

Abstract
This is the second of the two White Papers from the fourth UC Davis Cardiovascular Symposium Systems Approach to Understanding Cardiac Excitation–Contraction Coupling and Arrhythmias (3–4 March 2016), a biennial event that brings together leading experts in different fields of cardiovascular research. The theme of the 2016 symposium was ‘K+ channels and regulation’, and the objectives of the conference were severalfold: (1) to identify current knowledge gaps; (2) to understand what may go wrong in the diseased heart and why; (3) to identify possible novel therapeutic targets; and (4) to further the development of systems biology approaches to decipher the molecular mechanisms and treatment of cardiac arrhythmias. The sessions of the Symposium focusing on the functional roles of the cardiac K+ channel in health and disease, as well as K+ channels as therapeutic targets, were contributed by Ye Chen‐Izu, Gideon Koren, James Weiss, David Paterson, David Christini, Dobromir Dobrev, Jordi Heijman, Thomas O'Hara, Crystal Ripplinger, Zhilin Qu, Jamie Vandenberg, Colleen Clancy, Isabelle Deschenes, Leighton Izu, Tamas Banyasz, Andras Varro, Heike Wulff, Eleonora Grandi, Michael Sanguinetti, Donald Bers, Jeanne Nerbonne and Nipavan Chiamvimonvat as speakers and panel discussants. This article summarizes state‐of‐the‐art knowledge and controversies on the functional roles of cardiac K+ channels in normal and diseased heart. We endeavour to integrate current knowledge at multiple scales, from the single cell to the whole organ levels, and from both experimental and computational studies.
Keywords: atrial fibrillation, cardiac arrhythmia, cardiac K channels, cardiac repolarization, computational modeling, hERG channel, K channel blockers, long QT syndrome
Diversity and functional roles of K+ channels in cardiac repolarization: from single cell to whole organ levels
There are many types of K+ channels in mammalian cardiac cells, the expression of which varies greatly throughout the heart, from atria to ventricles, epi‐ to endocardium, and from apex to base. This remarkable diversity allows for precise and differential control of resting membrane potential (RMP), action potential (AP) duration (APD), and refractoriness throughout the heart (Bartos et al. 2015). Most cardiac cells express some combination of voltage‐gated transient outward (I to), voltage‐gated delayed rectifier, inward rectifier (I K1), and ligand‐gated (ATP‐sensitive (I K,ATP), acetylcholine‐activated (I K,ACh), and Ca2+‐activated (I K,Ca)) K+ channels. Three delayed rectifier K+ currents have been distinguished: ultra‐rapid (I Kur), rapid (I Kr) and slow (I Ks) (Zeng et al. 1995). The structure, function, and regulation of each channel type are discussed in detail in the companion White Paper by Grandi et al. (2017). Here, we will highlight the diversity of K+ channels throughout the heart and discuss how this diversity contributes to heterogeneities in repolarization and APD.
K+ channel diversity: atrio‐ventricular differences
There are several key differences between atrial and ventricular APs, with ventricular myocytes having a longer APD, a more hyperpolarized RMP, a longer plateau phase that reaches a more depolarized potential, and a faster rate of repolarization compared to atrial myocytes (Schram et al. 2002). Although differential expression of Na+ and Ca2+ channels, Na+/Ca2+ exchanger (NCX), and gap junctions contributes to atrioventricular differences, K+ channel diversity also plays a critical role. Most notably, I Kur and I K,ACh (corresponding to the Kv1.5 and Kir3.1:Kir3.4 proteins, respectively) are nearly absent in ventricular myocytes, whereas both of these K+ currents contribute to the atrial AP in humans and in several other mammals, including dogs, guinea pigs, and rats (Boyle & Nerbonne, 1991; Paulmichl et al. 1991; Fedida et al. 1993; Wang et al. 1993; Dobrzynski et al. 2001; Schram et al. 2002; Gaborit et al. 2007). Likewise, small‐conductance Ca2+‐activated K+ (SK) channels (KCa2.1, KCa2.2 and KCa2.3), which underlie I K,Ca, are predominantly expressed in the atria where they contribute to repolarization in mice and in humans (Xu et al. 2003; Tuteja et al. 2005). Therefore, targeting these atrial specific currents (I Kur, I K,ACh and I K,Ca) may represent a novel area for therapeutic intervention for atrial arrhythmias, the details of which are discussed in a later section entitled ‘Identification of novel therapeutic targets for cardiac arrhythmias’.
Ventricular myocytes exhibit a prominent I K1 and corresponding higher expression of Kir2.1 (KCNJ2), which probably contributes to the more hyperpolarized RMP in ventricular versus atrial myocytes (Giles & Imaizumi, 1988; Dhamoon et al. 2004; Gaborit et al. 2007). The plateau phase of the AP is longer in ventricular cells due to a lower density of K+ currents activated during the notch phase (smaller I to and lack of I Kur). This prolonged plateau phase allows for increased recovery of I Kr from inactivation and a faster rate of repolarization in the ventricular myocytes.
Ventricular K+ channel diversity: transmural differences
AP characteristics and corresponding K+ channel expression across the ventricular wall from epi‐ to endocardium have been well characterized in the canine heart (Antzelevitch et al. 1999; Antzelevitch, 2010). Three AP waveforms have been identified: epicardial, mid‐myocardial (M‐cells) and endocardial (Fig. 1). The key differences between these cell types include a large ‘spike and dome’ or notch phase in epicardial cells, and a significantly prolonged APD in M‐cells (Yan et al. 1998). Epicardial cells also have a shorter APD than endocardial cells, whereas endocardial cells have a less pronounced notch phase (Sicouri & Antzelevitch, 1991). In addition to a longer baseline APD, a key feature of M‐cells is a disproportionately prolonged APD in response to a slowing of rate and/or in response to APD‐prolonging agents (Antzelevitch et al. 1999) (Fig. 1). The ionic determinants of the unique features of canine M‐cells have been suggested to include a smaller I Ks and a larger late Na+ current (I Na,L) (Liu & Antzelevitch, 1995; Zygmunt et al. 2001). The disproportionate prolongation of APD in canine M‐cells can lead to increased transmural dispersion of repolarization and the likelihood for reentrant arrhythmias. Furthermore, a prolonged APD may also predispose M‐cells to early afterdepolarizations (EADs). Thus, if present in human ventricles, M‐cells may represent an important therapeutic target for the suppression of ventricular arrhythmias, especially in the setting of reduced repolarization reserve (Wilson et al. 2011). There is, however, considerable disagreement on the presence and functional importance of M cells, particularly in the human heart and the functional role of M cells in contributing to the dispersion of ventricular repolarization remains a topic of active debate (Antzelevitch et al. 1999; Antzelevitch, 2010; Glukhov et al. 2010; Houser et al. 2000; Jost et al. 2005; Taggart et al. 2014; Wilson et al. 2011).
Figure 1. Heterogeneity of canine ventricular repolarization and K+ current distribution.
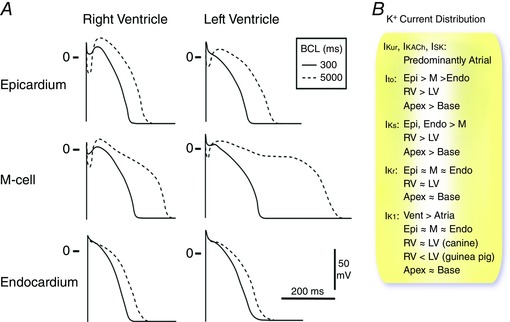
A, epicardial, M‐cell and endocardial APs from the RV and LV of the canine heart at basic cycle lengths (BCLs) of 300 and 5000 ms. Redrawn with permission from Antzelevitch et al. (1999). B, relative distribution of major K+ currents in the heart (see text for details and references).
In canine ventricles, the prominent notch phase of epicardial APs has been attributed to a large I to (Litovsky & Antzelevitch, 1988; Furukawa et al. 1990; Nabauer et al. 1996). In human and canine ventricles, Kv4.3 α‐subunits, together with the auxiliary subunit, KChIP2, generates I to. Interestingly, KChIP2, but not Kv4.3, mRNA expression is correlated with the gradient of I to and prominence of the AP notch (Rosati et al. 2001; Gaborit et al. 2007). I Kr and I K1 are also important for canine ventricular repolarization, although there is no clear evidence of transmural differences in these currents (Liu & Antzelevitch, 1995).
Ventricular K+ channel diversity: RV/LV differences
The transmural gradient of APD described above exists in canine right and left ventricles (RV and LV); however, the APD is typically longer in canine LV, compared to the RV (Sicouri & Antzelevitch, 1991). The shorter APD in the RV is associated with an increased I to and increased expression of both KChIP2 and Kv4.3 in both human and canine hearts (Di Diego et al. 1996; Volders et al. 1999; Boukens et al. 2009). In rodents, Kv4.2 α‐subunits conduct I to and Kv4.2 mRNA and protein expression are higher in the RV of rat hearts (Wickenden et al. 1999). Canine RV myocytes also have increased I Ks compared to LV, which correlates with increased protein expression of the accessory subunit, KCNE1, that modulates the Kv7.1 α‐subunits that underlie I Ks (Volders et al. 1999; Ramakers et al. 2003). No differences in other K+ currents, including I Kr or I K1, have been observed between the RV and LV of canine hearts (Volders et al. 1999); however, I K1 is higher in the LV than in the RV in guinea pig heart (Samie et al. 2001; Molina et al. 2016).
Ventricular K+ channel diversity: apico‐basal differences
In the canine heart, APDs are shorter in the apex, compared to the base, of the LV and these shorter APDs correlate with increased I to and I Ks, as well as increased protein expression of KChIP2, Kv7.1 and minK (Szentadrassy et al. 2005). No apico‐basal differences in I K1 or I Kr have been documented and expression of Kir2.1 (I K1), Kv11.1 (I Kr), and the K+ channel accessory subunit, MiRP1 are similar in the apex and base in the canine heart (Szentadrassy et al. 2005).
Repolarization in Purkinje fibre
Purkinje fibres form a specialized conduction system in the ventricles and have been shown to have unique electrophysiological properties and to play critical roles in the generation of cardiac arrhythmias (Boyden et al. 2010). Specifically, canine Purkinje cells exhibit different types and densities of repolarizing K+ currents compared to ventricular or atrial myocytes, as well as markedly different AP profiles (Vassalle & Bocchi, 2013). Moreover, Purkinje cells display spontaneous impulse initiation, similar to pacemaker cells. It has also been shown that I to is very large in canine Purkinje cells (Jeck et al. 1995) and that I to may play important roles in the generation of EADs in these cells (Zhao et al. 2012).
β‐Adrenergic regulation of K+ channels during cardiac AP
Extensive studies have shown that the cardiac K+ channels responsible for AP repolarization are intricately regulated by β‐adrenergic tone, and several K+ channels – I Ks, I Kr, I K1 – exhibit differential sensitivity to β‐adrenergic stimulation (Tromba & Cohen, 1990; Volders et al. 2003; Thomas et al. 2004; Harmati et al. 2011). I Ks is facilitated by β‐adrenergic stimulation (Sanguinetti et al. 1991; Marx et al. 2002; Volders et al. 2003; Harmati et al. 2011); increased I Ks contributes to the shortening of APD during rapid heart rates. When the I Ks channel is defective in long QT syndrome (LQTS), a lack of adrenergic response of I Ks could provide a substrate for arrhythmias.
The effects of β‐adrenergic stimulation on I Kr have been controversial. Harmati et al. (Harmati et al. 2011) and Heath & Terrar (2000) reported facilitation of I Kr by isoproterenol (isoprenaline) via PKA and PKC pathways in canine and guinea pig ventricular myocytes. In contrast, Karle et al. (2002) reported a reduction of I Kr amplitude following isoproterenol application in guinea pig ventricular myocytes and Sanguinetti et al. (1991) reported no measurable isoproterenol induced changes of I Kr. Studies of the effects of β‐adrenergic stimulation on I K1 have also reported controversial results. Both facilitation and reduction of I K1 by isoproterenol have also been reported (Gadsby, 1983; Tromba & Cohen, 1990; Koumi et al. 1995; Wischmeyer & Karschin, 1996; Fauconnier et al. 2005; Scherer et al. 2007).
Differential modulation of I Ks, I Kr and I K1 by β‐adrenergic stimulation has potentially important implications for cardiac AP repolarization and arrhythmogenesis. Banyasz et al. (2014) used the AP‐clamp sequential dissection technique to directly record I Ks, I Kr and I K1 during the AP under physiological conditions (internal and external solutions matching physiological milieu with preserved Ca2+ homeostasis). I Ks, I Kr and I K1 were systematically measured during APs at various adrenergic states using isoproterenol in a physiologically relevant concentration range (1–30 nm). Isoproterenol significantly enhanced I Ks, moderately increased I K1, but slightly decreased I Kr in a concentration‐dependent manner (Fig. 2). By recording the three K+ currents from the same cell, these investigators were able to dissect the relative contribution of each K+ current to repolarization. These analyses revealed that the dominant pattern of the K+ currents is I Kr > I K1 > I Ks under physiological conditions, but this pattern is reversed to I Ks > I K1 > I Kr following β‐adrenergic stimulation (Fig. 2). Therefore, β‐adrenergic stimulation fine‐tunes the cardiac AP morphology by shifting the functional importance of the different K+ currents in a concentration‐dependent manner. These findings clearly suggest an important role for sympathetic tone in determining the functional effects of K+ channel blockers.
Figure 2. β‐Adrenergic regulation of K+ channels during cardiac AP.
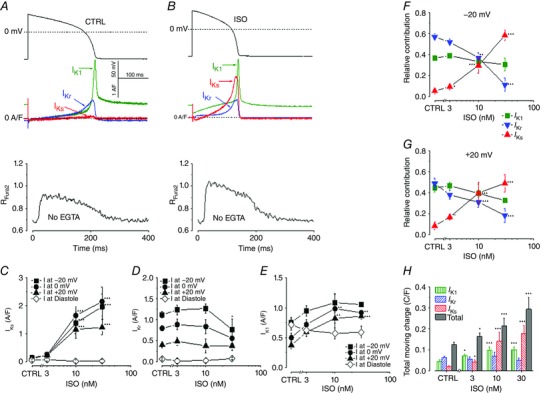
A, AP‐clamp Sequential Dissection experiments were performed as followed: steady‐state APs were recorded at a pacing rate of 1 Hz (upper panel), and used as the voltage command to obtain AP‐clamp recordings of the (three distinct) K+ currents in the same cell (middle panel) and the Ca2+ transients (lower panel) under physiological conditions. B, the effects of isoproterenol (ISO) on the AP waveform and the evoked K+ currents. At 30 nm, ISO significantly enhanced I Ks, moderately increased I K1, but slightly decreased I Kr. C, D and E, concentration‐dependent effects of ISO on the peak densities of I Ks, I Kr and I K1. F and G, the three K+ currents, measured in the same cell, were summed and each current was then normalized to this sum to calculate the relative contribution of each to the total K+ influx. The normalized current values at membrane potentials of +20 mV and −20 mV are shown in F and G, respectively. The ISO dose–response curves show how the relative contribution of each current shifts with various levels of β‐adrenergic stimulation. H, total K+ charge movement for individual K+ current and the sum of the currents during the AP. (Adapted from Banyasz et al. 2014 with permission.)
K+ channel diversity: what does it all mean?
As stated earlier, the remarkable diversity of K+ channel expression throughout the heart allows for precise and differential control of local RMP, APD and refractoriness. Indeed, computational AP models have shown that there are many possible combinations of ionic conductances that produce an equivalent RMP, APD and refractoriness. This is generally true of any input–output system in which the number of adjustable input parameters is large compared to the number of output constraints, leading to the concept that there are multiple ‘good enough solutions’ that all can produce physiologically robust function (Marder & Goaillard, 2006; Weiss et al. 2012). Why is this important? From an evolutionary biology standpoint, robustness is critical for any biological organism facing constantly changing environmental conditions. However, evolution also depends on the ability to adapt in response to changing environmental conditions. How, then, can robustness (the ability to maintain a stable phenotype) be compatible with adaptability (the ability to change phenotype)? A diverse range of good enough solutions among the individuals in a population resolves this paradox in the following manner: all good enough solutions confer robustness to the normal day‐to‐day environmental changes; however, when the environment changes in an unusual way, some ‘good enough solutions’ will adapt better than others (Marder & Goaillard, 2006; Weiss et al. 2012).
As previously demonstrated by Sarkar & Sobie, one AP model may strongly depend on I Kr for repolarization while the other is highly dependent on I Ks (Sarkar & Sobie, 2010). Both models yield physiological APDs and Ca2+ transients. Now we can consider the consequences of administering an I Kr‐blocking drug to two individuals whose good enough solutions correspond to the two ventricular APs illustrated. The individual whose ventricular repolarization has a high dependence on I Kr will exhibit much more significant APD and QT interval prolongation and consequently have a higher risk of Torsade de Pointes (TdP) than the other individual. Thus, in response to a potentially lethal perturbation, one perishes, but one survives. That is, robustness acts at the level of the individual, whereas adaptability operates at the level of the population. The ‘good enough solutions’ concept underlying genetic diversity provides a compelling explanation for why the pronounced (∼10‐fold) heterogeneity in K+ current amplitude between different cells (Banyasz et al. 2011) is more than just random biological variability. Rather, it serves a fundamental biological role.
The ‘good enough solutions’ concept also has major implications for mathematical modelling approaches to drug development and safety testing, in which off‐target cardiac K+ channel blocking effects are a major concern. At the present time, both non‐cardiac and cardiac drugs must undergo expensive animal testing to screen for K+ channel blocking and other effects that predispose to QT prolongation and TdP. Models which use average data to build ‘representative’ cell models of a specific type provide a binary yes or no answer to whether a drug will cause excessive APD prolongation, and there is often disagreement between specific models (Mirams et al. 2014). In biological populations, on the other hand, an I Kr‐blocking drug such as sotalol has a variable effect on APD and QT interval prolongation, posing an overall risk of TdP of < 5% to the population. With advances in high speed graphics processing unit (GPU)‐based computation, it is becoming increasingly feasible to create populations of models to simulate the genetic and phenotypic diversity of human populations (Britton et al. 2013). This approach starts with an AP model based on averaged data, and then randomly mutates the ionic conductances within appropriate experimentally determined ranges. The mutated model is then examined to define its AP and Ca2+ transient properties. Mutations which cause the model to exhibit properties outside the physiologically normal range are excluded. However, mutated models still falling within the physiologically normal range are retained, generating a diverse model population incorporating a range of electrophysiological parameter values, each one representing a good enough solution for a normal cardiac AP. The extent to which the model population realistically reflects a human population can be validated against existing clinical population data, for example the incidence with which known drugs cause QT prolongation and TdP. The ultimate goal is to be able to simulate the effects of a new drug on the clinically validated model population to yield a probabilistic, rather than a binary, estimate of adverse effects. A compelling futuristic strategy for integrating population‐based modelling into a three component pre‐clinical drug discovery and safety testing platform has recently been outlined by Gintant et al. (2016). The first component involves automated patch clamping to characterize in detail the biophysical effects of a drug on the major human cardiac ionic channels heterologously expressed in mammalian cells; the second component simulates these effects in silico in human population models to estimate the probability of adverse effects; and the third component, if the drug still looks promising, is to test the drug's effects in a population of genetically diverse human induced pluripotent stem cell (hiPSC)‐derived cardiac myocytes, perhaps even including the intended patient's iPSC‐derived cardiac myocytes.
In summary, the biological variability and genetic diversity embodied in the ‘good enough solutions’ concept apply to all aspects of human biology, including K+ channel diversity, and continue to play essential roles in the evolutionary success of the human race. We now have the tools to characterize the genetic diversity of human populations in unprecedented detail. The ability to incorporate this information into drug development and safety testing is an exciting new advance with tremendous potential to become a milestone in precision medicine (please see additional discussion in the later section ‘Identification of novel therapeutic targets for cardiac arrhythmias’).
Roles of K+ channels in cardiac arrhythmias
Cardiac arrhythmias can be divided into those resulting in abnormally fast electrical activity (tachyarrhythmias) or abnormally slow activity (bradyarrhythmias). Conceptually, tachyarrhythmias in both atria and ventricles are mediated by abnormal impulse formation, in particular ectopic (triggered) activity, and abnormal impulse propagation, notably reentry (Rosen, 1988; Weiss et al. 2015) (Fig. 3). Ectopic activity can result from secondary depolarizations occurring either during the AP, termed early afterdepolarizations (EADs), or during diastole, termed delayed afterdepolarizations (DADs), as well as from abnormal automaticity due to enhanced diastolic depolarization in normally quiescent tissue. When occurring repeatedly at a sufficiently rapid rate, ectopic activity can produce heterogeneous ‘fibrillatory’ conduction and sustain a tachyarrhythmia. Moreover, when ectopic activity encounters a vulnerable substrate characterized by short effective refractory periods (ERPs), large repolarization gradients and slow heterogeneous conduction, it can initiate reentry, which is considered the predominant tachyarrhythmia‐maintaining mechanism. Bradyarrhythmias, on the other hand, result from reduced automaticity in the sinoatrial node (e.g. in sick sinus syndrome) or impaired atrioventricular conduction. Numerous studies have shown that K+ channel dysregulation, resulting from genetic defects, drug effects, or disease‐related remodelling, can promote all of these fundamental arrhythmia mechanisms (Fig. 3) (Nerbonne & Kass, 2005; Schmitt et al. 2014).
Figure 3. Schematic representation of the role of K+ channels in inherited and acquired cardiac arrhythmias.
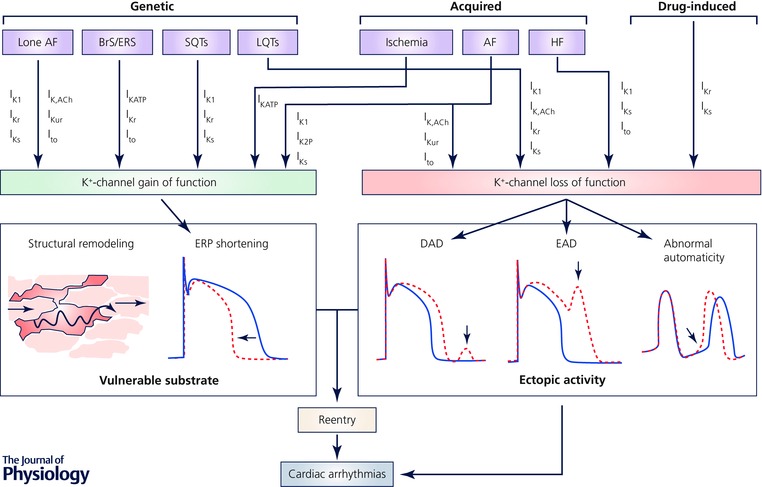
Numerous inherited (genetic), acquired and drug‐induced conditions involve alterations in a wide range of ion channels. Loss of K+ channel function can promote ectopic activity by reducing the repolarizing current offsetting delayed afterdepolarizations (DADs), thereby increasing DAD amplitude, and by promoting APD prolongation and associated early afterdepolarizations (EADs), or by accelerating phase‐4 depolarization and abnormal automaticity. K+ channel gain of function, on the other hand, produces a vulnerable substrate in which ectopic activity can initiate reentry, by shortening action potential duration (APD) and effective refractory period (ERP). Abbreviations: AF: atrial fibrillation; BrS: Brugada sydrome; ERS: early repolarization syndrome; HF: heart failure; LQTS: long‐QT syndrome; SQTS: short‐QT syndrome.
The critical role of K+ channel dysfunction in cardiac arrhythmias is particularly evident in congenital channelopathies such as long‐QT and short‐QT syndromes (LQTS and SQTS, respectively), Brugada or early repolarization syndrome (BrS), and familial (‘lone’) atrial fibrillation (AF), all of which have been associated with an increased likelihood of tachyarrhythmias. Genetic variants in more than 15 different genes, including loss‐of‐function mutations in the genes, KCNQ1 and KCNH2, encoding the pore‐forming α‐subunits, Kv7.1 and Kv11.1, of I Ks and I Kr channels, as well as in the genes encoding the accessory β‐subunits, KCNE1 and KCNE2, of I Ks and I Kr channels, have been associated with LQTS (Nakano & Shimizu, 2016). Given the critical role of I Kr and I Ks in ventricular repolarization and the aforementioned heterogeneous distribution of these channels in the heart, these mutations are expected to result in heterogeneous APD prolongation. APD prolongation provides a larger window for activation of the depolarizing L‐type Ca2+ current (I Ca,L), as well as the late Na+ current (I Na,L), thereby increasing the risk for EADs and ectopic activity. Loss‐of‐function mutations in I K1, which controls final AP repolarization and stabilizes the RMP, have also been associated with QT prolongation (Fodstad et al. 2004). In addition to prolonging APD, loss of I K1 can destabilize the RMP, resulting in abnormal automaticity (Miake et al. 2002) or in larger DAD amplitudes in response to a given transient‐inward current, thereby more readily achieving the threshold for activating Na+ channels and triggering an abnormal AP, all of which will increase the likelihood of ectopic activity.
There is also substantial evidence for K+ channel dysregulation in more common cardiovascular diseases. For example, down‐regulation of I K1, I Ks and I to contributes to APD prolongation in heart failure (Li et al. 2004). Similarly, excessive APD prolongation and EAD‐mediated triggered activity are probably also involved in the proarrhythmic side effects of numerous drugs, which predominantly result from effects on I Kr. Consequently, analysis of potential I Kr blocking effects is a mandatory element during the safety screening of every drug (Heijman et al. 2014 a).
Gain‐of‐function mutations in channel subunits generating I K1, I Kr, I Ks and I to, as well as I K,ATP, have been associated with ventricular arrhythmias in SQTS and BrS, and with familial AF (Lieve & Wilde, 2015; Brugada, 2016; Christophersen & Ellinor, 2016; Sarquella‐Brugada et al. 2016). Mechanistically, gain‐of‐function mutations in cardiac K+ channels will decrease APD and ERP, thereby increasing the likelihood of reentry. In addition, upregulation of I K1 may stabilize reentrant rotors through RMP hyperpolarization, promoting arrhythmia maintenance, as has been clearly demonstrated in the mouse (Noujaim et al. 2007). Similar to loss‐of‐function K+ channel disorders, increases in K+ currents have also been observed in acquired cardiovascular diseases. Activation of I K,ATP during the early phases of ischaemia, for example, shortens ERP and increases extracellular K+, thereby depolarizing the K+ reversal potential and the RMP, which slows conduction velocity. Both factors probably contribute to reentrant ventricular tachyarrhythmias observed under these conditions.
APD shortening is also a hallmark of AF‐related electrical remodelling, probably contributing to AF maintenance and progression (Heijman et al. 2014 b). Upregulation of I K1, I Ks, as well as the two‐pore K+ channel type 3.1 (K2P3.1) current, for example, plays a major role in AF‐related APD shortening and is sufficient to offset the downregulation of other K+ currents, such as I Kur and I to (Dobrev et al. 2005; Caballero et al. 2010; Schmidt et al. 2015). Upregulation of other K+ channels could also be involved in cardiac arrhythmogenesis, although most are incompletely understood. For example, in AF patients, I K,ACh develops constitutive, acetylcholine‐independent activity, contributing to the increase in total inward‐rectifier K+ current, despite reduction of agonist‐induced peak I K,ACh (Dobrev et al. 2005). The role of SK channels in atrial arrhythmias similarly remains a topic of active investigation (Xu et al. 2003; Zhang et al. 2015). Expression of SK channels is increased in the failing heart and Ca2+‐dependent activation of SK channels may shorten APD during fibrillation/defibrillation episodes, contributing to re‐induction of ventricular tachyarrhythmias (Chua et al. 2011). On the other hand, SK channels may constitute an important repolarization reserve and their inhibition has been associated with increased susceptibility to pacing‐induced ventricular arrhythmias during hypokalaemia (Chan et al. 2015).
Considerable evidence, therefore, demonstrates that K+ channel dysfunction plays a major role in cardiac arrhythmias. For most channels, both loss‐of‐function and gain‐of‐function have been associated with arrhythmias, albeit through distinct mechanisms. These findings suggest the existence of a Goldilocks zone in which a balance among the different K+ currents, as well as between repolarizing K+ currents and depolarizing Na+ and Ca2+ currents, ensures stability of cardiac electrophysiological activity. This complex interaction of ion channels, as well as the multitude of proarrhythmic mechanisms and diversity of K+ channel remodelling under different pathophysiological conditions, makes it challenging to predict the pro‐ or anti‐arrhythmic effects of a given alteration in K+ channel function. Of note, it has become clear that chronic modulation of K+ channel function (e.g. in a rabbit model of LQTS type 2) can produce extensive cardiac remodelling, which may further promote cardiac arrhythmias (Terentyev et al. 2014).
Differential roles of K+ currents in arrhythmogenesis
K+ currents affect APD by regulating the rate of repolarization. The proarrhythmic potential of alterations in different K+ channel types, however, are distinct, owing to differences in channel expression and/or biophysical properties.
Delayed rectifier K+ currents (I Kr and I Ks)
I Kr activates and inactivates rapidly (Spector et al. 1996), whereas I Ks activates slowly (Tristani‐Firouzi & Sanguinetti, 1998). Activation of I Ks is also Ca2+ dependent. In guinea pig, I Ks may have many closed states (Silva & Rudy, 2005), and thus, as the heart rates become slower, more channels gradually enter the more deeply closed states, and fewer channels are available for opening during the AP. At fast heart rates, the slow deactivation results in I Ks accumulation between beats and increased current during the AP plateau, resulting in APD shortening. In human and canine myocytes, however, I Ks is small, deactivation is fast, and I Ks accumulation does not occur between beats (Virag et al. 2001).
Because of marked differences in kinetics, I Ks and I Kr play distinct roles in regulating AP dynamics at different heart rates. As shown in experiments by Antzelevitch and colleagues (Antzelevitch et al. 1991, 1999), after I Kr is blocked, the APD of the M‐cells becomes very long and continues to prolong at very slow heart rates. Theoretical studies show that in the presence of prolonged APDs with reduced repolarization reserve, the rate dependence of APD at normal or slow heart rates and the slow activation and deactivation kinetics of a K+ current together with an inward window current of I Ca,L may culminate in APD alternans at normal and slow heart rates (Qu et al. 2010; Qu & Chung, 2012). This may form the basis for T‐wave alternans seen clinically in LQTS patients (Schwartz & Malliani, 1975).
The distinct properties and functional roles of I Kr and I Ks provide mechanistic insights into the clinical presentation of different LQTS types (Schwartz, 2006). In LQT2 and LQT3, arrhythmias tend to occur during bradycardia or after a pause (Viswanathan & Rudy, 1999; Viskin et al. 2000; Clancy & Rudy, 2002), whereas in LQT1, arrhythmias are often exercise‐induced (tachycardia related). During slow heart rates, there are fewer I Ks channels available for opening. Coupled with LQT2 and LQT3, this leads to disproportionately reduced repolarization reserve during bradycardia. In LQT1, I Kr and I to are the main repolarizing currents, and because these currents recover quickly, bradycardia does not preferentially reduce repolarization reserve. Instead, repolarization reserve is reduced during tachycardia, when Ca2+ elevation causing an increase in I NCX and adrenergic stimulation of I Ca,L is no longer counterbalanced by the adrenergic stimulation of I Ks.
Transient outward K+ current (I to)
A distinct feature of I to is that both activation and inactivation are fast, and I to is largely responsible for the spike‐and‐dome AP morphology in large animals. Unlike other K+ currents, increasing I to in canine ventricular myocytes first prolongs, then dramatically shortens (collapses) APD (Dong et al. 2006). I to also plays a somewhat unexpected role in the genesis of EADs. Recent studies in rabbit ventricular myocytes (Zhao et al. 2012; Nguyen et al. 2015), for example, showed that, under conditions of reduced repolarization reserve, no EADs occurred without I to and that the addition of I to increased the likelihood of EADs, probably reflecting an effect on the membrane voltage and I Ca,L re‐activation.
Inward rectifier K+ current (I K1)
In addition to stabilizing the RMP, I K1 contributes to phase 3 repolarization. Reducing I K1 destabilizes RMP and results in pacemaker‐like activity (Miake et al. 2002; Silva & Rudy, 2003) or DAD‐mediated triggered activity (Schlotthauer & Bers, 2000). Due to its strong inward rectification, I K1 is small during the AP plateau and probably will have only a small effect on phase‐2 EADs, although reducing I K1 may promote phase‐3 EADs (Maruyama et al. 2011).
Importantly, because AP dynamics of a single myocyte and the spatiotemporal conduction dynamics of three dimensional cardiac tissues reflect the repertoire of ion channels expressed, altering the properties/expression of one type of channel can be proarrhythmic or anti‐arrhythmic, depending on the status of all of the other ion channels present. As a result, the roles of individual K+ channel types in arrhythmia generation and maintenance can only be fully understood by using multi‐scale and systems approaches that integrate molecular scale behaviours with tissue and organ scale behaviours (Qu & Weiss, 2015), as well as behaviours at the population scale taking into account genetic diversities and complex environmental stress. Clearly, this is one of the grand challenges facing cardiac arrhythmia research today.
Mechanisms of cardiac electrical remodelling in acquired disease
Regulation of cardiac K+ channel expression both at the tissue and subcellular levels plays a critical role in normal cardiac excitability. Remodelling of K+ channel expression, which occurs in various disease states, can predispose to an increased risk of sudden cardiac death (Nass et al. 2008). Remodelling is the end result of multiple facets of cardiac disease. Several disease states provide stressors that cause an increased physical demand on the heart and higher strain on the individual myocytes of the heart. This strain becomes transduced to local signalling pathways, causing increased sympathetic tone and inflammatory activation, which stimulates altered protein expression (Armoundas et al. 2001). Despite a significant amount of investigation, the precise mechanisms that underlie remodelling and drive pathogenesis remain incompletely understood and there is considerable interest in understanding the mechanisms that underlie the regulation of functional myocardial K+ channel expression under both normal and pathological conditions.
Cells from diseased hearts often display a prolonged APD. Although increases in inward (depolarizing) (Undrovinas et al. 1999; Houser et al. 2000) or decreases in outward (repolarizing) (Nabauer & Kaab, 1998) currents can result in prolonged APs, remodelling of K+ currents is by far the most prevalent mechanism leading to AP prolongation. Numerous studies over the last 10–20 years have documented changes in K+ channel expression, including I to, I Kr, I Ks and I K1, that occur in heart failure and AF both in animal models as well as human tissues (Nass et al. 2008; Nattel et al. 2008).
In AF, different changes have been described for I Kr and I Ks while an increase in I K1 appears to be the consensus (Nattel et al. 2008; Yang & Nerbonne, 2016). I Kur is the most prominent repolarizing current in human atrium and demonstrates pronounced remodelling in AF patients (Van Wagoner et al. 1997; Van Wagoner, 2003). More recently, efforts have focused on the molecular mechanisms underlying these observations, as this is the key to determining whether it might be possible to modulate these processes for therapeutic benefit. Changes at the level of transcription, translation, assembly and biogenesis, post‐translational modifications, cellular localization and degradation have all been reported (Heijman et al. 2014 b; Nattel, 2015; Yang & Nerbonne, 2016), but how these changes are interrelated remains to be determined.
Recent technical developments in ‘omics’ technologies promise to be at the forefront of this field. At the gene level, next generation gene sequencing technologies have led to the identification of microRNAs and long non‐coding RNAs (lncRNAs) that play critical roles in regulating transcription and translation of many genes, including ion channel genes (Greco & Condorelli, 2015; Myers et al. 2015). At the protein level, new mass spectrometry methods have enabled global analyses of integrated responses rather than the analysis of individual components (Ferreira et al. 2015). Protein homeostasis or ‘proteostasis’ is another important factor in regulating gene expression involving a highly complex interconnection of pathways that influence the fate of a protein from synthesis to degradation (Balch et al. 2008). Further, it is likely that in stressed tissue, the overall cellular response may focus on maintaining certain subnetworks at the expense of others which can manifest as discrepancies between levels of transcripts, non‐coding RNAs and proteins, that in turn could contribute to remodelling without necessarily maintaining a direct correlation between protein and mRNA levels (Balch et al. 2008). Finally, developments in novel computer architectures have facilitated multi‐scale modelling approaches to integrate details of changes at multiple molecular levels to predict outputs at the whole tissue level for the expression and distributions of ion conductances (Weiss et al. 2012).
With these new levels of integration, we can now decipher not just what channels are altered, but how they change in terms of expression levels, post‐translational modifications, subcellular localization and how these changes evolve over time (Fig. 4). With such rich data sets, we will also be able to identify both the individual members, as well as networks of regulators, including microRNAs, lncRNAs, and transcription factors, that mediate electrical remodelling. Furthermore, we will be able to address questions such as how do related stresses result in such different outcomes, e.g. how does exercise result in adaptive hypertrophy whilst hypertension results in maladaptive remodelling. Importantly, the basis of these differences may be exploited for therapeutic benefits.
Figure 4. An integrated approach to studying K+ channel remodelling in heart disease.
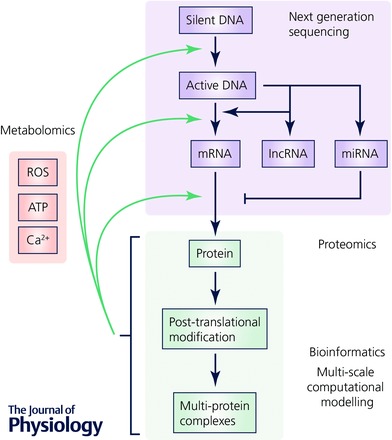
Information from next generation sequencing (lilac), proteomics (green) and metabolomics (pink) will need to be integrated using bioinformatics and multi‐scale computer modelling to establish a systems‐wide understanding of electrical remodelling.
Identification of novel therapeutic targets for cardiac arrhythmias
Multiple K+ currents have been targeted for arrhythmia therapy. Unfortunately, to date, these target‐specific drugs have not translated into new, safe, or effective anti‐arrhythmic therapy. Indeed, landmark studies of anti‐arrhythmic drugs have been shown to increase mortality in patients with myocardial infarction or left ventricular dysfunction compared to placebo (CAST Investigators, 1989; Waldo et al. 1996; Kober et al. 2008). The key reason is the very fact that arrhythmia mechanisms are dependent on a highly complex and dynamic multi‐scale system. Therefore, detailed mechanistic understanding of the molecular correlates, biophysical properties and interdependence of cardiac ion channels at subcellular, cellular and tissue levels are critical to the development of safe and effective anti‐arrhythmic drug therapy.
Atrial‐specific ion channel blockers
AF is the most common sustained arrhythmia clinically (Ferrari et al. 2016). Even though catheter ablation has been widely used for AF, the treatment is invasive and remains inadequate in a significant number of patients and development of new anti‐arrhythmic drugs for AF is highly desirable. Because atrial and ventricular myocytes express distinct repertoires of ion channels, there has been considerable interest in developing atrial‐specific ion channel blockers for atrial arrhythmias (Wettwer et al. 2007; Burashnikov et al. 2008; Antzelevitch & Burashnikov, 2010; Burashnikov & Antzelevitch, 2010; Schotten et al. 2016; Voigt & Dobrev, 2016). However, it is critical to note that the degree of atrial selectivity of anti‐arrhythmic drugs may be different in normal, compared to remodelled, hearts associated with different cardiac diseases.
Kv1.5, encoded by KCNA5 (Tamkun et al. 1991), underlies I Kur, which is expressed in human atria, but not ventricles (Fedida et al. 1993; Wang et al. 1993). Blockers of I Kur, AVE0118 and XEN‐D0101, prolong atrial APD and ERP in animal models and in humans (Wettwer et al. 2004; Schotten et al. 2007; Christ et al. 2008) and would be predicted to prevent AF in humans without risk of QT prolongation. However, Kv1.5 channels are down‐regulated in chronic AF (Van Wagoner et al. 1997; Van Wagoner, 2003). In addition, blockade of I Kur in human atrial tissue from patients in sinus rhythm elevates the AP plateau and shortens, rather than prolongs, APD, possibly by activating NCX in its reverse mode contributing to repolarizing current (Schotten et al. 2007). Block of I Kur, therefore, may provide the substrate for development of AF in healthy atria, via abbreviation of APD and ERP (Burashnikov & Antzelevitch, 2008). Moreover, evidence from humans has shown that both loss‐of‐function and gain‐of‐function mutations in KCN5A are associated with early onset lone AF (Christophersen et al. 2013).
Vernakalant was suggested as a potential I Kur blocker for the treatment of AF. However, it is a multichannel blocker, inhibiting not only I Kur, but also I to, I Kr, I K,ACh, and I K,ATP, as well as the peak and late I Na (Fedida, 2007; Burashnikov et al. 2008). The antifibrillatory effect of vernakalant may result from its blockade of the peak I Na which increases cardiac excitation threshold, slows conduction, and creates a period of refractoriness (Burashnikov et al. 2008). In addition, there are differences in the effects of vernakalant in canine and human atrial myocytes and there is a controversy about whether I Kur in dog is generated by Kv3.1 or Kv1.2, rather than Kv1.5, as in the human (Nattel et al. 1999; Fedida et al. 2003).
Another atrial‐specific ion channel that has received considerable attention is I K,ACh, encoded by the G‐protein activated inwardly rectifying K+ channel α‐subunits, Kir3.1/Kir3.4 (Ravens et al. 2013). The channels are more abundantly expressed in atrial than in ventricular myocytes (Krapivinsky et al. 1995; Dobrzynski et al. 2001; Schram et al. 2002; Gaborit et al. 2007). The current has been shown to mediate AF induced by vagal stimulation via activation of muscarinic M2 receptors. I K,ACh hyperpolarizes the membrane potential and shortens atrial APs, contributing to maintenance of AF by promoting reentry (Kovoor et al. 2001). More importantly, in chronic AF, the channels are constitutively active in the absence of any M2 receptor ligand (Dobrev et al. 2005), suggesting I K,ACh should be viewed as a promising target for AF (Voigt & Dobrev, 2016).
Several anti‐arrhythmic agents including azimilide, dofetilide, dronedarone, ibutilide, sotalol and terikalant block I K,ACh and may contribute to their efficacy in AF (Ravens et al. 2013). The benzopyrane derivative NIP‐142 selectively blocks I K,ACh and reverses the shortening effect of carbachol or adenosine on atrial APs (Matsuda et al. 2006) and inhibits vagally induced AF. The congener NIP‐151 blocks I K,ACh with a potency that is at least four orders of magnitude higher than I Kr and is highly effective in canine AF models (Hashimoto et al. 2008). Although many drugs have I K,ACh blocking properties, selective I K,ACh blockade has only recently been reported using the compound NTC‐801 that has been shown to be effective in AF models (Machida et al. 2011).
Novel therapeutic targets
Recent studies have provided evidence for possible novel therapeutic targets for K+ channels in the treatment of cardiac arrhythmias. Specifically, studies have demonstrated the important roles of SK channels in cardiac repolarization. Indeed, interests in cardiac SK channels are further fuelled by recent studies suggesting a possible role of SK channels in lone AF (Ellinor et al. 2010; Christophersen & Ellinor, 2016). Recently three different SK channel inhibitors, UCL1684, N‐(pyridin‐2‐yl)‐4‐(pyridin‐2‐yl)thiazol‐2‐amine (ICA) (Gentles et al. 2008) and NS8593, have been shown to have anti‐arrhythmic effects in models of AF in rat, guinea pig, rabbit and dog (Diness et al. 2010, 2011; Qi et al. 2014). The results from these studies suggest that SK channels may represent a potential therapeutic target for the treatment of atrial arrhythmias. However, there remain major gaps in our knowledge. Blockade of SK channels in cardiac arrhythmias has been shown to be both anti‐arrhythmic (Diness et al. 2010, 2011) and proarrhythmic (Hsueh et al. 2013; Wagner & Maier, 2013) in various models, possibly influenced by the state of other currents that integrate with the SK channel current to shape AP.
Combination drug therapy
One of the most commonly prescribed anti‐arrhythmic drugs is amiodarone, which has been shown to block several cardiac K+ currents, as well as I Na and I Ca,L. It is also a non‐competitive antagonist of α‐ and β‐adrenergic receptors. Amiodarone has been demonstrated to be effective and relatively safe without inducing TdP polymorphic ventricular tachycardia that is seen as a result of QT prolongation (Zimetbaum, 2007; Singh, 2008). In addition to amiodarone several anti‐arrhythmic drugs, including dronedarone, vernakalant and ranolazine, have been shown to be effective clinically for AF with low incidence of proarrhythmias (Burashnikov & Antzelevitch, 2010). These anti‐arrhythmic drugs inhibit I Na with relatively fast kinetics, as well as block I Kr and late I Na (Burashnikov et al. 2008). In addition, rapidly dissociating I Na blockers are found to be atrial selective in contrast to the slowly dissociating blockers (Burashnikov et al. 2008). A recent new line of investigation uses combinations of anti‐arrhythmic drugs that may allow the use of lower dosages with reduced side effects and better balance the inward and the outward currents in normalizing AP (Aguilar et al. 2015; Reiffel et al. 2015).
Drug‐induced arrhythmias
Drug‐induced arrhythmias can be caused by both cardiovascular and non‐cardiovascular drugs and can be life‐threatening. This is due to drug‐induced QT prolongation and TdP polymorphic ventricular tachycardia. Individual susceptibility includes pharmacokinetic risk factors and genetic predisposition. Additional risk factors include structural heart disease and electrolyte imbalance. Drug‐induced arrhythmias are discussed in more detail in the companion White Paper (see Grandi et al. 2017).
Inherited mutations in the Kv11.1 (hERG) α‐subunit of I Kr, encoded by KCNH2, are linked to type 2 LQTS. In addition, life‐threatening arrhythmia can also be induced by blockade of Kv11.1 channels in a surprisingly diverse group of drugs with vastly different chemical structures. Anti‐arrhythmic, antihistamine, antimicrobial, antipsychotic, and antidepressant drugs are important classes associated with risk of proarrhythmia. Indeed, an effect on I Kr is a common reason for drug failure in preclinical safety trials. Even though inherited LQTS and TdP can be caused by loss‐of‐function mutations in multiple cardiac K+ channels, drug‐induced LQTS and TdP are predominantly caused by direct blockade of Kv11.1 channels or disruption of Kv11.1 channel trafficking to the cell surface (Sanguinetti & Tristani‐Firouzi, 2006; Yang et al. 2014).
Previous studies have suggested that Kv11.1 channels have structural features that can more effectively accommodate the binding of drugs compared with other K+ channels. Specifically, two aromatic residues (Tyr‐652 and Phe‐656) located in the S6 domain of the Kv11.1 subunit are probably important for the binding of several classes of drugs. The side chains of the two residues are orientated towards the large central cavity of the channel. More importantly, these two residues are not conserved in other Kv channels in which an Ile and a Val are found in homologous positions. It was suggested that the eight aromatic side chains per channel are arranged in two concentric rings to accommodate drug–channel interactions (Sanguinetti & Tristani‐Firouzi, 2006).
Experimental models in the study of cardiac K+ channel function
A variety of experimental animal models have been used over the years in studies detailing the time‐ and voltage‐dependent properties and the physiological roles of the many K+ channels expressed in the mammalian heart (Barry & Nerbonne, 1996). Similar to functional analysis of other types of ion channels, the mouse models began to be used increasingly in the mid‐1990s to explore the molecular determinants of native myocardial K+ channels primarily owing to the ease and speed with which molecular genetic strategies can be exploited, functional assays can be completed, and molecular mechanisms can be probed in the mouse, particularly when compared with other mammalian species. These efforts quickly led to the detailed biophysical characterization of the voltage‐gated and non‐voltage‐gated K+ channels expressed in mouse myocardium, as well as the identification of the genes encoding the pore‐forming (α) subunits, as well as many of the accessory subunits of these channels (Nerbonne et al. 2001). There were also suggestions that the mouse might be used as a model system for investigating congenital and acquired arrhythmogenic cardiovascular disease mechanisms, particularly the ‘ion channelopathies’ (Ravens & Cerbai, 2008; Brenyo et al. 2012; Abriel & Zaklyazminskaya, 2013) linked to ion channel dysfunction (London, 2001; Charpentier et al. 2004), as well as for pre‐clinical testing of the anti‐arrhythmic efficacy and the proarrhythmic potential of drugs (Fabritz et al. 2007).
Although there are multiple repolarizing K+ currents in both human and mouse cardiac myocytes, there are clear differences in the properties and the molecular identities of the K+ channels expressed (Nerbonne & Kass, 2005). The two prominent delayed rectifier Kv currents, I Kr and I Ks, in human ventricular myocytes, for example, are not prominent in mouse cardiac myocytes and three distinct delayed rectifier Kv currents, I K,slow1, I K,slow2 and I ss, are expressed (Nerbonne & Kass, 2005). The genes, KCNQ1 and KCNH2, that encode the Kv α subunits, Kv7.1 and Kv11.1, that generate human cardiac I Kr and I Ks channels, and identified as loci of mutations in familial LQT1 and LQT2 (Ravens & Cerbai, 2008; Brenyo et al. 2012; Abriel & Zaklyazminskaya, 2013) and transgenic and targeted deletion strategies in mice have been used to probe the functioning of Kcnq1 and Kcnh2, as well as of the (Kcne1) gene, which encodes the I Ks channel accessory subunit, minK (Nerbonne et al. 2001). As might be expected, given that I Ks and I Kr are barely detectable in adult mouse cardiomyocytes, the cardiac effects of manipulating the Kcne1, Kcnq1 or Kcnh2 genes on myocardial K+ currents and myocardial functioning in the mouse heart are quite subtle (Nerbonne et al. 2001).
In several of the Kv channel transgenic and gene targeted mouse lines that have been generated, however, ECG abnormalities, including QT prolongation, as well as increased inducibility of ventricular arrhythmias, have been described and, in some cases, the observed abnormalities structurally resemble those seen in humans. Quite dramatic ECG phenotypes and spontaneous arrhythmias, for example, have been reported in some Kv channel transgenics (Nerbonne et al. 2001). In the Kcnq1‐isoform‐2‐expressing mouse, for example, QT prolongation, P wave abnormalities and TdP were reported. The severity of the cardiac phenotype, however, was correlated with the amount of the mutant protein expressed, observations that suggest non‐specific in vivo cardiac effects of ‘over‐expression’ of the Kcnq1‐isoform‐2 transgene. Overall, the incidence of spontaneous and/or inducible arrhythmias, particularly lethal arrhythmias that mimic those in humans, in mice with altered K+ channel expression is very low, observations that are interpreted as reflecting an inherent limitation of the mouse, perhaps owing to the small size of the mouse heart and the very rapid heart rate.
In spite of these limitations, important insights into the relationships between individual K+ channel subunits and functional K+ currents, as well as into the mechanisms underlying the electrical remodelling observed in association with alterations in the expression or functioning of individual K+ channel α and β subunits, have been provided through the application of in vivo molecular genetic strategies in the mouse. Together, these insights will guide the design, execution and interpretation of future experiments focused on delineating the molecular, cellular and systemic mechanisms underlying electrical remodelling and reprogramming in the myocardium. The mouse is expected to be a widely used model system in these efforts.
Large animal models of congenital and acquired cardiac arrhythmias
It has long been recognized that the electrophysiological properties of the hearts of large mammals, including the types of K+ channels expressed, more closely resemble those in the human heart, particularly compared with the mouse. Consistent with this, electrophysiological studies have detailed the distributions and the properties of the various K+ (and other) channels expressed in cardiac cells from cat, rabbit, canine and pig heart (Nerbonne & Kass, 2005). These large animal models are also more amenable (than the mouse) to studies focused on probing conduction, propagation and arrhythmias in the intact heart, although the inability to manipulate these systems genetically compromised their utility for studying human cardiac disease mechanisms.
This barrier to progress was broken with the development of the first transgenic rabbit models of LQT1 and LQT2, produced by Koren and colleagues with the cardiac specific expression of pore mutants of the human genes KCNQ1 (KvLQT1‐Y315S) and KCNH2 (HERG‐G628S), respectively (Brunner et al. 2008). These pore mutants were shown to function in a dominant negative fashion, eliminating I Ks and I Kr, and resulting in AP and QT prolongation (Brunner et al. 2008). Importantly and unexpectedly, the detailed electrophysiological characterization of ventricular myocytes from the transgenic KvLQT1‐Y315S and HERG‐G628S rabbits revealed co‐regulation of I Ks and I Kr (Brunner et al. 2008). In addition, the LQT2 rabbits show a high incidence of sudden death, attributed to increased dispersion of repolarization and polymorphic ventricular tachycardia. These transgenic rabbit models have enabled further studies focused on detailing arrhythmia mechanisms, as well as efforts to explore the effects of drugs and hormones on conduction, dispersion and arrhythmia susceptibility (Ziv et al. 2009; Odening et al. 2010, 2012, 2013; Ziupa et al. 2014; Kim et al. 2015). More recently, a novel transgenic rabbit LQT5 model, produced by cardiac specific expression of a KCNE1 mutant that functions as a dominant negative, was described with markedly altered repolarization reserve attributed to changes in both I Ks and I Kr (Major et al. 2016).
Although the idea of developing transgenic rabbit models to explore the functional effects of different mutations in the KCNQ1, KCNH2, KCNE1 and other ion channel subunit genes to explore genotype–phenotype relations is certainly a reasonable one, a clear limitation is the time and cost involved in generating and characterizing each transgenic rabbit line. In addition, phenotypic effects might well be variable across animals owing to genetic heterogeneity, a complication that can be avoided with transgenic mice, but not rabbits. Further limitations are expected to be realized given that, in spite of the similarities in K+ currents such as I Kr and I Ks, there are differences in the expression and properties of other repolarizing K+ currents in rabbit and human cardiomyocytes (Nerbonne & Kass, 2005). Given this, it seems reasonable to suggest that there should also be increased emphasis on electrophysiological studies of human myocardial K+ channels. Several previous reports have characterized native human cardiac K+ currents (Iost et al. 1998; Magyar et al. 2000; Virag et al. 2001; Jost et al. 2005; O'Hara et al. 2011); however, a significant number of studies are limited to atrial samples from patients undergoing open heart surgery (Van Wagoner et al. 1997) and ventricular samples from explanted hearts from end‐stage HF patients undergoing transplantation (Beuckelmann et al. 1993; Wettwer et al. 1994; Nabauer et al. 1996) reflecting the availability. One way to expand these efforts might be to acquire non‐failing human hearts deemed unsuitable for transplantation for non‐cardiac reasons (Glukhov et al. 2010). Even with expanded efforts, sample heterogeneity, reflecting genetics, prior history, health status, as well as previous and present medications, however, will still be a complicating factor to consider when analysing/interpreting electrophysiological data from human myocytes.
Cellular models to study cardiac arrhythmia mechanisms
Cell‐based systems have also been utilized to study the biophysical properties of genes/proteins involved in the generation of cardiac K+ channels, as well as functional effects of mutations in K+ channel subunit genes linked to congenital arrhythmias on channel expression, assembly, trafficking, targeting and biophysical properties. Heterologous expression systems offer several clear advantages in this regard, all of which are related to the ease and the speed with which constructs can be generated, expressed and functionally characterized. Expression systems can also be used for drug screening of potential channel blockers or activators (Haraguchi et al. 2015) because of the ease with which experimental manipulations can be made and high‐throughput screening methods can be applied. There are, however, also potential problems with interpreting results obtained in experiments conducted in heterologous cells for the simple reason that the detailed properties of K+ (and other) channels depend on the cellular environment in which they are expressed owing to cell type‐specific differences in RNA processing, protein–protein interactions, post‐translational modifications and membrane lipid composition, etc., all of which could influence the properties of expressed K+ channels.
An alternative cellular approach being used to detail the properties, functioning and regulation of human cardiac K+ (and other) channels emphasizes ‘native’ K+ channels expressed in human embryonic stem cell‐derived cardiac myocytes (hESC‐CMs) or human induced pluripotent stem cell‐derived cardiomyocytes (hiPSC‐CMs) (Itzhaki et al. 2011; Blazeski et al. 2012; Sallam et al. 2015). The clear advantages of using hESC‐CMs and hiPSC‐CMs is that the cells are of human origin, sample availability is not an issue, and, in addition, these preparations are amenable to electrophysiological and molecular manipulations and can also be used for high‐throughput screening. In addition, hiPSC‐CMs are generated from adult fibroblasts making it possible to generate patient specific hiPSC‐CM lines and to study the effect(s) of identified mutations in genes encoding K+ channels or other proteins linked to LQTS or SQTS, BrS, catecholaminergic polymorphic ventricular tachycardia, sick sinus syndrome, hypertrophic cardiomyopathy and other congenital arrhythmogenic cardiac disorders (Itzhaki et al. 2011; Dirschinger et al. 2012; Lahti et al. 2012; Sinnecker et al. 2013; Tanaka et al. 2015). Importantly, the fact that these cells can be readily modified means that gene mutations can be corrected, allowing direct comparisons between the properties of cells with and without mutations, in the same genetic background (Itzhaki et al. 2011; Sallam et al. 2015). One major limitation of hiPSC‐CMs at present is that the cells are immature both electrically and structurally and considerable effort is focused on developing methods to manipulate and improve cell maturation (Bett et al. 2013; Veerman et al. 2015).
Computational modelling and simulation approaches to study the mechanisms of K+ channel linked acquired cardiac arrhythmias
‘To evaluate the full impact of quantitative systems pharmacology, one must remember that the mechanisms of action of most drugs are not fully understood and the origins of patient‐to‐patient variability in therapeutic and adverse responses are often obscure …. And drugs that fail at late stages of clinical trials are rarely investigated further to determine the reasons for their failure.’, wrote the quantitative systems pharmacology workshop group in an NIH white paper (Sorger et al. 2011). These statements aptly describe the driving biomedical challenges facing scientists, regulators and clinicians in trying to determine safety and efficacy of drugs.
History has shown that the safety or toxicity of drug treatments cannot be observed or readily predicted via study of component elements alone. This is especially clear in the longstanding failure to anticipate cardiotoxicity in drug development (Roden, 2004). Cardiotoxicity is one of the most common risks for drugs in development, manifesting as prolongation of the QT interval and potential for fatal ventricular arrhythmias.
As described earlier, cardiac rhythm disturbances are most commonly a side effect from unintended block of the promiscuous drug target Kv11.1, the pore‐forming domain of I Kr in the heart. But not all Kv11.1 blockers are proarrhythmic. There is an urgent need to develop new approaches for selective and sensitive prediction of how drugs with complex interactions and multiple subcellular targets will alter the emergent electrical activity in the heart. Mathematical modelling and simulation constitute some of the most promising methodologies to reveal fundamental biological principles and mechanisms, model effects of interactions between system components and predict emergent treatment effects. There are no reasonable, efficient and cost‐effective experimental alternatives that can achieve these goals. Application of new computational and simulation methods may in the next decade usher in an era that allows for integration of data in physiological networks to reveal emergent drug effects at the cellular, tissue and organ levels and to facilitate prediction and development of safer therapeutic interventions. Quantitative systems pharmacology approaches are currently being developed in conjunction with high efficiency computational processes for probing the mechanisms of action of prototypical drugs in the setting of cardiac electrical disorders.
While the crystal structure of the Kv11.1 channel is not yet available, multiple in silico approaches including homology modelling, de novo protein design and molecular dynamics simulations have been undertaken to link structural determinants of Kv11.1 to its function (Stary et al. 2010). Sequence conservation is substantial between Kv11.1 and other Kv channels with known structures and new studies suggest that computer‐based homology models based on solved KV based on the available crystal structures of KcsA, MthK, Kv1.2 and KvAP pore domains can successfully be utilized to study the human K+ channels (Mitcheson et al. 2000; Perry et al. 2004; Stansfeld et al. 2007). The predictions from molecular modelling studies based bacterial KcsA and MthK (Perry et al. 2004; Stansfeld et al. 2007) channels or the mammalian channel Kv1.2 (Durdagi et al. 2010; Stary et al. 2010) concur with the experimental findings that have revealed two key residues responsible for drug stabilization in the Kv11.1 cavity, e.g. Y652 and F656 (Lees‐Miller et al. 2000; Mitcheson et al. 2000). Model studies reproduced this feature in studies of Kv11.1 blockers such as dofetilide, KN‐93 and other common high‐affinity blockers (Durdagi et al. 2011, 2012).
Because it is so difficult to predict how ion channel modification, studied in isolation, alters the functioning of the whole heart, computationally based modelling and simulation approaches have been developed and widely applied to link deviations in ion channel function to emergent electrical activity in cells, tissue and even simulated whole hearts. In the last 20 years, an explosion in the development of sophisticated models has occurred, concomitant with improved experimental techniques that have allowed identification and characterization of numerous ion channel subtypes and their regulation in the heart from multiple species (Romero et al. 2010, 2011; Bett et al. 2011; Moreno & Clancy, 2012; Qu et al. 2013; Verkerk & Wilders, 2013; Bueno‐Orovio et al. 2014; Glynn et al. 2014; Greenstein et al. 2014; Onal et al. 2014; Ramirez et al. 2014; Pathmanathan et al. 2015; Besse et al. 2011). Sophisticated ion channel models have even been developed that account for various genetic defects that alter the behaviour of ion channels. These complex ion channel representations have been incorporated into numerous cardiac model ‘cells’ from multiple species. The cellular level models have been widely replicated and coupled, creating mathematical representations of cardiac tissue in one, two or three dimensions, with the incorporation of complex anatomical heterogeneities including anisotropy, structural features and distinct cells with specifically associated electrophysiological characteristics (Trenor et al. 2007; Romero et al. 2009; Bers & Grandi, 2011; Niederer & Smith, 2012; Roberts et al. 2012; Sugiura et al. 2012; Trayanova et al. 2012; Zhang et al. 2012; Zhou & O'Rourke, 2012; Polakova & Sobie, 2013; Quail & Taylor, 2013; Romero et al. 2013; Sato & Clancy, 2013; Tobon et al. 2013; Ferrero et al. 2014; Gomez et al. 2014; Henriquez, 2014; Ramirez et al. 2014; Trayanova & Boyle, 2014; Duncker et al. 2015).
Simulation studies have revealed plausible experimentally testable mechanisms for how perturbations to ion channels and associated processes alter emergent electrical behaviour at higher system scales. For example, computer simulations have revealed that the APD prolongation exerted by most mutant channels (I Kr, I Ks and I Na) related to acquired LQTS (aLQTS) were shorter than those produced by mutations producing congenital LQTS (Itoh et al. 2006). In another study, the role of the R1047L polymorphisms in KCNH2 in dofetilide‐induced TdP was investigated (Sun et al. 2004). The R1047L missense mutation was linked to TdP in fibrillation patients treated with dofetilide (Sun et al. 2004). The mutation caused a positive shift of the activation curve and slowed the activation and inactivation kinetics. Simulation of these abnormalities resulted in prolongation of the APD, which suggests that 1047L may contribute to a higher incidence of TdP in the presence of I Kr blockers (Sun et al. 2004).
Investigation of drug‐induced, or aLQTS resulting from Kv11.1 block is critical to identify individuals who are susceptible to aLQTS, which is crucial for reducing the risk of cardiac arrhythmias. Experiments have shown that the functional changes of most mutations are mild and most drug sensitivities for mutant channels are similar to that of the WT channels (Itoh et al. 2006). Nevertheless, approximately 40% of patients with aLQTS have been shown to exhibit allelic variants that disrupt the function of cardiac ionic channels (Itoh et al. 2006).
More recently, a systematic and comprehensive computational study has been conducted to reveal new insights into the impact of latent I Kr channel kinetic dysfunction on the I Kr time course during the AP, susceptibility to aLQTS and the potential for adjunctive therapy with I Kr channel openers (Romero et al. 2014). Specifically, this study predicted the most potentially lethal combinations of kinetic anomalies and drug properties and the ideal inverse therapeutic properties of I Kr channel openers that would be expected to remedy a specific defect. The simulations predicted that drugs with disparate affinities to conformation states of the I Kr channel markedly enhanced the susceptibility to aLQTS, especially at slow pacing rates.
In the same study, Romero et al. simulated the M54T latent mutation in KCNE2, which has been related to aLQTS and arrhythmias, in the presence of dofetilide, which drastically prolonged the QT interval duration in the M54T mutation in KCNE2 compared to wild‐type. The study also predicted that application of a virtual potassium channel opener that only slows deactivation would be the ideal adjunctive therapy that could normalize the effect of dofetilide‐induced AP prolongation in the presence of the M54T hMiRP1 mutation. Simulation of the addition of the I Kr activator RPR260243, which slows the deactivation and increases the current magnitude by positively shifting the inactivation curve (Perry et al. 2007) was predicted to correct the APD and QT interval prolongation, but it introduced the risk of developing SQTS (Romero et al. 2014).
Finally, computational modelling has also been used to yield insights into the relationship between Kv11.1 1a/1b channels, drug sensitivity and arrhythmia proclivity (Sale et al. 2008). These simulations showed that altered channel kinetics may explain reduced rectification and an increase in current during repolarization. The model also predicted that drugs that block 1a homomers of Kv11.1 are more arrhythmogenic than those that block the heteromer.
Questions/controversies
-
‐
Can K+ channel diversity be exploited to target specific cell types for anti‐arrhythmic therapy?
-
‐
Are M‐cells present in human ventricle?
-
‐
During pathophysiological remodelling, are K+ channel sub‐types impacted similarly throughout the different regions of the heart?
-
‐
Given the proarrhythmic potential of both increased and decreased K+ channel function, what changes in K+ currents are adaptive and which are maladaptive in a given disease? Of note, the lack of highly selective blockers and activators for most of the myocardial K+ channels makes it difficult to assess their specific roles in arrhythmias.
-
‐
What can we learn from the species‐specific differences in the roles of K+ channels in cardiac arrhythmias?
-
‐
Through which mechanisms are different K+ currents co‐regulated with the counteracting depolarizing currents (i.e. Ca2+ currents) to ensure stable electrophysiological behaviour under a wide range of conditions? How are these mechanisms perturbed under diseases conditions?
-
‐
Which K+ channels, in addition to I K1, control the RMP in different regions of the heart?
-
‐
What are the (mal)adaptive responses to chronic modulation of K+ channel expression and/or function?
Summary
A variety of cellular and animal based model systems have been, and continue to be, used in studies focused on defining the functional properties of myocardial K+ channels and the impact of disease linked mutations in the genes that encode K+ channel subunits on cardiac myocyte membrane excitability and arrhythmia susceptibility. Nevertheless, the physiological roles of the various K+ channels expressed in human heart and the cellular, molecular and systemic mechanisms linking congenital and acquired changes in K+ expression and/or functioning to increased risk of arrhythmias and sudden death remain rather poorly understood. It seems clear that increased efforts, focused on delineating mechanisms that link changes in the expression and functioning of cardiac K+ (and other) channels to arrhythmogenic cardiovascular disease, are needed to provide new insights. Accomplishing this will, almost certainly, require the continued development and application of multiple model systems that allow multi‐scale and multidisciplinary experimental investigation and analyses.
Importantly, our understanding of how disruption in cardiac K+ channels leads to arrhythmias is continually improving due to the development and implementation of computational modelling and simulation approaches. These approaches span scales from the single atomistic ion channel scale to the high‐resolution reconstruction of the heart. These new computational strategies may be applied to improve preclinical screening of compounds to detect possible proarrhythmic effects and/or predisposition from underlying genetic causes. Computer‐based approaches can help to determine the mechanisms of drug actions. Expansion and development of new approaches may help to reveal why drugs that are non‐specific K+ channel blockers and interact with many targets, like amiodarone, seem to be less proarrhythmic than selective blockers like d‐sotalol. Finally, models might eventually be used to guide therapy for specific clinical situations and to identify optimal ‘polypharmacy’ to inform the common practice of clinical empirical mixing and matching of drugs to create multidrug therapeutic regimens (Yang et al. 2016). Central to the success of modelling and simulation approaches for predictive safety pharmacology is the development of models that are informed and validated via tightly planned integration of experiments and simulations at every stage (Clancy et al. 2016). It will be critical to demonstrate the usefulness of the frameworks and to validate their utility and reproducibility. Finally, although we have endeavoured to be as thorough and timely as possible in this review, this is a rapidly moving and expanding field. We have prioritized specific areas of emphasis at the expense of being comprehensive. We have, however, attempted to circumvent this limitation by including several published review articles in the references.
Additional information
Competing interests
None declared.
Funding
This conference was supported by a National Institutes of Health (NIH) grant (R13HL132515) to Y. Chen‐Izu, D. M. Bers, N. Chiamvimonvat and E. Grandi, and additional grants to other authors that allowed them to participate. We also thank the many sponsors that support the University of California Davis Cardiovascular Symposium Series. Please see the conference website for detailed information.
Acknowledgements
We thank the Organizing Committee, Consultants, and all conference participants for their contributions to the scientific exchange and discussions. For conference information, please refer to basicscience.ucdmc.ucdavis.edu/UCDavisCardiovascular Symposia/index.html.
Contributor Information
Nipavan Chiamvimonvat, Email: nchiamvimonvat@ucdavis.edu.
Jeanne M. Nerbonne, Email: jnerbonne@wustl.edu
References
- Abriel H & Zaklyazminskaya EV (2013). Cardiac channelopathies: genetic and molecular mechanisms. Gene 517, 1–11. [DOI] [PubMed] [Google Scholar]
- Aguilar M, Xiong F, Qi XY, Comtois P & Nattel S (2015). Potassium channel blockade enhances atrial fibrillation‐selective antiarrhythmic effects of optimized state‐dependent sodium channel blockade. Circulation 132, 2203–2211. [DOI] [PubMed] [Google Scholar]
- Antzelevitch C (2010). M cells in the human heart. Circ Res 106, 815–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antzelevitch C & Burashnikov A (2010). Atrial‐selective sodium channel block as a novel strategy for the management of atrial fibrillation. Ann NY Acad Sci 1188, 78–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antzelevitch C, Shimizu W, Yan GX, Sicouri S, Weissenburger J, Nesterenko VV, Burashnikov A, Di Diego J, Saffitz J & Thomas GP (1999). The M cell: its contribution to the ECG and to normal and abnormal electrical function of the heart. J Cardiovasc Electrophysiol 10, 1124–1152. [DOI] [PubMed] [Google Scholar]
- Antzelevitch C, Sicouri S, Litovsky SH, Lukas A, Krishnan SC, Di Diego JM, Gintant GA & Liu DW (1991). Heterogeneity within the ventricular wall. Electrophysiology and pharmacology of epicardial, endocardial, and M cells. Circ Res 69, 1427–1449. [DOI] [PubMed] [Google Scholar]
- Armoundas AA, Wu R, Juang G, Marban E & Tomaselli GF (2001). Electrical and structural remodeling of the failing ventricle. Pharmacol Ther 92, 213–230. [DOI] [PubMed] [Google Scholar]
- Balch WE, Morimoto RI, Dillin A & Kelly JW (2008). Adapting proteostasis for disease intervention. Science 319, 916–919. [DOI] [PubMed] [Google Scholar]
- Banyasz T, Horvath B, Jian Z, Izu LT & Chen‐Izu Y (2011). Sequential dissection of multiple ionic currents in single cardiac myocytes under action potential‐clamp. J Mol Cell Cardiol 50, 578–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banyasz T, Jian Z, Horvath B, Khabbaz S, Izu LT & Chen‐Izu Y (2014). Beta‐adrenergic stimulation reverses the I Kr–I Ks dominant pattern during cardiac action potential. Pflugers Arch 466, 2067–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry DM & Nerbonne JM (1996). Myocardial potassium channels: electrophysiological and molecular diversity. Annu Rev Physiol 58, 363–394. [DOI] [PubMed] [Google Scholar]
- Bartos DC, Grandi E & Ripplinger CM (2015). Ion channels in the heart. Compr Physiol 5, 1423–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers DM & Grandi E (2011). Human atrial fibrillation: insights from computational electrophysiological models. Trends Cardiovasc Med 21, 145–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besse IM, Mitchell CC, Hund TJ & Shibata EF (2011). A computational investigation of cardiac caveolae as a source of persistent sodium current. Front Physiol 2, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bett GC, Kaplan AD, Lis A, Cimato TR, Tzanakakis ES, Zhou Q, Morales MJ & Rasmusson RL (2013). Electronic ‘expression’ of the inward rectifier in cardiocytes derived from human‐induced pluripotent stem cells. Heart Rhythm 10, 1903–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bett GC, Zhou Q & Rasmusson RL (2011). Models of HERG gating. Biophys J 101, 631–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beuckelmann DJ, Nabauer M & Erdmann E (1993). Alterations of K+ currents in isolated human ventricular myocytes from patients with terminal heart failure. Circ Res 73, 379–385. [DOI] [PubMed] [Google Scholar]
- Blazeski A, Zhu R, Hunter DW, Weinberg SH, Boheler KR, Zambidis ET & Tung L (2012). Electrophysiological and contractile function of cardiomyocytes derived from human embryonic stem cells. Prog Biophys Mol Biol 110, 178–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boukens BJ, Christoffels VM, Coronel R & Moorman AF (2009). Developmental basis for electrophysiological heterogeneity in the ventricular and outflow tract myocardium as a substrate for life‐threatening ventricular arrhythmias. Circ Res 104, 19–31. [DOI] [PubMed] [Google Scholar]
- Boyden PA, Hirose M & Dun W (2010). Cardiac Purkinje cells. Heart Rhythm 7, 127–135. [DOI] [PubMed] [Google Scholar]
- Boyle WA & Nerbonne JM (1991). A novel type of depolarization‐activated K+ current in isolated adult rat atrial myocytes. Am J Physiol 260, H1236–H1247. [DOI] [PubMed] [Google Scholar]
- Brenyo AJ, Huang DT & Aktas MK (2012). Congenital long and short QT syndromes. Cardiology 122, 237–247. [DOI] [PubMed] [Google Scholar]
- Britton OJ, Bueno‐Orovio A, Van Ammel K, Lu HR, Towart R, Gallacher DJ & Rodriguez B (2013). Experimentally calibrated population of models predicts and explains intersubject variability in cardiac cellular electrophysiology. Proc Natl Acad Sci USA 110, E2098–E2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brugada P (2016). Brugada syndrome: More than 20 years of scientific excitement. J Cardiol 67, 215–220. [DOI] [PubMed] [Google Scholar]
- Brunner M, Peng X, Liu GX, Ren XQ, Ziv O, Choi BR, Mathur R, Hajjiri M, Odening KE, Steinberg E, Folco EJ, Pringa E, Centracchio J, Macharzina RR, Donahay T, Schofield L, Rana N, Kirk M, Mitchell GF, Poppas A, Zehender M & Koren G (2008). Mechanisms of cardiac arrhythmias and sudden death in transgenic rabbits with long QT syndrome. J Clin Invest 118, 2246–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bueno‐Orovio A, Sanchez C, Pueyo E & Rodriguez B (2014). Na/K pump regulation of cardiac repolarization: insights from a systems biology approach. Pflugers Arch 466, 183–193. [DOI] [PubMed] [Google Scholar]
- Burashnikov A & Antzelevitch C (2008). Can inhibition of I Kur promote atrial fibrillation? Heart Rhythm 5, 1304–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burashnikov A & Antzelevitch C (2010). New developments in atrial antiarrhythmic drug therapy. Nat Rev Cardiol 7, 139–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burashnikov A, Di Diego JM, Zygmunt AC, Belardinelli L & Antzelevitch C (2008). Atrial‐selective sodium channel block as a strategy for suppression of atrial fibrillation. Ann NY Acad Sci 1123, 105–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caballero R, de la Fuente MG, Gomez R, Barana A, Amoros I, Dolz‐Gaiton P, Osuna L, Almendral J, Atienza F, Fernandez‐Aviles F, Pita A, Rodriguez‐Roda J, Pinto A, Tamargo J & Delpon E (2010). In humans, chronic atrial fibrillation decreases the transient outward current and ultrarapid component of the delayed rectifier current differentially on each atria and increases the slow component of the delayed rectifier current in both. J Am Coll Cardiol 55, 2346–2354. [DOI] [PubMed] [Google Scholar]
- CAST Investigators (1989). Preliminary report: effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. The Cardiac Arrhythmia Suppression Trial (CAST) Investigators. N Engl J Med 321, 406–412. [DOI] [PubMed] [Google Scholar]
- Chan YH, Tsai WC, Ko JS, Yin D, Chang PC, Rubart M, Weiss JN, Everett TH 4th, Lin SF & Chen PS (2015). Small‐conductance calcium‐activated potassium current is activated during hypokalemia and masks short‐term cardiac memory induced by ventricular pacing. Circulation 132, 1377–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charpentier F, Demolombe S & Escande D (2004). Cardiac channelopathies: from men to mice. Ann Med 36, Suppl. 1, 28–34. [DOI] [PubMed] [Google Scholar]
- Christ T, Wettwer E, Voigt N, Hala O, Radicke S, Matschke K, Varro A, Dobrev D & Ravens U (2008). Pathology‐specific effects of the I Kur/I to/I K,ACh blocker AVE0118 on ion channels in human chronic atrial fibrillation. Br J Pharmacol 154, 1619–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christophersen IE & Ellinor PT (2016). Genetics of atrial fibrillation: from families to genomes. J Hum Genet 61, 61–70. [DOI] [PubMed] [Google Scholar]
- Christophersen IE, Olesen MS, Liang B, Andersen MN, Larsen AP, Nielsen JB, Haunso S, Olesen SP, Tveit A, Svendsen JH & Schmitt N (2013). Genetic variation in KCNA5: impact on the atrial‐specific potassium current I Kur in patients with lone atrial fibrillation. Eur Heart J 34, 1517–1525. [DOI] [PubMed] [Google Scholar]
- Chua SK, Chang PC, Maruyama M, Turker I, Shinohara T, Shen MJ, Chen Z, Shen C, Rubart‐von der Lohe M, Lopshire JC, Ogawa M, Weiss JN, Lin SF, Ai T & Chen PS (2011). Small‐conductance calcium‐activated potassium channel and recurrent ventricular fibrillation in failing rabbit ventricles. Circ Res 108, 971–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy CE, An G, Cannon WR, Liu Y, May EE, Ortoleva P, Popel AS, Sluka JP, Su J, Vicini P, Zhou X & Eckmann DM (2016). Multiscale modeling in the clinic: drug design and development. Ann Biomed Eng 44, 2591–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy CE & Rudy Y (2002). Na+ channel mutation that causes both Brugada and long‐QT syndrome phenotypes: a simulation study of mechanism. Circulation 105, 1208–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhamoon AS, Pandit SV, Sarmast F, Parisian KR, Guha P, Li Y, Bagwe S, Taffet SM & Anumonwo JM (2004). Unique Kir2.x properties determine regional and species differences in the cardiac inward rectifier K+ current. Circ Res 94, 1332–1339. [DOI] [PubMed] [Google Scholar]
- Di Diego JM, Sun ZQ & Antzelevitch C (1996). I to and action potential notch are smaller in left vs. right canine ventricular epicardium. Am J Physiol 271, H548–H561. [DOI] [PubMed] [Google Scholar]
- Diness JG, Skibsbye L, Jespersen T, Bartels ED, Sorensen US, Hansen RS & Grunnet M (2011). Effects on atrial fibrillation in aged hypertensive rats by Ca2+‐activated K+ channel inhibition. Hypertension 57, 1129–1135. [DOI] [PubMed] [Google Scholar]
- Diness JG, Sorensen US, Nissen JD, Al‐Shahib B, Jespersen T, Grunnet M & Hansen RS (2010). Inhibition of small‐conductance Ca2+‐activated K+ channels terminates and protects against atrial fibrillation. Circ Arrhythm Electrophysiol 3, 380–390. [DOI] [PubMed] [Google Scholar]
- Dirschinger RJ, Goedel A, Moretti A, Laugwitz KL & Sinnecker D (2012). Recapitulating long‐QT syndrome using induced pluripotent stem cell technology. Pediatr Cardiol 33, 950–958. [DOI] [PubMed] [Google Scholar]
- Dobrev D, Friedrich A, Voigt N, Jost N, Wettwer E, Christ T, Knaut M & Ravens U (2005). The G protein‐gated potassium current I K,ACh is constitutively active in patients with chronic atrial fibrillation. Circulation 112, 3697–3706. [DOI] [PubMed] [Google Scholar]
- Dobrzynski H, Marples DD, Musa H, Yamanushi TT, Henderson Z, Takagishi Y, Honjo H, Kodama I & Boyett MR (2001). Distribution of the muscarinic K+ channel proteins Kir3.1 and Kir3.4 in the ventricle, atrium, and sinoatrial node of heart. J Histochem Cytochem 49, 1221–1234. [DOI] [PubMed] [Google Scholar]
- Dong M, Sun X, Prinz AA & Wang HS (2006). Effect of simulated I to on guinea pig and canine ventricular action potential morphology. Am J Physiol Heart Circ Physiol 291, H631–H637. [DOI] [PubMed] [Google Scholar]
- Duncker DJ, Bakkers J, Brundel BJ, Robbins J, Tardiff JC & Carrier L (2015). Animal and in silico models for the study of sarcomeric cardiomyopathies. Cardiovasc Res 105, 439–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durdagi S, Deshpande S, Duff HJ & Noskov SY (2012). Modeling of open, closed, and open‐inactivated states of the hERG1 channel: structural mechanisms of the state‐dependent drug binding. J Chem Inf Model 52, 2760–2774. [DOI] [PubMed] [Google Scholar]
- Durdagi S, Duff HJ & Noskov SY (2011). Combined receptor and ligand‐based approach to the universal pharmacophore model development for studies of drug blockade to the hERG1 pore domain. J Chem Inf Model 51, 463–474. [DOI] [PubMed] [Google Scholar]
- Durdagi S, Subbotina J, Lees‐Miller J, Guo J, Duff HJ & Noskov SY (2010). Insights into the molecular mechanism of hERG1 channel activation and blockade by drugs. Curr Med Chem 17, 3514–3532. [DOI] [PubMed] [Google Scholar]
- Ellinor PT, Lunetta KL, Glazer NL, Pfeufer A, Alonso A, Chung MK, Sinner MF, de Bakker PI, Mueller M, Lubitz SA, Fox E, Darbar D, Smith NL, Smith JD, Schnabel RB, Soliman EZ, Rice KM, Van Wagoner DR, Beckmann BM, van Noord C, Wang K, Ehret GB, Rotter JI, Hazen SL, Steinbeck G, Smith AV, Launer LJ, Harris TB, Makino S, Nelis M, Milan DJ, Perz S, Esko T, Kottgen A, Moebus S, Newton‐Cheh C, Li M, Mohlenkamp S, Wang TJ, Kao WH, Vasan RS, Nothen MM, MacRae CA, Stricker BH, Hofman A, Uitterlinden AG, Levy D, Boerwinkle E, Metspalu A, Topol EJ, Chakravarti A, Gudnason V, Psaty BM, Roden DM, Meitinger T, Wichmann HE, Witteman JC, Barnard J, Arking DE, Benjamin EJ, Heckbert SR & Kaab S (2010). Common variants in KCNN3 are associated with lone atrial fibrillation. Nat Genet 42, 240–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabritz L, Breithardt G & Kirchhof P (2007). Preclinical testing of drug‐induced proarrhythmia: value of transgenic models. Cardiovasc Hematol Agents Med Chem 5, 289–294. [DOI] [PubMed] [Google Scholar]
- Fauconnier J, Lacampagne A, Rauzier JM, Vassort G & Richard S (2005). Ca2+‐dependent reduction of IK1 in rat ventricular cells: a novel paradigm for arrhythmia in heart failure? Cardiovasc Res 68, 204–212. [DOI] [PubMed] [Google Scholar]
- Fedida D (2007). Vernakalant (RSD1235): a novel, atrial‐selective antifibrillatory agent. Expert Opin Investig Drugs 16, 519–532. [DOI] [PubMed] [Google Scholar]
- Fedida D, Eldstrom J, Hesketh JC, Lamorgese M, Castel L, Steele DF & Van Wagoner DR (2003). Kv1.5 is an important component of repolarizing K+ current in canine atrial myocytes. Circ Res 93, 744–751. [DOI] [PubMed] [Google Scholar]
- Fedida D, Wible B, Wang Z, Fermini B, Faust F, Nattel S & Brown AM (1993). Identity of a novel delayed rectifier current from human heart with a cloned K+ channel current. Circ Res 73, 210–216. [DOI] [PubMed] [Google Scholar]
- Ferrari R, Bertini M, Blomstrom‐Lundqvist C, Dobrev D, Kirchhof P, Pappone C, Ravens U, Tamargo J, Tavazzi L & Vicedomini GG (2016). An update on atrial fibrillation in 2014: From pathophysiology to treatment. Int J Cardiol 203, 22–29. [DOI] [PubMed] [Google Scholar]
- Ferreira R, Moreira‐Goncalves D, Azevedo AL, Duarte JA, Amado F & Vitorino R (2015). Unraveling the exercise‐related proteome signature in heart. Basic Res Cardiol 110, 454. [DOI] [PubMed] [Google Scholar]
- Ferrero JM, Trenor B & Romero L (2014). Multiscale computational analysis of the bioelectric consequences of myocardial ischaemia and infarction. Europace 16, 405–415. [DOI] [PubMed] [Google Scholar]
- Fodstad H, Swan H, Auberson M, Gautschi I, Loffing J, Schild L & Kontula K (2004). Loss‐of‐function mutations of the K+ channel gene KCNJ2 constitute a rare cause of long QT syndrome. J Mol Cell Cardiol 37, 593–602. [DOI] [PubMed] [Google Scholar]
- Furukawa T, Myerburg RJ, Furukawa N, Bassett AL & Kimura S (1990). Differences in transient outward currents of feline endocardial and epicardial myocytes. Circ Res 67, 1287–1291. [DOI] [PubMed] [Google Scholar]
- Gaborit N, Le Bouter S, Szuts V, Varro A, Escande D, Nattel S & Demolombe S (2007). Regional and tissue specific transcript signatures of ion channel genes in the non‐diseased human heart. J Physiol 582, 675–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadsby DC (1983). β‐Adrenoceptor agonists increase membrane K+ conductance in cardiac Purkinje fibres. Nature 306, 691–693. [DOI] [PubMed] [Google Scholar]
- Gentles RG, Grant‐Young K, Hu S, Huang Y, Poss MA, Andres C, Fiedler T, Knox R, Lodge N, Weaver CD & Harden DG (2008). Initial SAR studies on apamin‐displacing 2‐aminothiazole blockers of calcium‐activated small conductance potassium channels. Bioorg Med Chem Lett 18, 5316–5319. [DOI] [PubMed] [Google Scholar]
- Giles WR & Imaizumi Y (1988). Comparison of potassium currents in rabbit atrial and ventricular cells. J Physiol 405, 123–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gintant G, Sager PT & Stockbridge N (2016). Evolution of strategies to improve preclinical cardiac safety testing. Nat Rev Drug Discov 15, 457–471. [DOI] [PubMed] [Google Scholar]
- Glukhov AV, Fedorov VV, Lou Q, Ravikumar VK, Kalish PW, Schuessler RB, Moazami N & Efimov IR (2010). Transmural dispersion of repolarization in failing and nonfailing human ventricle. Circ Res 106, 981–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glynn P, Unudurthi SD & Hund TJ (2014). Mathematical modeling of physiological systems: an essential tool for discovery. Life Sci 111, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez JF, Cardona K, Martinez L, Saiz J & Trenor B (2014). Electrophysiological and structural remodeling in heart failure modulate arrhythmogenesis. 2D simulation study. PLoS One 9, e103273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandi E, Sanguinetti MC, Bartos DC, Bers DM, Chen‐Izu Y, Chiamvimonvat N, Colecraft HM, Delisle BP, Heijman J, Navedo MF, Noskov S, Proenza C, Vandenberg JI & Yarov‐Yarovoy V (2017). Potassium channels in the heart: structure, function and regulation. J Physiol 595, 2209–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greco CM & Condorelli G (2015). Epigenetic modifications and noncoding RNAs in cardiac hypertrophy and failure. Nat Rev Cardiol 12, 488–497. [DOI] [PubMed] [Google Scholar]
- Greenstein JL, Foteinou PT, Hashambhoy‐Ramsay YL & Winslow RL (2014). Modeling CaMKII‐mediated regulation of L‐type Ca2+ channels and ryanodine receptors in the heart. Front Pharmacol 5, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haraguchi Y, Ohtsuki A, Oka T & Shimizu T (2015). Electrophysiological analysis of mammalian cells expressing hERG using automated 384‐well‐patch‐clamp. BMC Pharmacol Toxicol 16, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmati G, Banyasz T, Barandi L, Szentandrassy N, Horvath B, Szabo G, Szentmiklosi JA, Szenasi G, Nanasi PP & Magyar J (2011). Effects of β‐adrenoceptor stimulation on delayed rectifier K+ currents in canine ventricular cardiomyocytes. Br J Pharmacol 162, 890–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto N, Yamashita T & Tsuruzoe N (2008). Characterization of in vivo and in vitro electrophysiological and antiarrhythmic effects of a novel I KACh blocker, NIP‐151: a comparison with an I Kr‐blocker dofetilide. J Cardiovasc Pharmacol 51, 162–169. [DOI] [PubMed] [Google Scholar]
- Heath BM & Terrar DA (2000). Protein kinase C enhances the rapidly activating delayed rectifier potassium current, I Kr, through a reduction in C‐type inactivation in guinea‐pig ventricular myocytes. J Physiol 522, 391–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heijman J, Voigt N, Carlsson LG & Dobrev D (2014. a). Cardiac safety assays. Curr Opin Pharmacol 15, 16–21. [DOI] [PubMed] [Google Scholar]
- Heijman J, Voigt N, Nattel S & Dobrev D (2014. b). Cellular and molecular electrophysiology of atrial fibrillation initiation, maintenance, and progression. Circ Res 114, 1483–1499. [DOI] [PubMed] [Google Scholar]
- Henriquez CS (2014). A brief history of tissue models for cardiac electrophysiology. IEEE Trans Biomed Eng 61, 1457–1465. [DOI] [PubMed] [Google Scholar]
- Houser SR, Piacentino V 3rd, Mattiello J, Weisser J & Gaughan JP (2000). Functional properties of failing human ventricular myocytes. Trends Cardiovasc Med 10, 101–107. [DOI] [PubMed] [Google Scholar]
- Hsueh CH, Chang PC, Hsieh YC, Reher T, Chen PS & Lin SF (2013). Proarrhythmic effect of blocking the small conductance calcium activated potassium channel in isolated canine left atrium. Heart Rhythm 10, 891–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iost N, Virag L, Opincariu M, Szecsi J, Varro A & Papp JG (1998). Delayed rectifier potassium current in undiseased human ventricular myocytes. Cardiovasc Res 40, 508–515. [DOI] [PubMed] [Google Scholar]
- Itoh H, Horie M, Ito M & Imoto K (2006). Arrhythmogenesis in the short‐QT syndrome associated with combined HERG channel gating defects: a simulation study. Circ J 70, 502–508. [DOI] [PubMed] [Google Scholar]
- Itzhaki I, Maizels L, Huber I, Zwi‐Dantsis L, Caspi O, Winterstern A, Feldman O, Gepstein A, Arbel G, Hammerman H, Boulos M & Gepstein L (2011). Modelling the long QT syndrome with induced pluripotent stem cells. Nature 471, 225–229. [DOI] [PubMed] [Google Scholar]
- Jeck C, Pinto J & Boyden P (1995). Transient outward currents in subendocardial Purkinje myocytes surviving in the infarcted heart. Circulation 92, 465–473. [DOI] [PubMed] [Google Scholar]
- Jost N, Virag L, Bitay M, Takacs J, Lengyel C, Biliczki P, Nagy Z, Bogats G, Lathrop DA, Papp JG & Varro A (2005). Restricting excessive cardiac action potential and QT prolongation: a vital role for I Ks in human ventricular muscle. Circulation 112, 1392–1399. [DOI] [PubMed] [Google Scholar]
- Karle CA, Zitron E, Zhang W, Kathofer S, Schoels W & Kiehn J (2002). Rapid component I Kr of the guinea‐pig cardiac delayed rectifier K+ current is inhibited by β1‐adrenoreceptor activation, via cAMP/protein kinase A‐dependent pathways. Cardiovasc Res 53, 355–362. [DOI] [PubMed] [Google Scholar]
- Kim TY, Kunitomo Y, Pfeiffer Z, Patel D, Hwang J, Harrison K, Patel B, Jeng P, Ziv O, Lu Y, Peng X, Qu Z, Koren G & Choi BR (2015). Complex excitation dynamics underlie polymorphic ventricular tachycardia in a transgenic rabbit model of long QT syndrome type 1. Heart Rhythm 12, 220–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kober L, Torp‐Pedersen C, McMurray JJ, Gotzsche O, Levy S, Crijns H, Amlie J & Carlsen J; Dronedarone Study Group (2008). Increased mortality after dronedarone therapy for severe heart failure. N Engl J Med 358, 2678–2687. [DOI] [PubMed] [Google Scholar]
- Koumi S, Wasserstrom JA & Ten Eick RE (1995). β‐Adrenergic and cholinergic modulation of inward rectifier K+ channel function and phosphorylation in guinea‐pig ventricle. J Physiol 486, 661–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovoor P, Wickman K, Maguire CT, Pu W, Gehrmann J, Berul CI & Clapham DE (2001). Evaluation of the role of I KACh in atrial fibrillation using a mouse knockout model. J Am Coll Cardiol 37, 2136–2143. [DOI] [PubMed] [Google Scholar]
- Krapivinsky G, Gordon EA, Wickman K, Velimirovic B, Krapivinsky L & Clapham DE (1995). The G‐protein‐gated atrial K+ channel I KACh is a heteromultimer of two inwardly rectifying K+‐channel proteins. Nature 374, 135–141. [DOI] [PubMed] [Google Scholar]
- Lahti AL, Kujala VJ, Chapman H, Koivisto AP, Pekkanen‐Mattila M, Kerkela E, Hyttinen J, Kontula K, Swan H, Conklin BR, Yamanaka S, Silvennoinen O & Aalto‐Setala K (2012). Model for long QT syndrome type 2 using human iPS cells demonstrates arrhythmogenic characteristics in cell culture. Dis Model Mech 5, 220–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees‐Miller JP, Duan Y, Teng GQ & Duff HJ (2000). Molecular determinant of high‐affinity dofetilide binding to HERG1 expressed in Xenopus oocytes: involvement of S6 sites. Mol Pharmacol 57, 367–374. [PubMed] [Google Scholar]
- Li GR, Lau CP, Leung TK & Nattel S (2004). Ionic current abnormalities associated with prolonged action potentials in cardiomyocytes from diseased human right ventricles. Heart Rhythm 1, 460–468. [DOI] [PubMed] [Google Scholar]
- Lieve KV & Wilde AA (2015). Inherited ion channel diseases: a brief review. Europace 17, Suppl. 2, ii1–ii6. [DOI] [PubMed] [Google Scholar]
- Litovsky SH & Antzelevitch C (1988). Transient outward current prominent in canine ventricular epicardium but not endocardium. Circ Res 62, 116–126. [DOI] [PubMed] [Google Scholar]
- Liu DW & Antzelevitch C (1995). Characteristics of the delayed rectifier current (I Kr and I Ks) in canine ventricular epicardial, midmyocardial, and endocardial myocytes. A weaker IKs contributes to the longer action potential of the M cell. Circ Res 76, 351–365. [DOI] [PubMed] [Google Scholar]
- London B (2001). Cardiac arrhythmias: from (transgenic) mice to men. J Cardiovasc Electrophysiol 12, 1089–1091. [DOI] [PubMed] [Google Scholar]
- Machida T, Hashimoto N, Kuwahara I, Ogino Y, Matsuura J, Yamamoto W, Itano Y, Zamma A, Matsumoto R, Kamon J, Kobayashi T, Ishiwata N, Yamashita T, Ogura T & Nakaya H (2011). Effects of a highly selective acetylcholine‐activated K+ channel blocker on experimental atrial fibrillation. Circ Arrhythm Electrophysiol 4, 94–102. [DOI] [PubMed] [Google Scholar]
- Magyar J, Iost N, Kortvely A, Banyasz T, Virag L, Szigligeti P, Varro A, Opincariu M, Szecsi J, Papp JG & Nanasi PP (2000). Effects of endothelin‐1 on calcium and potassium currents in undiseased human ventricular myocytes. Pflugers Arch 441, 144–149. [DOI] [PubMed] [Google Scholar]
- Major P, Baczko I, Hiripi L, Odening KE, Juhasz V, Kohajda Z, Horvath A, Seprenyi G, Kovacs M, Virag L, Jost N, Prorok J, Ordog B, Doleschall Z, Nattel S, Varro A & Bosze Z (2016). A novel transgenic rabbit model with reduced repolarization reserve: long QT syndrome caused by a dominant‐negative mutation of the KCNE1 gene. Br J Pharmacol 173, 2046–2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marder E & Goaillard JM (2006). Variability, compensation and homeostasis in neuron and network function. Nat Rev Neurosci 7, 563–574. [DOI] [PubMed] [Google Scholar]
- Maruyama M, Lin SF, Xie Y, Chua SK, Joung B, Han S, Shinohara T, Shen MJ, Qu Z, Weiss JN & Chen PS (2011). Genesis of phase 3 early afterdepolarizations and triggered activity in acquired long‐QT syndrome. Circ Arrhythm Electrophysiol 4, 103–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx SO, Kurokawa J, Reiken S, Motoike H, D'Armiento J, Marks AR & Kass RS (2002). Requirement of a macromolecular signaling complex for β adrenergic receptor modulation of the KCNQ1‐KCNE1 potassium channel. Science 295, 496–499. [DOI] [PubMed] [Google Scholar]
- Matsuda T, Ito M, Ishimaru S, Tsuruoka N, Saito T, Iida‐Tanaka N, Hashimoto N, Yamashita T, Tsuruzoe N, Tanaka H & Shigenobu K (2006). Blockade by NIP‐142, an antiarrhythmic agent, of carbachol‐induced atrial action potential shortening and GIRK1/4 channel. J Pharmacol Sci 101, 303–310. [DOI] [PubMed] [Google Scholar]
- Miake J, Marban E & Nuss HB (2002). Biological pacemaker created by gene transfer. Nature 419, 132–133. [DOI] [PubMed] [Google Scholar]
- Mirams GR, Davies MR, Brough SJ, Bridgland‐Taylor MH, Cui Y, Gavaghan DJ & Abi‐Gerges N (2014). Prediction of thorough QT study results using action potential simulations based on ion channel screens. J Pharmacol Toxicol Methods 70, 246–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitcheson JS, Chen J, Lin M, Culberson C & Sanguinetti MC (2000). A structural basis for drug‐induced long QT syndrome. Proc Natl Acad Sci USA 97, 12329–12333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina CE, Heijman J & Dobrev D (2016). Differences in left versus right ventricular electrophysiological properties in cardiac dysfunction and arrhythmogenesis. Arrhythm Electrophysiol Rev 5, 14–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno JD & Clancy CE (2012). Pathophysiology of the cardiac late Na current and its potential as a drug target. J Mol Cell Cardiol 52, 608–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers R, Timofeyev V, Li N, Kim C, Ledford HA, Sirish P, Lau V, Zhang Y, Fayyaz K, Singapuri A, Lopez JE, Knowlton AA, Zhang XD & Chiamvimonvat N (2015). Feedback mechanisms for cardiac‐specific microRNAs and cAMP signaling in electrical remodeling. Circ Arrhythm Electrophysiol 8, 942–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabauer M, Beuckelmann DJ, Uberfuhr P & Steinbeck G (1996). Regional differences in current density and rate‐dependent properties of the transient outward current in subepicardial and subendocardial myocytes of human left ventricle. Circulation 93, 168–177. [DOI] [PubMed] [Google Scholar]
- Nabauer M & Kaab S (1998). Potassium channel down‐regulation in heart failure. Cardiovasc Res 37, 324–334. [DOI] [PubMed] [Google Scholar]
- Nakano Y & Shimizu W (2016). Genetics of long‐QT syndrome. J Hum Genet 61, 51–55. [DOI] [PubMed] [Google Scholar]
- Nass RD, Aiba T, Tomaselli GF & Akar FG (2008). Mechanisms of disease: ion channel remodeling in the failing ventricle. Nat Clin Pract Cardiovasc Med 5, 196–207. [DOI] [PubMed] [Google Scholar]
- Nattel S (2015). Changes in the atrial transcriptome and atrial fibrillation: susceptibility, persistence, causes, and consequences. Circ Arrhythm Electrophysiol 8, 5–7. [DOI] [PubMed] [Google Scholar]
- Nattel S, Burstein B & Dobrev D (2008). Atrial remodeling and atrial fibrillation: mechanisms and implications. Circ Arrhythm Electrophysiol 1, 62–73. [DOI] [PubMed] [Google Scholar]
- Nattel S, Yue L & Wang Z (1999). Cardiac ultrarapid delayed rectifiers: a novel potassium current family of functional similarity and molecular diversity. Cell Physiol Biochem 9, 217–226. [DOI] [PubMed] [Google Scholar]
- Nerbonne JM & Kass RS (2005). Molecular physiology of cardiac repolarization. Physiol Rev 85, 1205–1253. [DOI] [PubMed] [Google Scholar]
- Nerbonne JM, Nichols CG, Schwarz TL & Escande D (2001). Genetic manipulation of cardiac K+ channel function in mice: what have we learned, and where do we go from here? Circ Res 89, 944–956. [DOI] [PubMed] [Google Scholar]
- Nguyen TP, Singh N, Xie Y, Qu Z & Weiss JN (2015). Repolarization reserve evolves dynamically during the cardiac action potential: effects of transient outward currents on early afterdepolarizations. Circ Arrhythm Electrophysiol 8, 694–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niederer SA & Smith NP (2012). At the heart of computational modelling. J Physiol 590, 1331–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noujaim SF, Pandit SV, Berenfeld O, Vikstrom K, Cerrone M, Mironov S, Zugermayr M, Lopatin AN & Jalife J (2007). Up‐regulation of the inward rectifier K+ current (I K1) in the mouse heart accelerates and stabilizes rotors. J Physiol 578, 315–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odening KE, Choi BR, Liu GX, Hartmann K, Ziv O, Chaves L, Schofield L, Centracchio J, Zehender M, Peng X, Brunner M & Koren G (2012). Estradiol promotes sudden cardiac death in transgenic long QT type 2 rabbits while progesterone is protective. Heart Rhythm 9, 823–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odening KE, Jung BA, Lang CN, Cabrera Lozoya R, Ziupa D, Menza M, Relan J, Franke G, Perez Feliz S, Koren G, Zehender M, Bode C, Brunner M, Sermesant M & Foll D (2013). Spatial correlation of action potential duration and diastolic dysfunction in transgenic and drug‐induced LQT2 rabbits. Heart Rhythm 10, 1533–1541. [DOI] [PubMed] [Google Scholar]
- Odening KE, Kirk M, Brunner M, Ziv O, Lorvidhaya P, Liu GX, Schofield L, Chaves L, Peng X, Zehender M, Choi BR & Koren G (2010). Electrophysiological studies of transgenic long QT type 1 and type 2 rabbits reveal genotype‐specific differences in ventricular refractoriness and His conduction. Am J Physiol Heart Circ Physiol 299, H643–H655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Hara T, Virag L, Varro A & Rudy Y (2011). Simulation of the undiseased human cardiac ventricular action potential: model formulation and experimental validation. PLoS Comput Biol 7, e1002061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onal B, Unudurthi SD & Hund TJ (2014). Modeling CaMKII in cardiac physiology: from molecule to tissue. Front Pharmacol 5, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathmanathan P, Shotwell MS, Gavaghan DJ, Cordeiro JM & Gray RA (2015). Uncertainty quantification of fast sodium current steady‐state inactivation for multi‐scale models of cardiac electrophysiology. Prog Biophys Mol Biol 117, 4–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulmichl M, Nasmith P, Hellmiss R, Reed K, Boyle WA, Nerbonne JM, Peralta EG & Clapham DE (1991). Cloning and expression of a rat cardiac delayed rectifier potassium channel. Proc Natl Acad Sci USA 88, 7892–7895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry M, de Groot MJ, Helliwell R, Leishman D, Tristani‐Firouzi M, Sanguinetti, MC & Mitcheson J (2004). Structural determinants of HERG channel block by clofilium and ibutilide. Mol Pharmacol 66, 240–249. [DOI] [PubMed] [Google Scholar]
- Perry M, Sachse FB & Sanguinetti MC (2007). Structural basis of action for a human ether‐a‐go‐go‐related gene 1 potassium channel activator. Proc Natl Acad Sci USA 104, 13827–13832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polakova E & Sobie EA (2013). Alterations in T‐tubule and dyad structure in heart disease: challenges and opportunities for computational analyses. Cardiovasc Res 98, 233–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi XY, Diness JG, Brundel BJ, Zhou XB, Naud P, Wu CT, Huang H, Harada M, Aflaki M, Dobrev D, Grunnet M & Nattel S (2014). Role of small‐conductance calcium‐activated potassium channels in atrial electrophysiology and fibrillation in the dog. Circulation 129, 430–440. [DOI] [PubMed] [Google Scholar]
- Qu Z & Chung D (2012). Mechanisms and determinants of ultralong action potential duration and slow rate‐dependence in cardiac myocytes. PLoS One 7, e43587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Z, Nivala M & Weiss JN (2013). Calcium alternans in cardiac myocytes: order from disorder. J Mol Cell Cardiol 58, 100–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Z & Weiss JN (2015). Mechanisms of ventricular arrhythmias: from molecular fluctuations to electrical turbulence. Annu Rev Physiol 77, 29–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Z, Xie Y, Garfinkel A & Weiss JN (2010). T‐wave alternans and arrhythmogenesis in cardiac diseases. Front Physiol 1, 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quail MA & Taylor AM (2013). Computer modeling to tailor therapy for congenital heart disease. Curr Cardiol Rep 15, 395. [DOI] [PubMed] [Google Scholar]
- Ramakers C, Vos MA, Doevendans PA, Schoenmakers M, Wu YS, Scicchitano S, Iodice A, Thomas GP, Antzelevitch C & Dumaine R (2003). Coordinated down‐regulation of KCNQ1 and KCNE1 expression contributes to reduction of I Ks in canine hypertrophied hearts. Cardiovasc Res 57, 486–496. [DOI] [PubMed] [Google Scholar]
- Ramirez E, Saiz J, Romero L, Ferrero JM & Trenor B (2014). In silico ischaemia‐induced reentry at the Purkinje‐ventricle interface. Europace 16, 444–451. [DOI] [PubMed] [Google Scholar]
- Ravens U & Cerbai E (2008). Role of potassium currents in cardiac arrhythmias. Europace 10, 1133–1137. [DOI] [PubMed] [Google Scholar]
- Ravens U, Poulet C, Wettwer E & Knaut M (2013). Atrial selectivity of antiarrhythmic drugs. J Physiol 591, 4087–4097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiffel JA, Camm AJ, Belardinelli L, Zeng D, Karwatowska‐Prokopczuk E, Olmsted A, Zareba W, Rosero S & Kowey P; HARMONY Investigators (2015). The HARMONY trial: combined ranolazine and dronedarone in the management of paroxysmal atrial fibrillation: mechanistic and therapeutic synergism. Circ Arrhythm Electrophysiol 8, 1048–1056. [DOI] [PubMed] [Google Scholar]
- Roberts BN, Yang PC, Behrens SB, Moreno JD & Clancy CE (2012). Computational approaches to understand cardiac electrophysiology and arrhythmias. Am J Physiol Heart Circ Physiol 303, H766–H783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roden DM (2004). Drug‐induced prolongation of the QT interval. N Engl J Med 350, 1013–1022. [DOI] [PubMed] [Google Scholar]
- Romero L, Carbonell B, Trenor B, Rodriguez B, Saiz J & Ferrero JM (2010). Human and rabbit inter‐species comparison of ionic mechanisms of arrhythmic risk: A simulation study. Conf Proc IEEE Eng Med Biol Soc 2010, 3253–3256. [DOI] [PubMed] [Google Scholar]
- Romero L, Carbonell B, Trenor B, Rodriguez B, Saiz J & Ferrero JM (2011). Systematic characterization of the ionic basis of rabbit cellular electrophysiology using two ventricular models. Prog Biophys Mol Biol 107, 60–73. [DOI] [PubMed] [Google Scholar]
- Romero L, Trenor B, Alonso JM, Tobon C, Saiz J & Ferrero JM Jr (2009). The relative role of refractoriness and source‐sink relationship in reentry generation during simulated acute ischemia. Ann Biomed Eng 37, 1560–1571. [DOI] [PubMed] [Google Scholar]
- Romero L, Trenor B, Ferrero JM & Starmer CF (2013). Non‐uniform dispersion of the source‐sink relationship alters wavefront curvature. PLoS One 8, e78328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero L, Trenor B, Yang PC, Saiz J & Clancy CE (2014). In silico screening of the impact of hERG channel kinetic abnormalities on channel block and susceptibility to acquired long QT syndrome. J Mol Cell Cardiol 72, 126–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosati B, Pan Z, Lypen S, Wang HS, Cohen I, Dixon JE & McKinnon D (2001). Regulation of KChIP2 potassium channel β subunit gene expression underlies the gradient of transient outward current in canine and human ventricle. J Physiol 533, 119–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen MR (1988). Mechanisms for arrhythmias. Am J Cardiol 61, 2A–8A. [DOI] [PubMed] [Google Scholar]
- Sale H, Wang J, O'Hara T, Tester D, Phartiyal P, He J, Rudy Y, Ackerman M & Robertson G (2008). Physiological properties of hERG 1a/1b heteromeric currents and a hERG 1b‐specific mutation associated with long‐QT syndrome. Circ Res 103, E81–E95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallam K, Li Y, Sager PT, Houser SR & Wu JC (2015). Finding the rhythm of sudden cardiac death: new opportunities using induced pluripotent stem cell‐derived cardiomyocytes. Circ Res 116, 1989–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samie FH, Berenfeld O, Anumonwo J, Mironov SF, Udassi S, Beaumont J, Taffet S, Pertsov AM & Jalife J (2001). Rectification of the background potassium current: a determinant of rotor dynamics in ventricular fibrillation. Circ Res 89, 1216–1223. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC, Jurkiewicz NK, Scott A & Siegl PK (1991). Isoproterenol antagonizes prolongation of refractory period by the class III antiarrhythmic agent E‐4031 in guinea pig myocytes. Mechanism of action. Circ Res 68, 77–84. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC & Tristani‐Firouzi M (2006). hERG potassium channels and cardiac arrhythmia. Nature 440, 463–469. [DOI] [PubMed] [Google Scholar]
- Sarkar AX & Sobie EA (2010). Regression analysis for constraining free parameters in electrophysiological models of cardiac cells. PLoS Comput Biol 6, e1000914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarquella‐Brugada G, Campuzano O, Arbelo E, Brugada J & Brugada R (2016). Brugada syndrome: clinical and genetic findings. Genet Med 18, 3–12. [DOI] [PubMed] [Google Scholar]
- Sato D & Clancy CE (2013). Cardiac electrophysiological dynamics from the cellular level to the organ level. Biomed Eng Comput Biol 5, 69–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer D, Kiesecker C, Kulzer M, Gunth M, Scholz EP, Kathofer S, Thomas D, Maurer M, Kreuzer J, Bauer A, Katus HA, Karle CA & Zitron E (2007). Activation of inwardly rectifying Kir2.x potassium channels by β3‐adrenoceptors is mediated via different signaling pathways with a predominant role of PKC for Kir2.1 and of PKA for Kir2.2. Naunyn Schmiedebergs Arch Pharmacol 375, 311–322. [DOI] [PubMed] [Google Scholar]
- Schlotthauer K & Bers DM (2000). Sarcoplasmic reticulum Ca2+ release causes myocyte depolarization. Underlying mechanism and threshold for triggered action potentials. Circ Res 87, 774–780. [DOI] [PubMed] [Google Scholar]
- Schmidt C, Wiedmann F, Voigt N, Zhou XB, Heijman J, Lang S, Albert V, Kallenberger S, Ruhparwar A, Szabo G, Kallenbach K, Karck M, Borggrefe M, Biliczki P, Ehrlich JR, Baczko I, Lugenbiel P, Schweizer PA, Donner BC, Katus HA, Dobrev D & Thomas D (2015). Upregulation of K2P3.1 K+ current causes action potential shortening in patients with chronic atrial fibrillation. Circulation 132, 82–92. [DOI] [PubMed] [Google Scholar]
- Schmitt N, Grunnet M & Olesen SP (2014). Cardiac potassium channel subtypes: new roles in repolarization and arrhythmia. Physiol Rev 94, 609–653. [DOI] [PubMed] [Google Scholar]
- Schotten U, de Haan S, Verheule S, Harks EG, Frechen D, Bodewig E, Greiser M, Ram R, Maessen J, Kelm M, Allessie M & Van Wagoner DR (2007). Blockade of atrial‐specific K+‐currents increases atrial but not ventricular contractility by enhancing reverse mode Na+/Ca2+‐exchange. Cardiovasc Res 73, 37–47. [DOI] [PubMed] [Google Scholar]
- Schotten U, Dobrev D, Platonov PG, Kottkamp H & Hindricks G (2016). Current controversies in determining the main mechanisms of atrial fibrillation. J Intern Med 279, 428–438. [DOI] [PubMed] [Google Scholar]
- Schram G, Pourrier M, Melnyk P & Nattel S (2002). Differential distribution of cardiac ion channel expression as a basis for regional specialization in electrical function. Circ Res 90, 939–950. [DOI] [PubMed] [Google Scholar]
- Schwartz PJ (2006). The congenital long QT syndromes from genotype to phenotype: clinical implications. J Intern Med 259, 39–47. [DOI] [PubMed] [Google Scholar]
- Schwartz PJ & Malliani A (1975). Electrical alternation of the T‐wave: Clinical and experimental evidence of its relationship with the sympathetic nervous system and with the long Q‐T syndrome. Am Heart J 89, 45–50. [DOI] [PubMed] [Google Scholar]
- Sicouri S & Antzelevitch C (1991). A subpopulation of cells with unique electrophysiological properties in the deep subepicardium of the canine ventricle. The M cell. Circ Res 68, 1729–1741. [DOI] [PubMed] [Google Scholar]
- Silva J & Rudy Y (2003). Mechanism of pacemaking in I K1‐downregulated myocytes. Circ Res 92, 261–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva J & Rudy Y (2005). Subunit interaction determines I Ks participation in cardiac repolarization and repolarization reserve. Circulation 112, 1384–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh BN (2008). Amiodarone as paradigm for developing new drugs for atrial fibrillation. J Cardiovasc Pharmacol 52, 300–305. [DOI] [PubMed] [Google Scholar]
- Sinnecker D, Goedel A, Dorn T, Dirschinger RJ, Moretti A & Laugwitz KL (2013). Modeling long‐QT syndromes with iPS cells. J Cardiovasc Transl Res 6, 31–36. [DOI] [PubMed] [Google Scholar]
- Sorger PK, Allerheiligen SRB, Abernethy DR, Altman RB, Brouwer KLR, Califano A, D'Argenio DZ, Iyengar R, Jusko WJ, Lalonde R, Lauffenburger DA, Shoichet B, Stevens JL, Subramaniam S, Van der Graaf P & Vicini P (2011). Quantitative and systems pharmacology in the postgenomic era: new approaches to discovering drugs and understanding therapeutic mechanisms. In An NIH White Paper by the QSP Workshop Group – October 2011. NIH, Bethesda.
- Spector PS, Curran ME, Zou A, Keating MT & Sanguinetti MC (1996). Fast inactivation causes rectification of the IKr channel. J Gen Physiol 107, 611–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stansfeld PJ, Gedeck P, Gosling M, Cox B, Mitcheson JS & Sutcliffe MJ (2007). Drug block of the hERG potassium channel: Insight from modeling. Proteins 68, 568–580. [DOI] [PubMed] [Google Scholar]
- Stary A, Wacker SJ, Boukharta L, Zachariae U, Karimi‐Nejad Y, Aqvist J, Vriend G & de Groot BL (2010). Toward a consensus model of the HERG potassium channel. ChemMedChem 5, 455–467. [DOI] [PubMed] [Google Scholar]
- Sugiura S, Washio T, Hatano A, Okada J, Watanabe H & Hisada T (2012). Multi‐scale simulations of cardiac electrophysiology and mechanics using the University of Tokyo heart simulator. Prog Biophys Mol Biol 110, 380–389. [DOI] [PubMed] [Google Scholar]
- Sun Z, Milos PM, Thompson JF, Lloyd DB, Mank‐Seymour A, Richmond J, Cordes JS & Zhou J (2004). Role of a KCNH2 polymorphism (R1047 L) in dofetilide‐induced Torsades de Pointes . J Mol Cell Cardiol 37, 1031–1039. [DOI] [PubMed] [Google Scholar]
- Szentadrassy N, Banyasz T, Biro T, Szabo G, Toth BI, Magyar J, Lazar J, Varro A, Kovacs L & Nanasi PP (2005). Apico‐basal inhomogeneity in distribution of ion channels in canine and human ventricular myocardium. Cardiovasc Res 65, 851–860. [DOI] [PubMed] [Google Scholar]
- Taggart P, Orini M, Hanson B, Hayward M, Clayton R, Dobrzynski H, Yanni J, Boyett M, Lambiase PD (2014). Developing a novel comprehensive framework for the investigation of cellular and whole heart electrophysiology in the in situ human heart: historical perspectives, current progress and future prospects. Prog Biophys Mol Biol 115, 252–260. [DOI] [PubMed] [Google Scholar]
- Tamkun MM, Knoth KM, Walbridge JA, Kroemer H, Roden DM & Glover DM (1991). Molecular cloning and characterization of two voltage‐gated K+ channel cDNAs from human ventricle. FASEB J 5, 331–337. [DOI] [PubMed] [Google Scholar]
- Tanaka A, Yuasa S, Node K & Fukuda K (2015). Cardiovascular disease modeling using patient‐specific induced pluripotent stem cells. Int J Mol Sci 16, 18894–18922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terentyev D, Rees CM, Li W, Cooper LL, Jindal HK, Peng X, Lu Y, Terentyeva R, Odening KE, Daley J, Bist K, Choi BR, Karma A & Koren G (2014). Hyperphosphorylation of RyRs underlies triggered activity in transgenic rabbit model of LQT2 syndrome. Circ Res 115, 919–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas D, Kiehn J, Katus HA & Karle CA (2004). Adrenergic regulation of the rapid component of the cardiac delayed rectifier potassium current, I Kr, and the underlying hERG ion channel. Basic Res Cardiol 99, 279–287. [DOI] [PubMed] [Google Scholar]
- Tobon C, Ruiz‐Villa CA, Heidenreich E, Romero L, Hornero F & Saiz J (2013). A three‐dimensional human atrial model with fiber orientation. Electrograms and arrhythmic activation patterns relationship. PLoS One 8, e50883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trayanova NA & Boyle PM (2014). Advances in modeling ventricular arrhythmias: from mechanisms to the clinic. Wiley Interdiscip Rev Syst Biol Med 6, 209–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trayanova NA, O'Hara T, Bayer JD, Boyle PM, McDowell KS, Constantino J, Arevalo HJ, Hu Y & Vadakkumpadan F (2012). Computational cardiology: how computer simulations could be used to develop new therapies and advance existing ones. Europace 14 Suppl. 5, v82–v89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trenor B, Romero L, Ferrero JM Jr, Saiz J, Molto G & Alonso JM (2007). Vulnerability to reentry in a regionally ischemic tissue: a simulation study. Ann Biomed Eng 35, 1756–1770. [DOI] [PubMed] [Google Scholar]
- Tristani‐Firouzi M & Sanguinetti MC (1998). Voltage‐dependent inactivation of the human K+ channel KvLQT1 is eliminated by association with minimal K+ channel (minK) subunits. J Physiol 510, 37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tromba C & Cohen IS (1990). A novel action of isoproterenol to inactivate a cardiac K+ current is not blocked by beta and alpha adrenergic blockers. Biophys J 58, 791–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuteja D, Xu D, Timofeyev V, Lu L, Sharma D, Zhang Z, Xu Y, Nie L, Vazquez AE, Young JN, Glatter KA & Chiamvimonvat N (2005). Differential expression of small‐conductance Ca2+‐activated K+ channels SK1, SK2, and SK3 in mouse atrial and ventricular myocytes. Am J Physiol Heart Circ Physiol 289, H2714–H2723. [DOI] [PubMed] [Google Scholar]
- Undrovinas AI, Maltsev VA & Sabbah HN (1999). Repolarization abnormalities in cardiomyocytes of dogs with chronic heart failure: role of sustained inward current. Cell Mol Life Sci 55, 494–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Wagoner DR (2003). Electrophysiological remodeling in human atrial fibrillation. Pacing Clin Electrophysiol 26, 1572–1575. [DOI] [PubMed] [Google Scholar]
- Van Wagoner DR, Pond AL, McCarthy PM, Trimmer JS & Nerbonne JM (1997). Outward K+ current densities and Kv1.5 expression are reduced in chronic human atrial fibrillation. Circ Res 80, 772–781. [DOI] [PubMed] [Google Scholar]
- Vassalle M & Bocchi L (2013). Differences in ionic currents between canine myocardial and Purkinje cells. Physiol Rep 1, e00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veerman CC, Kosmidis G, Mummery CL, Casini S, Verkerk AO & Bellin M (2015). Immaturity of human stem‐cell‐derived cardiomyocytes in culture: fatal flaw or soluble problem? Stem Cells Dev 24, 1035–1052. [DOI] [PubMed] [Google Scholar]
- Verkerk AO & Wilders R (2013). Hyperpolarization‐activated current, If, in mathematical models of rabbit sinoatrial node pacemaker cells. Biomed Res Int 2013, 872454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virag L, Iost N, Opincariu M, Szolnoky J, Szecsi J, Bogats G, Szenohradszky P, Varro A & Papp JG (2001). The slow component of the delayed rectifier potassium current in undiseased human ventricular myocytes. Cardiovasc Res 49, 790–797. [DOI] [PubMed] [Google Scholar]
- Viskin S, Fish R, Zeltser D, Belhassen B, Heller K, Brosh D, Laniado S & Barron HV (2000). Arrhythmias in the congenital long QT syndrome: how often is torsade de pointes pause dependent? Heart 83, 661–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan PC & Rudy Y (1999). Pause induced early afterdepolarizations in the long QT syndrome: a simulation study. Cardiovasc Res 42, 530–542. [DOI] [PubMed] [Google Scholar]
- Voigt N & Dobrev D (2016). Atrial‐selective potassium channel blockers. Card Electrophysiol Clin 8, 411–421. [DOI] [PubMed] [Google Scholar]
- Volders P, Stengl M, van Opstal J, Gerlach U, Spatjens R, Beekman J, Sipido K & Vos M (2003). Probing the contribution of I Ks to canine ventricular repolarization: key role for β‐adrenergic receptor stimulation. Circulation 107, 2753–2760. [DOI] [PubMed] [Google Scholar]
- Volders PG, Sipido KR, Carmeliet E, Spatjens RL, Wellens HJ & Vos MA (1999). Repolarizing K+ currents I TO1 and I Ks are larger in right than left canine ventricular midmyocardium. Circulation 99, 206–210. [DOI] [PubMed] [Google Scholar]
- Wagner S & Maier LS (2013). Small conductance Ca‐activated K channel: small but powerful proarrhythmogenic? Heart Rhythm 10, 899–900. [DOI] [PubMed] [Google Scholar]
- Waldo AL, Camm AJ, deRuyter H, Friedman PL, MacNeil DJ, Pauls JF, Pitt B, Pratt CM, Schwartz PJ & Veltri EP (1996). Effect of d‐sotalol on mortality in patients with left ventricular dysfunction after recent and remote myocardial infarction. The SWORD Investigators. Survival With Oral d‐Sotalol. Lancet 348, 7–12. [DOI] [PubMed] [Google Scholar]
- Wang Z, Fermini B & Nattel S (1993). Sustained depolarization‐induced outward current in human atrial myocytes. Evidence for a novel delayed rectifier K+ current similar to Kv1.5 cloned channel currents. Circ Res 73, 1061–1076. [DOI] [PubMed] [Google Scholar]
- Weiss JN, Garfinkel A, Karagueuzian HS, Nguyen TP, Olcese R, Chen PS & Qu Z (2015). Perspective: a dynamics‐based classification of ventricular arrhythmias. J Mol Cell Cardiol 82, 136–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss JN, Karma A, MacLellan WR, Deng M, Rau CD, Rees CM, Wang J, Wisniewski N, Eskin E, Horvath S, Qu Z, Wang Y & Lusis AJ (2012). ‘Good enough solutions’ and the genetics of complex diseases. Circ Res 111, 493–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wettwer E, Amos GJ, Posival H & Ravens U (1994). Transient outward current in human ventricular myocytes of subepicardial and subendocardial origin. Circ Res 75, 473–482. [DOI] [PubMed] [Google Scholar]
- Wettwer E, Christ T, Dobrev D & Ravens U (2007). Novel anti‐arrhythmic agents for the treatment of atrial fibrillation. Curr Opin Pharmacol 7, 214–218. [DOI] [PubMed] [Google Scholar]
- Wettwer E, Hala O, Christ T, Heubach JF, Dobrev D, Knaut M, Varro A & Ravens U (2004). Role of I Kur in controlling action potential shape and contractility in the human atrium: influence of chronic atrial fibrillation. Circulation 110, 2299–2306. [DOI] [PubMed] [Google Scholar]
- Wickenden AD, Jegla TJ, Kaprielian R & Backx PH (1999). Regional contributions of Kv1.4, Kv4.2, and Kv4.3 to transient outward K+ current in rat ventricle. Am J Physiol 276, H1599–H1607. [DOI] [PubMed] [Google Scholar]
- Wilson LD, Jennings MM & Rosenbaum DS (2011). Point: M cells are present in the ventricular myocardium. Heart Rhythm 8, 930–933. [DOI] [PubMed] [Google Scholar]
- Wischmeyer E & Karschin A (1996). Receptor stimulation causes slow inhibition of IRK1 inwardly rectifying K+ channels by direct protein kinase A‐mediated phosphorylation. Proc Natl Acad Sci USA 93, 5819–5823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Tuteja D, Zhang Z, Xu D, Zhang Y, Rodriguez J, Nie L, Tuxson HR, Young JN, Glatter KA, Vazquez AE, Yamoah EN & Chiamvimonvat N (2003). Molecular identification and functional roles of a Ca2+‐activated K+ channel in human and mouse hearts. J Biol Chem 278, 49085–49094. [DOI] [PubMed] [Google Scholar]
- Yan GX, Shimizu W & Antzelevitch C (1998). Characteristics and distribution of M cells in arterially perfused canine left ventricular wedge preparations. Circulation 98, 1921–1927. [DOI] [PubMed] [Google Scholar]
- Yang KC & Nerbonne JM (2016). Mechanisms contributing to myocardial potassium channel diversity, regulation and remodeling. Trends Cardiovasc Med 26, 209–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang PC, Moreno JD, Miyake CY, Vaughn‐Behrens SB, Jeng MT, Grandi E, Wehrens XH, Noskov SY & Clancy CE (2016). In silico prediction of drug therapy in catecholaminergic polymorphic ventricular tachycardia. J Physiol 594, 567–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Chun YW, Stroud DM, Mosley JD, Knollmann BC, Hong C & Roden DM (2014). Screening for acute I Kr block is insufficient to detect torsades de pointes liability: role of late sodium current. Circulation 130, 224–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng J, Laurita KR, Rosenbaum DS & Rudy Y (1995). Two components of the delayed rectifier K+ current in ventricular myocytes of the guinea pig type. Theoretical formulation and their role in repolarization. Circ Res 77, 140–152. [DOI] [PubMed] [Google Scholar]
- Zhang P, Su J & Mende U (2012). Cross talk between cardiac myocytes and fibroblasts: from multiscale investigative approaches to mechanisms and functional consequences. Am J Physiol Heart Circ Physiol 303, H1385–H1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XD, Lieu DK & Chiamvimonvat N (2015). Small‐conductance Ca2+‐activated K+ channels and cardiac arrhythmias. Heart Rhythm 12, 1845–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Xie Y, Wen H, Xiao D, Allen C, Fefelova N, Dun W, Boyden PA, Qu Z & Xie LH (2012). Role of the transient outward potassium current in the genesis of early afterdepolarizations in cardiac cells. Cardiovasc Res 95, 308–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L & O'Rourke B (2012). Cardiac mitochondrial network excitability: insights from computational analysis. Am J Physiol Heart Circ Physiol 302, H2178–H2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimetbaum P (2007). Amiodarone for atrial fibrillation. N Engl J Med 356, 935–941. [DOI] [PubMed] [Google Scholar]
- Ziupa D, Beck J, Franke G, Perez Feliz S, Hartmann M, Koren G, Zehender M, Bode C, Brunner M & Odening KE (2014). Pronounced effects of HERG‐blockers E‐4031 and erythromycin on APD, spatial APD dispersion and triangulation in transgenic long‐QT type 1 rabbits. PLoS One 9, e107210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziv O, Morales E, Song YK, Peng X, Odening KE, Buxton AE, Karma A, Koren G & Choi BR (2009). Origin of complex behaviour of spatially discordant alternans in a transgenic rabbit model of type 2 long QT syndrome. J Physiol 587, 4661–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zygmunt AC, Eddlestone GT, Thomas GP, Nesterenko VV & Antzelevitch C (2001). Larger late sodium conductance in M cells contributes to electrical heterogeneity in canine ventricle. Am J Physiol Heart Circ Physiol 281, H689–H697. [DOI] [PubMed] [Google Scholar]