

Abstract
Previous studies have shown that increased parathyroid hormone (PTH) attributable to secondary hyperparathyroidism in chronic kidney disease accelerates the arteriosclerotic fibrosis and calcification. Although the underlying mechanisms remain largely unknown, endothelial cells (ECs) have recently been demonstrated to participate in calcification in part by providing chondrogenic cells via the endothelial-to-mesenchymal transition (EndMT). Therefore, this study aimed to investigate whether elevated PTH could induce endothelial-to-chondrogenic transition in aortic ECs and to determine the possible underlying signaling pathway. We found that treatment of ECs with PTH significantly upregulated the expression of EndMT-related markers. Accordingly, ECs treated with PTH exhibited chondrogenic potential. In vivo, lineage-tracing model-subjected mice with endothelial-specific green fluorescent protein fluorescence to chronic PTH infusion showed a marked increase in the aortic expression of chondrocyte markers, and confocal microscopy revealed the endothelial origin of cells expressing chondrocyte markers in the aorta after PTH infusion. Furthermore, this in vitro study showed that PTH enhanced the nuclear localization of β-catenin in ECs, whereas β-catenin siRNA or DKK1, an inhibitor of β-catenin nuclear translocation, attenuated the upregulation of EndMT-associated and chondrogenic markers induced by PTH. In summary, our study demonstrated that elevated PTH could induce the transition of ECs to chondrogenic cells via EndMT, possibly mediated by the nuclear translocation of β-catenin.
Keywords: parathyroid hormone, endothelial cells, chondrogenic transition, β-catenin
vascular calcification (VC) is a frequent complication of chronic kidney disease (CKD) and is associated with an increased risk of cardiovascular and all-cause mortality (27, 30). Numerous investigations have demonstrated that VC is a cell-regulated process with similarities to bone formation, driven by osteochondrogenic cells (27, 34). Recently, emerging evidence has further revealed that endochondral bone formation initiated by the presence of chondrocytes is a characteristic hallmark of VC in animal models with chronic renal failure (CRF) and in human aorta specimens from transplant donors (23, 24, 42). These findings showed that the accumulation of ectopic chondrogenic cells underlies the progression of VC in CKD.
Previously, smooth muscle cells, mesenchymal stem cells, and pericytes were considered to be the major precursors of ectopic chondrogenic cells in calcified vessels (5, 25, 31). In recent studies, endothelial cells (ECs) have been demonstrated to participate in tissue calcification by providing osteochondrogenic cells via the endothelial-to-mesenchymal transition (EndMT) (8, 21, 33, 38).
Elevated parathyroid hormone (PTH) levels are common in patients with CKD with the decline of renal function attributable to the development of secondary hyperparathyroidism (2). Although the importance of PTH in mineral metabolism is well recognized, a series of studies has recently suggested that PTH could directly induce endothelial dysfunction and contribute to VC (6, 26–29). Nevertheless, the underlying mechanisms remain largely unclear. Our recent experiments have found that PTH could induce EndMT and contribute to the progression of cardiac fibrosis in uremic rats (36). However, whether ECs that underwent EndMT could further be triggered into chondrogenic cells remains unknown. In this study, we investigated the effect of PTH-induced EndMT on chondrogenic potential and sought to determine its underlying signaling pathway.
MATERIALS AND METHODS
Cell culture.
Primary human aortic ECs were purchased from ScienCell Research Laboratories and cultured as previously described (33). All experiments were performed with cells at passages 3 to 5.
Cell differentiation.
After PTH (1–34) stimulation, the EC culture medium was changed to a chondrogenic differentiation medium (Sciencell MCDM medium containing 10 ng/ml TGF-β3 and 5 mmol/l β-glycerophosphate) for 7 days.
siRNA transfection.
The β-catenin siRNA was purchased from Cell Signaling Technology (no. 6225). The nontargeting scramble-sequence siRNA was used as a negative control. The siRNA transfection study was performed with the MACSfectin Reagent kit (130-098-411, Miltenyi Biotec) and corresponding protocol.
Real-time PCR analysis.
Total RNA from ECs was extracted and reverse transcribed. The real-time PCR amplification was performed using an ABI Prism 7300 Sequence Detection System (Applied Biosystems) with the SYBR Green real-time PCR Kit (TAKARA). The relative amount of mRNA was normalized to β-actin and calculated using the standard curve method as described previously (43). In brief, the pre-PCR product of each gene was used as the standard. The standard curve was established with a 10-fold serial dilution of the product and was included in all PCR runs. The ratio of target gene abundance/housekeeping gene abundance was used to evaluate the expression level of each gene. Controls consisting of ddH2O were negative in all runs.
Western blot analysis.
Western blot analysis was carried out as described previously (37). Immunoblotting was performed with primary antibodies against CD31 (sc-1506, Santa Cruz Biotechnology), fibroblast-specific protein 1 (FSP1) (ab27957, Abcam), α-smooth muscle actin (α-SMA) (ab5694, Abcam), β-catenin (ab32572, Abcam), sex-determining region Y-box 9 (SOX9) (ab185230, Abcam), and Col2A1 (sc-52658, Santa Cruz Biotechnology), followed by incubation with horseradish peroxidase-labeled secondary IgG antibodies (Santa Cruz Biotechnology). The signals were detected using an advanced enhanced chemiluminescence system (GE Healthcare).
Mice.
Endothelial tracing was achieved by tamoxifen administration to inducible Tie2-Cre mice (tamoxifen-inducible Cre activation under the control of endothelium-specific Tie2 promoter) crossed with membrane-targeted tdTomato (mT)/membrane-targeted EGFP (mG) mice (15, 23) at the Durham VA Medical Center. Study protocols were approved by the DVAMC Institutional Animal Care and Use Committee and conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals. In brief, experimental mice were randomly divided into 2 groups: the control group (CTL, n = 5) and the PTH-infusion group (PTH, n = 5). An Alzet model 2ML4 osmotic minipump was implanted subcutaneously in each mouse. In the PTH group, human recombinant PTH fragment 1–34 (P3796, Sigma) was continuously infused at an infusion rate (0.11 μg·100 g−1·h−1). In the CTL group, the minipumps were used to deliver vehicle only (PBS, Gibco). During the 8-wk experimental period, all mice were maintained on a 1.5% phosphorus diet (no. 113345, Dyets). By the end of the study period, aorta was collected for histological analysis.
Histology and immunohistochemical staining.
Paraffin-embedded aortic sections (3 μm thick) were prepared using a routine procedure. The sections were subjected to Alcian blue staining for assessing chondrocyte proteoglycans and immunohistochemical staining to detect the expression of SOX9 (ab185230, Abcam) and Col2A1 (sc-52658, Santa Cruz Biotechnology).
Immunofluorescent staining.
Frozen aortic sections or coverslips were processed and immunofluorescently stained as described previously (37). Images were captured using a laser scanning confocal microscope (LSM 510 META, Zeiss).
Statistical analysis.
Data are expressed as means ± SD and were analyzed with SPSS 16.0. Comparisons between two groups were done using the unpaired t-test when appropriate. Differences among groups were tested by one-way ANOVA followed by Bonferroni correction for normally distributed data and a Mann Whitney test for nonnormally distributed data. A value of P < 0.05 was considered statistically significant.
RESULTS
PTH induced EndMT in cultured aortic ECs.
Our previous dose-finding studies revealed that PTH induces EndMT in primary human aortic ECs in a concentration- and time-dependent manner. On the basis of this result, we treated aortic ECs with 10−7 mol/l PTH for 48 h to observe the expression of EndMT-related markers. The results of real-time PCR and Western blot showed that PTH treatment led to a significant decrement in the mRNA and protein expression of CD31 and an increase in the expression of FSP1 and α-SMA (Fig. 1, A–E). Confocal microscopy detected that cells treated with PTH acquired FSP1 staining and lost CD31 staining compared with control cells (Fig. 1F).
Fig. 1.

Exposure to parathyroid hormone (PTH) induces endothelial-to-mesenchymal transition (EndMT) in aortic endothelial cells (ECs). A–E: ECs were incubated with PTH (10−7 mol/l) for 48 h, as indicated. A and B: mRNA levels were assessed by real-time PCR. C–E: protein expression levels were determined by Western blot. The data are expressed as the means ± SDs (n = 6). *P < 0.05 vs. CTL group. F: immunofluorescence of CD31 (green) and fibroblast-specific protein 1 (FSP1) (red) was observed by confocal microscopy. Scale bar = 50 μm. α-SMA, α-smooth muscle actin.
EndMT induces chondrogenic potential in ECs.
We next assessed the differentiation capability of ECs that undergo EndMT. Western blot analysis indicated that the expressions of the chondrocyte-specific markers SOX9 and Col2A1 were significantly increased in PTH-treated cells compared with controls (Fig. 2, A and B).
Fig. 2.
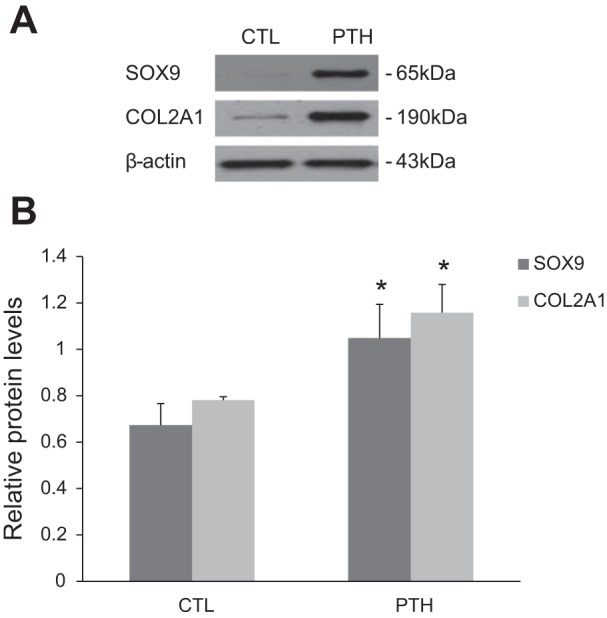
ECs that undergo PTH-induced EndMT exhibit chondrogenic potential. A and B: Western blot analysis for chondrocyte-specific markers sex-determining region Y-box 9 (SOX9) and Col2A1 in cells treated with PTH for 48 h followed by growth in chondrogenic culture medium for 7 days, as indicated. The data are expressed as the means ± SDs (n = 5). *P < 0.05 vs. CTL group.
Lineage tracing reveals the endothelial-to-chondrogenic transition in aortas following PTH infusion.
To further observe the effect of elevated PTH on the transition of the ECs to a chondrocyte phenotype, an in vivo lineage-tracing experiment was performed. We crossed inducible Tie2-Cre mice with mT/mG mice to obtain tamoxifen-inducible Tie2-Cre+ mT/mG mice in which enhanced green fluorescent protein (EGFP) is expressed under the control of the endothelial-specific Tie2 promoter. Because PTH (1–34) is the shortest fragment with the same biological effect as the intact PTH, we infused PTH (1–34) into these mice and controls and evaluated the expression of chondrocyte markers. As illustrated in Fig. 3, A and B, Alcian blue staining exhibited the conspicuously enhanced deposition of chondrocyte proteoglycans in aortic media from the PTH-infused group compared with controls (CTL) (arrows, Fig. 3B). As determined by immunohistochemical analysis, chondrocyte markers (SOX9 and Col2A1 protein) were dramatically increased in aortas from the PTH-infusion group compared with CTLs (Fig. 3, C–F). Furthermore, staining with anti-SOX9 antibody (blue) revealed EGFP-positive ECs (green) that coexpressed chondrocyte-specific markers SOX9 (arrows, cyan, Fig. 3, G and H). EGFP was only detected in the aortic endothelium in the control mice. These results suggested the endothelial origin of ectopic chondrocytes in the aorta from mice with continuous PTH infusion.
Fig. 3.

The expression of chondrocyte-related markers in aortas of Tie2-Cre+ mT/mG mice from CTL group (A, C, E, G) and PTH-infusion group (B, D, F, H). A and B: representative micrographs showing Alcian blue staining for chondrocyte proteoglycans in aorta samples of mice (original magnification is ×400). C–F: representative micrographs showing immunohistochemical staining for aortic expression of chondrocyte-specific markers SOX9 (C and D) and Col2A1 (E and F) in mice (original magnification is ×400). G and H: immunostaining of aortic tissues from Tie2-Cre+ mT/mG mice showed coexpression of Tie2-enhanced green fluorescent protein with chondrocyte-specific marker SOX9 (original magnification is ×600). DAPI was used to visualize nuclei.
Nuclear translocation of β-catenin mediates PTH-induced EndMT in aortic ECs.
To gain further insight into the mechanisms involved in EndMT triggered by PTH in ECs, we further examined the nuclear expression of β-catenin in cells treated with or without PTH. As shown in Fig. 4, A and B, the nuclear protein levels of β-catenin were induced in a concentration-dependent manner in ECs treated with PTH, suggesting the nuclear translocation of β-catenin. Immunofluorescence staining of β-catenin also showed a nuclear relocation of β-catenin in the cultured ECs stimulated with PTH (Fig. 4C). Because canonical Wnt signaling culminates in β-catenin nuclear translocation, these data suggest that PTH can activate the canonical Wnt/β-catenin pathway.
Fig. 4.

Effect of PTH on nuclear β-catenin abundances in aortic ECs. A: representative blots. B: graphic presentation. Lamin-B was used as internal control. The data are expressed as the means ± SDs (n = 4). *P < 0.05 vs. 0 group. C: immunofluorescent staining of β-catenin (red) in different groups of aortic ECs as indicated. DAPI was used for a nuclear stain (blue). Scale bar = 50 μm.
We next further evaluated whether canonical Wnt/β-catenin signaling activation is required for PTH-induced EndMT in our experiments. ECs were incubated with PTH in the presence or absence of β-catenin siRNA or the canonical Wnt/β-catenin signaling inhibitor Dickkopf-1 (DKK1). As shown in Fig. 5, downregulation of β-catenin by siRNA significantly reduced the PTH-triggered induction of EndMT-related markers. In addition, DKK1, the prototypical inhibitor of canonical Wnt signaling, also abolished the expression of EndMT-related markers in response to treatment with elevated PTH (Fig. 6).
Fig. 5.

β-Catenin siRNA blocked mRNA and protein expression of EndMT-related markers after PTH treatment in aortic ECs. ECs were transfected with siRNA for β-catenin. The nontargeting scramble-sequence siRNA (vehicle, vehi) was used as the control. A and B: real-time PCR analyses showed mRNA expression of CD31, FSP1, and α-SMA after various treatments for 48 h as indicated. C and D: Western blot analyses showed protein expression of CD31 and FSP1 after various treatments for 48 h. The data are expressed as the means ± SDs (n = 3). *P < 0.05 vs. Vehi group; #P < 0.05 vs. PTH + Vehi group. E: representative micrographs showing immunofluorescent staining of CD31 (green) and α-SMA (red) in ECs after various treatments, as indicated. Cells were counterstained with DAPI (blue) to demonstrate nuclei. Scale bar = 20 μm.
Fig. 6.

Canonical Wnt/β-catenin pathway inhibitor DKK1 blocked mRNA and protein expression of EndMT-related markers after PTH treatment in aortic ECs. A–C: real-time PCR analyses showed mRNA expression of CD31 (A), FSP1 (B), and α-SMA (C) after various treatments for 48 h with 10−7 mol/l PTH with or without 500 ng/ml DKK1 as indicated. D and E: Western blot analyses showed protein expression of CD31, FSP1, and α-SMA in different groups as indicated. Representative Western blots (D) and quantitative determinations of protein levels (E) are presented. The data are expressed as the means ± SDs (n = 3). *P < 0.05 vs. CTL group; #P < 0.05 vs. PTH group.
Blockade of EndMT by suppressing the canonical Wnt/β-catenin pathway prevents the expression of chondrocyte markers in cells.
We further examined whether β-catenin siRNA or DKK1 could prevent the expression of chondrocyte markers in cells undergoing EndMT induced by PTH. We found a significantly decreased protein level of chondrocyte markers in cells treated with β-catenin siRNA (Fig. 7, A and B) or DKK1 (Fig. 7, C and D) before growth in chondrogenic medium compared with control.
Fig. 7.

PTH-induced EndMT is necessary for the acquisition of chondrogenic potential. A and B: Western blot analysis showing the expression of chondrocyte-specific markers SOX9 and Col2A1 in cells exposed to PTH with or without β-catenin-specific siRNA. The data are expressed as the means ± SDs (n = 4). *P < 0.05 vs. Vehi group, #P < 0.05 vs. PTH + Vehi group, †P < 0.05 vs. siRNA group. C and D: Western blot analysis showing the protein expression of SOX9 and Col2A1 in cells with different treatment as indicated followed by growth in chondrogenic culture medium. The data are expressed as the means ± SDs (n = 3). *P < 0.05 vs. CTL group, #P < 0.05 vs. PTH group.
DISCUSSION
The mechanisms responsible for VC are complex. They typically require a decrease of calcification inhibitors with a concomitant increase in activators of biomineralization, both of which are tightly regulated at the cellular level (7). Vascular smooth muscle cells (VSMCs) are the best-characterized source of osteochondrogenic progenitors in the calcified vasculature. Numerous investigations have demonstrated that the transdifferentiation of VSMCs shares common molecular pathways with skeletal mineralization (32). During this process, VSMCs increase the expression of osteochondrogenic transcription factors and start the secretion of matrix vesicles, membrane microparticles that initiate hydroxyapatite crystallization. Notably, recent studies have suggested that cells of endothelial origin can also promote ectopic calcification (3, 14, 40). ECs isolated from solid tumors of the prostate, in which ectopic calcification was colocalized with endothelial marker CD31, clearly show ex vivo differentiation plasticity toward chondrocytes or osteoblasts (8). In the vascular system, experimental studies using lineage-tracing mice revealed that ECs contribute to the osteochondrogenic pool in aortic lesions in VC models associated with matrix ë-carboxyglutamic acid protein deficiency (Mgp knockout) and genetically determined diabetes (Akita mice) (39). In addition, clinical investigations have also revealed a correlation between circulating endothelial progenitor cells and vascular calcification in patients with atherosclerosis and diabetes (9–12, 42). It is recognized that the prevalence of VC is markedly higher in patients with CKD than in the general population. Experimental and clinical studies have revealed that heterotypic chondrocyte accumulation underlies uremic VC (23). However, whether endothelial phenotypic alterations contribute to chondrocyte accumulation in uremic calcified lesions has not been investigated yet.
The elevation of serum PTH concentration is a common condition in CKD with the decline of glomerular filtration rate. In this study, chronic infusion of PTH to mouse showed Tie2-positive chondrocytes in aortic samples by immunofluorescence analysis, suggesting an endothelial origin of ectopic cartilage in the aorta (Fig. 3). Moreover, the in vitro studies further showed that expression of chondrocyte markers SOX9 and Col2A1 were significantly increased in PTH-treated cells compared with the control (Fig. 2). These data clearly indicated that elevated PTH could induce the endothelial to chondrogenic phenotypical transition in ECs. Considering the involvement of PTH in the progression of uremic calcified lesions (6, 26), this PTH-induced phenotypic alteration in ECs might represent a novel mechanism contributing to VC in uremia.
Previous studies showed that animals receiving continuous PTH infusion developed media calcification (25). In our in vivo study, the cartilage proteoglycan is also mainly located in aortic media (Fig. 3). Recently, investigations have demonstrated that EndMT is a source of osteochondeogenic cells in media calcification (38, 39). Yao et al. (39) showed that the expression of endothelial markers was detected deep in the calcified media (39). Furthermore, they found that cells coexpressing endothelial and osteochondrogenic markers were highly abnormal and detached from the internal elastic lamina to penetrate into the media as visualized by phase contrast and transmission electron microscopy in calcified aortas. In lesions of aortic calcification in Mgp-deficient mice, the internal elastic lamina was gradually degraded, and ECs undergoing EndMT were oriented from the luminal side toward the aortic medial layer and calcifying lesional areas (38). Besides, numerous studies have disclosed that ECs could acquire the ability to migrate through EndMT (13, 35). Taken together, all these findings suggest that EndMTs are involved in media calcification in part by penetrating into the medial tissue and further transdifferentiating into osteochondrogenic cells. In the present study, we observed the coexpression of endothelial marker (EGFP-positive cells) and chondrocyte marker (SOX9) in tunica media in PTH-infusion mice. Although the exact mechanisms are unclear, it might be associated with the cellular motility endued by EndMT.
As shown by data from this study, cells undergoing EndMT acquire the transdifferentiation potential to become chondrocytes. Using siRNA or pharmacological approaches to inhibit PTH-induced EndMT in cultured aortic ECs attenuated the expression of chondrocyte markers SOX9 and Col2A1 in these cells (Fig. 7). Collectively, these results strongly suggest that EndMT enables cells to be further triggered into chondrocytes. This result was partly consistent with the previous work by Medici et al. (20), which demonstrated that constitutive ALK2 activation in mature ECs resulted in EndMT and the acquisition of a stem cell-like phenotype to differentiate into osteoblasts, chondrocytes, or adipocytes. As EndMT generates fibroblastic cells to participate in specific developmental processes, cancer progression, and tissue fibrosis (16, 41), our findings further broaden the scope for the role of EndMT in disease pathogenesis.
Because EndMT is the key phenotypic transition process for chondrogenic potential in ECs, the molecular mechanisms underlying EndMT are of great interest. Previous studies have clearly revealed a role of TGF-β signaling in EndMT (21). Recent investigations have further provided evidence of the canonical Wnt/β-catenin signaling pathway in this process (4). Using a Wnt-signaling reporter mouse strain, Liebner et al. (18) demonstrated that endocardial EndMT in heart cushion is accompanied by activation of β-catenin activity. Aisagbonhi et al. (1) reported the activation of canonical Wnt activation and the associated EndMT in response to myocardial infarction. In EndMT during the progression of cerebral cavernous malformation disease, increased β-catenin nuclear translocation and transcriptional signaling could also be detected (19). Moreover, Li et al. (17) found that Wnt/β-catenin pathway mediated EndMT in diabetic kidney disease. In this study, we showed that PTH-induced EndMT in aortic ECs was also accompanied by the nuclear translocation of β-catenin. Conversely, the knockdown of β-catenin with siRNA or blockade of its nuclear translocation with the specific inhibitor DKK1 significantly attenuated the expression of EndMT markers induced by PTH in ECs (Figs. 5 and 6). These findings indicate that canonical Wnt signaling mediates PTH-induced EndMT in aortic ECs. As endothelial dysfunction often initiates and propels the development and progression of cardiovascular disease, better understanding of the β-catenin-dependent Wnt pathway in EndMT might lead to the identification of novel therapeutic targets for VC.
In summary, this study demonstrated that elevated PTH could induce the phenotypic transition of ECs to chondrogenic cells via EndMT, partially mediated by the nuclear translocation of β-catenin. These findings suggest that strategies aimed at lowering PTH levels might prevent VC by preserving the integrity and phenotype of the vascular endothelium.
GRANTS
The work was supported by grants from the National Natural Science Foundation of China (81130010, 81390919, 81470997, 31571186, and 81500545), Natural Science Foundation of Jiangsu Province (BK20150640), Clinic Research Center of Jiangsu Province (BL2014080), National Institutes of Health (DK087893), and the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development, grant BX000893.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
M.W., R.-N.T., and B.-C.L. conception and design of research; M.W. and J.-d.z. performed experiments; M.W., R.-N.T., and H.L. analyzed data; M.W., R.-N.T., H.L., L.-L.L., K.-L.M., and B.-C.L. interpreted results of experiments; M.W. prepared figures; M.W. drafted manuscript; M.W., J.-d.z., S.D.C., and B.-C.L. edited and revised manuscript; M.W., J.-d.z., R.-N.T., S.D.C., H.L., L.-L.L., K.-L.M., and B.-C.L. approved final version of manuscript.
REFERENCES
- 1.Aisagbonhi O, Rai M, Ryzhov S, Atria N, Feoktistov Hatzopoulos AK. Experimental myocardial infarction triggers canonical Wnt signaling and endothelial-to-mesenchymal transition. Dis Model Mech 4: 469–483, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Asche CV, Marx SE, Kim J, Unni SK, Andress D. Impact of elevated intact parathyroid hormone on mortality and renal disease progression in patients with chronic kidney disease stage 3 and 4. Curr Med Res Opin 28: 1527–1536, 2012. [DOI] [PubMed] [Google Scholar]
- 3.Chen NX, O'Neill KD, Chen X, Moe SM. Annexin-mediated matrix vesicle calcification in vascular smooth muscle cells. J Bone Miner Res 23: 1798–1805, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell 149: 1192–1205, 2012. [DOI] [PubMed] [Google Scholar]
- 5.Crisan M, Yap S, Casteilla L, Chen CW, Corselli M, Park TS, Andriolo G, Sun B, Zhang B, Zhang L, Norotte C, Teng ON, Traas J, Schugar R, Deasy BM, Bradylak S, Buhring HJ, Giacobino JP, Lazzari L, Huard J, Peault B. A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell 3: 301–313, 2008. [DOI] [PubMed] [Google Scholar]
- 6.Custodio MR, Koike MK, Neves KR, dos Reis LM, Graciolli FG, Neves CL, Batista DG, Magalhaes AO, Hawlitschek P, Oliveira IB, Dominguez WV, Moyses RM, Jorgetti V. Parathyroid hormone and phosphorus overload in uremia: impact on cardiovascular system. Nephrol Dial Transplant 27: 1437–1445, 2012. [DOI] [PubMed] [Google Scholar]
- 7.Demer LL, Tintut Y. Inflammatory, metabolic, and genetic mechanisms of vascular calcification. Arterioscler Thromb Vasc Biol 34: 715–723, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dudley AC, Khan ZA, Shih SC, Kang SY, Zwaans BM, Bischoff J, Klagsbrun M. Calcification of multipotent prostate tumor endothelium. Cancer Cell 14: 201–211, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Flammer AJ, Gossl M, Widmer RJ, Reriani M, Lennon R, Loeffler D, Shonyo S, Simari RD, Lerman LO, Khosla S, Lerman A. Osteocalcin positive CD133+/CD34-/KDR+ progenitor cells as an independent marker for unstable atherosclerosis. Eur Heart J 33: 2963–2969, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Flammer AJ, Gossl M, Li J, Matsuo Y, Reriani M, Loeffler D, Simari RD, Lerman LO, Khosla S, Lerman A. Patients with an HbA1c in the prediabetic and diabetic range have higher numbers of circulating cells with osteogenic and endothelial progenitor cell markers. J Clin Endocrinol Metab 97: 4761–4768, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gossl M, Modder UI, Atkinson EJ, Lerman A, Khosla S. Osteocalcin expression by circulating endothelial progenitor cells in patients with coronary atherosclerosis. J Am Coll Cardiol 52: 1314–1325, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gossl M, Khosla S, Zhang X, Higano N, Jordan KL, Loeffler D, Enriquez-Sarano M, Lennon RJ, McGregor U, Lerman LO, Lerman A. Role of circulating osteogenic progenitor cells in calcific aortic stenosis. J Am Coll Cardiol 60: 1945–1953, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guo H, Zhang J, Liu T, Shi W. Rapamycin prevents endothelial migration by inhibiting the endothelial-to-mesenchymal transition and matrix metalloproteinase-2 and -9: An in vitro study. Mol Vis 17: 3406–3414, 2011. [PMC free article] [PubMed] [Google Scholar]
- 14.Kapustin AN, Davies JD, Reynolds JL, McNair R, Jones GT, Sidibe A, Schurgers LJ, Skepper JN, Proudfoot D, Mayr M, Shanahan CM. Calcium regulates key components of vascular smooth muscle cell-derived matrix vesicles to enhance mineralization. Circ Res 109: e1–e12, 2011. [DOI] [PubMed] [Google Scholar]
- 15.Korhonen H, Fisslthaler B, Moers A, Wirth A, Habermehl D, Wieland T, Schutz G, Wettschureck N, Fleming I, Offermanns S. Anaphylactic shock depends on endothelial Gq/G11. J Exp Med 206: 411–420, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li J, Qu X, Bertram JF. Endothelial-myofibroblast transition contributes to the early development of diabetic renal interstitial fibrosis in streptozotocin-induced diabetic mice. Am J Pathol 175: 1380–1388, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li L, Chen L, Zhang J, Tang X, Liu Y, Zhang J, Bai L, Yin Q, Lu Y, Cheng J, Fu P, Liu F. C3a and C5a receptor antagonists ameliorate endothelial-myofibroblast transition via the Wnt/β-catenin signaling pathway in diabetic kidney disease. Metabolism 64: 597–610, 2015. [DOI] [PubMed] [Google Scholar]
- 18.Liebner S, Cattelino A, Gallini R, Rudini N, Lurlaro M, Piccolo S, Dejana E. β-Catenin is required for endothelial-mesenchymal transformation during heart cushion development in the mouse. J Cell Biol 166: 359–367, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maddaluno L, Rudini N, Cuttano R, Bravi L, Giampietro C, Corada M, Ferrarini L, Orsenigo F, Papa E, Boulday G, Tournier-Lasserve E, Chapon F, Richichi C, Retta SF, Lampugnani MG, Dejana E. EndMT contributes to the onset and progression of cerebral cavernous malformations. Nature 498: 492–496, 2013. [DOI] [PubMed] [Google Scholar]
- 20.Medici D, Shore EM, Lounev VY, Kaplan FS, Kalluri R, Olsen BR. Conversion of vascular endothelial cells into multipotent stem-like cells. Nat Med 16: 1400–1406, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Medici D, Potenta S, Kalluri R. Transforming growth factor-β2 promotes Snail-mediated endothelial-mesenchymal transition through convergence of Smad-dependent and Smad-independent signaling. Biochem J 438: 515–520, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double-fluorescent Cre reporter mouse. Genesis 45: 593–605, 2007. [DOI] [PubMed] [Google Scholar]
- 23.Neven E, Persy V, Dauwe S, De Schutter T, De Broe ME, D'Haese PC. Chondrocyte rather than osteoblast conversion of vascular cells underlies medial calcification in uremic rats. Arterioscler Thromb Vasc Biol 30: 1741–1750, 2010. [DOI] [PubMed] [Google Scholar]
- 24.Neven E, Dauwe S, De Broe ME, D'Haese PC, Persy V. Endochondral bone formation is involved in media calcification in rats and in men. Kidney Int 72: 574–581, 2007. [DOI] [PubMed] [Google Scholar]
- 25.Neven E, De Schutter TM, De Broe ME, D'Haese PC. Cell biological and physicochemical aspects of arterial calcification. Kidney Int 79: 1166–1177, 2011. [DOI] [PubMed] [Google Scholar]
- 26.Neves KR, Graciolli FG, dos Reis LM, Graciolli RG, Neves CL, Magalhaes AO, Custodio MR, Batista DG, Jorgetti V, Moyses RM. Vascular calcification: Contribution of parathyroid hormone in renal failure. Kidney Int 71: 1262–1270, 2007. [DOI] [PubMed] [Google Scholar]
- 27.Paloian NJ, Giachelli CM. A current understanding of vascular calcification in CKD. Am J Physiol Renal Physiol 307: F891–F990, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rashid G, Bernheim J, Green J, Benchetrit S. Parathyroid hormone stimulates the endothelial nitric oxide synthase through protein kinase A and C pathways. Nephrol Dial Transplant 22: 2831–2837, 2007. [DOI] [PubMed] [Google Scholar]
- 29.Rashid G, Bernheim J, Green J, Benchetrit S. Parathyroid hormone stimulates endothelial expression of atherosclerotic parameters through protein kinase pathways. Am J Physiol Renal Physiol 292: F1215–F1218, 2007. [DOI] [PubMed] [Google Scholar]
- 30.Sage AP, Tintut Y, Demer LL. Regulatory mechanisms in vascular calcification. Nat Rev Cardiol 7: 528–536, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Speer MY, Yang HY, Brabb T, Leaf E, Look A, Lin WL, Frutkin A, Dichek D, Giachelli CM. Smooth muscle cells give rise to osteochondrogenic precursors and chondrocytes in calcifying arteries. Circ Res 104: 733–741, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Steitz SA, Speer MY, Curinga G, Yang HY, Haynes P, Aebersold R, Schinke T, Karsenty G, Giachelli CM. Smooth muscle cell phenotypic transition associated with calcification: Upregulation of Cbfa1 and downregulation of smooth muscle lineage markers. Circ Res 89: 1147–1154, 2001. [DOI] [PubMed] [Google Scholar]
- 33.Tang R, Gao M, Wu M, Liu H, Zhang X, Liu B. High glucose mediates endothelial-to-chondrocyte transition in human aortic endothelial cells. Cardivasc Diabetol 11: 113, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tompson B, Towler DA. Arterial calcification and bone physiology: Role of the bone-vascular axis. Nat Rev Endocrinol 8: 529–543, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang R, Wang Y, Liu N, Ren C, Jiang C, Zhang K, Yu S, Chen Y, Tang H, Deng Q, Fu C, Wang Y, Li R, Liu M, Pan W, Wang P. FBW7 regulates endothelial functions by targeting KLF2 for ubiquitination and degradation. Cell Res 23: 803–819, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu M, Tang RN, Liu H, Pan MM, Lv LL, Zhang JD, Crowley SD, Liu BC. Cinacalcet ameliorates cardiac fibrosis in uremic hearts through suppression of endothelial-to-mesenchymal transition. Int J Cardiol 171: e65–e69, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu M, Tang RN, Liu H, Xu M, Pan MM, Liu BC. Cinacalcet attenuates the renal endothelial-to-mesenchymal transition in rats with adenine-induced renal failure. Am J Physiol Renal Physiol 306: F138–F146, 2014. [DOI] [PubMed] [Google Scholar]
- 38.Yao J, Guihard PJ, Blazquez-Medela AM, Guo Y, Moon JH, Jumabay M, Bostrom KI, Yao Y. Serine protease activation essential for endothelial-mesenchymal transition in vascular calcification. Circ Res 117: 758–769, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yao Y, Jumabay M, Ly A, Radparvar M, Cubberly MR, Bostrom KI. A role for the endothelium in vascular calcification. Circ Res 113: 495–504, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yung LM, Sanchez-Duffhues G, Ten Dijke P, Yu PB. Bone morphogenetic protein 6 and oxidized low-density lipoprotein synergistically recruit osteogenic differentiation in endothelial cells. Cardiovasc Res 108: 278–287, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zeisberg EM, Potenta SE, Sugimoto H, Zeisberg M, Kalluri R. Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J Am Soc Nephrol 19: 2282–2287, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang H, Wang LJ, Si DL, Wang C, Yang JC, Jiang P, Du C, Wang JJ. Correlation between osteocalcin-positive endothelial progenitor cells and spotty calcification in patients with coronary artery disease. Clin Exp Pharmacol Physiol 42: 734–739, 2015. [DOI] [PubMed] [Google Scholar]
- 43.Zheng M, Lv LL, Ni J, Ni HF, Li Q, Ma KL, Liu BC. Urinary podocyte-associated mRNA profile in various stages of diabetic nephropathy. PLoS One 6: e20431, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]