

Abstract
Hyperpolarization-activated, cyclic nucleotide-gated (HCN) channels, the molecular correlate of the hyperpolarization-activated current (If/Ih), are membrane proteins which play an important role in several physiological processes and various pathological conditions. In the Sino Atrial Node (SAN) HCN4 is the target of ivabradine, a bradycardic agent that is, at the moment, the only drug which specifically blocks If. Nevertheless, several other pharmacological agents have been shown to modulate HCN channels, a property that may contribute to their therapeutic activity and/or to their side effects.
HCN channels are considered potential targets for developing drugs to treat several important pathologies, but a major issue in this field is the discovery of isoform-selective compounds, owing to the wide distribution of these proteins into the central and peripheral nervous systems, heart and other peripheral tissues. This survey is focused on the compounds that have been shown, or have been designed, to interact with HCN channels and on their binding sites, with the aim to summarize current knowledge and possibly to unveil useful information to design new potent and selective modulators.
Keywords: HCN channels, Hyperpolarization-activated current, Ivabradine, Drug design, Selectivity, Atrial and Ventricular arrhythmia, Atrial fibrillation, Parkinson’s disease
1. Introduction
Hyperpolarization-activated, cyclic nucleotide-gated (HCN) channels are a family of non-selective cation channels conducting sodium (and potassium) ions through the plasmalemma generating a current termed If (for “funny“ in the heart) or Ih or Iq (“queer” in neurons). The most peculiar feature of HCN/If, met with some skepticism in the early years after its description by Dario Di Francesco [1], is indeed the biophysical behavior, i.e., opening of the channel and flowing of a voltage- and time-dependent inward sodium current upon hyperpolarization. At that time, this tract unsettled the common belief based on well-known inward currents activating upon depolarization and, as for new challenging ideas, it took some time to be accepted by the scientific community [2]. The label “funny” current (If) is emblematic of its controversial initiation. Its functional role in cardiac and not-cardiac cells was even more debated and still stays so. Since its discovery in Purkinje fibers, If activation was associated to the presence of a diastolic depolarization phase in subsidiary and primary pacemaker cells [1-2]. While the role of other cation currents in setting the steepness of the diastolic phase has been increasingly recognized [3-4], the relevance of HCN as “sensor” of the autonomic control (thanks to its peculiar dependence on intracellular cAMP levels) is a fact [5].
Today, our appreciation of the contribution of If not only to pacemaking but also to impulse propagation has greatly increased [6], also thanks to knock-out mouse models [7]. The discovery of the molecular correlate of If/Ih in the late ‘90s gave impulse not only to the transgenic models, but also gained insights into channel tissue distribution beyond cardiac cells, as well as trafficking, plasticity [8], and genotype-phenotype correlation in human diseases [9]. Altogether, these studies ([10], also reviewed in papers quoted above) paved the way to new perspectives on HCN-based (dys)function, channelopathies, and possible therapies. The biology of the channel was also investigated by “evolutionary” and “ontogenetic” approaches proving the highly conserved and ancestral origin of this channel [11-12], as well as its premature functional and molecular expression in embryonic stem cells [13-14].
Despite the high interest on these proteins, to date the therapeutic value of specific modulators has been demonstrated only for cardiac pathologies. Indeed, the wide distribution of HCN channels in different tissues and their involvement in many physiological processes require the discovery of selective substances, to prove that these channels can be exploited as drug targets and to design safe drugs.
This survey is focused on the compounds that have been shown to affect Ih /If, starting from ivabradine (section 3), the only drug which specifically acts on HCN channels, with the aim to summarize current knowledge and to possibly unveil useful information to design new modulators. Therefore, some important aspects concerning structure, function, distribution and therapeutic potential of HCN channels will only briefly reported; the reader is referred to other excellent reviews in this field that deal with these issues in details [6, 8, 15-22].
2. Structure, function and distribution
Functional HCN channels result from the assembly of four alpha subunits. In all mammals studied so far, four genes coding for HCN subunits (1-4) have been cloned. All HCN monomers share the same fundamental structural scheme consisting of six alpha helices, the transmembrane domain, and two cytosolic domains at the NH and COOH termini [23]. The elements conferring ion selectivity and voltage sensitivity are located in the transmembrane core and show a remarkable degree of sequence homology (80-90%) within members of the HCN family [24]. Conversely, the NH and COOH termini largely vary in length and amino acidic composition. The COOH terminus harbors the Cyclic Nucleotide Binding Doman (CNBD), which is critical for the modulation by cyclic nucleotides [25-26].
Tissue and Cellular Expression Pattern of HCN Channels
The expression pattern of HCN channels has been studied in several species, both at the tissue and single-cell level. HCN subunits are strongly expressed in the Central and Peripheral Nervous System (CNS, PNS), with subunit-specific pattern [27-32]. In the CNS, HCN1 is highly expressed in the neocortex, hyppocampus, cerebellar cortex, brainstem and spinal cord. HCN2 is nearly ubiquitous across the CNS but especially abundant in thalamic and brainstem nuclei. Conversely, HCN4 is expressed strongly but in a limited number of areas such as the olfactory bulb and the thalamus, with a distribution pattern that appears complementary to that of HCN1. The expression of HCN3 is scattered throughout the brain and modest. All isoforms, except HCN3, are present in the retina [33]. In the PNS, all HCN subunits are expressed. HCN1 is the most abundant in dorsal root ganglia [34] although a prominent function of HCN2 in the transmission of painful stimuli has also been reported [19]. The expression of HCN channels has been found also in enteric neurons [35-36], and in the spiral [37] and trigeminal ganglion neurons [38].
In the heart, all the four isoforms have been detected, differently expressed according to the cardiac region. The amount of HCN channels is maximal in the sinoatrial node (SAN) region and in the conduction system (atrioventricular node and Purkinje fibers) and lower in the atria and ventricles. There is evidence that expression may vary also across species ([15, 22] and references therein). HCN4 is the main isoform in the SAN of humans, rabbits, mice and dogs, where also HCN1 (in rabbits), and HCN2 (in humans and mice) are substantially expressed [6, 15, 39-40]. In humans and rabbits HCN4 is the main isoform also in atrioventricular node and Purkinje fibers [15, 41], while HCN2 and HCN4 are the most abundant isoforms in most mammalian atria and ventricles [40, 42-43], which also display a minor presence of HCN1. HCN3 is expressed at low level all over the cardiac regions.
There is also evidence for the expression of HCN channels in cells outside of the NS and heart [44], such as kidney [45-46], pancreas [47] and bladder [48]. Outside of nervous and cardiac tissue, however, the physiological function of HCN channels is poorly understood.
Biophysical Properties and Modulation of Functional HCN Channels
HCN channels are voltage-dependent channels activated at potentials near or below average maximum diastolic or resting potential of excitable cells (-40/-70 depending on tissue and cells). In the cell membrane, HCN channels may form homo-tetramers with distinct activation kinetics, voltage dependence, and modulation by adenosine-3',5'-cyclic monophosphate (cAMP). In transfected cells, half activation voltage (V1/2) ranges from -70 mV for HCN1 to -100 mV for HCN4, with HCN2 and HCN3 lying in between, although these values slightly vary according to the cellular model and recording conditions [15].
Gating kinetics is also voltage dependent; the activation time constant (tau) is inversely related to the magnitude of the hyperpolarization. Moreover, values of tau are subunit specific, with HCN1 being the fastest (25 ms), HCN4 the slowest (hundreds of ms), HCN 2 and 3 positioned in between [49]. In vivo, different HCN subunits may form hetero-tetramers and interact with ancillary proteins, thus resulting in the generation of cell-type specific functional protein complexes with unique biophysical properties [50-52]. Relative permeability to sodium and potassium is 1:3/1:5, which leads to a calculated reversal potential between -25 and -40 mV [53]. However, due to the negative range of V1/2, HCN channels generate mainly a sodium current. HCN channels also have a modest calcium permeability (~0.5% of total current [54]) whose physiological significance has yet to be determined [15]. A host of ionic species, organic molecules as well as proteins also modulates HCN channels. The regulatory mechanism that probably better defines HCN channels is the direct sensitivity to cyclic nucleotides, cAMP in particular [26, 55-57]. This mechanism is harbored in the intracellular, COOH terminal CNBD domain. As a rule, binding of cAMP to the CNBD leads to a large (up to 20 mV) positive shift of activation curve and acceleration of activation tau. Once again, HCN subunits differ in this respect, as sensitivity to cAMP is highest in HCN2 and 4, weak in HCN1 and absent in HCN3 [15]. In addition, HCN channels are sensitive to intracellular pH [58], and are modulated by the interaction with a variety of organic molecules and proteins, such as membrane acidic lipids [59-60], caveolin 3 [61], MiRP1 [62-64], filamin A [65], Thy1 [66], Nedd4-2 [67] and the brain-specific auxiliary protein TRIP8b (tetratricopeptide repeat-containing Rab8b-interacting protein), reported to potently control HCN1 surface expression and subcellular distribution [68-69]. Each of these ancillary subunits are part of a cell/tissue specific modulatory network that interacts with HCN hetero-tetramers and, in addition to the distinct biophysical properties of HCN isofoms, contributes to widen the diversity of HCN-mediated current described in vivo. Phosphorylation by src tyrosine kynases [70], p38-MAPK serine/threonine kynases [71], PKC [72] and phosphatidylinositol-4,5-bisphosphate kinase [73] have been reported to diversely affect HCN channel function, independently of cAMP levels. More information on modulatory factors and interacting proteins that affect HCN channel properties can be found in [21, 74].
Functional Role of HCN Channels in Nervous and Cardiac Tissue
An extensive literature has accumulated over the past thirty years on the physiological role of HCN channels in nerve cells (for a recent comprehensive review see ref [21]). Here, we will briefly overview overall significance of HCN channels in neuronal and cardiac physiology and then focus on selected functions of greater relevance for the purpose of this review.
The impact of HCN channels on neuronal overall excitability is largely determined by two fundamental properties: 1) tonic activation at near-rest potentials and 2) an inversion potential lying at the low end of the activation curve. The first property depends on the activation curve of HCN channels and results in a tonic inward (mainly sodium) depolarizing current which maintains the membrane potential close to firing threshold. The second property results from the permeability to sodium and potassium and determines the effect of HCN opening and closing in response to perturbations of membrane potentials. HCN channels open (and depolarize) following hyperpolarization and vice-versa. By virtue of these two fundamental properties, HCN channels serve as feedback mechanism constantly opposing both negative and positive fluctuations of membrane potential (reviewed in refs [21, 75-76]). HCN basic properties result in a spectrum of diverse functions according to host cell type, expressed isoform and subcellular distribution. Somatic HCN channels control the intrinsic excitability and rythmogenesis in various neuronal types. In thalamic neurons, the activation state of HCN channels determines the activity pattern associate to circadian rhythm [77]. Ih current mediated by somatic HCN channels serves as a high-pass filter shaping the voltage response (“resonance”) to rhythmic oscillation in cortical [78-79] and subcortical neurons [80]. Functional HCN channels have been found in distal dendrites of several neuronal populations. In dendritic compartments, active HCN channels reduce input resistance and accelerate the decay phase of Excitatory Post-Synaptic Potentials (EPSP), thereby limiting the amplitude and duration of single EPSPs and dampening temporal summation of EPSP trains in cortical [76, 81], thalamic [82] and midbrain dopaminergic neurons [83]. Furthermore, thanks to an increasing soma-to-dendrite expression gradient found in CA1 hippocampal and cortical neurons, HCN channels normalize the duration of EPSPs originating at different distances from the soma, thus preventing loss of information [84-85].
Finally, HCN-mediated current has been reported to control synaptic efficacy with a presynaptic mechanism in many neurons. However, the role of HCN channels in this function is not univocal, as HCN channel blockade can either inhibit or facilitate neurotransmitter release depending on neuron type and location [21].
In the PNS, HCN (1 and 2) channels have been implicated in the intrinsic excitability of primary sensory neurons in Dorsal Root Ganglia (DRG) [19, 86].
In the heart, HCN channels are essential for normal heart impulse generation and conduction. In SAN cells they contribute to form a coordinated system with ryanodine receptors that drives spontaneous electrical activity [5, 87]. At maximum diastolic potentials (-70/-40 mV depending on SAN region) a substantial fraction of HCN channels are open and provides a steady-state inward current that slowly depolarize membrane during the diastolic phase toward the threshold required to generate a spontaneous action potential [56, 88-89]. In the atrioventricular node, although the function of HCN channels is less investigated, HCN channels have also been demonstrated to sustain generation of spontaneous electrical activity and conduction of stimuli from atria to ventricles [90-92]. The essential role of HCN channels in cardiac pacemaker centers has been further clarified in different transgenic models of cardiac deletion of HCN4. Constitutive deletion leads to mice embryonic lethality [89], whereas inducible deletion induces a fast and progressive bradycardia followed by atrioventricular block, cardiac arrest, and death [7].
The physiological role of HCN channels in the working healthy myocardium is still a matter of ongoing investigation. HCN mediated current in human atrial and ventricular cardiomyocytes is qualitatively similar to that retrieved in SAN cells, however its role is supposed to diverge since healthy atrial and ventricular cardiomyocytes have a stable resting membrane potential and do not generate spontaneous electrical activity. Indeed, at atrial and ventricular resting membrane potentials (-80/-70 mV) a small fraction of HCN channels are open, suggesting a minor contribution of HCN to resting potential and electrogenesis. Nonetheless, a clear-cut diastolic depolarization phase is observed in some human healthy atrial and ventricular myocytes (reviewed in [93]), raising questions on the local physiologic or pathologic conditions that may uncover a functional role of HCN channels in cardiomyocyte electrogenesis. Recently, using a HCN3-deficient mouse model, Fenske et al. hypothesized that HCN3 channels may contribute cardiomyocyte repolarization [94-95]. Such a mechanism relies on HCN3 slow-deactivation kinetic that leaves channels open during the course of action potential, and counterbalances repolarizing currents flowing during repolarization.
3. Ivabradine
Ivabradine (Fig. 1), developed by Servier, is the only drug on the market which specifically acts on HCN channels. It has been approved by the European Medical Agency in 2005 for the treatment of stable angina, and by the Food and Drug Administration (FDA) in 2015 to reduce hospitalization from worsening heart failure [96]; the drug is sold under different names, among which Procoralan, Corlentor and Corlanor. In this section only the interaction of ivabradine with ion channels is considered; its therapeutic applications is outlined in section 8, while the outcomes of clinical trials have been already extensively reviewed (see, for instance refs [6, 97-98]).
Fig. (1).
Ivabradine.
Ivabradine selectively inhibits If in rabbit SAN and in recombinant systems expressing hHCN4 in a use-dependent way, with IC50 in the micromolar range (Table 1), by interacting with the channels from the intracellular side [99-101] (for the structure of the binding site see section 7). The blockade of HCN4 by the drug require open channels, while on HCN1 the drug is a closed-channel blocker [101].
Table 1.
Potency of selected compounds for HCN channels blockade.
| Compound | IC50 or % inhibition of Ih | Isoform (cell) or tissue (species) | References |
|---|---|---|---|
| Acehytisine | 9.9 ±0.3 μM | Rabbit SAN | [188] |
| 64.9 ±8.6 μM | hHCN4 (Xenopus oocytes) | ||
| Amiodarone | 46.3 ± 11.7μM | hHCN1 (Xenopus oocytes) | [169] |
| 8.2 ± 4.2 μM | hHCN2 (Xenopus oocytes) | ||
| 2.1 ± 1.9 μM | hHCN4 (Xenopus oocytes) | ||
| 4.5 μMa | Rabbit HCN4 (HEK293) | [167] | |
| 4.9 ± 1.2 μM | SHR ventricular myocyte | [171] | |
| 6.9 ±1.3 μM | WKY rat ventricular myocyte | ||
| 0.8±0.1 μM | hHCN4 (CHO) | [173] | |
| Bupivacaine | 55±5 μM | rat DRG neurons | [165] |
| R- bupivacaine | 55±6 μM | rat DRG neurons | |
| S- bupivacaine | 67±8 μM | rat DRG neurons | |
| Capsazepine | 7.9±0.7 μM | hHCN1 (CHO) | [181] |
| 6.1±0.8 μM | hHCN2 (HEK293) | [182] | |
| 5.8±0.5 μM | hHCN4 (HEK293) | ||
| Clonidine | 8.2±1.4 μM | mHCN2 (HEK293) | [146] |
| 9.8±1.4 μM | hHCN4 (HEK293) | ||
| 3.1±0.5 μM | SAN (wt mice) | ||
| 2.8±0.7 μM | SAN (α2ABC KO mice) | ||
| Dexmedetomidine | 46 @10 μM | mHCN1 (HEK293) | [143] |
| 58 @10 μM | mHCN2 (HEK293) | ||
| Dronedarone | 1.0 ± 0.1 μM | hHCN4 (CHO) | [173] |
| Eugenol | 157 μM | rat TG neurons | [192] |
| Ketamine | ~32 @20 μM | mHCN1 (HEK293) | [164] |
| ~6 @20 μM | mHCN2 (HEK293) | ||
| ~ 16 μM | mHCN1-HCN2 (HEK293) | ||
| Lidocaine | 99±4 μM | rat DRG neurons | [165] |
| 31 @100 μM | mHCN1 (HEK293) | [166] | |
| 47 @100 μM | mHCN2 (HEK293) | ||
| 39 @100 μM | mHCN1-HCN2 (HEK293) | ||
| 30 @100 μM | mHCN4 (HEK293) | ||
| Loperamide | 4.9 ± 0.6 μM | large rat DRG | [147] |
| 11.0 ± 0.5 μM | small rat DRG | ||
| 13.5 ± 2.1 μM | HCN1 (HEK293) | [150] | |
| 37.1 ± 7.7 μM | hHCN4 (HEK293) | ||
| MEGX | 55 @100 μM | mHCN1 (HEK293) | [166] |
| 59 @100 μM | mHCN2 (HEK293) | ||
| 51 @100 μM | mHCN1-HCN2 (HEK293) | ||
| 48 @100 μM | mHCN4 (HEK293) | ||
| Mepivacaine | 190±15 μM | rat DRG neurons | [165] |
| MPP+ | 7.74 μMb | Rat SNc DA neurons | [83] |
| Niflumic acid | 10.64 μMb | Rabbit SAN | [176] |
| Nicotine | 62 nM | Mouse O-LM neurons | [183] |
| Propofol | 50 @20 μM | mHCN1 (Xenopus oocytes) | [157] |
| 70 @20 μM | mHCN2 (Xenopus oocytes) | ||
| 85 @20 μM | mHCN4 (Xenopus oocytes) | ||
| Tramadol | 13.6±2.7 μM | (rat anterior pituitary) GH3 cells | [151] |
| 2 | 0.32 μM | Guinea-pig SAN | [126] |
| 5 | 2.31 ±0.37 μM | mHCN1 (HEK293) | [135] |
| 17.22±1.74μM | mHCN2 (HEK293) | ||
| 7.23 ±2.60 μM | hHCN4 (HEK293) | ||
| 6 | 5.60 ±0.26 μM | mHCN1 (HEK293) | |
| 24.58±4.89μM | mHCN2 (HEK293) | ||
| 7.14 ±0.11 μM | hHCN4 (HEK293) | ||
| R-7 | 9.41±0.25 μM | mHCN1 (HEK293) | |
| 2.3±0.60 μM | mHCN2 (HEK293) | ||
| 24.94±0.10μM | hHCN4 (HEK293) | ||
| R-8 | 0.60±0.07 μM | mHCN1 (HEK293) | |
| 18.3±0.14 μM | mHCN2 (HEK293) | ||
| 103.78±29.8 μM | hHCN4 (HEK293) | ||
| 9 | 21±3.98 | mHCN1 (HEK293) | [137] |
| 19.35±4.48 | mHCN2 (HEK293) | ||
| 3.98±1.16 | hHCN4 (HEK293) | ||
| 11 | 0.4 μM | hHCN1 (HEK293) | [139] |
| 5.0 μM | hHCN2 (HEK293) | ||
| 3.2 μM | hHCN3 (HEK293) | ||
| 4.0 μM | hHCN4 (HEK293) | ||
| Cilobradine (DK-AH269) |
17.9 ± 4.4 μM | HCN1 (HEK293) | [150] |
| 21.8 ± 6.8 μM | hHCN4 (CHO) | ||
| 1.15 ±0.16 μM | hHCN1 (HEK293) | [106] | |
| 0.90 ±0.07 μM | hHCN2 (HEK293) | ||
| 0.99 ±0.16 μM | hHCN3 (HEK293) | ||
| 0.92 ±0.05 μM | hHCN4 (HEK293) | ||
| Ivabradine | 1.1±0.2 μM | hHCN4 (CHO) | [173] |
| 2.05 ±0.13 μM | hHCN1 (HEK293) | [106] | |
| 2.29 ±0.13 μM | hHCN2 (HEK293) | ||
| 2.51 ±0.13 μM | hHCN3 (HEK293) | ||
| 2.15 ±0.34 μM | hHCN4 (HEK293) | ||
| 0.54 μM | hHCN4 (CHO) | [133] | |
| 0.94 μM | mHCN1 (HEK293) | [101] | |
| 2.0 μM | hHCN4 (HEK293) | ||
| 1.5 μM | Rabbit SAN | [100] | |
| 2.18 μM | Rabbit SAN | [99] | |
| Zatebradine | 4.4 ± 0.4 μM | hHCN4 (Xenopus oocytes) | [188] |
| 1.83 ±0.39 μM | hHCN1 (HEK293) | [106] | |
| 2.21 ±0.21 μM | hHCN2 (HEK293) | ||
| 1.90 ±0.13 μM | hHCN3 (HEK293) | ||
| 1.88 ±0.12 μM | hHCN4 (HEK293) | ||
| ZD7288 | 23.8 ± 5.5 μM | SHR ventricular myocytes | [171] |
| 15.2 ± 2.5 μM | HCN1 (HEK293) | [150] | |
| 47.3 ±23.3 μM | hHCN4 (CHO) | ||
| 15 μM | rat DRG neurons | [116] | |
| ~ 0.3 μM | Guinea-pig SAN | [114] | |
| 41 μM | mHCN1 (Xenopus oocytes) | [205] | |
| 25.8±9.7 μM | mHCN1 (HEK293) | [206] |
Unless otherwise stated, the IC50 values were determined at potential ranging from -90 to -130 mV. a At -70 mV. b At -75 mV. WKY rats: Wistar-Kyoto rats. O-LM: Oriens-Lacunosum Moleculare. TG: Trigeminal Ganglion.
The absolute configuration of the stereogenic center of Ivabradine is S; no enantioselectivity has been found for the HCN blocking property, but the negligible activity on K+-currents of the S form, compared to that of its R-enantiomer, and also of zatebradine and cilobradine [99, 102-103], confers to this compound a safer pharmacological profile. As a matter of fact, Ivabradine is reported to be fairly selective for HCN channels compared to other ion channels. Delpon et al. found that the hKv1.5 was blocked by the drug in a concentration-dependent way with IC50 29.0 ± 1.9 μM, therefore at doses higher than those required to block HCN channels [103]. On rabbit SAN, Bois et al. [99] found that 10μM ivabradine had no detectable effect on T-type calcium current, while the drug (3 μM) produced a slight decreased (<20%) of L-type calcium current. On guinea-pig isolated cardiac preparations, Peres et al. found that ivabradine inhibited INa only at doses higher than those reducing heart rate [104]. More recently, Koncz et al. found that 10μM ivabradine did not affect the transient outward (Ito) and the inward rectifier (IK1) potassium currents in rabbit and dog ventricular myocytes, while it inhibited the rapid delayed rectifier (IKr) current in rabbit ventricular myocytes with an estimated IC50 value of 3.5 μM [105].
As far as selectivity among HCN channel isoforms, Stieber et al. found that ivabradine, as well as its close structural analogues cilobradine and zatebradine (see next section), is not selective for the HCN4 isoform, since it blocks the four isoforms with similar potency (Table 1) [106]. This lack of selectivity is probably the reason for one of the major adverse reactions of ivabradine administration at clinical dose, i.e. vision alterations (phosphenes): this effect has been related to the blockade of the neuronal HCN1 isoform in the retina, which occurs concurrently with that of HCN4 in SAN. Apart from this, other side effects due to the interaction with the HCN channels expressed in tissues other than heart have not been described. It is reported that ivabradine does not to cross the blood-brain barrier [107-108], and therefore it should not affect Ih in brain although its antiseizure activity in an animal models of epilepsy has been reported [109]. Nevertheless, the lack of selectivity may raise concern on its use.
A strategy to limit the occurrence of adverse reactions is to identify novel compounds able to block selectively single HCN channel isoforms, which are most abundant or which show the most relevant function in the target tissue.
4. Drug design projects
Intensive work has been made in the past on the “specific bradycardic agents”; unfortunately, the medicinal chemistry supporting them has been only partially described. In this section, we summarize the information found in the literature on drug design projects, which lead to the compounds reported in Figs. (2-4). It must be remembered that the most part of these studies have been performed before the cloning of HCN isoforms; therefore, they provide little useful information to design selective compounds.
Fig. (2).

Specific bradycardic agents.
Fig. (4).
Compounds developed at the University of Florence
The first drug discovery programs date back to 1980s, when the specific bradycardic agents have been discovered; the history of these compounds has been reviewed in ref [110]. These compounds can be divided in three different groups (imidazolines, aminopyrimidines and phenylalkylamines) according to their chemical structure (Fig. 2). Imidazolines, such as alinidine, are structural analogues of clonidine, but, at variance with the latter, have a substituent on the exocyclic nitrogen atom. Staehle et al. reported the bradycardic activity of a series of analogues with general formula I, measured in vivo in spinal rats [111]; these results can be related to the interaction with the HCN4 isoform in the SAN, although in these conditions heart rate may be affected by several mechanisms, excluding those mediated by the CNS. It was found that a double substitution with halogen in the ortho positions of the phenyl ring was optimal, with R1 being bromine and R2 being bromine or chlorine, suggesting that in the bioactive conformation the guanidine moiety and the aromatic ring should not be coplanar. The substituent on the exocyclic nitrogen atom (R3) may contain 2-5 carbon atoms; a double bond, as in alinidine, was not essential, and a productive substituent was the cyclopropylmethyl group. Opening the nitrogen-containing ring, or increasing its size, was detrimental for activity. An alkyl group (R4) only on the endocyclic guanidine nitrogen atoms (R3 = H), gave inactive compounds; however, derivative TH92:20 was four times more potent that alinidine in lowering heart rate [112]. Results of in vivo experiments are a combination of pharmacokinetic and pharmacodynamic properties, which makes difficult to derive sound structure-activity relationships; for this reason, physicochemical properties (pKa and logD7.4) were also taken into account [112]. The development of alinidine (ST-567) was stopped due to the many side effects of the compound, which were ascribed to the interaction with L-type Ca2+ channels and K+ channels, and to the metabolic transformation leading to clonidine [110, 113].
ZD7288 is a piridinium derivative, widely used as pharmacological tool to study HCN channels. Differently to alinidine, ZD7288 has no significant effect on the delayed rectifier current (IK) [114]; however, it has been reported to block T-type calcium channels in rat hippocampal pyramidal cells [115]. In rat DRG neurons it blocks Na+ currents with a potency (IC50 1.17μM) higher than that on hyperpolarization-activated current (IC50 15 μM) [116], raising questions on the selectivity of this compound for HCN channels. ZD7288 analogues have been reported only in a patent [117], with no indication of their bradycardic potency.
In the early nineties researchers at Boeringher described a series of phenylalkylamines, designed from verapamil with the aim of improving its negative chronotropic activity and suppress the other hemodynamic effects [118-119]. The successful modification was the replacement of the α-isopropyl-dimethoxyphenylacetonitrile group with various benzolactams rings, leading to the benzopyrrolidone falipamil (Fig. 1) and then, by increasing the size of the lactam ring, to the benzazepinone zatebradine. Several other analogues were synthesized and tested in vivo for their effect on heart rate by injection of a fixed dose (5 mg/kg i.v.) in anesthetized rats; this allowed to derive structure-activity relationships for the bradycardic activity, which can be useful, even if partially, to understand the interaction with HCN channels. On the benzazepinone ring, the best substitution (Fig. 1, R1, general formula II) was achieved with electron-donating groups (i.e. methoxy) in position 8 or in 7,8. Shortening, lengthening or branching the three-methylene chain, or adding a hydroxy group (R2) was detrimental for activity. R3 could be H or an alkyl group not longer than n-propyl; the nitrogen atom needs to be basic. Shortening the chain (Y) connecting the nitrogen atom to the second aromatic ring was detrimental, while the elongation up to five atoms and the insertion of a heteroatom to link the aromatic ring were well tolerated. This ring could carry a substituent in position 4 (R4) with both electron-donating and electron-withdrawing group, avoiding hydrophilic moieties such as hydroxy groups; ortho-substitution was detrimental for activity. Substitution with more than one methoxy group was productive in position 3,4, but not in 3,4,5. Replacement of the phenyl ring by heteroaromatic moieties such as thiophene or benzofuran also gave potent compounds. From this large number of derivatives, zatebradine was selected as drug candidate. It is remarkable that the simple replacement of the α-(3,4-dimethoxyphenyl)-α-isopropylacetonitrile group of verapamil with a 7,8-dimethoxy-benzazepin-3-one moiety can shift, even if not completely, the interaction from calcium channels to HCN channels [120].
Although the outcome of the modifications on the three-methylene chain were suggesting that this part of the molecule could not be modified, the same company disclosed another compound, cilobradine (DK-AH269), where the flexible chain and the basic nitrogen have been incorporated into a piperidine ring [121]. This modification introduced a stereogenic center into the molecule; cilobradine, the S enantiomer, has been tested in several experimental models, while the R enantiomer (DK-AH268) has been characterized on rat trigeminal ganglion neurons [38]. Cilobradine also entered clinical trials [122-123], but its development, as well as that of zatebradine, was discontinued.
In a series of papers published between 2003 and 2004 researchers at Yamanouchi described the discovery and optimization of a series of compounds ultimately yielding YM758 (Fig. 3) as drug candidate [124-128]. The screening of analogues has been made in vitro by measuring the decrease in heart rate on spontaneously-beating guinea-pig right atria, and in vivo by comparing the negative chronotropic activity and reduction of blood pressure in anesthetized rats. Starting from the known structure-activity relationships of zatebradine, 2-(1-benzylpiperidin-3-yl)-1,2,3,4-tetrahydroisoquinoline 1 was identified as suitable lead compound for structural optimization. To this aim, the basicity of the tetrahydroisoquinoline nitrogen atom was reduced by adding a carbonyl group, the basic piperidinyl nitrogen atom and the aromatic ring of the benzyl moiety were spaced away by elongating the chain and by introducing another amide moiety, and the substitution on both aromatic rings was suitably modulated, yielding YM758. YM758 is reported to be an If blocker, but its If blocking activity has never been described; only papers dealing with its pharmacokinetic have been published [129-131]. The development of this compound was discontinued, probably due to pharmacokinetic problems. Interestingly, in this series of compounds the If blocking properties have been reported only for 2 (Fig. 3), for which a submicromolar IC50 (0.32 μM) has been determined on guinea pig SAN [126].
Fig. (3).
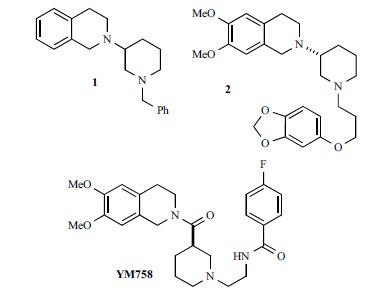
Phenylalkylamines developed at Yamanouchi.
The design leading to ivabradine, the rigid analogue of zatebradine obtained by constraining the phenylethyl moiety into a bicyclo[4.2.0]octa-1,3,5-triene structure, has not been disclosed. However, in a recent patent [132] some analogues have been described, characterized by the shifting of the carbonyl amide moiety from the lactam ring to the exocyclic chain (Fig. 2, general formula III). Activity has been determined by measuring the reduction of heart rate in rat spontaneously-beating right atria, and the IC30 values are provided for some compounds. Since ivabradine is fairly selective for HCN compared to other ion channels (see section 3), we can take the activity of these close structural analogues as a measure of the interaction of the compounds with the HCN4 isoform of the SAN. Under similar conditions, the IC30 of ivabradine varies from 0.19 μM to 0.28 μM [133], which suggests that the new analogues, whose IC30 values ranging between 0.4 μM and 2.0 μM, are less potent than the lead compound. Among the reported modifications, the best results are associated with R1 being a cyclopropylmethyl or cyclobutylmethyl group (R2, R3 = MeO, general formula III), and R2 or R3 being N-methyl-carbamate or N,N-dimethyl-carbamate (R1 = Me).
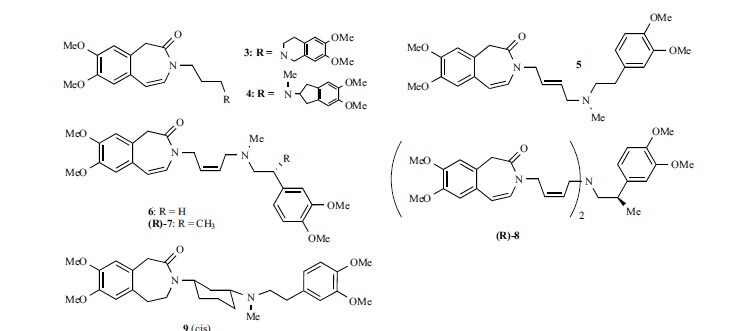
Other zatebradine analogues have been disclosed in 2005 at University of Florence (Fig. 4). Taking advantage of a long-lasting research on verapamil derivatives, aimed to change the pharmacological profile of the lead (see [134] and references therein), the authors introduced on zatebradine’s structure the same modifications which, on verapamil increased the negative chronotropic activity with respect to the other pharmacological properties (negative inotropism, smooth muscle relaxation, reversal of Multidrug Resistance). Structural changes were mainly done on the phenylethylamino group; the compounds were first screened in vitro for their chronotropic activity on spontaneously beating guinea-pig right atria, then the activity on If for some of them was assessed on isolated ventricular cardiomyocytes of old spontaneously-hypertensive rats (SHR). The tests on guinea-pig right atria showed that a 4,5 double bond in the benzazepinone ring gave compounds more potent than their saturated analogues, and that constraining the phenylethyl moiety into a tetrahydroisoquinoline or indane ring (3 and 4, respectively) gave compounds only 2-3 times less potent than zatebradine. Patch-clamp experiments for selected compounds on SHR myocytes confirmed for most of them that their bradycardic activity was actually due to If blockade. The most interesting compound was 5, the trans-butene analogue of zatebradine, which was equipotent with the lead: the EC30 values for the negative chronotropic activity were 11.3 μM and 13.4 μM for 5 and zatebradine, respectively, and the residual If was ~40% of control at a concentration 10 μM for both drugs.
These outcomes provided a basis for further manipulations, made by the same research group some years later [135-136]. A new screening model was used (HEK293 cells expressing recombinant mHCN1, mHCN2 and hHCN4 channels), in order to find derivatives endowed with isoform selectivity; IC50 values (Fig. 5) were determined at -120 mV. New structural elements and stereogenic centers were inserted into the structure of zatebradine, and the majority of the new derivatives carried the benzoazepin-2(3H)-one group. While 5 and it cis analogue 6 were endowed with comparable potency on HCN1, HCN2 and HCN4 channels, combining the cis-butene moiety with a stereogenic centre on the ethyl chain gave (R)-7, showing a 4-fold and 11-fold preference for HCN2 with respect to HCN1 and HCN4, respectively. The replacement of the small N-methyl group with a second benzazepinone/cis butene moiety gave (R)-8 endowed with selectivity for HCN1 channel vs HCN2 (30-fold) and HCN4 (173-fold). It was somehow unexpected that the eutomers (R)-7and (R)-8 had an opposite configuration with respect to ivabradine, since the stereogenic centres are placed on the phenethyl moiety in the same position. The insertion of the three-methylene chain of zatebradine into a cyclohexane ring gave cis-9, showing a 5-fold preference for HCN4 vs HCN2 and HCN1 [137]. Remarkably, the selectivity found in HEK cells for compounds (R)-8 and cis-9 was maintained also in native tissues (guinea pig SAN, mouse DRG neurons, and guinea pig spontaneously beating atria) [137-138].
Fig. (5).
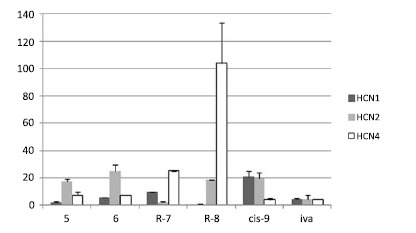
IC50 values (μM) of the HCN modulators reported in refs 135, 137.
A new series of HCN1 selective compounds was recently discovered at Johnson & Johnson Pharmaceutical (Fig. 6) [139]. The screening of a proprietary chemical library by means of a FRET-based voltage-sensitive dye system [140] yielded compound 10, endowed with micromolar potency on HCN1 (pIC50 5.9) and with 15-fold selectivity over HCN4. Several analogues were synthesized, finding that a basic piperazine ring, a quaternary carbon atom and a 2-alkoxybenzamide moiety were required for the activity of these analogues at HCN1 channel. Compound 11, endowed with reasonable metabolic stability, was selected for further studies: its potency on HCN1 was slightly increased (pIC50 6.4) with respect to the lead 10, but not the in vitro selectivity (pIC50(HCN4) 5.4; pIC50(HCN2) 5.3; pIC50(HCN3) 5.5). Nevertheless, 11 was able to relieve nerve injury-induced tactile allodynia in a spared nerve injury model (ED50 6 mg/kg), without affecting heart rate (ED20 for bradycardic activity 25 mg/kg). Further characterization of this compound has not been reported in the literature yet.
Fig. (6).
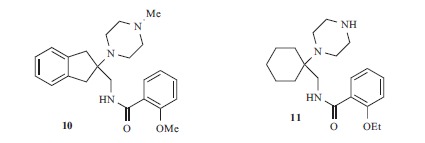
Compounds developed at Johnson & Johnson Pharmaceuticals.
5. Substances that have been shown to modulate HCN channel activity
This section reports a series of substances (Figs. 7-10) which have been tested on HCN channels, in native tissues and/or in recombinant systems. These compounds are mainly drugs, approved for different indications: in some instances, their Ih modulating activity can contribute to the therapeutic action, but to side effects in others.
Fig. (7).
Structure of adrenergic and opioid receptor agonists which modulate If/Ih
Fig. (10).
Structure of other HCN channels modulators.
The purpose of this section is to compare information on the potency and, when available, on the isoform selectivity of these structurally-diverse modulators, which are divided according to their main pharmacological activity.
Alpha2-Adrenergic and Opioid Receptors Agonists (Fig. 7)
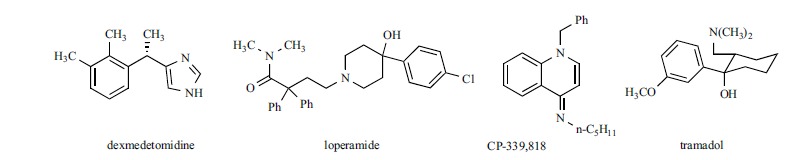
Dexmedetomidine, the dextrorotatory, S-isomer of medetomidine, is an alpha2 agonist used in the intensive care unit for sedation and analgesia [141]. It has been reported that a combination of dexmedetomidine and other local anaesthetics increases the duration of analgesia ([142] and references therein): using an in vivo model of a peripheral nerve block in rats, Brummett et al. showed that this effect is due to blockade of the Ih current and not to alpha2-adrenoceptor agonism [142]. Indeed, on HEK293 cells expressing mHCN1 or mHCN2 channels, dexmedetomidine blocked h-current with IC50 approximately 10 μM on both isoforms [143]. The analgesic activity of the drug was found to be largely reduced in HCN1 knocked-out mice, compared to wild type. Since in both models yohimbine significantly, but not completely, prevented the analgesic effect of the drug, the authors concluded that analgesia induced by dexmedetomidine was mediated by HCN inhibition, produced not only through alpha2-AR, but also through a direct channel blockade via alpha2-independent mechanism. Interestingly, bradycardia is a common side effect of dexmedetomidine [141], which is also reported to affect sinus function in children [144]: it is possible that this drug interacts also with HCN channel in heart, but to our knowledge, its selectivity for the HCN channel isoforms involved in pain has not be studied yet.
Another alpha2-agonist, clonidine (Fig. 2), is used in the clinic for several indications including analgesia. Clonidine has been tested on rat DRG neurons, where the reduction of Ih induced by this drug was prevented by the alpha2-AR antagonist yohimbine, suggesting that Ih inhibition was due to alpha2-adrenoceptor activation [145]. However, clonidine is able to interact directly with HCN channels: on HEK293 cells expressing recombinant mHCN2 and hHCN4 channels, the drug blocked f-current with IC50 of, respectively, 8.2±1.4 μM and 9.8±1.4 μM. On SAN of wild type mice and of mice where the three alpha2 adrenoceptors have been deleted, clonidine inhibited f-current: IC50 values at -100 mV were 3.1±0.5 μM and 2.8±0.7 μM for wild-type and α2ABC KO mice, respectively [146]. No data have been reported so far for clonidine on recombinant HCN1.
By screening a library of known ion channel modulators at Wyeth, Vasilyev et al. found that the opioid agonist loperamide is able to block HCN channels with a mechanism independent from opioid receptor activation [147]. On large rat DRG neurons loperamide blocked Ih with an IC50 of 4.9 ± 0.6 μM, while it was twice less potent in small DRG neurons (IC50 11.0 ± 0.5 μM). Loperamide shifted Ih activation toward more hyperpolarized potentials when applied in the bath, but not when applied in the pipette. The blockade was use-independent; the binding site was suggested to be in the extracellular region of the channel. Loperamide and CP-339,818 [148], a K+-channel modulator evaluated in the same screening program [149], were tested on recombinant HCN1 and HCN4 expressed in HEK293 cells. Both compounds were 2-3 times more potent on HCN1 than on HCN4. On HCN1 the blockade was voltage dependent, and more potent at less negative potential, confirming the findings for loperamide on rat DRG neurons [150].
The effect of tramadol, another opioid agonist, on Ih has been studied in GH3 cells, a cell line from a rat anterior pituitary [151]. It is not clear which isoform is functionally relevant in these cells: RT-PCR analysis have shown the presence of mRNA transcripts for HCN2, HCN3 and HCN4 subunits [152], and the characteristics of the current suggested that HCN2, HCN3, or mixed HCN2 - HCN3 channels are functionally expressed. Tramadol was able to reduce Ih amplitude in a dose dependent way, with an IC50 of 13.6±2.7 μM, and to shift activation curve to a more hyperpolarized potential. As for loperamide, the effect on Ih was independent from opioid receptor activation, since it was not affected by preincubation with naloxone. By analyzing the effect of the drug on activation kinetic at different potentials, the authors suggested that tramadol is a closed-state blocker, and that the binding site could be located at the extracellular region between S1 and S1–S2 linker [151].
General Anesthetics
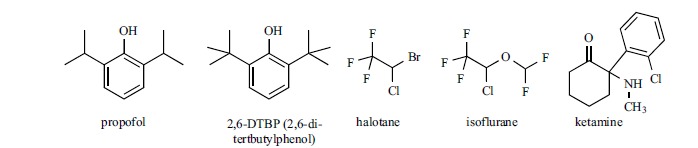
Propofol (Fig. 8) is an intravenous sedative-hypnotic agent for use in the induction and maintenance of anaesthesia; it activates GABA-A receptors, but it acts also on other targets, such as, for instance NMDA receptors [153]. Bradycardia is one of its common side effects [154]; moreover, it displays both pro- and anti-arrhythmic effects which are related to interaction with several ion channels, including HCN channels [155-156]. Propofol has been tested on mHCN1, mHCN2 and mHCN4 channel isoforms expressed in Xenopus leavis oocytes [157], where the drug (20 μM) decreased current amplitude to, roughly, 50%, 70% and 85% of control, respectively, showing that HCN1 is the most sensitive isoform. However, on guinea-pig SAN a 3 μM concentration was able to significantly reduce If conductance at physiological potentials [156]. Tibbs et al. [158] tested a series of commercially-available propofol analogues on HCN1 channels; the compounds show a double substitution on the aromatic ring (position 2,4 or 2,6) with tert- or sec-butyl groups, i.e. with higher steric hindrance with respect to the isopropyl groups that decorate propofol. The most active compound in the series was 2,6-di-tertbutylphenol (2,6-DTBP), which was 5, 2, 15 and 23 times more potent than, respectively, propofol, 2,4-DTBP, 2,6-di-secbutylphenol (2,6-DSBP) and 2,4-DSBP in inhibiting HCN1 channel gating. Moreover, HCN1 was more sensitive to the action of 2,6-DTBP compared to the other three isoforms: the shift in V1/2 of gating was evident at 3 μM concentration, while a statistically significant reduction on HCN2-4 was observed only at a 20 μM concentration. As a consequence of its HCN1 blocking property, 2,6-DTBP showed an antihyperalgesic effect in partial sciatic nerve ligation–induced neuropathic pain (mechanical and thermal insults). Other general anesthetics has been found to reduce Ih at therapeutically relevant concentrations: the HCN modulating properties of volatile anaesthetics such as halothane and isoflurane were studied on HCN1 and HCN2 channels expressed in HEK293 cells [159] and on cortical pyramidal neurons from wild-type and HCN1 knockout mice [160]; for both drugs the kind of inhibition (shift in the activation voltage, reduction of current amplitude) was found dependent on the subunit composition of the channel.
Fig. (8).
Structure of general anesthetics that modulate Ih.
Ketamine (Fig. 8) is a non-competitive antagonist of N-methyl-D-aspartate (NMDA) glutamate receptor [161], used as fast-acting anesthetic, as antidepressant and in pain management [162]. Some of the pharmacological properties of this drug are not linked to its activity on NMDA receptor, rather to the interaction with other receptors, transporters and ion channels [163], including HCN channels. When tested in HEK293 cells, on homomeric mHCN1 channels ketamine was able to shift the voltage of activation to more negative potential (ΔV1⁄2 = -14.7± 2.5 mV) and to reduce the maximal current amplitude (32% inhibition at 20 μM), but it had little effect on the HCN2 isoform [164]. On heteromeric mHCN1–mHCN2 channels, the shift in V1⁄2 (12 mV) was similar with respect to HCN1, but the maximal inhibition was higher (45%, estimated IC50 ~ 16 μM). Evidence from in vitro experiments on ketamine stereoisomers, and from HCN1 knocked out mice, suggests that the interaction with this channel is involved in the hypnotic activity of the drug.
Local Anesthetic and Antiarrhythmic Drugs
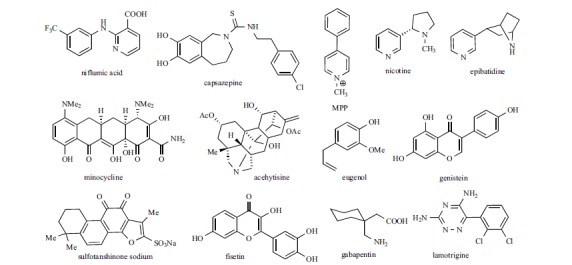
In addition to general anesthetics, also local anesthetics modulate hyperpolarization-activated current. The activity of lidocaine, mepivacaine and bupivacaine (Fig. 9) on Ih has been measured on rat DRG neurons; they have been found to reversibly block Ih with IC50 values (99±4 μM, 190±15 μM, and 55±5 μM, respectively) which are clinically relevant concentrations for spinal and epidural anesthesia. The authors suggested that Ih blockade plays an important role in the anesthetic activity of these drugs [165]. In the same study, it was shown that QX-314, the quaternary ammonium derivative of lidocaine, was devoid of activity. This finding supports an intracellular localization for the binding site of lidocaine and its structural analogues. In addition, the bupivacaine enantiomers showed a similar potency, the IC50 values being 55 ± 6 μM for the R-isomer, and 67 ± 8 μM for the S one, respectively.
Fig. (9).
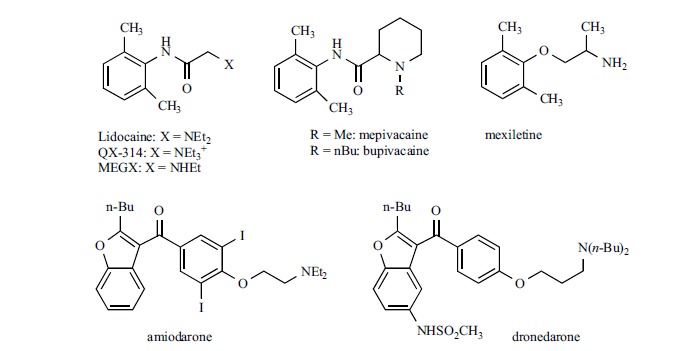
Structure of local anesthetic and antiarrhythmic drugs that modulate Ih.
More recently, lidocaine has been tested also on recombinant systems (HEK cells) [166], where a 100μM concentration of the drug reduced maximal current amplitude of 30.9%, 46.6% and 30.4% on, respectively, homomeric mHCN1, mHCN2 and mHCN4 at -120 mV; only on HCN1 the drug shifted the voltage of activation to more negative potential (ΔV1/2 = 11.8 ± 0.3 mV). Lidocaine was tested also on heteromeric HCN1-HCN2, where both the current amplitude (38.7% reduction) and the voltage of activation (-10.1mV shift of V v1/2) were affected. Estimated IC50 values (HCN1, 67.6 ± 9.6 μM; HCN2, 66.8 ± 15.3 μM; HCN1-HCN2, 51.6 ± 9.5 μM) were close to that previously found on DRG neurons (99±4 μM) [165]. Removing one ethyl group on the basic nitrogen gave monoethyl glycinexylidide (MEGX) which is one of lidocaine’s metabolites. MEGX, at the same 100 μM concentration produced a more pronounced decrease in current amplitude on all the tested isoforms (HCN1, 54.7 ± 2.8%; HCN2, 59.3 ± 2.8%; HCN1-HCN2, 51.0 ± 7.7%, HCN4, 48.8±6.6%) while the effect on the activation voltage was similar to that of lidocaine. Therefore, the secondary amine MEGX seemed slightly more potent than the parent compound.
Lidocaine and other antiarrhythmic drugs, including another local anesthetic, mexiletine, have been tested on rabbit HCN4 heterologously expressed in HEK293 cells; this study was made in order to assess the activity of antiarrhythmic compounds on ion channels, which are not their primary site of action, possibly leading to a more rational use of such drugs [167]. The blocking properties were measured at physiological voltage ranges (-70 mV), where the effect of lidocaine (30 μM) was minimal, leading to an IC50 value of 276 μM; the IC50 value for mexiletine was slightly higher (309 μM). The authors concluded that the inhibitory effect of these two compounds on HCN4 in the clinical setting would be small. Other antiarrhythmic drugs were tested in the same conditions, and a similar conclusion was drawn also for quinidine, disopyramide, cibenzoline, aprindine, flecainide, propranolol, sotalol, verapamil and propafenone; only for the latter drug the IC50 was in the low micromolar range (14.3 μM). On the contrary, two other compounds were found to inhibit HCN4 channels with IC50 values close to therapeutic concentrations: bepridil (IC50 4.9 μM) and amiodarone (IC50 4.5 μM). Interestingly, in a similar study made on myocytes from human right atria appendage, Hoppe and Beuckelmann did not find reduction of f-current size or change in activation kinetic after application of 10 μM flecainide, sotalol, verapamil and amiodarone; a blocking activity was found only for propafenone which, at the same concentration, shifted the half-maximal current activation voltage by –5.2±0.4 mV [168].
While the effect of bepridil on hyperpolarization activated current has not been further investigated, amiodarone (Fig. 9) has been tested also on human HCN1, 2 and 4 isoforms expressed in Xenopus oocytes [169]. The compound blocked the current mediated by the three isoforms with some preference for HCN4 (IC50 2.1±1.9 μM) with respect to HCN2 (IC50 8.2±4.2 μM) and HCN1 (IC50 46.3±11.7μM), without modifying the voltage dependence of activation. The blockade by amiodarone was use-dependent; further evidence prompted the authors to suggest that the binding site is located on the intracellular site. The blocking properties of amiodarone have been demonstrated also on native tissues, such as neonatal rat ventricular myocytes [170] and in hypertrophic ventricular myocytes from spontaneously hypertensive rats (SHR) [171]. In the latter study, the IC50 on cardiomyocytes from SHR and from normal rats (Wistar-Kyoto, WKY) (respectively, 4.9 ± 1.2 μM and 6.9 ± 1.3 μM at -120mV) were consistent with those found in oocytes: values are intermediate between those found on HCN2 and HCN4 isoforms, which mediate If in ventricular tissues. In addition, pretreatment of ventricular myocytes from SHR with amiodarone reduced If density by reducing the expression of both isoforms.
Dronedarone (Fig. 9) is a structural analog of amiodarone, approved in 2009 for the treatment of atrial fibrillation [172]. As well as its parent compound, dronedarone is able to block beta-adrenergic receptor, as well as Na+, K+, Ca2+ and HCN channels. On hHCN4 channel expressed in CHO cells, dronedarone is able to reduce hyperpolarization-activated current with an IC50 value of 1.0 ± 0.1 μM at – 110 mV. Under similar conditions, the IC50 values for amiodarone and ivabradine were 0.8±0.1 μM and 1.1±0.2 μM, respectively [173]. Further studies point to If blockade as the primary mechanism for the bradycardic effect of dronedarone, with no contribution due to the beta-adrenergic antagonistic activity [174]. Studies in anesthetized pigs confirmed that If inhibition is the mechanism underlining the reduction of AV conduction during atrial fibrillation, an effect found in clinical trials after administration of this drug [175].
Other Compounds (Fig. 10)
Niflumic acid is a non steroidal anti-inflammatory drug with a complex activity on several ion channels. Its ability to inhibit hyperpolarization-activated current has been demonstrated on rabbit SAN myocytes and on newt rod photoreceptors [176-177]: in both cases the inhibitory effect was voltage dependent, being weaker at hyperpolarized potentials and higher at potentials close to the physiological value. The activation curve was shifted to more negative voltages; on SAN, where the most abundant isoform is HCN4 [178], the calculated ED50 value was 10.64 μM and the maximal shift was around -8 mV. On rod photoreceptors, mainly expressing the HCN1 isoform [179], niflumic acid behaved as a closed state blocker; 100 μM niflumic acid shifted V1/2 about -11 mV. From these and other evidences, both studies suggested an extracellular localization of the interaction site. More recently the effect of this drug was tested on mouse HCN2 channel expressed in Xenopus laevis oocytes. This study confirmed the characteristics of blockade found on native tissues; the authors investigated also on the localization of the binding site by means of site-directed mutagenesis, finding evidence that the drug interacts with the extracellular end of the S4 voltage sensor domains [180].
The inhibition of Ih by Capsazepine, a TRPV1 antagonist, was first described in 2004 [181]: this compound was found able to block hHCN1 channel expressed in the CHO cell line with IC50 7.9±0.7 μM. Capsazepine is however not selective: when tested on recombinant hHCN2 and hHCN4 isoforms expressed in HEK293 cells, its IC50 values were comparable, being 6.1±0.8 and 5.8±0.5 μM, respectively [182].
1-Methyl-4-phenylpiperidinium iodide (MPP+), a neurotoxin usually used to induce Parkinson’s disease in experimental animals, has been found to block Ih in nigral dopaminergic neurons [83]. MPP+, used at concentrations similar to those used to induce nigrostriatal degeneration in rodents, blocked Ih in a dose- and voltage-dependent manner: 50 μM MPP+ shifted Ih activation curve towards negative potentials, and reduced Ih amplitude to 44% of maximum at – 75 mV, while the current was nearly unchanged at -135 mV. The IC50 at -75 mV was 7.74 μM, and the maximal inhibition was 47%. Interestingly, MPP+ did not block Ih in CA1 pyramidal neurons, suggesting selectivity for the isoform(s) present in the dopaminergic neurons and lack of effect on HCN1.
The activity of nicotine on HCN channels was first demonstrated in Oriens-lacunosum moleculare (O-LM) cells, a subpopulation of GABAergic interneurons in the CA1 hippocampal area [183], and later in lateral septum (LS), a nucleus that is modulated by excitatory afferents from the hippocampus [184]. In both cases the modulation of neuronal excitability induced by nicotine was not prevented by nicotinic antagonists, and it was characterized as a direct interaction with the HCN channel. On O-LM neurons nicotine dose-dependently blocked Ih, the maximal inhibition being 39%; the analysis of the dose response curve yielded an EC50 of 62 nM, with a Hill coefficient (7.3) suggesting cooperativity. The blockade of Ih was minimal at -70 mV, but higher at more negative potentials (-120 mV). Another nicotinic agonist, epibatidine, was also effective on these neurons, while acetylcholine was not. Docking studies using a homology model of the HCN2 channel in the open state [185] predicted that nicotine and epibatidine bind inside the inner cavity of the channel, close to the mid region of the S6 domain, in a position similar to that predicted for ZD7288 and cilobradine [186].
Several studies have shown that the antibiotic minocycline could be used as an antinociceptive agent in pain management (see ref. [187] and references therein). To see whether the mechanism of antinociceptive activity could be related to HCN modulation, Liu et al. tested this drug on substantia gelatinosa neurons in spinal dorsal horns. Minocycline shifted Ih current activation to more negative voltages and decreased Ih amplitude in a reversible and concentration-dependent manner (IC50 = 41 μM) [187]; maximal reduction was 40%. Several agents (TTX, CNQX, D-APV, bicuculine and strychnine) were unable to prevent the reduction of Ih induced by minocycline, indicating a direct interaction with the postsynaptic HCN channel. The isoform mainly involved in this activity could be HCN1 and HCN3, as inferred from experiments involving the cAMP activator forskolin. The drug was not effective when applied from the intracellular side, suggesting an extracellular location of the binding site.
Acehytisine is an alkaloid extracted from the root of Aconitum coreanum, a plant used in the traditional Chinese medicine to treat heart diseases; the compound has been approved in 2005 in China for the treatment of paroxysmal supraventricular tachycardia. Acehytisine is a multi-ion channel blocker endowed with antiarrhythmic properties, interacting with calcium, sodium and potassium channels [188]; on rabbit SAN, the drug slowed down spontaneous firing rate and the rate of diastolic depolarization [189]. More recently, the effect of acehytisine on If has been studied both in rabbit SAN and in Xenopus oocytes expressing hHCN4 channels. On SAN, acehytisine blocked f-current with a maximum of 89.2 ± 8.2% (at 100 µM) and with an IC50 9.9 ± 0.3 µM. On HCN4 expressed in Xenopus oocytes the IC50 was somehow higher (64.9 ± 8.6 µM). The drug shifted the activation curves to more negative potentials; the author demonstrated that the blockade was voltage-independent and not use-dependent. These experiments suggest that acehytisine is a close-channel blocker that binds to the extracellular domain of the channel [188].
Eugenol is a phenol used in dentistry to relief tooth pain; its analgesic activity has been associated to inhibition of sodium and calcium voltage gated channels. When tested on medium- and large-size trigeminal ganglion neuron, a tissue expressing mainly HCN1 and HCN2 channel isoforms [190-191], eugenol was able to inhibit Ih at concentration lower than those required to block voltage-gated sodium channels (IC50 157 μM), and to revert mechanical allodynia [192]. Interestingly, since the activity of eugenol was attenuated in presence of cAMP analogues (8-Br-cAMP, Sp-cAMPS and Rp-cAMPS), the authors suggested that these molecules competed for the same binding site on the CNBD.
Genistein is a phytoestrogen that affects f-current in different preparations; whether this effect is due to inhibition of phosphorylation (the drug is a tyrosine kinase inhibitor) or a direct interaction with the channel (reviewed in ref. [193]) is controversial. New evidence in favor to a direct binding of genistein to the channel comes from a recent paper by Rozario et al. [194], where the effect of genistein was measured on wt mHCN1 and mHCN2 and on mutants where a critical arginine in the CNBD has been replaced with glutamate (mHCN1-R538E and mHCN2-R591E). This modification reduces the interaction of cAMP with the channel [195]. The authors concluded that genistein exerts an allosteric effect on the channel which changes opening and cAMP coupling. This activity is independent from tyrosine kinase inhibition, but requires the presence of the intact C-linker domain. Unfortunately, this study could not provide evidence on the location of genistein binding site.
Tanshinone is a constituent of the lipophilic fraction of Danshen, the dried root of Salvia miltiorrhiza, widely used in China for the treatment of cardiovascular and cerebrovascular diseases. The activity of a water-soluble from of tanshinone IIA, the sodium sulphonate salt, has been assessed on mHCN2 and hHCN1 channels heterologously expressed in Xenopus oocytes, where the drug enhanced the initial instantaneous current and slowed channel activation and deactivation processes without affecting the voltage-dependent current. The effect was more pronounced on HCN2 compared to HCN1, but it was amplified by the co-expression of HCN2 with MiRP1. However, this enhancement of instantaneous current was thought not sufficient to increase the heart rate in vivo, possibly because the drug may interact with multiple targets [196]. Indeed, a decrease of sinus rhythm, among many other effects, has been reported for this drug [197].
Fisetin is a naturally occurring flavonoid [198], which potentiates HCN2 mediated current [199]. When tested on nHCN2 expressed in Xenopus oocytes, fisetin significantly shifted the V1⁄2 toward less negative potential, thus facilitating channel opening, with a EC50 1.8±0.3μM. Compared to cAMP, fisetin behaved as partial agonist, the shift of V1⁄2 of 30 μM fisetin being half of that produced by 10 mM cAMP (8.9mV and 17.5 mV, respectively). Moreover, the effects of the two compounds were not additive, indicating a common interaction site. Fluorescence-based assays confirmed that fisetin binds to the CNBD; [1H]-[13C] HSQC NMR spectroscopy suggested that the binding site was close to Met572, a residue located at the at the entrance of the cAMP binding pocket.
Gabapentin is an anticonvulsant drug, commonly used to treat different kinds of pain [200]. When tested on CA1 pyramidal cells, Gabapentin increased Ih amplitude in a concentration dependent way, by increasing conductance without affecting activation properties, with a cAMP independent mechanism [201]. Quantitatively, the increase of Ih was about 34% using 100 μM gabapentin. A similar effect was demonstrated also on hippocampal CA1 stratum oriens non-pyramidal neurons; interestingly, pretreatment with ZD7288 completely prevented the effect of the drug [202]. Ih activating properties have been demonstrated also for lamotrigine, another anticonvulsant, unrelated to gabapentin either structurally or regarding the mechanism of antiepileptic activity. As for gabapentin, the potentiation of Ih produced by lamotrigine in hippocampal CA1 stratum oriens non-pyramidal interneurons was due to an increase in HCN conductance without a change in the voltage dependence of activation [203]. On the contrary, on pyramidal neurons Poolos et al. found that lamotrigine shifted the activation curve to less negative potential (ΔV1⁄2 ~11 mV) without increasing maximal Ih at hyperpolarized potential (-115 mV) [204].
The potency of selected blockers reported in sections 4 and 5 are summarized in Table 1.
6. Cyclic nucleotides and analogues
cAMP is a physiological modulator of HCN channels; as reported in section 2, its interaction with the cyclic nucleotide binding domain (CNBD) shifts the voltage dependence of activation to more positive values, with different sensitivity according to the isoform. Other endogenous cyclic nucleotides can modulate HCN channels: when tested on recombinant channels expressed in HEK293 cells, cytidine-3',5'-cyclic monophosphate (cCMP) activates HCN2 and HCN4, but not HCN1 and HCN3 [55]. The maximal effect of cCMP was only 60% with respect to cAMP, and the EC50 value on HCN2 was about 30-fold higher; the activity of uridine-3',5'-cyclic monophosphate (cUMP) on HCN2 and HCN4 was comparable to that of cCMP. Guanosine-3',5'-cyclic monophosphate (cGMP) has the same efficacy as cAMP, but lower apparent affinity (see refs [15] and [21] and citations therein).
The structure of the carboxy terminal part of HCN channels, including the CNBD and the C-linker, alone and in complex with cAMP and cGMP, is known from X-ray crystallography. The two cyclic nucleotides bind with the purine ring in different orientations with respect to the ribose-cyclic phosphate moiety, being anti for cAMP and syn for cGMP. The affinity of a series of cAMP analogues has been studied by means of fluorescence polarization techniques in two different conditions: on a soluble protein construct, made by replacing the CNBD of the cAMP receptor protein (CRP) with that of the rat HCN2 ion channel [207] and, more recently, on the monomeric cytosolic C-terminal domain (comprising the CNBD and the C-linker) of mHCN1, hHCN2 and hHCN4 channels [208]. In the latter study, the affinity of 47 analogues, varied on the purine/pyrimidine moiety, the ribose ring and the cyclic phosphate, were determined, allowing the understanding of structure-activity relationship in this class of molecules. As expected, the affinity of cGMP for the CNBD of the three isoforms was found 3-7 times lower than that of cAMP; for other naturally occurring cyclic nucleotides (cCMP, cUMP, inosine-3’,5’-cyclic monophosphate cIMP) the EC50 values were further reduced (from 20- to 50-fold lower than cAMP), while it was not possible to detect interaction between cTMP (thymidine-3′,5′-cyclic monophosphate) and the HCN C-terminal domain. The replacement of an oxygen atom in the cyclic phosphate with sulfur, or the methylation of the 2’-hydroxy group in the ribose ring was detrimental for affinity on all the three isoforms, highlighting the crucial role of this part of the molecule in the interaction. Some modification on the purine ring gave compounds with improved affinity with respect to cAMP and cGMP: as expected from the high affinity of 8-Fluo-cAMP and 8-Fluo-cGMP, the 8 position can tolerate large substituents, such as a 6-aminohexylamino or 6-aminohexylthio group. As far as isoform selectivity is concerned, the compounds were generally not able to discriminate between HCN1, HCN2 and HCN4 CNBD, the only interesting molecule being 8-Br-cGMP, which was less potent than cAMP and cGMP, but which displayed a preference for HCN1 with respect to HCN2 and HCN4 (7-fold and 9-fold, respectively). The most interesting finding was that the 7-carba analogue of cAMP (7-CH-cAMP) binds with higher affinity (from 65-fold on HCN2 to >100-fold on HCN1 and HCN4) with respect to the physiological modulator. The interaction of 7-CH-cAMP with HCN4 was further characterized by means of X-ray crystallography, Isothermal Titration Calorimetry (ITC), Surface Plasmon Resonance (SPR), and electrophysiology. On hHCN4 expressed in HEK cells 7-CH-cAMP behaved as agonist, shifting the activation curve to more positive potential, with a potency 4 times higher than cAMP. 7-CH-cAMP binds with higher affinity to HCN channels with respect to PKA and Epac [208].
7. The location of the interaction site(s)
While the structure of the whole channel is not known at the moment, several crystal structures are available for the intracellular C-terminal portion of HCN1 [209], HCN2 [210-211] [209, 212], HCN4 [208-209, 213], and the sea urchin sperm SPIH channels [214]; this allowed detailed studies on the interaction of cyclic nucleotides and analogues, as outlined in the previous section. More important, in the cytosolic domain of the HCN4 channel an additional binding site has been recently discovered, which could be exploited for selective modulation.
A recent crystal structure of the cytosolic C-terminal fragment of HCN4 bound to cGMP allowed the discovery of a new interaction site [213], located at the interface between the CNBD and the C-linker. This cavity is wide enough to allow the binding of cyclic dinucleotides, such as c-di-GMP, which did not show activity when tested alone (100 μM), but which was able to completely revert the positive shift of the activation curve induced by 15 μM cAMP, behaving as an
antagonist. Docking studies using the X-ray structure of the C-terminal domain of HCN1 and HCN2 suggested that c-di-GMP would not have affinity for these two isoforms, indicating that this additional site is relevant only for the HCN4 channel; this hypothesis was further supported by electrophysiological studies. Virtual screening of a library of commercially-available compounds led to the discovery of a series of molecules, structurally unrelated to cyclic dinucleotides, which were predicted to interact with this site; one of them, called BPU, was found able to block the activity of cAMP at submicromolar doses (IC50 0.42±0.1 μM, hHCN4 expressed in HEK293 cells). Substances interacting with this site could represent a new class of isoform selective HCN4 blockers.
The cytosolic C-terminal domain harbors also the binding site of TRIP8b: this protein inhibits channel activation antagonizing the effect of cAMP. Saponaro et al. have recently studied the cAMP-free HCN2 CNBD by means of NMR spectroscopy and compared it to the cAMP-bound HCN2 CNBD crystal structure, reconstructing the movements induced by cAMP [215]. TRIP8b is proposed to stabilize the cAMP-free conformation by interacting with the terminal part of the channel formed by the C-helix and the N-bundle loop (helices E’ and A).
Since the compounds reported in sections 4 and 5 substantially differ in their chemical structure, it is reasonable to hypothesize that they bind to different sites. Indeed, for some of them the interaction site is suggested to be located on the extracellular side (niflumic acid [180], loperamide [147], tramadol [151], clonidine [17]), in the intracellular region of the pore (ZD7288 [186], cilobradine and ivabradine [186, 216], nicotine [183], lidocaine [165], amiodarone [169]) or within the cyclic nucleotide binding domain (eugenol [192], fisetin [199])
The location of the interaction site of ZD7288 and ivabradine has been studied by means of site-directed mutagenesis combined with electrophysiology. By comparing the different properties of ZD7288 in the blockade of HCN1 and SPIH channels, Shin et al. identified three residues in the S6 region (Y375, M377 and V379 in the mHCN1 channel) involved in ZD7288 interaction, which are likely to line the pore [205]. Some years later, Chan and co-workers performed, on the same isoform, mutations in the selectivity filter and the outer and the inner pore vestibules, finding that the three residues located in the inner S6 domain, MFV377–379, play a significant role in the mHCN1 channel blockade induced by ZD7288 [206].
By using an alanine scanning mutagenesis approach on mHCN2, Cheng et al. found that two mutations, A425G and I432A, significantly reduced the current block induced by ZD7288 or by cilobradine [186]. The mutation of I432 with alanine caused a 150-fold reduction of ZD7288 potency, while with leucine, valine or phenylalanine the drop was less than 3-fold, highlighting the importance of a hydrophobic residue in this position. Sequence alignment of mHCN1 and mHCN2 showed that I432 corresponds to V379 in mHCN1. The interaction of ZD7288 with the mHCN2 channel was visualized by means of docking studies on a homology model of the channel, built from the crystal coordinates of KscA K+ channel from Streptomyces lividans [217] and of the MthK channel [218], in a way similar to what reported by Giorgetti [185].These models predict that A425 and I432 face toward the central cavity of the pore; it has been also hypothesized [183] from docking studies, that they are involved also in the interaction of nicotine.
More recently, Di Francesco’s team used molecular modeling to explain the outcomes of a study in which they combined site directed mutagenesis and electrophysiology on hHCN4 expressed in HEK293 cells, with the aim to identify the interaction site of ivabradine [216]. The homology models of hHCN4 were built using the crystal coordinates of KscA K+ channel in the closed and open states, and several residues facing the internal cavity within the S6 segment and the lower part of the pore were mutated to alanine. As a confirmation of the previous studies on other isoforms, three residues were found to affect the activity of ivabradine: Y506, homologous to Y375 on HCN1, F509, homologous to Y378 on HCN1, and I510, homologous to V379 on HCN1 and I432 on HCN2. However, I510 is predicted not to interact with the ligand, but to stabilize the orientation of the close residue Y506.
The most interesting information came from the inspection of the binding mode of ivabradine. As expected, the dimensions of the cavity differ in the closed and open state, being, in the latter, approximately 11 Å wider. The conformation of the ligand was predicted to change from an extended conformation (closed state) to a bent conformation (open state) resembling the arrangement found in the crystal structure of ivabradine hydrochloride [219]. In addition, the ligand seems to be stabilized mainly by Van der Waals, hydrophobic and π stacking interactions; the authors suggest that a hydrogen bond can be dynamically formed between the protonated nitrogen atom and the carbonyl oxygen of one of the C478 residues of the selectivity filter, which are 3.1-3.7 Å away from the quaternary nitrogen atom in the channel closed state. This interaction likely could stop ion flux.
The location of the interaction sites is outlined in Fig. (11).
Fig. (11).

Schematic representation of the binding sites of different modulators. Left: structure of the transmembrane domain of the channel, with indication of the interaction sites of Cs+ and ivabradine. Right: structure of the HCN2 cytosolic C-terminal domain (PDB code 1Q5O) with indication on the binding sites of cyclic nucleotides and the ancillary protein TRIP8b.
8. HCN and pathologies
As previously discussed, HCN channels generate pacemaker activity and modulate cellular excitability in the brain and heart, thus potentially being responsible of cardiomyopathies/cardiac arrhythmias and neurological disorders following HCN altered expression and function. Indeed, in last few years, multiple connections between HCN channels dysfunction and pathological states have been made (Table 2) and HCN-related heart and central/peripheral nervous system disorders will be discussed in this section as well as its therapeutic potential.
Table 2.
Proposed implications of HCN channels in human disorders and supposed mechanism.
| Organ/Tissue | Region/Cell type | Disease | HCN subunit | Link with Disease | Ref. | |
|---|---|---|---|---|---|---|
| Brain | Cortex | Idiop. Gen. epilepsy | 2 | Polimorfism/Loss-of-function mutation | [255-257] | |
| Hippo. | CA1 | Depression/anxiety | 1 | Reduced expression levels | [262] | |
| Febrile seizures | 1, 2 | seizure-induced expression changes | [244-247] | |||
| EC | TLE | 1 | [251] | |||
| Thalamus | Absence epilepsy | 2 | Constitutive loss of function | [248] | ||
| Midbrain | SNc DA | Parkinson's | 2, 4 | Functional alteration | [83, 259] | |
| VTA DA | Addiction | 2, 4 | [266-270] | |||
| Depression | 2, 4 | [263-264] | ||||
| DRG | Nociceptors | Pain sensation | 1, 2 | Hyperactivation (HCN2); overexpression (HCN1) | [19, 271-275] | |
| Heart | SAN | Bradycardia | 4 | Inducible loss of function | [7] | |
| SAN | Sinus arrhythmias | 2 | Constitutive loss of function | [248] | ||
| Ventricle | Reduction of late ventricular repolarization | 3 | [94] | |||
Hippo, Hippocampus; EC, Entorhinal Cortex; TLE; Temporal Lobe Epilepsy; SNc, Substantia nigra pars compacta, DA, dopaminergic neurons; VTA, Ventral Tegmental Area; DRG, Dorsal Root Ganglia; SAN, Senoatrial node.
Therapeutic Value and Potential of HCN Channel Blockade in Cardiac Diseases
In the heart, HCN channels are considered major determinant of spontaneous electrical activity of SAN cells and contribute to generate and regulate heart rate. Since the discovery of this peculiar function, HCN channels have been considered suitable targets for pharmacological interventions aimed to reduce heart rate, a useful effect in patients with cardiovascular diseases. Following on from extensive preclinical testing, ivabradine emerged as promising candidate to selectively decrease HCN-mediated current in SAN preparations. Subsequent studies in both animal models and humans have documented that ivabradine decreases heart rate to an extent similar to that seen with clinical doses of beta-blockers [220-221]. Currently ivabradine is clinically used in the treatment of stable angina pectoris and cardiac failure as selective bradycardic agent [222-226]. Its use is recommended in patients unable to tolerate or with contra-indications to beta-blockers, because at variance with them, it does not directly affect myocardial systolic or diastolic function, nor it reduces blood pressure [227] or causes bronco-constriction in asthmatic or COPD subject. Ivabradine is also used in addition to beta-blockers in patients inadequately controlled with an optimal beta-blocker dose.
In line with the clinical benefit observed in patients with heart failure, experimental evidence in animal models have further investigated the effect of heart rate reduction with ivabradine on the functional remodeling of the hypertrophic ventricle, which is particularly prone to develop arrhythmias. In these settings chronic treatment with ivabradine ameliorated cardiac function and counteracted the global electrophysiological remodeling of the hypertrophic ventricle [228-229]. The mechanism(s) responsible for the benefit of heart rate lowering drug in heart failure and related pro-arrhythmic alterations remain elusive both in humans and experimental models and require additional investigations.
Recently, experimental data have shown that HCN channels are also functionally expressed in the atrioventricular node, suggesting an additional role of HCN channel in cardiac impulse conduction from atria to ventricle [90]. This observation led to hypothesize that HCN blockade in atrioventricular node could be useful in the setting of atrial fibrillation to slow cardiac conduction velocity and reduce ventricular rate. Subsequent investigation in animal models of atrial fibrillation have indeed demonstrated that ivabradine reduced cardiac conduction velocity without affecting cardiac contractility or mean arterial pressure [92]. Moreover, when ivabradine is co-administered with ranolazine, used to reduce atrial dominant frequency [230] in the fibrillating atria, reduction of conduction velocity is greater than the expected additive effects of the two drugs. Despite further studies are necessary to confirm and extend these results, ivabradine alone or in combination with ranolazine, has the potential to represent a novel pharmacological strategy to control heart rate in atrial fibrillation.
Atrial and Ventricular Arrhythmias
After the identification of HCN channel in the human atria, different studies have suggested that chronic atrial fibrillation alters channel function through intrinsic modifications of gating and/or stimulation by specific hormonal signals [231-233]. Indeed, in human chronic atrial fibrillation there is evidence showing that HCN current is significantly enhanced (+10%) at physiologically relevant voltages (-70 mV), thus strengthening the hypothesis of a contribution of HCN channels to atrial electrogenesis [234]. Conclusive evidence on the molecular mechanisms underlying HCN gain-of-function in atrial fibrillation still remains elusive, although the occurrence of transcriptional and post-transcriptional regulatory mechanisms has been demonstrated. In the context of atrial fibrillation, two physiologic mediators, e.g. serotonin release during platelet aggregation and adrenergic stimulation, both triggering factors of atrial arrhythmias, may enhance HCN channel function and exacerbate the consequences of HCN gain-of-function. Both signals increase current amplitude via activation of G protein coupled receptors and cAMP formation [232-233]. Importantly, serotoninergic and adrenergic stimulation remain operative in chronic atrial fibrillation, thus supporting the hypothesis that local condition in the atria may promote HCN channel involvement in electrogenesis [233, 235].
Expression and function of HCN channels in the hypertrophic ventricles are better defined and a multitude of studies aiming to assess the electrophysiological mechanisms of ventricular arrhythmias have proposed HCN channels as common marker of functional remodeling induced by hypertrophy and possibly associated to enhanced arrhythmogenesis (reviewed in [93]). Both rat and human hypertrophic ventricles express functional HCN channels, whose overall amount is related to the severity of myocardial hypertrophy [236-239] and to the disease etiology [240-242]. HCN overexpression in the ventricles occurs through transcriptional up-regulation of HCN4 and HCN2 isoforms which are then translated into functional proteins [43, 229, 237]. Interestingly, ventricular HCN channels share similar electrophysiological properties, sensitivity to autonomic transmitters and pharmacological blockade with sinoatrial node cells and atrial cardiomyocytes. However current activation threshold is much more negative (about -70 mV), a property that at ventricular resting membrane potentials (-80/-90 mV) favors a contribution of the HCN current to ventricular electrogenesis. Such a function would be further strengthened when other electrophysiological alterations, such as the reduced expression of inwardly rectifying potassium current, and the elevated β-adrenergic stimulation promote their contribution to electrogenesis. Although the arrhythmogenic role of HCN enhancement in the working myocardium is proposed based on the above reported studies, the hypothesis remains unproven in the clinical setting, since ivabradine administration in cardiovascular patients does not decrease cardiovascular deaths, including arrhythmic deaths [222-223, 226].
Epilepsy
Epilepsy is a group of neurological disorders characterized by epileptic seizures. Because of their role in pacemaker function, defective HCN channels are natural candidates for contributing to epileptogenesis. Rat model of childhood febrile seizures (hyperthermia model of febrile seizure) [243-244] was the first observation of HCN involvement in epilepsy. In this model, an enhanced Ih activity was linked in hippocampal CA1 neurons of epileptic rats to an increased probability of rebound depolarizations and action potential firing [244]. Expression of HCN channels revealed a decrease of the ratio of HCN1 vs. HCN2 protein [245-246] being HCN1 down-regulated while HCN2 up-regulated in hippocampal pyramidal neurons. Although the mechanism underlying changes in HCN channel expression levels in response to seizures is unclear, HCN1 channel expression is probably due to a transcriptionally regulated process involving activation of calmodulin (CaM) kinase II and Ca2+ entry via AMPA receptors while up-regulation of HCN2 was found to be CaM kinase II independent [247]. Recently, a co-immunoprecipitation study showed, along with altered hippocampal HCN1 and HCN2 expression levels, a long-lasting increase in the levels of heteromeric HCN1/2 channels in hippocampal neurons [246]. These data point out to a complex mixture of HCN1 and HCN2 homomers as well as of HCN1/2 heteromers in the generation and the long-term maintenance of hippocampal hyperexcitability. However, the strongest evidence linking Ih channels to epilepsy remains the HCN2-deficient mice which show absence epilepsy and spike-and-wave discharges [248]. Indeed, in thalamocortical and thalamic reticular neurons of these mice, Ih was found to be almost completely abolished [82, 248-250] thus shifting the resting membrane potential of these neurons to hyperpolarized potentials. Interestingly, at more hyperpolarized resting membrane potential, the fraction of T-type calcium channels present in the closed state (a state in which these channels are closed but activable) is higher than in wild-type cells where more T-type channels is in the inactivated state. Thus, the susceptibility of HCN2-deficient neurons to produce a Ca2+ spike is higher than that of wild-type neurons because the T-type channels present in these cells are easier to be activated by excitatory inputs thus firing in burst mode than wild-type neurons [248].
Moreover, down-regulation of Ih was also found in kainic acid rat model of temporal lobe epilepsy (TLE) [251] where layer III pyramidal neurons of the entorhinal cortex (EC) were found hyperexcitable, thus generating profound synchronous network activity through modulation of dendritic integration [85, 252-253]. Indeed, reduction of Ih densities in dendrites causes an increase of the dendritic input resistance which, in turn, increases dendritic EPSP summation and EPSP-spike coupling. Rat and mouse models clearly indicated that impaired HCN channel function or expression is associated with epileptiform activity. However, such a strong correlation, although likely, is so far missing in humans. Recently, changes in HCN channel expression have been found in the dentate gyrus from patients with temporal lobe epilepsy and severe hippocampal sclerosis [254]. Furthermore, mutations in HCN1 and HCN2 subunits have been discovered in patients with idiopathic generalized epilepsy [255-256] and with early-onset epileptic encephalopathy [257]. One such mutation, a recessive loss-of-function mutation in HCN2 that has been associated with idiopathic generalized epilepsy, has been shown to increase the excitability of rat cortical neurons [256].
Parkinson Disease
Parkinson's disease (PD) is caused by the progressive degeneration of nigrostriatal dopaminergic (DA) neurons and resulting fall of dopamine levels in the dorsal striatum, a subcortical brain formation involved in movement control. One of the many pending questions in the genesis of PD is the striking differential vulnerability among closely related midbrain DA neurons, with Substantia Nigra pars compacta (SNc) undergoing significantly greater degeneration compared to Ventral Tegmental Area (VTA) DA neurons. Midbrain DA neurons express HCN channels (mainly 2 and 4) to an extent that varies among subpopulations, with SNc DA neurons showing the highest expression levels, also in terms of current amplitude [27-28, 258]. Interestingly, it was recently reported that HCN current amplitude is reduced in SNc DA neurons from MitoPark mice, a genetic mouse model in which a DA-targeted mitochondrial defect leads to selective nigrostriatal degeneration and PD-like phenotype. According to the authors, this defect marks early, asymptomatic disease stages and is not due to HCN transcript down-regulation [259]. As mentioned in section 5, MPP+, a toxin capable of inducing PD-like selective nigrostriatal degeneration in primates and rodents, is a voltage-dependent inhibitor of HCN channels in SNc DA neurons in vitro. This block is effective at physiological potentials and results in increased dendritic excitability and overall responsiveness to excitatory synaptic inputs [83]. Altogether, these findings suggest that altered HCN channels expression or function in experimental PD models deserves in-depth scrutiny, especially within the context of differential vulnerability.
Depression
Recent studies have pointed out to the role of HCN in modulating motor learning deficits and enhancing resistance to multiple tasks of behavioral despair with high predictive validity for antidepressant efficacy of Ih current in mice. In particular, Lewis and co-workers generated a knock-out mouse lacking the HCN channel auxiliary subunit TRIP8b which significantly regulates the voltage gating and kinetics of Ih [68, 260-261]. Eliminating expression of TRIP8b dramatically reduced Ih expression in hippocampal pyramidal neurons showing that three different lines of knockout mice (HCN1, HCN2, and TRIP8b) with elimination or reduction of functional Ih displayed antidepressant-like behaviors [69].
Furthermore, in order to determine which brain regions are important for this antidepressant-like behavior, a lentiviral shRNA-HCN1 system that allowed for focused silencing of the HCN1 gene in a small population of neurons was created [262]. Indeed, knocking down of HCN1 channels increased cellular excitability and resulted in physiological changes consistent with a reduction of Ih. Moreover, rats infused with lentiviral shRNA-HCN1 in the dorsal hippocampal CA1 region displayed antidepressant- and anxiolytic-like behaviors associated with widespread enhancement of hippocampal activity and up-regulation of BDNF-mTOR signaling pathways thus suggesting an HCN1 dependent potential target for treatment of anxiety and depression disorders. Finally, following a social defeat stress model of depression, depressed mice displayed hyperactivity of VTA DA neurons, caused by an up-regulated hyperpolarization-activated current [263], a well-known excitatory driving force in these neurons [258, 264-265]. In addition, pharmacological reduction of the increased Ih in susceptible mice reversed depression-related symptoms and chronic antidepressant fluoxetine treatment reduced the hyperactivity and normalized Ih in these neurons [264]. Thus, hyperactivity and increased excitatory Ih in VTA DA neurons are both pathophysiological adaptations which could be promising for antidepressant action.
Addiction
VTA DA neurons in the mesolimbic/mescortical pathways are implicated in the mechanisms leading to positive reinforcement and addiction. The firing rate of these neurons is positively correlated to rewarding stimuli [266]. HCN channels have been detected in these neurons and reported to govern spontaneous firing [267]. Patch clamp recordings in rat midbrain slices have shown that ethanol reversibly enhances Ih by right-shifting its voltage dependence and increasing maximum conductance. This leads to an augmented spontaneous firing activity [268]. In an animal model of cocaine sensitization, it was been reported that behavioral changes are accompanied by reduced Ih current density, without changes in single-unit kinetic properties [269]. Although the literature on the real implication of HCN channels in the neurophysiological basis of addiction is still limited, the hypothesis that their function is a common target for different substances of abuse deserves careful consideration for the obvious clinical relevance [270].
Pain
HCN channels (in particular, HCN1) are expressed in larger DRG neurons [271-272]. Surprisingly, however, recent work by Dr. McNaughton’s team have found that HCN1-KO mice only exhibited subtle changes in chronic pain behaviors compared with wild type (WT) mice [273]. Indeed, another HCN subtype, HCN2, in a subset of small DRG neurons has been demonstrated to be essential for both inflammatory and neuropathic pain [274]. By knocking out HCN2 in NaV1.8 expressing DRG neurons (nociceptive sensory neurons), it was found that pain behaviors are largely relieved in NaV1.8-HCN2 KO mice. These data further pinpoint that the role of HCN2 in chronic pain is mediated through driving action potential firings in NaV1.8-expressing nociceptors and confirmed the importance of HCN channels in pain expecting that HCN2 may be an interesting target of analgesics for chronic pain treatment [19]. However, developing a selective HCN2 blocker would be crucial and, to our knowledge, such blocker is still not available.
9. Conclusion and perspectives
HCN are considered suitable drug target for several pathologies [17]. Indeed, the modulation of these proteins can be a therapeutic strategy for many diseases, as highlighted in the previous section. Further support to this idea is based on the results from transgenic animals [18, 276-277] and on the links that have been founds between HCN channels mutations and pathologies [278-280]. Despite all this evidence, there is only one drug on the market, which is specifically designed to block HCN channels. Ivabradine is an extremely valuable tool to probe the therapeutic potential of HCN blockade in cardiac pathologies, but its bradycardic activity may limit its use to treat diseases involving HCN channels in other tissues. Therefore, one of the main issue that need to be addressed in this field is the discovery of isoform selective compounds as tool to get novel understanding of the physiologic and pathologic implications of single HCN channel isoforms.
HCN isoforms can be studied by means of expression in recombinant systems, where the activity of old and new compounds can be tested on homomeric channels. Although the cellular context may alter to some extent the behavior of the channel [281], this simple experimental model can be exploited to discover selective modulators, providing that the selectivity found on recombinant channels is maintained also in native tissues, where these proteins may exist as heteromeric combinations (see [15] and references therein), or may be strongly regulated by auxiliary proteins, such as TRIP8b or Nedd4-2. Indeed, some recent reports show that this selectivity can be maintained. For instance, compound 9, which showed higher activity on recombinant HCN4 compared to HCN2 and HCN1, resulted to be the more potent on SAN cells and Purkinje fibers with respect to DRG neurons [137]; the activity of compound 11, more potent on recombinant HCN1 with respect to HCN4, showed in vivo antihyperalgesic activity without affecting heart rate [139].
The availability of recombinant systems can speed up the discovery of new modulators by using the high throughput screening technology. Recently, such methods have been set up [140, 149] allowing to search for molecules, structurally unrelated to known modulators. By this way compound 11 has been discovered [139]; in addition, the HCN blocking properties of some known substances (CP-339,818 and loperamide) have been disclosed [150]. It is conceivable that in the future this method will be exploited more and more to yield new drugs.
As a different approach, it could be possible to take one of the molecules reported in sections 4 and 5 as a lead compound for further optimization. In this respect, some points need to be considered. First, some compounds (i.e. eugenol, minocycline, nicotine, tramadol, MPP+, gabapentin, lamotrigine) have been tested only in tissues; therefore, their activity may be strongly influenced by other systems, by homo- or hetero-tetramerization, by the presence of ancillary proteins and/or by the interaction with cell specific intracellular organic modulators. Second, these compounds, which are endowed with activity in the micromolar range (nicotine is still the only modulator for which an IC50 value in the nanomolar range has been reported), must be modified in order to improve potency on HCN channels and at the same time to minimize the interaction with the original target(s). This may not be an easy task, since several compounds are reported to interact with different macromolecules. For instance, developing a HCN modulator carrying an imidazoline ring could be very difficult, since it is necessary to avoid interaction with adrenergic receptors (α2 and α1) and imidazoline binding sites (IBS, I1 and I2). Finally, only some compounds in section 5 have been tested on different isoforms, and even fewer were found able to block one isoform with some preference over the others, as it has been shown, for instance, for amiodarone, which is 4-fold and 23-fold more potent on hHCN4 than on hHCN2 and hHCN1, respectively (Table 1). Maybe, a more extensive screening of modulators on different isoforms could give suggestions for the design of selective compounds.
Although in recent times important progress has been made in the crystallization of membrane proteins, a high resolution crystal structure of the whole HCN channels is not available yet. Knowing the three-dimensional structure of the transmembrane region would allow performing structure-based drug design, at least for ivabradine and analogues, since the binding site of this drug has been recently located and studied (see section 7). A real possibility to design new compounds is to consider the cytosolic part of the channel for which several x-ray structures are available. The interaction of cyclic nucleotides with the cAMP binding site has been studied in details, providing information on the structural requirements for high affinity binding in this class of molecules [208]; however, it would be still difficult to design a selective compound with similar structure, since this site is highly conserved not only in HCN channels but also in other cAMP-binding proteins. Nevertheless, this domain could be studied to search for new modulators using docking methods, as it has been done with the additional site found in the C-linker domain of HCN4 [213]: the discovery that this site exist only in the HCN4 cytosolic domain opens the way to design HCN4-selective modulators.
While the potential of HCN blockers in cardiac pathologies and in the management of pain is well established, also compounds able to increase hyperpolarization activated current may have a therapeutic potential. Indeed, the evidence reported in Table 2 suggests that positive and negative Ih modulators may both be valuable tools to understand the involvement of HCN channels in (physio)pathological processes, providing that they are endowed with isoform selectivity.
Therefore, only the discovery of selective compounds will test the real potential of HCN channels as drug targets for non-cardiac pathologies.
ACKNOWLEDGEMENTS
Declared none.
List of abbreviations
- CHO
chinese hamster ovary
- cAMP
adenosine-3',5'-cyclic monophosphate
- cCMP
cytidine-3',5'-cyclic monophosphate
- cGMP
guanosine-3',5'-cyclic monophosphate
- cUMP
uridine-3',5'-cyclic monophosphate
- CNBD
cyclic nucleotide binding domain
- CNS
central nervous system
- DRG
dorsal root ganglion
- EC
enthorinal cortex
- EPSP
excitatory post-synaptic potentials
- FRET
fluorescence resonance energy transfer
- KO
knock-out
- HCN
hyperpolarization-activated cyclic nucleotide gated channels
- HEK
human embrionic kidney
- IBS
imidazoline binding sites
- PIP2
phosphatidyl inositole diphosphate
- NMDA
N-methyl D-aspartate
- PNS
peripheral nervous system
- SAN
sino atrial node
- SHR
spontaneously hypertensive rats
- SNc
substantia nigra pars compacta
- TRIP8b
tetratricopeptide repeat-containing Rab8b-interacting protein
- VTA
ventral tegmental area
- WT
wild type
CONFLICT OF INTEREST
The author(s) confirm that this article content has no conflict of interest.
REFERENCES
- 1.DiFrancesco D., Ojeda C. Properties of the current If in the sino-atrial node of the rabbit compared with those of the current IK, in Purkinje fibres. J. Physiol. 1980;308:353–367. doi: 10.1113/jphysiol.1980.sp013475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vassalle M., Kotake H., Lin C.I. Pacemaker current, membrane resistance, and K+ in sheep cardiac Purkinje fibres. Cardiovasc. Res. 1992;26:383–391. doi: 10.1093/cvr/26.4.383. [DOI] [PubMed] [Google Scholar]
- 3.Bogdanov K.Y., Vinogradova T.M., Lakatta E.G. Sinoatrial Nodal Cell Ryanodine Receptor and Na+-Ca2+ Exchanger: Molecular Partners in Pacemaker Regulation. Circ. Res. 2001;88(12):1254–1258. doi: 10.1161/hh1201.092095. [DOI] [PubMed] [Google Scholar]
- 4.Protas L., Robinson R.B. Mibefradil, an ICa,T blocker, effectively blocks ICa,L in rabbit sinus node cells. Eur. J. Pharmacol. 2000;401(1):27–30. doi: 10.1016/s0014-2999(00)00364-2. [DOI] [PubMed] [Google Scholar]
- 5.DiFrancesco D., Tortora P. Direct activation of cardiac pacemaker channels by intracellular cyclic AMP. Nature. 1991;351:145–147. doi: 10.1038/351145a0. [DOI] [PubMed] [Google Scholar]
- 6.Wahl-Schott C., Fenske S., Biel M. HCN channels: new roles in sinoatrial node function. Curr. Opin. Pharmacol. 2014;15:83–90. doi: 10.1016/j.coph.2013.12.005. [DOI] [PubMed] [Google Scholar]
- 7.Baruscotti M., Bucchi A., Viscomi C., Mandelli G., Consalez G., Gnecchi-Rusconi T., Montano N., Casali K.R., Micheloni S., Barbuti A., DiFrancesco D. Deep bradycardia and heart block caused by inducible cardiac-specific knockout of the pacemaker channel gene HCN4. Proc. Natl. Acad. Sci. USA. 2011;108(4):1705–1710. doi: 10.1073/pnas.1010122108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shah M.M. Cortical HCN channels: function, trafficking and plasticity. J. Physiol. 2014;592(13):2711–2719. doi: 10.1113/jphysiol.2013.270058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Milanesi R., Baruscotti M., Gnecchi-Ruscone T., DiFrancesco D. Familial Sinus Bradycardia Associated with a Mutation in the Cardiac Pacemaker Channel. N. Engl. J. Med. 2006;354(2):151–157. doi: 10.1056/NEJMoa052475. [DOI] [PubMed] [Google Scholar]
- 10.Cerbai E., Mugelli A. If in non-pacemaker cells: Role and pharmacological implications. Pharmacol. Res. 2006;53(5):416–423. doi: 10.1016/j.phrs.2006.03.015. [DOI] [PubMed] [Google Scholar]
- 11.Jackson H.A., Marshall C.R., Accili E.A. Evolution and structural diversification of hyperpolarization-activated cyclic nucleotide-gated channel genes. Physiol. Genomics. 2007;29(3):231–245. doi: 10.1152/physiolgenomics.00142.2006. [DOI] [PubMed] [Google Scholar]
- 12.Hegle A.P., Nazzari H., Roth A., Angoli D., Accili E.A. Evolutionary emergence of N-glycosylation as a variable promoter of HCN channel surface expression. Am. J. Physiol. Cell Physiol. 2010;298(5):C1066–C1076. doi: 10.1152/ajpcell.00389.2009. [DOI] [PubMed] [Google Scholar]
- 13.Sartiani L., Bettiol E., Stillitano F., Mugelli A., Cerbai E., Jaconi M.E. Developmental Changes in Cardiomyocytes Differentiated from Human Embryonic Stem Cells: A Molecular and Electrophysiological Approach. Stem Cells. 2007;25(5):1136–1144. doi: 10.1634/stemcells.2006-0466. [DOI] [PubMed] [Google Scholar]
- 14.Bosman A., Sartiani L., Spinelli V., Del Lungo M., Stillitano F., Nosi D., Mugelli A., Cerbai E., Jaconi M. Molecular and Functional Evidence of HCN4 and Caveolin-3 Interaction During Cardiomyocyte Differentiation from Human Embryonic Stem Cells. Stem Cells Dev. 2013;22(11):1717–1727. doi: 10.1089/scd.2012.0247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Biel M., Wahl-Schott C., Michalakis S., Zong X. Hyperpolarization-Activated Cation Channels: From Genes to Function. Physiol. Rev. 2009;89(3):847–885. doi: 10.1152/physrev.00029.2008. [DOI] [PubMed] [Google Scholar]
- 16.Wahl-Schott C., Biel M. HCN channels: structure, cellular regulation and physiological function. Cell. Mol. Life Sci. 2009;66(3):470–494. doi: 10.1007/s00018-008-8525-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Postea O., Biel M. Exploring HCN channels as novel drug targets. Nat. Rev. Drug Discov. 2011;10:903–913. doi: 10.1038/nrd3576. [DOI] [PubMed] [Google Scholar]
- 18.Herrmann S., Hofmann F., Stieber J., Ludwig A. HCN channels in the heart: lessons from mouse mutants. Br. J. Pharmacol. 2012;166(2):501–509. doi: 10.1111/j.1476-5381.2011.01798.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Emery E.C., Young G.T., McNaughton P.A. HCN2 ion channels: an emerging role as the pacemakers of pain. Trends Pharmacol. Sci. 2012;33(8):456–463. doi: 10.1016/j.tips.2012.04.004. [DOI] [PubMed] [Google Scholar]
- 20.Benarroch E.E. HCN channels. Function and clinical implications. Neurology. 2013;80(3):304–310. doi: 10.1212/WNL.0b013e31827dec42. [DOI] [PubMed] [Google Scholar]
- 21.He C., Chen F., Li B., Hu Z. Neurophysiology of HCN channels: From cellular functions to multiple regulations. Prog. Neurobiol. 2014;112:1–23. doi: 10.1016/j.pneurobio.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 22.Herrmann S., Schnorr S., Ludwig A. HCN Channels—Modulators of Cardiac and Neuronal Excitability. Int. J. Mol. Sci. 2015;16(1):1429–1447. doi: 10.3390/ijms16011429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu F.H., Yarov-Yarovoy V., Gutman G.A., Catterall W.A. Overview of Molecular Relationships in the Voltage-Gated Ion Channel Superfamily. Pharmacol. Rev. 2005;57(4):387–395. doi: 10.1124/pr.57.4.13. [DOI] [PubMed] [Google Scholar]
- 24.Kaupp U.B., Seifert R. Molecular diversity of pacemaker ion channels. Annu. Rev. Physiol. 2001;63:235–257. doi: 10.1146/annurev.physiol.63.1.235. [DOI] [PubMed] [Google Scholar]
- 25.Craven K.B., Zagotta W.N. CNG and HCN channels: Two Peas, One Pod. Annu. Rev. Physiol. 2006;68:375–401. doi: 10.1146/annurev.physiol.68.040104.134728. [DOI] [PubMed] [Google Scholar]
- 26.Wainger B.J., DeGennaro M., Santoro B., Siegelbaum S.A., Tibbs G.R. Molecular mechanism of cAMP modulation of HCN pacemaker channels. Nature. 2001;411(6839):805–810. doi: 10.1038/35081088. [DOI] [PubMed] [Google Scholar]
- 27.Santoro B., Chen S., Lüthi A., Pavlidis P., Shumyatsky G.P., Tibbs G.R., Siegelbaum S.A. Molecular and Functional Heterogeneity of Hyperpolarization-Activated Pacemaker Channels in the Mouse CNS. J. Neurosci. 2000;20(14):5264–5275. doi: 10.1523/JNEUROSCI.20-14-05264.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Notomi T., Shigemoto R. Immunohistochemical localization of Ih channel subunits, HCN1–4, in the rat brain. J. Comp. Neurol. 2004;471(3):241–276. doi: 10.1002/cne.11039. [DOI] [PubMed] [Google Scholar]
- 29.Dufour M.A., Woodhouse A., Goaillard J-M. Somatodendritic ion channel expression in substantia nigra pars compacta dopaminergic neurons across postnatal development. J. Neurosci. Res. 2014;92(8):981–999. doi: 10.1002/jnr.23382. [DOI] [PubMed] [Google Scholar]
- 30.Hughes D.I., Boyle K.A., Kinnon C.M., Bilsland C., Quayle J.A., Callister R.J., Graham B.A. HCN4 subunit expression in fast-spiking interneurons of the rat spinal cord and hippocampus. Neuroscience. 2013;237(100):7–18. doi: 10.1016/j.neuroscience.2013.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ramakrishnan N.A., Drescher M.J., Khan K.M., Hatfield J.S., Drescher D.G. HCN1 and HCN2 proteins are expressed in cochlear hair cells: HCN1 can form a ternary complex with protocadherin 15 CD3 and F-actin-binding filamin A or can interact with HCN2. J. Biol. Chem. 2012;287(45):37628–37646. doi: 10.1074/jbc.M112.375832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Doan T.N., Stephans K., Ramirez A.N., Glazebrook P.A., Andresen M.C., Kunze D.L. Differential Distribution and Function of Hyperpolarization-Activated Channels in Sensory Neurons and Mechanosensitive Fibers. J. Neurosci. 2004;24(13):3335–3343. doi: 10.1523/JNEUROSCI.5156-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fyk-Kolodziej B., Pourcho R.G. Differential distribution of hyperpolarization-activated and cyclic nucleotide-gated channels in cone bipolar cells of the rat retina. J. Comp. Neurol. 2007;501(6):891–903. doi: 10.1002/cne.21287. [DOI] [PubMed] [Google Scholar]
- 34.Chaplan S.R., Guo H-Q., Lee D.H., Luo L., Liu C., Kuei C., Velumian A.A., Butler M.P., Brown S.M., Dubin A.E. Neuronal Hyperpolarization-Activated Pacemaker Channels Drive Neuropathic Pain. J. Neurosci. 2003;23:1169–1178. doi: 10.1523/JNEUROSCI.23-04-01169.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Galligan J.J., Tatsumi H., Shen K-Z., Surprenant A., North A. Cation current activated by hyperpolarization (Ih) in guinea pig enteric neurons. Am. J. Physiol. 1990;259:G966–G972. doi: 10.1152/ajpgi.1990.259.6.G966. [DOI] [PubMed] [Google Scholar]
- 36.Xiao J., Nguyen T.V., Ngui K., Strijbos P.J., Selmer I.S., Neylon C.B., Furness J.B. Molecular and functional analysis of hyperpolarization-activated nucleotide-gated (HCN) channels in the enteric nervous system. Neuroscience. 2004;129(3):603–614. doi: 10.1016/j.neuroscience.2004.08.027. [DOI] [PubMed] [Google Scholar]
- 37.Chen C. Hyperpolarization-activated current (Ih) in primary auditory neurons. Hear. Res. 1997;110(1–2):179–190. doi: 10.1016/s0378-5955(97)00078-6. [DOI] [PubMed] [Google Scholar]
- 38.Janigro D., Martenson M.E., Baumann T.K. Preferential Inhibition of Ih in Rat Trigeminal Ganglion Neurons by an Organic Blocker. J. Membr. Biol. 1997;160:101–109. doi: 10.1007/s002329900299. [DOI] [PubMed] [Google Scholar]
- 39.Roubille F., Tardif J-C. New Therapeutic Targets in Cardiology: Heart Failure and Arrhythmia: HCN Channels. Circulation. 2013;127(19):1986–1996. doi: 10.1161/CIRCULATIONAHA.112.000145. [DOI] [PubMed] [Google Scholar]
- 40.Herrmann S., Layh B., Ludwig A. Novel insights into the distribution of cardiac HCN channels: An expression study in the mouse heart. J. Mol. Cell. Cardiol. 2011;51(6):997–1006. doi: 10.1016/j.yjmcc.2011.09.005. [DOI] [PubMed] [Google Scholar]
- 41.Shi W., Wymore R., Yu H., Wu J., Wymore R.T., Pan Z., Robinson R.B., Dixon J.E., McKinnon D., Cohen I.S. Distribution and Prevalence of Hyperpolarization-Activated Cation Channel (HCN) mRNA Expression in Cardiac Tissues. Circ. Res. 1999;85(1):e1–e6. doi: 10.1161/01.res.85.1.e1. [DOI] [PubMed] [Google Scholar]
- 42.Ludwig A., Zong X., Jeglitsch M., Hofmann F., Biel M. A family of hyperpolarization-activated mammalian cation channels. Nature. 1998;393:587–591. doi: 10.1038/31255. [DOI] [PubMed] [Google Scholar]
- 43.Stillitano F., Lonardo G., Zicha S., Varro A., Cerbai E., Mugelli A., Nattel S. Molecular basis of funny current (If) in normal and failing human heart. J. Mol. Cell. Cardiol. 2008;45(2):289–299. doi: 10.1016/j.yjmcc.2008.04.013. [DOI] [PubMed] [Google Scholar]
- 44.Calejo A., Reverendo M., Silva V., Pereira P., Santos M.S., Zorec R., Gonçalves P. Differences in the expression pattern of HCN isoforms among mammalian tissues: sources and implications. Mol. Biol. Rep. 2014;41(1):297–307. doi: 10.1007/s11033-013-2862-2. [DOI] [PubMed] [Google Scholar]
- 45.Bolívar J.J., Tapia D., Arenas G., Castañón-Arreola M., Torres H., Galarraga E. A hyperpolarization-activated, cyclic nucleotide-gated, (Ih-like) cationic current and HCN gene expression in renal inner medullary collecting duct cells. Am. J. Physiol. Cell Physiol. 2008;294(4):C893–C906. doi: 10.1152/ajpcell.00616.2006. [DOI] [PubMed] [Google Scholar]
- 46.Hurtado R., Bub G., Herzlinger D. The pelvis-kidney junction contains HCN3, a hyperpolarization-activated cation channel that triggers ureter peristalsis. Kidney Int. 2010;77(6):500–508. doi: 10.1038/ki.2009.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.El-Kholy W., MacDonald P.E., Fox J.M., Bhattacharjee A., Xue T., Gao X., Zhang Y., Stieber J., Li R.A., Tsushima R.G., Wheeler M.B. Hyperpolarization-Activated Cyclic Nucleotide-Gated Channels in Pancreatic β-Cells. Mol. Endocrinol. 2007;21(3):753–764. doi: 10.1210/me.2006-0258. [DOI] [PubMed] [Google Scholar]
- 48.He P., Deng J., Zhong X., Zhou Z., Song B., Li L. Identification of a Hyperpolarization-activated Cyclic Nucleotide-gated Channel and Its Subtypes in the Urinary Bladder of the Rat. Urology. 2012;79(6):1411.e7–1411.e13. doi: 10.1016/j.urology.2012.01.037. [DOI] [PubMed] [Google Scholar]
- 49.Baruscotti M., Bucchi A., DiFrancesco D. Physiology and pharmacology of the cardiac pacemaker (“funny”) current. Pharmacol. Ther. 2005;107(1):59–79. doi: 10.1016/j.pharmthera.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 50.Han Y., Noam Y., Lewis A.S., Gallagher J.J., Wadman W.J., Baram T.Z., Chetkovich D.M. Trafficking and Gating of Hyperpolarization-activated Cyclic Nucleotide-gated Channels Are Regulated by Interaction with Tetratricopeptide Repeat-containing Rab8b-interacting Protein (TRIP8b) and Cyclic AMP at Distinct Sites. J. Biol. Chem. 2011;286(23):20823–20834. doi: 10.1074/jbc.M111.236125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Altomare C., Terragni B., Brioschi C., Milanesi R., Pagliuca C., Viscomi C., Moroni A., Baruscotti M., DiFrancesco D. Heteromeric HCN1–HCN4 channels: a comparison with native pacemaker channels from the rabbit sinoatrial node. J. Physiol. 2003;549(2):347–359. doi: 10.1113/jphysiol.2002.027698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ulens C., Tytgat J. Functional Heteromerization of HCN1 and HCN2 Pacemaker Channels. J. Biol. Chem. 2001;276(9):6069–6072. doi: 10.1074/jbc.C000738200. [DOI] [PubMed] [Google Scholar]
- 53.Pape H-C. Queer Current and Pacemaker: The Hyperpolarization-Activated Cation Current in Neurons. Annu. Rev. Physiol. 1996;58(1):299–327. doi: 10.1146/annurev.ph.58.030196.001503. [DOI] [PubMed] [Google Scholar]
- 54.Yu X., Duan K-L., Shang C-F., Yu H-G., Zhou Z. Calcium influx through hyperpolarization-activated cation channels (Ih channels) contributes to activity-evoked neuronal secretion. Proc. Natl. Acad. Sci. USA. 2004;101(4):1051–1056. doi: 10.1073/pnas.0305167101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zong X., Krause S., Chen C-C., Krüger J., Gruner C., Cao-Ehlker X., Fenske S., Wahl-Schott C., Biel M. Regulation of Hyperpolarization-activated Cyclic Nucleotide-gated (HCN) Channel Activity by cCMP. J. Biol. Chem. 2012;287(32):26506–26512. doi: 10.1074/jbc.M112.357129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Alig J., Marger L., Mesirca P., Ehmke H., Mangoni M.E., Isbrandt D. Control of heart rate by cAMP sensitivity of HCN channels. Proc. Natl. Acad. Sci. USA. 2009;106(29):12189–12194. doi: 10.1073/pnas.0810332106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ulens C., Siegelbaum S.A. Regulation of hyperpolarization-activated HCN channels by cAMP through a gating switch in binding domain symmetry. Neuron. 2003;40:959–970. doi: 10.1016/s0896-6273(03)00753-0. [DOI] [PubMed] [Google Scholar]
- 58.Munsch T., Pape H-C. Modulation of the hyperpolarization-activated cation current of rat thalamic relay neurones by intracellular pH. J. Physiol. 1999;519(2):493–504. doi: 10.1111/j.1469-7793.1999.0493m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pian P., Bucchi A., Robinson R.B., Siegelbaum S.A. Regulation of Gating and Rundown of HCN Hyperpolarization-activated Channels by Exogenous and Endogenous PIP2. J. Gen. Physiol. 2006;128(5):593–604. doi: 10.1085/jgp.200609648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zolles G., Klöcker N., Wenzel D., Weisser-Thomas J., Fleischmann B.K., Roeper J., Fakler B. Pacemaking by HCN Channels Requires Interaction with Phosphoinositides. Neuron. 2006;52(6):1027–1036. doi: 10.1016/j.neuron.2006.12.005. [DOI] [PubMed] [Google Scholar]
- 61.Ye B., Balijepalli R.C., Foell J.D., Kroboth S., Ye Q., Luo Y-H., Shi N-Q. Caveolin-3 Associates with and Affects the Function of Hyperpolarization-Activated Cyclic Nucleotide-Gated Channel 4. Biochemistry. 2008;47(47):12312–12318. [PMC free article] [PubMed] [Google Scholar]
- 62.Yu H., Wu J., Potapova I., Wymore R.T., Holmes B., Zuckerman J., Pan Z., Wang H., Shi W., Robinson R.B., El-Maghrabi M.R., Benjamin W., Dixon J., McKinnon D., Cohen I.S., Wymore R. MinK-Related Peptide 1: A β Subunit for the HCN Ion Channel Subunit Family Enhances Expression and Speeds Activation. Circ. Res. 2001;88(12):e84–e87. doi: 10.1161/hh1201.093511. [DOI] [PubMed] [Google Scholar]
- 63.Decher N., Bundis F., Vajna R., Steinmeyer K. KCNE2 modulates current amplitudes and activation kinetics of HCN4: influence of KCNE family members on HCN4 currents. Pflugers Arch. 2003;446(6):633–640. doi: 10.1007/s00424-003-1127-7. [DOI] [PubMed] [Google Scholar]
- 64.Qu J., Kryukova Y., Potapova I.A., Doronin S.V., Larsen M., Krishnamurthy G., Cohen I.S., Robinson R.B. MiRP1 Modulates HCN2 Channel Expression and Gating in Cardiac Myocytes. J. Biol. Chem. 2004;279(42):43497–43502. doi: 10.1074/jbc.M405018200. [DOI] [PubMed] [Google Scholar]
- 65.Gravante B., Barbuti A., Milanesi R., Zappi I., Viscomi C., DiFrancesco D. Interaction of the Pacemaker Channel HCN1 with Filamin A. J. Biol. Chem. 2004;279(42):43847–43853. doi: 10.1074/jbc.M401598200. [DOI] [PubMed] [Google Scholar]
- 66.Partida G.J., Stradleigh T.W., Ogata G., Godzdanker I., Ishida A.T. Thy1 Associates with the Cation Channel Subunit HCN4 in Adult Rat Retina. Invest. Ophthalmol. Vis. Sci. 2012;53(3):1696–1703. doi: 10.1167/iovs.11-9307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wilkars W., Wollberg J., Mohr E., Han M., Chetkovich D.M., Bähring R., Bender R.A. Nedd4-2 regulates surface expression and may affect N-glycosylation of hyperpolarization-activated cyclic nucleotide-gated (HCN)-1 channels. FASEB J. 2014;28(5):2177–2190. doi: 10.1096/fj.13-242032. [DOI] [PubMed] [Google Scholar]
- 68.Lewis A.S., Schwartz E., Savio Chan C., Noam Y., Shin M., Wadman W.J., James Surmeier D., Baram T.Z., Macdonald R.L., Chetkovich D.M. Alternatively Spliced Isoforms of TRIP8b Differentially Control h Channel Trafficking and Function. J. Neurosci. 2009;29(19):6250–6265. doi: 10.1523/JNEUROSCI.0856-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lewis A.S., Vaidya S.P., Blaiss C.A., Liu Z., Stoub T.R., Brager D.H., Chen X., Bender R.A., Estep C.M., Popov A.B., Kang C.E., Van Veldhoven P.P., Bayliss D.A., Nicholson D.A., Powell C.M., Johnston D., Chetkovich D.M. Deletion of the Hyperpolarization-Activated Cyclic Nucleotide-Gated Channel Auxiliary Subunit TRIP8b Impairs Hippocampal Ih Localization and Function and Promotes Antidepressant Behavior in Mice. J. Neurosci. 2011;31(20):7424–7440. doi: 10.1523/JNEUROSCI.0936-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zong X., Eckert C., Yuan H., Wahl-Schott C., Abicht H., Fang L., Li R., Mistrik P., Gerstner A., Much B., Baumann L., Michalakis S., Zeng R., Chen Z., Biel M. A Novel Mechanism of Modulation of Hyperpolarization-activated Cyclic Nucleotide-gated Channels by Src Kinase. J. Biol. Chem. 2005;280(40):34224–34232. doi: 10.1074/jbc.M506544200. [DOI] [PubMed] [Google Scholar]
- 71.Poolos N.P., Bullis J.B., Roth M.K. Modulation of h-Channels in Hippocampal Pyramidal Neurons by p38 Mitogen-Activated Protein Kinase. J. Neurosci. 2006;26(30):7995–8003. doi: 10.1523/JNEUROSCI.2069-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fogle K.J., Lyashchenko A.K., Turbendian H.K., Tibbs G.R. HCN Pacemaker Channel Activation Is Controlled by Acidic Lipids Downstream of Diacylglycerol Kinase and Phospholipase A2. J. Neurosci. 2007;27(11):2802–2814. doi: 10.1523/JNEUROSCI.4376-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pian P., Bucchi A., DeCostanzo A., Robinson R., Siegelbaum S. Modulation of cyclic nucleotide-regulated HCN channels by PIP2 and receptors coupled to phospholipase C. Pflugers Arch. 2007;455(1):125–145. doi: 10.1007/s00424-007-0295-2. [DOI] [PubMed] [Google Scholar]
- 74.Lewis A.S., Estep C., Chetkovich D.M. The fast and slow ups and downs of HCN channel regulation. Channels. 2010;4(3):215–231. doi: 10.4161/chan.4.3.11630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Maccaferri G., Mangoni M., Lazzari A., DiFrancesco D. Properties of the hyperpolarization-activated current in rat hippocampal CA1 pyramidal cells. J. Neurophysiol. 1993;69(6):2129–2136. doi: 10.1152/jn.1993.69.6.2129. [DOI] [PubMed] [Google Scholar]
- 76.Nolan M.F., Dudman J.T., Dodson P.D., Santoro B. HCN1 Channels Control Resting and Active Integrative Properties of Stellate Cells from Layer II of the Entorhinal Cortex. J. Neurosci. 2007;27(46):12440–12451. doi: 10.1523/JNEUROSCI.2358-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.McCormick D.A., Pape H.C. Properties of a hyperpolarization-activated cation current and its role in rhythmic oscillation in thalamic relay neurones. J. Physiol. 1990;431(1):291–318. doi: 10.1113/jphysiol.1990.sp018331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ulrich D. Dendritic Resonance in Rat Neocortical Pyramidal Cells. J. Neurophysiol. 2002;87(6):2753–2759. doi: 10.1152/jn.2002.87.6.2753. [DOI] [PubMed] [Google Scholar]
- 79.Wang W.T., Wan Y.H., Zhu J.L., Lei G.S., Wang Y.Y., Zhang P., Hu S.J. Theta-frequency membrane resonance and its ionic mechanisms in rat subicular pyramidal neurons. Neuroscience. 2006;140(1):45–55. doi: 10.1016/j.neuroscience.2006.01.033. [DOI] [PubMed] [Google Scholar]
- 80.Xue W-N., Wang Y., He S-M., Wang X-L., Zhu J-L., Gao G-D. SK- and h-current contribute to the generation of theta-like resonance of rat substantia nigra pars compacta dopaminergic neurons at hyperpolarized membrane potentials. Brain Struct. Funct. 2012;217(2):379–394. doi: 10.1007/s00429-011-0361-6. [DOI] [PubMed] [Google Scholar]
- 81.Magee J.C. Dendritic Ih normalizes temporal summation in hippocampal CA1 neurons. Nat. Neurosci. 1999;2(6):508–514. doi: 10.1038/9158. [DOI] [PubMed] [Google Scholar]
- 82.Ying S-W., Jia F., Abbas S.Y., Hofmann F., Ludwig A., Goldstein P.A. Dendritic HCN2 Channels Constrain Glutamate-Driven Excitability in Reticular Thalamic Neurons. J. Neurosci. 2007;27(32):8719–8732. doi: 10.1523/JNEUROSCI.1630-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Masi A., Narducci R., Landucci E., Moroni F., Mannaioni G. MPP+-dependent inhibition of Ih reduces spontaneous activity and enhances EPSP summation in nigral dopamine neurons. Br. J. Pharmacol. 2013;169:130–142. doi: 10.1111/bph.12104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Magee J.C. Dendritic Hyperpolarization-Activated Currents Modify the Integrative Properties of Hippocampal CA1 Pyramidal Neurons. J. Neurosci. 1998;18(19):7613–7624. doi: 10.1523/JNEUROSCI.18-19-07613.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Williams S.R., Stuart G.J. Site Independence of EPSP Time Course Is Mediated by DendriticI h in Neocortical Pyramidal Neurons. J. Neurophysiol. 2000;83(5):3177–3182. doi: 10.1152/jn.2000.83.5.3177. [DOI] [PubMed] [Google Scholar]
- 86.Young G.T., Emery E.C., Mooney E.R., Tsantoulas C., McNaughton P.A. Inflammatory and neuropathic pain are rapidly suppressed by peripheral block of hyperpolarisation-activated cyclic nucleotide-gated ion channels. Pain. 2014;155:1708–1719. doi: 10.1016/j.pain.2014.05.021. [DOI] [PubMed] [Google Scholar]
- 87.Gao Z., Chen B., Joiner M-A., Wu Y., Guan X., Koval O.M., Chaudhary A.K., Cunha S.R., Mohler P.J., Martins J.B., Song L-S., Anderson M.E. If and SR Ca2+ release both contribute to pacemaker activity in canine sinoatrial node cells. J. Mol. Cell. Cardiol. 2010;49(1):33–40. doi: 10.1016/j.yjmcc.2010.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Herrmann S., Stieber J., Ludwig A. Pathophysiology of HCN channels. Pflugers Arch. 2007;454:517–522. doi: 10.1007/s00424-007-0224-4. [DOI] [PubMed] [Google Scholar]
- 89.Mangoni M.E., Nargeot J. Properties of the hyperpolarization-activated current (If) in isolated mouse sino-atrial cells. Cardiovasc. Res. 2001;52(1):51–64. doi: 10.1016/s0008-6363(01)00370-4. [DOI] [PubMed] [Google Scholar]
- 90.Liu J., Noble P.J., Xiao G., Abdelrahman M., Dobrzynski H., Boyett M.R., Lei M., Noble D. Role of pacemaking current in cardiac nodes: Insights from a comparative study of sinoatrial node and atrioventricular node. Prog. Biophys. Mol. Biol. 2008;96(1–3):294–304. doi: 10.1016/j.pbiomolbio.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 91.Marger L., Mesirca P., Alig J., Torrente A., Dübel S., Engeland B., Kanani S., Fontanaud P., Striessnig J., Shin H-S., Isbrandt D., Ehmke H., Nargeot J., Mangoni M.E. Functional roles of Cav1.3, Cav3.1 and HCN channels in automaticity of mouse atrioventricular cells. Channels. 2011;5(3):251–261. doi: 10.4161/chan.5.3.15266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Verrier R.L., Bonatti R., Silva A.F., Batatinha J.A., Nearing B.D., Liu G., Rajamani S., Zeng D., Belardinelli L. If inhibition in the atrioventricular node by ivabradine causes rate-dependent slowing of conduction and reduces ventricular rate during atrial fibrillation. Heart Rhythm. 2014;11(12):2288–2296. doi: 10.1016/j.hrthm.2014.08.007. [DOI] [PubMed] [Google Scholar]
- 93.Sartiani L., Cerbai E., Mugelli A. The funny current in cardiac non-pacemaker cells: functional role and pharmacological modulation. In: Das M.K., editor. Modern Pacemakers - Present and Future. InTech; 2011. pp. 595–610. [Google Scholar]
- 94.Fenske S., Mader R., Scharr A., Paparizos C., Cao-Ehlker X., Michalakis S., Shaltiel L., Weidinger M., Stieber J., Feil S., Feil R., Hofmann F., Wahl-Schott C., Biel M. HCN3 Contributes to the Ventricular Action Potential Waveform in the Murine Heart. Circ. Res. 2011;109(9):1015–1023. doi: 10.1161/CIRCRESAHA.111.246173. [DOI] [PubMed] [Google Scholar]
- 95.Fenske S., Krause S., Biel M., Wahl-Schott C. The Role of HCN Channels in Ventricular Repolarization. Trends Cardiovasc. Med. 2011;21:216–220. doi: 10.1016/j.tcm.2012.05.013. [DOI] [PubMed] [Google Scholar]
- 96.
- 97.Riccioni G. Ivabradine: from molecular basis to clinical effectiveness. Adv. Ther. 2010;27(3):160–167. doi: 10.1007/s12325-010-0014-9. [DOI] [PubMed] [Google Scholar]
- 98.Estep J.D. Chronic heart failure: what does the horizon look like? Curr. Opin. Cardiol. 2015;30:344–353. doi: 10.1097/HCO.0000000000000186. [DOI] [PubMed] [Google Scholar]
- 99.Bois P., Bescond J., Renaudon B., Lenfant J. Mode of action of bradycardic agent, S 16257, on ionic currents of rabbit sinoatrial node cells. Br. J. Pharmacol. 1996;118:1051–1057. doi: 10.1111/j.1476-5381.1996.tb15505.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bucchi A., Baruscotti M., DiFrancesco D. Current-dependent Block of Rabbit Sino-Atrial Node If Channels by Ivabradine. J. Gen. Physiol. 2002;120:1–13. doi: 10.1085/jgp.20028593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bucchi A., Tognati A., Milanesi R., Baruscotti M., DiFrancesco D. Properties of ivabradine-induced block of HCN1 and HCN4 pacemaker channels. J. Physiol. 2006;572(2):335–346. doi: 10.1113/jphysiol.2005.100776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Baruscotti M., Barbuti A., Bucchi A. The cardiac pacemaker current. J. Mol. Cell. Cardiol. 2010;48:55–64. doi: 10.1016/j.yjmcc.2009.06.019. [DOI] [PubMed] [Google Scholar]
- 103.Delpon E., Valenzuela C., Perez O., Franqueza L., Gay P., Snyders D.J., Tamargo J. Mechanism of block of a human cloned potassium channel by the enantiomers of a new bradycardic agent: S-16257-2 and S-16260-2. Br. J. Pharmacol. 1996;117:1293–1301. doi: 10.1111/j.1476-5381.1996.tb16728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Perez O., Gay P., Franqueza L., Carron R., Valenzuela C., Delpon E., Tamargo J. Effects of the two enantiomers, S-16257-2 and S-16260-2, of a new bradycardic agent on guinea-pig isolated cardiac preparations. Br. J. Pharmacol. 1995;115:787–794. doi: 10.1111/j.1476-5381.1995.tb15002.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Koncz I., Szél T., Bitay M., Cerbai E., Jaeger K., Fülöp F., Jost N., Virág L., Orvos P., Tálosi L., Kristóf A., Baczkó I., Papp J.G., Varró A. Electrophysiological effects of ivabradine in dog and human cardiac preparations: Potential antiarrhythmic actions. Eur. J. Pharmacol. 2011;668(3):419–426. doi: 10.1016/j.ejphar.2011.07.025. [DOI] [PubMed] [Google Scholar]
- 106.Stieber J., Wieland K., Stockl G., Ludwig A., Hofmann F. Bradycardic and Proarrhythmic Properties of Sinus Node Inhibitors. Mol. Pharmacol. 2006;69:1328–1337. doi: 10.1124/mol.105.020701. [DOI] [PubMed] [Google Scholar]
- 107.Savalieva I., Camm A.J. Novel If current inhibitor ivabradine: Safety considerations. Adv. Cardiol. 2006;43:79–96. doi: 10.1159/000095430. [DOI] [PubMed] [Google Scholar]
- 108.Riccioni G. Ivabradine: An Intelligent Drug for the Treatment of Ischemic Heart Disease. Molecules. 2012;17:13592–13604. doi: 10.3390/molecules171113592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Luszczki J.J., Prystupa A., Andres-Mach M., Marzêda E., Florek-Luszczki M. Ivabradine (a hyperpolarization activated cyclic nucleotide-gated channel blocker) elevates the threshold for maximal electroshock-induced tonic seizures in mice. Pharmacol. Rep. 2013;65:1407–1414. doi: 10.1016/s1734-1140(13)71500-7. [DOI] [PubMed] [Google Scholar]
- 110.Yusuf S., Camm A.J. Sinus tachyarrhythmias and the specific bradycardic agents: a marriage made in heaven? J. Cardiovasc. Pharmacol. Ther. 2003;8:89–105. doi: 10.1177/107424840300800202. [DOI] [PubMed] [Google Scholar]
- 111.Staehle H., Daniel H., Kobinger W., Lillie C., Pichler L. Chemistry, Pharmacology, and Structure-Activity Relationships with a New Type of Imidazolines Exerting a Specific Bradycardic Action at a Cardiac Site. J. Med. Chem. 1980;23:1217–1222. doi: 10.1021/jm00185a013. [DOI] [PubMed] [Google Scholar]
- 112.Challinor-Rogers J.L., Hay T.K., McPherson G.A. Comparison of the cromakalim antagonism and bradycardic actions of a series of novel alinidine analogues in the rat. Naunyn Schmiedebergs Arch. Pharmacol. 1994;350:158–166. doi: 10.1007/BF00241091. [DOI] [PubMed] [Google Scholar]
- 113.Harron D.W., Arndts D., Finch M., Shanks R.G. An assessment of the contribution of Clonidine metabolised from Alinidine to the cardiovascular effects of Alinidine. Br. J. Clin. Pharmacol. 1983;16:451–455. doi: 10.1111/j.1365-2125.1983.tb02196.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.BoSmith R. E., Briggs I., Sturgess N. C. Inhibitory action of ZENECA ZD7288 on whole-cell hyperpolarization activated inward current (If) in guinea-pig dissociated sinoatrial node cells. Br. J. Pharmacol. 1993;110:313–349. doi: 10.1111/j.1476-5381.1993.tb13815.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sanchez-Alonso J.L., Halliwell J.V., Colino A. ZD 7288 inhibits T-type calcium current in rat hippocampal pyramidal cells. Neurosci. Lett. 2008;439:275–280. doi: 10.1016/j.neulet.2008.05.016. [DOI] [PubMed] [Google Scholar]
- 116.Wu X., Liao L., Liu X., Luo F., Yang T., Li C. Is ZD7288 a selective blocker of hyperpolarization-activated cyclic nucleotide-gated channel currents? Channels. 2012;6:1–5. doi: 10.4161/chan.22209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hargreaves R. B., Marshall P. W., McLoughlin B. J., Mills S. D. Pyrimidine Derivatives. US5223505, 1993. [Google Scholar]
- 118.Reiffen M., Eberlien W., Müller P., Psiorz M., Noll K., Heider J., Lillie C., Kobinger W., Luger P. Specific Bradycardic Agents. Chemistry, Pharmacology, and Structure-Activity Relationships of Substituted Benzazepinones, a New Class of Compounds Exerting Antiischemic Properties. J. Med. Chem. 1990;33:1496–1504. doi: 10.1021/jm00167a033. [DOI] [PubMed] [Google Scholar]
- 119.Bomhard A., Reiffen M., Heider J., Psiorz M., Lillie C. Specific Bradycardic Agents. 2. Heteroaromatic Modifications in the Side Chain of Specific Bradycardic Benzazepinones: Chemistry, Pharmacology, and Structure-Activity Relationships. J. Med. Chem. 1991;34:942–947. doi: 10.1021/jm00107a011. [DOI] [PubMed] [Google Scholar]
- 120.Goethals M., Raes A., van Bogaert P.P. Use-dependent block of the pacemaker current I(f) in rabbit sinoatrial node cells by zatebradine (UL-FS 49). On the mode of action of sinus node inhibitors. Circulation. 1993;88:2389–2401. doi: 10.1161/01.cir.88.5.2389. [DOI] [PubMed] [Google Scholar]
- 121.Psiorz M., Heider J., Bomhard A., Reiffen M., Hauel N., Noll K., Narr B., Lillie C., Kobinger W., Dammgen J. Preparation and formulation of N-[(1-phenylethyl)piperidinylmethyl] benzazepinones and analogs for treating sinus tachycardia. US5175157 (CA 119:8698). [Google Scholar]
- 122.Vélez de Mendizábal N., Schäfer H.G., Staab A., Trommeshauser D., Döge C., Klüglich M., Roberts J., Trocóniz I.F. Joint population pharmacokinetic/pharmacodynamic model for the heart rate effects at rest and at the end of exercise for cilobradine. Pharm. Res. 2013;30(4):1110–1122. doi: 10.1007/s11095-012-0947-6. [DOI] [PubMed] [Google Scholar]
- 123.Fliss G., Staab A., Tillmann C., Trommeshauser D., Schaefer H.G., Kloft C. Population Pharmacokinetic Data Analysis of Cilobradine, an If Channel Blocker. Pharm. Res. 2008;25(2):359–368. doi: 10.1007/s11095-007-9351-z. [DOI] [PubMed] [Google Scholar]
- 124.Kubota H., Kakefuda A., Watanabe T., Taguchi Y., Ishii N., Masuda N., Sakamoto S., Tsukamoto S. (±)-2-(3-Piperidyl)-1,2,3,4-tetrahydroisoquinolines as a new class of specific bradycardic agents. Bioorg. Med. Chem. Lett. 2003;13(13):2155–2158. doi: 10.1016/s0960-894x(03)00349-4. [DOI] [PubMed] [Google Scholar]
- 125.Kakefuda A., Watanabe T., Taguchi Y., Masuda N., Tanaka A., Yanagisawa I. Synthesis and Pharmacological Evaluation of 2-(3-Piperidyl)-1,2,3,4-tetrahydroisoquinoline Derivatives as Specific Bradycardic Agents. Chem. Pharm. Bull. (Tokyo) 2003;51:390–398. doi: 10.1248/cpb.51.390. [DOI] [PubMed] [Google Scholar]
- 126.Kubota H., Kakefuda A., Watanabe T., Ishii N., Wada K., Masuda N., Sakamoto S., Tsukamoto S. Synthesis and Pharmacological Evaluation of 1-Oxo-2-(3-piperidyl)-1,2,3,4- tetrahydroisoquinolines and Related Analogues as a New Class of Specific Bradycardic Agents Possessing If Channel Inhibitory Activity. J. Med. Chem. 2003;46(22):4728–4740. doi: 10.1021/jm0301742. [DOI] [PubMed] [Google Scholar]
- 127.Kubota H., Watanabe T., Kakefuda A., Masuda N., Wada K., Ishii N., Sakamoto S., Tsukamoto S. Synthesis and pharmacological evaluation of piperidinoalkanoyl-1,2,3,4-tetrahydroisoqui-noline derivatives as novel specific bradycardic agents. Bioorg. Med. Chem. Lett. 2004;14(12):3049–3052. doi: 10.1016/j.bmcl.2004.04.031. [DOI] [PubMed] [Google Scholar]
- 128.Kubota H., Watanabe T., Kakefuda A., Masuda N., Wada K., Ishii N., Sakamoto S., Tsukamoto S. Synthesis and pharmacological evaluation of N-acyl-1,2,3,4-tetrahydroisoquinoline derivatives as novel specific bradycardic agents. Bioorg. Med. Chem. 2004;12(5):871–882. doi: 10.1016/j.bmc.2003.12.032. [DOI] [PubMed] [Google Scholar]
- 129.Umehara K-i., Iwai M., Adachi Y., Iwatsubo T., Usui T., Kamimura H. Hepatic Uptake and Excretion of (–)-N-{2-[(R)-3-(6,7-Dimethoxy-1,2,3,4-tetrahydroisoquinoline-2-carbonyl)piperidino]ethyl}-4-fluorobenzamide (YM758), a Novel If Channel Inhibitor, in Rats and Humans. Drug Metab. Dispos. 2008;36(6):1030–1038. doi: 10.1124/dmd.108.020669. [DOI] [PubMed] [Google Scholar]
- 130.Umehara K-i., Nakamata T., Suzuki K., Noguchi K., Usui T., Kamimura H. Pharmacokinetics of YK754, a novel If channel inhibitor in rats, dogs and humans. Eur. J. Drug Metab. Pharmacokinet. 2008;33:117–127. doi: 10.1007/BF03191028. [DOI] [PubMed] [Google Scholar]
- 131.Umehara K-i., Susaki Y., Van Teylingen R.H., Neat J.N., Ndikum-Moffor F., Noguchi K., Usui T., Parkinson A., Kamimura H. Evaluation of the inhibitory and induction potential of YM758, a novel If channel inhibitor, for human P450-mediated metabolism. Eur. J. Drug Metab. Pharmacokinet. 2008;33:211–223. doi: 10.1007/BF03190875. [DOI] [PubMed] [Google Scholar]
- 132.Peglion J. L., Goument B., Dessinges A., Caignard P., Vilaine J. P., Thollon C., Villeneuve N., Chimenti S. 1,2,4,5-Tetrahydro- 3H-benzazepine compounds, a process for their preparation and pharmaceutical composition containing them. US200900692296. [Google Scholar]
- 133.Thollon C., Bedut S., Villeneuve N., Coge F., Piffard L., Guillaumin J-P., Brunel-Jacquemin C., Chomarat P., Boutin J.A., Peglion J-P., Vilaine J-P. Use-dependent inhibition of hHCN4 by ivabradine and relationship with reduction in pacemaker Activity. Br. J. Pharmacol. 2007;150:37–46. doi: 10.1038/sj.bjp.0706940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Romanelli M.N., Cerbai E., Dei S., Guandalini L., Martelli C., Martini E., Scapecchi S., Teodori E., Mugelli A. Design, synthesis and preliminary biological evaluation of Zatebradine analogues as potential blockers of the hyperpolarization-activated current. Bioorg. Med. Chem. 2005;13:1211–1220. doi: 10.1016/j.bmc.2004.11.017. [DOI] [PubMed] [Google Scholar]
- 135.Melchiorre M., Del Lungo M., Guandalini L., Martini E., Dei S., Manetti D., Scapecchi S., Teodori E., Sartiani L., Mugelli A., Cerbai E., Romanelli M.N. Design, Synthesis, and Preliminary Biological Evaluation of New Isoform-Selective f-Current Blockers. J. Med. Chem. 2010;53(18):6773–6777. doi: 10.1021/jm1006758. [DOI] [PubMed] [Google Scholar]
- 136.Romanelli M. N., Mugelli A., Cerbai E., Sartiani L., Del Lungo M., Melchiorre M. New isoform-selective HCN blockers. WO2011000915 (A1). [Google Scholar]
- 137.Del Lungo M., Melchiorre M., Guandalini L., Sartiani L., Mugelli A., Koncz I., Szel T., Varro A., Romanelli M.N., Cerbai E. Novel blockers of hyperpolarization-activated current with isoform selectivity in recombinant cells and native tissue. Br. J. Pharmacol. 2012;166(2):602–616. doi: 10.1111/j.1476-5381.2011.01782.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Koncz I., Szel T., Jaeger I., Baczko I., Cerbai E., Romanelli M.N., Papp J.G., Varro A. Selective Pharmacological Inhibition of the Pacemaker Channel Isoforms (HCN1-4) as New Possible Therapeutical Targets. Curr. Pharm. Des. 2011;18:3662–3674. doi: 10.2174/092986711796642427. [DOI] [PubMed] [Google Scholar]
- 139.McClure K.J., Maher M.P., Wu N-T., Chaplan S.R., Eckert Iii W.A., Lee D.H., Wickenden A.D., Hermann M., Allison B., Hawryluk N., Breitenbucher J.G., Grice C.A. Discovery of a novel series of selective HCN1 blockers. Bioorg. Med. Chem. Lett. 2011;21(18):5197–5201. doi: 10.1016/j.bmcl.2011.07.051. [DOI] [PubMed] [Google Scholar]
- 140.Maher M.P., Wu N-T., Guo H-Q., Dubin A.E., Chaplan S.R., Wickenden A.D. HCN Channels as Targets for Drug Discovery. Comb. Chem. High Throughput Screen. 2009;12:64–72. doi: 10.2174/138620709787048028. [DOI] [PubMed] [Google Scholar]
- 141.Anger K.E. Dexmedetomidine: a review of its use for the management of pain, agitation, and delirium in the intensive care unit. Curr. Pharm. Des. 2013;19:4003–4013. doi: 10.2174/1381612811319220009. [DOI] [PubMed] [Google Scholar]
- 142.Brummett C.M., Hong E.K., Janda A.M., Amodeo F.S., Lydic R. Perineural Dexmedetomidine Added to Ropivacaine for Sciatic Nerve Block in Rats Prolongs the Duration of Analgesia by Blocking the Hyperpolarization-activated Cation Current. Anesthesiology. 2011;115(4):836–843. doi: 10.1097/ALN.0b013e318221fcc9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Yang Y-C., Meng Q-T., Pan X., Xia Z-Y., Chen X-D. Dexmedetomidine produced analgesic effect via inhibition of HCN currents. Eur. J. Pharmacol. 2014;740:560–564. doi: 10.1016/j.ejphar.2014.06.031. [DOI] [PubMed] [Google Scholar]
- 144.Hammer G.B., Drover D.R., Cao H., Jackson E., Williams G.D., Ramamoorthy C., Van Hare G.F., Niksch A., Dubin A.M. The effects of dexmedetomidine on cardiac electrophysiology in children. Anesth. Analg. 2008;106:79–83. doi: 10.1213/01.ane.0000297421.92857.4e. [DOI] [PubMed] [Google Scholar]
- 145.Yagi J., Sumino R. Inhibition of a Hyperpolarization-Activated Current by Clonidine in Rat Dorsal Root Ganglion Neurons. J. Neurophysiol. 1998;80(3):1094–1104. doi: 10.1152/jn.1998.80.3.1094. [DOI] [PubMed] [Google Scholar]
- 146.Knaus A., Zong X., Beetz N., Jahns R., Lohse M.J., Biel M., Hein L. Direct Inhibition of Cardiac Hyperpolarization-Activated Cyclic Nucleotide-Gated Pacemaker Channels by Clonidine. Circulation. 2007;115(7):872–880. doi: 10.1161/CIRCULATIONAHA.106.667675. [DOI] [PubMed] [Google Scholar]
- 147.Vasilyev D.V., Shan Q.J., Lee Y., Mayer S.C., Bowlby M.R., Strassle B.W., Kaftan E.J., Rogers K.E., Dunlop J. Direct Inhibition of Ih by Analgesic Loperamide in Rat DRG Neurons. J. Neurophysiol. 2007;97:3713–3721. doi: 10.1152/jn.00841.2006. [DOI] [PubMed] [Google Scholar]
- 148.Nguyen A., Kath J.C., Hanson D.C., Biggers M.S., Canniff P.C., Donovan C.B., Mather R.J., Bruns M.J., Rauer H., Aiyar J., Lepple-Wienhues A., Gutman G.A., Grissmer S., Cahalan M.D., Chandy K.G. Novel nonpeptide agents potently block the C-type inactivated conformation of Kv1.3 and suppress T cell activation. Mol. Pharmacol. 1996;50(6):1672–1679. [PubMed] [Google Scholar]
- 149.Vasilyev D.V., Shan Q.J., Lee Y.T., Soloveva V., Nawosghick S.P., Kaftan E.J., Dunlop J., Mayer S., Bowlby M.R. A Novel High-Throughput Screening Assay for HCN Channel Blocker Using Membrane Potential–Sensitive Dye and FLIPR. J. Biomol. Screen. 2009;14:1119–1128. doi: 10.1177/1087057109345526. [DOI] [PubMed] [Google Scholar]
- 150.Lee Y.T., Vasilyev D.V., Shan Q.J., Dunlop J., Mayer S., Bowlby M.R. Novel pharmacological activity of loperamide and CP-339,818 on human HCN channels characterized with an automated electrophysiology assay. Eur. J. Pharmacol. 2008;581:97–104. doi: 10.1016/j.ejphar.2007.11.058. [DOI] [PubMed] [Google Scholar]
- 151.Liu Y-C., Wang Y-J., Wu P-Y., Wu S-N. Tramadol-induced block of hyperpolarization-activated cation current in rat pituitary lactotrophs. Naunyn Schmiedebergs Arch. Pharmacol. 2009;379:127–135. doi: 10.1007/s00210-008-0353-0. [DOI] [PubMed] [Google Scholar]
- 152.Kretschmannova K., Gonzalez-Iglesias A.E., Tomić M., Stojilkovic S.S. Dependence of Hyperpolarisation-Activated Cyclic Nucleotide-Gated Channel Activity on Basal Cyclic Adenosine Monophosphate Production in Spontaneously Firing GH3 Cells. J. Neuroendocrinol. 2006;18(7):484–493. doi: 10.1111/j.1365-2826.2006.01438.x. [DOI] [PubMed] [Google Scholar]
- 153.Kotani Y., Shimazawa M., Yoshimura S., Iwama T., Hara H. The experimental and clinical pharmacology of propofol, an anesthetic agent with neuroprotective properties. CNS Neurosci. Ther. 2008;14:95–106. doi: 10.1111/j.1527-3458.2008.00043.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Lundström S., Twycross R., Mihalyo M., Wilcock A. Propofol. J. Pain Symptom Manage. 2010;40(3):466–470. doi: 10.1016/j.jpainsymman.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 155.Liu Q., Kong A-l., Chen R., Qian C., Liu S-w., Sun B-g., Wang L-x., Song L-s., Hong J. Propofol and arrhythmias: two sides of the coin. Acta Pharmacol. Sin. 2011;32(6):817–823. doi: 10.1038/aps.2011.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kojima A., Ito Y., Kitagawa H., Matsuura H. Ionic mechanisms underlying the negative chronotropic action of propofol on sinoatrial node automaticity in guinea-pig heart. Br. J. Pharmacol. 2015;172:799–814. doi: 10.1111/bph.12936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Cacheaux L.P., Topf N., Tibbs G.R., Schaefer U.R., Levi R., Harrison N.L., Abbott G.W., Goldstein P.A. Impairment of Hyperpolarization-Activated, Cyclic Nucleotide-Gated Channel Function by the Intravenous General Anesthetic Propofol. J. Pharmacol. Exp. Ther. 2005;315(2):517–525. doi: 10.1124/jpet.105.091801. [DOI] [PubMed] [Google Scholar]
- 158.Tibbs G.R., Rowley T.J., Sanford R.L., Herold K.F., Proekt A., Hemmings H.C., Andersen O.S., Goldstein P.A., Flood P.D. HCN1 Channels as Targets for Anesthetic and Nonanesthetic Propofol Analogs in the Amelioration of Mechanical and Thermal Hyperalgesia in a Mouse Model of Neuropathic Pain. J. Pharmacol. Exp. Ther. 2013;345(3):363–373. doi: 10.1124/jpet.113.203620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Chen X., Sirois J.E., Lei Q., Talley E.M., Lynch C.I., Bayliss D.A. HCN Subunit-Specific and cAMP-Modulated Effects of Anesthetics on Neuronal Pacemaker Currents. J. Neurosci. 2005;25:5803–5814. doi: 10.1523/JNEUROSCI.1153-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Chen X., Shu S., Kennedy D.P., Willcox S.C., Bayliss D.A. Subunit-Specific Effects of Isoflurane on Neuronal Ih in HCN1 Knockout Mice. J. Neurophysiol. 2009;101(1):129–140. doi: 10.1152/jn.01352.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Irifune M., Shimizu T., Nomoto M., Fukuda T. Ketamine-induced anesthesia involves the N-methyl-D-aspartate receptor-channel complex in mice. Brain Res. 1992;596:1–9. doi: 10.1016/0006-8993(92)91525-j. [DOI] [PubMed] [Google Scholar]
- 162.Persson J. Ketamine in Pain Management. CNS Neurosci. Ther. 2013;19(6):396–402. doi: 10.1111/cns.12111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Mion G., Villevieille T. Ketamine Pharmacology: An Update (Pharmacodynamics and Molecular Aspects, Recent Findings). CNS Neurosci. Ther. 2013;19(6):370–380. doi: 10.1111/cns.12099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Chen X., Shu S., Bayliss D.A. HCN1 Channel Subunits Are a Molecular Substrate for Hypnotic Actions of Ketamine. J. Neurosci. 2009;29:600–609. doi: 10.1523/JNEUROSCI.3481-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Bischoff U., Bräu M.E., Vogel W., Hempelmann G., Olschewski A. Local anaesthetics block hyperpolarization-activated inward current in rat small dorsal root ganglion neurones. Br. J. Pharmacol. 2003;139:1273–1280. doi: 10.1038/sj.bjp.0705363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Meng Q-T., Xia Z-Y., Liu J., Bayliss D.A., Chen X. Local Anesthetic Inhibits Hyperpolarization-Activated Cationic Currents. Mol. Pharmacol. 2011;79:866–873. doi: 10.1124/mol.110.070227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Tamura A., Ogura T., Uemura H., Reien Y., Kishimoto T., Nagai T., Komuro I., Miyazaki M., Nakaya H. Effects of Antiarrhythmic Drugs on the Hyperpolarization-Activated Cyclic Nucleotide–Gated Channel Current. J. Pharmacol. Sci. 2009;110:150–159. doi: 10.1254/jphs.08312fp. [DOI] [PubMed] [Google Scholar]
- 168.Hoppe U.C., Beuckelmann D.J. Modulation of the hyperpolarization-activated inward current (If) by antiarrhythmic agents in isolated human atrial myocytes. Naunyn Schmiedebergs Arch. Pharmacol. 1998;358:635–640. doi: 10.1007/pl00005305. [DOI] [PubMed] [Google Scholar]
- 169.Fan X., Chen Y., Wu P., Xing J., Chen H., Song T., Yang J., Zhang J., Huang C. Novel electropharmacological activity of amiodarone on human HCN channels heterologously expressed in the Xenopus oocytes. Eur. J. Pharmacol. 2011;669(1–3):15–23. doi: 10.1016/j.ejphar.2011.07.039. [DOI] [PubMed] [Google Scholar]
- 170.Li H.X., Yang X.J., Han L.H., Zhou Y.F., Zhao X., Jiang B., Dong N.Z., Song J.P., Liu Z.H., Jiang W.P. Effects of amiodarone on funny current If channel gene expression in neonatal rat ventricular myocytes. Zhonghua. Xin Xue Guan Bing Za Zhi. 2007;35:466–470. [PubMed] [Google Scholar]
- 171.Li H., Zhou Y., Jiang B., Zhao X., Li X., Yang X., Jiang W. Dual effects of amiodarone on pacemaker currents in hypertrophied ventricular myocytes isolated from spontaneously hypertensive rats. Clin. Exp. Pharmacol. Physiol. 2014;41(9):698–707. doi: 10.1111/1440-1681.12264. [DOI] [PubMed] [Google Scholar]
- 172.Patel C., Yan G-X., Kowey P.R. Dronedarone. Circulation. 2009;120:636–644. doi: 10.1161/CIRCULATIONAHA.109.858027. [DOI] [PubMed] [Google Scholar]
- 173.Bogdan R., Goegelein H., Ruetten H. Effect of dronedarone on Na+, Ca2+ and HCN channels. Naunyn Schmiedebergs Arch. Pharmacol. 2011;383:347–356. doi: 10.1007/s00210-011-0599-9. [DOI] [PubMed] [Google Scholar]
- 174.Sobrado L.F., Varone B.B., Machado A.D., Nearing B.D., Zeng D., Belardinelli L., Verrier R.L. Dronedarone’s Inhibition of If Current Is the Primary Mechanism Responsible for Its Bradycardic Effect. J. Cardiovasc. Electrophysiol. 2013;24:914–918. doi: 10.1111/jce.12155. [DOI] [PubMed] [Google Scholar]
- 175.Verrier R.L., Sobrado L.F., Pagotto V.P., Kanas A.F., Machado A.D., Varone B.B., Sobrado L.F., Nearing B.D., Zeng D., Belardinelli L. Inhibition of I(f) in the atrioventricular node as a mechanism for dronedarone'sreduction in ventricular rate during atrial fibrillation. Heart Rhythm. 2013;10(11):1692–1697. doi: 10.1016/j.hrthm.2013.08.007. [DOI] [PubMed] [Google Scholar]
- 176.Accili E.A., DiFrancesco D. Inhibition of the hyperpolarization-activated current (if) of rabbit SA node myocytes by niflumic acid. Pflugers Arch. 1996;431:757–762. doi: 10.1007/BF02253840. [DOI] [PubMed] [Google Scholar]
- 177.Satoh T-O., Yamada M. Niflumic acid reduces the hyperpolarization-activated current (Ih) in rod photoreceptor cells. Neurosci. Res. 2001;40:375–381. doi: 10.1016/s0168-0102(01)00252-8. [DOI] [PubMed] [Google Scholar]
- 178.Brioschi C., Micheloni S., Tellez J.O., Pisoni G., Longhi R., Moroni P., Billeter R., Barbuti A., Dobrzynski H., Boyett M.R., DiFrancesco D., Baruscotti M. Distribution of the pacemaker HCN4 channel mRNA and protein in the rabbit sinoatrial node. J. Mol. Cell. Cardiol. 2009;47(2):221–227. doi: 10.1016/j.yjmcc.2009.04.009. [DOI] [PubMed] [Google Scholar]
- 179.Moosmang S., Stieber J., Zong X., Biel M., Hofmann F., Ludwig A. Cellular expression and functional characterization of four hyperpolarization-activated pacemaker channels in cardiac and neuronal tissues. Eur. J. Biochem. 2001;268(6):1646–1652. doi: 10.1046/j.1432-1327.2001.02036.x. [DOI] [PubMed] [Google Scholar]
- 180.Cheng L., Sanguinetti M.C. Niflumic Acid Alters Gating of HCN2 Pacemaker Channels by Interaction with the Outer Region of S4 Voltage Sensing Domains. Mol. Pharmacol. 2009;75:1210–1221. doi: 10.1124/mol.108.054437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Gill C.H., Randall A., Bates S.A., Hill K., Owen D., Larkman P.M., Cairns W., Yusaf S.P., Murdock P.R., Strijbos P.J., Powell A.J., Benham C.D., Davies C.H. Characterization of the human HCN1 channel and its inhibition by capsazepine. Br. J. Pharmacol. 2004;143(3):411–421. doi: 10.1038/sj.bjp.0705945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Zuo G-F., Li M-H., Zhang J-X., Li B., Wang Z-M., Wang Q., Xiao H., Chen S-L. Capsazepine concentration dependently inhibits currents in HEK 293 cells mediated by human hyperpolarization-activated cyclic nucleotide-gated 2 and 4 channels. Exp. Biol. Med. 2013;238(9):1055–1061. doi: 10.1177/1535370213498973. [DOI] [PubMed] [Google Scholar]
- 183.Griguoli M., Maul A., Nguyen C., Giorgetti A., Carloni P., Cherubini E. Nicotine Blocks the Hyperpolarization-Activated Current Ih and Severely Impairs the Oscillatory Behavior of Oriens-Lacunosum Moleculare Interneurons. J. Neurosci. 2010;30(32):10773–10783. doi: 10.1523/JNEUROSCI.2446-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Kodirov S.A., Wehrmeister M., Colom L.V. Modulation of HCN channels in lateral septum by nicotine. Neuropharmacology. 2014;81(0):274–282. doi: 10.1016/j.neuropharm.2014.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Giorgetti A., Carloni P., Mistrik P., Torre V. A homology model of the pore region of HCN channels. Biophys. J. 2005;89:932–944. doi: 10.1529/biophysj.104.045286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Cheng L., Kinard K., Rajamani R., Sanguinetti M.C. Molecular Mapping of the Binding Site for a Blocker of Hyperpolarization-Activated, Cyclic Nucleotide-Modulated Pacemaker Channels. J. Pharmacol. Exp. Ther. 2007;322:931–939. doi: 10.1124/jpet.107.121467. [DOI] [PubMed] [Google Scholar]
- 187.Liu N., Zhang D., Zhu M., Luo S., Liu T. Minocycline inhibits hyperpolarization-activated currents in rat substantia gelatinosa neurons. Neuropharmacology. 2015;95:110–120. doi: 10.1016/j.neuropharm.2015.03.001. [DOI] [PubMed] [Google Scholar]
- 188.Fan X., Chen Y., Xing J., Wu P., Chen H., Yang J., Zhang J., Wang X., Huang C. Blocking effects of acehytisine on pacemaker currents (If) in sinoatrial node cells and human HCN4 channels expressed in Xenopus laevis oocytes. J. Ethnopharmacol. 2012;139(1):42–51. doi: 10.1016/j.jep.2011.10.039. [DOI] [PubMed] [Google Scholar]
- 189.Sun H., Zhang Q-Z., Wang R-B., Ji J-B. Effects of diacetyl guan-fu base A on pacemaker cells in sinoatrial node of rabbits. Acta Pharmacol. Sin. 2002;23(7):627–630. [PubMed] [Google Scholar]
- 190.Hatch R.J., Jennings E.A., Ivanusic J.J. Peripheral hyperpolarization-activated cyclic nucleotide-gated channels contribute to inflammation-induced hypersensitivity of the rat temporomandibular joint. Eur. J. Pain. 2013;17(7):972–982. doi: 10.1002/j.1532-2149.2012.00261.x. [DOI] [PubMed] [Google Scholar]
- 191.Cho H-j., Furness J.B., Jennings E.A. Postnatal maturation of the hyperpolarization-activated cation current, Ih, in trigeminal sensory neurons. J. Neurophysiol. 2011;106(4):2045–2056. doi: 10.1152/jn.00798.2010. [DOI] [PubMed] [Google Scholar]
- 192.Yeon K-Y., Chung G., Kim Y.H., Hwang J.H., Davies A.J., Park M-K., Ahn D.K., Kim J.S., Jung S.J., Oha S.B. Eugenol reverses mechanical allodynia after peripheral nerve injury by inhibiting hyperpolarization-activated cyclic nucleotide-gated (HCN) channels. Pain. 2011;152:2108–2116. doi: 10.1016/j.pain.2011.05.018. [DOI] [PubMed] [Google Scholar]
- 193.Bois P., Guinamard R., El Chemaly A., Faivre J-F., Bescond J. Molecular Regulation and Pharmacology of Pacemaker Channels. Curr. Pharm. Des. 2007;13:2338–2349. doi: 10.2174/138161207781368729. [DOI] [PubMed] [Google Scholar]
- 194.Rozario A.O., Turbendian H.K., Fogle K.J., Olivier N.B., Tibbs G.R. Voltage-dependent opening of HCN channels: facilitation or inhibition by the phytoestrogen, genistein, is determined by the activation status of the cyclic nucleotide gating ring. Biochim. Biophys. Acta. 2009;1788(9):1939–1949. doi: 10.1016/j.bbamem.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Chen S., Wang J., Siegelbaum S.A. Properties of Hyperpolarization-Activated Pacemaker Current Defined by Coassembly of HCN1 and HCN2 Subunits and Basal Modulation by Cyclic Nucleotide. J. Gen. Physiol. 2001;117(5):491–504. doi: 10.1085/jgp.117.5.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Shang Q., Xu H., Huang L. Tanshinone IIA: A Promising Natural Cardioprotective Agent. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Zhou L., Zuo Z., Chow M.S. Danshen: An Overview of Its Chemistry, Pharmacology, Pharmacokinetics, and Clinical Use. J. Clin. Pharmacol. 2005;45(12):1345–1359. doi: 10.1177/0091270005282630. [DOI] [PubMed] [Google Scholar]
- 198.Khan N., Syed D.N., Ahmad N., Mukhtar H. Fisetin: A Dietary Antioxidant for Health Promotion. Antioxid. Redox Signal. 2012;19(2):151–162. doi: 10.1089/ars.2012.4901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Carlson A.E., Rosenbaum J.C., Brelidze T.I., Klevit R.E., Zagotta W.N. Flavonoid Regulation of HCN2 Channels. J. Biol. Chem. 2013;228(46):33136–33145. doi: 10.1074/jbc.M113.501759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Yan P.Z., Butler P.M., Kurowski D., Perloff M.D. Beyond Neuropathic Pain: Gabapentin Use in Cancer Pain and Perioperative Pain. Clin. J. Pain. 2014;30(7):613–629. doi: 10.1097/AJP.0000000000000014. [DOI] [PubMed] [Google Scholar]
- 201.Surges R.Freiman, Thomas M., Feuerstein Thomas J. Gabapentin Increases the Hyperpolarization-activated Cation Current Ih in Rat CA1 Pyramidal Cells. Epilepsia. 2003;44(2):150–156. doi: 10.1046/j.1528-1157.2003.36802.x. [DOI] [PubMed] [Google Scholar]
- 202.Peng B-W., Justice J.A., Zhang K., Li J-x., He X-h., Sanchez R.M. Gabapentin promotes inhibition by enhancing hyperpolarization-activated cation currents and spontaneous firing in hippocampal CA1 interneurons. Neurosci. Lett. 2011;494(1):19–23. doi: 10.1016/j.neulet.2011.02.045. [DOI] [PubMed] [Google Scholar]
- 203.Peng B-W., Justice J.A., Zhang K., He X-h., Sanchez R.M. Increased Basal Synaptic Inhibition of Hippocampal Area CA1 Pyramidal Neurons by an Antiepileptic Drug that Enhances IH. Neuropsychopharmacology. 2010;35(2):464–472. doi: 10.1038/npp.2009.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Poolos N.P., Migliore M., Johnston D. Pharmacological upregulation of h-channels reduces the excitability of pyramidal neuron dendrites. Nat. Neurosci. 2002;5(8):767–774. doi: 10.1038/nn891. [DOI] [PubMed] [Google Scholar]
- 205.Shin K.S., Rothberg B.S., Yellen G. Blocker State Dependence and Trapping in Hyperpolarization-activated Cation Channels: Evidence for an Intracellular Activation Gate. J. Gen. Physiol. 2001;117:91–101. doi: 10.1085/jgp.117.2.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Chan Y-C., Wang K., Au K.W., Lau C-P., Tse H-F., Li R.A. Probing the bradycardic drug binding receptor of HCN-encoded pacemaker channels. Pflugers Arch. 2009;459:25–38. doi: 10.1007/s00424-009-0719-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Scott S-P., Shea P.W., Dryer S.E. Mapping Ligand Interactions with the Hyperpolarization Activated Cyclic Nucleotide Modulated (HCN) Ion Channel Binding Domain Using a Soluble Construct. Biochemistry. 2007;46(33):9417–9431. doi: 10.1021/bi6026049. [DOI] [PubMed] [Google Scholar]
- 208.Möller S., Alfieri A., Bertinetti D., Aquila M., Schwede F., Lolicato M., Rehmann H., Moroni A., Herberg F.W. Cyclic Nucleotide Mapping of Hyperpolarization-Activated Cyclic Nucleotide-Gated (HCN) Channels. ACS Chem. Biol. 2014;9(5):1128–1137. doi: 10.1021/cb400904s. [DOI] [PubMed] [Google Scholar]
- 209.Lolicato M., Nardini M., Gazzarrini S., Möller S., Bertinetti D., Herberg F.W., Bolognesi M., Martin H., Fasolini M., Bertrand J.A., Arrigoni C., Thiel G., Moroni A. Tetramerization Dynamics of C-terminal Domain Underlies Isoform-specific cAMP Gating in Hyperpolarization-activated Cyclic Nucleotide-gated Channels. J. Biol. Chem. 2011;286(52):44811–44820. doi: 10.1074/jbc.M111.297606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Zagotta W.N., Olivier N.B., Black K.D., Young E.C., Olson R., Gouaux E. Structural basis for modulation and agonist specificity of HCN pacemaker channels. Nature. 2003;425(6954):200–205. doi: 10.1038/nature01922. [DOI] [PubMed] [Google Scholar]
- 211.Craven K.B., Olivier N.B., Zagotta W.N. C-terminal Movement during Gating in Cyclic Nucleotide-modulated Channels. J. Biol. Chem. 2008;283(21):14728–14738. doi: 10.1074/jbc.M710463200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Taraska J.W., Puljung M.C., Olivier N.B., Flynn G.E., Zagotta W.N. Mapping the structure and conformational movements of proteins with transition metal ion FRET. Nat. Methods. 2009;6(7):532–537. doi: 10.1038/nmeth.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Lolicato M., Bucchi A., Arrigoni C., Zucca S., Nardini M., Schroeder I., Simmons K., Aquila M., DiFrancesco D., Bolognesi M., Schwede F., Kashin D., Fishwick C.W., Johnson A.P., Thiel G., Moroni A. Cyclic dinucleotides bind the C-linker of HCN4 to control channel cAMP responsiveness. Nat. Chem. Biol. 2014;10(6):457–462. doi: 10.1038/nchembio.1521. [DOI] [PubMed] [Google Scholar]
- 214.Flynn G.E., Black K.D., Islas L.D., Sankaran B., Zagotta W.N. Structure and Rearrangements in the Carboxy-Terminal Region of SpIH Channels. Structure. 2007;15:671–682. doi: 10.1016/j.str.2007.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Saponaro A., Pauleta S.R., Cantini F., Matzapetakis M., Hammann C., Donadoni C., Hu L., Thiel G., Banci L., Santoro B., Moroni A. Structural basis for the mutual antagonism of cAMP and TRIP8b in regulating HCN channel function. Proc. Natl. Acad. Sci. USA. 2014;111(40):14577–14582. doi: 10.1073/pnas.1410389111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Bucchi A., Baruscotti M., Nardini M., Barbuti A., Micheloni S., Bolognesi M., DiFrancesco D. Identification of the Molecular Site of Ivabradine Binding to HCN4 Channels. PLoS One. 2013;8:e53132. doi: 10.1371/journal.pone.0053132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Doyle D.A., Morais C.J., Pfuetzner R.A., Kuo A., Gulbis J.M., Cohen S.L., Chait B.T., MacKinnon R. The structure of the potassium channel: molecular basis of K1 conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 218.Jiang Y., Lee A., Chen J., Cadene M., Chait B.T., MacKinnon R. Crystal structure and mechanism of a calcium-gated potassium channel. Nature. 2002;417:515–522. doi: 10.1038/417515a. [DOI] [PubMed] [Google Scholar]
- 219.Duval D., Hennig P., Bouchet J.P., Vian J., Peglion J.L., Volland J.P., Platzer N., Guilhem J. Stereochemical Study of a Bradicardisant Benzazepine-Type Drug. X-Ray Structure of the Chloride Salt and High-Field NMR Study of the Stereochemistry in Solution. Magn. Reson. Chem. 1997;35:175–183. [Google Scholar]
- 220.Manz M., Reuter M., Lauck G., Omran H., Jung W. A single intravenous dose of ivabradine, a novel i(f) inhibitor, lowers heart rate but does not depress left ventricular function in patients with left ventricular dysfunction. Cardiology. 2003;100:149–155. doi: 10.1159/000073933. [DOI] [PubMed] [Google Scholar]
- 221.Joannides R., Moore N., Iacob M., Compagnon P., Lerebours G., Menard J.F., Thuillez C. Comparative effects of ivabradine, a selective heart ratelowering agent, and propranolol on systemic and cardiac haemodynamics at rest and during exercise. Br. J. Clin. Pharmacol. 2006;61:127–137. doi: 10.1111/j.1365-2125.2005.02544.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Fox K., Ford I., Steg P.G., Tendera M., Ferrari R. Ivabradine for patients with stable coronary artery disease and left-ventricular systolic dysfunction(BEAUTIFUL): a randomised, double-blind, placebo-controlled trial. Lancet. 2008;372(9641):807–816. doi: 10.1016/S0140-6736(08)61170-8. [DOI] [PubMed] [Google Scholar]
- 223.Swedberg K., Komajda M., Böhm M., Borer J.S., Ford I., Dubost-Brama A., Lerebours G., Tavazzi L., Investigators S. Ivabradine and outcomes in chronic heart failure(SHIFT): a randomised placebo-controlled study. Lancet. 2010;376:875–885. doi: 10.1016/S0140-6736(10)61259-7. [DOI] [PubMed] [Google Scholar]
- 224.McMurray J. J. V., Adamopoulos S., Anker S. D., Auricchio A., Böhm M., Dickstein K., Falk V., Filippatos G., Fonseca C., Gomez-Sanchez M. A., Jaarsma T., Køber L., Lip G. Y. H., Maggioni A. P., Parkhomenko A., Pieske B. M., Popescu B. A., Rønnevik P. K., Rutten F. H., Schwitter J., Seferovic P., Stepinska J., Trindade P. T., Voors A. A., Zannad F., Zeiher A., Bax J. J., Baumgartner H., Ceconi C., Dean V., Deaton C., Fagard R., Funck-Brentano C., Hasdai D., Hoes A., Kirchhof P., Knuuti J., Kolh P., McDonagh T., Moulin C., Reiner Ž., Sechtem U., Sirnes P. A., Tendera M., Torbicki A., Vahanian A., Windecker S., Bonet L. A., Avraamides P., Ben Lamin H. A., Brignole M., Coca A., Cowburn P., Dargie H., Elliott P., Flachskampf F. A., Guida G. F., Hardman S., Iung B., Merkely B., Mueller C., Nanas J. N., Nielsen O. W., Ørn S., Parissis J. T., Ponikowski P. ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2012. . 2012;33:1787–1847. doi: 10.1093/eurheartj/ehs104. [DOI] [PubMed] [Google Scholar]
- 225.Dierckx R., Cleland J.G., Parsons S., Putzu P., Pellicori P., Dicken B., Boyalla V., Clark A.L. Prescribing Patterns to Optimize Heart Rate: Analysis of 1,000 Consecutive Outpatient Appointments to a Single Heart Failure Clinic Over a 6-Month Period. JACC Heart Fail. 2015;3(3):224–230. doi: 10.1016/j.jchf.2014.11.003. [DOI] [PubMed] [Google Scholar]
- 226.Fox K., Ford I., Steg P.G., Tardif J-C., Tendera M., Ferrari R. Ivabradine in Stable Coronary Artery Disease without Clinical Heart Failure. N. Engl. J. Med. 2014;371(12):1091–1099. doi: 10.1056/NEJMoa1406430. [DOI] [PubMed] [Google Scholar]
- 227.DiFrancesco D., Camm A.J. Heart rate lowering by specific and selective I(f) current inhibition with ivabradine: a new therapeutic perspective in cardiovascular disease. Drugs. 2004;64:1757–1765. doi: 10.2165/00003495-200464160-00003. [DOI] [PubMed] [Google Scholar]
- 228.Ceconi C., Comini L., Suffredini S., Stillitano F., Bouly M., Cerbai E., Mugelli A., Ferrari R. Heart rate reduction with ivabradine prevents the global phenotype of left ventricular remodeling. Am. J. Physiol. Heart Circ. Physiol. 2011;300(1):H366–H373. doi: 10.1152/ajpheart.01117.2009. [DOI] [PubMed] [Google Scholar]
- 229.Suffredini S., Stillitano F., Comini L., Bouly M., Brogioni S., Ceconi C., Ferrari R., Mugelli A., Cerbai E. Long-term treatment with ivabradine in post-myocardial infarcted rats counteracts f-channel overexpression. Br. J. Pharmacol. 2012;165(5):1457–1466. doi: 10.1111/j.1476-5381.2011.01627.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Verrier R.L., Silva A.F., Bonatti R., Batatinha J.A., Nearing B.D., Liu G., Rajamani S., Zeng D., Belardinelli L. Combined Actions of Ivabradine and Ranolazine Reduce Ventricular Rate During Atrial Fibrillation. J. Cardiovasc. Electrophysiol. 2014;26:329–335. doi: 10.1111/jce.12569. [DOI] [PubMed] [Google Scholar]
- 231.Opthof T. The membrane current(If) in human atrial cells: implications for atrial arrhythmias. Cardiovasc. Res. 1998;38(3):537–540. doi: 10.1016/s0008-6363(98)00089-3. [DOI] [PubMed] [Google Scholar]
- 232.Pino R., Cerbai E., Calamai G., Alajmo F., Borgioli A., Braconi L., Cassai M., Montesi G.F., Mugelli A. Effect of 5-HT4 receptor stimulation on the pacemaker current If in human isolated atrial myocytes. Cardiovasc. Res. 1998;40(3):516–522. doi: 10.1016/s0008-6363(98)00198-9. [DOI] [PubMed] [Google Scholar]
- 233.Lonardo G., Cerbai E., Casini S., Giunti G., Bonacchi M., Battaglia F., Fiorani B., Stefano P.L., Sani G., Mugelli A. Pharmacological modulation of the hyperpolarization-activated current(If) in human atrial myocytes: focus on G protein-coupled receptors. J. Mol. Cell. Cardiol. 2005;38(3):453–460. doi: 10.1016/j.yjmcc.2004.12.010. [DOI] [PubMed] [Google Scholar]
- 234.Stillitano F., Lonardo G., Giunti G., Del Lungo M., Coppini R., Spinelli V., Sartiani L., Poggesi C., Mugelli A., Cerbai E. Chronic Atrial Fibrillation Alters the Functional Properties of If in the Human Atrium. J. Cardiovasc. Electrophysiol. 2013;24(12):1391–1400. doi: 10.1111/jce.12212. [DOI] [PubMed] [Google Scholar]
- 235.Lezoualc’h F., Steplewski K., Sartiani L., Mugelli A., Fischmeister R., Bril A. Quantitative mRNA analysis of serotonin 5-HT4 receptor isoforms, calcium handling proteins and ion channels in human atrial fibrillation. Biochem. Biophys. Res. Commun. 2007;357(1):218–224. doi: 10.1016/j.bbrc.2007.03.124. [DOI] [PubMed] [Google Scholar]
- 236.Cerbai E., Barbieri M., Mugelli A. Occurrence and properties of the hyperpolarization-activated current If in ventricular myocytes from normotensive and hypertensive rats during aging. Circulation. 1996;94:1674–1681. doi: 10.1161/01.cir.94.7.1674. [DOI] [PubMed] [Google Scholar]
- 237.Fernández-Velasco M., Goren N., Benito G., Blanco-Rivero J., Boscá L., Delgado C. Regional distribution of hyperpolarization-activated current(If) and hyperpolarization-activated cyclic nucleotide-gated channel mRNA expression in ventricular cells from control and hypertrophied rat hearts. J. Physiol. 2003;553(2):395–405. doi: 10.1113/jphysiol.2003.041954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Stilli D., Sgoifo A., Macchi E., Zaniboni M., De Iasio S., Cerbai E., Mugelli A., Lagrasta C., Olivetti G., Musso E. Myocardial remodeling and arrhythmogenesis in moderate cardiac hypertrophy in rats. Am. J. Physiol. Heart Circ. Physiol. 2001;280(1):H142–H150. doi: 10.1152/ajpheart.2001.280.1.H142. [DOI] [PubMed] [Google Scholar]
- 239.Sartiani L., De Paoli P., Stillitano F., Aimond F., Vassort G., Mugelli A., Cerbai E. Functional remodeling in post-myocardial infarcted rats: focus on beta-adrenoceptor subtypes. J. Mol. Cell. Cardiol. 2006;40(2):258–266. doi: 10.1016/j.yjmcc.2005.11.011. [DOI] [PubMed] [Google Scholar]
- 240.Cerbai E., Pino R., Porciatti F., Sani G., Toscano M., Maccherini M., Giunti G., Mugelli A. Characterization of the hyperpolarization-activated current, If, in ventricular myocytes from human failing heart. Circulation. 1997;95:568–571. doi: 10.1161/01.cir.95.3.568. [DOI] [PubMed] [Google Scholar]
- 241.Hoppe U.C., Jansen E., Südkamp M., Beuckelmann D.J. Hyperpolarization-Activated Inward Current in Ventricular Myocytes From Normal and Failing Human Hearts. Circulation. 1998;97(1):55–65. doi: 10.1161/01.cir.97.1.55. [DOI] [PubMed] [Google Scholar]
- 242.Cerbai E., Sartiani L., DePaoli P., Pino R., Maccherini M., Bizzarri F., DiCiolla F., Davoli G., Sani G., Mugelli A. The Properties of the Pacemaker Current IFin Human Ventricular Myocytes are Modulated by Cardiac Disease. J. Mol. Cell. Cardiol. 2001;33(3):441–448. doi: 10.1006/jmcc.2000.1316. [DOI] [PubMed] [Google Scholar]
- 243.Chen K., Baram T.Z., Soltesz I. Febrile seizures in the developing brain result in persistent modification of neuronal excitability in limbic circuits. Nat. Med. 1999;5(8):888–894. doi: 10.1038/11330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Chen K., Aradi I., Thon N., Eghbal-Ahmadi M., Baram T.Z., Soltesz I. Persistently modified h-channels after complex febrile seizures convert the seizure-induced enhancement of inhibition to hyperexcitability. Nat. Med. 2001;7(3):331–337. doi: 10.1038/85480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Brewster A., Bender R.A., Chen Y., Dube C., Eghbal-Ahmadi M., Baram T.Z. Developmental Febrile Seizures Modulate Hippocampal Gene Expression of Hyperpolarization-Activated Channels in an Isoform- and Cell-Specific Manner. J. Neurosci. 2002;22(11):4591–4599. doi: 10.1523/JNEUROSCI.22-11-04591.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Brewster A.L., Bernard J.A., Gall C.M., Baram T.Z. Formation of heteromeric hyperpolarization-activated cyclic nucleotide-gated(HCN) channels in the hippocampus is regulated by developmental seizures. Neurobiol. Dis. 2005;19(1–2):200–207. doi: 10.1016/j.nbd.2004.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Richichi C., Brewster A.L., Bender R.A., Simeone T.A., Zha Q., Yin H.Z., Weiss J.H., Baram T.Z. Mechanisms of seizure-induced ‘transcriptional channelopathy’ of hyperpolarization-activated cyclic nucleotide gated(HCN) channels. Neurobiol. Dis. 2008;29(2):297–305. doi: 10.1016/j.nbd.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Ludwig A., Budde T., Stieber J., Moosmang S., Wahl C., Holthoff K., Langebartels A., Wotjak C., Munsch T., Zong X., Feil S., Feil R., Lancel M., Chien K.R., Konnerth A., Pape H.C., Biel M., Hofmann F. Absence epilepsy and sinus dysrhythmia in mice lacking the pacemaker channel HCN2. EMBO J. 2003;22(2):216–224. doi: 10.1093/emboj/cdg032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Abbas S.Y., Ying S.W., Goldstein P.A. Compartmental distribution of hyperpolarization-activated cyclic-nucleotide-gated channel 2 and hyperpolarization-activated cyclic-nucleotide-gated channel 4 in thalamic reticular and thalamocortical relay neurons. Neuroscience. 2006;141(4):1811–1825. doi: 10.1016/j.neuroscience.2006.05.034. [DOI] [PubMed] [Google Scholar]
- 250.Rateau Y., Ropert N. Expression of a Functional Hyperpolarization-Activated Current(Ih) in the Mouse Nucleus Reticularis Thalami. J. Neurophysiol. 2006;95(5):3073–3085. doi: 10.1152/jn.00922.2005. [DOI] [PubMed] [Google Scholar]
- 251.Shah M.M., Anderson A.E., Leung V., Lin X., Johnston D. Seizure induced plasticity of h channels in entorhinal cortical layer III pyramidal neurons. Neuron. 2004;44:495–508. doi: 10.1016/j.neuron.2004.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Magee J.C. Dendritic integration of excitatory synaptic input. Nat. Rev. Neurosci. 2000;1(3):181–190. doi: 10.1038/35044552. [DOI] [PubMed] [Google Scholar]
- 253.Tsay D., Dudman J.T., Siegelbaum S.A. HCN1 Channels Constrain Synaptically Evoked Ca2+ Spikes in Distal Dendrites of CA1 Pyramidal Neurons. Neuron. 2007;56(6):1076–1089. doi: 10.1016/j.neuron.2007.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Bender R.A., Soleymani S.V., Brewster A.L., Nguyen S.T., Beck H., Mathern G.W., Baram T.Z. Enhanced Expression of a Specific Hyperpolarization-Activated Cyclic Nucleotide-Gated Cation Channel(HCN) in Surviving Dentate Gyrus Granule Cells of Human and Experimental Epileptic Hippocampus. J. Neurosci. 2003;23(17):6826–6836. doi: 10.1523/JNEUROSCI.23-17-06826.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Tang B., Sander T., Craven K.B., Hempelmann A., Escayg A. Mutation analysis of the hyperpolarization-activated cyclic nucleotide-gated channels HCN1 and HCN2 in idiopathic generalized epilepsy. Neurobiol. Dis. 2008;29(1):59–70. doi: 10.1016/j.nbd.2007.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.DiFrancesco J.C., Barbuti A., Milanesi R., Coco S., Bucchi A., Bottelli G., Ferrarese C., Franceschetti S., Terragni B., Baruscotti M., DiFrancesco D. Recessive loss-of-function mutation in the pacemaker HCN2 channel causing increased neuronal excitability in a patient with idiopathic generalized epilepsy. J. Neurosci. 2011;31:17327–17337. doi: 10.1523/JNEUROSCI.3727-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Nava C., Dalle C., Rastetter A., Striano P., de Kovel C.G., Nabbout R., Cances C., Ville D., Brilstra E.H., Gobbi G., Raffo E., Bouteiller D., Marie Y., Trouillard O., Robbiano A., Keren B., Agher D., Roze E., Lesage S., Nicolas A., Brice A., Baulac M., Vogt C., El Hajj N., Schneider E., Suls A., Weckhuysen S., Gormley P., Lehesjoki A-E., De Jonghe P., Helbig I., Baulac S., Zara F., Koeleman B.P., Euro E.R., Haaf T., LeGuern E., Depienne C. De novo mutations in HCN1 cause early infantile epileptic encephalopathy. Nat. Genet. 2014;46(6):640–645. doi: 10.1038/ng.2952. [DOI] [PubMed] [Google Scholar]
- 258.Neuhoff H., Neu A., Liss B., Roeper J. Ih Channels Contribute to the Different Functional Properties of Identified Dopaminergic Subpopulations in the Midbrain. J. Neurosci. 2002;22(4):1290–1302. doi: 10.1523/JNEUROSCI.22-04-01290.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Good C.H., Hoffman A.F., Hoffer B.J., Chefer V.I., Shippenberg T.S., Bäckman C.M., Larsson N-G., Olson L., Gellhaar S., Galter D., Lupica C.R. Impaired nigrostriatal function precedes behavioral deficits in a genetic mitochondrial model of Parkinson's disease. FASEB J. 2011;25(4):1333–1344. doi: 10.1096/fj.10-173625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Santoro B., Piskorowski R.A., Pian P., Hu L., Liu H., Siegelbaum S.A. TRIP8b splice variants form a family of auxiliary subunits that regulate gating and trafficking of HCN channels in the brain. Neuron. 2009;62(6):802–813. doi: 10.1016/j.neuron.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Zolles G., Wenzel D., Bildl W., Schulte U., Hofmann A., Muller C.S., Thumfart J.O., Vlachos A., Deller T., Pfeifer A., Fleischmann B.K., Roeper J., Fakler B., Klocker N. Association with the auxiliary subunit PEX5R/Trip8b controls responsiveness of HCN channels to cAMP and adrenergic stimulation. Neuron. 2009;62:814–825. doi: 10.1016/j.neuron.2009.05.008. [DOI] [PubMed] [Google Scholar]
- 262.Kim C.S., Chang P.Y., Johnston D. Enhancement of dorsal hippocampal activity by knockdown of HCN1 channels leads to anxiolytic- and antidepressant-like behaviors. Neuron. 2012;75(3):503–516. doi: 10.1016/j.neuron.2012.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Friedman A.K., Walsh J.J., Juarez B., Ku S.M., Chaudhury D., Wang J., Li X., Dietz D.M., Pan N., Vialou V.F., Neve R.L., Yue Z., Han M-H. Enhancing Depression Mechanisms in Midbrain Dopamine Neurons Achieves Homeostatic Resilience. Science. 2014;344(6181):313–319. doi: 10.1126/science.1249240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Cao J-L., Covington H.E., Friedman A.K., Wilkinson M.B., Walsh J.J., Cooper D.C., Nestler E.J., Han M-H. Mesolimbic Dopamine Neurons in the Brain Reward Circuit Mediate Susceptibility to Social Defeat and Antidepressant Action. J. Neurosci. 2010;30(49):16453–16458. doi: 10.1523/JNEUROSCI.3177-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Wanat M.J., Hopf F.W., Stuber G.D., Phillips P.E., Bonci A. Corticotropin-releasing factor increases mouse ventral tegmental area dopamine neuron firing through a protein kinase C-dependent enhancement of Ih. J. Physiol. 2008;586(8):2157–2170. doi: 10.1113/jphysiol.2007.150078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Ranaldi R. Dopamine and reward seeking: the role of ventral tegmental area. Rev. Neurosci. 2014;25(5):621–630. doi: 10.1515/revneuro-2014-0019. [DOI] [PubMed] [Google Scholar]
- 267.Inyushin M.U., Arencibia-Albite F., Vázquez-Torres R., Vélez-Hernández M.E., Jiménez-Rivera C.A. Alpha-2 noradrenergic receptor activation inhibits the hyperpolarization-activated cation current(Ih) in neurons of the ventral tegmental area. Neuroscience. 2010;167(2):287–297. doi: 10.1016/j.neuroscience.2010.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Okamoto T., Harnett M.T., Morikawa H. Hyperpolarization-Activated Cation Current(Ih) Is an Ethanol Target in Midbrain Dopamine Neurons of Mice. J. Neurophysiol. 2006;95(2):619–626. doi: 10.1152/jn.00682.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Arencibia-Albite F., Vázquez R., Velásquez-Martinez M.C., Jiménez-Rivera C.A. Cocaine sensitization inhibits the hyperpolarization-activated cation current Ih and reduces cell size in dopamine neurons of the ventral tegmental area. J. Neurophysiol. 2012;107(8):2271–2282. doi: 10.1152/jn.00818.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Chu H-Y., Zhen X. Hyperpolarization-activated, cyclic nucleotide-gated(HCN) channels in the regulation of midbrain dopamine systems. Acta Pharmacol. Sin. 2010;31(9):1036–1043. doi: 10.1038/aps.2010.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Kouranova E.V., Strassle B.W., Ring R.H., Bowlby M.R., Vasilyev D.V. Hyperpolarization-activated cyclic nucleotide-gated channel mRNA and protein expression in large versus small diameter dorsal root ganglion neurons: Correlation with hyperpolarization-activated current gating. Neuroscience. 2008;153(4):1008–1019. doi: 10.1016/j.neuroscience.2008.03.032. [DOI] [PubMed] [Google Scholar]
- 272.Tu H., Deng L., Sun Q., Yao L., Han J-S., Wan Y. Hyperpolarization-activated, cyclic nucleotide-gated cation channels: Roles in the differential electrophysiological properties of rat primary afferent neurons. J. Neurosci. Res. 2004;76(5):713–722. doi: 10.1002/jnr.20109. [DOI] [PubMed] [Google Scholar]
- 273.Momin A., Cadiou H., Mason A., McNaughton P.A. Role of the hyperpolarization-activated current Ih in somatosensory neurons. J. Physiol. 2008;586(24):5911–5929. doi: 10.1113/jphysiol.2008.163154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Emery E.C., Young G.T., Berrocoso E.M., Chen L., McNaughton P.A. HCN2 Ion Channels Play a Central Role in Inflammatory and Neuropathic Pain. Science. 2011;333(6048):1462–1466. doi: 10.1126/science.1206243. [DOI] [PubMed] [Google Scholar]
- 275.Descoeur J., Pereira V., Pizzoccaro A., Francois A., Ling B., Maffre V., Couette B., Busserolles J., Courteix C., Noel J., Lazdunski M., Eschalier A., Authier N., Bourinet E. Oxaliplatin-induced cold hypersensitivity is due to remodelling of ion channel expression in nociceptors. EMBO Mol. Med. 2011;3(5):266–278. doi: 10.1002/emmm.201100134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Ludwig A., Herrmann S., Hoesl E., Stieber J. Mouse models for studying pacemaker channel function and sinus node arrhythmia. Prog. Biophys. Mol. Biol. 2008;98(2–3):179–185. doi: 10.1016/j.pbiomolbio.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 277.Bucchi A., Barbuti A., DiFrancesco D., Baruscotti M. Funny current and cardiac rhythm: insights from HCN knockout and transgenic mouse models. Front. Physiol. 2012;3:240. doi: 10.3389/fphys.2012.00240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Verkerk A.O., Wilders R. Pacemaker Activity of the Human Sinoatrial Node: An Update on the Effects of Mutations in HCN4 on the Hyperpolarization-Activated Current. Int. J. Mol. Sci. 2015;16:3071–3094. doi: 10.3390/ijms16023071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Nakamura Y., Shi X., Numata T., Mori Y., Inoue R., Lossin C., Baram T.Z., Hirose S. Novel HCN2 Mutation Contributes to Febrile Seizures by Shifting the Channel's Kinetics in a Temperature-Dependent Manner. PLoS One. 2013;8(12):e80376. doi: 10.1371/journal.pone.0080376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Baruscotti M., Bottelli G., Milanesi R., DiFrancesco J., DiFrancesco D. HCN-related channelopathies. Pflugers Arch. 2010;460(2):405–415. doi: 10.1007/s00424-010-0810-8. [DOI] [PubMed] [Google Scholar]
- 281.Liao Z., Lockhead D., St. Clair J.R., Larson E.D., Wilson C.E., Proenza C. Cellular context and multiple channel domains determine cAMP sensitivity of HCN4 channels: Ligand-independent relief of autoinhibition in HCN4. J. Gen. Physiol. 2012;140(5):557–566. doi: 10.1085/jgp.201210858. [DOI] [PMC free article] [PubMed] [Google Scholar]