

Abstract
Background
Atlantic cod (Gadus morhua), is known to vary genetically across the North Atlantic, Greenland, and Newfoundland. This genetic variation occurs both spatially and temporally through decades of heavy fishing, and is concentrated in three linkage disequilibrium blocks, previously defined by pedigreed linkage mapping analysis. Variation within these genomic regions is correlated with both seawater temperature and behavioral ecotype. The full extent and nature of these linkage groups is important information for interpreting cod genetic structure as a tool for future fisheries management.
Results
We conducted whole genome sequencing for 31 individual cod from three sub-populations in the Gulf of Maine. Across the genome, we found 3,390,654 intermediate to high frequency Single Nucleotide Polymorphisms (SNPs). We show that pairwise linkage analysis among these SNPs is a powerful tool to detect linkage disequilibrium clusters by recovering the three previously detected linkage groups and identifying the 1031 genes contained therein. Across these genes, we found significant population differentiation among spawning groups in the Gulf of Maine and between Georges Bank and Gulf of Maine. Coordinated divergence among these genes and their differentiation at both short and long spatial scales suggests that they are acting as linked supergenes in local adaptation of cod populations.
Conclusions
Differentiation between SNPs in linkage disequilibrium blocks is the major signal of genetic differentiation between all groups tested within the Gulf of Maine. Our data provide a map of genes contained in these blocks, allowing an enhanced search for neutral genetic structure for demographic inference and fisheries modeling. Patterns of selection and the history of populations may be possible to identify in cod using this description of linkage disequilibrium blocks and future data sets to robustly separate neutral and selected genetic markers.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-017-3660-3) contains supplementary material, which is available to authorized users.
Keywords: Atlantic cod, Gadus morhua, Linkage disequilibrium, Linkage map, Genomic divergence, Supergene, Gulf of Maine
Background
Genomic islands of divergence are the result of selection on regions within the genome undergoing adaptive divergence between subgroups within a species [1, 2]. These islands can manifest as regions of strong linkage disequilibrium due to genetic hitchhiking, while other regions within the genome are recombined through gene flow [1, 3, 4]. Adaptive divergence in regions of the genome may be an important driving force in speciation, particularly when they create differences in critical biological processes such as reproductive behavior and timing [1, 5], although the conditions under which these processes can occur may be limited [6, 7]. A significant body of literature has been published on speciation with gene flow [8–11], and islands of divergence are often cited as a possible explanation of this phenomenon.
Within species, islands of divergence are associated with differential selection across variable environments. In the threespine stickleback within Lake Constance in Germany, for example, strong differentiation between migratory lake and resident stream ecotypes is associated with differentiation across a set of strongly linked genes [12]. Chromosomal inversions often lead to large regions of strong linkage disequilibrium [13], and studies of these inversion polymorphisms in various Drosophila species have shown that haplotype frequencies of these inversions change along environmental clines. For example, the In(3R)Payne inversion in D. melanogaster quickly evolved different frequencies across latitudes in Australia. Moreover, allele frequencies of the more equatorial haplotype of the inversion polymorphism have increased across the entire cline, suggesting a population adaptive response to increased temperature over 30 years [14–17]. A strikingly similar pattern occurs in D. subobscura, where the more southerly haplotype of the O inversion has increased in frequency over the last 15 to 30 years in all European populations tested [18]. In the yellow monkey flower (Mimulus guttatus), populations show differential fitness across coastal and inland habitats based on a set of genes linked within an inversion [19]. All in all, ‘supergenes’ are receiving more attention as a combination of genomic information and evolutionary theory shows how multi-locus selection and linkage can evolve to generate local adaptation [20–22].
The relationship between environmental conditions and genomic islands has also been studied in the Atlantic cod, Gadus morhua. Borza et al. (2010) and Hubert et al. (2010) developed a panel of ~1500 SNPs derived from expressed sequence tags (EST) and used them to create linkage maps from pedigreed cod populations [23, 24]. They discovered three genomic islands of divergence that were highly associated with mean ocean temperatures across the northern Atlantic [25, 26]. The same set of markers also describes ecotype divergence between migratory and stationary cod populations in Iceland and Norway [27], as well as small scale spatio-temporal patterns of population structure in Newfoundland [28, 29]. Using a custom SNP array from the recently published improved assembly of the cod genome [30], genomic differences were described between oceanic and coastal populations in the North Sea [31] and the Norwegian Sea [32]. In all these cases, genetic differences among populations at both oceanic and local scales are thought to be driven by environmental selection mediated by a suite of genes in several linkage groups, not by demographic barriers to gene flow. At smaller geographic and temporal scales, population genetic analyses of cod in the Gulf of Maine have focused on a suite of microsatellite and protein markers in a large number of individuals across many populations [33]. Most of the significant differences compiled in Zemeckis et al. [33] are from individual loci which appear to be under selection, rather than the result of neutral genetic structure between populations [34, 35].
To date, the loci within the cod linkage groups have been identified through breeding studies and classic quantitative trait loci (QTL) approaches. Moreover, the search for neutral genetic differentiation, outside the known linkage groups, has been based on relatively few loci compared to genome-level data sets. Both the searches for selected genes and those that might reflect neutral genetic differentiation have not yet taken advantage of full-genome approaches [36]. Here, we use high throughput sequencing and population genomic analyses to examine the degree and spatio-temporal distribution of linkage disequilibrium clusters throughout the genome of Atlantic cod in the Gulf of Maine and Georges Bank. We then use this information to eliminate linked genes from the analysis and use the remaining genes to test for patterns of neutral genetic differentiation.
Using complete genome sequencing, we describe a panel of 3,390,654 single nucleotide polymorphisms (SNPs) throughout the cod genome. Pairwise linkage analysis shows SNPs within 1031 genes falling into three cod ‘supergenes’, two of which show population divergence within the Gulf of Maine. Genes within these linkage blocks include DNA structural proteins and chromatin assembly genes, metabolic and catabolic genes, meiosis regulation and oocyte maturation genes, odorant receptors, egg coat structural proteins, heat shock proteins and many cell signaling genes that might be involved in environmental adaptation, habitat choice or mating. Once linkage disequilibrium blocks were identified and excluded from the analysis, our limited population data show no signs of neutral genetic differentiation in the three populations we sampled. Though our power to detect neutral structure is low with such few samples, comparison of many SNPs suggests that the signature of population differentiation is largely driven by shifts in the supergenes, possibly driven by present or past patterns of natural selection.
Methods
Sample collection
Fin-clip samples from adult Atlantic cod in spawning condition were collected and stored frozen prior to preparation of DNA libraries. A total of 31 individuals were sampled: 10 from Georges Bank, and 21 from the Gulf of Maine. Within the Gulf of Maine, the sample was subdivided into two groups, based on spawning season: 10 cod were sampled from a winter spawning group, and 11 cod from a spring spawning group (Fig. 1). These samples were taken from cod stocks in the northwestern part of Massachusetts Bay near Cape Ann, as studies of cod sampled from this region showed evidence of temporal population substructure based on breeding season [34, 35]. Samples were collected in March 2014 for the Georges Bank population, and in December 2013 and June 2014 for the Gulf of Maine winter and spring populations, respectively (Table 1).
Fig. 1.
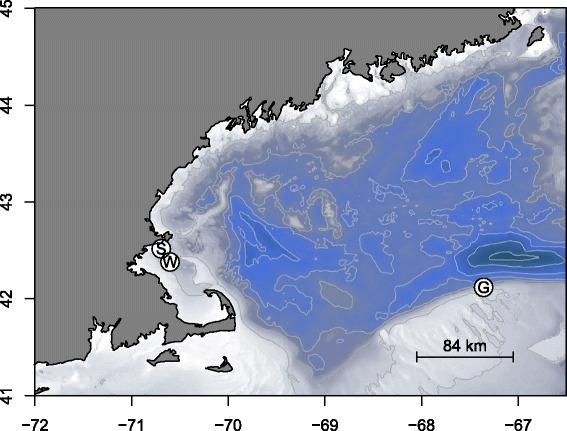
Sampling locations used in this study. Twenty one adults in spawning condition were sampled in northwestern Massachusetts Bay within the Gulf of Maine. These were subdivided into 11 spring spawning cod (site S) and 10 winter spawning cod (site W). Ten adults in spawning condition were sampled from Georges Bank (site G). Figure was generated using the marmap package in R
Table 1.
Cod sampling summary
| Stock | Date | Samples (n) | Latitude | Longitude |
|---|---|---|---|---|
| Gulf of Maine winter | Dec 2013 | 10 | 42.3813 | -70.6016 |
| Gulf of Maine spring | June 2014 | 11 | 42.5177 | -70.6921 |
| Georges Bank | Mar 2014 | 10 | 42.1192 | -67.3575 |
Locations, dates, and number of samples collected at each sampling site
DNA extraction, library preparation, sequencing
Genomic DNA (gDNA) from each fin clip was prepared by digesting entire fin clips (approximately 1 cm2) in 500 μL of Digestion Mixture (1 μg/μL Proteinase K, 50 mM EDTA, 5% Tween 20, 0.5% Triton-X 100, 800 mM GuHCl, 0.5% SDS) at 50 °C for 1 h. After digestion, the samples were centrifuged at 13,000 rpm for 10 min; the supernatant was recovered, and then mixed with 50 μL of 3 M potassium chloride to precipitate the SDS. An equal volume of isopropyl alcohol was added to precipitate nucleic acids, and the pellet was washed with 70% ethanol. DNA/RNA was resuspended in 100 μL of 10 mM Tris-HCl pH 8.0 and treated with 5 μL of 10 mg/mL RNase A. Another isopropyl alcohol precipitation was performed, then genomic DNA was isolated using AMPure XP beads. Total gDNA was resuspended and stored in TE buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8).
Sequencing libraries were prepared from 50 ng of template DNA using the Illumina Nextera DNA library preparation kit (Illumina, San Diego, CA) following their standard protocol. Samples were individually barcoded to allow for multiplexing during sequencing. Each library was independently size selected using Ampure XP beads (0.6 volume beads to 1.0 volume DNA) and checked for adequate sizing via gel electrophoresis. Libraries were quantified by qPCR (KAPA Illumina Quantification Kit), then normalized and mixed in equimolar fashion.
One set of samples, consisting of five individuals from each sampled location, was sequenced on an Illumina HiSeq 2000 using 100 base paired end reads. Each batch of five samples from the original sample groupings were mixed and multiplexed across a distinct eight-lane HiSeq flowcell, resulting in approximately 80x coverage across the genome (predicted size ~800 Mb). The remaining samples (five winter, six spring, and five Georges Bank) were pooled and sequenced to less depth of coverage (~25x) on three runs of an Illumina NextSeq 500 using 150 base paired end reads.
Read mapping and variant calling
Raw reads were trimmed of any residual adapter sequences, and low-quality base calls were removed using a baseline Phred score of 20 or less. Individual reads were retained if the length was > 70 bases, and reads were re-paired using Trimmomatic [37]. Read pairs were then mapped to the reference genome file gadMor2.fasta [30] using bowtie2, with end-to-end alignment, 22 base seed length, two re-seed attempts with no mismatches allowed in the seed, a seed interval of 17 bases, and all other settings at default values. Reads that became unpaired due to quality or trimming issues were mapped separately using the same parameters. The resulting mapped files were sorted and indexed using samtools [38] and duplicated reads were removed using Picard tools (http://broadinstitute.github.io/picard) for downstream SNP discovery and analysis.
SNP discovery was performed on individual samples using FreeBayes version 0.9.10-3-g47a713e, using the default settings [39]. Variants were filtered using vcftools to only include biallelic SNPs with a minimum minor allele frequency of 0.05 using vcftools version 0.1.12b [40]. Filtering by minor allele frequency ensures that SNP loci with extremely low diversity are removed from downstream analysis, as they are essentially uninformative. The resultant SNP list was further limited to those SNPs that were genotyped (Q > 30) in all 31 samples.
Data analysis
We limited our analysis of linkage disequilibrium to linkage groups (hereafter LG) 2, 7, and 12, as these regions contain the three “islands of divergence” studied in this species. Within each LG, we extracted every 250th SNP, and performed pairwise linkage disequilibrium analysis using the LD function in the ‘genetics’ package (version 1.3.8.1) in R version 3.03 (R development core team 2015). We determined the correlation coefficient (as r) between the paired genotypes of all 31 individuals, and reported the r 2 value to remove any arbitrary sign introduced by intermediate calculations. To examine the genomic extent of linkage disequilibrium effects within each LG, mean r 2 was calculated for every 250th SNP within each LG, as was pairwise FST. The edges of each linkage disequilibrium block (hereafter LD block) were located by finding the regions with high mean LD (mean r 2 > 0.1) and determining the first and last SNP with similar mean r 2 values to the block. As there are 250 SNPs between the first high LD SNP and its subset neighbor and untested linked loci are located within this space, we placed the LD block edges on the SNPs adjacent to the ones with the first and last high LD value.
Using the gene model information from the genome, we determined which genes were in each LD block, and performed enrichment tests of Gene Ontology (GO) categories of the genes within the resultant LD block using the weight01 algorithm within the ‘topGO’ package version 2.24.0 [41] in R. Additionally, all SNPs throughout the genome were binned into one of three classes; exon, intron, or intergenic. We determined the proportion of SNPs within each class inside each LD block and throughout the genome, to test if SNPs of any class were enriched within LD blocks.
To test for population differentiation, we filtered all SNPs for those found within exons and created subsets for analyses. For exonic SNPs on linkage groups 2, 7, and 12 we used all available exonic SNPs, each partitioned into two groups: those within the LD block and those outside of the block. For the remainder of the genome, we created a subset of every 10th exonic SNP. We focused solely on SNPs within exons for this analysis, as we are interested in both neutral population structure and adaptive divergence between spawning sites/times, and SNPs within the coding regions of genes can be used to examine both processes. From this subset of SNPs, Weir and Cockerham’s pairwise FST was calculated among the three populations using the wc function from the ‘hierfstat’ package [42, 43] within R. This was performed for exonic SNPs within each LD block, SNPs on the same linkage group but not in the LD block, and all exonic SNPs not on linkage groups 2, 7, or 12. Principal component analysis (PCA) was performed on SNPs both within and outside of each LD block using the ‘prcomp’ function within the standard release package of R.
To test for neutral population structure, we utilized the same subsets of exonic SNPs used for FST and PCA analysis above. To avoid potential confounding selection with population structure, SNPs within the linkage blocks on LG02, LG07, and LG12 were removed from this dataset and tested separately. With these sets of SNPs, we tested for population structure using the admixture algorithm NGSadmix [44]. For each admixture analysis, we tested the model against a number of subpopulation divisions (k = 2 to k = 5), and chose the k value that had the greatest likelihood among all k values tested using the Evanno method [45].
Results
Linkage disequilibrium block determination
After mapping reads to the genome assembly, we identified 3,390,654 biallelic SNP variants with a minor allele frequency greater than 0.05 that were confidently genotyped in all 31 samples. There were 147,356 SNPs located on LG02, 204,124 SNPs on LG07, and 139,785 SNPs on LG12. To find the edges of the LD blocks, pairwise linkage disequilibrium (as r2) and FST was calculated for every 250th SNP in LG02 (589 SNPs), LG07 (816 SNPs), and LG12 (559 SNPs). From these data, mean pairwise r 2 values were determined for each SNP (Fig. 2). Each LD block within a linkage group shows a highly contiguous and sharply defined region of elevated mean linkage disequilibrium (mean r 2 > 0.1) in comparison with the surrounding SNPs (mean r 2 ~ 0.03 to 0.04). FST was less useful in determining the edges of LD blocks, as this varied over wide ranges on SNPs within linkage groups 2 and 7. Interestingly, FST and LD appear correlated in linkage group 12 (Fig. 2, bottom right), perhaps due both to the large extent of the linkage region and the high degree of differentiation seen in this sample set between the winter spawning cod and Georges Bank populations in the linked loci.
Fig. 2.

Linkage and population differentiation for chromosomes 2, 7 and 12 inside and outside linkage blocks. Mean pairwise LD (as r 2) and FST are shown on the y-axis for every 250th SNP, with SNP position within its linkage group along the x-axis. The right hand figures show a regression of LD vs FST for each SNP
To capture potentially linked loci between the subset SNP loci, the starting base for each LD block was identified as the SNP immediately before the first SNP with a mean r 2 > 0.1 in the LD block, and the ending base was the first SNP after the last base with a mean r 2 > 0.1 in the LD block, resulting in LD blocks of 5.6 Mb (LG02), 9.3 Mb (LG07) and 11.6 Mb(LG12) in length (Table 2). There was a linkage gap of about 2 million bases in LG02, inside which SNPs were not linked to one another or to the SNPs in the rest of the linkage group (Fig. 2, top left). For LG02, 58 of the 174 SNPs within the LD block (~33%) were strongly linked (mean pairwise r 2 > 0.1), while 170 of the 312 SNPs (~54%) in LG07 and 42 of the 246 SNPs (~17%) in LG12 were strongly linked. Our findings agree with regions of linkage disequilibrium identified previously by EST-derived SNPs [23, 25–27] and Illumina SNP arrays [31, 32]. However, our analysis provides a higher degree of resolution of both the range and edges of these LD blocks, as previous work has described LD blocks in terms of relative centimorgans(cM) rather than actual base positions.
Table 2.
Linkage disequilibrium block extent, and approximate length of each block
| Starting base | Ending base | # genes | length | |
|---|---|---|---|---|
| LG02 | 18,442,392 | 24,053,701 | 294 | 5.6 Mb |
| LG07 | 13,743,341 | 23,039,143 | 306 | 9.3 Mb |
| LG21 | 2,039,256 | 13,611,225 | 437 | 11.6 Mb |
Genetic content of LD blocks
Using the available gene models from the cod genome and the start and stop base positions for each LD block described above, we determined which genes were within each LD block. Of the 1095 genes on LG02, 294 were found within the boundaries of the LD block. GO enrichment analysis on genes within this LD block showed several overlapping GO terms with significant enrichment (p-adj < 0.01 after false-discovery rate correction) centered on genes involved with DNA/chromatin structuring (Table 3). Genes within the significant GO terms consisted primarily of histone proteins involved in the assembly of the nucleosome: 14 of the 27 genes in the significant GO terms coded for histone proteins (Table 4). Additionally, two histone modification genes, histone-lysine N-methyltransferase and histone deacetylase are also located within this linkage block.
Table 3.
GO enrichment analysis of linkage disequilibrium block on LG02
| GO.ID | Term | Annotated | Significant | Fisher pval | padj |
|---|---|---|---|---|---|
| GO:0006325 | chromatin organization | 142 | 18 | 1.70E-14 | 2.52E-11 |
| GO:0065004 | protein-DNA complex assembly | 105 | 16 | 2.80E-14 | 2.52E-11 |
| GO:0071824 | protein-DNA complex subunit organization | 105 | 16 | 2.80E-14 | 2.52E-11 |
| GO:0051276 | chromosome organization | 178 | 19 | 7.10E-14 | 4.76E-11 |
| GO:0006334 | nucleosome assembly | 100 | 15 | 2.50E-13 | 9.39E-11 |
| GO:0031497 | chromatin assembly | 100 | 15 | 2.50E-13 | 9.39E-11 |
| GO:0034728 | nucleosome organization | 100 | 15 | 2.50E-13 | 9.39E-11 |
| GO:0006333 | chromatin assembly or disassembly | 101 | 15 | 2.90E-13 | 9.57E-11 |
| GO:0006323 | DNA packaging | 106 | 15 | 6.00E-13 | 1.77E-10 |
| GO:0071103 | DNA conformation change | 112 | 15 | 1.40E-12 | 3.65E-10 |
| GO:0034622 | cellular macromolecular complex assembly | 180 | 16 | 1.30E-10 | 3.26E-08 |
| GO:0043933 | macromolecular complex subunit organization | 324 | 20 | 3.90E-10 | 8.64E-08 |
| GO:0006461 | protein complex assembly | 244 | 17 | 1.50E-09 | 2.91E-07 |
| GO:0070271 | protein complex biogenesis | 244 | 17 | 1.50E-09 | 2.91E-07 |
| GO:0065003 | macromolecular complex assembly | 258 | 17 | 3.60E-09 | 6.38E-07 |
| GO:0071822 | protein complex subunit organization | 271 | 17 | 7.50E-09 | 1.24E-06 |
| GO:0022607 | cellular component assembly | 308 | 18 | 7.90E-09 | 1.24E-06 |
| GO:0006996 | organelle organization | 432 | 21 | 1.00E-08 | 1.50E-06 |
| GO:0044085 | cellular component biogenesis | 339 | 18 | 3.50E-08 | 4.95E-06 |
| GO:0016043 | cellular component organization | 636 | 22 | 1.70E-06 | 0.00023 |
| GO:0071840 | cellular component organization or biogenesis | 666 | 22 | 3.70E-06 | 0.00047 |
| GO:0042157 | lipoprotein metabolic process | 32 | 5 | 2.70E-05 | 0.0032 |
| GO:0044711 | single-organism biosynthetic-process | 295 | 9 | 0.0061 | 0.7060 |
| GO:0006897 | endocytosis | 35 | 3 | 0.0071 | 0.7895 |
| GO:0043543 | protein acylation | 12 | 2 | 0.0078 | 0.8318 |
| GO:0016568 | chromatin modification | 38 | 3 | 0.0089 | 0.8645 |
Using topGO, (in R), enrichment analysis shows multiple GO categories that may be enriched in the LD block from LG02, mostly centered on DNA/chromatin structuring. Total number of annotated genes in each GO category is shown, as well as those within the LD block that are significant (Fisher test p < 0.01). Multiple-test correction (calculated as false-discovery rate, ‘fdr’) is shown in p-adj column, p-values < 0.05 shown in bold
Table 4.
Genes in significant GO terms within LD block on LG02
| Gene | Starting base | Gene description |
|---|---|---|
| GAMO_00059458 | 18475327 | zgc:112234: Histone H2B 1/2 (Danio rerio) |
| GAMO_00059459 | 18476591 | Histone H2A (Oncorhynchus mykiss) |
| GAMO_00059461 | 18489582 | TGas006m08.1: Histone H4 (Xenopus tropicalis) |
| GAMO_00059462 | 18491420 | Histone H2A (Oncorhynchus mykiss) |
| GAMO_00059465 | 18506398 | Histone H2A (Oncorhynchus mykiss) |
| GAMO_00059556 | 19188916 | STRA13: Centromere protein X (Homo sapiens) |
| GAMO_00059593 | 19396062 | Crebbp: CREB-binding protein (Rattus norvegicus) |
| GAMO_00059696 | 20010135 | NMT1: Glycylpeptide N-tetradecanoyltransferase 1 (Homo sapiens) |
| GAMO_00059920 | 21260662 | Coro1b: Coronin-1B (Mus musculus) |
| GAMO_00059924 | 21287842 | suv420h2: Histone-lysine N-methyltransferase SUV420H2 (Xenopus laevis) |
| GAMO_00059998 | 21707418 | KCNA1: Potassium voltage-gated channel subfamily A member 1 (Homo sapiens) |
| GAMO_00060305 | 23130440 | tmem11: Transmembrane protein 11%2C mitochondrial (Danio rerio) |
| GAMO_00060314 | 23170545 | Histone H2A (Oncorhynchus mykiss) |
| GAMO_00060318 | 23177224 | Histone H2A (Oncorhynchus mykiss) |
| GAMO_00060319 | 23198635 | Histone H3 (Urechis caupo) |
| GAMO_00060320 | 23199855 | Histone H2A (Oncorhynchus mykiss) |
| GAMO_00060321 | 23201514 | TGas006m08.1: Histone H4 (Xenopus tropicalis) |
| GAMO_00060322 | 23202645 | Histone H3 (Urechis caupo) |
| GAMO_00060324 | 23204017 | Histone H2B (Salmo trutta) |
| GAMO_00060364 | 23359510 | Pigq: Phosphatidylinositol N-acetylglucosaminyltransferase subunit Q (Mus musculus) |
| GAMO_00060373 | 23393752 | Histone H2A (Oncorhynchus mykiss) |
| GAMO_00060375 | 23400842 | TGas006m08.1: Histone H4 (Xenopus tropicalis) |
| GAMO_00060376 | 23401993 | Histone H3 (Urechis caupo) |
| GAMO_00060390 | 23480761 | HDAC9: Histone deacetylase 9 (Homo sapiens) |
| GAMO_00060462 | 23855114 | APOL3: Apolipoprotein L3 (Homo sapiens) |
| GAMO_00060465 | 23866729 | APOL3: Apolipoprotein L3 (Homo sapiens) |
| GAMO_00060467 | 23891523 | APOL3: Apolipoprotein L3 (Homo sapiens) |
While several GO terms were significantly enriched, many of them collapsed to the same set of DNA structural genes, including several genes encoding all four major histone proteins that make up the nucleosome
Within the 1166 genes on LG07, 306 were located within its LD block, including genes involved with signaling and metabolic processes, though no GO categories remained significant after false-discovery rate correction (Table 5). Similarly, the LD block within LG12 contained 437 genes out of the 975 genes in that linkage group, and no significant enrichment of any GO categories remained after false-discovery rate correction (Table 6). Specific genic content within each block can be found in the Additional files 1, 2, 3 for this publication.
Table 5.
GO enrichment analysis of linkage disequilibrium block on LG07
| GO.ID | Term | Annotated | Significant | Fisher pval | p-adj (fdr) |
|---|---|---|---|---|---|
| GO:0006855 | drug transmembrane transport | 5 | 2 | 0.0014 | 1 |
| GO:0006066 | alcohol metabolic process | 24 | 3 | 0.0028 | 1 |
| GO:0015893 | drug transport | 8 | 2 | 0.0038 | 1 |
| GO:0042493 | response to drug | 8 | 2 | 0.0038 | 1 |
| GO:0044765 | single-organism transport | 1098 | 23 | 0.0044 | 1 |
| GO:0023051 | regulation of signalling | 275 | 9 | 0.0054 | 1 |
| GO:1902578 | single-organism localization | 1118 | 23 | 0.0055 | 1 |
| GO:1901615 | organic hydroxy compound metabolic process | 31 | 3 | 0.0058 | 1 |
| GO:0006071 | glycerol metabolic process | 10 | 2 | 0.0059 | 1 |
| GO:0019400 | alditol metabolic process | 10 | 2 | 0.0059 | 1 |
Using topGO, (in R), enrichment analysis shows multiple GO categories that may be enriched in the LD block from LG07, including genes involved with signal transport and metabolic processes. Total number of annotated genes in each GO category is shown, as well as those within the LD block that are significant (Fisher test p < 0.01). Multiple-test correction (calculated as false-discovery rate, ‘fdr’) is shown in p-adj column
Table 6.
GO enrichment analysis of linkage disequilibrium block on LG12
| GO.ID | Term | Annotated | Significant | Fisher pval | p-adj (fdr) |
|---|---|---|---|---|---|
| GO:0051648 | vesicle localization | 4 | 2 | 0.002 | 1 |
| GO:0051650 | establishment of vesicle localization | 4 | 2 | 0.002 | 1 |
| GO:0009057 | macromolecule catabolic process | 144 | 8 | 0.0051 | 1 |
| GO:0051656 | establishment of organelle localization | 7 | 2 | 0.0067 | 1 |
| GO:0051640 | organelle localization | 8 | 2 | 0.0088 | 1 |
Using topGO, (in R), enrichment analysis shows multiple GO categories that may be enriched in the LD block from LG12. Total number of annotated genes in each GO category is shown, as well as those within the LD block that are significant (Fisher test p < 0.01). Multiple-test correction (calculated as false-discovery rate ‘fdr’) is shown in p-adj column
In addition to the genic content within each LD block, we evaluated the distributions of SNP locations (classified as intergenic, exon, or intron) of all SNPs located on linkage groups 1 through 23 (n = 3,283,653 SNPs). This number is slightly smaller than the total number of SNPs passing our filters (n = 3,390,654) due to unused SNPs located on the few remaining unscaffolded contigs as well as mitochondrial DNA, yet still represents 97% of all identified SNPs. Proportions of each SNP type were calculated within each LD block, as well as the remaining SNPs not on linkage groups 2, 7, or 12 (Table 6). SNP density (as # SNPs/base) was also determined within each LD block and in the remainder of the genome as above. For the LD block on LG02, there was both an increase in overall SNP density (from 5.66e-03 to 7.72e-03, a 36% increase) and an increase in the proportion of exonic SNPs (from 0.065 to 0.089, a 37% increase) compared to genome-wide values. We also found a 50% increase over the genome-wide SNP density within the LD block on LG07 (from 5.66e-03 to 8.36e-03 SNPs/base), though there were no significant changes in the proportions of SNP type within this group and no deviations from expected polymorphism rates in LG12 (Table 7).
Table 7.
Comparison of genome contents and population divergence of cod chromosome linkage groups 2, 7 and 12
| Group | # SNPs | Block length (Mb) | SNP linked | # genes | Genes in LD | SNP density | proportion exonic SNPs | Fst in LD block | FST vs LD | FST outside LD block | Notes |
|---|---|---|---|---|---|---|---|---|---|---|---|
| LG02 | 43,341 | 5.6 | 33 | 1095 | 294 | 7.72E-03 | 0.089 | 0.098 | negative | 0.0025 | discontinuous block |
| LG07 | 77,751 | 9.3 | 54 | 1166 | 306 | 8.36E-03 | 0.064 | 0.089 | positive | 0.0004 | |
| LG12 | 61,251 | 11.6 | 17 | 975 | 437 | 5.29E-03 | 0.071 | 0.173 | higly positive | 0.0022 | marked lack of SNPs adjacent to linkage block |
| Not 2, 7, or 12 | 2,792,388 | 493.2 |
Differentiation among sampled populations
The subsets of exonic SNPs used for population structure analyses contained a total of 54,030 SNPs. FST analysis of linkage group 2 used 11,241 SNPs (3874 in LD block, 7367 outside), linkage group 7 used 13,839 SNPs (4920/8919) and linkage group 12 used 8835 SNPs (4343/4492). The remainder of the genome used a subset of exonic SNPs (n = 20,115 SNPs). Mean pairwise FST for all loci tested (n = 54,030) ranged between 0.0062 (spring spawners vs. Georges Bank) and 0.0297 (winter spawners vs. Georges Bank). Mean pairwise FST analysis of each SNP subset tested shows a trend of increased FST values within some LD blocks as compared to the exonic SNPs on the same linkage group. Mean pairwise FST for each comparison is shown in Table 8.
Table 8.
Mean pairwise FST comparisons between sampled populations
| Spring vs. Winter | Spring vs. GB | Winter vs. GB | |
|---|---|---|---|
| LG02 in LD | 0.1166 | -0.0189 | 0.0981 |
| LG02 out | 0.0025 | 0.0021 | 0.0016 |
| LG07 in LD | -0.0062 | 0.0121 | 0.0895 |
| LG07 out | 0.0004 | -7.43E-05 | 0.0001 |
| LG12 in LD | 0.0231 | 0.0766 | 0.1726 |
| LG12 out | 0.0022 | 0.0021 | 0.0036 |
| Genome | 0.0001 | -0.0003 | 0.0003 |
Mean pairwise FST calculations for each sampled population show some patterns of divergence between sampled populations, with high mean FST values between winter spawners and Georges Bank cod in all three LD blocks (shown in bold). LG02 is also highly divergent between spring and winter spawners
We also examined patterns of FST and LD along each of the chromosomes with linkage blocks. FST among all three populations was highly correlated with linkage disequilibrium in LG02 and LG12 (Fig. 2, right upper and lower), but was only moderately correlated in LG07 (Fig. 2, right middle). Linkage was highest in LG07, whereas FST was higher within the linkage groups LG07 and LG12. LG07 also shows a number of chromosomal areas outside the main region of linkage that have occasional high FST, and a low relationship overall between linkage and FST (Fig. 2, left middle). Variation in the relationship between linkage and FST suggests other patterns of selection acting on genes on LG07 that would warrant future research.
The LD block on linkage group 2 showed a significant increase in higher FST values when comparing spring spawning cod to the winter spawning cod (Fig. 3, A1, mean FST = 0.1166) as well as between winter spawners and Georges Bank cod (Fig. 3, A3, mean FST = 0.0981) using a Kolmogorov-Smirnov two-sided test on the FST distributions (p < 1e-10, both comparisons). The LD block on linkage group 7 was also genetically divergent between winter spawners and Georges Bank cod (Fig. 3, B3, mean FST = 0.0895, KS test p < 1e-10). However, the highest differentiation was found between winter spawners and Georges Bank cod within the LD block on LG12 (mean FST = 0.1726, KS test p < 1e-10), with a broader, longer-tailed distribution of FST values. Mean pairwise FST values for exonic SNPs on the same linkage groups but not within LD blocks ranged between 0.0001 and 0.0036, and mean FST values for the remainder of the genome ranged between 0 and 0.0003, which is consistent with one large, outbreeding population.
Fig. 3.

Distributions of pairwise FST within LD blocks between sampled populations. Pairwise FST was determined within LD blocks (blue), on the same LG but not in the LD block (red), as well as the remainder of the genome (black) for LD blocks on LG02 (top row) LG07 (middle row), and LG12 (bottom row). Column 1 is the pairwise FST comparison between spring and winter spawners, column 2 is spring vs. Georges Bank, and column 3 is winter vs. Georges Bank
Principal component analysis was used on SNPs within each LD block, and on SNPs on the same linkage group, but outside the LD block (Fig. 4). In all cases, PCA on SNPs within LD blocks showed three distinct clusters along principal component axis 1, correlating with the three possible genotypes of that LD block (Fig. 4, left). This is in contrast to the remaining exonic SNPs on the same linkage group (Fig. 4, right), where no real clustering is seen between sampled populations, suggesting that all significant divergence is centralized on the three main blocks of linkage disequilibrium.
Fig. 4.
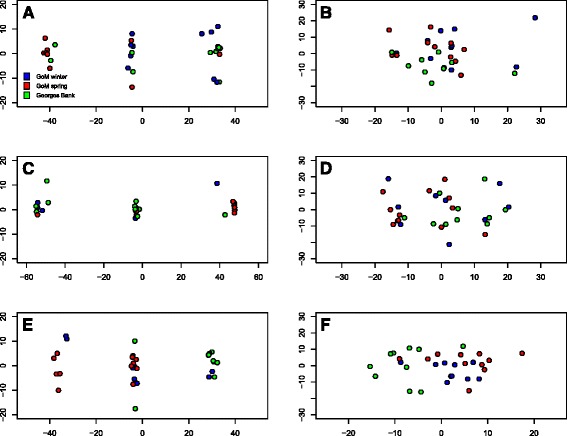
Principal component analyses. PCA was performed on exonic SNPs from LG02 (top row), LG07 (middle row), and LG12 (bottom row). SNPs within the LD block on that LG are shown on the left, SNPs outside the LD block on the right. In all cases, as expected, SNPs within each LD block separate into three distinct vertical clusters along PC axis 1, indicating the three genotypes that individuals might possess for each LD block. This separation is lost, however, for PCA on SNPs outside the LD block, but in the same linkage group. Note that SNPs outside of the LD block on LG07 (D, above) may signify the presence of another, undiscovered linkage disequilibrium cluster, as the pattern appears diffusely similar to that in C
Finally, we found no evidence for neutral population structure when using the population admixture analysis algorithm NGSadmix. When using k = 2 for SNPs within each linkage disequilibrium block, we found that individual admixture predictions perfectly matched the known genotypes of that individual in the linked loci with the LD block, with homozygous individuals binned into the two populations and heterozygotes having 50/50 admixture, as expected. However, all runs for the entire exonic SNP dataset predicted k = 1 as the most likely population model using the Evanno method of comparing model likelihood scores [45]. All models of k = 2 or higher predicted a high proportion of admixture for all samples and showed no clear differentiation between sampled locations.
Discussion
In this study, we used whole genome resequencing and single nucleotide polymorphism discovery to describe patterns of genome-wide linkage disequilibrium in the Atlantic cod (Gadus morhua) at small spatio-temporal scales within the Gulf of Maine and Georges Bank. Through sequencing and linkage disequilibrium analysis, we found three large clusters of linkage disequilibrium spanning multiple genes, in agreement with the known relative positions of these LD clusters from previously published studies of genomic “islands of divergence” in cod [23, 25–27, 31, 32]. Earlier work had identified such clusters through a set of pedigree-based linkage maps of EST-derived SNPs and microsatellites in controlled crosses. Using custom microarrays, gene lists have been developed to describe the genic content of these linkage regions [32]. Specifically, the LD blocks on LG02 and LG07 have been shown to contain 189 and 297 genes, respectively. Our analysis augments these gene lists to contain 294 and 306 genes, includes an additional 437 genes on LG12, and shows that the extent and genic content of linkage disequilibrium blocks can be detected de novo by deep sequencing of wild-caught individuals.
The Atlantic cod genomic islands of divergence are associated with strong natural selection across the Atlantic [25, 26], in the Atlantic Canadian region [27–29], and in the Norwegian Sea [31, 32, 46]. In our samples, large blocks of linkage disequilibrium within LG02, LG07, and LG12 showed a differential pattern of genetic differentiation, with the LD block in LG02 separating between spring and winter spawners within the Gulf of Maine, and the LD blocks within all three linkage groups separating between the winter spawners and Georges Bank cod. This is in direct contrast to SNPs on the same linkage group but outside the LD block, suggesting that the observed differentiation within LD blocks may be due to selection acting on the LD blocks as a unit.
Temperature and linkage clusters
Local adaptation is not uncommon in marine systems [47]. Studies typically examine local adaptation across large geographic scales, often working at ranges of thousands of kilometers [48–51]. However, evidence is accumulating that highly local adaptation in high gene flow species may be more common than previously thought in both terrestrial and marine systems [52, 53]. Indeed, within Atlantic cod, the genomic region in linkage map LG02 (at ~50 centimorgans) has been shown to have a clinal relationship to average water temperature across the Atlantic, with cod from both Georges Bank and Galway Bay, Ireland (“warmer” locations, mean bottom temp. ~8 °C) largely having one linkage haplotype, and cod from both Newfoundland and Norway (“colder” locations, mean bottom temp. ~0 °C) having the alternate haplotype [26]. We found the same LD block diverging between spring and winter spawning cod within the Gulf of Maine, where near-surface temperatures can vary from 3 °C in late winter to 10 °C in early autumn [54]. Such seasonal variation may facilitate the temporal partitioning of subpopulations of cod if they are adapted to spawning at certain temperature ranges [55].
Nature of selection in putative ‘supergenes’
Gene located within the LD block of linkage group 2 showed a significant enrichment for genes associated with DNA and chromatin structuring.
This cluster was most divergent between the spring and winter spawning cod within the Gulf of Maine, but was also divergent between winter spawners and Georges Bank. Historically, this linkage disequilibrium block has been shown to differentiate between stationary and migratory cod populations in Norway [31, 32], and have strong allelic associations with seawater temperatures across the Atlantic [26].
It is well understood that temperature plays a large role in the stress response of organisms, including the maintenance of DNA stability [56]. Chromatin plays an important role in the structuring and stability of the DNA molecule, controlling the regulation of gene expression either by relaxing and exposing regions of DNA for easier transcription or by tightening and shutting down transcription. Recently, expression changes in genes involved in chromatin organization and the regulation of DNA replication have been shown to be one of the earliest larval responses to temperature stress in larval zebrafish [57]. In our GO enrichment analysis for LG02, we found several of the major histone proteins (H2A/B, H3a, and H4A), as well as a histone methyltransferase, which methylates the chromatin to alter the ability to transcribe genes, and a histone deacetylase, which removes acetyl groups from chromatin which dampens gene expression. This clustering of histone structural proteins and associated histone methylation and deacetylation genes within the linkage block on LG02 fits the concept of a ‘supergene,’ having several linked loci, inherited as a single unit, with enrichment for related functional genes as a ‘complex phenotype.’
In addition to functional enrichment for histone-related genes, SNPs within the LD block on LG02 are also more likely to be found within exons, as compared to the rest of the genome. 8.9% of the 43,441 SNPs in this linkage block are found in exons, which is a 37% increase over the 6.5% of 2,792,388 SNPs in the rest of the genome. The increase in exonic SNPs, together with a 36% increase in overall SNP density within this LD block, suggests an accumulation and maintenance of genetic variation within this LD block. This is consistent with a balanced, older inversion polymorphism, as time has allowed mutation to accumulate, and selection along with protection from recombination maintains the entire region as a unit, protecting minor deleterious mutations from removal.
While no significant enrichment for GO terms was found in the LD blocks within LG07 or LG12, we know that these genomic regions have been shown to diverge strongly between coastal stationary and offshore migratory cod in Norway [31, 32]. The selection acting on these regions may be acting on one or a few genes of high importance within each LD block, and individuals inherit the region in its entirety due to the repressed recombination of inversion polymorphisms. In our study, we find differentiation between winter spawners and Georges Bank cod in LG12, but no differentiation in LG7 between any of our population samples. This may be due to either our reduced sampling size, or sampling solely from stationary, coastal cod populations.
It is interesting to note that, while these putative supergenes are likely to have been initially protected from recombination by chromosomal inversion [32], the expected result of decreased genetic diversity within inversions is not seen in our data. In fact we see both a larger density of SNPs and a greater proportion of exonic SNPs within the linkage block on LG02. In addition, several unlinked low allele frequency SNPs exist in between the highly linked loci, suggesting that time has allowed mutation to occur within the linkage blocks, as recombination is an unlikely source for the unlinked loci. All of these factors suggest that, if these supergenes arose from chromosomal inversions, they are likely to be older, stable, and adaptive inversion polymorphisms [58, 59].
Caution must be taken in describing potential selection factors within linkage disequilibrium clusters, however, because it is unclear as to whether the entire linkage unit is under selection as a true supergene, or is merely the effect of recent strong selection on one or a few genes with genomic hitchhiking of nearby genes. In the three major LD blocks we described here, the block on LG02 was only one with statistically significant within-cluster enrichment of Gene Ontology (GO) categories, suggesting that the LD blocks on linkage groups 7 and 12 may not have functionally linked genes within them.
Evidence for population structure
Previous work has shown that genetic differentiation is present within the Gulf of Maine and Georges Bank populations of cod [34, 35]. Most markers in these comparisons showed no structure, but two loci in both data sets showed significant differentiation (the microsatellite ‘Gmo132’ and the PanI locus). When Kovach et al. [35] removed these loci from their analysis, no patterns of genetic differentiation were found. Likewise, in our data, both principal component and FST analyses show that divergence between the sampled populations is limited solely to the linkage blocks within LG 2, 7, and 12, and not throughout the genome. This suggests that some level of adaptive divergence may be responsible for the amount of differentiation historically seen in these genomic regions, though caution must be employed in interpretations of selection in this study due to a low per-population sampling size.
Conclusions
Strong neutral genetic differentiation between populations implies very low genetic or demographic exchange between them, and as a result, neutral genetic boundaries in fisheries species have long been used to suggest the existence of different stocks [60, 61]. The lack of strong neutral differentiation does not necessarily mean strong gene flow between fish stocks, as large effective population sizes can reduce the effect of differential genetic drift [62]. However, if natural selection generates differentiation, local adaptation and minimal gene flow is often the interpretation. This may not be true, however, as demographic exchange between two populations may remain high [22]. For example, a strong cline in allele frequency at a single locus in the mussel Mytilus edulis in southern Long Island Sound is generated by natural selection each summer [63]. However, this genetic divergence does not imply different stocks: the southern population under selection is replenished each spring by strong immigration from the north, and there are few demographic barriers between them. Likewise, allele frequencies of multiple genes in linkage groups LG02, LG07 and LG12 shift over spatial scales of 100 s of miles or temporal scales of decades in cod in Newfoundland [28, 29], implying strong selective changes perhaps due to fishing or environmental shifts. As in the mussel example, such genetic differences do not by themselves imply demographic separation of these populations into separate stocks. They also do not imply that selection is ongoing. The action of strong selection in the past can be visible in current populations if past perturbations of gene flow have not yet been erased by migration.
Previous studies of cod within this system [33] have determined that population structure exists between spawning stocks of cod within the Gulf of Maine. However, these studies relied on markers whose physical locations within the genome were unknown, and often only one or a small number of the markers tested showed genetic divergence [34, 35]. It may be that these markers, which have been interpreted as evidence of population structure, are located within these linkage disequilibrium blocks and are in fact related to their adaptive divergence.
Our data set was designed to provide high coverage of whole genome sequences for relatively small numbers of individuals. Our analysis shows that large regions of select linkage groups appear to be under adaptive divergence between spawning groups. Furthermore, this genetic divergence does not extend to loci on the same linkage map groups within the Gulf of Maine. Together, these results suggest that extensive study of neutral gene divergence within the Gulf of Maine could be accomplished by choosing markers strictly outside known regions of linked polymorphisms. Furthermore, patterns of nucleotide differentiation among linked gene regions might suggest the way natural selection has acted on these regions, and if it continues to shape population genetics of modern cod.
Additional files
List of genes found within LD block of LG02. GFF file containing the gene ID, positions, and gene names of the 294 genes found within this linkage disequilibrium block. (TXT 108 kb)
List of genes found within LD block of LG07. GFF file containing the gene ID, positions, and gene names of the 306 genes found within this linkage disequilibrium block. (TXT 116 kb)
List of genes found within LD block of LG12. GFF file containing the gene ID, positions, and gene names of the 437 genes found within this linkage disequilibrium block. (TXT 166 kb)
Acknowledgements
We would like to thank the following people for their contributions to this work: Bill Hoffman (Massachusetts Division of Marine Fisheries) for the collection of specimens, Joe Mellor for the isolation of DNA and coordinating sequencing, Cindy Lawley (Illumina) for connecting the researchers at Gloucester Marine Genomics Institute and Stanford University, and Nina Therkildsen (Cornell University) for advice and background in cod genetics research.
Funding
The Massachusetts Division of Marine Fisheries funded the collection and DNA sequencing of samples.
Availability of data and materials
The critical datasets generated and analyzed during the current study are available within the Open Science Framework, at the following URL: http://osf.io/epydk/.
Authors’ contributions
DW and CM coordinated the collection of samples and sequencing. BTB and SRP performed all analyses and BTB wrote the final manuscript, with editorial assistance from SRP, CM, and DW. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval
All samples were collected by Bill Hoffman from the Massachusetts Division of Marine Fisheries, in accordance with their internal collection permissions and protocols. Samples consisted of 1 cm2 fin clips, an industry-standard non-lethal form of sampling fish for genetic analysis, and no ethics board approval was sought by the Massachusetts Division of Marine Fisheries.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- cM
Centimorgans
- EST
Expressed sequence tag
- GB
Georges Bank
- GO
Gene ontology
- GoM
Gulf of Maine
- LD
Linkage disequilibriuml LG, linkage map group
- PCA
Principal component analysis
- PCR
Polymerase chain reaction
- SNP
Single-nucleotide polymorphism
Contributor Information
Bryan T. Barney, Email: btbarney@stanford.edu
Christiane Munkholm, Email: chris.munkholm@gmgi.org.
David R. Walt, Email: David.Walt@tufts.edu
Stephen R. Palumbi, Email: spalumbi@stanford.edu
References
- 1.Turner TL, Hahn MW, Nuzhdin SV. Genomic islands of speciation in Anopheles gambiae. PLoS Biol. 2005;3:e285. doi: 10.1371/journal.pbio.0030285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nosil P, Feder JL. Genomic divergence during speciation: causes and consequences. Philos Trans R Soc B Biol Sci. 2012;367:332–42. doi: 10.1098/rstb.2011.0263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barton NH. Genetic hitchhiking. Philos Trans R Soc Lond B Biol Sci. 2000;355:1553–62. doi: 10.1098/rstb.2000.0716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nosil P, Funk DJ, Ortiz-Barrientos D. Divergent selection and heterogeneous genomic divergence. Mol Ecol. 2009;18:375–402. doi: 10.1111/j.1365-294X.2008.03946.x. [DOI] [PubMed] [Google Scholar]
- 5.Malinsky M, Challis RJ, Tyers AM, Schiffels S, Terai Y, Ngatunga BP, et al. Genomic islands of speciation separate cichlid ecomorphs in an East African crater lake. Science (80-) 2015;350:1493–8. doi: 10.1126/science.aac9927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Feder JL, Nosil P. The efficacy of divergence hitchhiking in generating genomic islands during ecological speciation. Evolution (N Y) 2010;64:1729–47. doi: 10.1111/j.1558-5646.2010.00943.x. [DOI] [PubMed] [Google Scholar]
- 7.Cruickshank TE, Hahn MW. Reanalysis suggests that genomic islands of speciation are due to reduced diversity, not reduced gene flow. Mol Ecol. 2014;23:3133–57. doi: 10.1111/mec.12796. [DOI] [PubMed] [Google Scholar]
- 8.Feder JL, Egan SP, Nosil P. The genomics of speciation-with-gene-flow. Trends Genet. 2012;28:342–50. doi: 10.1016/j.tig.2012.03.009. [DOI] [PubMed] [Google Scholar]
- 9.Flaxman SM, Feder JL, Nosil P. Genetic hitchhiking and the dynamic buildup of genomic divergence during speciation with gene flow. Evolution (N Y) 2013;67:2577–91. doi: 10.1111/evo.12055. [DOI] [PubMed] [Google Scholar]
- 10.Bird CE, Fernandez-Silva I, Skillings DJ, Toonen RJ. Sympatric speciation in the post “Modern Synthesis” era of evolutionary biology. Evol Biol. 2012;39:158–80. doi: 10.1007/s11692-012-9183-6. [DOI] [Google Scholar]
- 11.Butlin RK. Population genomics and speciation. Genetica. 2010;138:409–18. doi: 10.1007/s10709-008-9321-3. [DOI] [PubMed] [Google Scholar]
- 12.Marques DA, Lucek K, Meier JI, Mwaiko S, Wagner CE, Excoffier L, et al. Genomics of rapid incipient speciation in sympatric threespine stickleback. PLoS Genet. 2016;12:e1005887. doi: 10.1371/journal.pgen.1005887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Noor M a F, Bennett SM. Islands of speciation or mirages in the desert? Examining the role of restricted recombination in maintaining species. Heredity (Edinb) 2009;103:439–44. doi: 10.1038/hdy.2009.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ashburner M, Lemeunier F. Relationships within the melanogaster species subgroup of the Genus Drosophila (Sophophora). I.Inversion polymorphisms in drosophila melanogaster and drosophila simulans. Proc R Soc London B Biol Sci. 1976;193:137–57. doi: 10.1098/rspb.1976.0036. [DOI] [PubMed] [Google Scholar]
- 15.Knibb WR. Chromosome inversion polymorphisms in Drosophila melanogaster II. Geographic clines and climatic associations in Australasia, North America and Asia. Genetica. 1982;58:213–21. doi: 10.1007/BF00128015. [DOI] [Google Scholar]
- 16.Kennington WJ, Partridge L, Hoffmann AA. Patterns of diversity and linkage disequilibrium within the cosmopolitan inversion In(3R)Payne in Drosophila melanogaster are indicative of coadaptation. Genetics. 2006;172:1655–63. doi: 10.1534/genetics.105.053173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Umina PA, Weeks AR, Kearney MR, McKechnie SW, Hoffmann AA. A rapid shift in a classic clinal pattern in Drosophila reflecting climate change. Science (80-) 2005;308:691–3. doi: 10.1126/science.1109523. [DOI] [PubMed] [Google Scholar]
- 18.Balanya J, Sole E, Oller JM, Sperlich D, Serra L. Long-term changes in the chromosomal inversion polymorphism of Drosophila subobscura. II European populations. J Zool Syst Evol Res. 2004;42:191–201. doi: 10.1111/j.1439-0469.2004.00274.x. [DOI] [PubMed] [Google Scholar]
- 19.Lee YW, Fishman L, Kelly JK, Willis JH. A segregating inversion generates fitness variation in Yellow Monkeyflower (Mimulus guttatus) Genetics. 2016;202:1473–84. doi: 10.1534/genetics.115.183566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schwander T, Libbrecht R, Keller L. Supergenes and complex phenotypes. Curr Biol. 2014;24:R288–94. doi: 10.1016/j.cub.2014.01.056. [DOI] [PubMed] [Google Scholar]
- 21.Bay RA, Rose NH, Barrett RDH, Bernatchez L, Ghalambor CK, Lasky JR, et al. Predicting responses to contemporary environmental change using evolutionary response architectures. Am Nat. 2017;189. do:10.1086/691233. [DOI] [PubMed]
- 22.Tigano A, Friesen VL. Genomics of local adaptation with gene flow. Mol Ecol. 2016;25:2144–64. doi: 10.1111/mec.13606. [DOI] [PubMed] [Google Scholar]
- 23.Hubert S, Higgins B, Borza T, Bowman S. Development of a SNP resource and a genetic linkage map for Atlantic cod (Gadus morhua) BMC Genomics. 2010;11:191. doi: 10.1186/1471-2164-11-191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Borza T, Higgins B, Simpson G, Bowman S. Integrating the markers Pan I and haemoglobin with the genetic linkage map of Atlantic cod (Gadus morhua) BMC Res Notes. 2010;3:261–7. doi: 10.1186/1756-0500-3-261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bradbury IR, Hubert S, Higgins B, Bowman S, Borza T, Paterson IG, et al. Genomic islands of divergence and their consequences for the resolution of spatial structure in an exploited marine fish. Evol Appl. 2013;6:450–61. doi: 10.1111/eva.12026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bradbury IR, Hubert S, Higgins B, Borza T, Bowman S, Paterson IG, et al. Parallel adaptive evolution of Atlantic cod on both sides of the Atlantic Ocean in response to temperature. Proc Biol Sci. 2010;277:3725–34. doi: 10.1098/rspb.2010.0985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hemmer-Hansen J, Nielsen EE, Therkildsen NO, Taylor MI, Ogden R, Geffen AJ, et al. A genomic island linked to ecotype divergence in Atlantic cod. Mol Ecol. 2013;22:2653–67. doi: 10.1111/mec.12284. [DOI] [PubMed] [Google Scholar]
- 28.Therkildsen NO, Hemmer-Hansen J, Als TD, Swain DP, Morgan MJ, Trippel E a, et al. Microevolution in time and space: SNP analysis of historical DNA reveals dynamic signatures of selection in Atlantic cod. Mol Ecol. 2013;22:2424–40. doi: 10.1111/mec.12260. [DOI] [PubMed] [Google Scholar]
- 29.Therkildsen NO, Hemmer-Hansen J, Hedeholm RB, Wisz MS, Pampoulie C, Meldrup D, et al. Spatiotemporal SNP analysis reveals pronounced biocomplexity at the northern range margin of Atlantic cod Gadus morhua. Evol Appl. 2013;6:690–705. doi: 10.1111/eva.12055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tørresen OK, Star B, Jentoft S, Reinar WB, Grove H, Miller JR, et al. An improved genome assembly uncovers prolific tandem repeats in Atlantic cod. BMC Genomics. 2017;18:95. doi: 10.1186/s12864-016-3448-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sodeland M, Jorde PE, Lien S, Jentoft S, Berg PR, Grove H, et al. “Islands of divergence” in the Atlantic Cod genome represent polymorphic chromosomal rearrangements. Genome Biol Evol. 2016;8:1012–22. doi: 10.1093/gbe/evw057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berg PR, Star B, Pampoulie C, Sodeland M, Barth JMI. Three chromosomal rearrangements promote genomic divergence between migratory and stationary ecotypes of Atlantic cod. Sci Rep. 2016;6:23246. doi: 10.1038/srep23246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zemeckis DR, Martins D, Kerr LA, Cadrin SX. Stock identification of Atlantic cod (Gadus morhua) in US waters: an interdisciplinary approach. ICES J Mar Sci. 2014;71:1490–506. doi: 10.1093/icesjms/fsu032. [DOI] [Google Scholar]
- 34.Wirgin I, Kovach AI, Maceda L, Roy NK, Waldman J, Berlinsky DL. Stock identification of Atlantic cod in U.S. waters using microsatellite and single nucleotide polymorphism DNA analyses. Trans Am Fish Soc. 2007;136:375–91. doi: 10.1577/T06-068.1. [DOI] [Google Scholar]
- 35.Kovach AI, Breton TS, Berlinsky DL, Maceda L, Wirgin I. Fine-scale spatial and temporal genetic structure of Atlantic cod off the Atlantic coast of the USA. Mar Ecol Prog Ser. 2010;410:177–95. doi: 10.3354/meps08612. [DOI] [Google Scholar]
- 36.Therkildsen NO, Palumbi SR. Practical low-coverage genome-wide sequencing of hundreds of individually barcoded samples for population and evolutionary genomics in non-model species. Mol Ecol Resour. 2017;17:194–208. doi: 10.1111/1755-0998.12593. [DOI] [PubMed] [Google Scholar]
- 37.Bolger AM, Lohse M, Usadel B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:1–7. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Garrison E, Marth G. Haplotype-based variant detection from short-read sequencing. arXiv Prepr arXiv1207.3907. 2012;9:1.
- 40.Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–8. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alexa A, Rahnenfuhrer J. topGO: Enrichment Analysis for Gene Ontology. R package version 2.24.0.
- 42.Goudet J. FSTAT, a program to estimate and test gene diversities and fixation indices (version 2.9.3). 2001.
- 43.Weir BS, Cockerham CC. Estimating F-statistics for the analysis of population structure. Evolution (N Y) 1984;38:1358–70. doi: 10.1111/j.1558-5646.1984.tb05657.x. [DOI] [PubMed] [Google Scholar]
- 44.Skotte L, Korneliussen TS, Albrechtsen A. Estimating individual admixture proportions from next generation sequencing data. Genetics. 2013;195:693–702. doi: 10.1534/genetics.113.154138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol Ecol. 2005;14:2611–20. doi: 10.1111/j.1365-294X.2005.02553.x. [DOI] [PubMed] [Google Scholar]
- 46.Berg PR, Jentoft S, Star B, Ring KH, Knutsen H, Lien S, et al. Adaptation to low salinity promotes genomic divergence in Atlantic cod (Gadus morhua L.) Genome Biol Evol. 2015;7:1644–63. doi: 10.1093/gbe/evv093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sanford E, Kelly MW. Local adaptation in marine invertebrates. Annu Rev Mar Sci. 2011;3:509–35. doi: 10.1146/annurev-marine-120709-142756. [DOI] [PubMed] [Google Scholar]
- 48.Whiteley NM, Rastrick SPS, Lunt DH, Rock J. Latitudinal variations in the physiology of marine gammarid amphipods. J Exp Mar Bio Ecol. 2011;400:70–7. doi: 10.1016/j.jembe.2011.02.027. [DOI] [Google Scholar]
- 49.Pespeni MH, Garfield DA, Manier MK, Palumbi SR. Genome-wide polymorphisms show unexpected targets of natural selection. Proc R Soc B Biol Sci. 2011;279:1412–20. doi: 10.1098/rspb.2011.1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pespeni MH, Barney BT, Palumbi SR. Differences in the regulation of growth and biomineralization genes revealed through long-term common-garden acclimation and experimental genomics in the Purple Sea Urchin. Evolution (N Y) 2013;67:1901–14. doi: 10.1111/evo.12036. [DOI] [PubMed] [Google Scholar]
- 51.De Wit P, Palumbi SR. Transcriptome-wide polymorphisms of red abalone (Haliotis rufescens) reveal patterns of gene flow and local adaptation. Mol Ecol. 2012;22:2884–97. doi: 10.1111/mec.12081. [DOI] [PubMed] [Google Scholar]
- 52.Bay RA, Palumbi SR. Multilocus adaptation associated with heat resistance in reef-building corals. Curr Biol. 2014;24:2952–6. doi: 10.1016/j.cub.2014.10.044. [DOI] [PubMed] [Google Scholar]
- 53.Richardson JL, Urban MC. Strong selection barriers explain microgeographic adaptation in wild salamander populations. Evolution (N Y) 2013;67:1729–40. doi: 10.1111/evo.12052. [DOI] [PubMed] [Google Scholar]
- 54.True ED, Wiitala SA. Annual temperature curves in twelve regions of the Gulf of Maine. Northwest Atl Fish Organ Sci Counc Stud. 1977;14:21–7. [Google Scholar]
- 55.Hutchings JA, Myers RA. Timing of cod reproduction: interannual variability and the influence of temperature. Mar Ecol Prog Ser. 1994;108:21–31. doi: 10.3354/meps108021. [DOI] [Google Scholar]
- 56.Hochachka PW, Somero GN. Biochemical Adaptation: Mechanism and Process in Physiological Evolution. New York: Oxford University Press; 2002. [Google Scholar]
- 57.Long Y, Li L, Li Q, He X, Cui Z. Transcriptomic characterization of temperature stress responses in larval zebrafish. PLoS One. 2012;7:e37209. [DOI] [PMC free article] [PubMed]
- 58.McGaugh SE, Noor MAF. Genomic impacts of chromosomal inversions in parapatric Drosophila species. Philos Trans R Soc Lond B Biol Sci. 2012;367:422–9. doi: 10.1098/rstb.2011.0250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Guerrero RF, Rousset F, Kirkpatrick M. Coalescent patterns for chromosomal inversions in divergent populations. Philos Trans R Soc Lond B Biol Sci. 2012;367:430–8. doi: 10.1098/rstb.2011.0246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Grant WS, Milner GB, Krasnowski P, Utter FM. Use of biochemical genetic variants for identification of Sockeye salmon (Oncorhynchus nerka) stocks in Cook Inlet, Alaska. Can J Fish Aquat Sci. 1980;37:1236–47. doi: 10.1139/f80-159. [DOI] [Google Scholar]
- 61.Waples RS, Winans GA, Utter FM, Mahnken C. Genetic approaches to the management of Pacific salmon. Fisheries. 1990;15:19–25. doi: 10.1577/1548-8446(1990)015<0019:GATTMO>2.0.CO;2. [DOI] [Google Scholar]
- 62.Hauser L, Carvalho GR. Paradigm shifts in marine fisheries genetics: Ugly hypotheses slain by beautiful facts. Fish Fish. 2008;9:333–62. doi: 10.1111/j.1467-2979.2008.00299.x. [DOI] [Google Scholar]
- 63.Koehn RK, Newell RIE, Immermann F. Maintenance of an aminopeptidase allele frequency cline by natural selection. Proc Natl Acad Sci U S A. 1980;77:5385–9. doi: 10.1073/pnas.77.9.5385. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of genes found within LD block of LG02. GFF file containing the gene ID, positions, and gene names of the 294 genes found within this linkage disequilibrium block. (TXT 108 kb)
List of genes found within LD block of LG07. GFF file containing the gene ID, positions, and gene names of the 306 genes found within this linkage disequilibrium block. (TXT 116 kb)
List of genes found within LD block of LG12. GFF file containing the gene ID, positions, and gene names of the 437 genes found within this linkage disequilibrium block. (TXT 166 kb)
Data Availability Statement
The critical datasets generated and analyzed during the current study are available within the Open Science Framework, at the following URL: http://osf.io/epydk/.