

Abstract
The gastrointestinal barrier is - with approximately 400 m2 - the human body’s largest surface separating the external environment from the internal milieu. This barrier serves a dual function: permitting the absorption of nutrients, water and electrolytes on the one hand, while limiting host contact with noxious luminal antigens on the other hand. To maintain this selective barrier, junction protein complexes seal the intercellular space between adjacent epithelial cells and regulate the paracellular transport. Increased intestinal permeability is associated with and suggested as a player in the pathophysiology of various gastrointestinal and extra-intestinal diseases such as inflammatory bowel disease, celiac disease and type 1 diabetes. The gastrointestinal tract is exposed to high levels of endogenous and exogenous proteases, both in the lumen and in the mucosa. There is increasing evidence to suggest that a dysregulation of the protease/antiprotease balance in the gut contributes to epithelial damage and increased permeability. Excessive proteolysis leads to direct cleavage of intercellular junction proteins, or to opening of the junction proteins via activation of protease activated receptors. In addition, proteases regulate the activity and availability of cytokines and growth factors, which are also known modulators of intestinal permeability. This review aims at outlining the mechanisms by which proteases alter the intestinal permeability. More knowledge on the role of proteases in mucosal homeostasis and gastrointestinal barrier function will definitely contribute to the identification of new therapeutic targets for permeability-related diseases.
Keywords: Intestinal permeability, Intestinal barrier, Tight junction, Paracellular permeability, Proteases, Proteinase-activated receptor, Protease inhibitor, Antiproteases
Core tip: Increased intestinal permeability is a novel player in the pathophysiology of various intestinal and extra-intestinal diseases such as inflammatory bowel disease, celiac disease and type 1 diabetes. A dysregulated protease/antiproteases balance is suggested as a cause of intestinal barrier dysfunction, with a subsequent increase in permeability. Immune cells infiltrating in the lamina propria during inflammatory conditions provide a pro-inflammatory environment by the production of cytokines and proteases. Protease inhibition has therapeutic potential but more research is needed to elucidate the exact involvement of specific proteases in gut physiology and intestinal barrier function.
INTRODUCTION
The intestinal barrier represents the largest interface between the external environment and the internal milieu. Given the enormous intraluminal load of essential and noxious molecules, a selectively permeable barrier is indispensable for maintaining mucosal homeostasis[1]. The intestinal barrier serves a dual function: on the one hand limiting host contact with pathogens and antigens and on the other hand at the same time allowing the absorption of nutrients and water. Physical, biochemical and immune elements make up the heterogeneous intestinal barrier and collaborate to exert these functions. Firstly, the mucus layer covers the entire epithelial cell surface and consists of gel-forming mucins, produced by the goblet cells. This chemical barrier also contains defensins or antimicrobial peptides that are secreted by Paneth cells within the epithelial cell layer[2]. Secondly, the epithelial cell layer itself is a physical barrier that consists for 80% of enterocytes, regulating nutrient absorption via specific transporters, channels and receptors (transcellular transport)[3]. Finally, the immunological barrier consists of microfold (M) cells in the epithelial cell layer and patrolling antigen presenting cells (APC) in the lamina propria. The M cells constantly sample luminal antigens and deliver them to APC such as dendritic cells and macrophages. Innocuous antigens drive the APCs to create a tolerogenic environment with the production of immunosuppressive factors such as IL-10, TGF-β and nitric oxide. Further tolerance is thereby created through the induction of regulatory T cells[4]. Noxious antigens are also recognized by the APCs and trigger the activation of the inflammatory cascade, starting with T cell activation[5,6].
The movement of molecules, solutes and ions across the intestinal epithelial cell layer can take place by the trans- or paracellular pathway. Transcellular transport is the main route for nutrient absorption and is facilitated through size- and charge- selective channels and transporters. The paracellular pathway is less selective since it occurs through the intercellular space between neighboring intestinal epithelial cells. The capacity of this paracellular pathway is however low as cells are bound tightly together by junction proteins, with particularly the tight junctions (TJ) regulating transport in response to numerous stimuli[7].
In the last decade, a barrier defect is suggested as a common factor in the onset of various local and systemic diseases of an inflammatory, autoimmune or functional nature such as inflammatory bowel disease (IBD), celiac disease, irritable bowel syndrome (IBS), type 1 diabetes mellitus and multiple sclerosis[8-13]. For more detailed information on the relation between intestinal barrier function and these disease pathologies, we refer the readers to Odenwald and Turner who nicely reviewed this topic in 2013[14] and very recently updated their overview in 2016[15]. Although the literature data are rather scarce, proteases are believed to regulate the intestinal permeability. They can intervene directly by their proteolytic action on the junction proteins, both intra- and extracellularly, and indirectly through activation of proteinase-activated receptors (PARs). This review provides an overview of the proteases (Table 1) putting emphasis on their role as regulators of the intestinal paracellular permeability.
Table 1.
Overview of proteases affecting intestinal permeability
| Protease | Effect | Model | Mechanism of action | Ref. |
| Serine proteases | ||||
| Matriptase | Protective | ST14 hypomorphic mice | Genetic depletion of matriptase induces an increase in intestinal permeability (decreased TER and increased FITC-dextran flux) | [65, 66] |
| Epithelial cell monolayer | Inhibition of matriptase with silencing RNA and the synthetic inhibitor MI-432 increased the intestinal permeability (decreased TER and increased FITC-dextran flux) | [65, 68] | ||
| Granzyme M | Protective | GrzM-/- mice | GrzM-/- mice display a permeability increase (FITC-dexran method) | [73] |
| Zonulin, Zonula occludens toxin (Zot) | Harmful | Human epithelial cell monolayer | ↑ Permeability after exposure to gliadin (triggers zonulin release; disruption of occludin and ZO-1) | [76] |
| Ileal tissue of diabetes prone rats | Zonulin-dependent permeability increase in diabetic rats was abolished after oral treatment with zonulin inhibitor FZI/0 (AT1001/Larazotide) | [82] | ||
| PAR2 activation | ||||
| Trypsin, tryptase, chymase, synthetic SLIGRL | Harmful | WT mice, WT rats | ↑ Permeability due to PAR2 activation (confirmed by selective PAR2 agonist SLIGRL; increased 51Cr-EDTA flux) | [47, 48, 51] |
| PAR4 activation | ||||
| Cathepsin G | Harmful | Colonic biopsies from UC and healthy patients | ↑ Permeability in response to UC fecal supernatant was abolished by cathepsin G inhibition | [58] |
| PAR1 activation | ||||
| Thrombin, synthetic TFLLR-NH2 | Harmful | WT mice, epithelial cell monolayer | ↑ Permeability after PAR1 activation (caspase-3 mediated; disruption of ZO-1) | [62] |
| Endogenous inhibitors | ||||
| Elafin | Protective | Gluten sensitive mice | ↓ Permeability after elafin delivery by recombinant Lactococcus lactis (51Cr-EDTA flux) | [87] |
| Human epithelial cell monolayer | Treatment with elafin normalized the TNF-α-induced increase in paracellular permeability (FITC-dextran method) | [88] | ||
| Synthetic inhibitors | ||||
| Camostat mesilate | Protective | Rat IBS model | Treatment with camostat mesilate normalized the elevated permeability in the rats (51Cr-EDTA flux and ZO-1 expression) | [89] |
| Nafamostat mesilate | Protective | Rectal biopsies from IBS and healthy patients | Nafamostat abolished the trypsin-induced hyperpermeability (macromolecular flux in Ussing chambers) | [94] |
| Human epithelial cell monolayer | Treatment with nafamostat normalized the tryptase-induced permeability increase (TER and FITC-dextran method) | [95] | ||
| SPI | Protective | IBD mouse model | Treatment with SPI normalized the increased permeability in the T-cell transfer colitis model (FITC-dextran method) | [96] |
| Metalloproteases | ||||
| Meprin β | Protective | Mep1b-/- mice | Meprin β cleaves MUC2 and alters mucus composition | [128, 129] |
| Matrix metalloproteinases | ||||
| MMP-2 | Protective | MMP-2-/- mice | ↑ permeability in MMP-2-/- mice (FITC-dextran method) | [111] |
| MMP-9 | Harmful | MMP-9-/- mice | = Permeability in MMP-9-/- mice after DSS (FITC-dextran method; no increase in MLCK expression) | [114] |
| MMP-9-/- mice | ↑ Goblet cells and MUC2 expression in MMP-9-/- mice | [113] | ||
| MMP-9 transgenic mice | ↑ Permeability in mice overexpressing MMP-9 (FITC-dextran method) | [112] | ||
| MMP-3, MMP-7 | Harmful | Epithelial cell culture | MMP-7 cleaves E-cadherin | [121] |
| ADAM | ||||
| TACE/ADAM17 | Harmful | Human and mouse colon samples | ↑ TACE activity in IBD; ↑ TNF-α release; ↑ TNF-α-induced permeability increase | [131, 134, 135] |
| Caco-2 | ↓ Permeability after TACE inhibition (by TAPI-2 and GM6001) | [136] | ||
| Cysteine proteases | ||||
| Caspase-3, caspase-8 | Harmful | Human epithelial cell monolayer | ↓ Cell-cell adhesion (epithelial cell apoptosis; disruption of TJ proteins occludin and claudin-4) | [144] |
| Endogenous inhibitor | ||||
| Cystatin | No effect | WT mice | No effect on colonic paracellular permeability (51Cr-EDTA flux) | [51] |
| Luminal proteases | ||||
| Bacteroides fragilis | ||||
| Fragilysin | Harmful | Human epithelial cell monolayer | ↑ Permeability (decreased TER and increase in mannitol flux) | [149, 150] |
| Entamoeba histolytica | ||||
| Cysteine protease | Harmful | Mice transfected with E. histolytica trophozoites | ↑ Permeability (FITC-dextran method) | [151] |
| Enterococcus faecalis | ||||
| Gelatinases | Harmful | IL10-/- mice | ↑ Permeability (E-cadherin splicing) | [156] |
| Epithelial cell monolayers | ↑ Permeability (PAR2 signaling) | [155] | ||
| Dermatophagoides pteronyssinus | ||||
| Der p 1 | Harmful | Human colonic biopsies | ↑ Permeability (decreased TER in Ussing chambers; disruption of TJ proteins occludin and ZO-1 | [158] |
| Kiwifruit cysteine protease | ||||
| Act d1 | Harmful | Epithelial cell monolayer | ↑ Permeability (disruption of TJ proteins occludin and ZO-1) | [162] |
| WT mice | ↑ Permeability (FITC-dextran method) | [161] | ||
| Aspergillus | ||||
| Amano SD | Protective | WT rat | Improved mucosal homeostasis through alteration of the microbiome composition and SCFA induction | [163] |
TJ: Tight junction; PARs: Proteinase-activated receptors; MLC: Myosin light chain; MLCK: Myosin light chain kinase; PKC: Protein kinase C; ROCK: Rho-associated protein kinase; ZO-1: Zonula occludens 1.
INTESTINAL TIGHT JUNCTIONS REGULATE PARACELLULAR PERMEABILITY
The intercellular spaces between neighboring intestinal epithelial cells are sealed by the apical junction complex, which contains TJs and adherens junctions, and by the subjacent desmosomes (Figure 1). The selective paracellular permeability is mediated by the TJs, which encircle the apical end of the intercellular spaces[7]. Various proteins make up TJs, including the adhesive transmembrane proteins occludin, claudins and junctional adhesion molecules as well as cytoplasmic proteins such as zonula occludens (ZO) proteins (Figure 2). The latter act as scaffolding proteins that connect the transmembrane proteins at their cytoplasmic C-terminal strands with F-actin, a filamentous cytoskeleton component[16]. The adherens junction transmembrane protein, epithelial-cadherin (E-cadherin), is connected to F-actin via intracellular proteins of the catenin-family[17].
Figure 1.
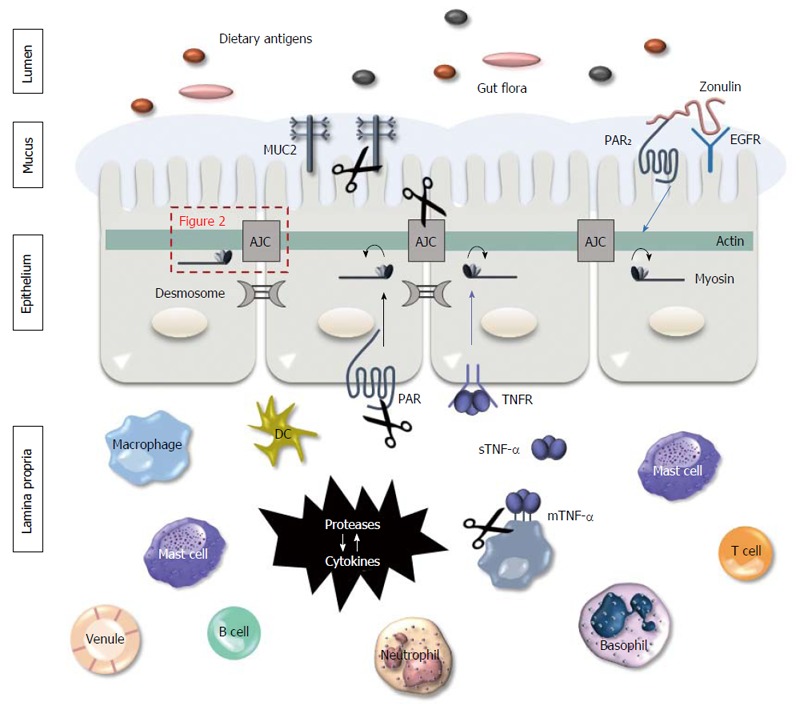
Proteases mediate gut barrier function. Intestinal epithelial cells are constantly exposed to proteases, both on their apical and basolateral side. Luminal proteases can be endogenous (e.g., pancreatic proteases) or can originate form bacteria or food particles present in the lumen. Their proteolytic activity can cause damage to the mucus layer and the junction proteins, affecting the barrier function. In the lamina propria, proteases are produced by various inflammatory cells and by the intestinal epithelial cells. In inflammatory conditions such as inflammatory bowel disease (IBD), immune cells infiltrate in the lamina propria where they produce various cytokines and proteases, contributing to the pro-inflammatory environment. Proteases stimulate immune cells to produce cytokines and vice versa. Besides, they alter the paracellular permeability by direct proteolytic cleaving of the junction proteins and by activation of the proteinase-activated receptors (PARs) on the epithelial cell surface, that induces a contraction of the actomyosin complex and subsequent opening of the apical junction complex (AJC; more in detail in Figure 2).
Figure 2.
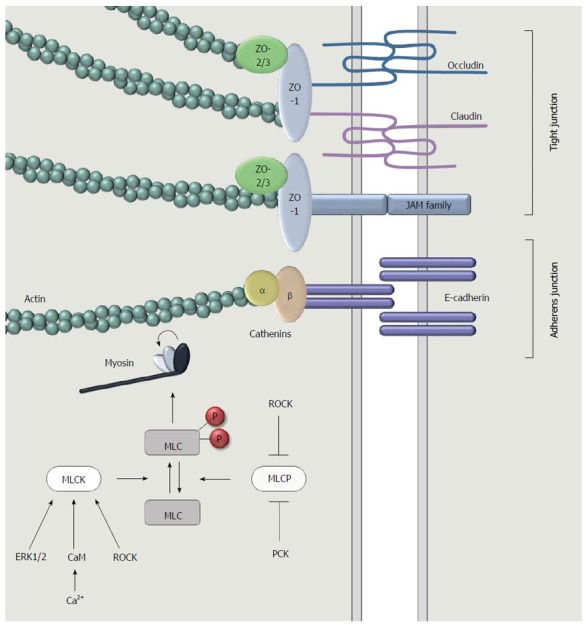
A more detailed representation of the apical junction complex at the intercellular surface between adjacent intestinal epithelial cells. Tight junctions are comprised of three types of transmembrane proteins: occludin, claudins and junctional adhesion molecules (JAMs). Adaptor proteins such as zonula occludens 1 (ZO-1), ZO-2 and ZO-3 connect the transmembrane proteins to filamentous actin. This cytoskeleton component interacts with myosin to induce a contraction, followed by the opening of the intercellular space. Myosin light chain (MLC) is the main regulator of this contractile machinery. Contraction occurs when MLC is phosphorylated. This is regulated through the activity of myosin light chain kinase (MLCK) and myosin light chain phosphatase (MLCP), which is on their turn regulated by intracellular signaling pathways involving for instance the extracellular signal-regulated kinases (ERK1/2), calcium, calmodulin, protein kinase C (PKC) or Rho-associated protein kinase (ROCK).
The opening of the intercellular spaces is achieved by contraction of the actomyosin microfilaments. Myosin is a motor protein that co-localizes with F-actin and converts chemical energy from adenosine triphosphate into mechanical energy. A crucial step in the induction of this mechanochemical contractile machinery is the phosphorylation of myosin light chain (MLC), the regulatory component of myosin (Figure 2). Myosin light chain kinase (MLCK) mediates the phosphorylation of MLC upon activation in response to Ca2+/calmodulin binding. However, there is evidence for other intracellular signaling pathways besides the calmodulin pathway to activate MLCK. The extracellular signal-regulated kinases (ERK1/2) have shown to induce MLCK activation[18]. Protein kinase C (PKC) on the other hand favors the phosphorylation of MLC by the inhibition of myosin light chain phosphatase (MLCP), the enzyme that dephosphorylates MLC[19]. Rho-associated protein kinase (ROCK) can increase contractility both by activating MLCK and inactivating MLCP, favoring MLC phosphorylation[20]. The phosphorylation status of myosin light chain induces a change in myosin tertiary structure causing myosin to “walk” along the actin filaments, increasing the tension in the cytoskeleton resulting in the disruption and cytosolic migration of TJ proteins[21,22]. This results in an impaired barrier function which is also referred to as a “leaky” barrier. Potentially noxious luminal proteins can now migrate to the underlying mucosal tissue and provoke a pro-inflammatory response. Even whole bacteria can cross the epithelial cells unrestricted at sites of epithelial damage caused by erosions and ulcers in GI disease. A “leaky” gut and epithelial damage often co-exist in disease state[14].
MUCOSAL IMMUNOLOGY AND BARRIER FUNCTION
In both physiological and pathological conditions, various mediators are able to affect the TJ conformation in order to control the paracellular permeability in epi- and endothelial cell layers throughout the body. Growth factors, cytokines, intestinal bacteria, dietary components and proteases are known to regulate the intestinal TJ opening[3,23-25]. Though the barrier-regulating capacity of pro-inflammatory cytokines is well studied, the effect of other mediators has received far less attention[24,26].
An inflamed mucosa -as seen in IBD patients- is characterized by the presence of cytokines amongst which TNF-α and IFN-γ, which are produced by a variety of cells including macrophages, T-cells and natural killer (NK) cells. The binding of these cytokines to specific receptors on the surface of infiltrating immune cells initiates a cascade of events starting with the activation of cell signaling pathways leading to the production of more inflammatory mediators (NFκB) or apoptosis maintaining on their turn the inflammatory process. Extensive reviews have been published on the regulation of TJs by cytokines[24,27]. It has been shown in cell culture experiments that TNF-α and IFN-γ regulate the paracellular permeability through the activation of MLCK, resulting in MLC hyperphosphorylation and opening of the TJs[28-30], while IL-4 and IL-13 increase paracellular permeability through the induction of the pore-forming claudin-2 and apoptotic pathways[31-33].
Next to cytokines also proteases are released into the mucosa by inflammatory cells such as macrophages, neutrophils and mast cells to regulate inflammation. On the one hand these proteases degrade the extracellular matrix, mucosal proteins and even live bacteria[34]. On the other hand, proteases act as signaling molecules via specific receptors, which will be discussed in the next section.
Intestinal epithelial cells also express receptors for cytokine and protease signaling. Since the apical and basolateral membranes of the intestinal epithelial cells are constantly exposed to large amounts of bacterial and endogenous proteases, the function of these proteolytic enzymes in intestinal barrier homeostasis should be further elucidated.
PROTEASES AND PROTEINASE-ACTIVATED RECEPTORS
Defined in general manner, proteases are enzymes that hydrolyze a peptide bond and in this respect they are best known for their digestive properties e.g., pancreatic proteases. However, also bacteria, epithelial cells, resident and infiltrating inflammatory cells produce luminal and mucosal proteases exerting various biological functions, both intra- and extracellularly. For instance, proteases are vital in inflammation, apoptosis, coagulation and cell growth and migration[35,36]. Since excessive proteolysis can cause tissue damage, a tight regulation of protease activity in order to prevent pathology is necessary. There are multiple mechanisms that control the protease activity such as the synthesis as inactive zymogens that require proteolytic cleaving for activation and on the other hand the termination of protease activity by endogenous inhibitors or antiproteases. A dysregulation in the protease balance with an increased protease activity has been observed in gastrointestinal diseases such as IBD and IBS, making protease inhibition by endogenous or synthetic inhibitors a potential therapeutic intervention[37]. Proteases are classified based on their mechanism of hydrolysis of the target peptide bond. This implicates that all proteases that belong to the same clan, share the same nucleophilic amino acid in their active site and are more likely to react with the same inhibitors. In mammals, five classes of proteases have been identified: serine-, metallo-, cysteine-, aspartate- and threonine proteases.
Proteases are not merely degrading enzymes. They can also act as signaling molecules by the proteolytic activation of the PARs. Four receptors have been identified in this family (PAR1, PAR2, PAR3, PAR4)[38]. Activation of these G-protein coupled receptors occurs after the proteolytic truncation of the N-terminal extracellular tail, releasing a new N-terminus that functions as a tethered ligand. This specific domain binds the second of three extracellular loops on the receptor and thereby generates an intracellular signal. PARs are expressed ubiquitously among tissues and cell types. In the gut, they are present on epithelial cells, endothelial cells, neurons, inflammatory cells, mast cells, smooth muscle cells and fibroblasts. Depending on the cell type, different signaling pathways have been described[39]. This also implicates that proteases mediate different GI physiological processes such as motility, cell proliferation and apoptosis, immune response, cytokine production, neurogenic inflammation, pain and epithelial barrier function through PAR activation[40]. For a more detailed description of this topic, we refer the reader to the companion review in (WJG Dec. 2016) from our group, illustrating this topic in detail[41].
SERINE PROTEASES
Of all proteolytic enzymes, serine proteases are by far the most abundant group[42,43]. Their successful mechanism of hydrolysis of peptide bonds occurs throughout the entire body in functionally diverse processes including digestion, immune response, blood coagulation, fibrinolysis, apoptosis and pro-hormone processing[42]. Serine proteases act both directly and indirectly as paracrine signaling effectors on PARs, provoking intracellular signals in order to mediate these vital processes. During the inflammatory cascade for example, proteases are released by infiltrating inflammatory cells and modulate the bioactivity of cytokines and chemokines by proteolytic cleavage[37]. For instance, the N-terminal truncation of CXCL-8 and CXCL-5 respectively by proteinase 3 and cathepsin G provides an increased chemotactic activity towards neutrophils[44,45]. Most proteases involved in PAR activation on the other hand belong to the serine clan of proteases[8]. In inflammatory cells, the activation of the G-protein coupled pathway leads to downstream activation and nuclear translocation of NFκB. In response, the cell synthesizes pro-inflammatory cytokines boosting inflammation[46]. In epithelial cells, paracrine signaling of proteases through PARs induces changes in paracellular permeability which will be discussed in the next paragraph.
The majority of the research in the field of intestinal permeability involves PAR2. In 2002, it was shown for the first time by Coelho et al[47] and Cenac et al[48] that PAR2 activation by the serine proteases trypsin, tryptase and chymase induced an increase in the colonic permeability of Cr51-EDTA, a marker of paracellular permeability[49,50]. The selective synthetic PAR2 agonist SLIGRL (H-serine-leucine-isoleucine-glycine-arginine-leucine-OH) mimicked the effect of endogenous serine proteases, thereby increasing intestinal permeability[47,51]. Although PAR2 receptors are expressed on both apical and basolateral membranes of epithelial cells, some authors suggest that only apical administration of PAR2 agonists - and not intraperitoneal (i.p.) administration- alters the intestinal barrier function[51]. Other authors however provided proof of direct basolateral PAR2 activation and a subsequent increase in paracellular permeability[18,52]. Further investigation into the mechanism of action revealed the involvement of calmodulin and MLCK in the PAR2-mediated alterations of paracellular permeability as intracolonic injection of SLIGRL increased the MLC phosphorylation on western blot. Pretreatment with ML-7, an MLCK inhibitor, abolished the elevated mucosal permeability caused by contraction of the epithelial cell cytoskeleton after phosphorylation of MLC[53]. In addition, MLCK was activated by the Ca2+-binding messenger protein calmodulin since precipitation of MLCK revealed an increased binding of calmodulin and the inhibition of calmodulin by chlorpromazine reduced the SLIGRL-induced increase in paracellular permeability[54,55]. Besides the calmodulin pathway, also ERK1/2 can activate MLCK which directly leads to the disruption of TJ composition and function[18]. The increased permeability induced by tryptase in cultured colonocytes was not only abolished by a tryptase inhibitor but also by the ERK1/2 inhibitor UO126. Finally, incubation of a human intestinal epithelial cell line (SCBN) with SLIGRL induced the disruption and migration into the cytoplasm of the TJ protein ZO-1[55].
The latest discovered member of the PAR-family, PAR4, is receiving increasing attention[56]. This is mainly due to the discovery that cathepsin G is a PAR4-selective neutrophil serine protease. As cathepsin G is a neutrophil protease (alongside with proteinase 3 and neutrophil elastase) it might represent an inflammatory mediator in conditions such as IBD, where neutrophil accumulation within the submucosa is considered a hallmark[57]. And indeed, ulcerative colitis (UC) patients express higher colonic levels of cathepsin G and PAR4, both involved in the increased paracellular permeability in UC patients[58]. This was proven by using the PAR4 inhibitor P4pal-10, a pepducin that selectively blocks PAR4 signaling[59]. In Ussing chamber experiments with mice colonic strips, fecal supernatant of UC and Crohn’s disease (CD) patients triggered a significant increase in FITC dextran permeability abolished by the pre-treatment with P4pal-10 in strips triggered with UC supernatant but interestingly not for the strips triggered with the supernatant of CD patients[58]. In addition, the PAR4 activating peptide AYPGKF-NH2 was able to induce an increased mucosal permeability with a similar effect as the UC fecal supernatant[60]. Finally, pre-incubation of UC fecal supernatant with the specific cathepsin G inhibitor completely normalized the elevated permeability effect[58]. These data suggest that cathepsin G plays a predominant role in the pathophysiology of UC by activating PAR4, while this is not shown in CD. However there is a lack of studies investigating the expression of cathepsin G and comparing this expression between UC vs CD patients. Other proteases are likely to play a key role in barrier function in CD.
Also PAR1-signaling induces epithelial barrier dysfunction, but in this case apoptosis seems to mediate the phenomenon. Treatment with PAR1 agonists (thrombin and selective PAR1 activating peptide TFLLR-NH2) increased the paracellular permeability in vitro in an epithelial cell line (SCBN) as well as in vivo in mice. This permeability increase depends on the disruption of ZO-1. Chromatin condensation and nuclear fragmentation, hallmarks for apoptosis, were induced in a caspase-3-dependent manner. Interestingly, pretreatment with a caspase-3 inhibitor (Z-DEVD-FMK), which irreversibly inhibits apoptosis[61], completely abolished the effect of the PAR1 agonists on permeability and apoptosis. Also the MLCK inhibitor ML-9 abolished the abnormalities[62]. Apart from PARs, also specific serine proteases were studied in the field of intestinal permeability.
A serine protease that is important for the intestinal barrier homeostasis is the transmembrane protein matriptase, or membrane type serine protease-1. A critical role in the epithelial barrier formation and the apical junction complex assembly is attributed to this trypsin-like serine protease[63,64]. Suppressor of tumorigenicity-14 (ST14) hypomorphic mice, which express less than 1% of the matriptase mRNA levels present in the intestine of control littermates, were found to have an impaired intestinal barrier as measured by transepithelial electrical resistance and FITC-dextran permeability[65,66]. This could explain the increased susceptibility of ST14 hypomorphic mice to DSS colitis, with a 30% survival rate after 7 d DSS vs 100% in control littermates[67]. Not only genetic depletion but also pharmacological inhibition (with MI-432) and RNAi silencing of matriptase modulate the TJ assembly, causing the opening of the paracellular gate[65,68]. In physiological conditions, matriptase regulates the expression pattern of the “pore-forming” TJ protein claudin-2. Since there is no evidence of direct proteolytic processing by matriptase, it is likely that matriptase enhances the claudin-2 protein turnover via activation of protein kinase C-zeta (PKC-ζ)[65]. Other studies also reported an association of matriptase with other TJ proteins. For instance in ST14 hypomorphic mice, multiple grades of TJ disruption were identified with expression patterns of occludin, ZO-1 and claudin-1 ranging from a decreased protein expression to areas of complete absence, whereas claudin-2 was upregulated[66,67]. E-cadherin levels however remained unaltered despite co-localisation of matriptase with E-cadherin[65]. In addition, matriptase expression in inflamed colonic tissues from CD and UC patients is significantly downregulated, making matriptase induction a potential therapeutic strategy[68]. Recently, Pászti-Gere et al[69] showed that reinforcement of the intestinal barrier is in fact possible by the induction of matriptase. Incubation of an intestinal epithelial cell line with the matriptase activator sphingosine-1-phosphate increased the TER and resulted in an upregulation of occludin at the apical junction. Also in vivo it was shown that matriptase restoration recovers the barrier integrity by decreasing permeability-associated claudin-2 protein levels and thereby protecting against DSS colitis[67]. Although it should be noted that a tight regulation of matriptase is necessary, since an overexpression could result in malignancies due to its involvement in epithelial proliferation[70-72].
Recently, serine protease granzyme M was also shown to be essential for normal barrier function[73]. In this study, mice deficient of granzyme M were more susceptible to DSS colitis and showed an elevated paracellular permeability compared to wild type (WT) mice. Furthermore, granzyme M expression was upregulated in the inflamed colon tissue samples from UC patients, suggesting that granzyme M acts to induce colonic protection during active disease[73].
In 2000, Fasano et al[74] discovered the serine protease analogue zonulin, the first known endogenous physiologic modulator of TJ proteins regulating the paracellular permeability. The human protein zonulin is similar to Zonula occludens toxin (Zot) that was discovered earlier in Vibrio cholerae. It increased intestinal paracellular permeability in a similar fashion[75]. Luminal exposure to bacteria and the gluten component gliadin are identified as the two most powerful triggers for zonulin release in the gut[76,77]. In addition, gliadin can cause celiac disease in genetically susceptible individuals, which is associated with increased paracellular permeability. The expression level of zonulin was shown to be increased in the intestinal submucosa of celiac disease patients[74]. Also in CD patients, serum zonulin levels are higher compared to their relatives and to healthy control subjects. Interestingly, 50% of the first degree relatives had serum zonulin levels that were increased tremendously (more than two times the standard deviation above the mean) whereas this large increase could only be observed in 4.9% of controls[78]. For both zonulin and Zot, the mechanism of opening intercellular TJs is likely to resemble the effect of certain serine proteases (cfr. supra) although it is not fully elucidated yet. It is suggested that zonulin and Zot cause TJ disassembly by activating the epidermal growth factor receptor (EGFR) on the epithelial cell through transactivation of PAR2 (Figure 1)[78,79]. The involvement of PAR2 in EGFR activation was confirmed by the findings that zonulin could not reduce the TER in ileal strips from PAR2-/- mice in contrast to strips of WT mice. Subsequently, intracellular signaling involves protein kinase C-α (PKC-α)-activation that leads to polymerization of F-actin, TJ disassembly and opening of the paracellular space[80]. Meanwhile, a synthetic octapeptide resembling the receptor-binding domain of zonulin was developed. This molecule, named Larazotide acetate or AT-1001, prevents the opening of TJs in response to zonulin by competitively antagonizing the zonulin receptors (EGFR and PAR2)[81,82]. Clinical trials are currently ongoing to assess its efficacy as a therapy for celiac disease, but there also is evidence for therapeutic potential in other pathologies such as IBD[83,84].
Serine protease inhibitors
Two families of endogenous protease inhibitors, the Serpins and Chelonianins, tightly regulate the activity of the serine proteases by binding to the target protease and largely adjusting its confirmation leading to an irreversible disruption of the active site[85]. A defect in the protease/antiprotease balance leads to tissue damage due to excessive proteolytic capacity causing gastrointestinal diseases.
Serpins are most studied in their role in controlling the coagulation cascade. However, in the gastrointestinal tract, secretory leucocyte protease inhibitor (SLPI) and elafin are of importance. Both are produced by intestinal epithelial cells or leucocytes and inhibit neutrophil elastase and proteinase-3. Besides, SLPI is also a potent inhibitor of trypsin, tryptase, chymotrypsin, chymase and cathepsin G[37]. Although elafin, or skin-derived antileukoproteinase, and SLPI are poorly investigated when it comes to barrier function disturbances, an elevated expression of these antiproteases was reported in “leaky gut”-related gastrointestinal diseases. For instance in CD and UC patients it was shown that the levels of elafin and SLPI are significantly higher in inflamed colonic tissue vs noninflamed and control tissues. Interestingly, the upregulation was less pronounced in inflamed CD samples vs inflamed UC samples[86]. In contrast, the elafin expression in small intestinal samples was lower in patients with active celiac disease compared to control patients. Local treatment of gluten-sensitive mice with elafin using a recombinant Lactococcus lactis vector, restored the intestinal barrier function and normalized the ZO-1 expression, which was disrupted in a mouse model of celiac disease[87]. In vitro experiments in a Caco-2 cell line confirm these barrier-protective effects of elafin. Cells treated with TNF-α to induce a barrier defect, showed a complete restoration in paracellular permeability after simultaneous treatment with elafin[88].
Restoring an impaired barrier by pharmacological protease inhibition has been proposed as a promising therapeutic treatment option for IBD, functional GI disorders such as IBS and for colorectal cancer[37] because epithelial barrier dysfunction is a common factor in these diverse pathologies. Studies with the synthetic broad specificity serine protease inhibitors nafamostat mesilate and camostat mesilate revealed positive effects on paracellular permeability. In an animal model for IBS, where the colonic permeability towards 51Cr-EDTA was elevated and ZO-1 disrupted, treatment with camostat mesilate not only normalized the fecal protease activity but also the colonic permeability significantly improved and the ZO-1 protein levels were restored[89]. Nafamostat mesilate, that has shown beneficial effects on disease outcome in animal models of colitis[90], IBS[91], acute pancreatitis[92] and colorectal cancer[93], also seems to restore the intestinal barrier function. The addition of tryptase to the basolateral side of human colonic strips mounted in Ussing chambers increased the permeability proportional to the tryptase concentration. This was abolished after simultaneous addition of nafamostat mesilate[94]. In an in vitro co-culture model, Caco-2 cells responded to mast cell degranulation with a disruption of epithelial integrity shown by a decrease in TER, an increase of FITC-dextran flux and a decrease in the expression of the TJ proteins claudin-1 and ZO-1. These effects were prevented by tryptase inhibition using nafamostat mesilate[95]. These positive findings of nafamostat on intestinal permeability could however not be confirmed in vivo in a chronic animal model of T-cell transfer colitis (unpublished results). However, in our own lab, a beneficial effect of a novel serine protease inhibitor (Di-(4-acetamidophenyl) 1-(benzyloxycarbonylamino)-2-[(4-guanidino)phenyl]ethanephosphonate trifluoroacetate; abbreviated as SPI in Table 1) was shown in a T-cell transfer colitis model. A curative i.p. treatment with this novel protease inhibitor abolished the elevated intestinal permeability that was seen in the vehicle-treated colitis animals while also exerting anti-inflammatory effects whereas nafamostat only showed the anti-inflammatory effects[96].
METALLOPROTEASES
Matrix metalloproteases
Matrix metalloproteases (MMPs) are generally known for their ability to degrade and remodel the extracellular matrix (ECM). But in addition to their role in ECM turnover, MMPs degrade or proteolytically activate a wide range of molecules such as chemokines, cytokines, growth factors, membrane receptors, cytoskeleton proteins and junctional proteins[97,98]. Under normal physiological conditions, their activity is tightly regulated by the tissue inhibitors of metalloproteases (TIMP-1-4). A dysregulation of the balance between MMPs and TIMPs is associated with inflammation and tissue damage[99,100]. Indeed, various studies have reported an upregulation of MMPs in inflamed IBD epithelium, suggesting that the inhibition of MMPs could be an interesting therapeutic intervention in IBD[99,101-104]. However, the failure of MMP inhibitors in cancer trials has led to rethink the clinical potential of these compounds[105,106]. Indirect inhibition of the effects of MMPs or intervening in the signal transduction pathways influenced by MMPs seems more likely to be successful. In this respect, the new physiological and pathological roles of MMPs in specific diseases are being further investigated, such as their effect on the epithelial barrier integrity, discussed below.
Previous studies have demonstrated the upregulation of the gelatinases MMP-2 and MMP-9 in IBD patients[103,107,108] as well as in animal models of colitis[109-111]. It was found that a specific overexpression of MMP-9 in the intestinal epithelium is associated with a defective barrier function and mucin production due to a decrease in goblet cell differentiation[112]. Supporting their role in barrier function, MMP-9-/- mice have an increased number of goblet cells and MUC-2 expression compared to WT mice[113]. Recently, Nighot et al[114] showed an attenuation of the DSS-induced increase in colonic permeability in MMP-9-/- mice. The protein levels of tight junctional occludin were elevated in MMP-9-/- mice, both DSS- and vehicle-treated, vs WT mice. It was also found that the protein expression of MLCK was upregulated in DSS colitis in WT mice but not in MMP-9-/- mice. Interestingly, MLCK is a key regulator of TJ permeability as described above[115], suggesting that MMP-9 is a key regulator of TJ permeability via MLC phosphorylation. The other gelatinase, MMP-2, plays a barrier protective role in contrast to MMP-9. Mice deficient from MMP-2 have an impaired intestinal barrier function, making them more susceptible to develop experimental colitis compared to their WT counterparts[111]. The authors suggest that MMP-2 exerts its barrier protective function by associating with TJ proteins, since multiple studies have shown close interaction between MMP-2 and claudins[116,117].
Besides MMP-2 and MMP-9, transcript and protein levels of MMP-7 (matrilysin) and MMP-3 (stromelysin-1) are shown to be upregulated in the mucosal tissues of active IBD patients vs healthy control tissue[118-120]. These MMPs are linked to the ectodomain shedding of E-cadherin and thereby releasing soluble E-cadherin in the interstitium and loss of E-cadherin function in cell-cell adhesio[121]. Although the majority of the research revolves around cancer, it is relevant to include the proteolytic splicing of E-cad in this review since loss of E-cadherin is associated with disturbances in barrier function during intestinal inflammatory diseases[122,123]. In cancerous cell lines the ability of matrilysin and stromelysin-1 to release soluble E-cadherin was proven with a loss of E-cad function in the cell-cell adhesion and a facilitation of tumor cell invasion as a consequence[121,124,125]. The released sE-cadherin works as a paracrine/autocrine signaling molecule, promoting MMP production and thereby worsening the disease progression[126,127].
Not all metalloproteases are however harmful. The transmembrane endopeptidase meprin β is found to be essential in mucus homeostasis. A loosely organized, nonattached mucus layer covers the epithelial cells on the luminal side, protecting them from digestive proteases thereby forming an important part of the physical barrier against infiltration of harmful substances. The mucin MUC2 is produced by goblet cells and detached by meprin β splicing. Mice lacking the gene encoding for meprin β (Mep1b-/-) display a more dense mucus fenotype[128]. In addition, the detached MUC2 is an important luminal factor promoting oral tolerance. The outer mucus layer inhabits commensal bacteria which bind MUC2 and are picked up by patrolling dendritic cells in the intestinal mucosa. After binding, MUC2 induces immunoregulatory signals such as IL-10 and retinoic acid, which are important co-stimulatory factors helping CD103+ dendritic cells to induce regulatory T cells, and thus oral tolerance[129].
A disintegrin and metalloproteinase
Another family of metalloproteases are A Disintegrin and Metalloproteinase (ADAM) involved in the release of the ectodomain from transmembrane proteins, a process that is referred to as “shedding”.
TNF-α converting enzyme (TACE), or ADAM17, generates the biologically active, soluble form of TNF-α by cleaving the transmembrane bound precursor at the cell surface[28,130,131]. Since TNF-α is a major contributor of the increased intestinal permeability in inflammatory conditions[28,131,132], TACE is a potential key player in the regulation of paracellular permeability. In the past, TACE inhibition (by the endogenous inhibitor TIMP-3 or synthetic inhibitors) has been investigated in diseases that benefit from anti-TNF-α therapy, such as IBD, showing promising results[133-135]. Recently, Al-Sadi et al[132] investigated the signaling pathways that mediate TNF-α-induced modulation of intestinal paracellular permeability showing MAP kinase ERK1/2 activation, on their turn phosphorylating and activating Elk-1 which subsequently leads to an increase in MLCK and opening of TJs.
The effect on intestinal permeability of pharmacological TACE-inhibition was investigated on a Caco-2 monolayer. Pretreatment with two synthetic inhibitors, the broad MMP-TACE inhibitor GM6001 or the TACE-specific inhibitor TAPI-2, suppressed the permeability increase induced by TACE[136]. In contrast, Fréour et al[137] demonstrated that TACE inhibition, by TIMP-3 or by TAPI-2, amplified the TNF-α-mediated increase in paracellular permeability in vitro. The authors suggest that this might be due to an autocrine effect of TIMP-3, triggering the release of pro-inflammatory cytokines that contribute to a hyperpermeable intestinal barrier. It should be noted however that no hard evidence to prove this statement was provided.
CYSTEINE PROTEASES
The vast majority of cysteine proteases reside intracellularly, where they mediate distinct signaling pathways affecting programmed cell death and inflammation[37,138]. Particularly the caspase family of cysteine proteases is well known in the field of cell death. For instance, caspase 1 and 5 take part in inflammasome activation, promoting IL1-β and IL-18 maturation. Caspase 8 plays a central role in both apoptotic and inflammatory pathways, activating respectively pro-apoptotic proteins and NF-κB[138]. Hence, cysteine proteases affect the epithelial barrier integrity for the most part indirectly via their effect on inflammation and cell death.
Intestinal epithelial cells undergo apoptosis in a tightly regulated fashion in order to renew the entire cell population every 72 to 96 h[139]. Under inflammatory conditions, such as during IBD, the apoptosis rate increases significantly, inducing morphologic changes in the intestinal barrier[140,141]. During apoptosis of the intestinal epithelial cells, TJ proteins undergo proteolytic cleavage and dislocate from the lateral cell surface[142]. In human colonic HT-29/B6 cells, induction of apoptosis resulted in a significant decrease of TER and an increase in macromolecular tracer permeability[143]. Caspases cleave the transmembrane protein occludin (at the cytoplasmic tail) and adaptor proteins ZO-1 and ZO-2. Although the chronologic sequence of events may not always be clear, there is evidence that a disruption of TJ proteins, caused by for instance bacterial infection or inflammation, activates caspase-8 and -3 and thus initiating cell death[144]. In an in vivo permeability assay however, no effect on Cr51-EDTA flux over the colonic barrier was measured after intraluminal administration of cystatin, a cysteine protease inhibitor, whereas serine- and metalloprotease inhibitors lowered the flux significantly[51].
LUMINAL PROTEASES
When it comes to research into the pathogenesis of IBD and IBS, both the gut microbiome and proteases are receiving increasing attention[37,145,146]. Surprisingly, the current knowledge on proteases focusses mostly on host-derived proteases while bacteria-derived proteases have been largely ignored[147]. As previously mentioned, IBD and IBS patients have elevated fecal protease levels. In IBS-D patients it was shown that the majority of the fecal protease activity is most likely due to human pancreatic enzymes and not bacteria. However, an oral antibiotic treatment in mice resulted in decreased fecal activity, supporting the hypothesis that bacteria contribute to the luminal protease content[148].
In the 1990s it was discovered that the metalloproteinase fragilysin, which is produced by the colonic commensal bacteroides fragilis, alters the intestinal permeability. Experiments with the purified fragilysin on a human colon cell line (HT-29) revealed the increase in permeability towards ions (shown by a decrease in TER) as well as towards macromolecules (increase in mannitol passage across the cell monolayer)[149,150]. Later, the role of Entamoeba histolytica (Eh)-derived cysteine proteases in the pathogenesis of amoebic colitis was investigated. The amoebic cysteine proteases induce inflammation by activating IL-1β in a way similar to the human caspases (cfr. supra), and damage to the intestinal epithelial barrier resulting in an increased paracellular permeability[151,152]. Two specific proteases mediating TJ disruption were identified; Eh cysteine protease A5 (EhCPA5) and EhCP112[152,153]. Recently, bacterial-derived gelatinases were investigated. Pruteanu et al[154] screened bacterial colonies in fecal samples of healthy controls and IBD patients for gelatinolytic activity. The researchers could link Clostridium perfringens to the majority of the proteolytic activity in the fecal samples. In addition, C. perfringens culture supernatant reduced the TER in Ussing chamber experiments performed on rat distal colon. Gelatinase (GelE) produced by the enteric microbe Enterococcus faecalis also has shown to induce a barrier defect in epithelial cell monolayers and in vivo in mice[155,156]. Originally, the degradation of E-cadherin by proteolytic activity of GelE was considered to be the cause of the permeability increase[156]. Later on, Maharshak et al[155] discovered the involvement of PAR2 activation in the GelE-induced permeability increase. Purified E. faecalis GelE failed to induce a permeability increase in PAR2-/- mice as well as in epithelial monolayers treated with a PAR2 antagonist.
The feces of the house dust mite (HDM; Dermatophagoides pteronyssinus) contains a cysteine protease, Der p 1, which is known to disrupt the lung epithelial TJ proteins occludin and ZO-1 and thereby facilitating allergen delivery and eventually provoking asthma[157]. Recently, Der p 1 was shown to be present in the human gut, affecting barrier function. Exposure of colonic biopsies to a HDM extract reduced the expression of TJ proteins occludin and ZO-1, reduced the mucus layer thickness, increased TNF-α and increased the paracellular permeability. Pre-incubation of the HDM-extract with the cysteine protease inhibitor E64 abolished the HDM-induced damage to the intestinal barrier[158].
Furthermore, there is an established link between food allergens and the degradation of the epithelial barrier[159,160]. In the digestion process, food particles are broken down by pancreatic and brush border proteases into tripeptides, dipeptides and single amino acids. When the intestinal barrier function is not impaired, oral tolerance is induced against these soluble peptides that can cross the epithelium in a selective and regulated manner. But in other -possibly genetically susceptible- individuals, partially or undigested proteins can still reach the mucosa where they provoke an inflammatory signal instead of tolerance. Plasma cells produce allergen-specific IgE which causes mast cell degranulation at contact. The secreted mast cell proteases and cytokines both contribute to the increased barrier dysfunction via their effect on the TJ configuration, leading to an opening of the paracellular route. The difficulty lies however in the “chicken and egg” paradigm. Until today there is evidence for only one food protease to affect the intestinal barrier directly. The kiwifruit cysteine protease actinidin (Act d1) has shown to induce a loss of barrier function in epithelial cell monolayers by the disruption of the occludin and ZO-1 network[161,162]. This effect could be confirmed in vivo. Mice gavaged with actinidin exerted an elevated permeability towards FITC-dextran compared to mice gavaged with the vehicle[161].
Dietary proteases can also contribute to intestinal health. Addition of Aspergillus-derived proteases (Amano SD) to the diet of rats improved intestinal health via the expansion of commensal colonic bacteria of the Bifidobacterium and Lactobacillus species. The altered microbiota composition enhanced the formation of short chain fatty (SCFA) acids such as butyrate, propionate and lactate[163]. SCFA with butyrate in particular promote mucosal homeostasis among other things by enhancing the intestinal barrier function through the upregulation of tight junction proteins[164].
CONCLUSION
The epithelial cell layer lining the gastrointestinal tract is the body’s largest surface area in contact with environmental antigens. The role of these intestinal epithelial cells in the continuous maintenance of intestinal homeostasis is indispensable, providing a physical and biochemical barrier against noxious luminal antigens as well as allowing nutrient absorption. The selective opening of the intercellular spaces, allowing paracellular transport of macromolecules, is regulated by the interplay between TJ proteins and the actomyosin contraction upon activation of intracellular signaling pathways. An increased intestinal permeability is suggested as an important player in the pathophysiology of various intestinal and extra-intestinal pathologies. Targeting the epithelial barrier is a tempting therapeutic approach, but so far no therapies have succeeded. Larazotide acetate showed promising results in preclinical trials, restoring the intestinal barrier function. But clinical trials failed to mirror these effects. This shows that more research is needed to define epithelial barrier function and dysfunction, underlying different pathologies and diseases. Proteases are important signaling molecules in this regard. With their proteolytic capacity they can cleave proteinase-activated receptors on the cell surface of intestinal epithelial cells, influencing the cytoskeleton contractile machinery and paracellular permeability. Also extracellular proteolytic cleavage of the junction proteins occurs, leading directly to epithelial damage and increased intestinal permeability. In homeostasis, the proteolytic activity is tightly regulated by antiproteases, but this balance is dysregulated in organic and functional GI disorders. As a result, protease inhibition has become a “hot topic” in a therapeutic point of view, mainly focusing on inflammation and hypersensitivity, ignoring the effect on permeability. But since a large array of proteases is involved and for many proteases no specific inhibitors are available yet, more research is needed to elucidate the exact involvement of specific proteases in gut physiology in general and intestinal permeability in particular, in order to become a therapeutic target.
Footnotes
Manuscript source: Invited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: Belgium
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B
Grade C (Good): C
Grade D (Fair): 0
Grade E (Poor): 0
Conflict-of-interest statement: No potential conflicts of interest.
Peer-review started: September 27, 2016
First decision: December 19, 2016
Article in press: March 2, 2017
P- Reviewer: Schnoor M, Tran CD S- Editor: Yu J L- Editor: A E- Editor: Wang CH
References
- 1.Groschwitz KR, Hogan SP. Intestinal barrier function: molecular regulation and disease pathogenesis. J Allergy Clin Immunol. 2009;124:3–20; quiz 21-22. doi: 10.1016/j.jaci.2009.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ayabe T, Satchell DP, Wilson CL, Parks WC, Selsted ME, Ouellette AJ. Secretion of microbicidal alpha-defensins by intestinal Paneth cells in response to bacteria. Nat Immunol. 2000;1:113–118. doi: 10.1038/77783. [DOI] [PubMed] [Google Scholar]
- 3.Ulluwishewa D, Anderson RC, McNabb WC, Moughan PJ, Wells JM, Roy NC. Regulation of tight junction permeability by intestinal bacteria and dietary components. J Nutr. 2011;141:769–776. doi: 10.3945/jn.110.135657. [DOI] [PubMed] [Google Scholar]
- 4.Li H, Shi B. Tolerogenic dendritic cells and their applications in transplantation. Cell Mol Immunol. 2015;12:24–30. doi: 10.1038/cmi.2014.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rautava S, Walker WA. Commensal bacteria and epithelial cross talk in the developing intestine. Curr Gastroenterol Rep. 2007;9:385–392. doi: 10.1007/s11894-007-0047-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pabst O, Mowat AM. Oral tolerance to food protein. Mucosal Immunol. 2012;5:232–239. doi: 10.1038/mi.2012.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Suzuki T. Regulation of intestinal epithelial permeability by tight junctions. Cell Mol Life Sci. 2013;70:631–659. doi: 10.1007/s00018-012-1070-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vogelsang H, Schwarzenhofer M, Oberhuber G. Changes in gastrointestinal permeability in celiac disease. Dig Dis. 1998;16:333–336. doi: 10.1159/000016886. [DOI] [PubMed] [Google Scholar]
- 9.Gerova VA, Stoynov SG, Katsarov DS, Svinarov DA. Increased intestinal permeability in inflammatory bowel diseases assessed by iohexol test. World J Gastroenterol. 2011;17:2211–2215. doi: 10.3748/wjg.v17.i17.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Michielan A, D’Incà R. Intestinal Permeability in Inflammatory Bowel Disease: Pathogenesis, Clinical Evaluation, and Therapy of Leaky Gut. Mediators Inflamm. 2015;2015:628157. doi: 10.1155/2015/628157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vaarala O. Leaking gut in type 1 diabetes. Curr Opin Gastroenterol. 2008;24:701–706. doi: 10.1097/MOG.0b013e32830e6d98. [DOI] [PubMed] [Google Scholar]
- 12.Camilleri M, Gorman H. Intestinal permeability and irritable bowel syndrome. Neurogastroenterol Motil. 2007;19:545–552. doi: 10.1111/j.1365-2982.2007.00925.x. [DOI] [PubMed] [Google Scholar]
- 13.Pastorelli L, De Salvo C, Mercado JR, Vecchi M, Pizarro TT. Central role of the gut epithelial barrier in the pathogenesis of chronic intestinal inflammation: lessons learned from animal models and human genetics. Front Immunol. 2013;4:280. doi: 10.3389/fimmu.2013.00280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Odenwald MA, Turner JR. The intestinal epithelial barrier: a therapeutic target? Nat Rev Gastroenterol Hepatol. 2017;14:9–21. doi: 10.1038/nrgastro.2016.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Odenwald MA, Turner JR. Intestinal permeability defects: is it time to treat? Clin Gastroenterol Hepatol. 2013;11:1075–1083. doi: 10.1016/j.cgh.2013.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Anderson JM, Van Itallie CM. Physiology and function of the tight junction. Cold Spring Harb Perspect Biol. 2009;1:a002584. doi: 10.1101/cshperspect.a002584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hartsock A, Nelson WJ. Adherens and tight junctions: structure, function and connections to the actin cytoskeleton. Biochim Biophys Acta. 2008;1778:660–669. doi: 10.1016/j.bbamem.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jacob C, Yang PC, Darmoul D, Amadesi S, Saito T, Cottrell GS, Coelho AM, Singh P, Grady EF, Perdue M, et al. Mast cell tryptase controls paracellular permeability of the intestine. Role of protease-activated receptor 2 and beta-arrestins. J Biol Chem. 2005;280:31936–31948. doi: 10.1074/jbc.M506338200. [DOI] [PubMed] [Google Scholar]
- 19.Weber LP, Seto M, Sasaki Y, Swärd K, Walsh MP. The involvement of protein kinase C in myosin phosphorylation and force development in rat tail arterial smooth muscle. Biochem J. 2000;352 Pt 2:573–582. [PMC free article] [PubMed] [Google Scholar]
- 20.Riento K, Ridley AJ. Rocks: multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol. 2003;4:446–456. doi: 10.1038/nrm1128. [DOI] [PubMed] [Google Scholar]
- 21.Shen Q, Rigor RR, Pivetti CD, Wu MH, Yuan SY. Myosin light chain kinase in microvascular endothelial barrier function. Cardiovasc Res. 2010;87:272–280. doi: 10.1093/cvr/cvq144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rigor RR, Shen Q, Pivetti CD, Wu MH, Yuan SY. Myosin light chain kinase signaling in endothelial barrier dysfunction. Med Res Rev. 2013;33:911–933. doi: 10.1002/med.21270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harhaj NS, Antonetti DA. Regulation of tight junctions and loss of barrier function in pathophysiology. Int J Biochem Cell Biol. 2004;36:1206–1237. doi: 10.1016/j.biocel.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 24.Capaldo CT, Nusrat A. Cytokine regulation of tight junctions. Biochim Biophys Acta. 2009;1788:864–871. doi: 10.1016/j.bbamem.2008.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nava P, Kamekura R, Nusrat A. Cleavage of transmembrane junction proteins and their role in regulating epithelial homeostasis. Tissue Barriers. 2013;1:e24783. doi: 10.4161/tisb.24783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Al-Sadi R, Boivin M, Ma T. Mechanism of cytokine modulation of epithelial tight junction barrier. Front Biosci (Landmark Ed) 2009;14:2765–2778. doi: 10.2741/3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee SH. Intestinal permeability regulation by tight junction: implication on inflammatory bowel diseases. Intest Res. 2015;13:11–18. doi: 10.5217/ir.2015.13.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ye D, Ma I, Ma TY. Molecular mechanism of tumor necrosis factor-alpha modulation of intestinal epithelial tight junction barrier. Am J Physiol Gastrointest Liver Physiol. 2006;290:G496–G504. doi: 10.1152/ajpgi.00318.2005. [DOI] [PubMed] [Google Scholar]
- 29.Zolotarevsky Y, Hecht G, Koutsouris A, Gonzalez DE, Quan C, Tom J, Mrsny RJ, Turner JR. A membrane-permeant peptide that inhibits MLC kinase restores barrier function in in vitro models of intestinal disease. Gastroenterology. 2002;123:163–172. doi: 10.1053/gast.2002.34235. [DOI] [PubMed] [Google Scholar]
- 30.Madara JL, Stafford J. Interferon-gamma directly affects barrier function of cultured intestinal epithelial monolayers. J Clin Invest. 1989;83:724–727. doi: 10.1172/JCI113938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ceponis PJ, Botelho F, Richards CD, McKay DM. Interleukins 4 and 13 increase intestinal epithelial permeability by a phosphatidylinositol 3-kinase pathway. Lack of evidence for STAT 6 involvement. J Biol Chem. 2000;275:29132–29137. doi: 10.1074/jbc.M003516200. [DOI] [PubMed] [Google Scholar]
- 32.Zünd G, Madara JL, Dzus AL, Awtrey CS, Colgan SP. Interleukin-4 and interleukin-13 differentially regulate epithelial chloride secretion. J Biol Chem. 1996;271:7460–7464. doi: 10.1074/jbc.271.13.7460. [DOI] [PubMed] [Google Scholar]
- 33.Wisner DM, Harris LR, Green CL, Poritz LS. Opposing regulation of the tight junction protein claudin-2 by interferon-gamma and interleukin-4. J Surg Res. 2008;144:1–7. doi: 10.1016/j.jss.2007.03.059. [DOI] [PubMed] [Google Scholar]
- 34.Pham CT. Neutrophil serine proteases: specific regulators of inflammation. Nat Rev Immunol. 2006;6:541–550. doi: 10.1038/nri1841. [DOI] [PubMed] [Google Scholar]
- 35.Rao MB, Tanksale AM, Ghatge MS, Deshpande VV. Molecular and biotechnological aspects of microbial proteases. Microbiol Mol Biol Rev. 1998;62:597–635. doi: 10.1128/mmbr.62.3.597-635.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Antalis TM, Shea-Donohue T, Vogel SN, Sears C, Fasano A. Mechanisms of disease: protease functions in intestinal mucosal pathobiology. Nat Clin Pract Gastroenterol Hepatol. 2007;4:393–402. doi: 10.1038/ncpgasthep0846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vergnolle N. Protease inhibition as new therapeutic strategy for GI diseases. Gut. 2016;65:1215–1224. doi: 10.1136/gutjnl-2015-309147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vergnolle N. Protease-activated receptors as drug targets in inflammation and pain. Pharmacol Ther. 2009;123:292–309. doi: 10.1016/j.pharmthera.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 39.Macfarlane SR, Seatter MJ, Kanke T, Hunter GD, Plevin R. Proteinase-activated receptors. Pharmacol Rev. 2001;53:245–282. [PubMed] [Google Scholar]
- 40.Amadesi S, Bunnett N. Protease-activated receptors: protease signaling in the gastrointestinal tract. Curr Opin Pharmacol. 2004;4:551–556. doi: 10.1016/j.coph.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 41.Ceuleers H, Van Spaendonk H, Hanning N, Heirbaut J, Lambeir AM, Joossens J, Augustyns K, De Man JG, De Meester I, De Winter BY. Visceral hypersensitivity in inflammatory bowel diseases and irritable bowel syndrome: The role of proteases. World J Gastroenterol. 2016;22:10275–10286. doi: 10.3748/wjg.v22.i47.10275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Di Cera E. Serine proteases. IUBMB Life. 2009;61:510–515. doi: 10.1002/iub.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Page MJ, Di Cera E. Serine peptidases: classification, structure and function. Cell Mol Life Sci. 2008;65:1220–1236. doi: 10.1007/s00018-008-7565-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Padrines M, Wolf M, Walz A, Baggiolini M. Interleukin-8 processing by neutrophil elastase, cathepsin G and proteinase-3. FEBS Lett. 1994;352:231–235. doi: 10.1016/0014-5793(94)00952-x. [DOI] [PubMed] [Google Scholar]
- 45.Nufer O, Corbett M, Walz A. Amino-terminal processing of chemokine ENA-78 regulates biological activity. Biochemistry. 1999;38:636–642. doi: 10.1021/bi981294s. [DOI] [PubMed] [Google Scholar]
- 46.Rothmeier AS, Ruf W. Protease-activated receptor 2 signaling in inflammation. Semin Immunopathol. 2012;34:133–149. doi: 10.1007/s00281-011-0289-1. [DOI] [PubMed] [Google Scholar]
- 47.Coelho AM, Vergnolle N, Guiard B, Fioramonti J, Bueno L. Proteinases and proteinase-activated receptor 2: a possible role to promote visceral hyperalgesia in rats. Gastroenterology. 2002;122:1035–1047. doi: 10.1053/gast.2002.32387. [DOI] [PubMed] [Google Scholar]
- 48.Cenac N, Coelho AM, Nguyen C, Compton S, Andrade-Gordon P, MacNaughton WK, Wallace JL, Hollenberg MD, Bunnett NW, Garcia-Villar R, et al. Induction of intestinal inflammation in mouse by activation of proteinase-activated receptor-2. Am J Pathol. 2002;161:1903–1915. doi: 10.1016/S0002-9440(10)64466-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bjarnason I, MacPherson A, Hollander D. Intestinal permeability: an overview. Gastroenterology. 1995;108:1566–1581. doi: 10.1016/0016-5085(95)90708-4. [DOI] [PubMed] [Google Scholar]
- 50.Groschwitz KR, Wu D, Osterfeld H, Ahrens R, Hogan SP. Chymase-mediated intestinal epithelial permeability is regulated by a protease-activating receptor/matrix metalloproteinase-2-dependent mechanism. Am J Physiol Gastrointest Liver Physiol. 2013;304:G479–G489. doi: 10.1152/ajpgi.00186.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Róka R, Demaude J, Cenac N, Ferrier L, Salvador-Cartier C, Garcia-Villar R, Fioramonti J, Bueno L. Colonic luminal proteases activate colonocyte proteinase-activated receptor-2 and regulate paracellular permeability in mice. Neurogastroenterol Motil. 2007;19:57–65. doi: 10.1111/j.1365-2982.2006.00851.x. [DOI] [PubMed] [Google Scholar]
- 52.Chin AC, Lee WY, Nusrat A, Vergnolle N, Parkos CA. Neutrophil-mediated activation of epithelial protease-activated receptors-1 and -2 regulates barrier function and transepithelial migration. J Immunol. 2008;181:5702–5710. doi: 10.4049/jimmunol.181.8.5702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Turner JR, Rill BK, Carlson SL, Carnes D, Kerner R, Mrsny RJ, Madara JL. Physiological regulation of epithelial tight junctions is associated with myosin light-chain phosphorylation. Am J Physiol. 1997;273:C1378–C1385. doi: 10.1152/ajpcell.1997.273.4.C1378. [DOI] [PubMed] [Google Scholar]
- 54.Cenac N, Garcia-Villar R, Ferrier L, Larauche M, Vergnolle N, Bunnett NW, Coelho AM, Fioramonti J, Bueno L. Proteinase-activated receptor-2-induced colonic inflammation in mice: possible involvement of afferent neurons, nitric oxide, and paracellular permeability. J Immunol. 2003;170:4296–4300. doi: 10.4049/jimmunol.170.8.4296. [DOI] [PubMed] [Google Scholar]
- 55.Cenac N, Chin AC, Garcia-Villar R, Salvador-Cartier C, Ferrier L, Vergnolle N, Buret AG, Fioramonti J, Bueno L. PAR2 activation alters colonic paracellular permeability in mice via IFN-gamma-dependent and -independent pathways. J Physiol. 2004;558:913–925. doi: 10.1113/jphysiol.2004.061721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kahn ML, Zheng YW, Huang W, Bigornia V, Zeng D, Moff S, Farese RV, Tam C, Coughlin SR. A dual thrombin receptor system for platelet activation. Nature. 1998;394:690–694. doi: 10.1038/29325. [DOI] [PubMed] [Google Scholar]
- 57.Shea-Donohue T, Thomas K, Cody MJ, Aiping Zhao LJ, Kopydlowski KM, Fukata M, Lira SA, Vogel SN. Mice deficient in the CXCR2 ligand, CXCL1 (KC/GRO-alpha), exhibit increased susceptibility to dextran sodium sulfate (DSS)-induced colitis. Innate Immun. 2008;14:117–124. doi: 10.1177/1753425908088724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dabek M, Ferrier L, Roka R, Gecse K, Annahazi A, Moreau J, Escourrou J, Cartier C, Chaumaz G, Leveque M, et al. Luminal cathepsin g and protease-activated receptor 4: a duet involved in alterations of the colonic epithelial barrier in ulcerative colitis. Am J Pathol. 2009;175:207–214. doi: 10.2353/ajpath.2009.080986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Covic L, Misra M, Badar J, Singh C, Kuliopulos A. Pepducin-based intervention of thrombin-receptor signaling and systemic platelet activation. Nat Med. 2002;8:1161–1165. doi: 10.1038/nm760. [DOI] [PubMed] [Google Scholar]
- 60.Houle S, Papez MD, Ferazzini M, Hollenberg MD, Vergnolle N. Neutrophils and the kallikrein-kinin system in proteinase-activated receptor 4-mediated inflammation in rodents. Br J Pharmacol. 2005;146:670–678. doi: 10.1038/sj.bjp.0706371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, Gareau Y, Griffin PR, Labelle M, Lazebnik YA. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- 62.Chin AC, Vergnolle N, MacNaughton WK, Wallace JL, Hollenberg MD, Buret AG. Proteinase-activated receptor 1 activation induces epithelial apoptosis and increases intestinal permeability. Proc Natl Acad Sci USA. 2003;100:11104–11109. doi: 10.1073/pnas.1831452100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bugge TH, List K, Szabo R. Matriptase-dependent cell surface proteolysis in epithelial development and pathogenesis. Front Biosci. 2007;12:5060–5070. doi: 10.2741/2448. [DOI] [PubMed] [Google Scholar]
- 64.List K, Haudenschild CC, Szabo R, Chen W, Wahl SM, Swaim W, Engelholm LH, Behrendt N, Bugge TH. Matriptase/MT-SP1 is required for postnatal survival, epidermal barrier function, hair follicle development, and thymic homeostasis. Oncogene. 2002;21:3765–3779. doi: 10.1038/sj.onc.1205502. [DOI] [PubMed] [Google Scholar]
- 65.Buzza MS, Netzel-Arnett S, Shea-Donohue T, Zhao A, Lin CY, List K, Szabo R, Fasano A, Bugge TH, Antalis TM. Membrane-anchored serine protease matriptase regulates epithelial barrier formation and permeability in the intestine. Proc Natl Acad Sci USA. 2010;107:4200–4205. doi: 10.1073/pnas.0903923107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.List K, Kosa P, Szabo R, Bey AL, Wang CB, Molinolo A, Bugge TH. Epithelial integrity is maintained by a matriptase-dependent proteolytic pathway. Am J Pathol. 2009;175:1453–1463. doi: 10.2353/ajpath.2009.090240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Netzel-Arnett S, Buzza MS, Shea-Donohue T, Désilets A, Leduc R, Fasano A, Bugge TH, Antalis TM. Matriptase protects against experimental colitis and promotes intestinal barrier recovery. Inflamm Bowel Dis. 2012;18:1303–1314. doi: 10.1002/ibd.21930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pászti-Gere E, McManus S, Meggyesházi N, Balla P, Gálfi P, Steinmetzer T. Inhibition of Matriptase Activity Results in Decreased Intestinal Epithelial Monolayer Integrity In Vitro. PLoS One. 2015;10:e0141077. doi: 10.1371/journal.pone.0141077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pászti-Gere E, Jerzsele Á, Balla P, Ujhelyi G, Székács A. Reinforced Epithelial Barrier Integrity via Matriptase Induction with Sphingosine-1-Phosphate Did Not Result in Disturbances in Physiological Redox Status. Oxid Med Cell Longev. 2016;2016:9674272. doi: 10.1155/2016/9674272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Förbs D, Thiel S, Stella MC, Stürzebecher A, Schweinitz A, Steinmetzer T, Stürzebecher J, Uhland K. In vitro inhibition of matriptase prevents invasive growth of cell lines of prostate and colon carcinoma. Int J Oncol. 2005;27:1061–1070. [PubMed] [Google Scholar]
- 71.Zoratti GL, Tanabe LM, Varela FA, Murray AS, Bergum C, Colombo É, Lang JE, Molinolo AA, Leduc R, Marsault E, Boerner J, List K. Targeting matriptase in breast cancer abrogates tumour progression via impairment of stromal-epithelial growth factor signalling. Nat Commun. 2015;6:6776. doi: 10.1038/ncomms7776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wadhawan V, Kolhe YA, Sangith N, Gautam AK, Venkatraman P. From prediction to experimental validation: desmoglein 2 is a functionally relevant substrate of matriptase in epithelial cells and their reciprocal relationship is important for cell adhesion. Biochem J. 2012;447:61–70. doi: 10.1042/BJ20111432. [DOI] [PubMed] [Google Scholar]
- 73.Souza-Fonseca-Guimaraes F, Krasnova Y, Putoczki T, Miles K, MacDonald KP, Town L, Shi W, Gobe GC, McDade L, Mielke LA, et al. Granzyme M has a critical role in providing innate immune protection in ulcerative colitis. Cell Death Dis. 2016;7:e2302. doi: 10.1038/cddis.2016.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fasano A, Not T, Wang W, Uzzau S, Berti I, Tommasini A, Goldblum SE. Zonulin, a newly discovered modulator of intestinal permeability, and its expression in coeliac disease. Lancet. 2000;355:1518–1519. doi: 10.1016/S0140-6736(00)02169-3. [DOI] [PubMed] [Google Scholar]
- 75.Fasano A, Fiorentini C, Donelli G, Uzzau S, Kaper JB, Margaretten K, Ding X, Guandalini S, Comstock L, Goldblum SE. Zonula occludens toxin modulates tight junctions through protein kinase C-dependent actin reorganization, in vitro. J Clin Invest. 1995;96:710–720. doi: 10.1172/JCI118114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Drago S, El Asmar R, Di Pierro M, Grazia Clemente M, Tripathi A, Sapone A, Thakar M, Iacono G, Carroccio A, D’Agate C, et al. Gliadin, zonulin and gut permeability: Effects on celiac and non-celiac intestinal mucosa and intestinal cell lines. Scand J Gastroenterol. 2006;41:408–419. doi: 10.1080/00365520500235334. [DOI] [PubMed] [Google Scholar]
- 77.El Asmar R, Panigrahi P, Bamford P, Berti I, Not T, Coppa GV, Catassi C, Fasano A. Host-dependent zonulin secretion causes the impairment of the small intestine barrier function after bacterial exposure. Gastroenterology. 2002;123:1607–1615. doi: 10.1053/gast.2002.36578. [DOI] [PubMed] [Google Scholar]
- 78.Fasano A. Zonulin, regulation of tight junctions, and autoimmune diseases. Ann N Y Acad Sci. 2012;1258:25–33. doi: 10.1111/j.1749-6632.2012.06538.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Soh UJ, Dores MR, Chen B, Trejo J. Signal transduction by protease-activated receptors. Br J Pharmacol. 2010;160:191–203. doi: 10.1111/j.1476-5381.2010.00705.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fasano A. Zonulin and its regulation of intestinal barrier function: the biological door to inflammation, autoimmunity, and cancer. Physiol Rev. 2011;91:151–175. doi: 10.1152/physrev.00003.2008. [DOI] [PubMed] [Google Scholar]
- 81.Di Pierro M, Lu R, Uzzau S, Wang W, Margaretten K, Pazzani C, Maimone F, Fasano A. Zonula occludens toxin structure-function analysis. Identification of the fragment biologically active on tight junctions and of the zonulin receptor binding domain. J Biol Chem. 2001;276:19160–19165. doi: 10.1074/jbc.M009674200. [DOI] [PubMed] [Google Scholar]
- 82.Watts T, Berti I, Sapone A, Gerarduzzi T, Not T, Zielke R, Fasano A. Role of the intestinal tight junction modulator zonulin in the pathogenesis of type I diabetes in BB diabetic-prone rats. Proc Natl Acad Sci USA. 2005;102:2916–2921. doi: 10.1073/pnas.0500178102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Arrieta MC, Madsen K, Doyle J, Meddings J. Reducing small intestinal permeability attenuates colitis in the IL10 gene-deficient mouse. Gut. 2009;58:41–48. doi: 10.1136/gut.2008.150888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Paterson BM, Lammers KM, Arrieta MC, Fasano A, Meddings JB. The safety, tolerance, pharmacokinetic and pharmacodynamic effects of single doses of AT-1001 in coeliac disease subjects: a proof of concept study. Aliment Pharmacol Ther. 2007;26:757–766. doi: 10.1111/j.1365-2036.2007.03413.x. [DOI] [PubMed] [Google Scholar]
- 85.Gettins PG. Serpin structure, mechanism, and function. Chem Rev. 2002;102:4751–4804. doi: 10.1021/cr010170+. [DOI] [PubMed] [Google Scholar]
- 86.Schmid M, Fellermann K, Fritz P, Wiedow O, Stange EF, Wehkamp J. Attenuated induction of epithelial and leukocyte serine antiproteases elafin and secretory leukocyte protease inhibitor in Crohn’s disease. J Leukoc Biol. 2007;81:907–915. doi: 10.1189/jlb.0906581. [DOI] [PubMed] [Google Scholar]
- 87.Galipeau HJ, Wiepjes M, Motta JP, Schulz JD, Jury J, Natividad JM, Pinto-Sanchez I, Sinclair D, Rousset P, Martin-Rosique R, et al. Novel role of the serine protease inhibitor elafin in gluten-related disorders. Am J Gastroenterol. 2014;109:748–756. doi: 10.1038/ajg.2014.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Motta JP, Magne L, Descamps D, Rolland C, Squarzoni-Dale C, Rousset P, Martin L, Cenac N, Balloy V, Huerre M, et al. Modifying the protease, antiprotease pattern by elafin overexpression protects mice from colitis. Gastroenterology. 2011;140:1272–1282. doi: 10.1053/j.gastro.2010.12.050. [DOI] [PubMed] [Google Scholar]
- 89.Zhao J, Wang J, Dong L, Shi H, Wang Z, Ding H, Shi H, Lu X. A protease inhibitor against acute stress-induced visceral hypersensitivity and paracellular permeability in rats. Eur J Pharmacol. 2011;654:289–294. doi: 10.1016/j.ejphar.2010.12.032. [DOI] [PubMed] [Google Scholar]
- 90.Isozaki Y, Yoshida N, Kuroda M, Handa O, Takagi T, Kokura S, Ichikawa H, Naito Y, Okanoue T, Yoshikawa T. Anti-tryptase treatment using nafamostat mesilate has a therapeutic effect on experimental colitis. Scand J Gastroenterol. 2006;41:944–953. doi: 10.1080/00365520500529470. [DOI] [PubMed] [Google Scholar]
- 91.Ceuleers H, Segaert E, Heirbaut J, Hanning N, Francque SM, Joossens J, De Man JG, De Winter BY. Su 1937 Two Serine Protease Inhibitors, Nafamostat Mesylate and the Newly Developed SPIx, Decrease Post-Inflammatory Visceral Hypersensitivity in Rats. Gastroenterology. 2016;150:S593–S594. [Google Scholar]
- 92.Chen CC, Wang SS, Lee FY. Action of antiproteases on the inflammatory response in acute pancreatitis. JOP. 2007;8:488–494. [PubMed] [Google Scholar]
- 93.Liu W, Hu D, Huo H, Zhang W, Adiliaghdam F, Morrison S, Ramirez JM, Gul SS, Hamarneh SR, Hodin RA. Intestinal Alkaline Phosphatase Regulates Tight Junction Protein Levels. J Am Coll Surg. 2016;222:1009–1017. doi: 10.1016/j.jamcollsurg.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lee JW, Park JH, Park DI, Park JH, Kim HJ, Cho YK, Sohn CI, Jeon WK, Kim BI. Subjects with diarrhea-predominant IBS have increased rectal permeability responsive to tryptase. Dig Dis Sci. 2010;55:2922–2928. doi: 10.1007/s10620-009-1094-8. [DOI] [PubMed] [Google Scholar]
- 95.Wilcz-Villega EM, McClean S, O’Sullivan MA. Mast cell tryptase reduces junctional adhesion molecule-A (JAM-A) expression in intestinal epithelial cells: implications for the mechanisms of barrier dysfunction in irritable bowel syndrome. Am J Gastroenterol. 2013;108:1140–1151. doi: 10.1038/ajg.2013.92. [DOI] [PubMed] [Google Scholar]
- 96.Van Spaendonk H, Nullens S, Ceuleers H, Schrijvers D, Francque SM, De Man J, De Winter BY. Tu1883 The Effect of a Protease Inhibitor in a Chronic Colitis Transfer Model. Gastroenterology. 2016;150:S967. [Google Scholar]
- 97.Rodríguez D, Morrison CJ, Overall CM. Matrix metalloproteinases: what do they not do? New substrates and biological roles identified by murine models and proteomics. Biochim Biophys Acta. 2010;1803:39–54. doi: 10.1016/j.bbamcr.2009.09.015. [DOI] [PubMed] [Google Scholar]
- 98.Cauwe B, Van den Steen PE, Opdenakker G. The biochemical, biological, and pathological kaleidoscope of cell surface substrates processed by matrix metalloproteinases. Crit Rev Biochem Mol Biol. 2007;42:113–185. doi: 10.1080/10409230701340019. [DOI] [PubMed] [Google Scholar]
- 99.Mäkitalo L, Kolho KL, Karikoski R, Anthoni H, Saarialho-Kere U. Expression profiles of matrix metalloproteinases and their inhibitors in colonic inflammation related to pediatric inflammatory bowel disease. Scand J Gastroenterol. 2010;45:862–871. doi: 10.3109/00365520903583863. [DOI] [PubMed] [Google Scholar]
- 100.Louis E, Ribbens C, Godon A, Franchimont D, De Groote D, Hardy N, Boniver J, Belaiche J, Malaise M. Increased production of matrix metalloproteinase-3 and tissue inhibitor of metalloproteinase-1 by inflamed mucosa in inflammatory bowel disease. Clin Exp Immunol. 2000;120:241–246. doi: 10.1046/j.1365-2249.2000.01227.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lawrance IC, Fiocchi C, Chakravarti S. Ulcerative colitis and Crohn’s disease: distinctive gene expression profiles and novel susceptibility candidate genes. Hum Mol Genet. 2001;10:445–456. doi: 10.1093/hmg/10.5.445. [DOI] [PubMed] [Google Scholar]
- 102.Rath T, Roderfeld M, Halwe JM, Tschuschner A, Roeb E, Graf J. Cellular sources of MMP-7, MMP-13 and MMP-28 in ulcerative colitis. Scand J Gastroenterol. 2010;45:1186–1196. doi: 10.3109/00365521.2010.499961. [DOI] [PubMed] [Google Scholar]
- 103.von Lampe B, Barthel B, Coupland SE, Riecken EO, Rosewicz S. Differential expression of matrix metalloproteinases and their tissue inhibitors in colon mucosa of patients with inflammatory bowel disease. Gut. 2000;47:63–73. doi: 10.1136/gut.47.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Pedersen G, Saermark T, Kirkegaard T, Brynskov J. Spontaneous and cytokine induced expression and activity of matrix metalloproteinases in human colonic epithelium. Clin Exp Immunol. 2009;155:257–265. doi: 10.1111/j.1365-2249.2008.03836.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Dufour A, Overall CM. Missing the target: matrix metalloproteinase antitargets in inflammation and cancer. Trends Pharmacol Sci. 2013;34:233–242. doi: 10.1016/j.tips.2013.02.004. [DOI] [PubMed] [Google Scholar]
- 106.Hu J, Van den Steen PE, Sang QX, Opdenakker G. Matrix metalloproteinase inhibitors as therapy for inflammatory and vascular diseases. Nat Rev Drug Discov. 2007;6:480–498. doi: 10.1038/nrd2308. [DOI] [PubMed] [Google Scholar]
- 107.Stallmach A, Chan CC, Ecker KW, Feifel G, Herbst H, Schuppan D, Zeitz M. Comparable expression of matrix metalloproteinases 1 and 2 in pouchitis and ulcerative colitis. Gut. 2000;47:415–422. doi: 10.1136/gut.47.3.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bailey CJ, Hembry RM, Alexander A, Irving MH, Grant ME, Shuttleworth CA. Distribution of the matrix metalloproteinases stromelysin, gelatinases A and B, and collagenase in Crohn’s disease and normal intestine. J Clin Pathol. 1994;47:113–116. doi: 10.1136/jcp.47.2.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ishida K, Takai S, Murano M, Nishikawa T, Inoue T, Murano N, Inoue N, Jin D, Umegaki E, Higuchi K, et al. Role of chymase-dependent matrix metalloproteinase-9 activation in mice with dextran sodium sulfate-induced colitis. J Pharmacol Exp Ther. 2008;324:422–426. doi: 10.1124/jpet.107.131946. [DOI] [PubMed] [Google Scholar]
- 110.Tarlton JF, Whiting CV, Tunmore D, Bregenholt S, Reimann J, Claesson MH, Bland PW. The role of up-regulated serine proteases and matrix metalloproteinases in the pathogenesis of a murine model of colitis. Am J Pathol. 2000;157:1927–1935. doi: 10.1016/S0002-9440(10)64831-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Garg P, Rojas M, Ravi A, Bockbrader K, Epstein S, Vijay-Kumar M, Gewirtz AT, Merlin D, Sitaraman SV. Selective ablation of matrix metalloproteinase-2 exacerbates experimental colitis: contrasting role of gelatinases in the pathogenesis of colitis. J Immunol. 2006;177:4103–4112. doi: 10.4049/jimmunol.177.6.4103. [DOI] [PubMed] [Google Scholar]
- 112.Liu H, Patel NR, Walter L, Ingersoll S, Sitaraman SV, Garg P. Constitutive expression of MMP9 in intestinal epithelium worsens murine acute colitis and is associated with increased levels of proinflammatory cytokine Kc. Am J Physiol Gastrointest Liver Physiol. 2013;304:G793–G803. doi: 10.1152/ajpgi.00249.2012. [DOI] [PubMed] [Google Scholar]
- 113.Garg P, Ravi A, Patel NR, Roman J, Gewirtz AT, Merlin D, Sitaraman SV. Matrix metalloproteinase-9 regulates MUC-2 expression through its effect on goblet cell differentiation. Gastroenterology. 2007;132:1877–1889. doi: 10.1053/j.gastro.2007.02.048. [DOI] [PubMed] [Google Scholar]
- 114.Nighot P, Al-Sadi R, Rawat M, Guo S, Watterson DM, Ma T. Matrix metalloproteinase 9-induced increase in intestinal epithelial tight junction permeability contributes to the severity of experimental DSS colitis. Am J Physiol Gastrointest Liver Physiol. 2015;309:G988–G997. doi: 10.1152/ajpgi.00256.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Cunningham KE, Turner JR. Myosin light chain kinase: pulling the strings of epithelial tight junction function. Ann N Y Acad Sci. 2012;1258:34–42. doi: 10.1111/j.1749-6632.2012.06526.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Agarwal R, D’Souza T, Morin PJ. Claudin-3 and claudin-4 expression in ovarian epithelial cells enhances invasion and is associated with increased matrix metalloproteinase-2 activity. Cancer Res. 2005;65:7378–7385. doi: 10.1158/0008-5472.CAN-05-1036. [DOI] [PubMed] [Google Scholar]
- 117.Miyamori H, Takino T, Kobayashi Y, Tokai H, Itoh Y, Seiki M, Sato H. Claudin promotes activation of pro-matrix metalloproteinase-2 mediated by membrane-type matrix metalloproteinases. J Biol Chem. 2001;276:28204–28211. doi: 10.1074/jbc.M103083200. [DOI] [PubMed] [Google Scholar]
- 118.Meijer MJ, Mieremet-Ooms MA, van der Zon AM, van Duijn W, van Hogezand RA, Sier CF, Hommes DW, Lamers CB, Verspaget HW. Increased mucosal matrix metalloproteinase-1, -2, -3 and -9 activity in patients with inflammatory bowel disease and the relation with Crohn’s disease phenotype. Dig Liver Dis. 2007;39:733–739. doi: 10.1016/j.dld.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 119.O’Sullivan S, Gilmer JF, Medina C. Matrix metalloproteinases in inflammatory bowel disease: an update. Mediators Inflamm. 2015;2015:964131. doi: 10.1155/2015/964131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Rath T, Roderfeld M, Graf J, Wagner S, Vehr AK, Dietrich C, Geier A, Roeb E. Enhanced expression of MMP-7 and MMP-13 in inflammatory bowel disease: a precancerous potential? Inflamm Bowel Dis. 2006;12:1025–1035. doi: 10.1097/01.mib.0000234133.97594.04. [DOI] [PubMed] [Google Scholar]
- 121.Noë V, Fingleton B, Jacobs K, Crawford HC, Vermeulen S, Steelant W, Bruyneel E, Matrisian LM, Mareel M. Release of an invasion promoter E-cadherin fragment by matrilysin and stromelysin-1. J Cell Sci. 2001;114:111–118. doi: 10.1242/jcs.114.1.111. [DOI] [PubMed] [Google Scholar]
- 122.Schneider MR, Dahlhoff M, Horst D, Hirschi B, Trülzsch K, Müller-Höcker J, Vogelmann R, Allgäuer M, Gerhard M, Steininger S, et al. A key role for E-cadherin in intestinal homeostasis and Paneth cell maturation. PLoS One. 2010;5:e14325. doi: 10.1371/journal.pone.0014325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Muise AM, Walters TD, Glowacka WK, Griffiths AM, Ngan BY, Lan H, Xu W, Silverberg MS, Rotin D. Polymorphisms in E-cadherin (CDH1) result in a mis-localised cytoplasmic protein that is associated with Crohn’s disease. Gut. 2009;58:1121–1127. doi: 10.1136/gut.2008.175117. [DOI] [PubMed] [Google Scholar]
- 124.Davies G, Jiang WG, Mason MD. Matrilysin mediates extracellular cleavage of E-cadherin from prostate cancer cells: a key mechanism in hepatocyte growth factor/scatter factor-induced cell-cell dissociation and in vitro invasion. Clin Cancer Res. 2001;7:3289–3297. [PubMed] [Google Scholar]
- 125.Wheelock MJ, Buck CA, Bechtol KB, Damsky CH. Soluble 80-kd fragment of cell-CAM 120/80 disrupts cell-cell adhesion. J Cell Biochem. 1987;34:187–202. doi: 10.1002/jcb.240340305. [DOI] [PubMed] [Google Scholar]
- 126.Hu QP, Kuang JY, Yang QK, Bian XW, Yu SC. Beyond a tumor suppressor: Soluble E-cadherin promotes the progression of cancer. Int J Cancer. 2016;138:2804–2812. doi: 10.1002/ijc.29982. [DOI] [PubMed] [Google Scholar]
- 127.Nawrocki-Raby B, Gilles C, Polette M, Bruyneel E, Laronze JY, Bonnet N, Foidart JM, Mareel M, Birembaut P. Upregulation of MMPs by soluble E-cadherin in human lung tumor cells. Int J Cancer. 2003;105:790–795. doi: 10.1002/ijc.11168. [DOI] [PubMed] [Google Scholar]
- 128.Schütte A, Ermund A, Becker-Pauly C, Johansson ME, Rodriguez-Pineiro AM, Bäckhed F, Müller S, Lottaz D, Bond JS, Hansson GC. Microbial-induced meprin β cleavage in MUC2 mucin and a functional CFTR channel are required to release anchored small intestinal mucus. Proc Natl Acad Sci USA. 2014;111:12396–12401. doi: 10.1073/pnas.1407597111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Shan M, Gentile M, Yeiser JR, Walland AC, Bornstein VU, Chen K, He B, Cassis L, Bigas A, Cols M, et al. Mucus enhances gut homeostasis and oral tolerance by delivering immunoregulatory signals. Science. 2013;342:447–453. doi: 10.1126/science.1237910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Moss ML, Jin SL, Milla ME, Bickett DM, Burkhart W, Carter HL, Chen WJ, Clay WC, Didsbury JR, Hassler D, et al. Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-alpha. Nature. 1997;385:733–736. doi: 10.1038/385733a0. [DOI] [PubMed] [Google Scholar]
- 131.Ma TY, Iwamoto GK, Hoa NT, Akotia V, Pedram A, Boivin MA, Said HM. TNF-alpha-induced increase in intestinal epithelial tight junction permeability requires NF-kappa B activation. Am J Physiol Gastrointest Liver Physiol. 2004;286:G367–G376. doi: 10.1152/ajpgi.00173.2003. [DOI] [PubMed] [Google Scholar]
- 132.Al-Sadi R, Guo S, Ye D, Ma TY. TNF-α modulation of intestinal epithelial tight junction barrier is regulated by ERK1/2 activation of Elk-1. Am J Pathol. 2013;183:1871–1884. doi: 10.1016/j.ajpath.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Moss ML, White JM, Lambert MH, Andrews RC. TACE and other ADAM proteases as targets for drug discovery. Drug Discov Today. 2001;6:417–426. doi: 10.1016/s1359-6446(01)01738-x. [DOI] [PubMed] [Google Scholar]
- 134.Colón AL, Menchén LA, Hurtado O, De Cristóbal J, Lizasoain I, Leza JC, Lorenzo P, Moro MA. Implication of TNF-alpha convertase (TACE/ADAM17) in inducible nitric oxide synthase expression and inflammation in an experimental model of colitis. Cytokine. 2001;16:220–226. doi: 10.1006/cyto.2001.0969. [DOI] [PubMed] [Google Scholar]
- 135.Brynskov J, Foegh P, Pedersen G, Ellervik C, Kirkegaard T, Bingham A, Saermark T. Tumour necrosis factor alpha converting enzyme (TACE) activity in the colonic mucosa of patients with inflammatory bowel disease. Gut. 2002;51:37–43. doi: 10.1136/gut.51.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Forsyth CB, Banan A, Farhadi A, Fields JZ, Tang Y, Shaikh M, Zhang LJ, Engen PA, Keshavarzian A. Regulation of oxidant-induced intestinal permeability by metalloprotease-dependent epidermal growth factor receptor signaling. J Pharmacol Exp Ther. 2007;321:84–97. doi: 10.1124/jpet.106.113019. [DOI] [PubMed] [Google Scholar]
- 137.Fréour T, Jarry A, Bach-Ngohou K, Dejoie T, Bou-Hanna C, Denis MG, Mosnier JF, Laboisse CL, Masson D. TACE inhibition amplifies TNF-alpha-mediated colonic epithelial barrier disruption. Int J Mol Med. 2009;23:41–48. [PubMed] [Google Scholar]
- 138.Man SM, Kanneganti TD. Converging roles of caspases in inflammasome activation, cell death and innate immunity. Nat Rev Immunol. 2016;16:7–21. doi: 10.1038/nri.2015.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Leblond CP. The life history of cells in renewing systems. Am J Anat. 1981;160:114–158. doi: 10.1002/aja.1001600202. [DOI] [PubMed] [Google Scholar]
- 140.Fiocchi C. Inflammatory bowel disease: etiology and pathogenesis. Gastroenterology. 1998;115:182–205. doi: 10.1016/s0016-5085(98)70381-6. [DOI] [PubMed] [Google Scholar]
- 141.Ruemmele FM, Seidman EG, Lentze MJ. Regulation of intestinal epithelial cell apoptosis and the pathogenesis of inflammatory bowel disorders. J Pediatr Gastroenterol Nutr. 2002;34:254–260. doi: 10.1097/00005176-200203000-00005. [DOI] [PubMed] [Google Scholar]
- 142.Bojarski C, Weiske J, Schöneberg T, Schröder W, Mankertz J, Schulzke JD, Florian P, Fromm M, Tauber R, Huber O. The specific fates of tight junction proteins in apoptotic epithelial cells. J Cell Sci. 2004;117:2097–2107. doi: 10.1242/jcs.01071. [DOI] [PubMed] [Google Scholar]
- 143.Bojarski C, Gitter AH, Bendfeldt K, Mankertz J, Schmitz H, Wagner S, Fromm M, Schulzke JD. Permeability of human HT-29/B6 colonic epithelium as a function of apoptosis. J Physiol. 2001;535:541–552. doi: 10.1111/j.1469-7793.2001.00541.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Beeman N, Webb PG, Baumgartner HK. Occludin is required for apoptosis when claudin-claudin interactions are disrupted. Cell Death Dis. 2012;3:e273. doi: 10.1038/cddis.2012.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kostic AD, Xavier RJ, Gevers D. The microbiome in inflammatory bowel disease: current status and the future ahead. Gastroenterology. 2014;146:1489–1499. doi: 10.1053/j.gastro.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Motta JP, Martin L, Vergnolle N. Proteases/Antiproteases in Inflammatory Bowel Disease. In: Vergnolle N, Chignard M, SpringerLink (Online service), editors. Proteases and Their Receptors in Inflammation. Basel: Springer Basel AG; 2011. p. 1 online resource. [Google Scholar]
- 147.Steck N, Mueller K, Schemann M, Haller D. Bacterial proteases in IBD and IBS. Gut. 2012;61:1610–1618. doi: 10.1136/gutjnl-2011-300775. [DOI] [PubMed] [Google Scholar]
- 148.Róka R, Rosztóczy A, Leveque M, Izbéki F, Nagy F, Molnár T, Lonovics J, Garcia-Villar R, Fioramonti J, Wittmann T, et al. A pilot study of fecal serine-protease activity: a pathophysiologic factor in diarrhea-predominant irritable bowel syndrome. Clin Gastroenterol Hepatol. 2007;5:550–555. doi: 10.1016/j.cgh.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 149.Obiso RJ, Azghani AO, Wilkins TD. The Bacteroides fragilis toxin fragilysin disrupts the paracellular barrier of epithelial cells. Infect Immun. 1997;65:1431–1439. doi: 10.1128/iai.65.4.1431-1439.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Wells CL, van de Westerlo EM, Jechorek RP, Feltis BA, Wilkins TD, Erlandsen SL. Bacteroides fragilis enterotoxin modulates epithelial permeability and bacterial internalization by HT-29 enterocytes. Gastroenterology. 1996;110:1429–1437. doi: 10.1053/gast.1996.v110.pm8613048. [DOI] [PubMed] [Google Scholar]
- 151.Zhang Z, Wang L, Seydel KB, Li E, Ankri S, Mirelman D, Stanley SL. Entamoeba histolytica cysteine proteinases with interleukin-1 beta converting enzyme (ICE) activity cause intestinal inflammation and tissue damage in amoebiasis. Mol Microbiol. 2000;37:542–548. doi: 10.1046/j.1365-2958.2000.02037.x. [DOI] [PubMed] [Google Scholar]
- 152.Kissoon-Singh V, Moreau F, Trusevych E, Chadee K. Entamoeba histolytica exacerbates epithelial tight junction permeability and proinflammatory responses in Muc2(-/-) mice. Am J Pathol. 2013;182:852–865. doi: 10.1016/j.ajpath.2012.11.035. [DOI] [PubMed] [Google Scholar]
- 153.Betanzos A, Javier-Reyna R, García-Rivera G, Bañuelos C, González-Mariscal L, Schnoor M, Orozco E. The EhCPADH112 complex of Entamoeba histolytica interacts with tight junction proteins occludin and claudin-1 to produce epithelial damage. PLoS One. 2013;8:e65100. doi: 10.1371/journal.pone.0065100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Pruteanu M, Hyland NP, Clarke DJ, Kiely B, Shanahan F. Degradation of the extracellular matrix components by bacterial-derived metalloproteases: implications for inflammatory bowel diseases. Inflamm Bowel Dis. 2011;17:1189–1200. doi: 10.1002/ibd.21475. [DOI] [PubMed] [Google Scholar]
- 155.Maharshak N, Huh EY, Paiboonrungruang C, Shanahan M, Thurlow L, Herzog J, Djukic Z, Orlando R, Pawlinski R, Ellermann M, et al. Enterococcus faecalis Gelatinase Mediates Intestinal Permeability via Protease-Activated Receptor 2. Infect Immun. 2015;83:2762–2770. doi: 10.1128/IAI.00425-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Steck N, Hoffmann M, Sava IG, Kim SC, Hahne H, Tonkonogy SL, Mair K, Krueger D, Pruteanu M, Shanahan F, et al. Enterococcus faecalis metalloprotease compromises epithelial barrier and contributes to intestinal inflammation. Gastroenterology. 2011;141:959–971. doi: 10.1053/j.gastro.2011.05.035. [DOI] [PubMed] [Google Scholar]
- 157.Wan H, Winton HL, Soeller C, Tovey ER, Gruenert DC, Thompson PJ, Stewart GA, Taylor GW, Garrod DR, Cannell MB, et al. Der p 1 facilitates transepithelial allergen delivery by disruption of tight junctions. J Clin Invest. 1999;104:123–133. doi: 10.1172/JCI5844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Tulic MK, Vivinus-Nébot M, Rekima A, Rabelo Medeiros S, Bonnart C, Shi H, Walker A, Dainese R, Boyer J, Vergnolle N, et al. Presence of commensal house dust mite allergen in human gastrointestinal tract: a potential contributor to intestinal barrier dysfunction. Gut. 2016;65:757–766. doi: 10.1136/gutjnl-2015-310523. [DOI] [PubMed] [Google Scholar]
- 159.Yu LC. Intestinal epithelial barrier dysfunction in food hypersensitivity. J Allergy (Cairo) 2012;2012:596081. doi: 10.1155/2012/596081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Perrier C, Corthésy B. Gut permeability and food allergies. Clin Exp Allergy. 2011;41:20–28. doi: 10.1111/j.1365-2222.2010.03639.x. [DOI] [PubMed] [Google Scholar]
- 161.Grozdanovic MM, Čavić M, Nešić A, Andjelković U, Akbari P, Smit JJ, Gavrović-Jankulović M. Kiwifruit cysteine protease actinidin compromises the intestinal barrier by disrupting tight junctions. Biochim Biophys Acta. 2016;1860:516–526. doi: 10.1016/j.bbagen.2015.12.005. [DOI] [PubMed] [Google Scholar]
- 162.Cavic M, Grozdanovic MM, Bajic A, Jankovic R, Andjus PR, Gavrovic-Jankulovic M. The effect of kiwifruit (Actinidia deliciosa) cysteine protease actinidin on the occludin tight junction network in T84 intestinal epithelial cells. Food Chem Toxicol. 2014;72:61–68. doi: 10.1016/j.fct.2014.07.012. [DOI] [PubMed] [Google Scholar]
- 163.Yang Y, Sitanggang NV, Kato N, Inoue J, Murakami T, Watanabe T, Takafumi Iguchi, Yukako Okazaki. Beneficial effects of protease preparations derived from Aspergillus on the colonic luminal environment in rats consuming a high-fat diet. Biomed Rep. 2015;3:715–720. doi: 10.3892/br.2015.490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Morrison DJ, Preston T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes. 2016;7:189–200. doi: 10.1080/19490976.2015.1134082. [DOI] [PMC free article] [PubMed] [Google Scholar]