

Abstract
Histone deacetylase 6 (HDAC6) inhibition has been reported to protect against ischemic stroke and prolong survival after sepsis in animal models. However, it remains unknown whether HDAC6 inhibition offers a renoprotective effect after acute kidney injury (AKI). In this study, we examined the effect of tubastatin A (TA), a highly selective inhibitor of HDAC6, on AKI in a murine model of glycerol (GL) injection-induced rhabdomyolysis. Following GL injection, the mice developed severe acute tubular injury as indicated by renal dysfunction; expression of neutrophil gelatinase-associated lipocalin (NGAL), an injury marker of renal tubules; and an increase of TdT-mediated dUTP nick-end labeling (TUNEL)-positive tubular cells. These changes were companied by increased HDAC6 expression in the cytoplasm of renal tubular cells. Administration of TA significantly reduced serum creatinine and blood urea nitrogen levels as well as attenuated renal tubular damage in injured kidneys. HDAC6 inhibition also resulted in decreased expression of NGAL, reduced apoptotic cell, and inactivated caspase-3 in the kidney after acute injury. Moreover, injury to the kidney increased phosphorylation of nuclear factor (NF)-κB and expression of multiple cytokines/chemokines including tumor necrotic factor-α and interleukin-6 and monocyte chemoattractant protein-1, as well as macrophage infiltration. Treatment with TA attenuated all those responses. Finally, HDAC6 inhibition reduced the level of oxidative stress by suppressing malondialdehyde (MDA) and preserving expression of superoxide dismutase (SOD) in the injured kidney. Collectively, these data indicate that HDAC6 contributes to the pathogenesis of rhabdomyolysis-induced AKI and suggest that HDAC6 inhibitors have therapeutic potential for AKI treatment.
Keywords: rhabdomyolysis, acute kidney injury, HDAC6, inflammation, oxidative stress
acute kidney injury (AKI) characterized by a rapid decline of the glomerular filtration rate is a serious clinical problem. It is not only linked with high rates of mortality and but also an increased risk of chronic kidney diseases (CKD) (12). Rhabdomyolysis, one of the major causes of community-acquired AKI accounting for 5–15% of cases, is induced by different conditions, including crush injuries, severe trauma, intense physical exercise, and some medications and illicit drugs like cocaine (21, 30, 53).
Although the detailed mechanism responsible for pathogenesis of rhabdomyolysis-induced AKI is poorly understood, rhabdomyolysis produces multiple deleterious effects on the kidney, including apoptosis, inflammation, oxidative stress, vasoconstriction, and tubular obstruction (15, 30). During these pathological processes, multiple signaling pathways and numerous genes involved in cell death and inflammatory responses are activated and/or upregulated (13). For example, inhibition of caspase-3 has been shown to ameliorate renal damage in an experimental animal model of rhabdomyolysis, while activation of the NF-κB pathway is essential for the progression of rhabdomyolysis-induced AKI (13). In addition, multiple cytokines/chemokines, including tumor necrosis factor-α (TNF-α), and interleukin-1 (IL-1), interleukin-6 (IL-6), and monocyte chemoattractant protein-1 (MCP-1), are upregulated and produced in the injured kidney after glycerol (GL) injection (30). Moreover, oxidative stress also contributes to the development of AKI. Previous studies demonstrated that oxidative stress, which occurred in the condition of excessive pro-oxidants or reactive oxygen species (ROS) in the body, was a predominant cause involved in the pathogenesis of AKI (35). Malondialdehyde (MDA) and superoxide dismutase (SOD), two sensitive biomarkers for oxidative stress, can largely reflect the level of oxidative stress (35). Currently, the mechanisms that control gene expression and signaling pathway activation in rhabdomyolysis-induced AKI remain unclear. Understanding the molecular basis of these processes will aid in devising therapeutic strategies to treat AKI.
Lysine acetylation, a critical posttranslational modification in regulation of protein function, is a key component of many signaling networks (26, 37). An acetyl group can be added to a lysine residue by histone acetyl transferases (HATs) and removed by histone deacetylases (HDACs). Based on their structure and homology to yeast histone deacetylases, HDACs are classified into four classes: class I HDACs (HDAC1, 2, 3, and 8); class II HDACs (HDAC4, 5, 6, 7, 9, and 10); class III HDACs (SIRT1–7); and class IV (HDAC11) (8, 9). HDACs have been shown to be associated with many cell functions including cell growth, dedifferentiation, and proliferation and apoptosis (24, 41) and have become a target for drug development to treat cancer, including acute myeloid leukemia, acute lymphocytic leukemia, multiple myeloma, and breast cancer (43).
Of the 11 isoforms of HDACs, HDAC6 is unique among histone deacetylases: existing predominantly in the cytoplasm, it is able to deacetylate nonhistone proteins in the cytoplasm (23). Increasing evidence has indicated that the function of HDAC6 is not limited to tumorgenesis but also implicated in some chronic diseases such as neurodegenerative diseases, cardiovascular disease, fibrosis, and cystogenesis (3, 4, 8, 11, 51). Interestingly, recent studies have shown that HDAC6 activation is also associated with pathogenesis of acute organ/tissue injury (9, 25, 38, 46). For example, postischemic treatment with tubastatin A (TA), a highly selective HDAC6 inhibitor, improved functional recovery, reduced brain infarct volume, and ameliorated neuronal cell death in a rat model of transient middle cerebral artery occlusion (38). TA also significantly improves survival in a mouse model of cecal ligation and puncture-induced lethal sepsis (25) and in a rat model of hemorrhagic shock (9). Furthermore, TA was able to block TNF-α-induced lung endothelial cell hyperpermeability, which is associated with reduced pulmonary edema (46). These data indicate that targeting HDAC6 with selective pharmacological inhibitors is protective against acute injury to tissues and organs (9, 25, 38, 46). However, it remains unclear whether HDAC6 inhibition also offers a renoprotective effect in AKI.
In this study, we examined the effect of HDAC6 inhibition with TA on rhabdomyolysis-induced AKI and the mechanisms involved.
MATERIALS AND METHODS
Antibodies and reagents.
Tubastatin A and MG132 were purchased from Selleckchem (Houston, TX). Antibodies against HDAC6, acetylation histone H3, cleaved caspase-3, p-NF-κB (p65), and NF-κB (p65) were purchased from Cell Signaling Technology (Dancers, MA). Antibodies to F4/80, MCP-1, IL-6, and TNF-α were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Antibody to neutrophil gelatinase-associated lipocalin (NGAL) was purchased from R&D Systems (Minneapolis, MN). Texas red-labeled or FITC green-labeled secondary antibodies for immunofluorescent staining were purchased from Invitrogen (Grand Island, NY). Malondialdehyde (MDA), superoxide dismutase (SOD), and creatine kinase (CK) biochemical reagent kits were purchased from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). Antibody to β-actin, secondary antibodies for Western blot, and all other chemicals were from Sigma (St. Louis, MO).
Creation of rhabdomyolysis-induced AKI mouse model and TA treatment.
Male C57/black mice (Shanghai Super–B&K Laboratory Animal) that weighed 20–25 g were used for the study. To establish GL-induced acute kidney injury, the animals were injected with 50% GL at 6 ml/kg body wt to the two hind legs intramuscularly. To examine the effect of the inhibitor (TA) on AKI, TA (70 mg/kg) in 50 μl of DMSO was given intraperitoneally immediately after GL injection and then administered daily. The sham group was injected with DMSO as a control. Six mice were used in each group. At 48 h after GL injection, the animals were killed and kidneys were collected for protein analysis and histological examination. Blood was taken for the measurement of serum creatinine and blood urea nitrogen (BUN). The animal protocol was reviewed and approved by the Institutional Animal Care and Use Committee at Tongji University (Shanghai, China).
Measurement of renal function.
Renal function was evaluated by serum creatinine and BUN, which were determined by using automatic biochemistry assay (P800; Modular).
Assay of oxidative stress index.
The concentration of malondialdehyde (MDA) and superoxide dismutase (SOD) in kidney tissues were detected by commercial kits according to the manufacturer’s instruction, and the final levels of MDA and SOD were normalized to the protein concentration of kidney tissue homogenate.
Measurement of creatine kinase.
At 48 h after GL injection, blood samples (1 ml/mouse) were collected and serum was obtained by centrifugation. Commercial assay kits from Nanjing Jiancheng were used to detect the activities of serum CK. After the color developing agent was added, mixture was measured spectrophotometrically at 660 nm. According to the standard curve, serum CK activity was calculated and expressed as U/ml.
Cell culture and treatment.
Human tubular epithelial cells (HK2) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) with F-12 containing 5% fetal bovine serum (FBS) and 0.5% penicillin and streptomycin in an atmosphere of 5% CO2-95% air at 37°C. To determine the effect of MG132, HK2 cells were starved for 24 h with DMEM containing 0.5% FBS and then exposed to TA (10 μM) and cisplatin (20 μg/ml) for 24 h in the presence or absence of MG132 (5 μM). All of the in vitro experiments were repeated four times.
Assessment of tubular injury.
The degree of tubular injury was determined using a semiquantitative grade scale that tubular injury was scored on a scale from 0 to 3, where 0 = normal, 1 = injury <30%, 2 = injury, 30–60%, and 3 = injury>60%. Two sections were randomly selected from each sample of at least 3 for every group and 10 fields were randomly selected at a magnification of ×200 from each section in periodic acid-Schiff staining. To determine apoptosis, kidneys were removed at 48 h after GL injection. TdT-mediated dUTP nick-end labeling (TUNEL) staining was performed according to the protocol provided by Roche Molecular System (Branchburg, NJ).
Immunoblot analysis.
Immunoblot analysis for tissue samples was performed according to our previous protocols (29). Densitometry analysis of immunoblot results was conducted with ImageJ software (National Institutes of Health, Bethesda, MD).
Immunofluorescent staining.
Immunofluorescent staining was carried out according to the procedure described in our previous studies (29). Briefly, renal tissues were fixed in 4.5% buffered formalin, dehydrated, and embedded in paraffin. For immunofluorescent staining, the tissue sections were rehydrated and labeled with antibodies, including primary antibodies NGAL (1:100) and HDAC6 (1:100) and then exposed to Texas red-labeled or FITC green-labeled secondary antibodies (Invitrogen).
Immunohistochemical staining.
The 3-μm thick sections were deparaffinized and rehydrated, quenched with 3% H2O2, immersed in citrate buffer, and heated in microwave, and periodic acid-Schiff staining was performed according to the protocol provided by the manufacturer (Sigma-Aldrich). Immunohistochemical staining was performed as previously described (29).
Statistical analysis.
All the experiments were conducted at least three times. Data depicted in graphs represent the means ± SE for each group. Intergroup comparisons were made using one-way ANOVA. Multiple means were compared using Tukey’s test. The differences between two groups were determined by Student t-test. Statistical significant difference between mean values was marked in each graph. P < 0.05 is considered significant.
RESULTS
Inhibition of HDAC6 with TA alleviates rhabdomyolysis-induced AKI.
Recent studies have reported that HDAC6 inhibition offers a neuroprotective effect and prolongs survival in an animal model of sepsis (9, 25, 38). To determine whether targeting HDAC6 may also have a renal protective effect, we examined the effect of TA, a selective HDAC6 inhibitor, on renal function and pathological changes in a mouse model of rhabdomyolysis-induced AKI. As demonstrated in Fig. 1, A–C, at 48 h after GL injection, both serum creatinine and BUN were markedly elevated and tubular damage (as evidenced by increase of tubular dilatation, swelling, necrosis, luminal congestion) was clearly observed. Administration of TA significantly improved renal dysfunction and alleviated renal pathologic changes. The tubular injury score of kidney sections showed a significant decrease in mice injected with GL followed by TA treatment compared with those injected with GL alone (Fig. 1D). Administration of TA neither affected renal function nor caused renal damage in mice without GL injection. In addition, serum CK levels were markedly increased in mice at 48 h after GL injection and TA treatment significantly reduced this response (Fig. 1E). These data suggest that inhibition of HDAC6 activity protects against rhabdomyolysis-induced AKI.
Fig. 1.

Inhibition of histone deacetylase 6 (HDAC6) with tubastatin A (TA) alleviates glycerol (GL)-induced acute kidney injury. A: serum creatinine. B: serum blood urea nitrogen. C: photomicrographs (×200) illustrate periodic acid-Schiff (PAS) staining of the kidney tissues in control or GL mice with/without TA. D: morphological changes were scored based on the scale described in materials and methods. E: serum creatine kinase (CK). Data are represented as the means ± SE (n = 6). Means with different superscript letters are significantly different from one another (P < 0.05).
TA inhibits expression of HDAC6 and enhances acetylation of histone H3 in the kidney of rhabdomyolysis-induced AKI.
Inhibition of histone deacetylase is reflected by increased expression of acetyl histone H3. To understand the inhibitory effect of TA on HDAC6, we examined expression of acetyl histone H3 and HDAC6 by immunoblot analysis and immunofluorescence staining, respectively. In the sham kidney of mice, a low level of acetylated histone H3 was detected and its expression level was slightly increased after TA treatment. In comparison, GL injection increased the expression of acetylated histone H3 to the extent that was observed in the sham kidneys treated with TA. Injection of mice with GL followed by TA administration resulted in a remarkable increase in histone H3 acetylation, which was 10-fold higher than in mice injected with GL alone (Fig. 2, A and B). Sham kidneys exhibited a minimal level of renal HDAC6, while GL injection slightly increased its expression (Fig. 2, C and D). Administration of TA reduced levels of HDAC6 in both sham and GL-injected mice. Densitometry analysis displays more than an 80% reduction of HDAC6 in GL-induced AKI mice treated with TA compared with those treated with vehicle (Fig. 2, C and D). Consistent with our immunobot results, immunofluorescence staining also showed increased expression of HDAC6 in the kidney of GL-induced AKI. Notably, HDAC6 was most distributed in the cytoplasm of renal tubules. Taken together, these data indicate that TA is a potent HDAC6 inhibitor at the dose administered. TA also induces HDAC6 downregulation.
Fig. 2.

TA inhibits expression of HDAC6 and enhances acetylation histone H3. The kidney tissue lysates were subjected to immunoblot analysis with specific antibodies against acetylated histone H3 (A) and β-actin and HDAC6 (C). Expression level of acetyl-H3 (B) and HDAC6 (D) was quantified by densitometry and normalized with β-actin. E: photomicrographs illustrating HDAC6 immunofluorescence staining of kidney tissue collected at day 2 after GL administration with/without TA. Data are represented as the means ± SE (n = 6). Means with different superscript letters are significantly different from one another (P < 0.05).
TA attenuates tubular damage in the murine model of rhabdomyolysis-induced AKI.
NGAL has been recognized as a hallmark of renal tubular injury (1). To examine the role of HDAC6 in renal injury, we first analyzed the effect of TA on the expression of NGAL by Western blotting. As expected, GL-induced rhabdomyolysis resulted in an increase in the expression of NGAL in the kidney of mice. TA treatment dramatically reduced its expression. NGAL was not detected in sham kidneys with/without administration of TA. To confirm these results, we also performed the immunofluorescence staining. NGAL was localized in the injured tubules in rhabdomyolysis-induced AKI kidney and was largely diminished by TA treatment. NGAL expression was not observed in the sham kidneys either subjected to vehicle (DMSO) or TA (Fig. 3, B and D). These data suggest that inhibition of HDAC6 reduces renal tubular damage from rhabdomyolysis-induced AKI injury. TA itself does not injure renal tubular cells.
Fig. 3.

TA attenuates tubular damage in GL-induced acute kidney injury (AKI). A: the kidney tissue lysates were subjected to immunoblot analysis with specific antibodies against neutrophil gelatinase-associated lipocalin (NGAL) and β-actin. B: photomicrographs illustrating NGAL immunofluorescence staining of kidney tissue collected at 48 h after sham and GL administration with/without TA treatment. C: expression level of NGAL was quantified by densitometry and normalized with β-actin. D: positive NGAL staining cells were counted. Data are represented as the means ± SE (n = 6). Means with different superscript letters are significantly different from one another (P < 0.05).
Inhibition of HDAC6 activity decreases renal tubular cell apoptosis in rhabdomyolysis-induced AKI.
It has been documented that apoptosis is a primary feature of death in renal tubular cells after AKI (50). Thus we examined the effect of HDAC6 inhibition on tubular cell apoptosis by the TUNEL staining. As shown Fig. 4, A and C, there were no TUNEL-positive cells in sham mice with or without treatment of TA, but a large number of TUNEL-positive renal tubular cells were observed in the kidney after GL injection. TA administration reduced TUNEL positive cells by ~73% in the kidney of mice with GL injection compared with those injected with GL alone (Fig. 4C).
Fig. 4.

Inhibition of HDAC6 activity decreases renal tubular cell apoptosis in GL-induced AKI. A: photomicrographs illustrate TdT-mediated dUTP nick-end labeling (TUNEL) staining of kidney tissue collected at 48 h after sham and GL administration with/without TA treatment. B: the kidney tissue lysates were subjected to immunoblot analysis with specific antibodies against cleaved caspase-3 and β-actin. C: positive TUNEL staining cells were counted and expressed as means ± SE. D: expression level of cleaved caspase-3 was quantified by densitometry and normalized with β-actin. Data are represented as the means ± SE (n = 6). Means with different superscript letters are significantly different from one another (P < 0.05).
Caspase-3 is a primary mediator in apoptosis induced by a variety of stimuli (34). We further examined expression of cleaved caspase-3 in the kidney after injury with/without administration of TA by immunoblot analysis. Consistent with TUNEL staining results, expression of cleaved caspase-3 was observed in the kidney of mice after GL injection and TA treatment largely diminished this response. Expression of cleaved caspase-3 was not observed in the sham kidneys with or without administration with TA as indicated in Fig. 4, B and D.
Taken together, our data illustrate that HDAC6 may be coupled to the molecular machinery that leads to renal tubular cell apoptosis in GL-induced AKI. TA is not toxic to the kidney.
HDAC6 inhibition suppresses NF-κB phosphorylation in rhabdomyolysis-induced AKI.
It has been documented that the inflammatory response contributes to AKI initiation and progression (28). Activation of the NF-κB signaling pathway is the critical mechanism for inducing expression of proinflammatory chemokine/chemokines (16). To evaluate if HDAC6 participates in AKI-associated NF-κB signaling pathway activation, we further examined expression of phospho-NF-κB and total NF-κB by immunoblot analysis. Western blotting results show that phospho-NF-κB was minimally expressed in the sham kidneys administrated with or without TA (Fig. 5, A and B). Of note, phospho-NF-κB expression was markedly upregulated in the kidney after GL injection and suppressed in the injured kidney with TA administration. Total NF-κB was equally expressed in the kidney of every group (Fig. 5, A and B). Thus our data indicate that blockade of HDAC6 activity inhibits activation of the NF-κB signaling pathway in GL-induced AKI.
Fig. 5.
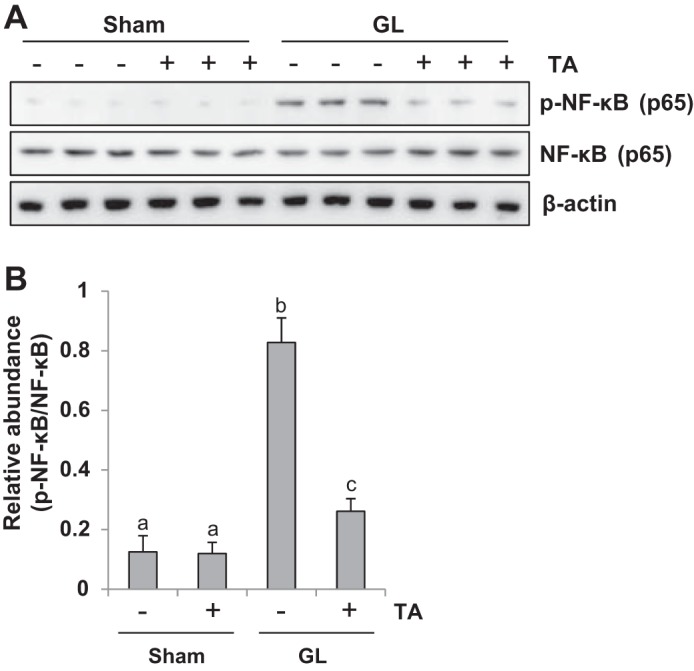
Treatment with TA significantly inhibited NF-κB signaling pathway activation. A: the kidney tissue lysates were subjected to immunoblot analysis with specific antibodies against phospho-NF-κB (p65), NF-κB (p65), and β-actin. B: expression level of phospho-NF-κB (p65) was quantified by densitometry and normalized with NF-κB (p65). Data are represented as the means ± SE (n = 6). Means with different superscript letters are significantly different from one another (P < 0.05).
HDAC6 inhibition reduces expression of proinflammatory cytokines/chemokines in rhabdomyolysis-induced AKI.
AKI triggers progressive activation of selected proinflammatory genes, leading to expression of proinflammatory cytokines that mediate organ dysfunction (28). To examine the effect of TA treatment on proinflammatory cytokines, we conducted immunohistochemistry staining for TNF-α, IL-6, and MCP-1. GL injection-induced upregulation of TNF-α (Fig. 6), IL-6 (Fig. 7), and MCP-1 (Fig. 8), which was significantly suppressed by TA administration. As indicated in Figs. 6–8, TNF-α, IL-6, and MCP-1 were predominantly expressed in the renal tubules. These data indicated that blocking of HDAC6 with TA suppresses expression of multiple proinflammatory cytokines/chemokines.
Fig. 6.

TA significantly reduced expression of TNF-α in GL-induced AKI injured kidneys. A: photomicrographs (×200) illustrating TNF-α-stained sections of kidney tissue on 48 h after GL treatment as indicated. B: TNF-α staining graphic presentation of quantitative data. Data are represented as the means ± SE (n = 6). Means with different superscript letters are significantly different from one another (P < 0.05).
Fig. 7.

TA significantly reduced expression of IL-6 in GL-induced AKI injured kidneys. A: photomicrographs (×200) illustrating IL-6-stained sections of kidney tissue on 48 h after GL treatment as indicated. B: IL-6 staining graphic presentation of quantitative data. Data are represented as the means ± SE (n = 6). Means with different superscript letters are significantly different from one another (P < 0.05).
Fig. 8.

TA significantly reduced expression of monocyte chemoattractant protein-1 (MCP-1) in GL-induced AKI injured kidneys. A: photomicrographs (×200) illustrating MCP-1-stained sections of kidney tissue on 48 h after GL treatment as indicated. B: MCP-1 staining graphic presentation of quantitative data. Data are represented as the means ± SE (n = 6). Means with different superscript letters are significantly different from one another (P < 0.05).
HDAC6 inhibition reduces infiltrating macrophage in the kidney of rhabdomyolysis-induced AKI.
It has been demonstrated that macrophage accumulation stands out as a significant pathologic feature in AKI progression (28). F4/80 is a marker of macrophage. Thus we performed F4/80 expression by immunohistochemistry. A low level of F4/80 was detected in the sham kidneys with or without TA treatment (Fig. 9, A and B). However, abundant expression of F4/80 was detected in the kidney after GL injection, primarily localized in interstitial area (Fig. 9, A and B). Suppression of HDAC6 activity remarkably reduced the number of F4/80-positive cells (Fig. 9, A and B). Thus TA also inhibits macrophage infiltration in GL-induced AKI.
Fig. 9.

TA significantly suppressed macrophage infiltration in GL-induced AKI injured kidneys. A: photomicrographs (×200) illustrating F4/80-stained sections of kidney tissue on 48 h after GL treatment as indicated. B: F4/80 staining graphic presentation of quantitative data. Data are represented as the means ± SE (n = 6). Means with different superscript letters are significantly different from one another (P < 0.05).
HDAC6 inhibition reduces oxidative stress in the kidney of rhabdomyolysis-induced AKI.
Production of ROS and lipid peroxidation products in the kidney and suppression of antioxidant systems are regarded as the major oxidative stress mechanism of AKI (2). To further explore the mechanisms of HDAC6-regulated AKI following rhabdomyolysis, we measured the content of MDA and SOD, two important biomarkers of oxidative stress, in the kidney. As indicated in Fig. 10, TA significantly suppressed MDA level and preserved expression of SOD in the kidney of mice with rhabdomyolysis-induced AKI. Therefore, HDAC6 inhibition may also alleviate AKI through reducing oxidative stress.
Fig. 10.

Treatment with TA reduced oxidative stress in GL-induced AKI. SOD and malondialdehyde (MDA) in kidney tissues were measured according to the protocol as indicated in materials and methods. A: SOD. B: MDA. Data are represented as the means ± SE (n = 6). Means with different superscript letters are significantly different from one another (P < 0.05).
Ubiquitin plays an essential role in TA-elicited HDAC6 degradation.
It has been documented that MG132 is a specific inhibitor that can reduce the degradation of ubiquitin-conjugated proteins in mammalian cells (44), we examined the effect of MG132 (5 μM) on HDAC6 expression in HK2 cells, a human tubular epithelial cell line, treated with cisplatin and TA treatment. As shown in Fig. 11, exposure of HK2 cells to TA alone led to reduction of HDAC6 expression, this was partially inhibited by additional treatment of MG132 to the culture, suggesting that ubiquitination at least in part contributes to downregulation of HDAC6.
Fig. 11.
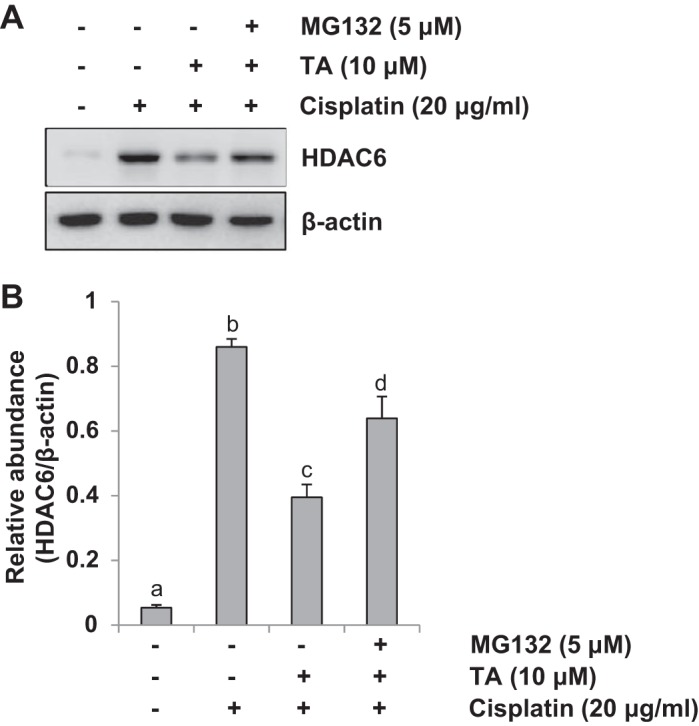
MG132 significantly upregulated expression of HDAC6 in human tubular epithelial cell. HK2 cells were exposed to cisplatin and TA in the absence or presence of MG132 (5 μM) for 24 h. A: cell lysates were subjected to immunoblot analysis with specific antibodies against HDAC6 and β-actin. B: expression level of HDAC6 was quantified by densitometry and normalized with β-actin. Data are represented as the means ± SE (n = 4). Means with different superscript letters are significantly different from one another (P < 0.05). The results indicated that TA treatment reduces HDAC6 expression and in the presence of MG132 HDAC6 expression levels are partially resumed.
Mechanisms of HDAC6 inhibition-elicited attenuation of rhabdomyolysis-induced AKI.
As shown in Fig. 12, we have concluded the mechanisms of HDAC6 inhibition-elicited protective role in rhabdomyolysis-induced AKI.
Fig. 12.
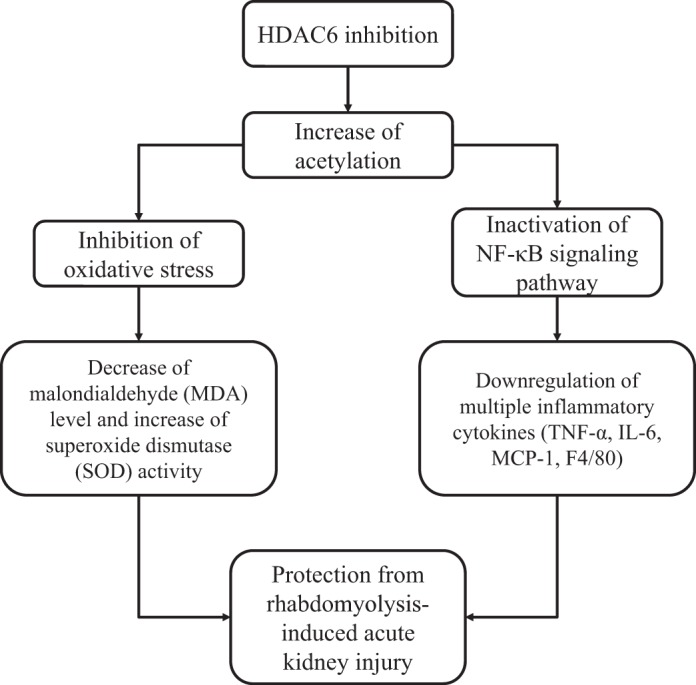
Mechanisms of HDAC6 inhibition-elicited attenuation of rhabdomyolysis-induced AKI. HDAC6 inhibition protects against rhabdomyolysis-induced acute kidney injury through at least 2 mechanisms: inhibition of NF-κB signaling pathway and subsequent production of multiple inflammatory cytokines/chemokines and reduction of oxidative stress by decreasing MDA expression levels and increasing SOD activity.
DISCUSSION
In this study, we explored the role of HDAC6 in AKI. Our results showed that inhibition of HDAC6 by TA resulted in improved renal dysfunction and attenuated tubular cell damage and apoptosis in a mouse model of rhabdomyolysis-induced AKI by using GL intramuscular injection. TA was also effective in alleviating expression of multiple cytokines/chemokines and infiltration of macrophages in the injured kidney. These results provide strong evidence that HDAC6 activity is critically involved in the renal epithelial tubular cell injury and death as well as inflammatory response in the kidney after acute injury. Therefore, targeting HDAC6 may be a potential therapeutic approach for AKI.
HDAC6 inhibition may protect the kidney against injury via multiple mechanisms. One of the possibilities is that the HDAC6 inhibition blocks renal tubular cell death by targeting the key pathway(s) leading to cell death. It is well known that myoglobulin released from the lysed striated muscle cells is critical for development of rhabdomyolysis-induced AKI by inducing apoptosis of renal tubular cells (13). Further studies showed that caspase-3 activation is the predominant mechanism responsible for renal damage under this pathological condition. In this study, we found that HDAC6 inhibition blocked cleavage of caspase-3 in the injured kidney, suggesting that HDAC6 mediates caspase-3 activation. In support of this hypothesis, HDAC6 inhibition also prevents TNF-α-induced caspase-3 activation in lung endothelial cells (45). Currently, it remains unclear how HDAC6 is coupled to the caspase pathway to initiate renal tubular cells death. However, since caspase-3 activation can be induced through intrinsic and extrinsic apoptotic pathways, it is possible that HDAC6 may be involved in the regulation of the key enzymes or machinery in these pathways. In addition, there is the possibility that HDAC inhibitors may provide renal tubular survival advantage through activation of AKT and other survival signaling molecules. In this context, HDAC6 inhibition has been reported to stimulate AKT activation in tumor cells (17). Further studies are required to determine whether HDAC6 also stimulates AKT phosphorylation in renal tubular cells.
On the other hand, HDAC6 inhibition may mediate renal protection through suppression of inflammation. In response to myoglobin-derived heme moiety, endothelial and renal tubular cells release numerous proinflammatory mediators such as TNF-α, IL-6, and MCP-1 (30). Increased chemokines can recruit various inflammatory cells including macrophages to the injured kidney. Infiltrated macrophages will further produce cytokines such as TNF-α and IL-1β, resulting in a positive feedback that perpetuates inflammatory response and worsens renal injury (30). Therefore, inhibition of inflammatory response is an attractive approach to reduce renal injury. In this study, we observed that administration of TA significantly reduced expression of TNF-α, IL-6, and MCP-1 and decreased the number of macrophages in the kidney of rhabdomyolysis-induced AKI. Therefore, the HDAC6 inhibitor’s renoprotective effect may be partially attributable to suppression of inflammatory response.
There are at least two potential mechanisms contributing to the anti-inflammatory effect of HDAC6 inhibition in the kidney after acute injury. TA may exert such an effect via attenuation of renal cell damage. Injury to the kidney results in the damage of renal tubular and endothelial cells, which release a large amount of injury mediators, including CD14, in rhabdomyolysis-induced AKI (42). In response to damage-associated molecular patterns, CD14 facilitates pro- and anti-inflammatory cytokine production (42). Therefore, attenuation of renal injury by TA could reduce the release of these inflammatory mediators. Another possibility is that HDAC6 inhibition suppresses the transcriptional factors that mediate upregulation of proinflammatory cytokines/chemokines. In this regard, it has been reported that activation of the NF-κB pathway plays a critical role in the progression of rhabdomyolysis-induced AKI (13). In this study, we showed that TA treatment significantly inhibited phosphorylation of p65, the prototypical heterodimer complex partner of NF-κB. The p65 can interact with various proteins in both the cytoplasm and in the nucleus during the process of classical NF-κB activation and nuclear translocation. Since we observed that HDAC6 is predominantly expressed in the cytoplasm and TA treatment also induces histone H3 acetylation, HDAC6 may act in both compartments to regulate activation of NF-κB pathways and gene expression. Future studies are needed to investigate the detailed mechanism by which HDAC6 regulates activation of the NF-κB signaling pathway.
It is unlikely that NF-κB is the sole pathway mediating regulation of cytokines/chemokines and renal injury in rhabdomyolysis-induced AKI. Microarray analysis indicates that in addition to the NF-κB pathway, three mitogen-activated protein kinase (MAPK) signaling pathways including extracellular signal-regulated kinases-1 and -2 (ERK), c-Jun NH2-terminal kinases (JNK), and p38 are also upregulated in rhabdomyolysis-induced AKI (13). Our group and others have shown that activation of the ERK1/2 pathway contributes to apoptosis of renal tubular cells in response to oxidant stress in vitro and in a murine model of cisplantin-induced AKI (5, 27, 33, 52). Furthermore, activation of JNK is also involved in the experimental renal ischemia-reperfusion injury (18). Interestingly, MAPKs are linked to HDAC6 regulation. For example, ERK1/2 can phosphorylate HDAC6 at serine 1035 (40), and HDAC6 can induce deacetylation of p38 and subsequent phosphorylation of p38 in response to proteasome inhibition (19). Moreover, JNK1 is able to sustain HDAC6 expression (48). Therefore, it will be interesting to further identify the role of MAPKs in HDAC6’s function as a mediator in rhabdomyolysis-induced AKI in the future.
In addition, HDAC6 inhibition may also protect against AKI through reduction of oxidative stress. It has been reported that oxidative stress is involved in the rhabdomyolysis-induced AKI. After muscle injury, released myoglobin from cells is accumulated in the renal tubules where it leads to the formation of reactive oxygen species that causes renal injury (47). ROS can promote lipid peroxidation of membrane fatty acids (6, 20) and induce MDA synthesis, which mediates alterations of proteins and DNA (31, 39). In contrast, rhabdomyolysis can result in decreased SOD activity in kidney tissues (14). To determine whether HDAC6 inhibition would interfere with oxidative stress triggered in the kidney during rhabdomyolysis, we measured the MDA and SOD, two important biomarkers of oxidative stress in the kidney tissue, and found that blocking HDAC6 with TA significantly suppressed MDA and preserved SOD in the kidney of mice with rhabdomyolysis-induced AKI. Therefore, HDAC6 inhibition has a protective effect against oxidative stress. Further studies are needed to explore the mechanism of HDAC6-mediated oxidative stress.
It should be noted that the functional significance of HDACs in acute tissue injury is still controversial (36, 37). Whereas the present study clearly shows that TA-elicited HDAC6 inhibition offers a protective effect to the kidney in rhabdomyolysis, blocking class I HDACs by MS-275 results in worsening renal dysfunction, potentiating renal tubule injury and enhancing apoptosis and caspase-3 activation in the same model of AKI (36). In support of our observations, TA administration was reported to significantly improve long-term survival of animals whereas treatment with MS-275 is not protective against septic shock (25). A possible explanation is that different classes of HDACs may play a distinct role in activating the molecular machinery that leads to development of AKI or other tissue injury. Another possibility is that the function of individual HDACs may be directly related to their unique molecular substrates. Unlike class I HDACs, HDAC6 is predominantly localized in the cytoplasm, where it associates with many nonhistone substrates and deacetylates them. Moreover, HDAC6 is able to bind ubiquitin and is directly involved in protein degradation via the ubiquitin-proteasome system (22). In this study, we found that TA not only increases histone H3 acetylation but also reduces expression of HDAC6, suggesting that HDAC6-mediated protein degradation may also regulate expression of itself. In support of this hypothesis, treatment with MG132, a highly specific ubiquitin inhibitor, in part, reversed TA-elicited downregulation of HDAC6 in HK2 cells exposed to cisplatin. HDAC6 has been designated as a potential therapeutic target, and a number of specific HDAC6 inhibitors (HDAC6i) have been developed (7). Preclinical data suggest that small molecule-specific HDAC6i may play a role in treatment of certain cancers, neurodegenerative diseases, and autoimmune disorders (3, 4, 8, 11, 51). In addition, HDAC6 inhibitors have also been tested in some acute injury models. For example, HDAC6 inhibitors improved long-term survival in a lethal septic model and alleviated stroke-induced brain infarction and functional deficits (38). Selective knockdown of HDAC6 by shRNA has been shown to protect mouse cortical neurons from oxygen and glucose deprivation in an in vitro cerebral ischemia model (10). Specific inhibition of HDAC6 probably confers more beneficial effects than pan-HDAC inhibitors or other specific HDAC inhibitors since HDAC6 knockout mice are viable and fertile (32). TA is a safe agent that has very low toxicity. Daily intraperitoneal injection of 25 mg/kg TA for 20 days in mice does not affect gross brain morphology, liver enzyme expression, and kidney function (49). Strikingly, the time window of TA treatment has been reported to be at least 24 h after the ischemic onset in a rat model of focal cerebral ischemia (38); thus TA may be promising agent for the treatment of AKI and other acute organ injury.
In summary, our studies demonstrated that inhibition of HDAC6 activity protects against rhabdomyolysis-induced AKI. HDAC6 blockage-elicited renal protection is associated with inhibition of renal tubular cell apoptosis, suppression of inflammatory responses, and reduction of oxidative stress. Thus selective inhibition of HDAC6 may be a novel strategy for the treatment of AKI.
GRANTS
This study was supported by the National Nature Science Foundation of China Grants 81670690, 81470991, and 81200492 (to N. Liu) and 81270778, 81470920, and 81670623 (to S. Zhuang), National Institute of Diabetes and Digestive and Kidney Diseases Grant 2R01-DK-08506505A1 (to S. Zhuang), and Key Discipline Construction Project of Pudong Health Bureau of Shanghai Grant PWZx2014–06 (to S. Zhuang). This work was also supported by National Natural Science Foundation of Youth Science Fund Project of China Grant 81400693 (to J. Tang) and Program for Young Excellent Talents in Tongji University Grant 2014KJ079 (to J. Tang).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
N.L. and S.Z. conceived and designed the experiments; Y.S., L.X., J.T., L.F., S.M., X.M., A.Q. and J.N. performed experiments; Y.S., L.X., J.T., X.P., A.Q., N.L. and S.Z. analyzed data; Y.S., L.X., N.L. and S.Z. wrote the paper. S.Z., Y.S., L.X., J.T., L.F., S.M., X.M., J.N., X.P.,A.Q. and N.L. approved final version of paper.
REFERENCES
- 1.Abassi Z, Sagi O, Armaly Z, Bishara B. [Neutrophil gelatinase-associated lipocalin (NAGL): a novel biomarker for acute kidney injury]. Harefuah 150: 111–116, 2011. [PubMed] [Google Scholar]
- 2.Aydinoz S, Uzun G, Cermik H, Atasoyu EM, Yildiz S, Karagoz B, Evrenkaya R. Effects of different doses of hyperbaric oxygen on cisplatin-induced nephrotoxicity. Ren Fail 29: 257–263, 2007. doi: 10.1080/08860220601166487. [DOI] [PubMed] [Google Scholar]
- 3.Bai J, Lei Y, An GL, He L. Down-regulation of deacetylase HDAC6 inhibits the melanoma cell line A375.S2 growth through ROS-dependent mitochondrial pathway. PLoS One 10: e0121247, 2015. doi: 10.1371/journal.pone.0121247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Batchu SN, Brijmohan AS, Advani A. The therapeutic hope for HDAC6 inhibitors in malignancy and chronic disease. Clin Sci (Lond) 130: 987–1003, 2016. doi: 10.1042/CS20160084. [DOI] [PubMed] [Google Scholar]
- 5.Bocanegra V, Gil Lorenzo AF, Cacciamani V, Benardón ME, Costantino VV, Vallés PG. RhoA and MAPK signal transduction pathways regulate NHE1-dependent proximal tubule cell apoptosis after mechanical stretch. Am J Physiol Renal Physiol 307: F881–F889, 2014. doi: 10.1152/ajprenal.00232.2014. [DOI] [PubMed] [Google Scholar]
- 6.Boutaud O, Roberts LJ II. Mechanism-based therapeutic approaches to rhabdomyolysis-induced renal failure. Free Radic Biol Med 51: 1062–1067, 2011. doi: 10.1016/j.freeradbiomed.2010.10.704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brindisi M, Cavella C, Brogi S, Nebbioso A, Senger J, Maramai S, Ciotta A, Iside C, Butini S, Lamponi S, Novellino E, Altucci L, Jung M, Campiani G, Gemma S. Phenylpyrrole-based HDAC inhibitors: synthesis, molecular modeling and biological studies. Future Med Chem 8: 1573–1587, 2016. doi: 10.4155/fmc-2016-0068. [DOI] [PubMed] [Google Scholar]
- 8.Cebotaru L, Liu Q, Yanda MK, Boinot C, Outeda P, Huso DL, Watnick T, Guggino WB, Cebotaru V. Inhibition of histone deacetylase 6 activity reduces cyst growth in polycystic kidney disease. Kidney Int 90: 90–99, 2016. doi: 10.1016/j.kint.2016.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang Z, Li Y, He W, Liu B, Halaweish I, Bambakidis T, Liang Y, Alam HB. Selective inhibition of histone deacetylase 6 promotes survival in a rat model of hemorrhagic shock. J Trauma Acute Care Surg 79: 905–910, 2015. doi: 10.1097/TA.0000000000000784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen YT, Zang XF, Pan J, Zhu XL, Chen F, Chen ZB, Xu Y. Expression patterns of histone deacetylases in experimental stroke and potential targets for neuroprotection. Clin Exp Pharmacol Physiol 39: 751–758, 2012. doi: 10.1111/j.1440-1681.2012.05729.x. [DOI] [PubMed] [Google Scholar]
- 11.Choi SY, Ryu Y, Kee HJ, Cho SN, Kim GR, Cho JY, Kim HS, Kim IK, Jeong MH. Tubastatin A suppresses renal fibrosis via regulation of epigenetic histone modification and Smad3-dependent fibrotic genes. Vascul Pharmacol 72: 130–140, 2015. doi: 10.1016/j.vph.2015.04.006. [DOI] [PubMed] [Google Scholar]
- 12.Coca SG. Long-term outcomes of acute kidney injury. Curr Opin Nephrol Hypertens 19: 266–272, 2010. doi: 10.1097/MNH.0b013e3283375538. [DOI] [PubMed] [Google Scholar]
- 13.Geng X, Wang Y, Hong Q, Yang J, Zheng W, Zhang G, Cai G, Chen X, Wu D. Differences in gene expression profiles and signaling pathways in rhabdomyolysis-induced acute kidney injury. Int J Clin Exp Pathol 8: 14087–14098, 2015. [PMC free article] [PubMed] [Google Scholar]
- 14.Gu H, Yang M, Zhao X, Zhao B, Sun X, Gao X. Pretreatment with hydrogen-rich saline reduces the damage caused by glycerol-induced rhabdomyolysis and acute kidney injury in rats. J Surg Res 188: 243–249, 2014. doi: 10.1016/j.jss.2013.12.007. [DOI] [PubMed] [Google Scholar]
- 15.Humphreys BD, Valerius MT, Kobayashi A, Mugford JW, Soeung S, Duffield JS, McMahon AP, Bonventre JV. Intrinsic epithelial cells repair the kidney after injury. Cell Stem Cell 2: 284–291, 2008. doi: 10.1016/j.stem.2008.01.014. [DOI] [PubMed] [Google Scholar]
- 16.Ivashkiv LB. Inflammatory signaling in macrophages: transitions from acute to tolerant and alternative activation states. Eur J Immunol 41: 2477–2481, 2011. doi: 10.1002/eji.201141783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaliszczak M, Trousil S, Ali T, Aboagye EO. AKT activation controls cell survival in response to HDAC6 inhibition. Cell Death Dis 7: e2286, 2016. doi: 10.1038/cddis.2016.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kanellis J, Ma FY, Kandane-Rathnayake R, Dowling JP, Polkinghorne KR, Bennett BL, Friedman GC, Nikolic-Paterson DJ. JNK signalling in human and experimental renal ischaemia/reperfusion injury. Nephrol Dial Transplant 25: 2898–2908, 2010. doi: 10.1093/ndt/gfq147. [DOI] [PubMed] [Google Scholar]
- 19.Kästle M, Woschee E, Grune T. Histone deacetylase 6 (HDAC6) plays a crucial role in p38MAPK-dependent induction of heme oxygenase-1 (HO-1) in response to proteasome inhibition. Free Radic Biol Med 53: 2092–2101, 2012. doi: 10.1016/j.freeradbiomed.2012.09.023. [DOI] [PubMed] [Google Scholar]
- 20.Kim SH, Chang JW, Kim SB, Park SK, Park JS, Lee SK. Myoglobin induces vascular cell adhesion molecule-1 expression through c-Src kinase-activator protein-1/nuclear factor-kappaB pathways. Nephron, Exp Nephrol 114: e48–e60, 2010. doi: 10.1159/000254391. [DOI] [PubMed] [Google Scholar]
- 21.Komada T, Usui F, Kawashima A, Kimura H, Karasawa T, Inoue Y, Kobayashi M, Mizushina Y, Kasahara T, Taniguchi S, Muto S, Nagata D, Takahashi M. Role of NLRP3 inflammasomes for rhabdomyolysis-induced acute kidney injury. Sci Rep 5: 10901, 2015. doi: 10.1038/srep10901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leyk J, Goldbaum O, Noack M, Richter-Landsberg C. Inhibition of HDAC6 modifies tau inclusion body formation and impairs autophagic clearance. J Mol Neurosci 55: 1031–1046, 2015. doi: 10.1007/s12031-014-0460-y. [DOI] [PubMed] [Google Scholar]
- 23.Li M, Zhuang Y, Shan B. Analysis of expression and functions of histone deacetylase 6 (hdac6). Methods Mol Biol 1436: 85–94, 2016. doi: 10.1007/978-1-4939-3667-0_6. [DOI] [PubMed] [Google Scholar]
- 24.Li S, Liu X, Chen X, Zhang L, Wang X. Histone deacetylase 6 promotes growth of glioblastoma through inhibition of SMAD2 signaling. Tumour Biol 36: 9661–9665, 2015. 10.1007/s13277-015-3747-x. [DOI] [PubMed] [Google Scholar]
- 25.Li Y, Zhao T, Liu B, Halaweish I, Mazitschek R, Duan X, Alam HB. Inhibition of histone deacetylase 6 improves long-term survival in a lethal septic model. J Trauma Acute Care Surg 78: 378–385, 2015. doi: 10.1097/TA.0000000000000510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu N, Zhuang S. Treatment of chronic kidney diseases with histone deacetylase inhibitors. Front Physiol 6: 121, 2015. doi: 10.3389/fphys.2015.00121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Malik S, Suchal K, Gamad N, Dinda AK, Arya DS, Bhatia J. Telmisartan ameliorates cisplatin-induced nephrotoxicity by inhibiting MAPK mediated inflammation and apoptosis. Eur J Pharmacol 748: 54–60, 2015. doi: 10.1016/j.ejphar.2014.12.008. [DOI] [PubMed] [Google Scholar]
- 28.Markó L, Szijártó IA, Filipovic MR, Kaßmann M, Balogh A, Park JK, Przybyl L, N’diaye G, Krämer S, Anders J, Ishii I, Müller DN, Gollasch M. Role of cystathionine gamma-lyase in immediate renal impairment and inflammatory response in acute ischemic kidney injury. Sci Rep 6: 27517, 2016. doi: 10.1038/srep27517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pang M, Kothapally J, Mao H, Tolbert E, Ponnusamy M, Chin YE, Zhuang S. Inhibition of histone deacetylase activity attenuates renal fibroblast activation and interstitial fibrosis in obstructive nephropathy. Am J Physiol Renal Physiol 297: F996–F1005, 2009. doi: 10.1152/ajprenal.00282.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Panizo N, Rubio-Navarro A, Amaro-Villalobos JM, Egido J, Moreno JA. Molecular mechanisms and novel therapeutic approaches to rhabdomyolysis-induced acute kidney injury. Kidney Blood Press Res 40: 520–532, 2015. doi: 10.1159/000368528. [DOI] [PubMed] [Google Scholar]
- 31.Petejova N, Martinek A. Acute kidney injury due to rhabdomyolysis and renal replacement therapy: a critical review. Crit Care 18: 224, 2014. doi: 10.1186/cc13897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ran J, Yu F, Qin J, Zhang Y, Yang Y, Li D, Zhou J, Liu M. Functional interplay between cylindromatosis and histone deacetylase 6 in ciliary homeostasis revealed by phenotypic analysis of double knockout mice. Oncotarget 7: 27527–27537, 2016. doi: 10.18632/oncotarget.8374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Romanov V, Whyard TC, Waltzer WC, Grollman AP, Rosenquist T. Aristolochic acid-induced apoptosis and G2 cell cycle arrest depends on ROS generation and MAP kinases activation. Arch Toxicol 89: 47–56, 2015. doi: 10.1007/s00204-014-1249-z. [DOI] [PubMed] [Google Scholar]
- 34.Savitskaya MA, Onishchenko GE. Mechanisms of apoptosis. Biochemistry (Mosc) 80: 1393–1405, 2015. doi: 10.1134/S0006297915110012. [DOI] [PubMed] [Google Scholar]
- 35.Sureshbabu A, Ryter SW, Choi ME. Oxidative stress and autophagy: crucial modulators of kidney injury. Redox Biol 4: 208–214, 2015. doi: 10.1016/j.redox.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tang J, Yan Y, Zhao TC, Gong R, Bayliss G, Yan H, Zhuang S. Class I HDAC activity is required for renal protection and regeneration after acute kidney injury. Am J Physiol Renal Physiol 307: F303–F316, 2014. doi: 10.1152/ajprenal.00102.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tang J, Zhuang S. Epigenetics in acute kidney injury. Curr Opin Nephrol Hypertens 24: 351–358, 2015. doi: 10.1097/MNH.0000000000000140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Z, Leng Y, Wang J, Liao HM, Bergman J, Leeds P, Kozikowski A, Chuang DM. Tubastatin A, an HDAC6 inhibitor, alleviates stroke-induced brain infarction and functional deficits: potential roles of α-tubulin acetylation and FGF-21 up-regulation. Sci Rep 6: 19626, 2016. doi: 10.1038/srep19626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wijayanti N, Katz N, Immenschuh S. Biology of heme in health and disease. Curr Med Chem 11: 981–986, 2004. doi: 10.2174/0929867043455521. [DOI] [PubMed] [Google Scholar]
- 40.Williams KA, Zhang M, Xiang S, Hu C, Wu JY, Zhang S, Ryan M, Cox AD, Der CJ, Fang B, Koomen J, Haura E, Bepler G, Nicosia SV, Matthias P, Wang C, Bai W, Zhang X. Extracellular signal-regulated kinase (ERK) phosphorylates histone deacetylase 6 (HDAC6) at serine 1035 to stimulate cell migration. J Biol Chem 288: 33156–33170, 2013. doi: 10.1074/jbc.M113.472506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Woan KV, Lienlaf M, Perez-Villaroel P, Lee C, Cheng F, Knox T, Woods DM, Barrios K, Powers J, Sahakian E, Wang HW, Canales J, Marante D, Smalley KS, Bergman J, Seto E, Kozikowski A, Pinilla-Ibarz J, Sarnaik A, Celis E, Weber J, Sotomayor EM, Villagra A. Targeting histone deacetylase 6 mediates a dual anti-melanoma effect: enhanced antitumor immunity and impaired cell proliferation. Mol Oncol 9: 1447–1457, 2015. doi: 10.1016/j.molonc.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wooten RM, Weis JJ. Host-pathogen interactions promoting inflammatory Lyme arthritis: use of mouse models for dissection of disease processes. Curr Opin Microbiol 4: 274–279, 2001. doi: 10.1016/S1369-5274(00)00202-2. [DOI] [PubMed] [Google Scholar]
- 43.Yoon S, Eom GH. HDAC and HDAC inhibitor: from cancer to cardiovascular diseases. Chonnam Med J 52: 1–11, 2016. doi: 10.4068/cmj.2016.52.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.You J, Lee E, Bonilla L, Francis J, Koh J, Block J, Chen S, Hansen PJ. Treatment with the proteasome inhibitor MG132 during the end of oocyte maturation improves oocyte competence for development after fertilization in cattle. PLoS One 7: e48613, 2012. doi: 10.1371/journal.pone.0048613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu J, Ma M, Ma Z, Fu J. HDAC6 inhibition prevents TNF-α-induced caspase 3 activation in lung endothelial cell and maintains cell-cell junctions. Oncotarget 7: 54714–54722, 2016. doi: 10.18632/oncotarget.10591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yu J, Ma Z, Shetty S, Ma M, Fu J. Selective HDAC6 inhibition prevents TNF-α-induced lung endothelial cell barrier disruption and endotoxin-induced pulmonary edema. Am J Physiol Lung Cell Mol Physiol 311: L39–L47, 2016. doi: 10.1152/ajplung.00051.2016. [DOI] [PubMed] [Google Scholar]
- 47.Zager RA, Foerder CA. Effects of inorganic iron and myoglobin on in vitro proximal tubular lipid peroxidation and cytotoxicity. J Clin Invest 89: 989–995, 1992. doi: 10.1172/JCI115682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang D, Li J, Costa M, Gao J, Huang C. JNK1 mediates degradation HIF-1alpha by a VHL-independent mechanism that involves the chaperones Hsp90/Hsp70. Cancer Res 70: 813–823, 2010. doi: 10.1158/0008-5472.CAN-09-0448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang L, Liu C, Wu J, Tao JJ, Sui XL, Yao ZG, Xu YF, Huang L, Zhu H, Sheng SL, Qin C. Tubastatin A/ACY-1215 improves cognition in Alzheimer’s disease transgenic mice. J Alzheimers Dis 41: 1193–1205, 2014. [DOI] [PubMed] [Google Scholar]
- 50.Zhang YL, Zhang J, Cui LY, Yang S. Autophagy activation attenuates renal ischemia-reperfusion injury in rats. Exp Biol Med (Maywood) 240: 1590–1598, 2015. doi: 10.1177/1535370215581306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhu T, Zhao D, Song Z, Yuan Z, Li C, Wang Y, Zhou X, Yin X, Hassan MF, Yang L. HDAC6 alleviates prion peptide-mediated neuronal death via modulating PI3K-Akt-mTOR pathway. Neurobiol Aging 37: 91–102, 2016. doi: 10.1016/j.neurobiolaging.2015.09.021. [DOI] [PubMed] [Google Scholar]
- 52.Zhuang S, Kinsey GR, Yan Y, Han J, Schnellmann RG. Extracellular signal-regulated kinase activation mediates mitochondrial dysfunction and necrosis induced by hydrogen peroxide in renal proximal tubular cells. J Pharmacol Exp Ther 325: 732–740, 2008. doi: 10.1124/jpet.108.136358. [DOI] [PubMed] [Google Scholar]
- 53.Zimmerman JL, Shen MC. Rhabdomyolysis. Chest 144: 1058–1065, 2013. doi: 10.1378/chest.12-2016. [DOI] [PubMed] [Google Scholar]