

Abstract
This paper is the outcome of the fourth UC Davis Systems Approach to Understanding Cardiac Excitation–Contraction Coupling and Arrhythmias Symposium, a biannual event that aims to bring together leading experts in subfields of cardiovascular biomedicine to focus on topics of importance to the field. The theme of the 2016 symposium was ‘K+ Channels and Regulation’. Experts in the field contributed their experimental and mathematical modelling perspectives and discussed emerging questions, controversies and challenges on the topic of cardiac K+ channels. This paper summarizes the topics of formal presentations and informal discussions from the symposium on the structural basis of voltage‐gated K+ channel function, as well as the mechanisms involved in regulation of K+ channel gating, expression and membrane localization. Given the critical role for K+ channels in determining the rate of cardiac repolarization, it is hardly surprising that essentially every aspect of K+ channel function is exquisitely regulated in cardiac myocytes. This regulation is complex and highly interrelated to other aspects of myocardial function. K+ channel regulatory mechanisms alter, and are altered by, physiological challenges, pathophysiological conditions, and pharmacological agents. An accompanying paper focuses on the integrative role of K+ channels in cardiac electrophysiology, i.e. how K+ currents shape the cardiac action potential, and how their dysfunction can lead to arrhythmias, and discusses K+ channel‐based therapeutics. A fundamental understanding of K+ channel regulatory mechanisms and disease processes is fundamental to reveal new targets for human therapy.
Keywords: gating, heterogeneity, phosphorylation, trafficking
Abbreviations
- AF
atrial fibrillation
- AP
action potential
- APD
action potential duration
- CaMKII
Ca2+/calmodulin‐dependent protein kinase II
- COP
coatamer associated protein
- cryo‐EM
cryo‐electron microscopy
- ER
endoplasmic reticulum
- HF
heart failure
- hiPSC‐CM
human induced pluripotent stem cell‐derived cardiomyocyte
- IPC
ischaemic preconditioning
- KV
voltage‐gated K+
- LQTS
long QT syndrome
- MD
molecular dynamics
- PIP2
phosphatidylinositol‐4,5‐bisphosphate
- REFER
rate‐equilibrium free energy
- RMP
resting membrane potential
- SR
sarcoplasmic reticulum
- SUR
sulfonylurea receptor
- VSD
voltage sensor domain
Voltage‐gated K+ channel structure and function
In this session Toby Allen, Francisco Bezanilla, Jianmin Cui, Peter Larsson, Daniel Minor, Sergei Noskov, Jon Sack, Matthew Trudeau, Jamie Vandenberg and Vladimir Yarov‐Yarovoy participated as speakers, discussion leaders, or panelists.
Voltage‐gated K+ (KV) channels are gated pores in the membranes of excitable cells that open and close in response to changes in transmembrane voltage to allow the selective passage of K+ ions across the cell membrane (Bezanilla, 2000). The existence of KV channels was first inferred over 60 years ago through a combination of functional and theoretical studies in the squid giant axon (Hodgkin & Huxley, 1952). Many of the principles underlying how these channels sense change in membrane voltage and then transduce that into the opening and closing of the voltage‐dependent gates are now well established, but there is still a way to go before we will understand voltage sensing and voltage sensor to pore coupling at the same atomic level resolution that we understand the basis of ion selectivity (Zhou et al. 2001; Medovoy et al. 2016).
Our current understanding of the molecular basis of cardiac K+ channel function, including selective ion permeation, voltage sensing and gating, is derived from a combination of experimental observations and modelling. Heterologous expression of cloned channels has greatly facilitated quantitative biophysical and structural studies of K+ channels. Static structures of prokaryotic K+ channels (e.g. KcsA, MthK, KVAP, KirBac1.1, Kir3.1/KirBac1.3 chimera) and eukaryotic K+ channels (e.g. Kir2.2, KV1.2) determined by X‐ray crystallography and more recently by cryo‐electron microscopy (cryo‐EM) for Slo2.2 (Hite et al. 2015) have provided crucial information for constructing homology models of cardiac K+ channels whose structures have yet to be solved. These structures have also enabled molecular dynamics (MD) simulations that provide insights into the mechanisms of ion selectivity and permeation, channel gating and ligand binding at an atomic level of detail. Structures have not yet been determined for most of the cardiac K+ channels. However, structures of highly related mammalian K+ channels (i.e. KV1.2, Kir3.2 and Slo2.2) as well as the cytoplasmic domains of Kir2.1, Kir3.1 and KV11.1 are available and provide useful templates to construct homology models.
Questions, controversies, challenges:
What do we know about the mechanisms of voltage sensing?
In a seminal series of papers published in 1952, Hodgkin and Huxley identified many of the critical features of voltage‐gated channels; however, we are still trying to work out the precise details of the structural basis of channel function. Perhaps the most prescient prediction made by Hodgkin and Huxley, was that ‘changes in ionic permeability depend on the movement of some component of the membrane which behaves as though it had a large charge or dipole moment’ (Hodgkin & Huxley, 1952). Twenty years passed before the small transient current associated with the movement of this charge, the gating current, was first recorded (Armstrong & Bezanilla, 1973). Measuring gating currents remains to this day at the centre of ongoing efforts to understand how voltage sensors work (see e.g. Lacroix et al. 2014). It was another decade before cloning of the first members of the voltage‐gated ion channel family led to the identification of the S4 transmembrane domain, with its multiple arginine residues, as the principal component of the voltage‐sensing domain in these channels (Noda et al. 1984; Papazian et al. 1987). In the last 20 years, the development of methods to attach chemical probes ranging from H+ (Starace & Bezanilla, 2001) to fluorophores (Mannuzzu et al. 1996) to electron paramagnetic resonance probes (Cuello et al. 2004) has allowed indirect monitoring of the motion of the voltage‐sensor domain. These spectroscopic methods have complemented gating current measurements in our quest to understand the extraordinary molecular gymnastics that underlie the function of these channels. Recent studies utilizing these techniques have revealed the gating mechanisms for several cardiac KV channels, including KV11.1 (ERG1) (Es‐Salah‐Lamoureux et al. 2010), KV1.5 (Vaid et al. 2009), and I Ks channels formed by co‐assembly of KV7.1 with KCNE1 (minK) accessory subunits (Barro‐Soria et al. 2014; Zaydman et al. 2014).
In most KV channels that have been investigated, the voltage dependence of the voltage sensor domain (VSD) movement occurs over a more negative voltage range than that required for channel activation. Channel opening only occurs after all the VSDs have been activated, and involves a final highly cooperative state transition. In a process called electromechanical coupling, S4–S5 linkers act as mechanical levers that couple outward movement of the S4 segments to opening of the pore domain (Long et al. 2005 b; Jensen et al. 2012). Recent studies have revealed that gating of KV7.1 and KV11.1 channels differs from this canonical model. Quite unexpectedly, channel gating is relatively normal when KV11.1 subunits are split at the S4/S5 linker into separate VSD and pore domains and heterologously expressed in oocytes (Lorinczi et al. 2015). Split KV1 or KV7.1 channels are non‐functional. Thus, a rigid covalent linkage between the S4 and S5 segments is not required for normal gating of all KV channels. Given the widely accepted canonical model of KV channel gating, recent insights into the unusual gating mode adopted by KV7.1 channels and its modulation by KCNE1 subunits (described below) were equally unexpected.
Compared to current conducted by KV7.1 homotetrameric channels, KV7.1–KCNE1 heteromeric channel currents (i.e. cardiac I Ks) are much larger, activate more slowly and at more positive potentials, do not exhibit inactivation and have altered pharmacology (Fig. 1 A). A plethora of site‐directed mutagenesis and cross‐linking studies have guided structural modelling that strongly suggests that the single transmembrane domain of KCNE1 subunits nestles between the VSDs of adjacent KV7.1 subunits. This arrangement would permit a 4:4 KV7.1–KCNE1 stoichiometry. The naturally occurring stoichiometry and the mechanisms by which KCNE1 alters the gating of KV7.1 have been hotly debated. In heterologous expression systems, KV7.1–KCNE1 stoichiometry has been shown to be either fixed at 4:2 (Plant et al. 2014), or variable from 4:1 to 4:4 (Nakajo et al. 2010; Murray et al. 2016), although the predominant KV7.1–KCNE1 stoichiometry in native myocytes remains unclear. Ion channel gating can be quantified by measuring both gating and ionic currents; however, it is not possible to quantitatively measure both signals at the same time. As summarized at the UC Davis meeting by Jianmin Cui, voltage clamp fluorometry of heterologously expressed channels that permits the simultaneous monitoring of ionic currents and VSD movement has recently revealed new and unexpected insights into the molecular mechanisms underlying KCNE1 modulation of KV7.1 channel gating. For these studies, a Cys residue was introduced into the S3–S4 linker of the cloned channel and labelled with a thiol‐reactive fluorophore. As the VSD moves across the membrane in response to a stepped change in voltage, the local environment surrounding the tethered fluorophore is altered. The resulting change in the fluorescence emission of the fluorophore can be monitored with a photodiode at the same time ionic current is measured. Unlike a typical KV channel, the voltage dependence of VSD movement (fluorescence–voltage or F–V relationship) and ionic currents (conductance–voltage, or G–V relationship) of KV7.1 channels essentially overlap (Fig. 1 B). In striking contrast to KV1, movement of individual VSDs can lead to channel opening of KV7.1. The F–V relationship of KCNQ1 splits into two clearly separable components, F1 and F2. The large and fast F1 component represents movement of the VSDs at quite negative potentials and a smaller and slower F2 component can be measured at more positive voltages. The G–V relationship of Kv7.1 nearly superimposes with the F1 component of F–V (Fig. 1 B). KCNE1 β‐subunits drastically alter the F1 component of VSD movement in KV7.1, but not the F2 component, and change the G–V relationship from being superimposed with F1 to overlapping with F2 (Fig. 1 B; Osteen et al. 2010; Barro‐Soria et al. 2014; Zaydman et al. 2014). A gating model for KV7.1–KCNE1 channels that includes independent movements of all four VSDs (giving rise to the F1 component) and a final concerted conformational change in the S4 segments that corresponds to channel opening and the F2 component from the voltage clamp fluorometry experiments (Barro‐Soria et al. 2014) can account for most experimental findings. The model is an oversimplification because it does not account for the many subconductance states of KV7.1–KCNE1 channels that may correspond to partial openings in response to single VSD movements (Werry et al. 2013). This is in contrast to the Zaydman model that is simplified to transitions between a closed, intermediate‐open and activated‐open states (C ↔ IO ↔ AO) and where F1 represents the C to IO transition and F2 represents the IO to AO transition (Fig. 1 C). In this model, KCNE1 suppresses channel opening when VSDs are in the IO state such that KV7.1–KCNE1 channels are only open in the AO state. Although the two gating models differ in details, both attribute KCNE1 effects to altered coupling between the VSDs and the pore domain. Recent findings have also shown that VSD–pore domain coupling in KV7.1 is dependent on phosphatidylinositol‐4,5‐bisphosphate (PIP2) and ATP. PIP2 binds to basic residues located between the VSD and pore domain (Zaydman et al. 2013), whereas ATP binds to basic residues located in the cytoplasmic C linker close to the S6 gate (Li et al. 2013).
Figure 1. KCNE1 β‐subunits alter the gating of Kv7.1 channels.
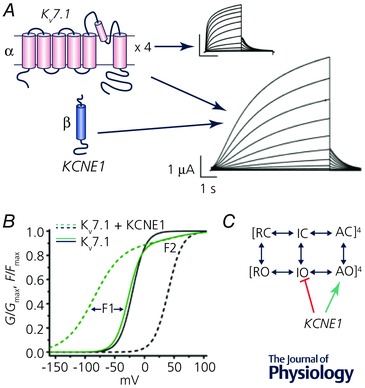
A, Kv7.1 channels are formed by co‐assembly of four identical α‐subunits, each composed of six transmembrane segments. Co‐assembly of Kv7.1 channels with KCNE1 β‐subunits (single transmembrane domain) form slowly activating channels that conduct cardiac I Ks in cardiac myocytes. B, KCNE1 subunits alter the voltage dependence of voltage sensor movement. The F–V relationship is shifted to more negative potentials, whereas the G–V relationship is shifted to more positive potentials. C, simplified kinetic model of Kv7.1 and IKs channel gating. Fourth power notation symbolizes four VSDs per channel. KCNE1 subunits suppresses channel opening when the VSDs are in the intermediate‐open (IO) state and stabilizes the activated‐open (AO) state. Modified from Zaydman et al. (2014).
Questions and challenges:
What remains to be elucidated about the mechanisms of voltage sensing?
Closed state structures of KV channels remain elusive
Can a combination of new approaches and MD simulations provide an atomic level view of voltage‐dependent VSD movement?
Using spectroscopic methods it has been established that the VSD of KV1 channels moves outwards in response to a depolarization and that this outward movement is likely to involve at least a 6.5 Å outward displacement combined with rotation of the S4 helix, which facilitates charge–charge pairing of successive arginine side‐chains with relatively fixed counter charges in the S1–S3 segments of the VSD (Bezanilla, 2000). However, the flexibility of attached spectroscopic tags means that it is unlikely that these methods are going to enable us to get a more accurate measure of the extent of this motion. In theory we could obtain more accurate estimates if we had atomic resolution structures of both the up and down states of the VSD in intact KV channels. We have crystal structures of voltage gated K+ channels in the activated (likely to be the open and inactivated) state (Long et al. 2005 a, 2007). However, given the high entropy of the closed state, which can be inferred from the very shallow voltage dependence of gating current measurements at negative voltages, obtaining a crystal structure of a closed state voltage sensor conformation will be extremely difficult. We suggest that it will be slightly less challenging to get insights into the structure and location of the voltage sensor in the closed state using cryo‐EM. First, the use of cryo‐EM eliminates the need to crystalize the channel, which is one of the most challenging aspects of the structural analysis of membrane proteins using X‐ray crystallography (Liao et al. 2013). Second, provided one can capture a sufficient number of images of single particles in the cryo‐electron microscope then in theory it should be possible to capture channels with voltage sensors in the open (likely to be > 99.9%) and closed (< 0.1%) states at 0 mV (Kowal et al. 2014). Introduction of mutations that stabilize the down state conformation of the VSD would make it more feasible to capture a sufficient number of particles in the closed state.
Ultimately, even if we were to obtain static structures of the stable end states of VSDs we ideally would want to know the pathway by which these domains transit between the closed and open channel state conformations. In particular, how do the positively charged arginines move through the ‘hydrophobic’ core to transfer charges from the intracellular to extracellular sides? One promising candidate to study the motion of arginine residues is the use of the sulfhydryl‐reactive fluorophore monobromo‐trimethylammonio‐bimane (qBBr). qBBr fluorescence is quenched by tryptophan and, to a lesser extent, tyrosine – so one can monitor movement of qBBr relative to Trp/Tyr residues by monitoring changes in fluorescence (Semenova et al. 2009). Another possibility is applying the technique of rate‐equilibrium free energy (REFER) analysis where one can infer the motion of a two‐state gating transition by studying how mutations perturb the energetics of the transition state (proportional to the logarithm of perturbation to the kinetics of the reaction) relative to the energetics of the equilibrium distribution of the stable end states. This technique has been applied to study the gating of acetylcholine receptor channels (Auerbach, 2010) as well as inactivation gating in KV11.1 channels (Wang et al. 2011), but never before applied to the motion of voltage sensors. In theory this technique can achieve residue level resolution. A more likely scenario, however, is that REFER analysis studies will be used for obtaining constraints for MD simulations.
Challenges:
Few cardiac K+ channels structures have been solved
MD simulations of channel function rarely include interactions with membrane lipids or other regulatory molecules
Recent progress in MD simulation, particularly with the introduction of the ANTON chips (Dror et al. 2012, Jensen et al. 2012) and structural modelling of ion channels using Rosetta (Yarov‐Yarovoy et al. 2006, 2012; Pathak et al. 2007), suggests that we will soon be able to simulate the motion of the voltage sensor at atomic resolution. The movement of charged species through variable membrane electrical fields is however a more complex phenomenon than the movement of ions in aqueous solution and through the selectivity filter. Thus, it is possible that we will also need improvements in incorporation of the membrane potential into molecular simulations (Raval et al. 2016) before we will be able to achieve accurate simulations of the motion of VSDs in response to a changing membrane electric field. With the availability of two end‐points in the VSD (or any other) gating pathway, one can construct and relax the transition pathway using one of several developed techniques – meta‐dynamics, string or multi‐dimensional umbrella sampling simulations to name a few (Lindahl & Sansom, 2008). The Roux and Perozo groups (Ostmeyer et al. 2013) used free energy sampling to investigate molecular mechanisms responsible for slow recovery from inactivation in the bacterial channel KcsA. This technique allows detailed mapping of the equilibrium free energy surfaces underlying complex conformational transitions. The equilibrium surfaces extracted from free energy simulations can be used for various kinetic modelling schemes to model voltage‐ or concentration‐dependent effects in dynamics of K+ channels, hence bringing modelling and experiments together (Perissinotti et al. 2015).
Several challenges remain before we can connect molecular modelling of cardiac K+ channels to experimental data and use this information for developing better therapeutic interventions. The most obvious challenge relates to the lack of molecular structures for most relevant cardiac channels. A viable strategy to refine 3D structures of cardiac K+ channel targets is to combine de novo structural modelling driven by experimental constrains from toxin binding, electrophysiology, cryo‐EM and other techniques with MD simulations enabled by the ANTON platform. For example, the structural modelling of peptide toxin interaction with KV channel VSDs allows the refinement of toxin–channel complex in the activated state of the voltage sensor (Gupta et al. 2015). Using the Rosetta molecular modelling suite and MD simulations, an integrated approach for modelling of the KV11.1 channel based on homologous Kv channel templates was developed (Lees‐Miller et al. 2009; Subbotina et al. 2010).
Several recent studies have emphasized an important role for lipid‐specific modulation in the normal function of several cardiac channels. The functional implications of PIP2 regulation of I KATP were firmly established by Hilgemann and Ball 20 years ago (Hilgemann & Ball, 1996), and specific binding pockets for lipids were recently established (Lee et al. 2013). Other examples of lipids modulating ion channel function include cholesterol regulation of currents conducted by Kir2.1 (Romanenko et al. 2004; Rosenhouse‐Dantsker et al. 2013) and polyunsaturated fatty acids in the cardiac membrane that may exert anti‐arrhythmic activity by attenuating Kv7.1 channel function (Liin et al. 2015). However, most (if not all) MD simulations of various cardiac K+ channels are performed in model one‐component lipid bilayers. To start to address the role of lipid–channel interactions using molecular modelling, a model membrane with a more realistic lipid composition is desperately needed. The most recent developments in MD techniques and force‐field parameters have yielded simulations of complex membranes with up to 65 different lipid types, including asymmetric component distribution in leaflets. These models provide a high‐resolution view of the lipid organization of plasma membranes at an unprecedented level of complexity (Ingolfsson et al. 2014). The membrane simulations revealed a complex and dynamic interplay of lipid components with emergence of transient domains varying in size of composition enriched in cholesterol, gangliosides or phosphatidylinositol mono‐, bis‐ and trisphosphate panel (Ingolfsson et al. 2016). The next logical step for the field would be to investigate cardiac ion channel dynamics in the mixed lipid bilayers under a wide variety of conditions, including electrical fields and binding of peripheral membrane proteins. Taken together, simulations of cardiac ion channels with soluble domains in a complex bilayer will open an avenue to understanding specific lipid regulation of cardiac function at the supra‐molecular level.
Can we build a complete model of an arbitrary K+ channel, including its regulation, in a cardiac myocyte? The answer is conceptually ‘yes’, but we are not there yet. A rapidly emerging area for future research is the development of better approaches for integration of multi‐scale processes. For example, lipid peroxidation or residue ionization in ion channels in response to changes in intracellular pH can result in significantly altered functional dynamics, but MD simulation is limited in its ability to model bond‐formation and bond‐breaking events. The incorporation of various cellular signals such as phosphorylation, glycosylation and binding of short regulatory peptides to KV channels into simulations of conformational dynamics and corresponding responses to perturbation remains challenging. More generally, understanding of complex interactions between KV channels and various peripheral membrane or soluble proteins requires the development of supra‐molecular (non‐atomistic) models of protein–protein interactions. The coupling between the cytoskeleton and lipid‐modulated function of various K+ channels (e.g. KATP) while established experimentally, has remained underexplored and the mechanisms responsible for large macro‐molecular assembly organization and conformational dynamics are largely unknown. A strategy that incorporates future experimental, structural and modelling studies will be a fundamental test of our understanding of the physical and chemical basis of molecular interactions between KV channel modulators, lipid environment and channel at the atomic level. Importantly, this research will enable targeting of any KV channel subtype for treatment of cardiac disorders and for monitoring the activity of specific KV channel types.
Cardiac K+ channel regulation
In the sessions related to K+ channel regulation, Tamas Banyasz, Nipavan Chiamvimonvat, Laszlo Csernoch, Isabelle Deschenes, Dobromir Dobrev, Eleonora Grandi, Jordi Heijman, Leighton Izu, Robert Kass, Jonathan Lederer, Yu‐Fung Lin, Jon Makielski, Manuel F. Navedo, Jeanne Nerbonne, Colin Nichols, Brian O'Rourke, Catherine Proenza, Zhilin Qu, Randall Rasmusson, Ursula Ravens, Crystal Ripplinger, Gail Robertson, Fernando Santana, Daisuke Sato, James Trimmer, Stefan Wagner, Heike Wulff and Antonio Zaza participated as speakers, discussion leaders, or panelists.
K+ channels play a critical role in cardiac electrophysiology, and their dysfunction links intracellular signalling, metabolism, remodelling and arrhythmogenesis in many cardiovascular diseases. Below, we discuss our current understanding of cardiac K+ channel types and mechanisms for their (dys)regulation. The role of K+ channels in the normal and diseased heart is discussed in more detail in a second white paper (see Chiamvimonvat et al. in this issue).
Types of K+ channels in heart
Numerous K+ channel types are expressed in the sarcolemmal membrane of the heart (Table 1), where they exert precise control over action potential (AP) duration (APD) (Fig. 2) (Bartos et al. 2015). Each K+ channel type has distinct kinetics and regulation, allowing control over particular phases of the AP. KV channels include the slow, rapid and ultra‐rapid delayed rectifiers (I Ks, I Kr and I Kur), and the components that rapidly and slowly recover from inactivation of the transient outward current (I to,f and I to,s, respectively). Inward‐rectifier K+ current (I K1) is indirectly voltage‐dependent, a consequence of channel block by intracellular polyamines and Mg2+. Total I to rapidly activates during the AP upstroke and produces a fast and early phase of repolarization. Subsequent activation of I Kur, I Kr and I Ks controls the AP plateau and APD in large mammals. Activation of I K1 at negative membrane potentials is responsible for the final repolarization and the resting membrane potential (RMP; Fig. 2) (Nerbonne & Kass, 2005, Schmitt et al. 2014). Ligand‐gated K+ currents in the heart include the acetylcholine‐dependent inward‐rectifier K+ current (I K,ACh), the ATP‐sensitive K+ current (I KATP), the small‐conductance Ca2+‐activated K+ (SK) current (I SK), the intracellular Na+‐activated K+ current (I KNa) and the cAMP‐activated pacemaker or ‘funny’ current (I f), which is carried by both K+ and Na+ (Bartos et al. 2015, Nerbonne & Kass, 2005, Schmitt et al. 2014). Several members of the large family of two‐pore‐domain K+ (K2P) channels, including TREK‐1 (K2P2.1) and TASK‐1 (K2P3.1), are expressed in the heart and contribute to the background plateau current I KP and the steady‐state K+ current (I SS) in rodent hearts. K2P channels are regulated by many factors, including temperature, pH, oxygen and membrane stretch (Schmitt et al. 2014). Besides these sarcolemmal K+ channels, several ion channels are located on subcellular membranes, including mitochondrial and nuclear KATP channels and sarcoplasmic reticulum (SR) K+ channels involved in normalizing SR potential during Ca2+ release (Takeshima et al. 2015).
Table 1.
Cardiac K+ currents and their molecular identities (α‐subunits)
| Current | Protein | Gene | Location in humans | Notes |
|---|---|---|---|---|
| I Kr | Kv11.1 | hERG (KCNH2) | 7q36.1 | |
| I Ks | Kv7.1 | KCNQ1 | 11p15.5 | |
| I to,f | Kv4.2/4.3 | KCND2/3 | 7q311p13.3 | Gene and protein names depend on species; significant expression differences among species and regions of the heart |
| I to,s | Kv1.4 | KCNA4 | 11p14 | |
| I Kur | Kv1.5 | KCNA5 | 12p13 | Mostly expressed in atrial cells, and in ventricles for some species (e.g. rodents) |
| I K,slow | Kv2.1 | KCNB1 | 20q13.2 | |
| I K1 | Kir2.1 | KCNJ2 | 17q24.3 | |
| I f | HCN4 | HCN4 | 15q24.1 | Pacemaker cells |
| I K,ACh | Kir3.1/3.4 | GIRK1 (KCNJ3)/GIRK4 (KNCJ5) | 2q24.1 | Both homo‐ and heteromers |
| 11q24 | ||||
| I KATP | Kir6.2 | KCNJ11 | 11p15.1 | |
| I K,Ca | SK1, SK2, SK3 | KCNN1, KCNN2, KCNN3 | 19p13.1 | SK2 predominant isoform |
| 5q22.3 | ||||
| 1q21.3 |
Figure 2. Cardiac K+ channel diversity.
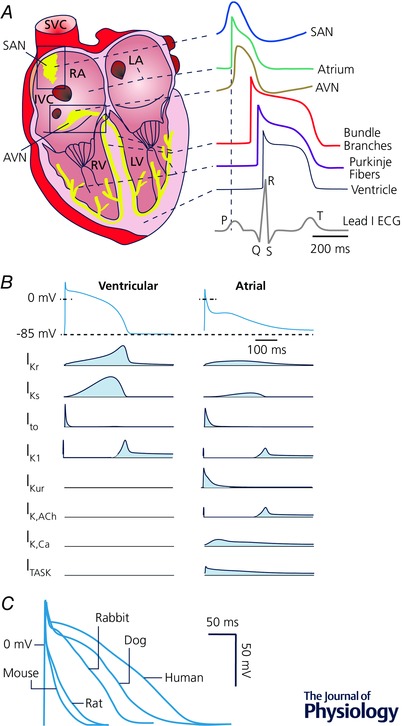
A, AP regional heterogeneity; redrawn from Bartos et al. (2015). B, K+ currents during ventricular and atrial APs; redrawn from Schmitt et al. (2014). C, species differences in ventricular AP; redrawn from Kaese & Verheule (2012).
Challenges:
Difficulties in isolating individual K+ current types
Incomplete knowledge of the molecular basis of K+ channel types and their dynamic role during the AP
Detailed vs. simplified mathematical models of K+ channel gating
Inconsistent and incomplete datasets for modelling
Several challenges complicate the experimental identification of the precise contribution to repolarization of each K+‐current type, even in isolated cardiomyocytes. During patch‐clamp experiments currents are often separated by use of drugs or toxins to block specific currents. However, many of these compounds are not as selective as commonly assumed. Ion channels are also often studied in heterologous expression systems, which facilitates detailed characterization of their biophysical properties independent of the availability of specific blockers; however, incomplete understanding of the macromolecular complexes that produce native currents and the absence of cofactors endogenous to cardiomyocytes make it difficult to recapitulate all regulatory mechanisms in such systems (see below). Experimental protocols for myocyte isolation and culture and the absence of electrical interactions with the surrounding myocardium can alter ion channel expression and function, further complicating the identification of the role of individual K+ currents in cardiac electrophysiology and arrhythmogenesis in vivo. Ex vivo tissue preparations, such as the isolated arterially perfused left ventricular wedge and the Langendorff‐perfused heart, also suffer from technical limitations (e.g. hypoxia) and are devoid of neurohumoral inputs, so direct extrapolation of some findings to in vivo effects is difficult. Further improvements in molecular biological approaches to down‐regulate (e.g. siRNA, CRISPR/Cas9) or overexpress (e.g. lentiviral infection) specific proteins in intact cardiomyocytes and living animals may help to clarify the molecular correlates of individual K+ currents and facilitate the analysis of their contribution to cardiac electrophysiology and arrhythmogenesis independent from pharmacological tools.
Because of the complexity of the cardiac system, mathematical models and simulation are powerful tools to investigate the role of K+ channels under normal and pathological conditions. The Hodgkin–Huxley formalism is widely used in cellular electrophysiological models for quantitative characterization of K+ channels. However, gating parameters in this formulation do not explicitly represent channel kinetic states and are sometimes unable to accurately describe certain channel behaviours or channel–drug interactions (Liu & Rasmusson, 1997; Rudy & Silva, 2006; Bett et al. 2011). Markov model transitions typically represent specific channel movements that have been characterized experimentally (e.g. subunit cooperativity) and can represent the dependence of a given transition on the occupancy of different states of the channel. Many recent computational studies have highlighted the importance of accurately describing state‐dependent drug binding, instead of relying on steady‐state concentration–response curves or EC50 values (Moreno et al. 2011; Aguilar & Nattel, 2015; Lee et al. 2016, Morotti et al. 2016). The choice between Hodgkin–Huxley and Markov models is a balancing act between need for detail and practicality of simplified models, particularly for multi‐scale simulations.
Formulations for individual currents and validation of the whole‐cell system require many experiments, which are typically performed in different laboratories for purposes other than model building. This situation often leads to incomplete (or non‐physiological) datasets, with large variances that reflect differences in experimental conditions (e.g. temperature, solutions, species) rather than true physiological variability (Krogh‐Madsen et al. 2016). The re‐use of model components based on data from other species often masks the link between model parameters and the original data source (Niederer et al. 2009). These inconsistences might be accentuated in disease models, due for example to different severities and/or extents of disease‐induced remodelling. Thus, use of consistent, well‐defined data sets and dedicated experiments would be ideal to fit model parameters and validate model responses over the full range of physiological conditions. Moreover, commonly used voltage‐clamp protocols predominantly provide information about steady‐state ion‐channel properties (e.g. activation, inactivation), or assess a single dynamic parameter for a specific condition (e.g. recovery from inactivation at a given membrane potential). Such protocols do not always provide sufficient information to fully characterize channel kinetics during the complex dynamics of the AP. A new approach to build cell‐specific cardiac models was recently developed by coupling genetic algorithm parameter fitting with complex electrophysiology protocols, which allow extracting dynamically rich information inherent to arrhythmias (Groenendaal et al. 2015).
Diversity of cardiac K+ currents
The pronounced heterogeneity in AP morphology and duration among different species and regions of the heart is in large part due to differential expression of multiple K+ channel types (Fig. 2 A and B). APD is much shorter and more triangular in rodents compared to humans (Fig. 2 C). However, even large mammals such as the dog, which appear to have reasonably comparable electrophysiological characteristics to humans, respond differently to pharmacological interventions targeting K+‐channels (O'Hara & Rudy, 2012; Jost et al. 2013), indicating a species‐dependent contribution of individual currents.
In all species, sinoatrial and atrioventricular cells show spontaneous diastolic depolarization mediated by a complex system, which includes the HCN channel‐mediated I f in combination with a low I K1 expression. Atrial and ventricular cardiomyocytes have a stable RMP, but distinct AP morphology (Fig. 2 A and B). On average, atrial cardiomyocytes have a more depolarized RMP than ventricular cardiomyocytes due to a smaller I K1, and a more triangular AP shape due to the atrial‐specific expression of KV1.5 (I Kur). Further differences have been identified between the left and right sides of the heart, including a larger I to and I Ks in the right compared to the left ventricle (Molina et al. 2016), and between the different layers of the ventricular wall, with a much larger I to near the epicardium than near the endocardium (Nerbonne & Guo, 2002).
Challenges:
Cell‐to‐cell variability
Differential regulation of individual K+ currents
Limitations of model systems to study K+ channel‐based therapy
Regulation of K+ current in other coupled systems
Sequential measurements of I K1, I Ks and I Kr using AP‐clamp in ventricular cardiomyocytes suggest that there is pronounced (∼10‐fold) heterogeneity in K+ current amplitude between different cells (Banyasz et al. 2011). Because the action potential is shaped by a balancing act of all ionic currents, larger/smaller outward K+ currents can be counterbalanced by larger/smaller inward currents to generate normal action potentials. Modelling has predicted that similar electrophysiological behaviour can result from different sets of underlying conductances (Golowasch et al. 2002; Sarkar & Sobie, 2010), and experimental work suggests that compensatory changes in current densities maintain relatively stable function in response to changes in activity or overexpression of ion channels (MacLean et al. 2003). Variable cell‐to‐cell channel expression is often not adequately reported or taken into account in traditional cardiac electrophysiological experiments and models, which instead mostly use averaged data and build ‘representative’ cell models of a specific type. This might be problematic because averages over multiple samples can fail to characterize a system whose behaviour depends on interactions involving a number of highly variable components (Golowasch et al. 2002). Thus, range or variance of datasets should be reported, instead of mean values and standard errors. Population‐based computational approaches, calibrated by dynamically rich datasets, allow simulating ‘good enough solutions’ (Weiss et al. 2012) and determining those which will adapt better to pathophysiological regulation. Indeed, they have proven valuable for understanding variability in cardiotoxicity (Sarkar & Sobie, 2010) and identifying drug targets with favourable anti‐arrhythmic properties (Cummins et al. 2014).
The regional and cell‐to‐cell differences in K+ channels are dynamic and the contribution of a specific channel type may only become apparent after modulation of other channels. For example, I Ks is small under basal conditions in ventricular cardiomyocytes of large mammals, with I Kr providing most of the repolarizing current. However, I Ks is significantly increased by sympathetic stimulation, reversing the relative dominance of I Ks vs. I Kr controlling AP repolarization (Banyasz et al. 2014). I Ks also constitutes an important repolarization reserve that is activated upon loss of other repolarizing currents (Jost et al. 2013). Similarly, I f does not have an important role in the ventricles under normal conditions. However, during pathological conditions, or after inhibition of I K1, ventricular cardiomyocytes can show abnormal automaticity, which may be partly mediated by I f (Miake et al. 2002). Differential regulation of individual K+ channels makes drug sensitivity dependent on the specific (patho)physiological conditions, and thus is a confounding variable in the study and quantitative description of channel regulation.
Differences in K+ channel expression have important implications for K+ channel‐based pharmacological therapy in patients. On the one hand, the heterogeneity in K+ channel expression can be exploited to increase the selectivity of pharmacological interventions. For example, targeting channels predominantly expressed in the atria (e.g. K2P3.1, Kv1.5, Kir3.1, Kir3.4 channels) for rhythm control of atrial fibrillation (AF) may avoid unwanted ventricular proarrhythmic side effects (Heijman et al. 2016). On the other hand, these differences raise questions about which model system is the most appropriate to study novel potential K+ channel‐based therapies in patients. Mouse models have the advantages of low cost, ease of genetic manipulation, multi‐scale and multi‐system analyses, and a homogeneous genetic background for detection of small effects. However, the electrophysiological differences between mice and men (Fig. 2 C) make these models less suitable to assess antiarrhythmic efficacy or proarrhythmic risk (Kaese & Verheule, 2012). Large animal models more closely resemble the human K+ channel distribution and cardiac repolarization time course (Fig. 2 C), making them more suitable to evaluate pro‐ or antiarrhythmic effects. However, their application is restricted by costs, availability of clinically relevant disease models, and limited options for genetic manipulation. Of note are the long QT syndrome (LQTS) type 1 and 2 transgenic rabbits, LQT1 and LQT2 (Brunner et al. 2008). Recently, CRISPR/Cas9 was used successfully to genetically modify genomes in various species (Reggio et al. 2001; Hai et al. 2014; Ni et al. 2014), and has the potential to establish large animal models that can more faithfully mimic human cardiac physiology. Human hearts and isolated cardiomyocytes might represent the most clinically relevant samples to study K+ channel (dys)function. However, the limited amount of available tissue and the highly selected patient population (e.g. explanted hearts from end‐stage heart failure (HF) patients, or atrial samples from patients undergoing open heart surgery), as well as the very limited control over confounding clinical variables make their routine application challenging. Human induced pluripotent stem cell‐derived cardiomyocytes (hiPSC‐CMs) are a relatively novel option providing potentially unlimited supplies of human cardiomyocytes for any desired pathology. Unfortunately, the phenotype of hiPSC‐CMs remains generally immature, including pronounced abnormal automaticity due to the relatively small I K1. Electronic ‘expression’ of I K1 using an in silico interface and dynamic clamp has proven useful to stabilize the electrical behaviour of hiPSC‐CMs, by maintaining a physiological RMP, eliminating automaticity and reducing AP variability (Bett et al. 2013). Such increased stability enables the use of this preparation for quantitative analysis of electrophysiological properties. There are encouraging examples in which application of physical cues (electrical and mechanical, such as oriented growth, stimulation, relevant stiffness matrices, co‐culture with fibroblasts and extracellular matrix scaffolds) can push maturation of hiPSC‐CMs in vitro (Lundy et al. 2013; Zhu et al. 2014). The unknown genetic variation between different hiPSC‐CM lines may also increase electrophysiological variability and limit reproducibility. However, recent advances in gene editing allow the creation of isogenic controls to study the effect of specific genetic variants on K+ channel regulation and APD (Bellin et al. 2013).
Cell types other than myocytes in the heart also express K+ channels. Cardiac (myo)fibroblasts express I K1, KV channel currents and several Ca2+‐activated K+ currents (Yue et al. 2011). Their regulation may directly, via electrotonic interactions between fibroblasts and cardiomyocytes, or indirectly, by regulating fibroblast proliferation, fibrosis and paracrine signalling, modulate cardiac electrophysiology and arrhythmogenesis in vivo. Several K+ channels (or their regulatory subunits) are also expressed outside the heart, resulting in a variety of interconnected disease manifestations by their dysfunction. For example, KV and Ca2+‐activated K+ channels in vascular smooth muscle regulate vascular tone and smooth muscle cell migration and proliferation (Ko et al. 2008). Similarly, although KCNE2 was first primarily considered a KV β‐subunit in the heart and implicated in LQTS, it is now known to have a wide array of functions in the heart, stomach, thyroid and choroid plexus (Abbott, 2015). These findings highlight the need for a multi‐scale systems biology approach to understanding K+‐channel regulation. A combination of different approaches, from heterologous expression systems to in vitro and in vivo animal studies, experiments in human samples, and computational models, is likely to be needed to obtain a complete understanding of the regulation of individual K+ channels and their (patho)physiological relevance.
Mechanisms for regulation of cardiac K+ channels
Given the critical role for K+ channels in determining repolarization rate, it is hardly surprising that K+ channel function is exquisitely regulated in cardiomyocytes (Fig. 3). Overlapping transcriptional, translational and post‐translational mechanisms provide nuanced control over K+ currents across multiple time scales, ranging from milliseconds to days, or even longer. Regulatory mechanisms modify K+ currents by controlling channel expression levels, gating sensitivity and kinetics, ligand availability, and the structural states that determine conduction and drug binding. The complex web of regulation allows for fine control of multiple K+ currents during different phases of the AP, and within different cardiac regions and cell types. Different (patho)physiological circumstances change the balance of regulatory mechanisms, and hence the predominant currents in a given cell type or region, resulting in highly context‐dependent responses to pharmacological agents.
Figure 3. Mechanisms for regulation of cardiac K+ channels.
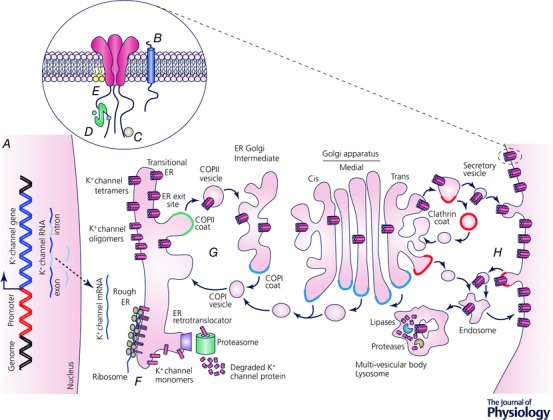
A, transcriptional regulation; B, interacting proteins; C, post‐translational modifications; D, ligands, Ca2+ and calmodulin; E, membrane lipids; F, co‐assembly of subunits; G, forward trafficking, including packaging into COPII‐coated vescicles, transport to ER‐Golgi intermediate compartament and Golgi apparatus, and trafficking to specific membrane destinations into clathrin‐coated vescicles; H, internalization.
Transcriptional control
Transcriptional control of multiple K+ channels regulates their spatial and temporal profiles to modulate cardiac function during (patho)physiological challenges. For example, the transcription factors Irx5 (Costantini et al. 2005) and NFATc3 (Rossow et al. 2006) differentially regulate KV4 expression across the mouse ventricular wall, contributing to the I to,f transmural gradient. Both Irx5 and NFATc3 have expression and activity profiles opposite that of KV4‐encoded I to,f, and their independent deletion in the heart completely abrogates the KV4‐encoded I to,f gradient in some species (Costantini et al. 2005; Rossow et al. 2006). An expression and activity gradient has also been described for several I KATP subunits in different cardiac compartments that is established by differential activity of the Forkhead transcription factors FoxO and FoxF2 (Philip‐Couderc et al. 2008).
Non‐coding RNAs (ncRNAs), including long non‐coding RNAs and microRNAs, are emerging as important regulators of gene expression in cardiomyocytes (Greco & Condorelli, 2015; Tao et al. 2015) and are also associated with K+ channel remodelling in humans with AF and cardiac hypertrophy (Girmatsion et al. 2009; Matkovich et al. 2010). Although our understanding of the mechanisms by which ncRNAs regulate cardiac K+ channel expression and function is in its infancy, these regulatory mechanisms may provide the basis for development of novel therapies.
Questions:
How do physiological and pathophysiological remodelling differ, and how do they interact with K+ channel regulatory mechanisms?
Aberrant K+ channel expression and function contributes to electrical remodelling and arrhythmias in several disease states. In HF, AP prolongation is linked to reduced I to,f, I Ks, I Kr and I K1 (Yang & Nerbonne, 2016), due in part to transcriptional downregulation of the underlying genes (Nass et al. 2008). Similarly, suppressed expression of KV4.3 and KV1.5 is correlated with reduced I to,f and I Kur density during AF (Van Wagoner et al. 1997; Yue et al. 1999). Downregulation in gene and protein expression levels of KV1.5, KV2.1 (I K,slow) and KV4.2/4.3, resulting from Ca2+‐dependent activation of the calcineurin–NFATc3 signalling pathway (Rossow et al. 2009), leads to AP prolongation after myocardial infarction in mice (Rossow et al. 2004). A similar decrease in K+ current density was observed during pathological cardiac hypertrophy caused by pressure overload (Marionneau et al. 2008), but was associated with cellular hypertrophy rather than changes in I to,f and I K1 gene expression.
These observations distinctly contrast with physiological cardiac hypertrophy induced by chronic exercise, where cellular hypertrophy is accompanied by a concomitant increase in the expression of repolarizing (I K) and depolarizing (I Na, I Ca) currents that normalize the ionic conductances and AP waveform, leading to protection from arrhythmogenic events (Yang et al. 2010). These results also highlight the exquisite balance that must exist between K+, Na+ and Ca2+ conductances to generate a ‘normal’ APD. Evidently, distinct signalling pathways contribute to physiological (insulin‐like growth factor 1/phosphatidylinositol‐3‐kinase α signalling, independent of protein kinase B (Akt), leading to upregulation of ion channels, including K+ channels) and pathological (Gαq signalling downstream of G‐protein coupled receptors activated by humoral factors) cardiac hypertrophy (Bernardo et al. 2010; Yang et al. 2012). However, the precise mechanisms underlying differential K+ channel regulation in physiological vs. pathological hypertrophy remain poorly understood. A further understanding of these mechanisms and associated cardioprotective signalling pathways and genes could reveal targets for development of novel therapeutic options.
Interacting proteins
Questions, controversies, or challenges:
What are the interacting proteins in complex with cardiac K+ channels, and how do they modulate function?
Stoichiometry of channel complexes
An emerging concept is that formation of heterogeneous macromolecular channel complexes formed by pore‐forming α‐subunit heteromultimers, auxiliary subunits and variable interacting proteins contribute to distinct biophysical properties, differential regulation, and regional‐ or species‐dependent differences in K+ current diversity.
KVβ subunits are co‐assembled with multiple KVα subunits to promote trafficking, accelerate inactivation and even act as redox sensors. In ventricular myocytes, KVβ1 influences the functional expression of KV4.3‐encoded I to,f and KV2.1‐encoded I K,slow via direct and indirect mechanisms (Aimond et al. 2005). Differential expression of KChIP2, which facilitates trafficking and function of KV4‐encoded channels, is believed to underlie the epi‐ to endocardial I to,f transmural gradient in large mammals (Rosati et al. 2001). KCNE subunits are single membrane‐spanning proteins that associate with several KVα subunits to regulate channel activity. For example, co‐assembly of KCNE1 and KCNQ1 slows KCNQ1 activation and generates I Ks (Sanguinetti et al. 1996). Mutations in KCNE1 have been associated with LQTS and AF (Crump & Abbott, 2014). Surprisingly, a role for the voltage‐gated Na+ channel accessory subunit, Navβ1, in the functioning of Kv4.2‐encoded channels has also been demonstrated (Deschenes & Tomaselli, 2002).
Scaffolding proteins provide spatiotemporal targeting of regulatory molecules (e.g. kinases and phosphatases) to ion channels with important implications for cardiac function (Langeberg & Scott, 2015). Prototypes are A‐kinase anchoring proteins (AKAPs), which dynamically regulate cardiac K+ channel function via multiple mechanisms. Indeed, the AKAP Yotiao co‐assembles with KCNQ1 channels to mediate protein kinase A (PKA)‐dependent phosphorylation of KCNQ1 during sympathetic nerve stimulation (Marx et al. 2002). Conversely, the anchoring protein AKAP79/150 is required for calcineurin/NFATc3‐mediated transcriptional downregulation and functional suppression of KV channels following myocardial infarction (Nieves‐Cintron et al. 2016). These observations underscore the extensive functional diversity that may coexist within a cell to contribute to a specific phenotype.
The stoichiometry of different channel subunits and splice variants within channel complexes can also profoundly alter the response to regulatory factors. However, the exact composition of subunit isoforms or variants within native channel complexes is difficult to determine experimentally and can vary considerably between different species and cardiac cell types. The mechanisms that determine the stoichiometric balance of different subunits are poorly understood, are often not recapitulated in heterologous expression systems, and may not be specified by amino acid sequence alone. For example, subunit expression can be regulated by co‐translational association with RNA (Liu et al. 2016).
Post‐translational modifications
Several post‐translational mechanisms can alter properties or expression levels of cardiac K+ channels. The most common and best characterized of these mechanisms is phosphorylation by protein kinases such as cAMP‐dependent PKA, which phosphorylates K+ channels as part of the flight‐or‐flight response, and Ca2+/calmodulin‐dependent protein kinase II (CaMKII; Nerbonne, 2011; Mustroph et al. 2014), which is overexpressed and hyperactive in HF and persistent AF (Anderson et al. 2011). CaMKII acutely regulates I to by increasing current density and hastening recovery from inactivation (Sergeant et al. 2005; Wagner et al. 2009; Zhang et al. 2015) via a mechanism that may involve direct phosphorylation of KV4.2/KV4.3 (I to,f; Varga et al. 2004) and KV1.4 (I to,s; Roeper et al. 1997). CaMKII co‐immunoprecipitates with both KV4.2 and KV4.3 (Colinas et al. 2006), suggesting that it may be a component of native cardiac I to,f channel complexes. These complexes are composed of several cytosolic (KChIP/Kvβ) and transmembrane (DPP6/10, KCNEs) accessory subunits that determine the time‐ and voltage‐dependent properties and surface expression of native cardiac I to,f channels (Niwa & Nerbonne, 2010). Each of the protein components might also be a target for CaMKII‐mediated regulation. In fact, there are multiple putative CaMKII phosphorylation sites on both KChIP2 and KVβ2 (Nerbonne, 2011). Furthermore, KV4.2/KV4.3 channels form complexes with SAP97 and CaMKII (El‐Haou et al. 2009), and in neurons CaMKII‐dependent phosphorylation of SAP97 enhances KV4.2 trafficking (Gardoni et al. 2007). Intriguingly, regional heterogeneity in I to,f expression and function might be associated with heterogeneous regulation of I to,f complexes. In contrast, chronic CaMKII overexpression that leads to HF development results in a reduction of I to (Wagner et al. 2009), but this appears to be a secondary effect.
CaMKII may also regulate cardiac I K1 channels. CaMKII enhances I K1 acutely, but in failing ventricular myocytes overexpressing CaMKIIδ I K1 is attenuated and expression of KCNJ2 (which encodes Kir2.1) is markedly reduced (Wagner et al. 2009). What are the mechanisms involved in the acute and chronic CaMKII‐mediated regulation of native cardiac I K1 channels? Experiments focused on characterizing endogenous I K1 channel complexes and identifying CaMKII phosphorylation sites on Kir2.1, as well as of other contributors to I K1, are needed.
Calcium
Ca2+‐activated K+ channels provide a direct link between intracellular Ca2+ and membrane voltage with every heartbeat. Whether or not Ca2+‐activated K+ (or Na+‐activated Slo2.1) channels are important for normal cardiac electrophysiology has long been debated. Recent evidence suggests that only SK channels contribute to cardiac repolarization. SK α‐subunits are constitutively bound to calmodulin and activate upon intracellular Ca2+ binding to the calmodulin EF hand (K d ∼500 nm), which also regulates membrane expression (Zhang et al. 2015). While initial experiments revealed an atrial dominant expression pattern for SK channels in normal or non‐diseased cardiomyocytes (with the specific blocker apamin prolonging atrial but not ventricular APD), subsequent studies also described SK currents in nodal, pulmonary vein and ventricular myocytes. Furthermore, in ventricular myocytes from diseased animal models or failing human myocytes, SK channel expression is upregulated, suggesting a role for these channels in pathological ventricular repolarization (Chua et al. 2011; Chang et al. 2013).
SK channel interaction with cytoskeletal proteins such as α‐actinin2 and filamin A is critical for cellular localization, which indirectly links SK channels to their intracellular Ca2+ source (Lu et al. 2007; Rafizadeh et al. 2014). Specifically, SK2 channels colocalize with α‐actinin2 and L‐type Cav channels (Cav1.2/Cav1.3) along the Z‐line in atrial myocytes (Lu et al. 2009), suggesting that SK channels may be controlled by subsarcolemmal [Ca2+] via activation of L‐type Cav channels (Marrion & Tavalin, 1998; Zhang et al. 2015). Interestingly, SR Ca2+ has been suggested as a source for activation of overexpressed SK channels (Terentyev et al. 2014), although overexpression can change subcellular localization, and thus could alter regulation compared to native SK channels.
Intracellular Ca2+ also indirectly regulates many other K+ channels in cardiomyocytes. For example, native and expressed KV7.1/KCNE1 (I Ks) channels are activated by intracellular Ca2+ in a CaM‐dependent manner (Tohse, 1990; Ghosh et al. 2006). Many findings over the years have improved our understanding of how Ca2+ regulates K+ channels in the heart. Yet determining the precise mechanisms of Ca2+ sensitivity of K+ channels in cardiomyocytes remains technically challenging, in part because intracellular [Ca2+] greater than 500 nm can cause unwanted cellular contraction during recordings.
Membrane lipids
Lipids may interact directly and dynamically with ion channels to alter properties such as channel conformation or gating, sarcolemma surface charge, and molecular affinity. The regulation of ion channels by the acidic phospholipid PIP2 is one of the best understood.
In the heart, PIP2 is necessary for proper function of numerous K+ channels via distinct mechanisms. Several Kir channels are highly PIP2 dependent, only conducting current when PIP2 is bound. Studies of KATP channels in guinea pig ventricular myocytes were among the first to show rapid current rundown upon PIP2 depletion and reactivation when PIP2 is replenished (Hilgemann & Ball, 1996; Fan & Makielski, 1997). Kir2.1 affinity for PIP2 is important for normal ventricular repolarization in humans. Many KCNJ2 mutations associated with Andersen–Tawil syndrome reduce Kir2.1's affinity for PIP2 and decrease I K1 (Donaldson et al. 2003). Other cardiac KV channels such as KV11.1 (hERG/I Kr) and KV7.1 (KCNQ1/I Ks) also rely on PIP2 for proper function. Activation of voltage‐sensitive phosphatases or Gq/11‐coupled receptors causes PIP2 depletion at the membrane, thus reducing PIP2 association with KV7.1, and decreasing I Ks (Zaydman et al. 2013).
Ligands
Ligand‐gated K+ cardiac channels that conduct I KATP, I K,ACh and I f are also subject to the regulatory mechanisms described above, but their regulation is largely via control of ligand availability in response to physiological and pathophysiological conditions, such as metabolic status, autonomic nervous system activity and ischaemia. Cardiac KATP channels are of particular translational interest, because they contribute to ischaemic preconditioning (IPC), in which short bouts of ischaemia protect the myocardium against subsequent more severe insults. I KATP activation shortens and may even prevent APs and Ca2+ transients in myocytes that are ATP depleted, allowing energetically compromised cells to conserve energy (Lederer et al. 1989). They might synergize with gap junction channels, which are closed by acidosis and elevated diastolic [Ca2+]i (Spray et al. 1982). KATP channels and their regulators have long been investigated as targets for development of new treatments for ischaemic heart disease.
KATP channels are composed of four pore‐forming Kir6 α‐subunits plus four regulatory sulfonylurea receptor (SUR) subunits. KATP channels are inhibited by ATP, which binds to Kir6 subunits, and activated by Mg2+‐ADP, which binds to the SUR subunits. Channels are closed at physiological ATP concentrations, but open as the ATP/ADP ratio decreases during hypoxia or ischaemia. This regulation of channel activity is thought to be critical for IPC, which is prevented by KATP blockers and mimicked by KATP channel openers. However, different KATP channels with distinct properties are expressed in the sarcolemma (sarc‐KATP) and the mitochondrial inner membrane (mito‐KATP) of cardiomyocytes. The location of KATP channels that mediate IPC remains controversial (Garlid & Halestrap, 2012).
Sarc‐KATP channels clearly incorporate Kir6.2 and SUR2A subunits, and Kir6.1 and SUR1 may also contribute (Zhang et al. 2010). Sarc‐KATP channels are highly expressed in cardiomyocytes and have a large single channel conductance (∼75 pS). Hence opening of even a small percentage of the channels on the sarcolemma is predicted to shorten APD. However, this simple model is belied by the observation of fairly normal ECGs with no QT shortening in patients with Cantu syndrome, a multi‐organ syndrome associated with gain‐of‐function mutations in SUR2 or Kir6.1 (Cooper et al. 2015; Levin et al. 2015). The lack of pronounced effects of sarc‐KATP activation on the ECG appears to be associated with increased L‐type Ca2+ current via a complex compensatory remodelling process, a situation that underscores how co‐regulation of different types of ion channels poses challenges to the description of regulatory effects.
Mito‐KATP channels were first identified in giant fused mitoplasts (Inoue et al. 1991) and their primary function appears to be regulation of mitochondrial matrix volume. Identification of subunits that comprise mito‐KATP channels has been challenging owing to the difficulty of isolating pure mitochondrial membrane fractions and the lack of specific probes (i.e. drugs or antibodies). However, recent evidence suggests that ROMK2 subunits form the pore and SUR2 serve as the regulatory subunits in mito‐KATP channels (Foster et al. 2012). Mitochondrial large conductance Ca2+‐activated K+ (BK) channels have also been identified in the heart, where they may improve cardiac energetics (Singh et al. 2012).
Trafficking and sequestration of K+ channels
Presenters, discussion leaders and panelists for this session included Geoffrey Abbott, Fadi Akar, Ravi Balijepalli, Nipavan Chiamvimonvat, Henry Colecraft, Brian Delisle, Isabelle Deschenes and Aldrin Gomes.
Questions:
Are we using appropriate cell types (heterologous expression systems, cardiomyocytes, or hiPSC‐CMs) to characterize channel trafficking?
What possible roles do regulatory proteins (e.g. chaperones) play in the trafficking of wild‐type or mutant K+ channels?
What is the impact of unfolded protein accumulation in the endoplasmic reticulum and how does this affect cellular physiology?
How do we quantify the relative physiological importance of multiple isoforms encoded by the same gene?
What are the roles that promiscuous interacting proteins have in regulating K+ channel trafficking and channel stability?
As noted in the above sections, K+ channel structure, function, diversity and regulation have been studied extensively. Much less is known about protein folding and trafficking events. A generalized trafficking scheme for the biogenesis and targeting of K+ channels to the cell surface membrane is shown in Fig. 3. K+ channel α‐subunit helices are inserted in the endoplasmic reticulum (ER) membrane during translation. The K+ channel α‐subunit proteins co‐assemble, and they are recruited into ER exit sites located in the smooth or transitional ER compartment. K+ channels traffic out of the ER in coat protein (COP) II vesicles; undergo processing as they transit through the Golgi apparatus; and are then inserted into the cell surface membrane. The turnover of K+ channels from the cell surface membrane is likely to involve clathrin‐ or caveolin‐mediated endocytosis and degradation by proteases in the lysosomes.
Defects in K+ channel folding can negatively affect co‐assembly, trafficking and the membrane stability of K+ channels. The meeting presentations centred on the mechanisms by which different LQTS‐linked mutations in the genes encoding KV11.1 (KCNH2) and KV7.1 (KCNQ1) channels decrease the number of K+ channels at the cell surface membrane.
An important finding is that the trafficking‐deficient phenotype and functional expression for many mutant KV11.1 channels is improved by incubating cells with drugs that bind to KV11.1 channels (pharmacological correction) (Anderson et al. 2014). This finding indicates that some KV11.1 mutant channels are promising targets for therapeutic intervention. Heterologous expression studies in mammalian cell lines or hIPSC‐CMs show that these trafficking‐deficient KV11.1 channels are selectively retained in the transitional ER compartment (Smith et al. 2011; Smith et al. 2013). Although the transitional ER compartment contains ER exit sites from which COPII vesicles bud off for transport to the Golgi apparatus, the mutant KV11.1 channels do not undergo ER export because they are excluded from the ER exit sites. Pharmacological correction directly facilitates the trafficking of mutant KV11.1 channels sequestered in the transitional ER compartment (Smith et al. 2011, 2013). Drug treatment for as little as 30 min is sufficient to improve the functional expression of mutant KV11.1 channels at the cell surface membrane for many hours.
Several LQTS‐linked KV11.1 mutations that do not undergo pharmacological correction disrupt the co‐assembly of KV11.1 α‐subunits (Ficker et al. 2002). Heterologous expression of these mutant proteins in cell lines or in hiPSC‐CMs show that they exhibit a distinctive immunostaining pattern that consists of several aggregates around the cell nucleus. The KV11.1 aggregates do not strongly co‐localize with several ER marker proteins, but their immunostaining partially overlaps with ER retrotranslocator channels and cytosolic proteins. These studies suggest mutant KV11.1 proteins that disrupt KV11.1 α‐subunit co‐assembly are retrotranslocated into the cytosol and targeted for degradation via the ubiquitin proteasome pathway.
Heterologous expression studies indicate that KV7.1 α‐subunits expressed alone assemble and traffic well to the cell surface. On the other hand, whether KCNE1 channel subunits expressed by themselves can target to the plasma membrane appears to be cell type dependent, and may be influenced by the presence of endogenous KV7.1 subunits in particular cells used for heterologous expression (Krumerman et al. 2004; Chandrasekhar et al. 2006). In cardiomyocytes, it has been proposed that KV7.1 channels and KCNE1 subunits traffic separately and assemble at the surface sarcolemma to form channels that conduct I Ks (Wang et al. 2013). Development of optical tools and approaches to visually monitor KV7.1/KCNE1 assembly and trafficking is important to decipher molecular mechanisms of subunit transport and the features that are affected by specific LQT1 mutations. Aromolaran et al. (2014) used an optical approach that simultaneously monitors surface (using quantum dot) and total (using yellow fluorescent protein (YFP) fluorescence) KV7.1–YFP channels exogenously expressed in rat cardiomyocytes. The surface density of channels remained relatively constant over a fourfold range of KV7.1–YFP expression, suggesting the existence of homeostatic mechanisms in cardiomyocytes that maintain KV7.1 surface density within tight limits over a wide range of protein expression level. This approach also showed that an LQTS‐linked KV7.1 mutation in the C‐terminus significantly diminishes cell surface channel density in both cardiomyocytes and cells used for heterologous expression (Aromolaran et al. 2014).
As noted above, it is well recognized that K+ channels are heterogeneous macromolecular complexes. Different experimental systems have been utilized to study trafficking mechanisms, each with its own set of advantages and limitations. Heterologous expression systems allow ease of transfection, the ability to precisely determine channel components, and favourable geometry for in‐depth analyses. However, they lack the cyto‐architecture, intracellular milieu and physiologically important protein–protein interactions present in cardiomyocytes. This could contribute to the phenomenon where there is an inconsistent correlation between severity of biophysical defects in heterologous cells and risk of arrhythmias in patients (Priori et al. 1999). Heterologous expression of K+ channels in primary adult cardiomyocytes offers a more physiologically relevant environment to explore trafficking mechanisms (Aromolaran et al. 2014). At the whole‐animal level, transgenic rabbits have been useful in providing in vivo models of LQTS (Brunner et al. 2008). A drawback of transgenic animals is the time and cost required for screening functional consequences of many different K+ channel mutations. Finally, hiPSC‐CMs from LQTS patients provide a model system that is amenable to in vitro drug testing to potentially advance personalized therapy (Zhang et al. 2014). Overall, an all‐of‐the‐above strategy with respect to the various experimental systems discussed could be useful in providing a holistic molecular‐, cellular‐ and organ‐level view of arrhythmia mechanisms and K+ channel trafficking.
Additional information
Competing interests
The authors have no conflicts to disclose.
Author contributions
All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This conference was supported by the National Institutes of Health (NIH) R13HL132515 grant to Y.C.‐I., D.M.B., N.C. and E.G., R01HL131517 grant to E.G., American Heart Association (15SDG24910015 to E.G.) and additional grants to other authors that allowed them to participate. We also thank many sponsors that support the University of California Davis Cardiovascular Symposium Series. See the conference website for detailed information.
Acknowledgements
We thank the Organizing Committee, Consultants, and all conference participants for their contributions to the scientific exchange and discussions. For conference information see basicscience.ucdmc.ucdavis.edu/UCDavisCardiovascularSymposia/index.html.
Linked articles This article is highlighted by a Perspective by Grandi. To read this Perspective, visit http://dx.doi.org/10.1113/JP273665.
References
- Abbott GW (2015). The KCNE2 K+ channel regulatory subunit: Ubiquitous influence, complex pathobiology. Gene 569, 162–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilar M & Nattel S (2015). The past, present, and potential future of sodium channel block as an atrial fibrillation suppressing strategy. J Cardiovasc Pharmacol 66, 432–440. [DOI] [PubMed] [Google Scholar]
- Aimond F, Kwak SP, Rhodes KJ & Nerbonne JM (2005). Accessory Kvβ1 subunits differentially modulate the functional expression of voltage‐gated K+ channels in mouse ventricular myocytes. Circ Res 96, 451–458. [DOI] [PubMed] [Google Scholar]
- Anderson CL, Kuzmicki CE, Childs RR, Hintz CJ, Delisle BP & January CT (2014). Large‐scale mutational analysis of Kv11.1 reveals molecular insights into type 2 long QT syndrome. Nat Commun 5, 5535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson ME, Brown JH & Bers DM (2011). CaMKII in myocardial hypertrophy and heart failure. J Mol Cell Cardiol 51, 468–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong CM & Bezanilla F (1973). Currents related to movement of the gating particles of the sodium channels. Nature 242, 459–461. [DOI] [PubMed] [Google Scholar]
- Aromolaran AA, Subramanyam P, Chang DD, Kobertz WR & Colecraft HM (2014). LQT1 mutations in KCNQ1 C‐terminus assembly domain suppress I Ks using different mechanisms. Cardiovasc Res 104, 501–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerbach A (2010). The gating isomerization of neuromuscular acetylcholine receptors. J Physiol 588, 573–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banyasz T, Horvath B, Jian Z, Izu LT & Chen‐Izu Y (2011). Sequential dissection of multiple ionic currents in single cardiac myocytes under action potential‐clamp. J Mol Cell Cardiol 50, 578–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banyasz T, Jian Z, Horvath B, Khabbaz S, Izu LT & Chen‐Izu Y (2014). Beta‐adrenergic stimulation reverses the IKr‐IKs dominant pattern during cardiac action potential. Pflugers Arch 466, 2067–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barro‐Soria R, Rebolledo S, Liin SI, Perez ME, Sampson KJ, Kass RS & Larsson HP (2014). KCNE1 divides the voltage sensor movement in KCNQ1/KCNE1 channels into two steps. Nat Commun 5, 3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartos DC, Grandi E & Ripplinger CM (2015). Ion channels in the heart. Compr Physiol 5, 1423–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellin M, Casini S, Davis RP, D'Aniello C, Haas J, Ward‐van Oostwaard D, Tertoolen LG, Jung CB, Elliott DA, Welling A, Laugwitz KL, Moretti A & Mummery CL (2013). Isogenic human pluripotent stem cell pairs reveal the role of a KCNH2 mutation in long‐QT syndrome. EMBO J 32, 3161–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardo BC, Weeks KL, Pretorius L & McMullen JR (2010). Molecular distinction between physiological and pathological cardiac hypertrophy: experimental findings and therapeutic strategies. Pharmacol Ther 128, 191–227. [DOI] [PubMed] [Google Scholar]
- Bett GC, Kaplan AD, Lis A, Cimato TR, Tzanakakis ES, Zhou Q, Morales MJ & Rasmusson RL (2013). Electronic “expression” of the inward rectifier in cardiocytes derived from human‐induced pluripotent stem cells. Heart Rhythm 10, 1903–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bett GC, Zhou Q & Rasmusson RL (2011). Models of HERG gating. Biophys J 101, 631–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezanilla F (2000). The voltage sensor in voltage‐dependent ion channels. Physiol Rev 80, 555–592. [DOI] [PubMed] [Google Scholar]
- Brunner M, Peng X, Liu GX, Ren XQ, Ziv O, Choi BR, Mathur R, Hajjiri M, Odening KE, Steinberg E, Folco EJ, Pringa E, Centracchio J, Macharzina RR, Donahay T, Schofield L, Rana N, Kirk M, Mitchell GF, Poppas A, Zehender M & Koren G (2008). Mechanisms of cardiac arrhythmias and sudden death in transgenic rabbits with long QT syndrome. J Clin Invest 118, 2246–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrasekhar KD, Bas T & Kobertz WR (2006). KCNE1 subunits require co‐assembly with K+ channels for efficient trafficking and cell surface expression. J Biol Chem 281, 40015–40023. [DOI] [PubMed] [Google Scholar]
- Chang PC, Turker I, Lopshire JC, Masroor S, Nguyen BL, Tao W, Rubart M, Chen PS, Chen Z & Ai T (2013). Heterogeneous upregulation of apamin‐sensitive potassium currents in failing human ventricles. J Am Heart Assoc 2, e004713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua SK, Chang PC, Maruyama M, Turker I, Shinohara T, Shen MJ, Chen Z, Shen C, Rubart‐von der Lohe M, Lopshire JC, Ogawa M, Weiss JN, Lin SF, Ai T & Chen PS (2011). Small‐conductance calcium‐activated potassium channel and recurrent ventricular fibrillation in failing rabbit ventricles. Circ Res 108, 971–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colinas O, Gallego M, Setien R, Lopez‐Lopez JR, Perez‐Garcia MT & Casis O (2006). Differential modulation of Kv4.2 and Kv4.3 channels by calmodulin‐dependent protein kinase II in rat cardiac myocytes. Am J Physiol Heart Circ Physiol 291, H1978–H1987. [DOI] [PubMed] [Google Scholar]
- Cooper PE, Sala‐Rabanal M, Lee SJ & Nichols CG (2015). Differential mechanisms of Cantu syndrome‐associated gain of function mutations in the ABCC9 (SUR2) subunit of the KATP channel. J Gen Physiol 146, 527–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantini DL, Arruda EP, Agarwal P, Kim KH, Zhu Y, Zhu W, Lebel M, Cheng CW, Park CY, Pierce SA, Guerchicoff A, Pollevick GD, Chan TY, Kabir MG, Cheng SH, Husain M, Antzelevitch C, Srivastava D, Gross GJ, Hui CC, Backx PH & Bruneau BG (2005). The homeodomain transcription factor Irx5 establishes the mouse cardiac ventricular repolarization gradient. Cell 123, 347–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crump SM & Abbott GW (2014). Arrhythmogenic KCNE gene variants: current knowledge and future challenges. Front Genet 5, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuello LG, Cortes DM & Perozo E (2004). Molecular architecture of the KvAP voltage‐dependent K+ channel in a lipid bilayer. Science 306, 491–495. [DOI] [PubMed] [Google Scholar]
- Cummins MA, Dalal PJ, Bugana M, Severi S & Sobie EA (2014). Comprehensive analyses of ventricular myocyte models identify targets exhibiting favorable rate dependence. PLoS Comput Biol 10, e1003543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deschenes I & Tomaselli GF (2002). Modulation of Kv4.3 current by accessory subunits. FEBS Lett 528, 183–188. [DOI] [PubMed] [Google Scholar]
- Donaldson MR, Jensen JL, Tristani‐Firouzi M, Tawil R, Bendahhou S, Suarez WA, Cobo AM, Poza JJ, Behr E, Wagstaff J, Szepetowski P, Pereira S, Mozaffar T, Escolar DM, Fu YH & Ptacek LJ (2003). PIP2 binding residues of Kir2.1 are common targets of mutations causing Andersen syndrome. Neurology 60, 1811–1816. [DOI] [PubMed] [Google Scholar]
- Dror RO, Dirks RM, Grossman JP, Xu H & Shaw DE (2012). Biomolecular simulation: a computational microscope for molecular biology. Annu Rev Biophys 41, 429–452. [DOI] [PubMed] [Google Scholar]
- El‐Haou S, Balse E, Neyroud N, Dilanian G, Gavillet B, Abriel H, Coulombe A, Jeromin A & Hatem SN (2009). Kv4 potassium channels form a tripartite complex with the anchoring protein SAP97 and CaMKII in cardiac myocytes. Circ Res 104, 758–769. [DOI] [PubMed] [Google Scholar]
- Es‐Salah‐Lamoureux Z, Fougere R, Xiong PY, Robertson GA & Fedida D (2010). Fluorescence‐tracking of activation gating in human ERG channels reveals rapid S4 movement and slow pore opening. PLoS One 5, e10876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Z & Makielski JC (1997). Anionic phospholipids activate ATP‐sensitive potassium channels. J Biol Chem 272, 5388–5395. [DOI] [PubMed] [Google Scholar]
- Ficker E, Obejero‐Paz CA, Zhao S & Brown AM (2002). The binding site for channel blockers that rescue misprocessed human long QT syndrome type 2 ether‐a‐gogo‐related gene (HERG) mutations. J Biol Chem 277, 4989–4998. [DOI] [PubMed] [Google Scholar]
- Foster DB, Ho AS, Rucker J, Garlid AO, Chen L, Sidor A, Garlid KD & O'Rourke B (2012). Mitochondrial ROMK channel is a molecular component of mitoKATP . Circ Res 111, 446–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardoni F, Mauceri D, Marcello E, Sala C, Di Luca M & Jeromin A (2007). SAP97 directs the localization of Kv4.2 to spines in hippocampal neurons: regulation by CaMKII. J Biol Chem 282, 28691–28699. [DOI] [PubMed] [Google Scholar]
- Garlid KD & Halestrap AP (2012). The mitochondrial KATP channel–fact or fiction? J Mol Cell Cardiol 52, 578–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Nunziato DA & Pitt GS (2006). KCNQ1 assembly and function is blocked by long‐QT syndrome mutations that disrupt interaction with calmodulin. Circ Res 98, 1048–1054. [DOI] [PubMed] [Google Scholar]
- Girmatsion Z, Biliczki P, Bonauer A, Wimmer‐Greinecker G, Scherer M, Moritz A, Bukowska A, Goette A, Nattel S, Hohnloser SH & Ehrlich JR (2009). Changes in microRNA‐1 expression and IK1 up‐regulation in human atrial fibrillation. Heart Rhythm 6, 1802–1809. [DOI] [PubMed] [Google Scholar]
- Golowasch J, Goldman MS, Abbott LF & Marder E (2002). Failure of averaging in the construction of a conductance‐based neuron model. J Neurophysiol 87, 1129–1131. [DOI] [PubMed] [Google Scholar]
- Greco CM & Condorelli G (2015). Epigenetic modifications and noncoding RNAs in cardiac hypertrophy and failure. Nat Rev Cardiol 12, 488–497. [DOI] [PubMed] [Google Scholar]
- Groenendaal W, Ortega FA, Kherlopian AR, Zygmunt AC, Krogh‐Madsen T & Christini DJ (2015). Cell‐specific cardiac electrophysiology models. PLoS Comput Biol 11, e1004242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta K, Zamanian M, Bae C, Milescu M, Krepkiy D, Tilley DC, Sack JT, Vladimir YY, Kim JI & Swartz KJ (2015). Tarantula toxins use common surfaces for interacting with Kv and ASIC ion channels. Elife 4, e06774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hai T, Teng F, Guo R, Li W & Zhou Q (2014). One‐step generation of knockout pigs by zygote injection of CRISPR/Cas system. Cell Res 24, 372–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heijman J, Algalarrondo V, Voigt N, Melka J, Wehrens XH, Dobrev D & Nattel S (2016). The value of basic research insights into atrial fibrillation mechanisms as a guide to therapeutic innovation: a critical analysis. Cardiovasc Res 109, 467–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgemann DW & Ball R (1996). Regulation of cardiac Na+,Ca2+ exchange and KATP potassium channels by PIP2 . Science 273, 956–959. [DOI] [PubMed] [Google Scholar]
- Hite RK, Yuan P, Li Z, Hsuing Y, Walz T & MacKinnon R (2015). Cryo‐electron microscopy structure of the Slo2.2 Na+‐activated K+ channel. Nature 527, 198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin AL & Huxley AF (1952). A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol 117, 500–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolfsson HI, Arnarez C, Periole X & Marrink SJ (2016). Computational ‘microscopy’ of cellular membranes. J Cell Sci 129, 257–268. [DOI] [PubMed] [Google Scholar]
- Ingolfsson HI, Melo MN, van Eerden FJ, Arnarez C, Lopez CA, Wassenaar TA, Periole X, de Vries AH, Tieleman DP & Marrink SJ (2014). Lipid organization of the plasma membrane. J Am Chem Soc 136, 14554–14559. [DOI] [PubMed] [Google Scholar]
- Inoue I, Nagase H, Kishi K & Higuti T (1991). ATP‐sensitive K+ channel in the mitochondrial inner membrane. Nature 352, 244–247. [DOI] [PubMed] [Google Scholar]
- Jensen MO, Jogini V, Borhani DW, Leffler AE, Dror RO & Shaw DE (2012). Mechanism of voltage gating in potassium channels. Science 336, 229–233. [DOI] [PubMed] [Google Scholar]
- Jost N, Virag L, Comtois P, Ordog B, Szuts V, Seprenyi G, Bitay M, Kohajda Z, Koncz I, Nagy N, Szel T, Magyar J, Kovacs M, Puskas LG, Lengyel C, Wettwer E, Ravens U, Nanasi PP, Papp JG, Varro A & Nattel S (2013). Ionic mechanisms limiting cardiac repolarization reserve in humans compared to dogs. J Physiol 591, 4189–4206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaese S & Verheule S (2012). Cardiac electrophysiology in mice: a matter of size. Front Physiol 3, 345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko EA, Han J, Jung ID & Park WS (2008). Physiological roles of K+ channels in vascular smooth muscle cells. J Smooth Muscle Res 44, 65–81. [DOI] [PubMed] [Google Scholar]
- Kowal J, Chami M, Baumgartner P, Arheit M, Chiu PL, Rangl M, Scheuring S, Schroder GF, Nimigean CM & Stahlberg H (2014). Ligand‐induced structural changes in the cyclic nucleotide‐modulated potassium channel MloK1. Nat Commun 5, 3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh‐Madsen T, Sobie EA & Christini DJ (2016). Improving cardiomyocyte model fidelity and utility via dynamic electrophysiology protocols and optimization algorithms. J Physiol 594, 2525–2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krumerman A, Gao X, Bian JS, Melman YF, Kagan A & McDonald TV (2004). An LQT mutant minK alters KvLQT1 trafficking. Am J Physiol Cell Physiol 286, C1453–C1463. [DOI] [PubMed] [Google Scholar]
- Lacroix JJ, Hyde HC, Campos FV & Bezanilla F (2014). Moving gating charges through the gating pore in a Kv channel voltage sensor. Proc Natl Acad Sci USA 111, E1950–E1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langeberg LK & Scott JD (2015). Signalling scaffolds and local organization of cellular behaviour. Nat Rev Mol Cell Biol 16, 232–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lederer WJ, Nichols CG & Smith GL (1989). The mechanism of early contractile failure of isolated rat ventricular myocytes subjected to complete metabolic inhibition. J Physiol 413, 329–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, Wang S, Borschel W, Heyman S, Gyore J & Nichols CG (2013). Secondary anionic phospholipid binding site and gating mechanism in Kir2.1 inward rectifier channels. Nat Commun 4, 2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W, Mann SA, Windley MJ, Imtiaz MS, Vandenberg JI & Hill AP (2016). In silico assessment of kinetics and state dependent binding properties of drugs causing acquired LQTS. Prog Biophys Mol Biol 120, 89–99. [DOI] [PubMed] [Google Scholar]
- Lees‐Miller JP, Subbotina JO, Guo J, Yarov‐Yarovoy V, Noskov SY & Duff HJ (2009). Interactions of H562 in the S5 helix with T618 and S621 in the pore helix are important determinants of hERG1 potassium channel structure and function. Biophys J 96, 3600–3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin MD, Zhang H, Uchida K, Grange DK, Singh GK & Nichols CG (2015). Electrophysiologic consequences of KATP gain of function in the heart: Conduction abnormalities in Cantu syndrome. Heart Rhythm 12, 2316–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Gao J, Lu Z, McFarland K, Shi J, Bock K, Cohen IS & Cui J (2013). Intracellular ATP binding is required to activate the slowly activating K+ channel IKs . Proc Natl Acad Sci USA 110, 18922–18927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao M, Cao E, Julius D & Cheng Y (2013). Structure of the TRPV1 ion channel determined by electron cryo‐microscopy. Nature 504, 107–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liin SI, Silvera Ejneby M, Barro‐Soria R, Skarsfeldt MA, Larsson JE, Starck Harlin F, Parkkari T, Bentzen BH, Schmitt N, Larsson HP & Elinder F (2015). Polyunsaturated fatty acid analogs act antiarrhythmically on the cardiac IKs channel. Proc Natl Acad Sci USA 112, 5714–5719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl E & Sansom MS (2008). Membrane proteins: molecular dynamics simulations. Curr Opin Struct Biol 18, 425–431. [DOI] [PubMed] [Google Scholar]
- Liu F, Jones DK, de Lange WJ & Robertson GA (2016). Cotranslational association of mRNA encoding subunits of heteromeric ion channels. Proc Natl Acad Sci USA 113, 4859–4864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S & Rasmusson RL (1997). Hodgkin‐Huxley and partially coupled inactivation models yield different voltage dependence of block. Am J Physiol Heart Circ Physiol 272, H2013–H2022. [DOI] [PubMed] [Google Scholar]
- Long SB, Campbell EB & Mackinnon R (2005. a). Crystal structure of a mammalian voltage‐dependent Shaker family K+ channel. Science 309, 897–903. [DOI] [PubMed] [Google Scholar]
- Long SB, Campbell EB & Mackinnon R (2005. b). Voltage sensor of Kv1.2: structural basis of electromechanical coupling. Science 309, 903–908. [DOI] [PubMed] [Google Scholar]
- Long SB, Tao X, Campbell EB & MacKinnon R (2007). Atomic structure of a voltage‐dependent K+ channel in a lipid membrane‐like environment. Nature 450, 376–382. [DOI] [PubMed] [Google Scholar]
- Lorinczi E, Gomez‐Posada JC, de la Pena P, Tomczak AP, Fernandez‐Trillo J, Leipscher U, Stuhmer W, Barros F & Pardo LA (2015). Voltage‐dependent gating of KCNH potassium channels lacking a covalent link between voltage‐sensing and pore domains. Nat Commun 6, 6672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L, Timofeyev V, Li N, Rafizadeh S, Singapuri A, Harris TR & Chiamvimonvat N (2009). α‐Actinin2 cytoskeletal protein is required for the functional membrane localization of a Ca2+‐activated K+ channel (SK2 channel). Proc Natl Acad Sci USA 106, 18402–18407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L, Zhang Q, Timofeyev V, Zhang Z, Young JN, Shin HS, Knowlton AA & Chiamvimonvat N (2007). Molecular coupling of a Ca2+‐activated K+ channel to L‐type Ca2+ channels via α‐actinin2. Circ Res 100, 112–120. [DOI] [PubMed] [Google Scholar]
- Lundy SD, Zhu WZ, Regnier M & Laflamme MA (2013). Structural and functional maturation of cardiomyocytes derived from human pluripotent stem cells. Stem Cells Dev 22, 1991–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean JN, Zhang Y, Johnson BR & Harris‐Warrick RM (2003). Activity‐independent homeostasis in rhythmically active neurons. Neuron 37, 109–120. [DOI] [PubMed] [Google Scholar]
- Mannuzzu LM, Moronne MM & Isacoff EY (1996). Direct physical measure of conformational rearrangement underlying potassium channel gating. Science 271, 213–216. [DOI] [PubMed] [Google Scholar]
- Marionneau C, Brunet S, Flagg TP, Pilgram TK, Demolombe S & Nerbonne JM (2008). Distinct cellular and molecular mechanisms underlie functional remodeling of repolarizing K+ currents with left ventricular hypertrophy. Circ Res 102, 1406–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrion NV & Tavalin SJ (1998). Selective activation of Ca2+‐activated K+ channels by co‐localized Ca2+ channels in hippocampal neurons. Nature 395, 900–905. [DOI] [PubMed] [Google Scholar]
- Marx SO, Kurokawa J, Reiken S, Motoike H, D'Armiento J, Marks AR & Kass RS (2002). Requirement of a macromolecular signaling complex for β adrenergic receptor modulation of the KCNQ1‐KCNE1 potassium channel. Science 295, 496–499. [DOI] [PubMed] [Google Scholar]
- Matkovich SJ, Wang W, Tu Y, Eschenbacher WH, Dorn LE, Condorelli G, Diwan A, Nerbonne JM & Dorn GW 2nd (2010). MicroRNA‐133a protects against myocardial fibrosis and modulates electrical repolarization without affecting hypertrophy in pressure‐overloaded adult hearts. Circ Res 106, 166–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medovoy D, Perozo E & Roux B (2016). Multi‐ion free energy landscapes underscore the microscopic mechanism of ion selectivity in the KcsA channel. Biochim Biophys Acta 1858, 1722–1732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miake J, Marban E & Nuss HB (2002). Biological pacemaker created by gene transfer. Nature 419, 132–133. [DOI] [PubMed] [Google Scholar]
- Molina CE, Heijman J & Dobrev D (2016). Differences in left versus right ventricular electrophysiological properties in cardiac dysfunction and arrhythmogenesis. Arrhythm Electrophysiol Rev 5, 14–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno JD, Zhu ZI, Yang PC, Bankston JR, Jeng MT, Kang C, Wang L, Bayer JD, Christini DJ, Trayanova NA, Ripplinger CM, Kass RS & Clancy CE (2011). A computational model to predict the effects of class I anti‐arrhythmic drugs on ventricular rhythms. Sci Transl Med 3, 98ra83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morotti S, McCulloch AD, Bers DM, Edwards AG & Grandi E (2016). Atrial‐selective targeting of arrhythmogenic phase‐3 early afterdepolarizations in human myocytes. J Mol Cell Cardiol 96, 63–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray CI, Westhoff M, Eldstrom J, Thompson E, Emes R & Fedida D (2016). Unnatural amino acid photo‐crosslinking of the IKs channel complex demonstrates a KCNE1:KCNQ1 stoichiometry of up to 4:4. Elife 5, e11815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustroph J, Maier LS & Wagner S (2014). CaMKII regulation of cardiac K channels. Front Pharmacol 5, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajo K, Ulbrich MH, Kubo Y & Isacoff EY (2010). Stoichiometry of the KCNQ1 ‐ KCNE1 ion channel complex. Proc Natl Acad Sci USA 107, 18862–18867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nass RD, Aiba T, Tomaselli GF & Akar FG (2008). Mechanisms of disease: ion channel remodeling in the failing ventricle. Nat Clin Pract Cardiovasc Med 5, 196–207. [DOI] [PubMed] [Google Scholar]
- Nerbonne JM (2011). Repolarizing cardiac potassium channels: multiple sites and mechanisms for CaMKII‐mediated regulation. Heart Rhythm 8, 938–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nerbonne JM & Guo W (2002). Heterogeneous expression of voltage‐gated potassium channels in the heart: roles in normal excitation and arrhythmias. J Cardiovasc Electrophysiol 13, 406–409. [DOI] [PubMed] [Google Scholar]
- Nerbonne JM & Kass RS (2005). Molecular physiology of cardiac repolarization. Physiol Rev 85, 1205–1253. [DOI] [PubMed] [Google Scholar]
- Ni W, Qiao J, Hu S, Zhao X, Regouski M, Yang M, Polejaeva IA & Chen C (2014). Efficient gene knockout in goats using CRISPR/Cas9 system. PLoS One 9, e106718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niederer SA, Fink M, Noble D & Smith NP (2009). A meta‐analysis of cardiac electrophysiology computational models. Exp Physiol 94, 486–495. [DOI] [PubMed] [Google Scholar]
- Nieves‐Cintron M, Hirenallur‐Shanthappa D, Nygren PJ, Hinke SA, Dell'Acqua ML, Langeberg LK, Navedo M, Santana LF & Scott JD (2016). AKAP150 participates in calcineurin/NFAT activation during the down‐regulation of voltage‐gated K currents in ventricular myocytes following myocardial infarction. Cell Signal 28, 733–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa N & Nerbonne JM (2010). Molecular determinants of cardiac transient outward potassium current (Ito) expression and regulation. J Mol Cell Cardiol 48, 12–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda M, Shimizu S, Tanabe T, Takai T, Kayano T, Ikeda T, Takahashi H, Nakayama H, Kanaoka Y, Minamino N et al (1984). Primary structure of Electrophorus electricus sodium channel deduced from cDNA sequence. Nature 312, 121–127. [DOI] [PubMed] [Google Scholar]
- O'Hara T & Rudy Y (2012). Quantitative comparison of cardiac ventricular myocyte electrophysiology and response to drugs in human and nonhuman species. Am J Physiol Heart Circ Physiol 302, H1023–H1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osteen JD, Gonzalez C, Sampson KJ, Iyer V, Rebolledo S, Larsson HP & Kass RS (2010). KCNE1 alters the voltage sensor movements necessary to open the KCNQ1 channel gate. Proc Natl Acad Sci USA 107, 22710–22715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostmeyer J, Chakrapani S, Pan AC, Perozo E & Roux B (2013). Recovery from slow inactivation in K+ channels is controlled by water molecules. Nature 501, 121–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papazian DM, Schwarz TL, Tempel BL, Jan YN & Jan LY (1987). Cloning of genomic and complementary DNA from Shaker, a putative potassium channel gene from Drosophila . Science 237, 749–753. [DOI] [PubMed] [Google Scholar]
- Pathak MM, Yarov‐Yarovoy V, Agarwal G, Roux B, Barth P, Kohout S, Tombola F & Isacoff EY (2007). Closing in on the resting state of the shaker K+ channel. Neuron 56, 124–140. [DOI] [PubMed] [Google Scholar]
- Perissinotti LL, Guo J, De Biase PM, Clancy CE, Duff HJ & Noskov SY (2015). Kinetic model for NS1643 drug activation of WT and L529I variants of Kv11.1 (hERG1) potassium channel. Biophys J 108, 1414–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philip‐Couderc P, Tavares NI, Roatti A, Lerch R, Montessuit C & Baertschi AJ (2008). Forkhead transcription factors coordinate expression of myocardial KATP channel subunits and energy metabolism. Circ Res 102, e20–e35. [DOI] [PubMed] [Google Scholar]
- Plant LD, Xiong D, Dai H & Goldstein SA (2014). Individual IKs channels at the surface of mammalian cells contain two KCNE1 accessory subunits. Proc Natl Acad Sci USA 111, E1438–E1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priori SG, Napolitano C & Schwartz PJ (1999). Low penetrance in the long‐QT syndrome: clinical impact. Circulation 99, 529–533. [DOI] [PubMed] [Google Scholar]
- Rafizadeh S, Zhang Z, Woltz RL, Kim HJ, Myers RE, Lu L, Tuteja D, Singapuri A, Bigdeli AA, Harchache SB, Knowlton AA, Yarov‐Yarovoy V, Yamoah EN & Chiamvimonvat N (2014). Functional interaction with filamin A and intracellular Ca2+ enhance the surface membrane expression of a small‐conductance Ca2+‐activated K+ (SK2) channel. Proc Natl Acad Sci USA 111, 9989–9994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raval A, Piana S, Eastwood MP & Shaw DE (2016). Assessment of the utility of contact‐based restraints in accelerating the prediction of protein structure using molecular dynamics simulations. Protein Sci 25, 19–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reggio BC, James AN, Green HL, Gavin WG, Behboodi E, Echelard Y & Godke RA (2001). Cloned transgenic offspring resulting from somatic cell nuclear transfer in the goat: oocytes derived from both follicle‐stimulating hormone‐stimulated and nonstimulated abattoir‐derived ovaries. Biol Reprod 65, 1528–1533. [DOI] [PubMed] [Google Scholar]
- Roeper J, Lorra C & Pongs O (1997). Frequency‐dependent inactivation of mammalian A‐type K+ channel KV1.4 regulated by Ca2+/calmodulin‐dependent protein kinase. J Neurosci 17, 3379–3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanenko VG, Fang Y, Byfield F, Travis AJ, Vandenberg CA, Rothblat GH & Levitan I (2004). Cholesterol sensitivity and lipid raft targeting of Kir2.1 channels. Biophys J 87, 3850–3861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosati B, Pan Z, Lypen S, Wang HS, Cohen I, Dixon JE & McKinnon D (2001). Regulation of KChIP2 potassium channel β subunit gene expression underlies the gradient of transient outward current in canine and human ventricle. J Physiol 533, 119–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenhouse‐Dantsker A, Noskov S, Durdagi S, Logothetis DE & Levitan I (2013). Identification of novel cholesterol‐binding regions in Kir2 channels. J Biol Chem 288, 31154–31164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossow CF, Dilly KW & Santana LF (2006). Differential calcineurin/NFATc3 activity contributes to the Ito transmural gradient in the mouse heart. Circ Res 98, 1306–1313. [DOI] [PubMed] [Google Scholar]
- Rossow CF, Dilly KW, Yuan C, Nieves‐Cintron M, Cabarrus JL & Santana LF (2009). NFATc3‐dependent loss of Ito gradient across the left ventricular wall during chronic β adrenergic stimulation. J Mol Cell Cardiol 46, 249–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossow CF, Minami E, Chase EG, Murry CE & Santana LF (2004). NFATc3‐induced reductions in voltage‐gated K+ currents after myocardial infarction. Circ Res 94, 1340–1350. [DOI] [PubMed] [Google Scholar]
- Rudy Y & Silva JR (2006). Computational biology in the study of cardiac ion channels and cell electrophysiology. Q Rev Biophys 39, 57–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL & Keating MT (1996). Coassembly of KVLQT1 and minK (IsK) proteins to form cardiac IKs potassium channel. Nature 384, 80–83. [DOI] [PubMed] [Google Scholar]
- Sarkar AX & Sobie EA (2010). Regression analysis for constraining free parameters in electrophysiological models of cardiac cells. PLoS Comput Biol 6, e1000914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt N, Grunnet M & Olesen SP (2014). Cardiac potassium channel subtypes: new roles in repolarization and arrhythmia. Physiol Rev 94, 609–653. [DOI] [PubMed] [Google Scholar]
- Semenova NP, Abarca‐Heidemann K, Loranc E & Rothberg BS (2009). Bimane fluorescence scanning suggests secondary structure near the S3‐S4 linker of BK channels. J Biol Chem 284, 10684–10693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergeant GP, Ohya S, Reihill JA, Perrino BA, Amberg GC, Imaizumi Y, Horowitz B, Sanders KM & Koh SD (2005). Regulation of Kv4.3 currents by Ca2+/calmodulin‐dependent protein kinase II. Am J Physiol Cell Physiol 288, C304–C313. [DOI] [PubMed] [Google Scholar]
- Singh H, Stefani E & Toro L (2012). Intracellular BKCa (iBKCa) channels. J Physiol 590, 5937–5947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JL, McBride CM, Nataraj PS, Bartos DC, January CT & Delisle BP (2011). Trafficking‐deficient hERG K+ channels linked to long QT syndrome are regulated by a microtubule‐dependent quality control compartment in the ER. Am J Physiol Cell Physiol 301, C75–C85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JL, Reloj AR, Nataraj PS, Bartos DC, Schroder EA, Moss AJ, Ohno S, Horie M, Anderson CL, January CT & Delisle BP (2013). Pharmacological correction of long QT‐linked mutations in KCNH2 (hERG) increases the trafficking of Kv11.1 channels stored in the transitional endoplasmic reticulum. Am J Physiol Cell Physiol 305, C919–C930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spray DC, Stern JH, Harris AL & Bennett MV (1982). Gap junctional conductance: comparison of sensitivities to H and Ca ions. Proc Natl Acad Sci USA 79, 441–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starace DM & Bezanilla F (2001). Histidine scanning mutagenesis of basic residues of the S4 segment of the shaker K+ channel. J Gen Physiol 117, 469–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbotina J, Yarov‐Yarovoy V, Lees‐Miller J, Durdagi S, Guo J, Duff HJ & Noskov SY (2010). Structural refinement of the hERG1 pore and voltage‐sensing domains with ROSETTA‐membrane and molecular dynamics simulations. Proteins 78, 2922–2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeshima H, Venturi E & Sitsapesan R (2015). New and notable ion‐channels in the sarcoplasmic/endoplasmic reticulum: do they support the process of intracellular Ca2+ release? J Physiol 593, 3241–3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao L, Bei Y, Zhou Y, Xiao J & Li X (2015). Non‐coding RNAs in cardiac regeneration. Oncotarget 6, 42613–42622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terentyev D, Rochira JA, Terentyeva R, Roder K, Koren G & Li W (2014). Sarcoplasmic reticulum Ca2+ release is both necessary and sufficient for SK channel activation in ventricular myocytes. Am J Physiol Heart Circ Physiol 306, H738–H746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tohse N (1990). Calcium‐sensitive delayed rectifier potassium current in guinea pig ventricular cells. Am J Physiol Heart Circ Physiol 258, H1200–H1207. [DOI] [PubMed] [Google Scholar]
- Vaid M, Horne A, Claydon T & Fedida D (2009). Rapid outer pore movements after opening in a KV1 potassium channel are revealed by TMRM fluorescence from the S3‐S4 linker, and modulated by extracellular potassium. Channels (Austin) 3, 3–5. [DOI] [PubMed] [Google Scholar]
- Van Wagoner DR, Pond AL, McCarthy PM, Trimmer JS & Nerbonne JM (1997). Outward K+ current densities and Kv1.5 expression are reduced in chronic human atrial fibrillation. Circ Res 80, 772–781. [DOI] [PubMed] [Google Scholar]
- Varga AW, Yuan LL, Anderson AE, Schrader LA, Wu GY, Gatchel JR, Johnston D & Sweatt JD (2004). Calcium‐calmodulin‐dependent kinase II modulates Kv4.2 channel expression and upregulates neuronal A‐type potassium currents. J Neurosci 24, 3643–3654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner S, Hacker E, Grandi E, Weber SL, Dybkova N, Sossalla S, Sowa T, Fabritz L, Kirchhof P, Bers DM & Maier LS (2009). Ca/calmodulin kinase II differentially modulates potassium currents. Circ Arrhythm Electrophysiol 2, 285–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang DT, Hill AP, Mann SA, Tan PS & Vandenberg JI (2011). Mapping the sequence of conformational changes underlying selectivity filter gating in the Kv11.1 potassium channel. Nat Struct Mol Biol 18, 35–41. [DOI] [PubMed] [Google Scholar]
- Wang Y, Zankov DP, Jiang M, Zhang M, Henderson SC & Tseng GN (2013). [Ca2+]i elevation and oxidative stress induce KCNQ1 protein translocation from the cytosol to the cell surface and increase slow delayed rectifier (IKs) in cardiac myocytes. J Biol Chem 288, 35358–35371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss JN, Karma A, MacLellan WR, Deng M, Rau CD, Rees CM, Wang J, Wisniewski N, Eskin E, Horvath S, Qu Z, Wang Y & Lusis AJ (2012). “Good enough solutions” and the genetics of complex diseases. Circ Res 111, 493–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werry D, Eldstrom J, Wang Z & Fedida D (2013). Single‐channel basis for the slow activation of the repolarizing cardiac potassium current, IKs . Proc Natl Acad Sci USA 110, E996–E1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang KC, Foeger NC, Marionneau C, Jay PY, McMullen JR & Nerbonne JM (2010). Homeostatic regulation of electrical excitability in physiological cardiac hypertrophy. J Physiol 588, 5015–5032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang KC & Nerbonne JM (2016). Mechanisms contributing to myocardial potassium channel diversity, regulation and remodeling. Trends Cardiovasc Med 26, 209–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang KC, Tseng YT & Nerbonne JM (2012). Exercise training and PI3Kα‐induced electrical remodeling is independent of cellular hypertrophy and Akt signaling. J Mol Cell Cardiol 53, 532–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarov‐Yarovoy V, Baker D & Catterall WA (2006). Voltage sensor conformations in the open and closed states in ROSETTA structural models of K+ channels. Proc Natl Acad Sci USA 103, 7292–7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarov‐Yarovoy V, Decaen PG, Westenbroek RE, Pan CY, Scheuer T, Baker D & Catterall WA (2012). Structural basis for gating charge movement in the voltage sensor of a sodium channel. Proc Natl Acad Sci USA 109, E93–E102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue L, Melnyk P, Gaspo R, Wang Z & Nattel S (1999). Molecular mechanisms underlying ionic remodeling in a dog model of atrial fibrillation. Circ Res 84, 776–784. [DOI] [PubMed] [Google Scholar]
- Yue L, Xie J & Nattel S (2011). Molecular determinants of cardiac fibroblast electrical function and therapeutic implications for atrial fibrillation. Cardiovasc Res 89, 744–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaydman MA, Kasimova MA, McFarland K, Beller Z, Hou P, Kinser HE, Liang H, Zhang G, Shi J, Tarek M & Cui J (2014). Domain‐domain interactions determine the gating, permeation, pharmacology, and subunit modulation of the IKs ion channel. Elife 3, e03606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaydman MA, Silva JR, Delaloye K, Li Y, Liang H, Larsson HP, Shi J & Cui J (2013). Kv7.1 ion channels require a lipid to couple voltage sensing to pore opening. Proc Natl Acad Sci USA 110, 13180–13185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Flagg TP & Nichols CG (2010). Cardiac sarcolemmal KATP channels: Latest twists in a questing tale! J Mol Cell Cardiol 48, 71–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, D'Aniello C, Verkerk AO, Wrobel E, Frank S, Ward‐van Oostwaard D, Piccini I, Freund C, Rao J, Seebohm G, Atsma DE, Schulze‐Bahr E, Mummery CL, Greber B & Bellin M (2014). Recessive cardiac phenotypes in induced pluripotent stem cell models of Jervell and Lange‐Nielsen syndrome: disease mechanisms and pharmacological rescue. Proc Natl Acad Sci USA 111, E5383–E5392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XD, Lieu DK & Chiamvimonvat N (2015). Small‐conductance Ca2+‐activated K+ channels and cardiac arrhythmias. Heart Rhythm 12, 1845–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Morais‐Cabral JH, Kaufman A & MacKinnon R (2001). Chemistry of ion coordination and hydration revealed by a K+ channel‐Fab complex at 2.0 Å resolution. Nature 414, 43–48. [DOI] [PubMed] [Google Scholar]
- Zhu R, Blazeski A, Poon E, Costa KD, Tung L & Boheler KR (2014). Physical developmental cues for the maturation of human pluripotent stem cell‐derived cardiomyocytes. Stem Cell Res Ther 5, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]