secondary antibodies (Santa Cruz).
qRT-PCR
Total RNA was isolated using GenElute Kit (Sigma). Reverse transcription was performed with ImProm-II™ (Promega). mRNA levels were determined using SYBR Green Mix (BioRad) on an ABI 7900HT using 50°C (2 min), 95°C (10 min), followed by 95°C (15sec) and 60°C (1 min) for 40 cycles. The results were normalized to ACTB. Relative expression was calculated by the ΔΔCt method. PCR primers were: MYCN sense, CGACCACAAGGCCCTCAGTA; MYCN anti-sense, CAGCCTTGGTGTTGGAGGAG; ACTB sense, GCAAGCAGGAGTATGACGAGTC; ACTB anti-sense, CAAGAAAGGGTGTAACGCAACTAAG.
Protein stability and translation assays
Cells treated with 30 μM CHX (EMD) were harvested at various times for Western analysis. Signal intensity was quantified by ImageStudioLite. After normalization to α-tubulin, Log2 values were plotted exponentially in Excel. Half-life (t1/2) was determined using the Excel function, t1/2=ln(2)/λ (where λ is the decay constant).
Nascent protein was examined using Click-iT protein detection kit (Life Technology). Cells were incubated for 20 min in RB medium with methionine-free IMDM (Life Technology), followed by labeling with 50 μM of L-azidohomoalanine AHA (Life Technology) for 1h. The cells were lysed in RIPA (Millipore) with inhibitors as above for 10 min, and sonicated. The lysate was centrifuged at 14,000 rpm for 10 min at 4 °C. The supernatant was incubated with 2 μg of MYCN antibody with rotation for 2 hours, incubated with 30 μl of Protein A/G beads at 4°C overnight, and washed as above. Azide-modified protein were labeled with TAMRA and purified following manufacture’s protocol, separated by electrophoresis, and detected by laser scanner (Biorad-FX Pro) or by Western blot using anti-TAMRA antibody (Life Technology).36
Xenografts
Xenografts were performed on 8-week-old male athymic (Foxn1−/−) mice (Taconic). RB176-luc cells were established by transduction of RB176 with lentiviral vector expressing thymidine kinase-GFP-luciferase (TGL) under the EF1α promoter.53 At two days after lentivirus-mediated transductions with shMDM2, shMDM4, shMYCN, or shScr, infected cells were suspended at 1×105 cells/ml and 2 μl (2×105 cells/eye) injected into sub-retinal space.3 Tumor formation was examined by bioluminescent imaging with IVIS and LivingImage V.2.11 (Xenogen). Mice were injected with 100 μl of 30 mg/ml D-Luciferin (GoldBio) via retro-orbital route and imaged 5 min following injection. Sample size was as needed to assess tumor phenotypes. Mice were randomly assigned to different xenograft regimens and the investigator blinded to the assignment until the tumor analyses. Three mice with early death prior to the first observed tumor in the cohort were excluded from the analyses. All mouse experiments were approved and performed according to the guidelines of the Institutional Animal Care and Usage Committee of Children’s Hospital Los Angeles.
Statistical analyses
Measurements were performed in triplicate and differences between means assessed for significance using unpaired Student’s t-tests.
Supplementary Material
Acknowledgments
D.–L. Qi and D. Cobrinik designed the study. D.–L. Qi conducted all the experiments. D.–L. Qi and D. Cobrinik wrote the manuscript. We thank Xiaoliang Leon Xu for early passage retinoblastoma cell preparations, Narine Harutyunyan and Jennifer Aparicio for CHLAVC-RB43, Pat Reynolds and A. Linn Murphree for CHLA-RB215 cells, and the Saban Research Institute Research Imaging Core for assistance. This study was supported by NIH grants 1R01CA137124 and P30CA014089, by a Saban Research Institute Research Career Development Fellowship to D.–L. Qi, by Research to Prevent Blindness (New York, New York), by the Larry & Celia Moh Foundation, and by the charitable support of the Nautica Malibu Triathlon event produced by MESP, Inc.
This study was supported by NIH grants 1R01CA137124 and P30CA014089, by a Saban Research Institute Research Career Development Fellowship (D.–L. Qi), by Research to Prevent Blindness (New York, New York), by the Larry & Celia Moh Foundation, and by the charitable support of the Nautica Malibu Triathlon event produced by MESP, Inc.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
References
- 1.Dimaras H, Corson TW, Cobrinik D, White A, Zhao J, Munier FL, et al. Retinoblastoma. Nat Rev Dis Primers. 2015;1:15021. doi: 10.1038/nrdp.2015.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xu XL, Singh HP, Wang L, Qi DL, Poulos BK, Abramson DH, et al. Rb suppresses human cone-precursor-derived retinoblastoma tumours. Nature. 2014;514:385–388. doi: 10.1038/nature13813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xu XL, Fang Y, Lee TC, Forrest D, Gregory-Evans C, Almeida D, et al. Retinoblastoma has properties of a cone precursor tumor and depends upon cone-specific MDM2 signaling. Cell. 2009;137:1018–1031. doi: 10.1016/j.cell.2009.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kooi IE, Mol BM, Moll AC, van der Valk P, de Jong MC, de Graaf P, et al. Loss of photoreceptorness and gain of genomic alterations in retinoblastoma reveal tumor progression. EBioMedicine. 2015;2:660–670. doi: 10.1016/j.ebiom.2015.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cobrinik D. Retinoblastoma Progression. EBioMedicine. 2015;2:623–624. doi: 10.1016/j.ebiom.2015.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee TC, Almeida D, Claros N, Abramson DH, Cobrinik D. Cell cycle-specific and cell type-specific expression of Rb in the developing human retina. Invest Ophthalmol Vis Sci. 2006;47:5590–5598. doi: 10.1167/iovs.06-0063. [DOI] [PubMed] [Google Scholar]
- 7.Wang H, Bauzon F, Ji P, Xu X, Sun D, Locker J, et al. Skp2 is required for survival of aberrantly proliferating Rb1-deficient cells and for tumorigenesis in Rb1+/− mice. Nat Genet. 2010;42:83–88. doi: 10.1038/ng.498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lowe SW, Cepero E, Evan G. Intrinsic tumour suppression. Nature. 2004;432:307–315. doi: 10.1038/nature03098. [DOI] [PubMed] [Google Scholar]
- 9.Cobrinik D. Pocket proteins and cell cycle control. Oncogene. 2005;24:2796–2809. doi: 10.1038/sj.onc.1208619. [DOI] [PubMed] [Google Scholar]
- 10.Viatour P, Sage J. Newly identified aspects of tumor suppression by RB. Dis Model Mech. 2011;4:581–585. doi: 10.1242/dmm.008060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chicas A, Wang X, Zhang C, McCurrach M, Zhao Z, Mert O, et al. Dissecting the unique role of the retinoblastoma tumor suppressor during cellular senescence. Cancer Cell. 2010;17:376–387. doi: 10.1016/j.ccr.2010.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bates S, Phillips AC, Clark PA, Stott F, Peters G, Ludwig RL, et al. p14ARF links the tumour suppressors RB and p53. Nature. 1998;395:124–125. doi: 10.1038/25867. [DOI] [PubMed] [Google Scholar]
- 13.Aslanian A, Iaquinta PJ, Verona R, Lees JA. Repression of the Arf tumor suppressor by E2F3 is required for normal cell cycle kinetics. Genes Dev. 2004;18:1413–1422. doi: 10.1101/gad.1196704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Komori H, Enomoto M, Nakamura M, Iwanaga R, Ohtani K. Distinct E2F-mediated transcriptional program regulates p14ARF gene expression. EMBO J. 2005;24:3724–3736. doi: 10.1038/sj.emboj.7600836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lowe SW, Sherr CJ. Tumor suppression by Ink4a-Arf: progress and puzzles. Curr Opin Genet Dev. 2003;13:77–83. doi: 10.1016/s0959-437x(02)00013-8. [DOI] [PubMed] [Google Scholar]
- 16.Sherr CJ, McCormick F. The RB and p53 pathways in cancer. Cancer Cell. 2002;2:103–112. doi: 10.1016/s1535-6108(02)00102-2. [DOI] [PubMed] [Google Scholar]
- 17.Kato MV, Shimizu T, Ishizaki K, Kaneko A, Yandell DW, Toguchida J, et al. Loss of heterozygosity on chromosome 17 and mutation of the p53 gene in retinoblastoma. Cancer Lett. 1996;106:75–82. doi: 10.1016/0304-3835(96)04305-4. [DOI] [PubMed] [Google Scholar]
- 18.Guo Y, Pajovic S, Gallie BL. Expression of p14ARF, MDM2, and MDM4 in human retinoblastoma. Biochem Biophys Res Commun. 2008;375:1–5. doi: 10.1016/j.bbrc.2008.07.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harbour JW, Worley L, Ma D, Cohen M. Transducible peptide therapy for uveal melanoma and retinoblastoma. Arch Ophthalmol. 2002;120:1341–1346. doi: 10.1001/archopht.120.10.1341. [DOI] [PubMed] [Google Scholar]
- 20.Elison JR, Cobrinik D, Claros N, Abramson DH, Lee TC. Small molecule inhibition of HDM2 leads to p53-mediated cell death in retinoblastoma cells. Arch Ophthalmol. 2006;124:1269–1275. doi: 10.1001/archopht.124.9.1269. [DOI] [PubMed] [Google Scholar]
- 21.Laurie NA, Donovan SL, Shih CS, Zhang J, Mills N, Fuller C, et al. Inactivation of the p53 pathway in retinoblastoma. Nature. 2006;444:61–66. doi: 10.1038/nature05194. [DOI] [PubMed] [Google Scholar]
- 22.McEvoy J, Flores-Otero J, Zhang J, Nemeth K, Brennan R, Bradley C, et al. Coexpression of normally incompatible developmental pathways in retinoblastoma genesis. Cancer Cell. 2011;20:260–275. doi: 10.1016/j.ccr.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McEvoy J, Ulyanov A, Brennan R, Wu G, Pounds S, Zhang J, et al. Analysis of MDM2 and MDM4 single nucleotide polymorphisms, mRNA splicing and protein expression in retinoblastoma. PLoS One. 2012;7:e42739. doi: 10.1371/journal.pone.0042739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wade M, Wang YV, Wahl GM. The p53 orchestra: Mdm2 and Mdmx set the tone. Trends Cell Biol. 2010;20:299–309. doi: 10.1016/j.tcb.2010.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brennan RC, Federico S, Bradley C, Zhang J, Flores-Otero J, Wilson M, et al. Targeting the p53 pathway in retinoblastoma with subconjunctival Nutlin-3a. Cancer Res. 2011;71:4205–4213. doi: 10.1158/0008-5472.CAN-11-0058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thériault BL, Dimaras H, Gallie BL, Corson TW. The genomic landscape of retinoblastoma: a review. Clin Experiment Ophthalmol. 2014;42:33–52. doi: 10.1111/ceo.12132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Efeyan A, Serrano M. p53: guardian of the genome and policeman of the oncogenes. Cell Cycle. 2007;6:1006–1010. doi: 10.4161/cc.6.9.4211. [DOI] [PubMed] [Google Scholar]
- 28.Wang SP, Wang WL, Chang YL, Wu CT, Chao YC, Kao SH, et al. p53 controls cancer cell invasion by inducing the MDM2-mediated degradation of Slug. Nat Cell Biol. 2009;11:694–704. doi: 10.1038/ncb1875. [DOI] [PubMed] [Google Scholar]
- 29.Hu B, Gilkes DM, Farooqi B, Sebti SM, Chen J. MDMX overexpression prevents p53 activation by the MDM2 inhibitor Nutlin. J Biol Chem. 2006;281:33030–33035. doi: 10.1074/jbc.C600147200. [DOI] [PubMed] [Google Scholar]
- 30.Komarov PG, Komarova EA, Kondratov RV, Christov-Tselkov K, Coon JS, Chernov MV, et al. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science. 1999;285:1733–1737. doi: 10.1126/science.285.5434.1733. [DOI] [PubMed] [Google Scholar]
- 31.Sohn D, Graupner V, Neise D, Essmann F, Schulze-Osthoff K, Janicke RU. Pifithrin-alpha protects against DNA damage-induced apoptosis downstream of mitochondria independent of p53. Cell Death Differ. 2009;16:869–878. doi: 10.1038/cdd.2009.17. [DOI] [PubMed] [Google Scholar]
- 32.Liu DP, Song H, Xu Y. A common gain of function of p53 cancer mutants in inducing genetic instability. Oncogene. 2010;29:949–956. doi: 10.1038/onc.2009.376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Terzian T, Suh YA, Iwakuma T, Post SM, Neumann M, Lang GA, et al. The inherent instability of mutant p53 is alleviated by Mdm2 or p16INK4a loss. Genes Dev. 2008;22:1337–1344. doi: 10.1101/gad.1662908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cobrinik D, Francis RO, Abramson DH, Lee TC. Rb induces a proliferative arrest and curtails Brn-2 expression in retinoblastoma cells. Mol Cancer. 2006;5:72. doi: 10.1186/1476-4598-5-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Evans L, Chen L, Milazzo G, Gherardi S, Perini G, Willmore E, et al. SKP2 is a direct transcriptional target of MYCN and a potential therapeutic target in neuroblastoma. Cancer Lett. 2015;363:37–45. doi: 10.1016/j.canlet.2015.03.044. [DOI] [PubMed] [Google Scholar]
- 36.Dieterich DC, Link AJ, Graumann J, Tirrell DA, Schuman EM. Selective identification of newly synthesized proteins in mammalian cells using bioorthogonal noncanonical amino acid tagging (BONCAT) Proc Natl Acad Sci U S A. 2006;103:9482–9487. doi: 10.1073/pnas.0601637103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miles WO, Korenjak M, Griffiths LM, Dyer MA, Provero P, Dyson NJ. Post-transcriptional gene expression control by NANOS is up-regulated and functionally important in pRb-deficient cells. EMBO J. 2014;33:2201–2215. doi: 10.15252/embj.201488057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Efeyan A, Garcia-Cao I, Herranz D, Velasco-Miguel S, Serrano M. Tumour biology: Policing of oncogene activity by p53. Nature. 2006;443:159. doi: 10.1038/443159a. [DOI] [PubMed] [Google Scholar]
- 39.Christophorou MA, Ringshausen I, Finch AJ, Swigart LB, Evan GI. The pathological response to DNA damage does not contribute to p53-mediated tumour suppression. Nature. 2006;443:214–217. doi: 10.1038/nature05077. [DOI] [PubMed] [Google Scholar]
- 40.Robanus-Maandag E, Dekker M, van der Valk M, Carrozza ML, Jeanny JC, Dannenberg JH, et al. p107 is a suppressor of retinoblastoma development in pRb-deficient mice. Genes Dev. 1998;12:1599–1609. doi: 10.1101/gad.12.11.1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ajioka I, Martins RA, Bayazitov IT, Donovan S, Johnson DA, Frase S, et al. Differentiated horizontal interneurons clonally expand to form metastatic retinoblastoma in mice. Cell. 2007;131:378–390. doi: 10.1016/j.cell.2007.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gratias S, Schuler A, Hitpass LK, Stephan H, Rieder H, Schneider S, et al. Genomic gains on chromosome 1q in retinoblastoma: consequences on gene expression and association with clinical manifestation. Int J Cancer. 2005;116:555–563. doi: 10.1002/ijc.21051. [DOI] [PubMed] [Google Scholar]
- 43.Wang YV, Wade M, Wong E, Li YC, Rodewald LW, Wahl GM. Quantitative analyses reveal the importance of regulated Hdmx degradation for p53 activation. Proc Natl Acad Sci U S A. 2007;104:12365–12370. doi: 10.1073/pnas.0701497104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li Q, Lozano G. Molecular pathways: targeting Mdm2 and Mdm4 in cancer therapy. Clin Cancer Res. 2013;19:34–41. doi: 10.1158/1078-0432.CCR-12-0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.To KH, Pajovic S, Gallie BL, Thériault BL. Regulation of p14ARF expression by miR-24: a potential mechanism compromising the p53 response during retinoblastoma development. BMC Cancer. 2012;12:69. doi: 10.1186/1471-2407-12-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rushlow DE, Mol BM, Kennett JY, Yee S, Pajovic S, Thériault BL, et al. Characterisation of retinoblastomas without RB1 mutations: genomic, gene expression, and clinical studies. Lancet Oncol. 2013;14:327–334. doi: 10.1016/S1470-2045(13)70045-7. [DOI] [PubMed] [Google Scholar]
- 47.Huang M, Weiss WA. Neuroblastoma and MYCN. Cold Spring Harb Perspect Med. 2013;3:a014415. doi: 10.1101/cshperspect.a014415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gu L, Zhang H, He J, Li J, Huang M, Zhou M. MDM2 regulates MYCN mRNA stabilization and translation in human neuroblastoma cells. Oncogene. 2012;31:1342–1353. doi: 10.1038/onc.2011.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carr-Wilkinson J, O’Toole K, Wood KM, Challen CC, Baker AG, Board JR, et al. High Frequency of p53/MDM2/p14ARF Pathway Abnormalities in Relapsed Neuroblastoma. Clin Cancer Res. 2010;16:1108–1118. doi: 10.1158/1078-0432.CCR-09-1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cobrinik D, Ostrovnaya I, Hassimi M, Tickoo SK, Cheung IY, Cheung NK. Recurrent pre-existing and acquired DNA copy number alterations, including focal TERT gains, in neuroblastoma central nervous system metastases. Genes Chromosomes Cancer. 2013;52:1150–1166. doi: 10.1002/gcc.22110. [DOI] [PubMed] [Google Scholar]
- 51.Embade N, Fernandez-Ramos D, Varela-Rey M, Beraza N, Sini M, Gutierrez de Juan V, et al. Murine double minute 2 regulates Hu antigen R stability in human liver and colon cancer through NEDDylation. Hepatology. 2012;55:1237–1248. doi: 10.1002/hep.24795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Candeias MM, Malbert-Colas L, Powell DJ, Daskalogianni C, Maslon MM, Naski N, et al. P53 mRNA controls p53 activity by managing Mdm2 functions. Nat Cell Biol. 2008;10:1098–1105. doi: 10.1038/ncb1770. [DOI] [PubMed] [Google Scholar]
- 53.Barberi T, Bradbury M, Dincer Z, Panagiotakos G, Socci ND, Studer L. Derivation of engraftable skeletal myoblasts from human embryonic stem cells. Nat Med. 2007;13:642–648. doi: 10.1038/nm1533. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.