

Abstract
Following vascular injury medial smooth muscle cells dedifferentiate and migrate through the internal elastic lamina where they form a neointima. The goal of the current study was to identify changes in gene expression that occur before the development of neointima and are associated with the early response to injury. Vascular injury was induced in C57BL/6 mice and in Myh11-creER(T2) mTmG reporter mice by complete ligation of the left carotid artery. Reporter mice were used to visualize cellular changes in the injured vessels. Total RNA was isolated from control carotid arteries or from carotid arteries 3 days following ligation of C57BL/6 mice and analyzed by Affymetrix microarray and quantitative RT-PCR. This analysis revealed decreased expression of mRNAs encoding smooth muscle-specific contractile proteins that was accompanied by a marked increase in a host of mRNAs encoding inflammatory cytokines following injury. There was also marked decrease in molecules associated with BMP, Wnt, and Hedgehog signaling and an increase in those associated with B cell, T cell, and macrophage signaling. Expression of a number of noncoding RNAs were also altered following injury with microRNAs 143/145 being dramatically downregulated and microRNAs 1949 and 142 upregulated. Several long noncoding RNAs showed altered expression that mirrored the expression of their nearest coding genes. These data demonstrate that following carotid artery ligation an inflammatory cascade is initiated that is associated with the downregulation of coding and noncoding RNAs that are normally required to maintain smooth muscle cells in a differentiated state.
Keywords: vascular smooth muscle, neointima, smooth muscle differentiation, inflammation, long noncoding RNA, microRNA
vascular smooth muscle cells (VSMCs), the major contractile components of the vascular system, are critically important for regulating blood pressure and flow throughout the vascular system. In disease states such as atherosclerosis, hypertension, stenosis following injury, and restenosis following vascular interventions, VSMCs dedifferentiate and proliferate and migrate toward the lumen of the vessel leading to the formation of a neointima that may impair blood flow (16, 19). Classically these diseases are described as being associated with dedifferentiated VSMCs that have decreased expression of proteins required for the normal contractile function, increased expression of extracellular matrix proteins, and increased cell proliferation (11). Many different stimuli are known to be able to induce VSMC dedifferentiation. For example, inflammatory cytokines and growth factors such as interleukin-1β (IL-1β) and platelet-derived growth factor b (PDGFb) have been shown to drive VSMCs into a dedifferentiated, proliferative state (6, 18, 60, 61). It is less clear how mechanical injury or cessation of blood flow mediates these changes. In the current report a carotid artery occlusion was utilized to identify the genetic responses of carotid artery vascular cells to a ligation injury in mice. The results of this study demonstrate that before the development of a neointima there is a local inflammatory response that is associated with dedifferentiation of medial VSMCs.
MATERIALS AND METHODS
Transgenic mice and carotid ligation.
All animal procedures were performed using procedures approved by the Indiana University School of Medicine Institutional Animal Care and Use Committee under protocol number 10310. For mRNA expression studies control male C57BL/6J mice underwent carotid artery ligation at 8–10 wk of age as described previously (27). Prior to surgery, mice were anesthetized with intraperitoneal injection of ketamine/xylazine (88 mg/kg, 12 mg/kg). Following surgery mice were given a single subcutaneous injection of carprofen analgesic (0.5 mg). Three days following ligation mice were killed under anesthesia (intraperitoneal injection of ketamine/xylazine (88 mg/kg, 12 mg/kg), and the injured and control contralateral carotid arteries were harvested. Carotid arteries were also harvested from an additional control group of male C57BL/6J mice that were not subject to carotid ligation. To visualize VSMCs and other cell types in carotid arteries after ligation, smooth muscle myosin heavy chain (Myh11) creER(T2)−/+ mice (54) (on a C57BL/6J background) were crossed with mTomato/mEGFP (mTmG) reporter mice (Jackson strain: B6.129(Cg)-Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J) (19). The reporter mice express membrane-associated Tomato fluorescent protein in all cell types in the absence of cre-mediated recombination and membrane-associated enhanced green fluorescent protein (EGFP) following cre-mediated recombination. Crossing reporter mice with the Myh11 creER(T2) generates mice in which the tamoxifen-inducible cre deletes the mTomato cassette, thereby resulting in mEGFP expression specifically from smooth muscle cells. The creER(T2) transgene was used under license from the Institut de Génétique et de Biologie Moléculaire et Cellulaire. Double heterozygous male transgenic mice at 6 wk of age were treated with tamoxifen (1 mg/mouse intraperitoneal) or corn oil control once a day for 5 days. At 2 wk following the last treatment the left carotid artery was ligated as described above for the C57BL/6J mice. The injured and contralateral control carotid arteries were isolated at 3 days following ligation. In addition, right and left carotid arteries from nonsurgical, uninjured mice were obtained as controls where indicated and the tissues processed for RNA isolation or imaging.
Confocal imaging.
For confocal imaging of mTmG mice, carotid tissues were excised and immediately fixed in 4% paraformaldehyde for 2 h on ice without dissection. Following fixation, vessels were washed with rotation in phosphate buffered saline for 3 × 5 min and then frozen in OCT tissue freezing media (Tissue-Tek) on a bed of dry ice and 2-methylbutane. Cryosections (8 µm) were cut and stored at −80°C. For immunofluorescent staining cryosections were permeabilized in 0.2% Triton X-100 in Tris-buffered saline (TBS) for 5 min, washed in TBS for 5 min, incubated with 5% horse serum diluted in TBS at room temperature for 1 h to block nonspecific antibody binding. Blocked sections were incubated overnight at 37°C with primary antibodies to CD68 (1:200, clone FA-11, AbD Serotec) or CD31(1:100, clone 390, E Bioscience), diluted in 5% horse serum/TBS. Sections were incubated with an irrelevant primary antibody as a negative control. Following washing in TBS, primary antibodies were detected by incubation with anti-rat Alexa Fluor 647 (1:10,000, Jackson ImmunoResearch). For Mac3 and F4/80 immunostaining, sections were not permeabilized and after blocking they were incubated with Mac3 (clone M3/84) or F4/80 (clone BM8) antibodies that were directly conjugated to Alexa Fluor 647 (Biolegend). After extensive washing slides were mounted in Prolong Gold containing 4',6-diamidino-2-phenylindole (Invitrogen) and visualized by confocal microscopy using a Zeiss LSM700 laser scanning confocal microscope. Images shown are single planes obtained by sequential scanning of each channel using a ×40/1.2 objective.
Primary aortic smooth muscle cells.
Mouse aortic smooth muscle cells were prepared by enzymatic digestion of aorta isolated from 3 wk old mice, as described previously (20). Briefly, aorta were isolated from 8 mice, cleaned of adventitia and blood, and minced in 3 ml of digestion mix (0.6 U/ml Liberase TM (Roche), 0.25 mg/ml DNase1, in Hanks' balanced salt solution). The tissue was digested for 30 min at 37°C, filtered through a 100 µm sieve, and plated into a six-well dish in Dulbecco’s modified eagle medium containing 10% fetal calf serum (FCS). Once cells reached confluence they were trypsinized and replated at 5×104 cells per well in a 12-well plate. Cells were switched to media containing 0.5% FCS 24 h before treatment with IL-1β (10 ng/ml).
RNA preparation and Affymetrix arrays.
For analysis of mRNA expression carotids were dissected free of adventitia, the clotted blood was extracted, and the tissues were rapidly frozen in liquid N2. Total RNA was isolated from pools of six carotid arteries from six injured mice or three control, uninjured mice (using both left and right carotids) by homogenizing in 1 ml Trizol using a Polytron (Kinematica). RNA was purified using a Purelink Micro RNA isolation kit (Invitrogen). RNA integrity was validated using an Agilent Bioanalyzer and samples with a relative integrity number <7 were rejected. A total of four separate pooled RNA preparations from injured vessels and control nonsurgical, uninjured vessels were used to probe Affymetrix Mouse Gene 2.0ST arrays. Total RNA samples were labeled using the standard protocol for the Ambion WT Expression kit combined with the Affymetrix GeneChip WT Terminal Labeling and Controls Kit. Individual labeled samples were hybridized to the Mouse Gene 2.0 ST GeneChips for 17 h, then washed, stained, and scanned with the standard protocol using Affymetrix GeneChip Command Console Software to generate data (CEL files). Arrays were visually scanned for abnormalities or defects, and none were found. CEL files were imported into Partek Genomics Suite (Partek, St. Louis, MO). Robust multiarray average (RMA) signals were generated for the core probe sets using the RMA background correction, quantile normalization, and summarization by Median Polish (23). Summarized signals for each probe set were log2 transformed. These log-transformed signals were used for Principal Components Analysis, hierarchical clustering, and signal histograms to determine if there were any outlier arrays and no outliers were detected. Untransformed RMA signals were used for fold change calculations. Data was analyzed by a t-test with the log2-transformed signals comparing the injured vessels to the controls. Fold changes were calculated using the untransformed RMA signals (34). Probe sets with log2 expression levels < 4.0 are very close to background and were removed before the false discovery rate (FDR) was calculated using the Storey method (46). Genes differentially expressed at FDR< 0.05 and absolute fold ≥ 2.0 were imported into QIAGEN's Ingenuity Pathway Analysis 2015 Winter Release (IPA, Qiagen Redwood City, http://www.qiagen.com/ingenuity).
Quantitative RT-PCR.
Total RNA was isolated as described above. For reverse transcriptase reactions, 100 ng of RNA was used (High Capacity RT-cDNA Kit, Life Technologies). Real-time PCR was performed using Syber Green (Roche). Levels of mRNA expression were normalized to an Hprt internal control and are expressed relative to levels seen in control nonsurgical uninjured carotid arteries. Statistical significance was determined using Students T-tests. The primers used for quantitative (q)RT-PCR are listed in Supplemental Table S1. (The online version of this article contains supplemental material.) These primers match mouse sequences, span introns, and were screened to confirm similar reaction efficiencies.
Western blotting.
Pools of three carotid arteries were first dissected to remove adventitia and clotted blood and then homogenized in RIPA lysis buffer supplemented with protease inhibitors. Protein concentrations were determined using a BCA Protein Assay kit (Pierce). Proteins (1 or 5 µg) were separated on 7.5 and 15% SDS-polyacrylamide gels and transferred to nitrocellulose or polyvinyl difluoride membranes, respectively. Membranes were blocked in 5% bovine serum albumen in 10 mM Tris pH 7.5, 150 mM NaCl, 0.05% Tween 20, and incubated with antibodies diluted in the same blocking buffer. Antibodies used for Western blotting were, Myh11 (SM2, 1:2,000) (17), Mylk1 (Sigma, clone K36, 1:5,000), Acta2 (Sigma, clone 3A1, 1:10,000), Tagln (SM22α, generated in rabbits against the synthetic peptide CGPDVGRPDRGRLGFQVW, Proteintech, 1:10,000), Spp1 (Osteopontin, Santa Cruz Sc10593, 1:1,000), vinculin (Sigma, clone VIN-11-5, 1:5,000). Primary antibodies were then detected using horseradish peroxidase-conjugated secondary antibodies and visualized using chemiluminescence. Chemiluminescent signals were collected and analyzed on a G Box imager (Syngene).
RESULTS
Induction of inflammatory cytokines following carotid ligation.
Previous studies utilizing a dual color reporter transgenic mouse line (mTmG: B6.129(Cg)-Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J) together with a tamoxifen-regulated smooth muscle-specific cre transgene (Myh11 or Acta2 promoter driven) revealed that the majority of the neointimal cells that arise following mouse carotid ligation come from previously differentiated medial VSMCs (19). These studies also showed that the neointima begins to develop between 7 and 14 days postligation in this model (19). To expand on these results and identify the early changes in medial VSMCs that lead to their dedifferentiation, migration, and formation of the neointima, we conducted a genome-wide comparative analysis of RNA expression that occurs 3 days following carotid ligation. This time point was chosen to assess the early changes that occur in the vessel wall before the development of any significant neointima (19). Results from an Affymetrix microarray analysis identified over 1,100 RNAs whose expression was increased over twofold following carotid ligation and ~700 RNAs whose expression was decreased over twofold (at a P < 0.05, FDR < 0.05; Supplemental Table S2). Ingenuity Pathway analysis revealed that among the RNAs that were upregulated more than twofold following injury, a significant enrichment in mRNAs encoding proteins associated with inflammation occurred (Table 1, Supplemental Table S3). Notably, there was a marked increase in expression of RNAs encoding proteins associated with inflammatory blood cells as well as cell adhesion and diapedesis (Supplemental Table S3). Using qRT-PCR we confirmed the increase in expression of several of these upregulated RNAs, including Secreted phosphoprotein 1 (Spp1, also known as osteopontin), interleukin-1 beta (IL1β), Interleukin-6 (IL6), chemokine ligand 2 (Ccl2, also known as monocyte chemoattractant protein 1 or MCP-1), Triggering receptor expressed on Myeloid Cells 1 and 2 (Trem1 and Trem2) (Fig. 1). To determine if this represented an infiltration of immune cells into the vessel wall we analyzed tissue sections from ligation injured carotid vessels isolated from Myh11creER(T2) mTmG reporter mice. Tamoxifen treatment of these reporter mice 2 wk before carotid ligation results in cre-mediated excision of the mTomato cassette specifically from smooth muscle cells, resulting in these cells expressing membrane-localized EGFP while all other cell types maintain expression of mTomato (36). Thus, all VSMC that were differentiated at the time of tamoxifen treatment are labeled green and remain green, even if they subsequently dedifferentiate, while all other cell types are labeled red. This permits us to readily identify inflammatory cells infiltrating the injured vessel as red cells among the green VSMCs. As shown in Fig. 2, there were numerous nonsmooth muscle cells (red membrane) within the lumen of injured vessels; very few were observed within the medial and adventitial layers at 3 days following carotid injury. Immunostaining with anti-CD68, Mac3, F4/80, and Lys6G antibodies confirmed that these luminal, nonsmooth muscle cells include monocytes/macrophages and neutrophils (Fig. 2). This analysis further confirmed the lack of formation of neointima 3 days following carotid ligation and the presence of residual endothelial cells detected by their expression of CD31 (Fig. 2).
Table 1.
Twenty most upregulated genes 3 days following carotid ligation
| P Value | FDR | Fold | Gene | Gene Name |
|---|---|---|---|---|
| 3.55E-09 | 6.74E-06 | 90 | Saa3 | serum amyloid A 3 |
| 6.05E-08 | 3.56E-05 | 57 | Spp1 | secreted phosphoprotein 1 |
| 3.05E-07 | 8.01E-05 | 52 | Il6 | interleukin 6 |
| 5.42E-04 | 3.58E-03 | 29 | Ccl3 | chemokine (C-C motif) ligand 3 |
| 1.63E-04 | 1.81E-03 | 28 | Arg1 | arginase, liver |
| 2.33E-05 | 6.59E-04 | 27 | Il1b | interleukin 1 beta |
| 5.21E-11 | 6.42E-07 | 23 | Ccl2 | chemokine (C-C motif) ligand 2 |
| 5.97E-03 | 1.72E-02 | 23 | Cxcl3 | chemokine (C-X-C motif) ligand 3 |
| 5.07E-10 | 2.17E-06 | 21 | Prg4 | proteoglycan 4 |
| 8.52E-04 | 4.78E-03 | 20 | Clec4e | C-type lectin domain family 4e |
| 5.57E-06 | 3.12E-04 | 20 | Slfn4 | schlafen 4 |
| 3.00E-10 | 1.71E-06 | 19 | Ccl7 | chemokine (C-C motif) ligand 7 |
| 3.45E-04 | 2.76E-03 | 19 | Clec4d | C-type lectin domain family 4d |
| 8.84E-05 | 1.32E-03 | 19 | Chi3l3 | chitinase 3-like 3 |
| 6.95E-05 | 1.17E-03 | 18 | LOC100038947 | signal-regulatory protein beta 1-like |
| 5.52E-05 | 1.02E-03 | 17 | Serpinb2 | serine peptidase inhibitor, clade B2 |
| 7.03E-07 | 1.28E-04 | 17 | Ccr5 | chemokine (C-C motif) receptor 5 |
| 8.44E-06 | 3.90E-04 | 16 | Gp49a | glycoprotein 49 A |
| 1.71E-04 | 1.85E-03 | 16 | Clec4n | C-type lectin domain family 4n |
| 1.95E-08 | 1.85E-05 | 16 | Ccr2 | chemokine (C-C motif) receptor 2 |
The most upregulated RNAs excerpted from Supplemental Table S2 are indicated.
Fig. 1.
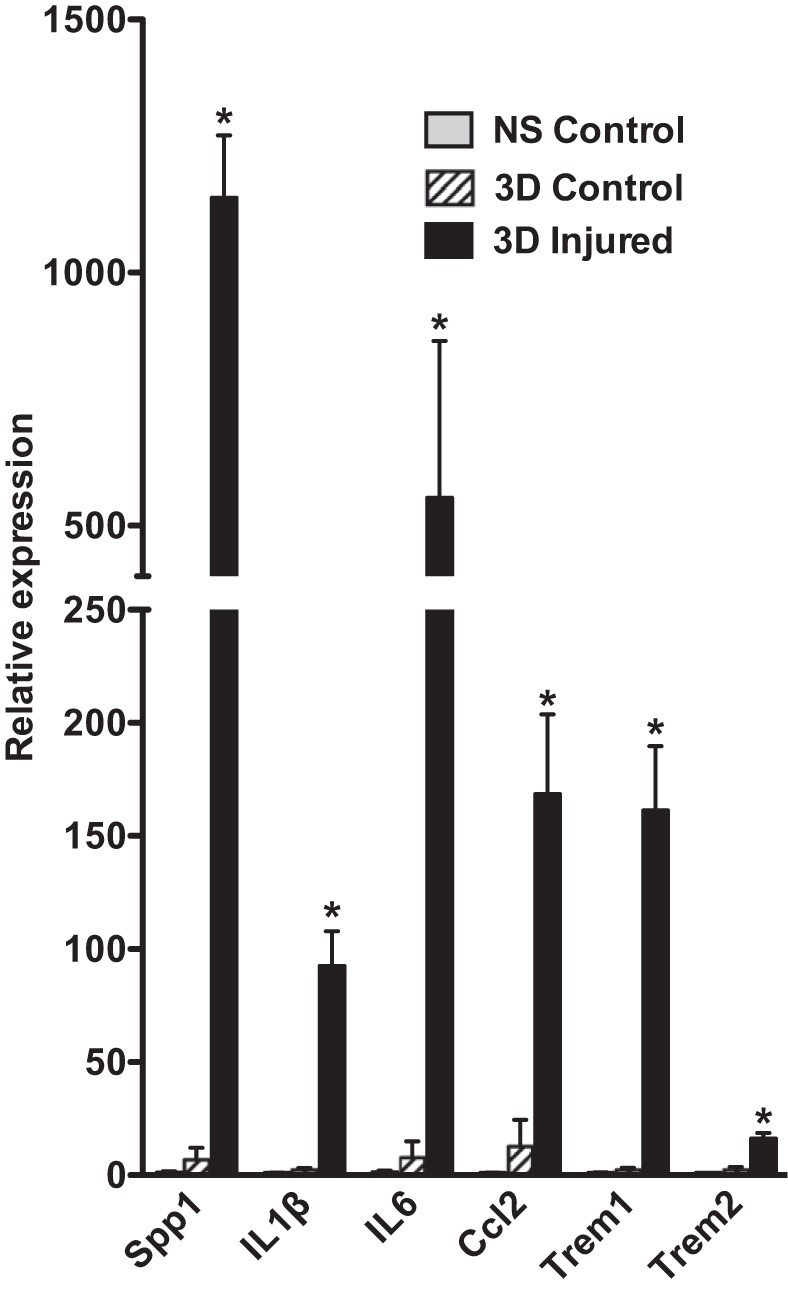
mRNAs encoding proteins associated with inflammation are upregulated following injury. The left carotid artery was ligated in 8–10 wk old male C57BL/6J mice, and tissues were harvested 3 days later. Total RNA was isolated from the left injured artery and the right uninjured control artery as well as from control carotid arteries obtained from mice that had not undergone carotid ligation. For each sample 6 injured, 6 contralateral control, and 6 uninjured, control arteries were pooled before RNA extraction as described in materials and methods. mRNA expression levels were analyzed by quantitative (q)RT-PCR, and data presented are expressed relative to vessels obtained from control mice after normalization to an Hprt internal control (RQ = 2-ΔΔCt, where ΔΔCt = (Experimental; Ct gene – Ct hprt) – (control nonsurgical; Ct gene – Ct hprt); n = 3–5. *P < 0.05. NS, nonsurgical control; 3D control, contralateral control carotid; 3D injured, ligated carotid 3 days following ligation.
Fig. 2.

Inflammatory cells are recruited into injured vessels. We treated 5 wk old male Myh11 creER(T2)−/+ mTmG−/+ mice with tamoxifen (1 mg ip) once a day for 5 days. Two weeks following the last tamoxifen injection the left carotid artery was ligated and tissues were harvested 3 days later. A–C: cryosection (8 µm) obtained from injured artery stained with antibodies to CD68. A: smooth muscle cells are visualized by their membrane localized green EGFP fluorescence. Other cell types are red due to expression of membrane localized Tomato. Nuclei were visualized with DAPI (blue) and CD68 expression visualized with Alexifluor 647 antibodies is indicated in white. B: white (CD68) and blue (DAPI) channels from the image shown in A. C: red (non-SMC), green (VSMC), and blue (DAPI) channels from the image shown in A. D–G: sections from injured (D, E) and contralateral control carotid arteries (F, G) stained with antibodies to CD31 to visualize endothelial cells (white), mEGFP-positive VSMC are green, mTomato positive non-VSMCs are red, and nuclei are blue (DAPI). E and G are blue (DAPI) and white (CD31) channels of the images shown in D and F, respectively. H, I: sections stained with Ly6G antibodies to detect neutrophils (white). J: parallel section reacted with antibodies to an irrelevant protein [HA epitope tag (Bethyl laboratories), white] showing no background white staining. K: section stained with Mac3 (white; only white and blue channels shown). L: section stained with F4/80 (white; only white and blue channels shown) M, N: sections from 2 different mice stained with antibodies to Ki67 to detect proliferating cells (white). All images are representative of data obtained from 3 animals and shown at the same magnification with the scale bar shown representing 20 µm. Adv, adventitia; mTom, membrane-localized Tomato; mEGFP, membrane-localized EGFP; MC, monocytes/macrophages; EC, endothelial cells; N, neutrophils; DAPI, 4',6-diamidino-2-phenylindole.
Decreased expression of mRNAs encoding contractile proteins following IL-1β stimulation in vitro and carotid ligation in vivo.
Although many of the elevated inflammatory cytokines may arise from the adhered immune cells, these cytokines can also be released directly from VSMCs and endothelial cells. For example, VSMCs have been reported to increase IL-6 expression in response to IL-1β and TNF-α stimulation (32). Moreover, VSMCs have been shown to secrete inflammatory cytokines before the infiltration of immune cells in a rat model of vascular injury (15). Results shown in Fig. 3 demonstrate that treatment of primary VSMCs with IL-1β (10 ng/ml) causes a 40-fold increase in IL-6 mRNA. In addition, there is a corresponding decrease in expression of mRNA encoding transcription factors (myocardin, SRF) that are required to sustain a differentiated contractile phenotype of VSMCs (Fig. 3). Consistent with these observations, IL-1β treatment also resulted in a fourfold repression of mRNA encoding a smooth muscle cell-specific differentiation marker, Myh11, which encodes smooth muscle myosin heavy chain, in the cultured VSMCs (Fig. 3). Paralleling these in vitro observations, we observed decreases in expression of mRNAs encoding smooth muscle contractile proteins and several transcription factors linked to VSMC differentiation including myocardin, GATA6, Foxf1, and Brm following carotid ligation in vivo (Fig. 4). Importantly, qRT-PCR analysis revealed that this is not a consequence of elevated systemic levels of inflammatory cytokines as uninjured contralateral carotid arteries obtained from the same mice as the injured left carotid arteries did not display this decreased expression of mRNAs encoding contractile proteins (Fig. 4A; compare 3-day contralateral control samples with 3-day injured samples) or myogenic transcription factors (Fig. 4C). Supporting these findings, Ingenuity Pathway analysis of the mRNAs downregulated twofold or more following vascular injury showed enrichment of mRNAs encoding proteins that are part of the pathway associated with SMC contraction (e.g., proteins whose activity is regulated following inhibition of phosphodiesterase 5 by Sildenafil, Tables 2, 3). Despite these significant changes in mRNAs encoding proteins characteristic of differentiated VSMCs, we observed very little change in the expression of the proteins themselves by Western blotting (Fig. 5). Although some individual samples had decreased expression, overall these changes were not significant following normalization to vinculin levels. mRNAs encoding components of several signaling pathways associated with sustaining VSMC differentiation, including Wnt, Hedgehog, Notch, and TGF-β/BMP, were also downregulated following injury (Table 4; Fig. 4, B and C). We observed decreased expression of mRNAs encoding Notch 3 (Table 4) and the Notch effector Hey2 (Fig. 4C). mRNAs encoding the Hedgehog effectors Gli1 and Gli2 and the hedgehog regulatory proteins Hhip were downregulated, whereas Hhipl1 and Hhat were upregulated (Fig. 4C, Table 4). mRNAs encoding many components of the TGF-β/BMP signaling pathways were also altered following vascular injury with several ligands, including BMP2, 4, 6 and TGF-β2, 3 being downregulated together with receptors Bmpr2, Bmpr1a,b, Tgfbr1, Acvr1, and Acvrl1 (Table 4).
Fig. 3.
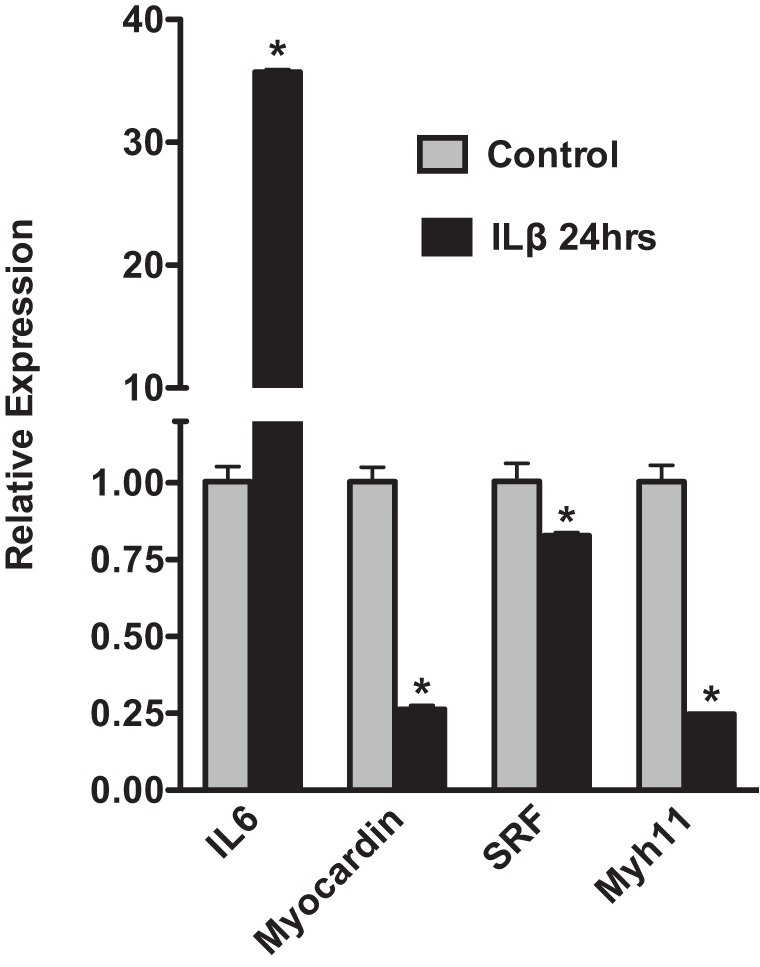
IL-1β induces a marked upregulation of IL-6 expression in vascular smooth muscle cells (VSMCs). Mouse aortic smooth muscle cells were isolated as described in materials and methods. First-passage cells were serum-starved to promote differentiation for 24 h and then treated with IL-1β (10 ng/ml) or vehicle control in serum-free media for 24 h. After 24 h RNA expression levels were analyzed by qRT-PCR and data presented are expressed relative to cells treated with vehicle control after normalization to an Hprt internal control (RQ = 2-ΔΔCt, where ΔΔCt = (IL-1β treated; Ct gene – Ct hprt) – (control; Ct gene – Ct hprt); n = 4 *P < 0.05.
Fig. 4.

Downregulation of mRNAs encoding contractile proteins, transcription factors, and signaling molecules following injury. RNA was isolated from carotid arteries at 3 days following carotid ligation injury or from control uninjured mice and analyzed by qRT-PCR as described in Fig. 1. A: smooth muscle contractile proteins. Myh11, smooth muscle myosin heavy chain; Tagln, SM22α; Cnn1, calponin 1; Acta2, smooth muscle α-actin; Actg2, smooth muscle γ-actin; Mylk1, smooth muscle myosin light chain kinase. B: signaling molecules. C: transcription factors. n = 3–5 *P < 0.05 NS, nonsurgical control; 3D control, contralateral control carotid; 3D injured, ligated carotid 3 days following ligation.
Table 2.
Most significantly enriched downregulated pathways 3 days following carotid ligation
| Ingenuity Canonical Pathways | FDR | Ratio | z-Score | Molecules |
|---|---|---|---|---|
| Cellular effects of sildenafil (Viagra) | 4.37E-04 | 0.11 | NaN | MYH10, PLCB4, PRKG1, PPP1R12A, GUCY1A3, ADCY5, PDE3A, PDE5A, MYLK, ACTG2, ITPR1, MYH11, PLCL1, GUCY1B3 |
| Gap junction signaling | 5.62E-03 | 0.08 | NaN | PLCB4, PRKG1, NOV, GUCY1A3, ADCY5, NPR1, GNAI1, ACTG2, ITPR1, PLCL1, NPR2, GUCY1B3, HTR2A |
| Human embryonic stem cell pluripotency | 5.62E-03 | 0.09 | NaN | FZD8, NTF3, BMP4, NTRK3, BMP3, TGFB3, SMAD6, FGFR2, TCF7L1, PDGFD, BMP6, WNT5A |
Ingenuity analysis of RNAs downregulated ≥2-fold. The ratio indicates the proportion of genes in the pathway whose expression was altered. The z-score indicates the relative activation (positive numbers) or repression (negative numbers) of the pathway. FDR, false discovery rate; NaN, not able to be determined.
Table 3.
Twenty most downregulated genes 3 days following carotid ligation
| P Value | FDR | Fold Change | Gene | Gene Name |
|---|---|---|---|---|
| 2.59E-05 | 6.92E-04 | −9.3 | Ptprz1 | protein tyrosine phosphatase, receptor type Z, polypeptide 1 |
| 1.97E-07 | 6.47E-05 | −7.9 | Klk10 | kallikrein related-peptidase 10 |
| 5.13E-04 | 3.47E-03 | −7.0 | Omd | osteomodulin |
| 6.24E-04 | 3.90E-03 | −6.6 | Hhip | Hedgehog-interacting protein |
| 9.00E-04 | 4.95E-03 | −6.1 | LOC621549 | uncharacterized LOC621549 |
| 4.20E-05 | 8.85E-04 | −6.0 | 4930429F24Rik | RIKEN cDNA 4930429F24 gene |
| 3.48E-03 | 1.19E-02 | −5.9 | Sost | sclerostin |
| 2.95E-04 | 2.51E-03 | −5.8 | ORF63 | open reading frame 63 |
| 1.61E-03 | 7.22E-03 | −5.7 | E030013I19Rik | RIKEN cDNA E030013I19 gene |
| 1.42E-04 | 1.67E-03 | −5.4 | AI507597 | expressed sequence AI507597 |
| 1.11E-03 | 5.66E-03 | −5.3 | Npy1r | neuropeptide Y receptor Y1 |
| 8.39E-04 | 4.73E-03 | −5.1 | Myom1 | myomesin 1 |
| 3.17E-03 | 1.12E-02 | −4.9 | Mylk4 | myosin light chain kinase family, member 4 |
| 3.06E-04 | 2.57E-03 | −4.8 | Slc38a11 | solute carrier family 38, member 11 |
| 1.60E-03 | 7.20E-03 | −4.7 | Synpo2 | synaptopodin 2 |
| 4.07E-03 | 1.33E-02 | −4.7 | Cytl1 | cytokine-like 1 |
| 2.53E-03 | 9.68E-03 | −4.7 | Pkhd1l1 | polycystic kidney and hepatic disease 1-like 1 |
| 5.09E-04 | 3.46E-03 | −4.7 | Fmod | fibromodulin |
| 5.77E-04 | 3.72E-03 | −4.6 | C7 | complement component 7 |
| 1.52E-03 | 6.95E-03 | −4.6 | Scube3 | signal peptide, CUB domain, EGF-like 3 |
The most downregulated RNAs excerpted from Supplemental Table S2 are indicated.
Fig. 5.

Changes in contractile protein expression following injury. Western blot analysis of protein expression in extracts from nonsurgical control arteries (NS Control) and contralateral (3D Control) and injured (3D injured) arteries 3 days after ligation. Each lane represents a pool of 3 vessels. The positions of molecular mass markers are indicated to the left of the blots. The asterisk on the vinculin blot indicates the band corresponding to vinculin.
Table 4.
Altered signaling pathways associated with VSMC differentiation
| Wnt | Fold Change | TGFB | Fold Change |
| Wnt2 | 1.67 | Bmp2k | 1.92 |
| Wnt2b | −1.57 | Bmper | 1.70 |
| Wnt5a | −2.43 | Bmp1 | 1.49 |
| Wif1 | −2.83 | Bmpr2 | −1.44 |
| Fzd5 | −1.33 | Crim1 | −1.72 |
| Fzd7 | −1.95 | Bmpr1a | −1.84 |
| Fzd8 | −2.62 | Bmp4 | −2.35 |
| Pygo1 | −1.95 | Bmp6 | −2.53 |
| Hedgehog | Fold Change | Bmpr1b | −2.72 |
| Hhipl1 | 1.91 | Bmp3 | −3.15 |
| Hhat | 1.32 | Tab1 | 1.07 |
| Hhip | −6.62 | Tgfbr1 | −1.78 |
| Gli2 | −1.85 | Tgfb1i1 | −1.79 |
| Gli1 | −1.89 | Tgfb2 | −1.85 |
| Tgfb3 | −2.55 | ||
| Notch | Fold Change | Acvr1 | −1.16 |
| Notch3 | −1.92 | Acvrl1 | −1.25 |
| Jag 1 | −1.50 | SMAD4 | −1.20 |
| Hey 2 | −2.76 | SMAD6 | −2.40 |
| SMAD9 | −2.60 |
Examples of changes mRNAs encoding signaling molecules that are known to play a role in regulating the differentiation of VSMCs are indicated. Data are excerpted from Supplemental Table S2.
Altered expression of noncoding RNAs following vascular injury.
Among the 10 most dramatically downregulated mRNAs only five represent annotated protein-coding RNAs (Table 3). These five genes encode protein tyrosine phosphatase receptor type Z-polypeptide 1 (Ptprz1), kallikrein-related peptidase 10 (Klk10), osteomodulin (Omd), Hedgehog-interacting protein (Hhip), and Sclerostin (Sost). The downregulation of all of these RNAs was confirmed by qRT-PCR analysis except for Ptprz1, which was expressed at such low levels in control vessels that it was near the threshold of detection by qRT-PCR analysis and could not be reliably quantitated (Figs. 4, 6). In addition to changes in expression of coding mRNAs, we observed significant changes in several microRNAs and long noncoding RNAs (lncRNAs). The most significantly up- and downregulated microRNAs are shown in Table 5. As anticipated from other studies, microRNAs 143/145 were the most significantly downregulated microRNAs following vascular injury (7, 14, 56). Notably, five microRNAs (1949, 142, 511, 5104, 1948, and 692-1) were upregulated greater than twofold following injury. We also noted significant changes in expression of many lncRNAs, a recently appreciated class of regulatory RNA molecules that control expression of coding genes through multiple mechanisms (58, 59). For example, the most downregulated RNA that does not encode a known protein, accession #4940429F24Rik, is an lncRNA located between the Tmem30a and Filip1 loci. In many cases, lncRNAs have been found to affect the activity of the coding genes that are physically closest to the lncRNA on chromosomes (58, 59). Examination of unknown RNAs annotated as lncRNAs on the UCSC Genome Browser, whose change in expression paralleled that of the nearest protein-coding gene, led to the identification of several lncRNAs whose downregulation paralleled the downregulation of nearby coding genes. For example, we saw downregulation of the coding and lncRNA gene pairs, integrin alpha 8 (Itga8) and lncRNA E030013I19Rik, thrombospondin 4 (Thsd4) and lncRNA 9230112J17Rik, protein kinase G (Prkg1) and lncRNA 8430431K14Rik, sclerostin (Sost) and lncRNA 4930417O22Rik, and Gata6 (Gata6) and lncRNA 1010001N08Rik (Fig. 6A, Table 6). Consistent with these observations, the lncRNAs adjacent to Gata6 and Prkg1 were also decreased in mouse aortic VSMCs treated with IL-1β in parallel with decreases in Gata6 and Prkg1 mRNAs (Fig. 6B).
Fig. 6.

Several long noncoding RNAs and their nearest coding mRNAs are downregulated following injury. A: RNA was isolated at 3 days following carotid ligation. and analyzed by qRT-PCR as described in Fig. 1. NS, nonsurgical control; 3D control, contralateral control carotid; 3D injured, ligated carotid 3 days following ligation. B: RNA was isolated from mouse aortic smooth muscle cells following treatment with IL-1β or vehicle, and RNA expression was analyzed by qRT-PCR as described in Fig. 2. n = 3–5 *P < 0.05.
Table 5.
Most up- or downregulated microRNAs following carotid ligation
| P Value | FDR | Fold Change | Gene Symbol |
|---|---|---|---|
| 1.08E-03 | 5.58E-03 | 4.5 | microRNA 1949 |
| 5.85E-04 | 3.75E-03 | 3.7 | microRNA 142 |
| 1.63E-05 | 5.56E-04 | 3.2 | microRNA 511 |
| 2.24E-03 | 8.91E-03 | 2.9 | microRNA 5104 |
| 1.49E-04 | 1.72E-03 | 2.0 | microRNA 1948 |
| 3.04E-04 | 2.56E-03 | 1.9 | microRNA 692-1 |
| 8.41E-03 | 2.19E-02 | −1.9 | microRNA 504 |
| 5.96E-04 | 3.79E-03 | −1.9 | microRNA let7d |
| 9.81E-03 | 2.43E-02 | −2.0 | microRNA let7a-2 |
| 1.55E-04 | 1.76E-03 | −2.0 | microRNA 421 |
| 5.65E-04 | 3.68E-03 | −2.0 | microRNA 568 |
| 5.86E-03 | 1.70E-02 | −2.0 | microRNA 466n |
| 1.52E-03 | 6.95E-03 | −2.1 | microRNA 130a |
| 3.92E-05 | 8.61E-04 | −2.1 | microRNA 1941 |
| 1.93E-04 | 1.99E-03 | −2.2 | microRNA 301 |
| 2.06E-05 | 6.22E-04 | −2.2 | microRNA 126 |
| 8.83E-05 | 1.32E-03 | −2.3 | microRNA 1192 |
| 4.12E-03 | 1.34E-02 | −2.5 | microRNA 30a |
| 5.03E-04 | 3.43E-03 | −2.5 | microRNA 24-1 |
| 7.87E-04 | 4.57E-03 | −2.5 | microRNA 28c |
| 8.55E-04 | 4.78E-03 | −2.6 | microRNA 27b |
| 1.98E-04 | 2.01E-03 | −2.7 | microRNA 193b |
| 1.92E-05 | 6.12E-04 | −2.8 | microRNA 28 |
| 1.39E-05 | 5.06E-04 | −2.8 | microRNA 29b-2 |
| 1.07E-03 | 5.56E-03 | −2.9 | microRNA 105 |
| 3.45E-03 | 1.19E-02 | −3.0 | microRNA 140 |
| 5.86E-03 | 1.70E-02 | −3.1 | microRNA 701 |
| 2.67E-03 | 1.01E-02 | −3.6 | microRNA 224 |
| 7.88E-05 | 1.25E-03 | −3.6 | microRNA 23b |
| 1.97E-04 | 2.01E-03 | −3.6 | microRNA 365-1 |
| 1.29E-03 | 6.26E-03 | −4.3 | microRNA 143 |
| 4.87E-04 | 3.37E-03 | −4.4 | microRNA 145 |
Data are excerpted from Supplemental Table S2.
Table 6.
Most altered lncRNAs following carotid ligation
| lncRNA Accession # | Fold Change | Nearest Coding Gene | Fold Change |
|---|---|---|---|
| F630028O10Rik | 5.56 | miR223 | ns |
| 6330407A03Rik | 4.66 | Lyn | 4.76 |
| C430002N11Rik | 4.63 | Zbtb38 | −1.52 |
| E230029C05Rik | 3.79 | Eed | ns |
| 2700099C18Rik | 2.61 | Mett14 | −1.29 |
| 1110038B12Rik | 2.24 | Hspa1b | ns |
| 1700048M11Rik | −2.01 | Smim11 | ns |
| C230004F18Rik | −2.09 | Cdr1 | −2.57 |
| A730098P11Rik | −2.11 | Lpp | −1.91 |
| 4930562F07Rik | −2.19 | Tnn | ns |
| E130006D01Rik | −2.19 | none close | |
| 5031426D15Rik | −2.21 | Celf2 | −1.34 |
| 0610043K17Rik | −2.22 | Rp9 | ns |
| 1700120C14Rik | −2.24 | Mcrs1 | ns |
| 1010001N08Rik | −2.35 | Gata6 | −2.01 |
| 9530026P05Rik | −2.38 | Adamts9 | ns |
| 2310015D24Rik | −2.38 | Parn | −1.40 |
| 5930403L14Rik | −2.46 | Prdm16 | −2.33 |
| 1190005I06Rik | −2.59 | Emc8, Gins2 | ns, 1.81 |
| 4833412C05Rik | −2.62 | Synm | −1.28 |
| 4930417O22Rik | −3.00 | Sost | −5.88 |
| E030003E18Rik | −3.07 | Itpkb | −1.27 |
| 9230112J17Rik | −3.08 | Thsd4 | −3.77 |
| 4930520O04Rik | −3.27 | Crtap | ns |
| 8430431K14Rik | −3.65 | Prkg1 | −2.86 |
| 2900055J20Rik | −4.10 | Kctd16 | −2.62 |
| E030013I19Rik | −5.68 | Itga8 | −2.84 |
| 4930429F24Rik | −6.01 | Tmem30a, Filip1 | ns, −3.37 |
RNAs identified from Supplemental Table S2 that are annotated as encoding long noncoding (lnc)RNAs on the UCSC Genome Browser and whose expression changed >2-fold are indicated. ns, not significant.
DISCUSSION
One of the most striking observations from our study is that the mouse carotid ligation model is associated with marked increases in local inflammatory markers at an early time point, 3 days following ligation (Table 1; Supplemental Tables S2, S3). This finding is consistent with previous studies, which also showed marked inflammation in a rat carotid artery injury model before leukocyte infiltration (15). It is likely that the elevated levels of inflammatory cytokines that are observed following injury arise both directly from injured VSMCs and endothelial cells as well as from adhered blood cells and platelets (15, 25, 35, 37, 44). VSMCs have been shown to be able to secrete Spp1 (Osteopontin), Saa3, CCL2 (MCP-1), CCL3 (Mip1α), TNF-α, IL-6, and IL-1β when challenged with proinflammatory stimuli such as cigarette smoke or cytokines released by activated blood cells (24, 32, 43–45, 53). VSMCs not only secrete these cytokines but can also respond to them in an autocrine fashion, leading to further increases in their secretion in a positive feedback loop (Fig. 3). Many of these cytokines are also well known to recruit monocytes, which is consistent with the observed elevated levels of TREM 1 and 2, which are IgG type receptors specifically expressed on neutrophils, monocytes, and macrophages (Fig. 1). This is further supported by immunohistological analysis using antibodies to CD68, Mac3, and F4/80, which revealed an accumulation of monocytes/macrophages in the lumen of injured vessels (Fig. 2). Activation of TREM receptors on monocytes can lead to further increases of MCP-1 (CCL2), IL-6, and TNF-α. These inflammatory cytokines, together with PDGF-B, which was elevated almost twofold following injury (Supplemental Table S2), are known to repress transcription of contractile proteins in VSMCs (42). These findings provide a possible mechanism that could explain the dramatically decreased expression of mRNAs encoding contractile proteins found 3 days following carotid ligation or in vitro following treatment with IL-1β (Figs. 3, 4, 7). As inflammation is well known to contribute to the development of atherosclerosis (5, 29), these data further suggest that inflammation may be a common trigger for changes in VSMCs that occur following acute vessel injury and in atherosclerosis, a prolonged disease process. Despite the significant repression of mRNAs encoding contractile proteins observed 3 days following ligation (Fig. 4), there was less of a decrease in expression of the proteins themselves (Fig. 5). This may reflect the relatively long half-life of many contractile proteins, as previous studies have shown that Myh11 protein (SM2) decreases in expression by 7 days following ligation (17, 19).
Fig. 7.

Model summarizing injury induced VSMC dedifferentiation. The schematic summarizes data from the current study together with previously published studies discussed in the text. In the control, uninjured vessel, Wnt, Hedgehog, Notch, and TGF-β signaling pathways promote the activity of transcription factors (Myocardin, Foxf1, Brm, Hey2, Gli, GATA6) required to drive the expression of smooth muscle contractile proteins (Myh11, Tagln, Cnn1, Acta2, Actg2, Mylk1, Prkg1) and promote differentiation of VSMCs. Our data suggest that these pathways may also regulate expression of microRNAs (miR 143/145, miR 29b-2) and lncRNAs (lnc101, lnc843) to further promote the differentiated state of VSMCs, while inhibiting factors that would promote dedifferentiation and proliferation (KLF4, Sp1). Following carotid ligation there is a marked increase in inflammatory cytokines such as IL-6 and IL-1β as well as osteopontin (SPP1) and Macrophage chemoattractant protein (MCP1/CCL2) that parallels a decrease in signaling molecules associated with Wnt, Hedgehog, Notch, and TGF-β signaling. We propose that the inhibition of expression of key transcription factors, such as myocardin and GATA6, by inflammatory cytokines acts together with decreased prodifferentiation signaling to coordinate the downregulation of smooth muscle contractile proteins leading to dedifferentiation of VSMCs following injury. Light gray symbols indicate downregulated pathways or molecules. Myh11, smooth muscle myosin heavy chain; Tagln, SM22α; Cnn1, calponin 1; Acta2, smooth muscle α-actin; Actg2, smooth muscle γ-actin; Mylk1, smooth muscle myosin light chain kinase; Prkg1, protein kinase G.
Another significant finding is the number of signaling pathways that have been previously shown to promote the differentiation state of VSMC such as Wnt, Hedgehog, Notch, and TGF-β were altered following carotid ligation injury (Table 4) (2, 22, 30, 47, 50). In particular, we observed a significant decrease in expression of many mRNAs encoding proteins associated with signaling from the TGF-β superfamily (Table 4). We observed decreased expression of mRNAs encoding several ligands, including BMP3, 4, 6 and TGF-β2, 3, and receptors Bmpr1 and Tgfbr1. These changes are also consistent with the observed decreased expression of mRNAs associated with differentiated VSMCs (Fig. 7). A downregulation of mRNAs encoding Notch 3, Hey 2, and Jagged 1, suggestive of altered notch signaling following carotid ligation, was also observed (Table 4). Notch signaling has been previously shown to be important for VSMC differentiation and for the proliferation and migration of neointimal VSMCs (4, 13, 28, 40). Studies have suggested that Notch 3 signaling is important for VSMC differentiation, whereas Notch 1 signaling promotes neointimal VSMC proliferation (13, 28). Consistent with this, 3 days following carotid ligation we observed decreased expression of mRNAs encoding Notch3, Jagged 1, and Hey 2 coincident with decreased differentiation of medial VSMCs and the decreased expression of the Notch target gene mylk1 (Fig. 4A) (4). At 3 days following injury there are no detectable neointimal VSMCs, providing an explanation for the lack of a significant increase in mRNA encoding Notch 1, which was previously observed in neointimal VSMCs 14 days following injury (28). Although there is no neointima present at 3 days postligation it has been reported that significant medial VSMC proliferation begins between 2 and 5 days postligation (27). Consistent with this, we observed a marked increase in expression of mRNAs encoding several proteins related to cell cycle progression, including cyclins A2, B1, B2, E1, E2, and F, as well as the cyclin-dependent kinase Cdk1 and myc. However, we did not detect any Ki67- or PCNA-positive cells in the medial layer of injured vessels on the sections we analyzed (Fig. 2). We did observe Ki67-positive cells in the adventitia and on the luminal side of the internal elastic lamina. The lack of detectable proliferation of medial VSMCs would be consistent with a recent study that showed that the neointima arises from the proliferation of only a very small number of medial VSMCs (8).
In addition to protein-coding genes, we also identified noncoding RNAs that are significantly altered in response to carotid ligation injury. It is now well established that noncoding RNAs such as microRNAs and lncRNAs also play important roles in the pathogenesis of cardiovascular disease (1, 38, 39, 48, 49). Data in this report show that microRNAs 143/5, which are critical regulators of the differentiated state of VSMCs, were the most highly downregulated (greater than fourfold) microRNAs observed 3 days following carotid ligation (Table 5) (7, 14, 56). This is consistent with previous studies that showed downregulation of these microRNAs in other models of vascular injury (14, 31). These microRNAs promote the differentiation state of VSMCs while inhibiting proliferation and migration (Fig. 7). This occurs through their ability to increase myocardin expression coupled with their repression of KLF4/5, versican, Sp1, Rtkn, Fscn1, and p70S6K (9, 12, 41, 51, 52, 57). Vascular injury can result in the downregulation of these microRNAs by multiple mechanisms, including decreased expression of myocardin leading to impaired transcription and impaired processing of the primary microRNA transcripts (31, 56). The decreased expression of microRNA-29b-2 we observed following vascular injury could also be contributing to the dedifferentiation of the medial VSMCs, as downregulation of this microRNA has been shown to promote VSMC proliferation and migration and decrease expression of smooth muscle contractile proteins (21). In addition to these characterized microRNAs, there are a number of others that are more than twofold downregulated, requiring further investigation to elucidate their potential roles in the response to ligation injury.
We also identified significant changes in expression of several lncRNAs that parallel changes in juxtaposed coding genes. LncRNAs are known to regulate gene expression through many different pathways; however, there is currently little information how these molecules may affect vascular disease (48). LncRNAs have been shown to inhibit VSMC migration, proliferation, and apoptosis (3, 55, 62, 63). In the current study, we identified several lncRNAs whose expression levels changed following injury in a pattern concomitant with the nearest protein-coding genes (Fig. 6, Table 6). Although lncRNAs can function by different mechanisms it is not uncommon for these RNAs to regulate the expression of nearby protein-coding genes (58). Interestingly, some of the protein-coding genes adjacent to the downregulated lncRNAs shown in Fig. 6 are known to play important roles in VSMCs. Protein kinase G (Prkg1) and GATA6 both promote VSMC differentiation and sustained expression of GATA6 has been shown to inhibit neointima formation (10, 33). The decreased expression of the lncRNAs within these loci may thus be contributing to the decreased expression of GATA6 and Prkg1 mRNAs, thereby promoting the dedifferentiation of VSMCs following injury (Fig. 7). In further support of this idea, both of these lncRNAs are also repressed following IL-1β treatment of VSMCs in vitro (Fig. 6B). Another downregulated lncRNA was identified adjacent to the Itga8 gene, which encodes integrin alpha 8, a novel integrin expressed at high levels in VSMCs (26). In addition to these downregulated lncRNAs, we also observed a large increase in expression of an lncRNA located in an intron of the Lyn gene (Table 6). As Lyn is abundantly expressed in myeloid cells, the increased expression of this lncRNA together with the almost fivefold increased expression of Lyn (Supplemental Table S2) likely reflects recruitment of monocytes/macrophages or other myeloid cells into the injured vessel. Future studies will be required to determine if the Lyn-associated lncRNA regulates Lyn expression in myeloid cells.
In summary, the mouse carotid ligation injury model induces a rapid local inflammatory response associated with dramatic increases in cytokines such as IL-1β, CCL2, and IL-6. This early inflammatory response correlates with a downregulation of mRNAs encoding proteins characteristic of differentiated VSMCs, including contractile proteins as well as the transcription factors and signaling pathways that regulate them (Fig. 7). We propose that the inhibitory action of inflammatory cytokines acts together with decreased activity of prodifferentiation signaling to coordinate changes in transcription of coding and noncoding genes to mediate the alterations in VSMCs that occur following vascular injury.
GRANTS
This research was funded by research support funds from Indiana University Purdue University Indianapolis to B. P. Herring and with support from the Indiana Clinical and Translational Sciences Institute funded, in part, by National Center for Advancing Translational Sciences Grant UL1TR-001108 Clinical and Translational Sciences Award to P. J. Gallagher. The microarray experiments were carried out using the facilities of the Center for Medical Genomics at Indiana University School of Medicine, which was funded in part by a grant from the Indiana 21st Century Research and Technology Fund, and by the Indiana Genomics Initiative (INGEN). (INGEN is supported in part by the Lilly Endowment.).
DISCLOSURES
The authors declare that they have no competing interests.
AUTHOR CONTRIBUTIONS
B.P.H., A.M.H., S.L.G., and J.N.M. performed experiments; B.P.H., A.M.H., and J.N.M. analyzed data; B.P.H., J.N.M., and P.J.G. interpreted results of experiments; B.P.H. prepared figures; B.P.H. drafted manuscript; B.P.H., A.M.H., J.N.M., and P.J.G. edited and revised manuscript; B.P.H., A.M.H., S.L.G., J.N.M., and P.J.G. approved final version of manuscript.
Supplementary Material
REFERENCES
- 1.Albinsson S, Suarez Y, Skoura A, Offermanns S, Miano JM, Sessa WC. MicroRNAs are necessary for vascular smooth muscle growth, differentiation, and function. Arterioscler Thromb Vasc Biol 30: 1118–1126, 2010. doi: 10.1161/ATVBAHA.109.200873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Azhar M, Schultz JJ, Grupp I, Dorn GW II, Meneton P, Molin DG, Gittenberger-de Groot AC, Doetschman T. Transforming growth factor beta in cardiovascular development and function. Cytokine Growth Factor Rev 14: 391–407, 2003. doi: 10.1016/S1359-6101(03)00044-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ballantyne MD, Pinel K, Dakin R, Vesey AT, Diver L, Mackenzie R, Garcia R, Welsh P, Sattar N, Hamilton G, Joshi N, Dweck MR, Miano JM, McBride MW, Newby DE, McDonald RA, Baker AH. Smooth muscle enriched long noncoding RNA (SMILR) regulates cell proliferation. Circulation 133: 2050–2065, 2016. doi: 10.1161/CIRCULATIONAHA.115.021019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Basu S, Srinivasan DK, Yang K, Raina H, Banerjee S, Zhang R, Fisher SA, Proweller A. Notch transcriptional control of vascular smooth muscle regulatory gene expression and function. J Biol Chem 288: 11191–11202, 2013. doi: 10.1074/jbc.M112.442996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bennett MR, Sinha S, Owens GK. Vascular smooth muscle cells in atherosclerosis. Circ Res 118: 692–702, 2016. doi: 10.1161/CIRCRESAHA.115.306361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blank RS, Owens GK. Platelet-derived growth factor regulates actin isoform expression and growth state in cultured rat aortic smooth muscle cells. J Cell Physiol 142: 635–642, 1990. doi: 10.1002/jcp.1041420325. [DOI] [PubMed] [Google Scholar]
- 7.Boettger T, Beetz N, Kostin S, Schneider J, Krüger M, Hein L, Braun T. Acquisition of the contractile phenotype by murine arterial smooth muscle cells depends on the Mir143/145 gene cluster. J Clin Invest 119: 2634–2647, 2009. doi: 10.1172/JCI38864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chappell J, Harman JL, Narasimhan VM, Yu H, Foote K, Simons BD, Bennett MR, Jørgensen HF. Extensive proliferation of a subset of differentiated, yet plastic, medial vascular smooth muscle cells contributes to neointimal formation in mouse injury and atherosclerosis models. Circ Res 119: 1313–1323, 2016. doi: 10.1161/CIRCRESAHA.116.309799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chiyomaru T, Enokida H, Tatarano S, Kawahara K, Uchida Y, Nishiyama K, Fujimura L, Kikkawa N, Seki N, Nakagawa M. miR-145 and miR-133a function as tumour suppressors and directly regulate FSCN1 expression in bladder cancer. Br J Cancer 102: 883–891, 2010. doi: 10.1038/sj.bjc.6605570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choi C, Sellak H, Brown FM, Lincoln TM. cGMP-dependent protein kinase and the regulation of vascular smooth muscle cell gene expression: possible involvement of Elk-1 sumoylation. Am J Physiol Heart Circ Physiol 299: H1660–H1670, 2010. doi: 10.1152/ajpheart.00677.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dandré F, Owens GK. Platelet-derived growth factor-BB and Ets-1 transcription factor negatively regulate transcription of multiple smooth muscle cell differentiation marker genes. Am J Physiol Heart Circ Physiol 286: H2042–H2051, 2004. doi: 10.1152/ajpheart.00625.2003. [DOI] [PubMed] [Google Scholar]
- 12.Davis-Dusenbery BN, Chan MC, Reno KE, Weisman AS, Layne MD, Lagna G, Hata A. down-regulation of Kruppel-like factor-4 (KLF4) by microRNA-143/145 is critical for modulation of vascular smooth muscle cell phenotype by transforming growth factor-beta and bone morphogenetic protein 4. J Biol Chem 286: 28097–28110, 2011. doi: 10.1074/jbc.M111.236950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Domenga V, Fardoux P, Lacombe P, Monet M, Maciazek J, Krebs LT, Klonjkowski B, Berrou E, Mericskay M, Li Z, Tournier-Lasserve E, Gridley T, Joutel A. Notch3 is required for arterial identity and maturation of vascular smooth muscle cells. Genes Dev 18: 2730–2735, 2004. doi: 10.1101/gad.308904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elia L, Quintavalle M, Zhang J, Contu R, Cossu L, Latronico MV, Peterson KL, Indolfi C, Catalucci D, Chen J, Courtneidge SA, Condorelli G. The knockout of miR-143 and -145 alters smooth muscle cell maintenance and vascular homeostasis in mice: correlates with human disease. Cell Death Differ 16: 1590–1598, 2009. doi: 10.1038/cdd.2009.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fedorov A, Kostareva A, Raud J, Roy J, Hedin U, Razuvaev A. Early changes of gene expression profiles in the rat model of arterial injury. J Vasc Interv Radiol 25: 789– 796, 2014. doi: 10.1016/j.jvir.2013.11.031. [DOI] [PubMed] [Google Scholar]
- 16.Feil S, Fehrenbacher B, Lukowski R, Essmann F, Schulze-Osthoff K, Schaller M, Feil R. Transdifferentiation of vascular smooth muscle cells to macrophage-like cells during atherogenesis. Circ Res 115: 662–667, 2014. doi: 10.1161/CIRCRESAHA.115.304634. [DOI] [PubMed] [Google Scholar]
- 17.Gallagher PJ, Jin Y, Killough G, Blue EK, Lindner V. Alterations in expression of myosin and myosin light chain kinases in response to vascular injury. Am J Physiol Cell Physiol 279: C1078–C1087, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heldin CH, Westermark B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol Rev 79: 1283–1316, 1999. [DOI] [PubMed] [Google Scholar]
- 19.Herring BP, Hoggatt AM, Burlak C, Offermanns S. Previously differentiated medial vascular smooth muscle cells contribute to neointima formation following vascular injury. Vasc Cell 6: 21, 2014. doi: 10.1186/2045-824X-6-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoggatt AM, Kim JR, Ustiyan V, Ren X, Kalin TV, Kalinichenko VV, Herring BP. The transcription factor Foxf1 binds to serum response factor and myocardin to regulate gene transcription in visceral smooth muscle cells. J Biol Chem 288: 28477–28487, 2013. doi: 10.1074/jbc.M113.478974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iaconetti C, De Rosa S, Polimeni A, Sorrentino S, Gareri C, Carino A, Sabatino J, Colangelo M, Curcio A, Indolfi C. Down-regulation of miR-23b induces phenotypic switching of vascular smooth muscle cells in vitro and in vivo. Cardiovasc Res 107: 522–533, 2015. doi: 10.1093/cvr/cvv141. [DOI] [PubMed] [Google Scholar]
- 22.Inamoto S, Kwartler CS, Lafont AL, Liang YY, Fadulu VT, Duraisamy S, Willing M, Estrera A, Safi H, Hannibal MC, Carey J, Wiktorowicz J, Tan FK, Feng XH, Pannu H, Milewicz DM. TGFBR2 mutations alter smooth muscle cell phenotype and predispose to thoracic aortic aneurysms and dissections. Cardiovasc Res 88: 520–529, 2010. doi: 10.1093/cvr/cvq230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res 31: e15, 2003. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jalvy S, Renault MA, Leen LL, Belloc I, Bonnet J, Gadeau AP, Desgranges C. Autocrine expression of osteopontin contributes to PDGF-mediated arterial smooth muscle cell migration. Cardiovasc Res 75: 738–747, 2007. doi: 10.1016/j.cardiores.2007.05.019. [DOI] [PubMed] [Google Scholar]
- 25.Kawasaki T, Dewerchin M, Lijnen HR, Vreys I, Vermylen J, Hoylaerts MF. Mouse carotid artery ligation induces platelet-leukocyte-dependent luminal fibrin, required for neointima development. Circ Res 88: 159–166, 2001. doi: 10.1161/01.RES.88.2.159. [DOI] [PubMed] [Google Scholar]
- 26.Kitchen CM, Cowan SL, Long X, Miano JM. Expression and promoter analysis of a highly restricted integrin alpha gene in vascular smooth muscle. Gene 513: 82–89, 2013. doi: 10.1016/j.gene.2012.10.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kumar A, Lindner V. Remodeling with neointima formation in the mouse carotid artery after cessation of blood flow. Arterioscler Thromb Vasc Biol 17: 2238–2244, 1997. doi: 10.1161/01.ATV.17.10.2238. [DOI] [PubMed] [Google Scholar]
- 28.Li Y, Takeshita K, Liu PY, Satoh M, Oyama N, Mukai Y, Chin MT, Krebs L, Kotlikoff MI, Radtke F, Gridley T, Liao JK. Smooth muscle Notch1 mediates neointimal formation after vascular injury. Circulation 119: 2686–2692, 2009. doi: 10.1161/CIRCULATIONAHA.108.790485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Libby P. Inflammation in atherosclerosis. Arterioscler Thromb Vasc Biol 32: 2045–2051, 2012. doi: 10.1161/ATVBAHA.108.179705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin DW, Chang IC, Tseng A, Wu ML, Chen CH, Patenaude CA, Layne MD, Yet SF. Transforming growth factor beta up-regulates cysteine-rich protein 2 in vascular smooth muscle cells via activating transcription factor 2. J Biol Chem 283: 15003–15014, 2008. doi: 10.1074/jbc.M801621200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu X, Cheng Y, Yang J, Qin S, Chen X, Tang X, Zhou X, Krall TJ, Zhang C. Flank sequences of miR-145/143 and their aberrant expression in vascular disease: mechanism and therapeutic application. J Am Heart Assoc 2: e000407, 2013. doi: 10.1161/JAHA.113.000407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Loppnow H, Libby P. Proliferating or interleukin 1-activated human vascular smooth muscle cells secrete copious interleukin 6. J Clin Invest 85: 731–738, 1990. doi: 10.1172/JCI114498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mano T, Luo Z, Malendowicz SL, Evans T, Walsh K. Reversal of GATA-6 downregulation promotes smooth muscle differentiation and inhibits intimal hyperplasia in balloon-injured rat carotid artery. Circ Res 84: 647–654, 1999. doi: 10.1161/01.RES.84.6.647. [DOI] [PubMed] [Google Scholar]
- 34.McClintick JN, Edenberg HJ. Effects of filtering by Present call on analysis of microarray experiments. BMC Bioinformatics 7: 49, 2006. doi: 10.1186/1471-2105-7-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McPherson JA, Barringhaus KG, Bishop GG, Sanders JM, Rieger JM, Hesselbacher SE, Gimple LW, Powers ER, Macdonald T, Sullivan G, Linden J, Sarembock IJ. Adenosine A(2A) receptor stimulation reduces inflammation and neointimal growth in a murine carotid ligation model. Arterioscler Thromb Vasc Biol 21: 791–796, 2001. doi: 10.1161/01.ATV.21.5.791. [DOI] [PubMed] [Google Scholar]
- 36.Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double-fluorescent Cre reporter mouse. Genesis 45: 593–605, 2007. doi: 10.1002/dvg.20335. [DOI] [PubMed] [Google Scholar]
- 37.Myers DL, Harmon KJ, Lindner V, Liaw L. Alterations of arterial physiology in osteopontin-null mice. Arterioscler Thromb Vasc Biol 23: 1021–1028, 2003. doi: 10.1161/01.ATV.0000073312.34450.16. [DOI] [PubMed] [Google Scholar]
- 38.Ohtani K, Dimmeler S. Control of cardiovascular differentiation by microRNAs. Basic Res Cardiol 106: 5–11, 2011. doi: 10.1007/s00395-010-0139-7. [DOI] [PubMed] [Google Scholar]
- 39.Port JD, Sucharov C. Role of microRNAs in cardiovascular disease: therapeutic challenges and potentials. J Cardiovasc Pharmacol 56: 444–453, 2010. doi: 10.1097/FJC.0b013e3181f605b6. [DOI] [PubMed] [Google Scholar]
- 40.Proweller A, Pear WS, Parmacek MS. Notch signaling represses myocardin-induced smooth muscle cell differentiation. J Biol Chem 280: 8994–9004, 2005. doi: 10.1074/jbc.M413316200. [DOI] [PubMed] [Google Scholar]
- 41.Qiu T, Zhou X, Wang J, Du Y, Xu J, Huang Z, Zhu W, Shu Y, Liu P. MiR-145, miR-133a and miR-133b inhibit proliferation, migration, invasion and cell cycle progression via targeting transcription factor Sp1 in gastric cancer. FEBS Lett 588: 1168–1177, 2014. doi: 10.1016/j.febslet.2014.02.054. [DOI] [PubMed] [Google Scholar]
- 42.Raines EW. PDGF and cardiovascular disease. Cytokine Growth Factor Rev 15: 237–254, 2004. doi: 10.1016/j.cytogfr.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 43.Renault MA, Jalvy S, Potier M, Belloc I, Genot E, Dekker LV, Desgranges C, Gadeau AP. UTP induces osteopontin expression through a coordinate action of NFkappaB, activator protein-1, and upstream stimulatory factor in arterial smooth muscle cells. J Biol Chem 280: 2708–2713, 2005. doi: 10.1074/jbc.M411786200. [DOI] [PubMed] [Google Scholar]
- 44.Shen J, Yang M, Ju D, Jiang H, Zheng JP, Xu Z, Li L. Disruption of SM22 promotes inflammation after artery injury via nuclear factor kappaB activation. Circ Res 106: 1351–1362, 2010. doi: 10.1161/CIRCRESAHA.109.213900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Starke RM, Ali MS, Jabbour PM, Tjoumakaris SI, Gonzalez F, Hasan DM, Rosenwasser RH, Owens GK, Koch WJ, Dumont AS. Cigarette smoke modulates vascular smooth muscle phenotype: implications for carotid and cerebrovascular disease. PLoS One 8: e71954, 2013. doi: 10.1371/journal.pone.0071954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci USA 100: 9440–9445, 2003. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tagliafico E, Brunelli S, Bergamaschi A, De Angelis L, Scardigli R, Galli D, Battini R, Bianco P, Ferrari S, Cossu G, Ferrari S. TGFbeta/BMP activate the smooth muscle/bone differentiation programs in mesoangioblasts. J Cell Sci 117: 4377–4388, 2004. doi: 10.1242/jcs.01291. [DOI] [PubMed] [Google Scholar]
- 48.Uchida S, Dimmeler S. Long noncoding RNAs in cardiovascular diseases. Circ Res 116: 737–750, 2015. doi: 10.1161/CIRCRESAHA.116.302521. [DOI] [PubMed] [Google Scholar]
- 49.van Rooij E, Olson EN. MicroRNA therapeutics for cardiovascular disease: opportunities and obstacles. Nat Rev Drug Discov 11: 860–872, 2012. doi: 10.1038/nrd3864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang D, Prakash J, Nguyen P, Davis-Dusenbery BN, Hill NS, Layne MD, Hata A, Lagna G. Bone morphogenetic protein signaling in vascular disease: anti-inflammatory action through myocardin-related transcription factor A. J Biol Chem 287: 28067–28077, 2012. doi: 10.1074/jbc.M112.379487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang S, Bian C, Yang Z, Bo Y, Li J, Zeng L, Zhou H, Zhao RC. miR-145 inhibits breast cancer cell growth through RTKN. Int J Oncol 34: 1461–1466, 2009. [PubMed] [Google Scholar]
- 52.Wang X, Hu G, Zhou J. Repression of versican expression by microRNA-143. J Biol Chem 285: 23241–23250, 2010. doi: 10.1074/jbc.M109.084673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wilson TC, Bachurski CJ, Ikegami M, Jobe AH, Kallapur SG. Pulmonary and systemic induction of SAA3 after ventilation and endotoxin in preterm lambs. Pediatr Res 58: 1204–1209, 2005. doi: 10.1203/01.pdr.0000185269.93228.29. [DOI] [PubMed] [Google Scholar]
- 54.Wirth A, Benyó Z, Lukasova M, Leutgeb B, Wettschureck N, Gorbey S, Orsy P, Horváth B, Maser-Gluth C, Greiner E, Lemmer B, Schütz G, Gutkind JS, Offermanns S. G12-G13-LARG-mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat Med 14: 64–68, 2008. doi: 10.1038/nm1666. [DOI] [PubMed] [Google Scholar]
- 55.Wu G, Cai J, Han Y, Chen J, Huang ZP, Chen C, Cai Y, Huang H, Yang Y, Liu Y, Xu Z, He D, Zhang X, Hu X, Pinello L, Zhong D, He F, Yuan GC, Wang DZ, Zeng C. LincRNA-p21 regulates neointima formation, vascular smooth muscle cell proliferation, apoptosis, and atherosclerosis by enhancing p53 activity. Circulation 130: 1452–1465, 2014. doi: 10.1161/CIRCULATIONAHA.114.011675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xin M, Small EM, Sutherland LB, Qi X, McAnally J, Plato CF, Richardson JA, Bassel-Duby R, Olson EN. MicroRNAs miR-143 and miR-145 modulate cytoskeletal dynamics and responsiveness of smooth muscle cells to injury. Genes Dev 23: 2166–2178, 2009. doi: 10.1101/gad.1842409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xu Q, Liu LZ, Qian X, Chen Q, Jiang Y, Li D, Lai L, Jiang BH. MiR-145 directly targets p70S6K1 in cancer cells to inhibit tumor growth and angiogenesis. Nucleic Acids Res 40: 761–774, 2012. doi: 10.1093/nar/gkr730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang L, Froberg JE, Lee JT. Long noncoding RNAs: fresh perspectives into the RNA world. Trends Biochem Sci 39: 35–43, 2014. doi: 10.1016/j.tibs.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yoon JH, Abdelmohsen K, Gorospe M. Posttranscriptional gene regulation by long noncoding RNA. J Mol Biol 425: 3723–3730, 2013. doi: 10.1016/j.jmb.2012.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yoshida T, Gan Q, Shang Y, Owens GK. Platelet-derived growth factor-BB represses smooth muscle cell marker genes via changes in binding of MKL factors and histone deacetylases to their promoters. Am J Physiol Cell Physiol 292: C886–C895, 2007. doi: 10.1152/ajpcell.00449.2006. [DOI] [PubMed] [Google Scholar]
- 61.Yoshida T, Yamashita M, Horimai C, Hayashi M. Smooth muscle-selective inhibition of nuclear factor-κB attenuates smooth muscle phenotypic switching and neointima formation following vascular injury. J Am Heart Assoc 2: e000230, 2013. doi: 10.1161/JAHA.113.000230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhao J, Zhang W, Lin M, Wu W, Jiang P, Tou E, Xue M, Richards A, Jourd’heuil D, Asif A, Zheng D, Singer HA, Miano JM, Long X. MYOSLID is a novel serum response factor-dependent long noncoding RNA that amplifies the vascular smooth muscle differentiation program. Arterioscler Thromb Vasc Biol 36: 2088–2099, 2016. doi: 10.1161/ATVBAHA.116.307879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhao Y, Feng G, Wang Y, Yue Y, Zhao W. Regulation of apoptosis by long non-coding RNA HIF1A-AS1 in VSMCs: implications for TAA pathogenesis. Int J Clin Exp Pathol 7: 7643–7652, 2014. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.