

Abstract
Background
Astragaloside IV (AS-IV) has been shown to prevent ischemia-induced acute kidney injury (AKI) in rat models of ischemia and reperfusion. However, the effects of AS-IV on AKI during sepsis and endotoxinemia is unclear. The current study aimed to investigate the effects and molecular mechanisms of AS-IV on lipopolysaccharide (LPS)-induced AKI.
Material/Methods
Adult male CD-1 mice were randomly assigned into 6 groups (n=8/group): control group: mice were intraperitoneally (i.p.) injected with normal saline; LPS group (10 mg/kg, i.p.); low-dose AS-IV (25 mg/kg; gavage for 7 days) + LPS (i.p., 1 hour after last gavage) group; medial-dose AS-IV (50 mg/kg) + LPS group; high-dose AS-IV (100 mg/kg) + LPS group; high-dose AS-IV alone (100 mg/kg; gavage for 7 days) group. Blood samples were collected at 24 hours after LPS injection, and plasma uric acid and BUN were measured with colorimetric detection kits. The concentration of plasma tumor necrosis factor (TNF)-α and interleukin 1β, renal p-extracellular signal-regulated kinases, and urinary albumin were evaluated by ELISA. The expression of CCR5 in renal tissue was evaluated by PCR and Western blotting. Concentrations of glutathione (GSH) and reactive oxygen species (ROS) in renal tissue were also measured.
Results
AS-IV decreased LPS-stimulated production of blood TNF-α and IL-6, LPS-induced the expression of CCR5, and activation of ERK in the kidneys in a rodent model of endotoxinemia. AS-IV attenuated LPS-caused decreased GSH and increased ROS. It also attenuated LPS-induced increases in plasma uric acid, BUN, and urinary albumin.
Conclusions
AS-IV protects against AKI during bacterial endotoxinemia by attenuating expression of cytokines, CCR5, and p-ERK, and elevating anti-oxidative ability.
MeSH Keywords: Acute Kidney Injury, Inflammation, Oxidative Stress
Background
Sepsis is a systemic inflammatory response induced by infection that can result in organ dysfunction and mortality. It causes millions of deaths per year worldwide, and is the most common cause of death in hospitalized patients [1,2]. Bacteria accounts for 90% of sepsis, and Gram-negative bacteria endotoxin lipopolysaccharide (LPS) plays an important role in the pathogenesis of septic shock and endotoxinemia. Acute kidney injury (AKI) is one of the most frequent complications of sepsis and endotoxinemia. It increases the cost and complexity of care, and is associated with a very high mortality rate [3].
Developing new therapies for sepsis, endotoxinemia, and sepsis-induced AKI is particularly challenging; there have been more than 25 failed clinical drug trials for sepsis therapy [4]. Candidate therapies for sepsis and endotoxinemia include hemoperfusion with polymyxin B coated fiber devices, and modulation of the immune system with treatments such as mesenchymal stem cells, histone modification, and granulocyte-macrophage colony stimulating factor (GM-CSF) [5–8]. In addition, several pharmacologic investigations aimed at limiting or reversing sepsis-induced AKI. Anti-tumor necrosis factor (anti-TNF) therapy has displayed promising results in animal models [9–11]. However, a number of large clinical trials with neutralizing anti-TNF antibodies and soluble TNF receptor fusion proteins failed to show survival benefits in septic patients [11]. Similarly, inhibition of platelet-activating factor, endothelin, anti-thrombin, tissue factor pathway, leukocyte adhesion, and administration of natriuretic peptides and growth factors are all novel therapeutic methods that either failed to make it to clinical trials or did not produce the desired effects in large clinical trials [12]. Therefore, medical research is greatly needed to generate new therapeutic approaches to treatment of sepsis and endotoxinemia-induced AKI.
Astragaloside IV (AS-IV) was reported to have a protective effect on cardiovascular, immune, digestive, and nervous systems due to its regulation of calcium balance, and its anti-oxidant and anti-apoptotic properties [13]. In rat models of ischemia and reperfusion, AS-IV prevented ischemia-induced AKI through inhibiting nuclear factor-kappa B (NF-κB)-mediated expression of inflammatory genes and upregulating p53-upregulated modulator of apoptosis (PUMA) [14,15]. Studies also found that AS-IV protected against ischemia-induced AKI by inhibiting oxidative stress and apoptosis pathways [16]. However, it is unclear what the effects are of AS-IV on sepsis and endotoxinemia-induced acute renal injury.
Studies have highlighted possible mechanisms underlying sepsis and endotoxinemia-induced AKI, including inflammation, renal ischemia-reperfusion injury and oxidative damage, and systemic hypotension [17–19]. Therefore, inflammatory and oxidative pathways could be potential drug targets to treat sepsis and endotoxinemia-induced AKI. Inflammatory cytokines are important mediators that impair cellular and organ function during inflammatory responses; and C-C chemokine receptor type 5 (CCR5) plays an essential role in inflammatory responses to infection. CCR5 is a protein on the surface of T cells, macrophages, dendritic cells, eosinophils, and microglia. It acts as a receptor for chemokines, and is involved in the process by which T cells are attracted to specific tissue and organs [20]. Inflammatory responses is also associated with the activation of extracellular signal-regulated kinase (ERK) [21]. Moreover, Glutathione (GSH) is an important antioxidant in plants, animals, fungi, and some bacteria. GSH prevents damage to important cellular components caused by reactive oxygen species (ROS) including free radicals, peroxides and heavy metals [22]. The current study aimed to investigate the effects of AS-IV on LPS-induced AKI, and explore the molecular mechanisms in inflammatory and oxidative pathways.
Material and Methods
Reagents
Reagents included: Escherichia coli LPS (serotype O127: B8; Sigma Chemical Co., St. Louis, MO, USA); Astragaloside IV (Sigma Chemical Co., St. Louis, MO, USA); anti- C-C chemokine receptor type 5 (CCR5) antibody (Abcam Inc., Cambridge, MA, USA); anti-p-extracellular signal-regulated kinases (p-ERK; Cell Signaling Technology Inc., Danvers, MA, USA); uric acid testing kit (Sigma Chemical Co., St. Louis, MO, USA); blood urea nitrogen (BUN; Sigma Chemical Co., St. Louis, MO, USA); urinary albumin test kit (Bethyl Laboratories Inc., Montgomery, TX, USA); tumor necrosis factor-alpha (TNF-α) enzyme-linked immunosorbent assay (ELISA) kit (Bethyl Laboratories Inc., Montgomery, TX, USA); interleukin (IL)-1β ELISA kit (Bethyl Laboratories Inc., Montgomery, TX, USA); p-ERK ELISA kit (ThermoFisher Scientific Inc., Waltham, MA, USA); GSH kit (Beyotime Inc., Shanghai, China); ROS kit (Beyotime Inc., Shanghai, China); RNALater (Ambion, Austin, TX, USA); TRIzol Reagent (Invitrogen, Carlsbad, CA, USA); First Strand cDNA Synthesis Kit (Fermentas, Hanover, MD, USA).
Experimental design
Adult male CD-1 mice (8 week-old, 28–32 g) were purchased from Animal Center of Chinese Academy of Sciences. Mice were randomly divided into 6 groups (n=8/group): (1) control group: mice were intraperitoneally (i.p) injected with normal saline; (2) LPS group: mice were injected with LPS (10 mg/kg, i.p); (3) low-dose AS-IV (LA) + LPS group: mice were pretreated with 25 mg/kg of AS-IV by gavage for 7 days; (4) medial-dose AS-IV (MA) + LPS group: mice were pretreated with 50 mg/kg of AS-IV by gavage for 7 days; (5) high-dose AS-IV (HA) + LPS group: mice were pretreated with 100 mg/kg of AS-IV by gavage for 7 days. LPS was injected (i.p.) 1 hour after last gavage; (6) high-dose AS-IV alone (HA) group: mice were pretreated with 100 mg/kg of AS-IV by gavage for 7 days. All mice were euthanized with carbon dioxide and cervical dislocation. Mice were sacrificed at 24 hour after LPS injection for LPS-subjected animals, and at day 7 for control group and high-dose AS-IV only group. Blood samples were drawn by cardiac puncture. Kidneys were harvested and kept at −80°C for subsequent experiments.
ELISA
Blood samples were collected in each group, and renal homogenates were prepared. The concentration of plasma TNF-α and IL-1β, renal p-extracellular signal-regulated kinases (p-ERK), and urinary albumin were evaluated by ELISA according to the ELISA kit instructions. Signals were then detected by microplate reader.
Western blot
Protein expression of CCR5 in renal tissue was detected by Western blot. Total proteins were extracted. Proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) at 120 V, and transferred to polyvinylidene fluoride (PVDF) membranes at 100 V for 120 minutes. Membranes were blocked with 5% non-fat milk powder for 1 hour at room temperature, and then incubated with primary antibodies for CCR5 (1: 1,000) at 4°C overnight. Then membranes were incubated with goat anti-rabbit secondary antibody labeled with HRP at room temperature for 1 hour. Membranes were incubated with electro-chemiluminescence (ECL) solution, and films were exposed in a dark room.
Reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA was extracted from kidney tissue stored in RNAlater with TRIzol reagent following manufacturer’s instructions. First strand cDNA for PCR amplification was synthesized from 3 μg of total RNA by First Strand cDNA Synthesis Kit. Each reaction contained 1 μL of random hexamer primers (0.2 μg/μL) and 40 U M-MuLV reverse transcriptase (20 U/μL). No reverse transcriptase was added in the negative control. The specific primers for detection of CCR-5 genes were designed. PCR conditions for CCR-5 were as follows: denaturing at 94°C for 1 minute; annealing at 55°C for 45 seconds; and polymerization at 72°C for 1 minute. PCR was performed in a GeneAmp PCR System 9700 thermocycler, and the optimal cycle numbers were 25 for CCR-5 and 22 for β-actin. PCR products were separated on 2% agarose gel at 150 V for 40 minutes. The intensity of bands were quantified by GEL DOC1000/2000 Quantity One program software (Bio-Rad Laboratories, Hercules, CA, USA). Gene expression was determined as the ratio of relative optical density of target genes to β-actin. The primers used were: CCR5-forward, 5′-CAAAAAGAAGGTCTTCATTACACC-3′; CCR5-reverse, 5′-CCTGTGCCTCTTCTTCTCATTTCG-3′; actin-forward, 5′-GCA CCA CAC CTT CTA CAA TG-3′ and actin-reverse, 5′-TGC TTG CTG ATC CAC ATC TG-3′.
Biochemical analysis
Blood samples were collected at 24 hours after LPS injection. Plasma uric acid and BUN were measured with colorimetric detection kits. The level of urinary albumin was measured using ELISA. For preparation of renal homogenates, 100 mg of kidney tissue was homogenized on ice in 1 mL of homogenization buffer (50 mM Tris–HCl, 180 mM KCl, 10 mM EDTA, pH 7.4). Renal GSH contents and ROS were measured based upon the standard kit instructions.
Statistical analysis
Results were presented as mean ±SEM. One way analysis of variance (ANOVA) was used to compare differences among three or more groups, followed by Bonferroni post hoc testing for multiple comparisons. Student’s t-test was used to compare differences between two groups; p values of 0.05 or less were regarded as significant. Statistical analysis and figures and were made using GraphPad Prism 5.0 software (GraphPad Software Inc., La Jolla, CA, USA).
Results
Astragaloside IV decreased LPS-stimulated production of inflammatory cytokines in a murine model of endotoxinemia
Animals were pretreated with 3 doses of AS-IV by gavage for 7 days, and LPS was injected intraperitoneally 1 hour after last gavage. Mice were sacrificed at 24 hours after LPS injection for LPS-subjected animals, and at day 7 for the control group and AS-IV only group. Blood samples were collected for each group, and concentrations of TNF-α and IL-1β in the plasma were detected by ELISA. LPS induced production of TNF-α and IL-1β in the plasma was comparable to the control group, whereas AS-IV attenuated the increase in a dose-dependent manner (TNF-α: p<0.0001; IL-1β: p<0.0001; Figure 1). Medial and high dose-AS-IV markedly decreased plasma TNF-α and IL-1β levels in LPS-subjected animals (p<0.05; Figure 1). AS-IV alone did not alter TNF-α and IL-1β when compared to the control group.
Figure 1.

Astragaloside IV decreased LPS-stimulated production of inflammatory cytokines in a murine model of endotoxinemia. (A) Concentration of plasma TNF-α; (B) Concentration of plasma IL-1β. Concentrations of TNF-α and IL-1β in the plasma were detected by ELISA. LPS induced production of TNF-α and IL-1β in the plasma compared to the control group, whereas AS-IV attenuated the increase in a dose-dependent manner (mean ±SEM, n=8/group). * Indicates that p<0.05 when compared to the LPS group. LPS – lipopolysaccharide; LA – low-dose AS-IV; MA – medial-dose AS-IV; HA – high-dose AS-IV; TNF – tumor necrosis factor; IL – interleukin.
Astragaloside IV decreased LPS-induced expression of CCR5 in renal tissue during endotoxinemia
The protein expression of CCR5 in renal tissue was detected by Western blotting, and mRNA of CCR5 was detected by RT-PCR. LPS increased protein and mRNA levels of CCR5, while AS-IV decreased levels of CCR5 as its concentration increased (protein: p=0.0028; mRNA: p=0.0007; Figure 2). High-dose AS-IV attenuated the increase of protein and mRNA of CCR5 significantly in LPS-subjected animals (p<0.05; Figure 2). Compared to the control group, AS-IV did not alter the protein and mRNA levels of CCR5 in the absence of LPS.
Figure 2.

Astragaloside IV decreased LPS-induced expression of CCR5 in renal tissue during endotoxinemia. (A) Representative Western Blot image of CCR5, and the relative ratio of protein expression in each group compared to the control group; (B) The relative ratio of mRNA levels in each group compared to the control group. The protein expression of CCR5 was detected by Western Blot, and mRNA of CCR5 was detected by RT-PCR. LPS increased protein and mRNA levels of CCR5. High-dose AS-IV attenuated the increase of protein and mRNA of CCR5 in LPS-subjected animals (mean ±SEM, n=8/group). * Indicates that p<0.05 when compared to the LPS group. LPS – lipopolysaccharide; LA – low-dose AS-IV; MA – medial-dose AS-IV; HA – high-dose AS-IV; CCR5 – C-C chemokine receptor type 5.
Astragaloside IV decreased LPS-stimulated activation of ERK in renal tissue during endotoxinemia
The concentration of p-ERK in renal tissue was detected by ELISA. Levels of p-ERK increased in LPS-treated animals. AS-IV decreased the protein expression of p-ERK as its dose increased in LPS-subjected animals (p=0.0002; Figure 3). High-dose AS-IV markedly decreased the concentration of p-ERK in LPS-subjected animals compared to the LPS group (p<0.05; Figure 3), whereas it did not alter p-ERK levels in animals without LPS insult.
Figure 3.
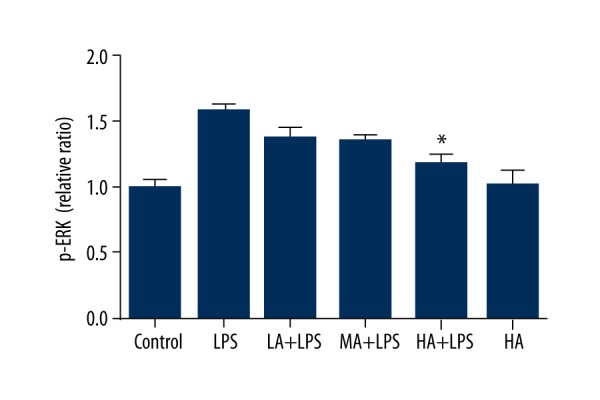
Astragaloside IV decreased LPS-stimulated activation of ERK in renal tissue during endotoxinemia. The concentration of p-ERK in renal tissue was detected by ELISA. Levels of p-ERK increased in LPS-treated animals. AS-IV decreased the concentration of p-ERK as the dose increased in LPS-subjected animals. High-dose AS-IV markedly decreased the concentration of p-ERK in LPS-subjected animals compared to the LPS group (mean ±SEM, n=8/group). * Indicates that p<0.05 when compared to the LPS group. LPS – lipopolysaccharide; LA – low-dose AS-IV; MA – medial-dose AS-IV; HA – high-dose AS-IV; ERK – extracellular signal-regulated kinases.
Astragaloside IV attenuated LPS-induced decrease in GSH and increase in ROS in renal tissue during endotoxinemia
GSH and ROS levels were measured in renal homogenates. LPS-subjected animals displayed decreased GSH levels and increased ROS levels. AS-IV attenuated the changes in a dose-dependent manner (GSH: p=0.0008; ROS: p<0.0001; Figure 4). High-dose AS-IV increased GSH levels and decreased ROS levels markedly in LPS-subjected animals compared to the LPS group (p<0.05; Figure 4).
Figure 4.

Astragaloside IV attenuated LPS-induced decrease in GSH and increase in ROS in renal tissue during endotoxinemia. (A) Concentration of GSH in renal tissue; (B) Concentration of ROS in renal tissue. Concentration of GSH and ROS was measured in renal homogenates. LPS-subjected animals displayed decreased GSH levels and increased ROS levels. AS-IV attenuated the changes in a dose-dependent manner. High-dose AS-IV increased GSH levels and decreased ROS levels markedly compared to the LPS group (mean ±SEM, n=8/group). * Indicates that p<0.05 when compared to the LPS group. LPS – lipopolysaccharide; LA – low-dose AS-IV; MA – medial-dose AS-IV; HA – high-dose AS-IV; GSH – glutathione; ROS – reactive oxygen species.
Astragaloside IV attenuated LPS-induced increase in plasma uric acid, BUN, and urinary albumin
Blood samples were collected, and plasma uric acid and BUN were measured with colorimetric detection kits. The concentration of urinary albumin was measured by ELISA. LPS increased plasma uric acid, BUN, and urinary albumin, whereas AS-IV attenuated the increase in a dose-dependent manner (plasma uric acid: p<0.0001; BUN: p<0.0001; urinary albumin: p<0.0001; Figure 5). AS-IV decreased plasma uric acid and BUN at medial and high doses, and decreased urinary albumin at low, medial and high doses significantly in LPS-subjected animals (p<0.05; Figure 5). AS-IV alone did not alter uric acid, BUN, and urinary albumin significantly compared to the control group.
Figure 5.

Astragaloside IV attenuated LPS-induced increase in plasma uric acid, BUN, and urinary albumin. (A) Concentration of plasma uric acid; (B) Concentration of blood urea nitrogen; (C) Concentration of urinary albumin. Blood samples were collected, and plasma uric acid and BUN were measured with colorimetric detection kits. The concentration of urinary albumin was measured by ELISA. LPS increased plasma uric acid, BUN, and urinary albumin, whereas AS-IV attenuated the increase in a dose-dependent manner (mean ±SEM, n=8/group). * Indicates that p<0.05 when compared to the LPS group. LPS – lipopolysaccharide; LA – low-dose AS-IV; MA – medial-dose AS-IV; HA – high-dose AS-IV.
Discussion
We have demonstrated that AS-IV decreased LPS-induced production of inflammatory cytokines, expression of CCR5, and activation of ERK in renal tissue in a murine model of endotoxinemia. In addition, AS-IV attenuated LPS-stimulated decrease in GSH and increase in ROS in renal tissue, and attenuated LPS-induced increase in plasma uric acid, BUN and urinary albumin.
AS-IV is the chief ingredient of Radix Astragali, which has been used in traditional Chinese medicine for tissue and organ repair and regeneration. AS-IV has been shown to inhibit allergic inflammation by regulating Th1/Th2 cytokine and enhancing CD4+ CD25+ Foxp3 T cells in ovalbumin-induced asthma. The rise of IL-4 and the decrease of IL-10 and interferon gamma (IFN-γ) were reduced after AS-IV treatment in an asthma model [23]. The protective effects of AS-IV against ovalbumin-induced lung inflammation were shown to be regulated by T-cell-specific transcription factor T-bet and GATA-3. AS-IV modulated the key switches GATA-3 and T-bet, committing helper T cells to a Th1 phenotype [24]. In addition, AS-IV suppressed joint inflammation, and inhibited IL-1β, TNF-α, and nitric oxide (NO) production in macrophages in a rat model of adjuvant-induced arthritis [25]. AS-IV also decreased the swelling induced by intraarticular injection of IL-1β, and protected against IL-1β-induced damage of cartilage proteoglycan and chondrocyte proliferation [25]. We demonstrated in the current study that AS-IV reduced LPS-increased inflammatory cytokines in a murine model of endotoxinemia, providing more evidence for the anti-inflammatory properties of AS-IV.
CCR5 is a protein on the surface of white blood cells. It is involved in the immune system as it acts as a receptor for chemokines. This regulates the activation and chemotaxis of leukocytes. Variations in CCR5 are associated with susceptibility to kidney disease in patients with type 2 diabetes. CCR5 AA genotype is over-represented in subjects with kidney disease due to type 2 diabetes [26]. Research has also shown that blockade of CCR5 improved renal function after ischemia-reperfusion injury of the kidney [27]. In our current study, we demonstrated that AS-IV decreased LPS-induced expression of CCR5 in renal tissue during endotoxinemia. The anti-inflammatory properties of AS-IV may reduce the activation and mobilization of white blood cells in tissue and organs. This may protect the kidney from damage caused by inflammatory cytokine release and enzymatic reactions.
ERK is part of the mammalian mitogen-activated protein kinase (MAPK) family. It plays a crucial role as a transducer of extracellular stimuli in intracellular phosphorylation cascades, which ultimately results in cell differentiation, proliferation, survival, and death [28,29]. ERK is activated by inflammatory cytokines and extracellular stressors including UV light, heat, and glutamate [13,30]. ERK signaling pathway has been implicated in the protective effects of AS-IV in different models. AS-IV exerted potent inhibitory effects on receptor activator for nuclear factor-κB ligand (RANKL)-induced osteoclastogenesis by suppressing ERK signaling pathway in vitro [31]. AS-IV also prevented glutamate-induced apoptosis in PC12 neuroblastic cells by blocking the phosphorylation of ERK [32]. In our current study, we showed that AS-IV decreased the activation of ERK2 in renal tissue in an endotoxinemic model. This effect may promote cell survival in renal cells, and account for better renal function after AS-IV treatment.
The effects of AS-IV on oxidative damage have been revealed in hyperglycemia and nervous system models. AS-IV inhibited hyperglycemia-induced mesangial cell proliferation and glomerular contractile dysfunction through inhibition of NADPH oxidase/ROS/Akt/NF-κB pathway [33]. AS-IV also prevented 1-methyl-4-phenylpyridnium ion (MPP+)-induced cell death in SH-SY5Y neuron cells via inhibiting ROS production and Bax-mediated pathways [34]. We have shown in our current study that AS-IV protected the kidney from oxidative injury caused by LPS. This finding broadens current understanding of the application of anti-oxidant properties of AS-IV.
In our current study, we found that AS-IV attenuated LPS-induced increases in plasma uric acid, BUN, and urinary albumin. The better renal function can be explained by the decreased cytokines and CCR5, decreased activation of ERK2, and reduced oxidative injury to the kidney, which may promote cell survival and protect again AKI. AS-IV has been proven to be protected against ischemia/reperfusion-induced AKI in a rat model by suppressing inflammatory cytokines and MPO activity [14]. Our current study revealed for the first time that AS-IV was protective against LPS-induced endotoxinemia and AKI by suppressing inflammatory cytokine production and signal transduction, and reducing oxidative injury in a murine model.
Conclusions
We demonstrated that AS-IV protects against AKI during bacterial endotoxinemia by attenuating the expression of inflammatory cytokines, CCR5, and p-ERK, and elevating anti-oxidative ability. Although research is needed to elucidate more underlying mechanisms, AS-IV may represent a novel treatment for endotoxinemia and sepsis-associated AKI.
Footnotes
Conflict of interest
None.
Source of support: Departmental sources
References
- 1.Deutschman CS, Tracey KJ. Sepsis: Current dogma and new perspectives. Immunity. 2014;40(4):463–75. doi: 10.1016/j.immuni.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 2.Hicks P, Cooper DJ, Webb S, et al. The Surviving Sepsis Campaign: International guidelines for management of severe sepsis and septic shock: 2008. An assessment by the Australian and New Zealand intensive care society. Anaesth Intensive Care. 2008;36(2):149–51. doi: 10.1177/0310057X0803600202. [DOI] [PubMed] [Google Scholar]
- 3.Bagshaw SM1, George C, Bellomo R ANZICS Database Management Committee. Early acute kidney injury and sepsis: A multicentre evaluation. Crit Care. 2008;12(2):R47. doi: 10.1186/cc6863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boomer JS, To K, Chang KC, et al. Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA. 2011;306(23):2594–605. doi: 10.1001/jama.2011.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Antonelli M, Ronco C. Polymyxin B hemoperfusion in sepsis: growing body of evidence and occasional conflicting results. Blood Purif. 2015;39(1–3):I–II. doi: 10.1159/000431018. [DOI] [PubMed] [Google Scholar]
- 6.Lombardo E, van der Poll T, DelaRosa O, Dalemans W. Mesenchymal stem cells as a therapeutic tool to treat sepsis. World J Stem Cells. 2015;7(2):368–79. doi: 10.4252/wjsc.v7.i2.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao T, Li Y, Liu B, et al. Novel pharmacologic treatment attenuates septic shock and improves long-term survival. Surgery. 2013;154(2):206–13. doi: 10.1016/j.surg.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mathias B, Szpila BE, Moore FA, et al. A review of GM-CSF therapy in sepsis. Medicine. 2015;94(50):e2044. doi: 10.1097/MD.0000000000002044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fiedler VB, Loof I, Sander E, et al. Monoclonal antibody to tumor necrosis factor-alpha prevents lethal endotoxin sepsis in adult rhesus monkeys. J Lab Clin Med. 1992;120(4):574–88. [PubMed] [Google Scholar]
- 10.Cunningham PN, Dyanov HM, Park P, et al. Acute renal failure in endotoxemia is caused by TNF acting directly on TNF receptor-1 in kidney. J Immunol. 2002;168(11):5817–23. doi: 10.4049/jimmunol.168.11.5817. [DOI] [PubMed] [Google Scholar]
- 11.Reinhart K, Karzai W. Anti-tumor necrosis factor therapy in sepsis: update on clinical trials and lessons learned. Crit Care Med. 2001;29(7 Suppl):S121–25. doi: 10.1097/00003246-200107001-00037. [DOI] [PubMed] [Google Scholar]
- 12.De Vriese AS, Bourgeois M. Pharmacologic treatment of acute renal failure in sepsis. Curr Opin Crit Care. 2003;9(6):474–80. doi: 10.1097/00075198-200312000-00003. [DOI] [PubMed] [Google Scholar]
- 13.Ren S, Zhang H, Mu Y, et al. Pharmacological effects of Astragaloside IV: A literature review. J Tradit Chin Med. 2013;33(3):413–16. doi: 10.1016/s0254-6272(13)60189-2. [DOI] [PubMed] [Google Scholar]
- 14.Tan S, Wang G, Guo Y, et al. Preventive effects of a natural anti-inflammatory agent, Astragaloside IV, on ischemic acute kidney injury in rats. Evidence-based complementary and alternative medicine: eCAM. 2013;2013:284025. doi: 10.1155/2013/284025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xin Y, Li G, Liu H, Ai D. AS-IV protects against kidney IRI through inhibition of NF-kappaB activity and PUMA upregulation. Int J Clin Exp Med. 2015;8(10):18293–301. [PMC free article] [PubMed] [Google Scholar]
- 16.Gui D, Huang J, Liu W, et al. Astragaloside IV prevents acute kidney injury in two rodent models by inhibiting oxidative stress and apoptosis pathways. Apoptosis. 2013;18(4):409–22. doi: 10.1007/s10495-013-0801-2. [DOI] [PubMed] [Google Scholar]
- 17.Gomez H, Ince C, De Backer D, et al. A unified theory of sepsis-induced acute kidney injury: Inflammation, microcirculatory dysfunction, bioenergetics, and the tubular cell adaptation to injury. Shock. 2014;41(1):3–11. doi: 10.1097/SHK.0000000000000052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Langenberg C, Bagshaw SM, May CN, Bellomo R. The histopathology of septic acute kidney injury: A systematic review. Crit Care. 2008;12(2):R38. doi: 10.1186/cc6823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Langenberg C, Wan L, Egi M, et al. Renal blood flow in experimental septic acute renal failure. Kidney Int. 2006;69(11):1996–2002. doi: 10.1038/sj.ki.5000440. [DOI] [PubMed] [Google Scholar]
- 20.Martin-Blondel G, Brassat D, Bauer J, et al. CCR5 blockade for neuroinflammatory diseases – beyond control of HIV. Nat Rev Neurol. 2016;12(2):95–105. doi: 10.1038/nrneurol.2015.248. [DOI] [PubMed] [Google Scholar]
- 21.Adwanikar H, Karim F, Gereau RWt. Inflammation persistently enhances nocifensive behaviors mediated by spinal group I mGluRs through sustained ERK activation. Pain. 2004;111(1–2):125–35. doi: 10.1016/j.pain.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 22.Pompella A, Visvikis A, Paolicchi A, et al. The changing faces of glutathione, a cellular protagonist. Biochem Pharmacol. 2003;66(8):1499–503. doi: 10.1016/s0006-2952(03)00504-5. [DOI] [PubMed] [Google Scholar]
- 23.Huang X, Tang L, Wang F, Song G. Astragaloside IV attenuates allergic inflammation by regulation Th1/Th2 cytokine and enhancement CD4(+)CD25(+)Foxp3 T cells in ovalbumin-induced asthma. Immunobiology. 2014;219(7):565–71. doi: 10.1016/j.imbio.2014.03.005. [DOI] [PubMed] [Google Scholar]
- 24.Qiu YY, Zhu JX, Bian T, et al. Protective effects of astragaloside IV against ovalbumin-induced lung inflammation are regulated/mediated by T-bet/GATA-3. Pharmacology. 2014;94(1–2):51–59. doi: 10.1159/000362843. [DOI] [PubMed] [Google Scholar]
- 25.Wang B, Chen MZ. Astragaloside IV possesses antiarthritic effect by preventing interleukin 1beta-induced joint inflammation and cartilage damage. Arch Pharm Res. 2014;37(6):793–802. doi: 10.1007/s12272-014-0336-2. [DOI] [PubMed] [Google Scholar]
- 26.Yadav AK, Kumar V, Dutta P, et al. Variations in CCR5, but not HFE, ELMO1, or SLC12A3, are associated with susceptibility to kidney disease in north Indian individuals with type 2 diabetes. J Diabetes. 2014;6(6):547–55. doi: 10.1111/1753-0407.12128. [DOI] [PubMed] [Google Scholar]
- 27.Ko GJ, Linfert D, Jang HR, et al. Transcriptional analysis of infiltrating T cells in kidney ischemia-reperfusion injury reveals a pathophysiological role for CCR5. Am J Physiol Renal Physiol. 2012;302(6):F762–73. doi: 10.1152/ajprenal.00335.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410(6824):37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 29.Junttila MR, Li SP, Westermarck J. Phosphatase-mediated crosstalk between MAPK signaling pathways in the regulation of cell survival. FASEB J. 2008;22(4):954–65. doi: 10.1096/fj.06-7859rev. [DOI] [PubMed] [Google Scholar]
- 30.Mielke K, Brecht S, Dorst A, Herdegen T. Activity and expression of JNK1, p38 and ERK kinases, c-Jun N-terminal phosphorylation, and c-jun promoter binding in the adult rat brain following kainate-induced seizures. Neuroscience. 1999;91(2):471–83. doi: 10.1016/s0306-4522(98)00667-8. [DOI] [PubMed] [Google Scholar]
- 31.Li M, Wang W, Geng L, et al. Inhibition of RANKL-induced osteoclastogenesis through the suppression of the ERK signaling pathway by astragaloside IV and attenuation of titanium-particle-induced osteolysis. Int J Mol Med. 2015;36(5):1335–44. doi: 10.3892/ijmm.2015.2330. [DOI] [PubMed] [Google Scholar]
- 32.Yue R, Li X, Chen B, et al. Astragaloside IV Attenuates Glutamate-Induced Neurotoxicity in PC12 Cells through Raf-MEK-ERK Pathway. PloS One. 2015;10(5):e0126603. doi: 10.1371/journal.pone.0126603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun L, Li W, Li W, et al. Astragaloside IV prevents damage to human mesangial cells through the inhibition of the NADPH oxidase/ROS/Akt/NFkappaB pathway under high glucose conditions. Int J Mol Med. 2014;34(1):167–76. doi: 10.3892/ijmm.2014.1741. [DOI] [PubMed] [Google Scholar]
- 34.Zhang ZG, Wu L, Wang JL, et al. Astragaloside IV prevents MPP(+)-induced SH-SY5Y cell death via the inhibition of Bax-mediated pathways and ROS production. Mol Cell Biochem. 2012;364(1–2):209–16. doi: 10.1007/s11010-011-1219-1. [DOI] [PubMed] [Google Scholar]