

Abstract
Background
The mass spectrometry based technical pipeline has provided a high-throughput, high-sensitivity and high-resolution platform for post-genomic biology. Varied models and algorithms are implemented by different tools to improve proteomics data analysis. The target-decoy searching strategy has become the most popular strategy to control false identification in peptide and protein identifications. While this strategy can estimate the false discovery rate (FDR) within a dataset, it cannot directly evaluate the false positive matches in target identifications.
Results
As a supplement to target-decoy strategy, the entrapment sequence method was introduced to assess the key steps of mass spectrometry data analysis process, database search engines and quality control methods. Using the entrapment sequences as the standard, we evaluated five database search engines for both the origanal scores and reprocessed scores, as well as four quality control methods in term of quantity and quality aspects. Our results showed that the latest developed search engine MS-GF+ and percolator-embeded quality control method PepDistiller performed best in all tools respectively. Combined with efficient quality control methods, the search engines can improve the low sensitivity of their original scores. Moreover, based on the entrapment sequence method, we proved that filtering the identifications separately could increase the number of identified peptides while improving the confidence level.
Conclusion
In this study, we have proved that the entrapment sequence method could be an useful strategy to assess the key steps of the mass spectrometry data analysis process. Its applications can be extended to all steps of the common workflow, such as the protein assembling methods and data integration methods.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-017-3491-2) contains supplementary material, which is available to authorized users.
Keywords: Proteomics, Tandem mass spectrometry, Entrapment sequence method, Target-decoy search, Quality control
Background
The development of mass spectrometry has provided a high-throughput, high-sensitivity and high-resolution analysis platform for proteomics. Tandem mass spectrometry has become one of the most powerful technologies for protein identification, making possible the global protein profiling. Meanwhile, using the database searching strategy allows high-throughput identification of peptides and proteins in shotgun proteomics. Varied models and algorithms are implemented by different search engines, including the early produced engines SEQUEST [1], Mascot [2] and X!Tandem [3] as well as some newly developed engines, such as Comet [4], Tide [5], MS-GF+[6] and MS Amanda [7]. Then such quality control methods have been applied to achieve high reliability identifications as PeptideProphet [8–10], PepDistiller [11], Mfs [12], RockerBox [13], FDRAnalysis [14] and BuildSummary [15].
The target-decoy database search strategy is the most commonly used strategy to estimate false identifications in target database with the assumption that the number of false identifications in target database is equal to that in decoy database [16]. However, this strategy can estimate the false discovery rate (FDR) within a dataset rather than directly evaluate the false positive matches in target identifications.
In our previous work, we used the protein sequences from Archaea species as appended database for standard dataset analysis to avoid the ambiguous matches caused by the sequence similarity between control protein sequences and searched database sequences [17, 18]. Similar work had been published in Granholm et al.’s [19] and Vaudel et al.’s paper [20]. Granholm et al. suggested a semi-labeled method for evaluating the calibration of a given score function using dataset of known protein sample by searching the database composed of a small number of sample sequences and a large number of entrapment sequences. Vaudel et al. proposed constructing a database that contained both the sample sequences (true positive) and entrapment sequences (false positive) and proved that the Pyrococcu furiosus proteome can provide a method for detecting random hits (comparable to the decoy database).
All the above-mentioned work reminds us to introduce the entrapment sequence to target-decoy search strategy as a good supplement. By using different labels, we can separate the PSMs into different kinds and calculate the false matches in target identifications directly. Using the entrapment sequence as the objective standard (pure false positive), we assessed five database search engines and four quality control methods in terms of both quantity and quality. On the basis of the results of two datasets, the entrapment sequence method is proved to be a useful strategy to assess the mass spectrometry data analysis workflow.
Methods
Datasets
Two previously published datasets were used in this study. The Pfu dataset was produced by analyzing Pyrococcus furiosus sample on LTQ Orbitrap Velos (Thermo Scientific) [20], and used as a standard dataset here. The LM3 dataset was generated from a shotgun analysis of the metastatic human hepatocellular carcinoma cell line (HCCLM3) using Q-Exactive (Thermo Scientific) [21].
Protein Sequence Database
Three protein sequences were downloaded from UniProt database [22]: (1) Pyrococcus furiosus protein sequences (Pfu2045, containing 2,045 sequences, downloaded on January 5, 2016). (2) Homo sapiens protein sequences (Homo20187, containing 20,187 sequences, downloaded on January 5, 2016). (3) Archaea protein sequences (Arc20825, containing 20,825 sequences, downloaded on September 21, 2016). We randomized the Archaea protein sequences ten times to get (4) The large entrapment sequences for LM3 dataset (Arc208250, containing 208,250 sequences). The composition of the three target databases is shown in Table 1. For Pfu dataset, the Pfu2045 was used as sample sequences and the Homo20187 was used as entrapment sequences. For LM3 dataset, the Homo20187 was used as sample sequences, while the Arc208250 and the Arc20825 were used as two different entrapment sequences. Then all target sequences were reversed to create the decoy database for target-decoy search strategy. The general view of the construction of searched databases is shown in Fig. 1.
Table 1.
Construction of the target database for Pfu and LM3 datasets
| DataSets | Sample sequences | Entrapment sequences | Sample tryptic peptides | Entrapment tryptic peptides | Shared tryptic peptide | Shared/Sample tryptic peptides (%) |
|---|---|---|---|---|---|---|
| Pfu | Pfu2045 | Homo20187 | 145358 | 2338004 | 102 | 0.070 |
| LM3 | Homo20187 | Arc208250 | 2338004 | 15344503 | 4864 | 0.208 |
| LM3 | Homo20187 | Arc20825 | 2338004 | 1479773 | 1333 | 0.057 |
Fig. 1.
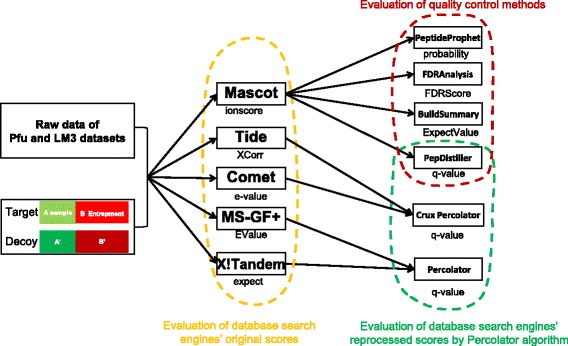
Workflow for evaluation of database search engines and quality control methods using the entrapment sequence method. A total of five search engines (Mascot, X!Tandem, Comet, MS-GF+ and Tide) and four quality control methods (PepDistiller, BuildSummary, PeptideProphet and FDRAnalysis) were studied on the basis of a standard Pfu dataset and a complex LM3 dataset
Both Granholm et al.’s [19] and Vaudel et al.’s [20] work suggested sufficiently that large entrapment sequences should be used, and that the probability that a random match hits the sample database is negligible, but the best size hasn’t been examined. Here, about ten times as many entrapment sequences were used as sample sequences, which is a similar ratio to Vaudel et al.’s work. Also, we compared the tryptic peptides of all sample sequences and entrapment sequences. As shown in Table 1, the ratios of shared peptides are respectively low for three constructed databases (0.07%, 0.21% and 0.06%). Thus, very few positive PSMs should hit the entrapment sequences. A spectrum that matches both sample and entrapment sequences is considered a sample identification.
Database Searching
All mzML and MGF files were converted from raw files using the msconvert module [23] in the Trans-Proteomic Pipeline (TPP v4.7.0) [24]. The MS/MS peak list files were searched against the combined database using Mascot [2] (local server v2.3.2), Comet [4] (in Curx v2.1.16833 [25, 26]), Tide [5] (in Curx v2.1.16833), MS-GF+[6] (v10089) and X!Tandem [3] (TPP v4.7.0) [24]. The monoisotopic mass was used for both peptide and fragment ions with fixed modification (Carbamidomethyl, +57 Da) on Cys and variable modification (Oxidation, +16 Da) on Met. Tryptic cleavage at only Lys or Arg was selected. The miss cleavage number was set to be 1.
Quality control and protein assembling
Four commonly used quality control methods were used in this study, including BuildSummary [15], PeptideProphet [8–10], FDRAnalysis [14] and PepDistiller [11], all of which produced a rescore of Mascot results for each PSM: BuildSummary’s ExpectValue, PeptideProphet’s probability, FDRAnalysis’s FDRScore and PepDistiller’s q-value. Comet and Tide results were processed by Percolator integrated in Crux, which gave a rescore of q-value. MS-GF+ and X!Tandem results were processed by percolator-converters (v3-00) followed by percolator (v2-08) for further quality control. The percolator tools can be downloaded from (https://github.com/percolator/percolator) [27]. In this study, we used MAYU for protein assembling [28]. Peptides less than 7 amino acids were not taken into account.
False Discovery Rate and False Match Rate
There are two formulas commonly used for false discovery rate estimation in target-decoy search strategy. One is for seperated database search (formula (1)), and the other is for concatenated database search (formula (2)). N target and N decoy are the number of target and decoy matches, respectively.
| 1 |
| 2 |
As we introduced the entrapment sequences in the target database, the entrapment hits in filtered target identifications can be considered as false positive results. Thus, we defined a false match rate (FMR) to approximately estimate the false positive identifications under given FDR. The FMR can be calculated by formula (3), where Ntrap is the number of identifications matched the entrapment sequences in target hits.
| 3 |
Results and discussion
With the advance of proteome research, a growing number of database search engines as well as the subsequent quality control methods have emerged and played the key roles in the whole process of MS/MS data analysis. As shown in Fig. 1, using the entrapment sequences as a standard, we performed the evaluation of five database search engines’ original scores and reprocessed scores and four quality control methods in the two important aspects, quantity and quality.
Evaluation of different database search engines based on both the original scores and reprocessed scores
First, we used the Pfu dataset as a standard dataset to compare five search engines based on their original scores, Mascot’s ionscore, X!Tandem’s expect, Comet’s e-value, MS-GF + 's EValue and Tide’s XCorr. As shown in Additional file 1 Figure S1A-C, the MS-GF+ far outperforms the other search engines, and the use of the MS-GF + 's EValue allows significantly more identifications at all PSM, peptide and protein levels with the pre-defined FDR. The same trend has also been observed in the large LM3 dataset (Additional file 1: Figure S1D-F).
The original scores of search engines are usually of very low sensitivity. However, this flaw can be overcome when combined with the followed quality control methods by considering the distributions of target and decoy hits and reanalyzing the original scores. The SVM-based percolator algorithm has been proved to be an ideal QC method [11, 29]. Thus, we further analyzed Mascot’s results by PepDistiller [11] (a bulit-in Percolator classifier), X!Tandem’s results and MS-GF+’s results by Percolator [27, 30], Tide’s results and Comet’s results by Percolator intergrated in Crux [25, 26]. All the above combinations can produce a q-value for each identification and be used for FDR calculation. As shown in Fig. 2, although the MS-GF+ combined percolator still performs best, other search engines can also perform quite well, especially in the large dataset (LM3 dataset) and at the stringent quality control level (protein level).
Fig. 2.
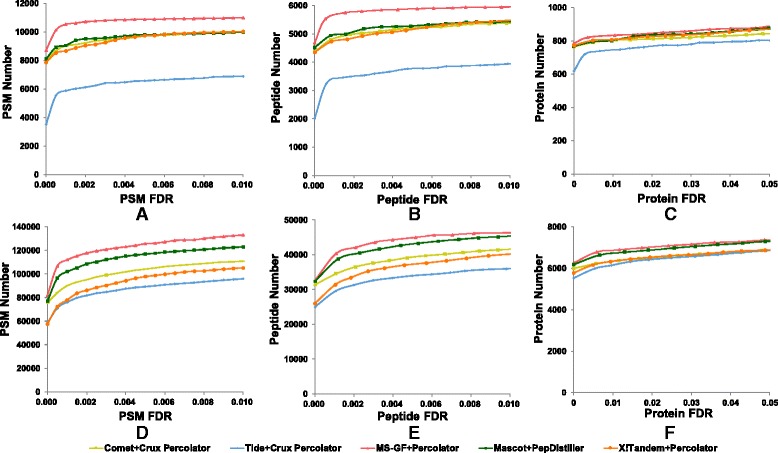
Plot figures of the numbers of PSM, peptide and protein identified by five search engines under the estimated FDRs on Pfu dataset (a-c) and LM3 dataset (d-f). The reprocessed scores of all five search engines are used
In previous studies, the performance of different search engines and quality control methods were assessed by the number of results identified with fixed FDR (e.g. 1% or 5%) estimated by target-decoy hits. The most productive tool or method is usually consider the best one, which is because only quantity is used as the criterion. Here, we introduced the entrapment sequence method as a complement to the target-decoy search strategy. Thus, we can use the entrapment hits to calculate the false march rate (FMR) to assess the quality of the results. As shown in Fig. 3 (also refer to Additional file 1: Figure S2), obviously, the FDRs determined by decoy hits remain stable (0.01 FDR of PSM, peptede and protein level respectively), while the FMRs vary with search engines and confident levels.
Fig. 3.
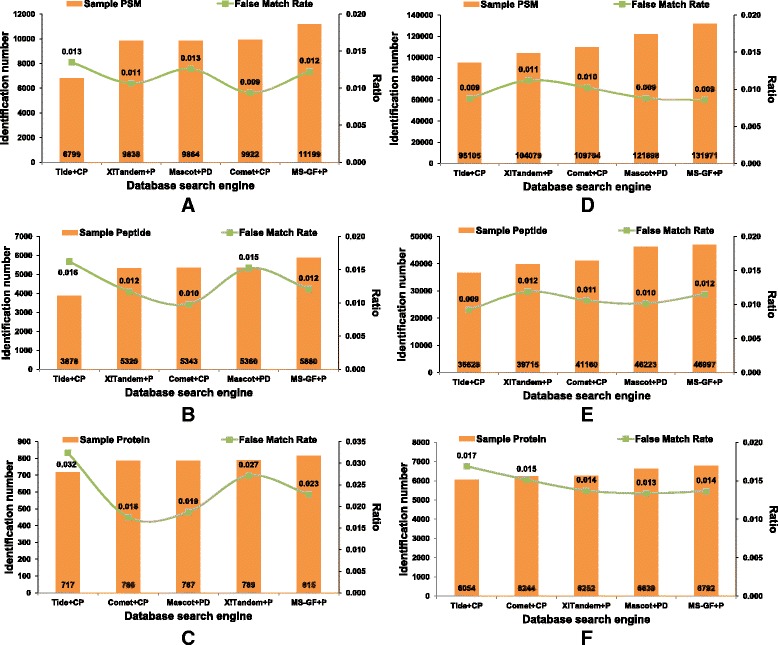
Distribution figures of the identification numbers and FMRs under 0.01 FDR of spectrum, peptide and protein level for five search engines on Pfu dataset (a-c) and LM3 dataset (d-f). The reprocessed scores of all five search engines are used
In gerenal, fewer entrapment hits occur in PSM and peptide identificaitons and in large dataset (LM3 dataset) than those in protein identificaitons and in small dataset (Pfu dataset). In most cases, the FMRs estimated by entrapment hits are roughly equal to those of FDRs estimated by decoy hits. But in some cases, the false matches represented by entrapment hits would far outnumber the expected ones, such as the Tide (FMR = 3.2%) and X!Tandem (FMR = 2.7%) searched results in Pfu dataset in 0.01 protein FDR condition (Fig. 3c), which would remind the researcher that more strict QC should be applied. Thus, we concluded that the entrapment sequence can be used as an internal scale for reseachers to monitor their peptide or protein identifications at any time.
Evaluation of four quality control methods
As Mascot is one of the most widely used search engines, it has been improved to accommodate the MS/MS data generated by different instruments with different accuracy, and most quality control methods can handle its output result files. Here, Mascot’s searched files were used as inputs and reprocessed by four QC methods, including PepDistiller, BuildSummary, PeptideProphet and FDRAnalysis. As shown in Fig. 4, the percolator based QC method PepDistiller identified the most PSMs, peptides and proteins in both Pfu and LM3 datasets, and other three methods were not significantly different. The trends of FMRs of filtered results by differet QC methods are close to the predefined FDR than those of search engines (Fig. 5). Using MAYU as the protein assembling tool can help four QC methods to keep confident at the peptide and protein level, especially in the large LM3 dataset.
Fig. 4.
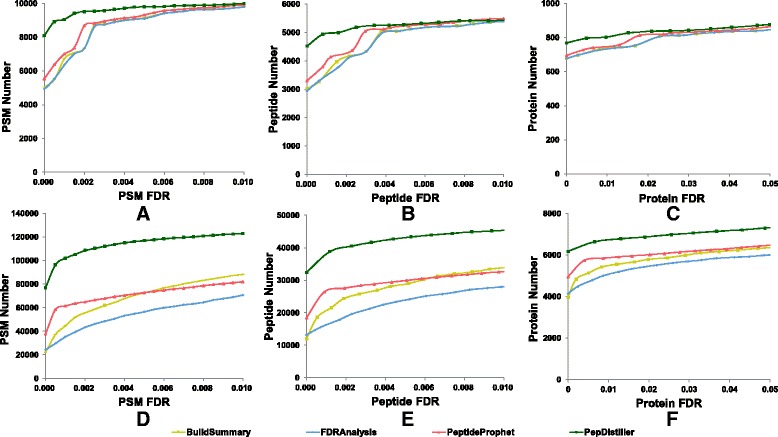
Plot figures of the numbers of PSM, peptide and protein filtered by four quality control methods under the estimated FDRs on Pfu dataset (a-c) and LM3 dataset (d-f)
Fig. 5.
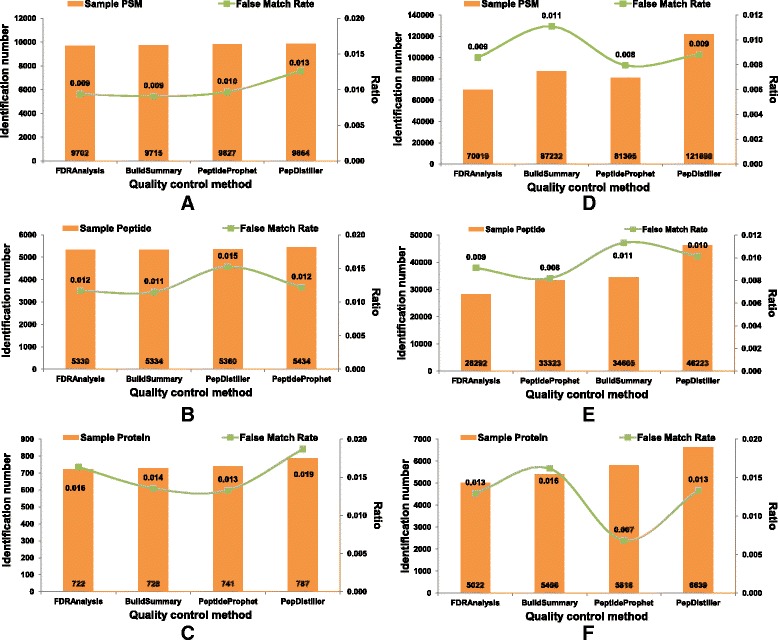
Distribution figures of the identification numbers and FMRs under 0.01 FDR of spectrum, peptide and protein level for four quality control methods on Pfu dataset (a-c) and LM3 dataset (d-f)
Combining identificaitons of different search engines and quality control methods with an appropriate framework
Varied models and algorithms that are implemented by different search engines and quality control methods, which make themselves mutually complementary and well-performing for different subsets of mass spectrometry data. Each search engine and QC method can uniquely identify some spectra (Additional file 1: Figure S3). Indeed, combining the results of multiple database search engines or QC methods can increase identifications, however, more false positive hits will be produced by uniquely identified results.
Take LM3 dataset as an example. Under 1% PSM FDR, the distributions of PSMs identified by one or several search engines or QC tools are shown in Fig. 6 and Additional file 1: Figure S4. Obviously, the FDRs and FMRs of peptides identified by one or two tools are much higher than those identified by three or more tools (Fig. 6a and Additional file 1: Figure S4A). If all these PSMs of five search engine are directly put together, there are 167,259 PSMs in total, resulting in 25.88% ~ 74.61% more hits than any single engine, but the FDR increases to 2.66% with the FMR of 2.61% too. Here we proposed an alternative intergrated method in which further filterings were applied to the identificaitons according to their overlap conditions. We seperated the PSMs into subgroups by the number of identified tools, and then filtered each subgroup hits to keep their sub-FDR lower than the pre-defined one (Fig. 6b and Additional file 1: Figure S4B). There are total 137,342 PSMs identified by this intergrated method, resulting in 3.37% ~ 43.38% more hits than any single engine, but the FDR decrease to 0.40% with the FMR of 0.48%.
Fig. 6.
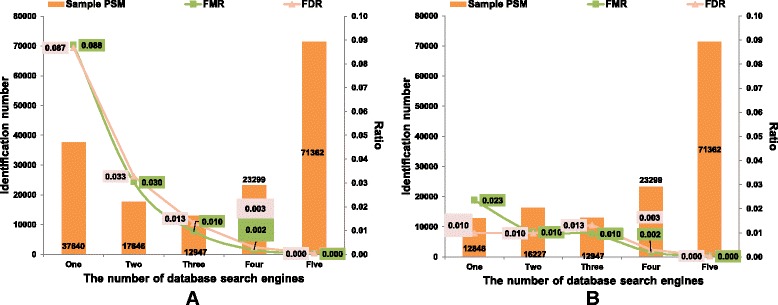
Distribution figures of the PSMs, FDRs and FMRs identified by different number of search engines on LM3 dataset. a Distributions of original FDRs and FMRs under 0.01 spectrum FDR. b Distributions of refined FDRs and FMRs under 0.01 spectrum FDR, as the PSMs in each subgroup are further filtered to keep their sub-FDRs lower than the pre-defined one
Thus, combining the results of multiple database search engines and QC methods with an appropriate framework would benefit the data analysis process, increase the numbers of identified peptides and improve the confidence level of identifications.
Using a small size of entrapment sequences to evaluate the search engines and tools in large dataset
As mentioned in Granholm et al.’s [19] and Vaudel et al.’s [20] papers, to efficiently separate correct PSMs from incorrect ones, the size of the entrapment sequences is supposed to be many times larger than the size of the sample sequences. However, the oversize database would greatly increase the search time while decreasing the total positive identifications. Thus, an appropriate size database is preferable in practical use. Here, we used the original Archaea protein sequences (Arc20825) as a small size entrapment sequence and reprocessed the LM3 dataset. Then the similar results are gained as with large size entrapment sequence search (details are shown in Additional file 1: Figure S5 and S6). Thus, an easy way to use the entrapment sequence method is to randomize the sample sequences, label them and combine them with the sample sequence to construct a routine target-decoy database search, so that the entrapment hits included in each step can be used to provide a rough estimation of the confidence of the intermediate or final results.
Conclusions
In this study, we proposed a complementary use of target-decoy search strategy for evaluation of proteomics data analysis workflow. The labeled entrapment sequences are combined with the sample sequences to construct the target database for search, then the entrapment hits can be considered as false positive results and used to access the quality of proteomics data analysis tools. Based on this method, we assessed the two key steps of the mass spectrometry data analysis process, database search engines and quality control methods. Tested by both standard and experimental datasets, we found that the new search engine MS-GF+ and the support vector machine model based quality control method PepDistiller performed best in all evaluated tools, and the performance of search engines can be improved after the combination with efficient quality control methods. We also proposed an alternative intergrated method for results from different tools. Filtering the identificaitons according to their overlap conditions, we can increase the number of identifications and improve the confidence level at the same time.
Moreover, the entrapment sequence method could be an excellent strategy to assess all steps of the mass spectrometry data analysis process. Its applications can be extended to protein assembling methods, data integration methods and so on. By objective assessment of all steps of the common MS data analysis, we can standardize the analysis pipeline of mass spectrometry data.
Acknowledgements
We thank Dong-sheng Li for his help and support. We would like to acknowledge all members of the bioinformatics lab in Beijing Proteome Research Center for helpful discussion.
Funding
This study was financially supported by the Special Project of National Science and Technology Cooperation (2014DFB30010) and National Natural Science Foundation of China (21275160, 21475150). Work in J.M.’s laboratory was supported by National High Technology Research and Development Program of China (2015AA020108) and National Basic Research Program of China (2013CB910800). X.F. was supported by Chong Qing postgraduate scientific research and innovation project (CYS14154).
Availability of data and material
The datasets analyzed during the study are available in iProX with the identifier IPX0000812000 (www.iprox.org).
Authors’ contributions
JM and KS conceived and designed the project. XF, LL and JZ collected the datasets, constructed the protein sequence database and implemented the evaluation workflow. CC assisted with the analysis of LM3 dataset. JM, KS and YZ made intellectual contributions to the whole project. JM and XF wrote the manuscript. All authors contributed to the editing of the manuscript, and all authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
About this supplement
This article has been published as part of BMC Genomics Volume 18 Supplement 2, 2017. Selected articles from the 15th Asia Pacific Bioinformatics Conference (APBC 2017): genomics. The full contents of the supplement are available online http://bmcgenomics.biomedcentral.com/articles/supplements/volume-18-supplement-2.
Abbreviations
- FDR
False discovery rate
- FMR
False match rate
- MS
Mass spectrometry
- PSM
Peptide spectrum match
- QC
Quality control
Additional file
This file contains supplementary figures, including Figure S1-S6. (PDF 477 kb)
Contributor Information
Kun-xian Shu, Phone: 8623-62468319, Email: shukx@cqupt.edu.cn.
Jie Ma, Phone: 8610-61777053, Email: majie@hupo.org.cn.
References
- 1.Eng JK, McCormack AL, Yates JR. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. Journal of the American Society for Mass Spectrometry. 1994;5(11):976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 2.Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20(18):3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 3.Craig R, Beavis RC. TANDEM: matching proteins with tandem mass spectra. Bioinformatics. 2004;20(9):1466–1467. doi: 10.1093/bioinformatics/bth092. [DOI] [PubMed] [Google Scholar]
- 4.Eng JK, Jahan TA, Hoopmann MR. Comet: an open-source MS/MS sequence database search tool. Proteomics. 2013;13(1):22–24. doi: 10.1002/pmic.201200439. [DOI] [PubMed] [Google Scholar]
- 5.Diament BJ, Noble WS. Faster SEQUEST searching for peptide identification from tandem mass spectra. Journal of proteome research. 2011;10(9):3871–3879. doi: 10.1021/pr101196n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim S, Pevzner PA. MS-GF+ makes progress towards a universal database search tool for proteomics. Nature communications. 2014;5:5277. doi: 10.1038/ncomms6277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dorfer V, Pichler P, Stranzl T, Stadlmann J, Taus T, Winkler S, Mechtler K. MS Amanda, a universal identification algorithm optimized for high accuracy tandem mass spectra. Journal of proteome research. 2014;13(8):3679–3684. doi: 10.1021/pr500202e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Analytical chemistry. 2002;74(20):5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 9.Choi H, Nesvizhskii AI. Semisupervised model-based validation of peptide identifications in mass spectrometry-based proteomics. Journal of proteome research. 2008;7(1):254–265. doi: 10.1021/pr070542g. [DOI] [PubMed] [Google Scholar]
- 10.Ding Y, Choi H, Nesvizhskii AI. Adaptive discriminant function analysis and reranking of MS/MS database search results for improved peptide identification in shotgun proteomics. Journal of proteome research. 2008;7(11):4878–4889. doi: 10.1021/pr800484x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li N, Wu S, Zhang C, Chang C, Zhang J, Ma J, Li L, Qian X, Xu P, Zhu Y, et al. PepDistiller: A quality control tool to improve the sensitivity and accuracy of peptide identifications in shotgun proteomics. Proteomics. 2012;12(11):1720–1725. doi: 10.1002/pmic.201100167. [DOI] [PubMed] [Google Scholar]
- 12.Jian L, Xia Z, Niu X, Liang X, Samir P, Link A. l2 multiple kernel fuzzy SVM-based data fusion for improving peptide identification. IEEE/ACM Trans Comput Biol Bioinform. 2016;13(4):804-9. [DOI] [PubMed]
- 13.van den Toorn HW, Munoz J, Mohammed S, Raijmakers R, Heck AJ, van Breukelen B. RockerBox: analysis and filtering of massive proteomics search results. Journal of proteome research. 2011;10(3):1420–1424. doi: 10.1021/pr1010185. [DOI] [PubMed] [Google Scholar]
- 14.Wedge DC, Krishna R, Blackhurst P, Siepen JA, Jones AR, Hubbard SJ. FDRAnalysis: a tool for the integrated analysis of tandem mass spectrometry identification results from multiple search engines. Journal of proteome research. 2011;10(4):2088–2094. doi: 10.1021/pr101157s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sheng Q, Dai J, Wu Y, Tang H, Zeng R. BuildSummary: using a group-based approach to improve the sensitivity of peptide/protein identification in shotgun proteomics. Journal of proteome research. 2012;11(3):1494–1502. doi: 10.1021/pr200194p. [DOI] [PubMed] [Google Scholar]
- 16.Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nature methods. 2007;4(3):207–214. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- 17.Zhang J, Ma J, Dou L, Wu S, Qian X, Xie H, Zhu Y, He F. Bayesian nonparametric model for the validation of peptide identification in shotgun proteomics. Molecular & cellular proteomics : MCP. 2009;8(3):547–557. doi: 10.1074/mcp.M700558-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ma J, Zhang J, Wu S, Li D, Zhu Y, He F. Improving the sensitivity of MASCOT search results validation by combining new features with Bayesian nonparametric model. Proteomics. 2010;10(23):4293–4300. doi: 10.1002/pmic.200900668. [DOI] [PubMed] [Google Scholar]
- 19.Granholm V, Noble WS, Kall L. On using samples of known protein content to assess the statistical calibration of scores assigned to peptide-spectrum matches in shotgun proteomics. Journal of proteome research. 2011;10(5):2671–2678. doi: 10.1021/pr1012619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vaudel M, Burkhart JM, Breiter D, Zahedi RP, Sickmann A, Martens L. A complex standard for protein identification, designed by evolution. Journal of proteome research. 2012;11(10):5065–5071. doi: 10.1021/pr300055q. [DOI] [PubMed] [Google Scholar]
- 21.Wu S, Li N, Ma J, Shen H, Jiang D, Chang C, Zhang C, Li L, Zhang H, Jiang J, et al. First proteomic exploration of protein-encoding genes on chromosome 1 in human liver, stomach, and colon. Journal of proteome research. 2013;12(1):67–80. doi: 10.1021/pr3008286. [DOI] [PubMed] [Google Scholar]
- 22.Apweiler R, Bairoch A, Wu CH, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, Lopez R, Magrane M, et al. UniProt: the Universal Protein knowledgebase. Nucleic acids research. 2004;32(Database issue):D115–119. doi: 10.1093/nar/gkh131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kessner D, Chambers M, Burke R, Agus D, Mallick P. ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics. 2008;24(21):2534–2536. doi: 10.1093/bioinformatics/btn323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deutsch EW, Mendoza L, Shteynberg D, Farrah T, Lam H, Tasman N, Sun Z, Nilsson E, Pratt B, Prazen B, et al. A guided tour of the Trans-Proteomic Pipeline. Proteomics. 2010;10(6):1150–1159. doi: 10.1002/pmic.200900375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Park CY, Klammer AA, Kall L, MacCoss MJ, Noble WS. Rapid and accurate peptide identification from tandem mass spectra. Journal of proteome research. 2008;7(7):3022–3027. doi: 10.1021/pr800127y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McIlwain S, Tamura K, Kertesz-Farkas A, Grant CE, Diament B, Frewen B, Howbert JJ, Hoopmann MR, Kall L, Eng JK, et al. Crux: rapid open source protein tandem mass spectrometry analysis. Journal of proteome research. 2014;13(10):4488–4491. doi: 10.1021/pr500741y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kall L, Canterbury JD, Weston J, Noble WS, MacCoss MJ. Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nature methods. 2007;4(11):923–925. doi: 10.1038/nmeth1113. [DOI] [PubMed] [Google Scholar]
- 28.Reiter L, Claassen M, Schrimpf SP, Jovanovic M, Schmidt A, Buhmann JM, Hengartner MO, Aebersold R. Protein identification false discovery rates for very large proteomics data sets generated by tandem mass spectrometry. Molecular & cellular proteomics : MCP. 2009;8(11):2405–2417. doi: 10.1074/mcp.M900317-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tu C, Sheng Q, Li J, Ma D, Shen X, Wang X, Shyr Y, Yi Z, Qu J. Optimization of Search Engines and Postprocessing Approaches to Maximize Peptide and Protein Identification for High-Resolution Mass Data. Journal of proteome research. 2015;14(11):4662–4673. doi: 10.1021/acs.jproteome.5b00536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Granholm V, Kim S, Navarro JC, Sjolund E, Smith RD, Kall L. Fast and accurate database searches with MS-GF + Percolator. Journal of proteome research. 2014;13(2):890–897. doi: 10.1021/pr400937n. [DOI] [PMC free article] [PubMed] [Google Scholar]