

Graphical abstract
Abstract
Xylanases catalyze the hydrolysis of xylan, an abundant carbon and energy source with important commercial ramifications. Despite tremendous efforts devoted to the catalytic improvement of xylanases, success remains limited because of our relatively poor understanding of their molecular properties. Previous reports suggested the potential role of atomic-scale residue dynamics in modulating the catalytic activity of GH11 xylanases; however, dynamics in these studies was probed on time scales orders of magnitude faster than the catalytic time frame. Here, we used nuclear magnetic resonance titration and relaxation dispersion experiments (15N-CPMG) in combination with X-ray crystallography and computational simulations to probe conformational motions occurring on the catalytically relevant millisecond time frame in xylanase B2 (XlnB2) and its catalytically impaired mutant E87A from Streptomyces lividans 66. Our results show distinct dynamical properties for the apo and ligand-bound states of the enzymes. The apo form of XlnB2 experiences conformational exchange for residues in the fingers and palm regions of the catalytic cleft, while the catalytically impaired E87A variant displays millisecond dynamics only in the fingers, demonstrating the long-range effect of the mutation on flexibility. Ligand binding induces enhanced conformational exchange of residues interacting with the ligand in the fingers and thumb loop regions, emphasizing the potential role of residue motions in the fingers and thumb loop regions for recognition, positioning, processivity, and/or stabilization of ligands in XlnB2. To the best of our knowledge, this work represents the first experimental characterization of millisecond dynamics in a GH11 xylanase family member. These results offer new insights into the potential role of conformational exchange in GH11 enzymes, providing essential dynamic information to help improve protein engineering and design applications.
Cellulosic biomass is considered an inexhaustible energy source for a number of renewable energy applications relying on enzymatic bioconversion, including fuel production, food preparation, pulp and paper transformation, and medical treatment. Cellulosic polysaccharides account for more than 50% of all plant biomass, representing one of the most abundant organic carbon sources on the planet.1,2 Xylanases are involved in the degradation of heteroxylan (hemicellulose), one of the major components of plant biomass. On the basis of amino acid sequence similarity and hydrophobic cluster analysis, the Carbohydrate-Active Enzyme Database (CAZy, http://cazy.org/) classifies xylanases into six glycoside hydrolase (GH) families: GH5, GH8, GH10, GH11, GH30, and GH43. Among them, xylanases from the GH11 family have primarily been used in biobleaching applications.3 This family has been thoroughly characterized and is considered a true xylanase family because of its high substrate specificity toward xylan.1,4,5 As a result, several GH11 members have been structurally and functionally characterized as β-glycosidase models.6–15
The typical structural feature of the GH11 family is the overall conservation of the β-jellyroll domain, which shows a closed right-handed architecture and an active site cleft covered by a “thumb loop”.1 Like many bacteria and fungi that concomitantly secrete a number of hydrolytic enzymes to degrade plant cell walls, Streptomyces lividans 66 secretes two isoforms of the GH11 xylanase B (XlnB), namely, XlnB1 and XlnB2. XlnB1 is composed of two discrete structural and functional units, an N-terminal catalytic domain and a C-terminal xylan-binding domain, while XlnB2 contains only the β-jellyroll catalytic domain. Both forms have the same specific activity against xylan and retain endoacting activity profiles toward this substrate, generating large amounts of oligoxylo-sides with degrees of polymerization ranging from one to eight xylose units.16,17 Despite the high degree of structural homology within the family, GH11 xylanases display significant biophysical variability, including an optimal catalytic pH ranging from 1 to 9, and distinctive thermostability.1 Interestingly, few structural differences account for these vast functional disparities, emphasizing the importance of understanding the molecular and mechanistic determinants responsible for the diverse properties defining optimal catalytic efficiency among GH11 xylanases.8
Dynamic properties of xylanases were previously observed in crystal structures and from molecular dynamic (MD) simulations performed on a number of GH11 enzymes, suggesting the existence of a thumb loop motion that may play a major role in substrate binding, product release, and/or catalysis.18–22 Connelly et al. showed that GH11 xylanase from Bacillus circulans (BCX) is rigid on the picosecond-to-nanosecond time scale in the absence and presence of ligand,23 while Havukainen et al. illustrated that the width of the active site cavity of the Trichoderma reesei xylanase II (XYNII) is reduced upon binding to epoxy-alkyl-β-d-xyloside analogues.24 Using novel in silico modeling approaches combined with in vitro studies, Paës et al. showed the important role of thumb loop residues in product release and (de)glycosylation.25 While these studies provided important information about protein flexibility, they nevertheless report on motions occurring on time scales much faster than that of the catalytic reaction. Indeed, crystal structure investigation provides access to limited snapshots of possible xylanase conformations, while classical MD simulations explore motions on a much faster time scale (nanoseconds), which is far from the millisecond time frame of catalytic turnover in these enzymes.
In the study presented here, we characterized the conformational dynamics of XlnB2 and a catalytically impaired variant (XlnB2-E87A) from S. lividans in the apo and ligand-bound states over a broad range of time scales using 15N-CPMG NMR relaxation dispersion measurements and molecular dynamics (MD) simulations to probe conformational fluctuations on the catalytic time frame. Mutation of the general acid/base residue E87 (which performs hydrolysis by adding or removing a proton) to an alanine blocks the first step of the hydrolytic reaction,26 resulting in a catalytically impaired enzyme variant. This mutation prevents distortion of the substrate conformation in the active site cleft,26 providing information about the structural complex between xylanase and longer xyloside ligands. The XlnB2-E87A variant was, therefore, also used to investigate the conformational exchange experienced by a Michaelis–Menten complex between the enzyme and a typical oligomeric substrate. Our results show that XlnB2 and its catalytically impaired E87A variant experience distinct conformational exchange behavior in the apo and ligand-bound states. Enhanced conformational dynamics in the thumb loop region was observed upon binding of the xylobiose (X2) product and xylopentaose (X5) substrate. Analysis of the conformational substates of the top dynamic modes associated with the interconversion between the free and substrate-bound states further revealed a “twist type” motion involving the thumb loop and fingers of XlnB2. These observations provide important insights into the potential role of functionally relevant millisecond time-scale dynamics in XlnB2.
Experimental Procedures
Cloning, Mutagenesis, and Protein Purification
The gene encoding XlnB2 was cloned as described previously.27 The XlnB2-E87A variant was generated by site-directed mutagenesis using standard procedures. The sequence encoding the XlnB2-E87A variant (without the signal sequence) was amplified from vector pIAF935, which contained the xlnB2 gene as a template, using the following DNA primers: 5′-CCGGTACCGACACGGTCGTCACGA-3′ (forward primer, with the KpnI site underlined) and 5′-CCGAGCTCTCACCCGCCGACGTTG-ATGCT-3′ (reverse primer, with the SacI site underlined). The point mutation E87A was introduced by overlap extension polymerase chain reaction (PCR),28 using the following DNA primers: 5′-CCGCTCGTCGCGTACTACATCGTCG-3′ (forward) and 5′-CGACGATGTAGTACGCGACGAGCGG-3′ (reverse). The 593 bp KpnI–SacI-digested PCR product containing the E87A mutation was cloned into the same restriction sites in plasmid pC109 (NEB, Ipswich, MA), which carries a thiostrepton resistance marker. The resulting construct was subsequently transformed into S. lividans 66 10-164 (American Type Culture Collection, Manassas, VA), a xylanase-and cellulose-negative protoplast, and plated on R5 medium without an antibiotic for 16 h at 34 °C. The agar plates were then flooded with a thiostrepton solution and further incubated for 3–4 days at 34 °C. The resulting plasmid was isolated, and positive clones were confirmed by DNA sequencing.
XlnB2 and XlnB2-E87A were enriched by growing the transformed S. lividans strains in M14 medium supplemented with (15NH4)2SO4 (Sigma-Aldrich, St. Louis, MO) and 1% (w/v) xylose as the sole nitrogen and carbon sources, respectively. For multidimensional NMR experiments, xylose was replaced with [;13C]glucose (w/v) (Sigma-Aldrich). It is known that glucose represses the secretion of extracellulosic enzymes in S. lividans.16 However, expression yields of the targeted enzymes with [;13C]glucose were shown to be sufficient for NMR investigation. After incubation for 72 h, proteins were obtained with final yields of 63 and 36 mg/L for 13C- and 15N-labeled XlnB2 and XlnB2-E87A, respectively. The proteins were concentrated by ultrafiltration and dialyzed against 20 mM citric buffer (pH 4.5), prior to being loaded on a CM FF column (GE Healthcare, Baie d'Urfé, QC) pre-equilibrated with 20 mM citrate buffer (pH 4.5). The enzymes were eluted with a linear gradient of 20 mM citrate buffer (pH 4.5) and 1 M NaCl. Fractions containing XlnB2 or XlnB2-E87A were collected, dialyzed in water, and lyophilized. Mass spectrometry was also employed to confirm sequence identity and integrity. The protein concentration was determined by UV–vis spectroscopy (between 250 and 350 nm) using a predicted extinction coefficient value (ε280) of 63830 M−1 cm−1 (ExPASy ProtParam). Protein purity was assessed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis using ImageJ (National Institute of Health, Bethesda, MD).
Crystallization and Structure Determination of XlnB2
Crystals of XlnB2 were obtained by vapor diffusion by mixing equal volumes of the purified XlnB2 and a mother liquor composed of 0.2 M zinc acetate dehydrate, 0.1 M sodium acetate (pH 4.5), and 10% (w/v) PEG 3000 at room temperature. Rod-shaped crystals were harvested and soaked in the mother liquor supplemented with 20% glycerol. A single diffraction data set was collected by the Life-Science-Collaborative Access Team at Argonne National Laboratory (Lemont, IL). The structure was determined by molecular replacement using Xyl1 from Streptomyces sp. S38 as a search model [;Protein Data Bank (PDB) entry 1HIX].29 The structure of XlnB2 was completed using iterative rounds of model building and refinement using Coot30 and Phaser,31 respectively. The quality of the structure was assessed using MolProbity.32
Resonance Assignments
NMR samples were prepared by solubilizing lyophilized, uniformly 15N-labeled or 15N- and 13C-labeled proteins in 20 mM MES (pH 6.5), 20 mM NaCl, 0.01% NaN3, and 10% 2H2O. All NMR experiments were recorded at 298 K on samples containing 0.6–0.8 mM protein in 5 mm Shigemi NMR tubes. NMR experiments were performed on 600 and 800 MHz Inova NMR spectrometers (Agilent, Santa Clara, CA) equipped with triple-resonance cold probes and pulsed-field gradients. For backbone assignments of XlnB2, a series of standard Agilent BioPack 1H, 15N two-dimensional (HSQC) and 1H, 13C, 15N three-dimensional experiments were performed.33 All spectra were processed with NMRPipe34 and analyzed using NMRViewJ version 8 and Sparky, in addition to a series of in-house scripts. The backbone assignments of XlnB2-E87A were achieved by overlaying the 1H–15N HSQC spectrum with the corresponding assigned spectrum of wild type (WT) XlnB2. Peaks showing chemical shift variations were confirmed using three-dimensional (3D) assignment experiments.
NMR Titration Experiments
All NMR titration experiments were performed in 20 mM MES (pH 6.5), 20 mM NaCl, and 0.01% NaN3. Concentrations of 657 μM (661 μM) of 15N-labeled XlnB2 (XlnB2-E87A) were used for the titration of unlabeled xylobiose (xylopentaose). 1H–15N HSQC experiments were performed at 800 MHz with 256 and 8192 points in the t1 and t2 dimensions, respectively. 1H–15N HSQC spectra of XlnB2 were recorded by gradually increasing the ligand:enzyme molar ratios to 0.3, 1.2, 3, 6, 12, 20, 30, 40, 60, 80, and 100. Similarly, 1H–15N HSQC spectra of XlnB2-E87A were recorded with increasing ligand:enzyme molar ratios of 0.3, 1.2, 3, 6, 12, 20, 30, 60, 80, 90, and 100. Chemical shift variations (Δδ) were calculated using the equation described by Grzesiek et al.:35 Δδ (ppm) = [;(ΔδHN2 + ΔδN2/25)/2]. The dissociation constant (Kd) was determined for each residue showing a Δδ of ≥0.05 ppm and averaged, as previously described.36 Residues Trp20, Thr120, Glu128, Thr130, and Tyr171 were used for determining the Kd, which corresponds to the intercept on the x-axis (ligand concentration) at 50% the total variation of Δδ.
15N Spin Relaxation
15N T1, T2, and 15N{1H} steady-state NOE spectra for free WT XlnB2 were acquired with water-selective pulse sequences described by Farrow et al.37 1H – 15N HSQC spectra recorded to measure the T1 and T2 series were collected with 216 complex points in the 15N dimension and 1666 points in the proton dimension and zero-filled to 6664. Sixteen transients were recorded for each measurement. Relaxation delays of 0.01, 0.08, 0.16, 0.32, 0.64, 0.96, 1.28, and 1.92 s and 0.01, 0.03, 0.05, 0.07, 0.09, 0.11, and 0.15 s were used to measure T1 and T2, respectively. A 15N{1H} NOE spectrum with and without a 5.0 s proton presaturation period following a 5.0 s relaxation delay was used compared to a single 10 s relaxation delay in the reference spectrum. NOE spectra were recorded with 192 complex points in the 15N dimension but were otherwise identical to the T1 and T2 spectra. The sample was dissolved in potassium phosphate buffer (pH 6.5), and all spectra were recorded at 800 MHz and 25 °C.
NMR Relaxation Dispersion Experiments (millisecond motions)
Backbone amide 15N-CPMG relaxation dispersion experiments were performed at 25 °C for XlnB2 (657 μM) and XlnB2-E87A (661 μM) in the apo and saturated ligand-bound forms (100 molar equivalent ligand concentration) with B0 fields of 14.1 T (600 MHz) and 18.77 T (800 MHz) using published sequences.38 The constant time delay was set to 20 ms, and interleaved two-dimensional spectra were recorded in a constant time manner with τcp CPMG repetition delays of 0.625, 0.714 (×2), 1.0, 1.25, 1.67, 2.0, 2.50 (×2), 3.33, 5.0, and 10 ms. Global residue fits and model analyses were performed by fitting 600 and 800 MHz CPMG dispersion data to the single-quantum CPMG equation33,39,40 using GraphPad Prism 5. Global residue fits were performed using the following residues from the fingers region: Gln8, Phe19, Trp20, Val48, Gly71, Asn72, Thr176, and Gly178 (XlnB2) and Gly71, Asn72, Thr176, Gly178, and Tyr179 (XlnB2-E87A). Residue selection for global kex calculation was based on (1) the quality of dual-field individual fits (600 and 800 MHz) for residues showing similar kex values (within error) and (2) similar residue position correspondence between the two proteins (XlnB2 and XlnB2-E87A).
Chemical Shift Projection Analysis (CHESPA)
A CHESPA was used to characterize the effect of binding of two ligands to XlnB2-E87A using the protocol described by Axe et al.41 For each selected residue, the fractional and were calculated as described by Selvaratnam et al.42 using the 1H and 15N coordinates of the peak in its initial state (apoenzyme) and at saturation upon binding two ligands, X2 and X5.
Computational Modeling of the Apo and Substrate-Bound States of XlnB2
Molecular dynamics (MD) simulations were performed to model XlnB2 in the free and substrate-bound (binary complex) forms, both with explicit water solvent. Xylopentaose (X5), xylohexaose (X6), and xylononaose (X9) substrates were used for the ligand-bound simulations. Model preparation and simulations were performed using the AMBER version 1443 suite of programs for biomolecular simulations. AMBER's 99SB_ildn force field was used for all simulations. MD simulations were performed using NVIDIA graphical processing units (GPUs) and AMBER's pmemd.cuda simulation engine (with the default SPFP precision).
For the free form, the coordinates of the XlnB2 enzyme were taken from the crystal structure determined in this study (PDB entry 5EJ3). The system was neutralized by addition of counterions (two Cl− ions), and the resulting system was then immersed in a rectangular box of SPC/E water, with a 10 Å minimum distance between the protein and the edge of the periodic box A similar protocol was used to prepare the system for the binary complex with a model substrate. The crystal structure of substrate-bound xylanase II from T. reesei (PDB entry 4HK8) was used to prepare the substrate-bound conformations of XlnB2. The longer chain substrate (X9) was created by extending the coordinates of the X6 substrate through duplication of three xylose units (+3, +2, +1) and translating them to attach at the end of the chain (−3 xylose unit) using PyMOL visualization software. Analysis of MD trajectories showed that X6 and X9 were most stable in the binding pocket during the long MD runs and were therefore selected for further structural and dynamic characterization.
The prepared systems were equilibrated using a protocol described previously.44 The equilibrated systems were then used to run 1 μs of production MD under constant energy conditions (NVE ensemble). Coordinates stored every 10 ps for a total of 100000 conformational snapshots were used for analysis, collected for each of the two systems (free form and binary complex).
Quasi-Anharmonic Analysis (QAA)
The 200000 conformational snapshots obtained from the (apo and substrate-bound complex) MD simulations were used for QAA The top QAA-independent component vectors were analyzed in detail to characterize the primary dynamics associated with the substrate binding process in XlnB2. Note that QAA with 100000 (and more) conformational snapshots was used to provide reproducible and accurate conformational fluctuations, and only the protein C\α atom positions were used for the analysis. To obtain an accurate description of the conformational dynamics associated with the substrate binding process, good sampling is required. We therefore used the X6 and X9 substrates with six and nine xylose units, respectively, for the simulations and subsequent characterization of conformational dynamics in XlnB2.
Enzyme–Substrate Interactions
The energy for the enzyme–substrate interactions was calculated as a sum of electrostatic and van der Waals energies between atom pairs. We developed this protocol previously to investigate other enzyme systems.45
where Eel is the electrostatic contribution, Evdw is the van der Waals term, and the summation runs over all atom pairs for the enzyme–substrate complex The Eel and Evdw terms were computed as follows
where qi values are partial charges and Aij and Bij are Lennard-Jones parameters. These parameters were obtained from the AMBER force field. A distance-dependent dielectric function was used:
where B = εo – A, εo = 78.4 for water, A = −8.5525, λ = 0.003627, and k = 7.7839.
All xylanase and substrate atom pairs were included in the calculations, and resulting interaction energies were summed up per residue pair. The energies were calculated for 1000 snapshots, every 1 ns, sampled during the full 1 μs simulation and were averaged over these 1000 snapshots.
Results
Crystal Structure of XlnB2
To generate a working model for XlnB2, we determined the crystal structure of WT XlnB2 (PDB entry 5EJ3) by molecular replacement using Xyl1 as a search model (Table 1). Two molecules were placed in the asymmetric unit and refined to 1.31 Å. The final model was composed of two chains, each comprising residues 1–191 of XlnB2. The structure folds as a β-jellyroll domain displaying two β-sheets of five and nine β-strands (termed sheets A and B, respectively). The single α-helix is positioned under β-sheet B. Comparison of the XlnB2 crystal structure with two canonical GH11 xylanases from T. reesei8 and B. circulans46 shows a high degree of structural homology, with root-mean-square deviation (rmsd) values of 0.8 and 0.4 Å for all atoms, respectively. These results demonstrate that XlnB2 shares the common structural properties of other xylanases, such as a closed right-handed architecture and a substrate-binding site covered by a β-hairpin (termed the thumb loop) formed by β-strands B8 and B7 (Figure 1A). A set of six β-strands, also termed fingers, form the other side of the substrate-binding cleft. The cord loop, which joins β-strands B6 and B9, is 12 residues long in XlnB2.
Table 1. Data Collection and Refinement Statistics for the Crystal Structure of WT XlnB2 from S. lividans 66a.
| Data Collectionb | |
| space group | P21 |
| cell dimensions | |
| a, b, c (Å) | 41.0, 66.9, 65.4 |
| β (deg) | 94.1 |
| resolution (Å) | 46.7–1.3 (1.44–1.3) |
| Rsym | 6.7 (55.2) |
| I/σI | 12.2 (2.6) |
| completeness (%) | 97.5 (96.4) |
| redundancy | 4.2 (4.2) |
| Refinement | |
| resolution (Å) | 46.7–1.3 |
| no. of reflections | 81872 |
| Rwork,Rfree | 12.9–16.6 |
| no. of atoms | |
| XlnB2 | 3055 |
| water | 410 |
| B factor (Å2) | |
| XlnB2 | 15.5 |
| water | 31.8 |
| rmsd | |
| bond lengths (Å) | 0.014 |
| bond angles (deg) | 1.42 |
| Molprobity score | 0.98 |
| Ramachandran plot (%) | |
| outliers | 0 |
| favored | 98 |
The data set has been collected on a single crystal.
Data for the highest-resolution shell are given in parentheses.
Figure 1.
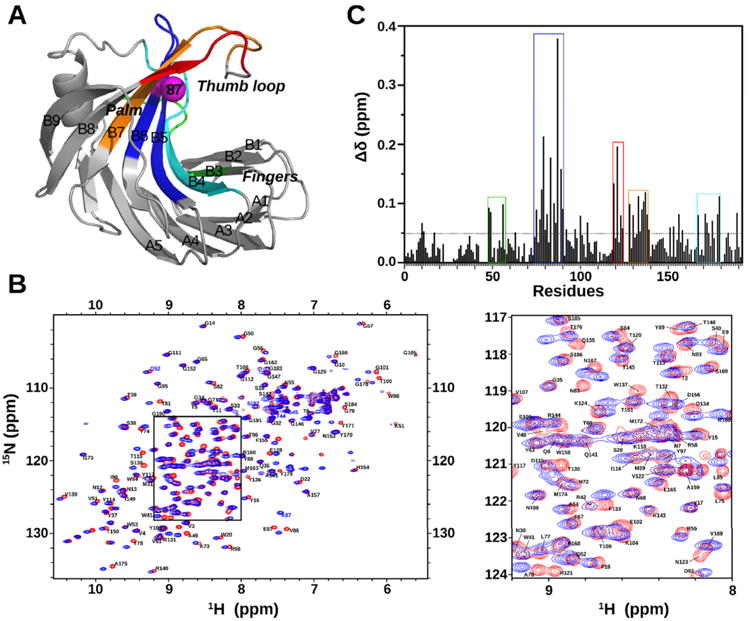
Chemical shift variations experienced by XlnB2 upon introduction of the E87A mutation. (A) Cartoon representation of the crystal structure of XlnB2 (PDB entry 5EJ3). The E87A point mutation is displayed as a magenta sphere. Residue clusters presenting the most significant chemical shift changes are color-coded on the structure of XlnB2. (B) 1H–15N HSQC overlay of WT XlnB2 (red) and XlnB2-E87A (blue). A close-up view of the central spectral region is shown on the right. Amino acids are identified by the single-letter code followed by sequential residue numbers. (C) Bar graph representing chemical shift variations (Δδ) between XlnB2 and its E87A variant. The dotted line indicates the altered threshold of chemical shift variations considered significant (Δδ > 0.05 ppm). Δδ values were calculated using the equation Δδ (ppm) = [;(ΔδHN2 + ΔδN2/25)/2]1/2 as described by Gresziek et al.35 (see Experimental Procedures).
NMR Assignments and Spectral Comparison of XlnB2 and the E87A Variant
The 1H–15N HSQC (heteronuclear single quantum coherence) spectra of XlnB2 and XlnB2-E87A show well-dispersed resonances, indicating well-folded proteins (Figure 1B). We used a combination of two-dimensional (1H–15N HSQC) and three-dimensional [;3D HNCO, HNCACB, CBCA(CO)NH, and HN(CA)CO] NMR experiments to sequentially assign the 1H, 13C, and 15N backbone resonances of XlnB2 and XlnB2-E87A. Peak assignments were confirmed with a 15N-NOESY experiment. A total of 172 (171) backbone 1H–15N peaks were assigned of the 186 peaks expected for XlnB2 (XlnB2-E87A), excluding the five prolines and the N-terminal residue, corresponding to 92.5% (91.9%) of all non-proline residues. Resonance assignments were obtained for 84.4% (84.4%) of non-proline Cα atoms and 69.9% (69.9%) of non-proline and non-glycine Cβ atoms. Most of the unassigned resonances were located on finger loops, suggesting that these residues experience signal broadening and potential dynamics in solution. In both enzymes, the NH cross peaks from arginine residues and the indole NH cross peaks from the tryptophan side chains were not assigned. Chemical shift assignments have been deposited in the BioMagResBank (http://www.bmrb.wisc.edu) as entry 26761.
A comparison of the chemical shift changes and predicted order parameters of the WT and E87A variant shows that the point mutation does not significantly perturb the three-dimensional structure of the enzyme (Figure S1). The chemical environment of residues affected by the mutation was characterized by quantifying the chemical shift variations (Δδ) observed between XlnB2 and the E87A variant. An overlay of the 1H–15N HSQC spectra of XlnB2 and the E87A variant showed several residues with Δδ values of >0.05 ppm between the two proteins (Figure 1B,C). As expected, Glu87 (Δδ = 0.37 ppm) and the surrounding residues in β-strands B5 and B6 showed the largest chemical shift variations (blue box, Figure 1A,C). Sizable chemical shift changes were also observed for residues of β-strands B3, B4, and B7 (and associated loops), as well as in the thumb (red and orange boxes, Figure 1A,C).
Comparison of the Effect of Ligand Binding on XlnB2 and the E87A Variant
Changes in the chemical environment upon ligand binding can provide valuable information about potential binding site(s), as well as the kinetics and ligand affinity for the enzyme. We performed NMR chemical shift titration measurements to characterize and compare the binding properties of XlnB2 and its E87A variant with two xyloside ligands. XlnB2 is an endoxylanase that hydrolyzes xylan to oligoxylosides with varying lengths, ranging from one to eight xylose units.16 Xylotriose (X3) is the smallest substrate of this endocleavage reaction, releasing xylose (X1) and xylobiose (X2) as uncleavable products. Here, we titrated XlnB2 and the E87A variant with incremental concentrations of X2 and X5 (E87A only), up to saturation, to determine ligand binding effects on the chemical environment of the enzyme. All residues displaying Δδ values of >0.05 ppm are highlighted on the 3D structure of XlnB2 and the E87A variant (Figure 2A–C). A total of four residues (Gln8, Gly10, Thr11, and Tyr17) and 11 residues (Gly10, Thr11, Phe19, Trp20, Gly79, Glu128, Thr130, Lys143, Leu165, Tyr171, and Ile173) displayed Δδ values of >0.1 ppm, defined as 2σ, upon binding to X2 in XlnB2 and the E87A variant, respectively (Figure 2D). Both proteins exhibited weak binding affinities (Kd) of 7.07 and 8.71 mM, respectively, toward X2, suggesting that the mutation did not significantly perturb binding to product X2. Although the number of residues with a Δδ value of >0.1 ppm is larger for XlnB2-E87A, most residues affected by the presence of the ligand were located in the vicinity of the active site.
Figure 2.
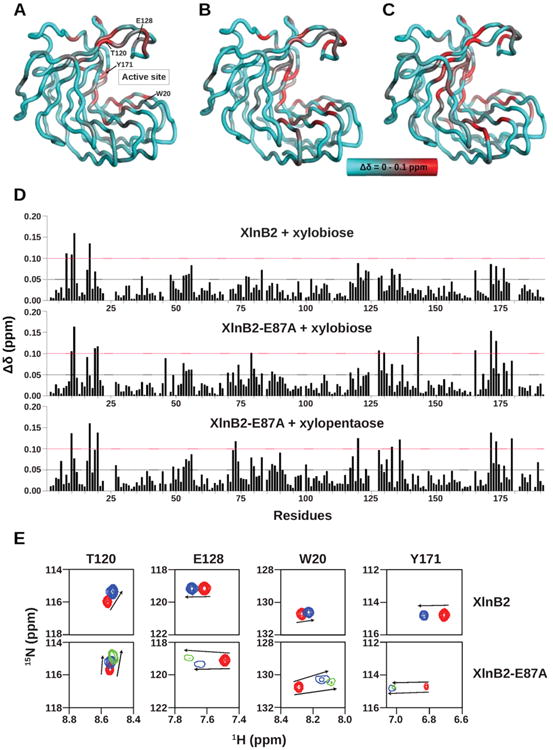
1H–15N composite chemical shift variations experienced by XlnB2 and XlnB2-E87A upon oligoxyloside binding. Structural mapping of chemical shift variations (Δδ) observed for (A) XlnB2 induced upon xylobiose (X2) binding, (B) XlnB2-E87A induced upon xylobiose binding, and (C) XlnB2-E87A induced upon xylopentaose (X5) binding. The Δδ was calculated between free and ligand-saturated enzymes as described by the equation Δδ (ppm) = [;(ΔδHN2 + ΔδN2/25)/2]1/2 and reported on the three-dimensional structure of XlnB2 using a cyan-to-red color scale with a 0.0–0.1 ppm range.35 The locations of the active site and key residues are reported on the 3D structure in panel A. (D) Bar graph representation of the chemical shift variations observed for XlnB2 and XlnB2-E87A upon binding to xylobiose (X2) and xylopentaose (X5). Δδ > 0.05 ppm and Δδ > 0.1 ppm chemical shift changes are represented by black and red dotted lines, respectively. (E) 1H–15N chemical shift direction and magnitude for residues Trp20, Thr120, Glu128, and Tyr171 in XlnB2 and XlnB2-E87A observed upon ligand binding (see the text for details). The 1H–15N peak position of the free enzyme and that upon saturation with X2 (X5) is shown in red and blue (green), respectively. All images were prepared with PyMOL, and graphs were produced with GraphPad Prism.
Previous studies suggested that the active site of GH11 family members could accommodate linear xylan substrates ranging from five to six xylose units. On the basis of these observations, we titrated XlnB2-E87A with a xylopentaose (X5) ligand (five xylose units), effectively mimicking a substrate-bound Michaelis–Menten reaction complex. A total of 10 residues (Gly10, Tyr17, Trp20, Ala73, Thr120, Phe133, Tyr136, Tyr171, Ile173, and Tyr179) showed Δδ values of >0.1 ppm in the E87A variant upon X5 binding (Figure 2D). Residues displaying the largest Δδ were located in the active site region (Figure 2C), in addition to residues in the thumb loop hinge (Thr120 and Tyr136) and fingers (Tyr17 and Tyr179). Despite titration with a longer ligand, binding of X5 to the E87A variant did not induce significantly different chemical shift variations relative to X2, both in terms of the number and magnitude shift of affected residues, and showed a similar low binding affinity for X5 (Kd = 7.93 mM). Our chemical shift analyses do not suggest the presence of a secondary binding site (SBS), as was observed in xylanases from other organisms to enhance xylan hydrolysis and substrate binding.47,48 These results suggest that the enzyme may primarily bind and stabilize a limited number of xylose units in the active site, regardless of xylose substrate unit length.
Recent studies showed that the direction and magnitude of the 1H and 15N chemical shift perturbations could provide valuable information about the nature and behavior of ligand binding events between protein variants.41,42,49 Using this CHESPA approach, we investigated whether the E87A mutation in XlnB2 affected thumb loop behavior with respect to ligand binding. Although the E87A mutation did not extensively perturb the general chemical shift profile induced by X2 binding relative to XlnB2, Glu128 and Thr130 showed some of the most significant thumb loop Δδ variations upon ligand binding in both proteins (Figure 1). Interestingly, these two variable residues of the thumb loop also showed significant 1H and 15N chemical shift variations upon introduction of the E87A mutation (Figure 1A,C). We determined that the 1H and 15N chemical shift magnitude and direction for Glu128 were identical between XlnB2 and its mutational variant upon binding to X2 (Figure 2E). Furthermore, the direction and intensity of the movement were also similar upon binding to both X2 and X5, suggesting that the length of the ligand did not affect the magnetic environment or direction of the thumb loop movement. When ligand binds to XlnB2 and XlnB2-E87A, we also observed chemical shift variations for Thr120 (Figure 2D), a residue close to the hinge of the thumb loop. Much like Glu128, Thr120 showed coordinated chemical shift changes upon binding to X2 and X5 (Figure 2E), further suggesting that the movement and orientation of the thumb loop are the same in XlnB2 and its E87A variant. Two additional residues, Trp20 and Tyr171, located on opposite sides of the active site, were also highly affected upon ligand binding. These conserved residues play an important role in substrate binding in GH11 family members.1,46,50 Our results demonstrate a similar behavior, showing comparable Kd values upon ligand binding, and coordinated chemical shift responses (Figure 2E). These results suggest that the catalytically inactive XlnB2-E87A variant preserves a similar WT thumb loop movement and orientation and that these conformational properties may be important for substrate positioning, discrimination, or processivity. However, because the direction and magnitude of chemical shifts are both preserved in WT XlnB2 and the catalytically inactive E87A variant upon ligand binding, these results suggest that thumb loop movement is not involved in the actual chemical step of XlnB2 catalysis.
Conformation Exchange on the Millisecond Time Scale
It was previously demonstrated that protein flexibility and coordinated residue motions play an important role in catalysis for a variety of enzyme systems, such as dihydrofolate reductase,51 RNA-dependent RNA polymerase,52 and ribonuclease.53 A number of studies have also investigated the fast time-scale dynamics in GH11 xylanases.1,18,22,24,25 For comparison with homologous members of the family, we also acquired 15N T1, T2, and 15N{1H} NOE data to investigate fast time-scale dynamics in apo WT XlnB2 (Figure S2). Much like previous observations for the B. circulans xylanase,23 we found that the measured T1, T2, and NOE parameters for the backbone amides do not vary significantly. A number of 15N{1H} heteronuclear NOEs still show significantly smaller values relative to the protein average, including one thumb loop residue (G129) and three residues of the cord (T96, Y97, and G101), indicating that these positions undergo larger-amplitude fluctuations on the picosecond-to-nanosecond time scale. While a full Lipari–Szabo investigation or reduced spectral density mapping analysis in the presence and absence of ligand(s) could potentially yield information about fast time-scale dynamics in this enzyme, our XlnB2 relaxation parameters are relatively uniform across the sequence, suggesting an organized tertiary structure on this fast time scale. However, conformational fluctuations in XlnB2 occurring on the millisecond time scale could potentially be coupled to substrate recognition, stabilization, processivity (substrate sliding), and/or a direct catalytic event in GH11 xylanases. To characterize these dynamic events in solution, and to further understand the functional importance of conformational fluctuations occurring on the catalytic time scale in GH11 xylanases, we performed 15N-Carr–Purcell–Meiboom–Gill (15N-CPMG) relaxation dispersion measurements of the apo and ligand-bound forms of XlnB2 and the catalytically impaired XlnB2-E87A variant. This methodology is particularly suited to identifying residues experiencing conformational exchange (motions occurring on the millisecond time scale) in enzymes, overlapping with the rate of catalysis.54 While Connelly et al. reported on potential microsecond-to-millisecond motions extracted from the rate of chemical exchange (Rex) determined using the Lipari–Szabo model-free formalism on the B. circulans xylanase,23 to the best of our knowledge, the work presented here represents the first dedicated experimental validation of the conformational fluctuations experienced by a GH11 xylanase on the catalytically relevant millisecond time frame.
All residues experiencing significant conformational exchange [;R2(1/τCP) > 1.8 s–1 (Figure S3)] are shown as spheres on the structure of XlnB2 and its XlnB2-E87A variant (Figure 3). In the apo forms (Figure 3A,B), a total of 22 (12) residues showed significant millisecond motions in XlnB2 (XlnB2-E87A). In WT XlnB2 (Figure 3A), residues experiencing conformational exchange are predominantly clustered in the fingers, and few others in the palm region of the enzyme. These results demonstrate that other regions of the protein, including the thumb loop, are rigid in the apo form on this particular time scale. While the global profile remains similar in the free XlnB2-E87A variant (with visible loss of millisecond dynamics in the B1–B2–B3 sheets), the global rate of conformational exchange (kex) in the fingers and palm region is 2-fold greater for the mutant (for WT XlnB2, kex = 1220 ± 392 s−1; for XlnB2-E87A, kex = 2794 ± 399 s−1).
Figure 3.
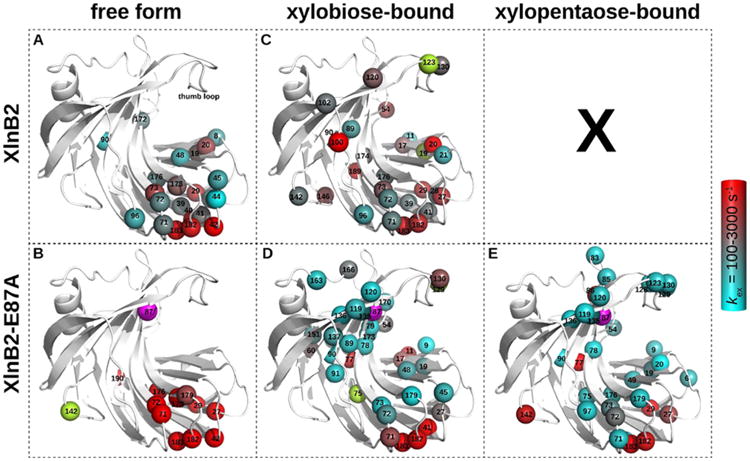
Residues experiencing conformational exchange in the free and ligand-bound forms of XlnB2 and XlnB2-E87A. Conformational exchange was probed using 15N-CPMG NMR relaxation dispersion for XlnB2 and its catalytically impaired variant E87A in the (A and B) free, (C and D) xylobiose-saturated (X2), and (E) xylopentaose-saturated (X5) forms. Residues with a ΔR2(1/τcp) of >1.8 s–1 are reported as spheres on the 3D structure of the enzyme. The color-graded scale represents the rate of conformational exchange (kex) experienced by the backbone 1H–15N vector, calculated on a per residue basis using the single-quantum 15N-CPMG equation.63 Residues with a ΔR2(1/τcp) of >1.8 s–1 for which a kex could not be extracted are colored light green. The mutation site (E87A) is represented as a magenta sphere. All images were prepared with PyMOL. Representative relaxation dispersion curves for each system are shown in Figure S2.
The product-bound enzyme complex between XlnB2 and X2 showed a total of 29 and 32 residues in XlnB2 and XlnB2-E87A, respectively, demonstrating an increased level of conformational exchange relative to the apo state (Figure 3C,D). Residues in the thumb loop and the region facing the active site cleft acquire conformational fluctuations upon binding to X2. We note that more residues in the palm region facing the active site cleft experience millisecond dynamics in the XlnB2-E87A mutant relative to WT XlnB2. The substrate-bound complex with X5 in the XlnB2-E87A variant showed 33 residues displaying millisecond conformational exchange (Figure 3E), with enhanced conformational motions in the thumb loop and distal regions in the vicinity of this loop. Residues in the finger loop regions, such as Ser182 and Gly183, showed significantly greater conformational exchange rates (kex > 3000 s−1) for free and ligand-bound states, consistent with line broadening observations for some residues of this region (Figure 1B). A comparison of the chemical shift variations (ΔδN) from titration experiments with the Δω obtained from 15N-CPMG showed no significant correlation, suggesting that the chemical shift variations observed upon ligand binding do not exclusively report on the existence of interconverting conformations between a ground and excited state in apo XlnB2 in solution (Figure S4).
Interestingly, while the thumb loop and the palm region facing the active site cleft are practically devoid of millisecond dynamics in the free enzyme, ligand binding (substrate and product) induces considerable conformational exchange in these regions for both XlnB2 and the E87A variant. Previous studies, based on crystal structures and MD simulations probing the nanosecond time scale, showed a dynamic shift of the thumb loop upon ligand binding.24,25 In contrast, our results reveal that the thumb loop region is rigid on the millisecond time scale in the free form of both XlnB2 and the E87A variant, while enhanced conformational exchange (including the thumb loop) was observed upon ligand binding in both enzymes.
Computational Simulations of the Free and Substrate-Bound Forms of XlnB2
To obtain insights into the potential relevance of longer time-scale residue dynamics in the function of XlnB2, we performed molecular dynamics (MD) simulations of XlnB2 in the free form (apoenzyme), and as a binary complex with substrates of different lengths (see Experimental Procedures). Conformations of the enzyme and enzyme–substrate complexes collected during a 1 μs MD simulation were analyzed using quasi-anharmonic analysis (QAA) to identify the conformational substates associated with the interconversion between the apo and binary complex forms. This type of analysis has been used previously for other enzyme systems to provide unique insights into the role of conformational substates and longer time-scale fluctuations. 44,46 MD simulation results showed that the X5 substrate displayed highly dynamic behavior, where the substrate moved around the active site and adopted different orientations. This behavior made it difficult to focus on the functionally relevant motions. Therefore, we characterized the conformational behavior of X6 and X9 bound to XlnB2.
The all-atom root mean square deviations (rmsd), calculated using the first structure as reference (Figure S5), was stable throughout the simulation, indicating the stability of the proteins and convergence of the simulations. Average enzyme–substrate interaction energies (van der Waals and electrostatic) for X6- and X9-bound XlnB2 are shown in panels A and B of Figure 4, respectively, where the corresponding binary complex structures are shown in the bottom panel. Regions displaying negative average interaction energies (blue end of the spectrum) correspond to those showing the strongest favorable interactions between the enzyme and substrate. Table S1 shows the top residues with the most favorable enzyme–substrate interaction energy for X6- and X9-bound enzyme (averaged over 1000 conformations). Not surprisingly, catalytic residues E87 and E177 showed strong interactions with the substrate. Other residues with favorable interaction energies encompass the fingers, thumb loop, and palm regions of XlnB2, highlighting the role of residues in these regions in ligand binding and catalysis, consistent with observations from our NMR titration experiments. Interestingly, the most favorable interactions with the enzyme are observed for substrate units occupying the −2 to +2 docking sites in both the X6- and X9-bound XlnB2 systems (Figure 4 and Table S1), consistent with previous studies indicating the importance of these docking subsites in this enzyme family.1
Figure 4.
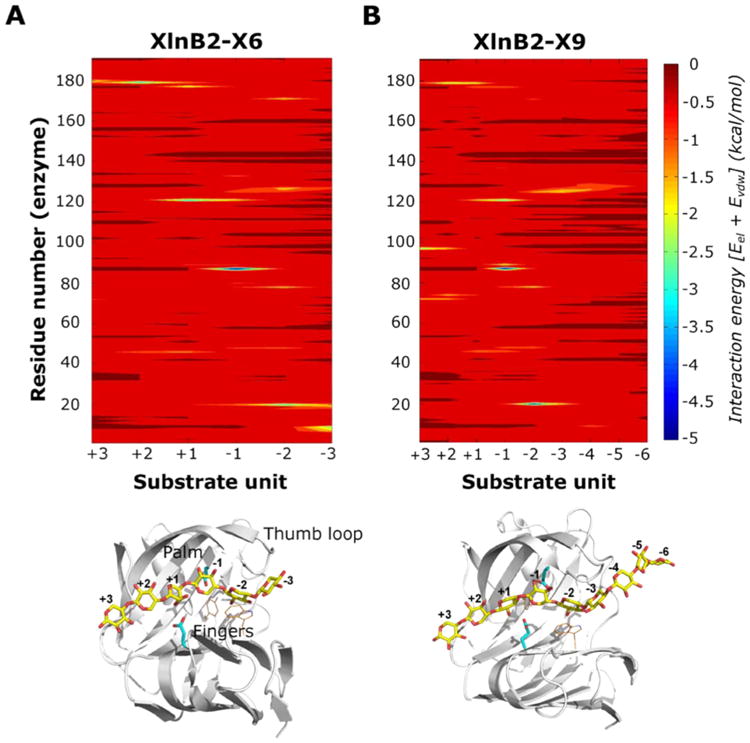
Protein–ligand interactions of the XlnB2–substrate binary complexes. Protein–substrate interaction energy maps for (A) XlnB2-X6 and (B) XlnB2-X9 binary complexes displaying the average interaction energies between XlnB2 and the substrate. Interaction energies were calculated as the sum of van der Waals and electrostatic energies averaged over all simulation snapshots (see Experimental Procedures). Favorable interactions correspond to the blue end of the spectrum, while unfavorable interaction energies correspond to the red end of the spectrum. The bottom panel shows the structure of the binary complex between XlnB2 and X6/X9 substrate (yellow sticks) used for the MD simulations. Residues within 4 Å of the ligand that display millisecond conformational exchange in the apo and ligand-bound states, determined from NMR relaxation dispersion experiments, are displayed as orange lines. Catalytic residues are shown as cyan sticks. The individual sugar units of the substrate are numbered according to the standard nomenclature for the reducing and nonreducing units.
We used QAA to characterize the conformational substates associated with the interconversion between the apo and substrate-bound states. QAA is a computational methodology that allows accurate identification of conformational fluctuations associated with protein function, particularly at longer time scales.57 In particular, the use of higher-order statistics has allowed the identification of conformational dynamics (and functionally related substates) associated with enzyme catalysis and biomolecular recognition for a number of protein systems. More details regarding QAA are available in ref 57. Note that the preference for use of QAA over second-order methods (such as normal-mode analysis, principal component analysis, or quasi-harmonic analysis) is due to its important advantage of providing quantitatively accurate and reproducible conformational motions at longer time scales. As has been previously discussed, methods such as normal-mode analysis depend on the starting structure and are known to provide only qualitative results that can be difficult to match with experimental data.58 Our QAA results show large-scale conformational fluctuations associated with the interconversion between the apo and binary complex of XlnB2 (Figure 5A,C). These fluctuations correspond to the transition between conformational substates corresponding to populations in lower- and higher-energy regions of the conformational landscape. The apo form of XlnB2 was observed to sample conformations that are also visible in the substrate-bound state (represented by the red dashed region in Figure 5A,C), indicating that conformations associated with substrate recognition and binding are presampled in the apo form on the microsecond time scale. The top three QAA-independent component vectors (or modes) were analyzed in detail to characterize the primary dynamics associated with substrate binding in the X6 and X9 XlnB2 systems (panels B and D of Figure 5, respectively). These modes correspond to the intrinsic motions or conformational fluctuations associated with protein function56 and correspond to the interconversion between the apo and substrate-bound states in this case. For the interconversion between free and X6-bound states (Figure 5B), the top three modes displayed large amplitudes of motions in the thumb loop, with the β-strands and loops in the fingers showing significant motions in modes 1 and 2. Specifically, modes 1 and 2 showed significant dynamics in the thumb loop, β-strands B1–4 and A2–4 in the fingers region of XlnB2, and B5–8 of the palm region, while mode 3 showed dynamics predominantly in the thumb loop (the β-strand numbering follows that in Figure 1A). Similar large amplitude motions of the thumb loop and fingers regions were observed for the interconversion between free and X9-bound states (Figure 5D). Interestingly, modes 1 and 2 in both X6 and X9 systems showed a twisting motion (as seen from the shift from the red to blue spectrum of the conformations that track the first to last conformations, respectively, in Figure 5B,D). As has been demonstrated using QAA for a number of other proteins,56,57 these motions are an intrinsic property or are the built-in motions of the protein. Therefore, these motions provide insights into how the movement of the thumb and palm regions may favor substrate binding. Overall, our computational results illustrate the potential role of conformational dynamics of the β-strands (and associated loops) of the fingers and thumb loop regions in the transition between the free and substrate-bound complex forms.
Figure 5.
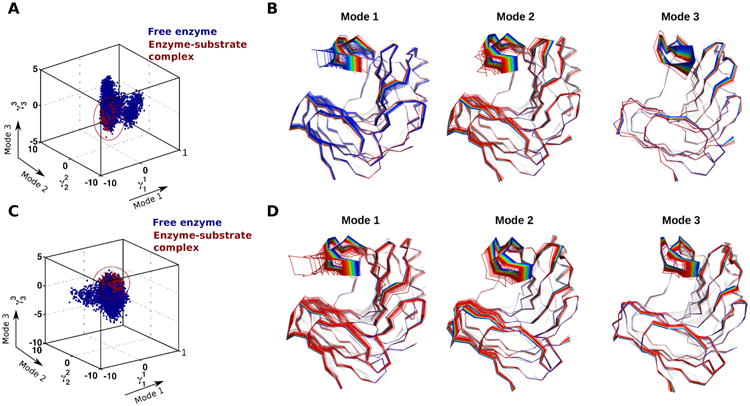
Conformational substates of XlnB2 determined using computational simulations and QAA. Representative conformations along the top three modes 1–3 for the interconversion between free and (A–B) X6-bound and (C–D) X9-bound XlnB2 binary complexes. A total of 200000 conformational snapshots obtained from the MD simulations were used for QAA to identify the top QAA-independent component vectors for characterizing the primary dynamics associated with the substrate binding process in XlnB2 (see Experimental Procedures).
Discussion
The use of enzyme biocatalysts in industrial chemical processes provides a sustainable and environmentally friendly alternative to a number of chemical transformations. However, the narrow range of desired characteristics such as substrate specificity and thermal stability, among others, currently limits their extensive commercial usage. The field of protein engineering has been devoted to developing strategies for designing structural and biochemical features in enzymes to impart them with new and/or improved function. While the theoretical principles of protein engineering remain simple, significant challenges remain with respect to targeting functionally relevant residues and characterizing the role of flexibility in enzyme activity. With advances in biocomputational approaches and NMR spectroscopy methodologies, the role of conformational fluctuations in substrate binding and product release, either directly or indirectly involved in the catalytic step, has been partially characterized.59,60 In the work presented here, we aimed to characterize conformational flexibility on the catalytically relevant millisecond time scale for XlnB2 and its catalytically impaired E87A variant.
The role of conformational flexibility in modulating enzyme catalysis has been well established for a variety of proteins.51,61 Previous studies of GH11 family enzymes have shown the existence of fast nanosecond time-scale motions in some xylanase members.1 MD studies based on 1 ns simulations of xylanase in T. reesei suggested that flexibility of the thumb loop may be important for allowing access of substrate to the active site,55 while simulations of xylanase from Bacillus subtilis showed that the extent of thumb loop movement is reduced in the presence of X2, which restricts access to the active site.18 However, these studies explored the dynamical properties of xylanases on the nanosecond time scale, which is several orders of magnitude faster than the catalytically relevant millisecond time scale of substrate turnover in these enzymes. On the basis of observations of fitting to the model-free formalism, Connelly et al.23 suggested that several residues in the apo form of BCX may display longer time-scale microsecond-to-millisecond mobility.
In this study, we provide the first experimental validation of conformational exchange processes occurring on the catalytically relevant millisecond time scale in xylanases, using NMR relaxation dispersion experiments to probe the potential influence of dynamics on ligand binding. Our results show that ligand binding enhances millisecond dynamics in the thumb loop and fingers of XlnB2. The apo form of the enzyme displayed a rigid thumb loop on the millisecond time scale, as previously suggested on the basis of fits from model-free analyses performed on the B. circulans xylanase.23 Ligand binding enhanced millisecond dynamics in the thumb loop region, in contrast to previous studies in which binding of X2 was shown to reduce mobility of the thumb loop on the fast nanosecond time scale, restricting access to the active site.18 Interestingly, the number of residues displaying dynamics increased upon ligand binding, in contrast to previous model-free observations.23 The enhanced conformational exchange in the thumb loop region upon ligand binding indicates that this motion may play an important role in ligand processing, consistent with observations from mutations in the thumb loop region leading to decreased activity of the xylanase from Thermobacillus xylanilyticus.19,22 The thumb loop has been shown to play important roles in substrate selectivity,19 product release, and (de)glycosylation.25 Residues of the thumb loop, including Glu128 and Thr120 near the hinge region of the loop, show significant chemical shift variations upon ligand binding in XlnB2. Further, enhanced conformational exchange in residues Asn123, Gly128, and Thr130 of the thumb loop upon ligand binding may allow the enzyme to adopt conformations that (destabilize) stabilize the binary complex with the (product) substrate. Changes in the conformational motions and chemical shift variations upon ligand binding are also observed for residues, such as Trp20 and Tyr171, in the fingers regions. Modifications of these residues through point mutations will provide important insights into their role in substrate binding and conformational fluctuations affecting function in XlnB2.
Our results reveal the presence of similar millisecond motions in the fingers and palm regions, triggered by binding of ligands of different sizes (Figure 3), suggesting that dynamics does not play a significant role in substrate selectivity. The length of the ligand does not seem to play a significant role in enhanced conformational exchange observed in the fingers and palm regions, as illustrated by similar rates of conformational exchange observed in the presence of X2 and X5 ligands (Figure 3). Similar conformational exchange rates for the catalytically impaired XlnB2-E87A variant suggest that the mutation did not affect ligand binding in XlnB2, as evidenced from similar Kd values and comparable thumb loop CHESPA behavior (see above). Analysis of the interconversion between the free and ligand-bound states of XlnB2, using quasi-anharmonic analysis of conformations obtained from MD simulations, shows substates that display enhanced dynamics in the fingers and thumb loop regions, consistent with our results from NMR relaxation experiments (Figure 3). Some of the residues displaying dynamics in the apo form and enhanced dynamics upon ligand binding are located within 4 Å of the ligand in the active site (orange sticks in the 3D structures shown in Figure 4), suggesting a direct effect of these conformational fluctuations upon ligand binding. QAA further revealed a twisting motion of the thumb loop and fingers regions in the top dynamic modes 1 and 2. We speculate that this twisting movement presents an alternate robust motion compared to the open–close motion for a processive enzyme like XlnB2. We note that the open–close motion has been observed for the thumb loop in the presence of substrate in the B. circulans xylanase.1
In summary, we characterized the broad dynamic profile experienced by the S. lividans 66 XlnB2 and its catalytically impaired E87A variant on the slow (millisecond) time scale using MD simulations and NMR relaxation dispersion experiments, in addition to determining its crystal structure. Our results show a distinct millisecond dynamic behavior for these enzymes in the apo and ligand-bound states. A comparison of these two states revealed that the ligand-bound protein experiences enhanced conformational exchange in the fingers and thumb loop regions, suggesting a potential role of these fluctuations for optimal ligand recognition, positioning, discrimination, processivity, and/or catalysis. This observation suggests a ligand-induced conformational shift in line with a potential induced fit mechanism of molecular recognition in GH11 xylanases, which was previously suggested for GH8 xylanases.62 This provides an avenue to further investigate the relative importance of conformational selection and induced fit in the catalytic mechanism of GH11 xylanases.
Supplementary Material
Acknowledgments
The authors thank Tara Sprules and Sameer Al-Abdul-Wahid of the Quebec/Eastern Canada High Field NMR Facility (McGill University, Montreal, QC) for their excellent technical assistance, in addition to Claude Dupont (INRS-Institut Armand-Frappier, Université du Québec) for providing the original XlnB2 construct and for transferring his vast Streptomyces knowledge to our lab.
Funding: This work was supported by a Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery Grant via Grant RGPIN-2016-05557 (to N.D.) and the National Institute of General Medical Sciences (NIGMS) of the National Institutes of Health (NIH) via Grant R01GM105978 (to N.D. and P.K.A.). N.D. holds a Fonds de Recherche Québec-Santé (FRQS) Research Scholar Junior 2 Career Award, and D.G. held an NSERC Alexander Graham Bell Canada Graduate Scholarship.
Footnotes
Supporting Information: The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.6b00130. Comparisons between the structural properties of WT XlnB2 and XlnB2-E87A (Figure S1), 15N T1, T2, and 15N{1H} NOEs for free XlnB2 (Figure S2), representative relaxation dispersion curves (Figure S3), a comparison between Δω and Δδ (Figure S4), the convergence of MD simulations (Figure S5), and average interaction energies between XlnB2 and the xylohexaose (X6) and xylononaose (X9) substrates (Table S1) (PDF)
Notes: The authors declare no competing financial interest.
References
- 1.Paës G, Berrin JG, Beaugrand J. GH11 xylanases: Structure/function/properties relationships and applications. Biotechnol Adv. 2012;30:564–592. doi: 10.1016/j.biotechadv.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 2.Uffen RL. Xylan degradation: a glimpse at microbial diversity. J Ind Microbiol Biotechnol. 1997;19:1–6. [Google Scholar]
- 3.Xia T, Wang Q. Directed evolution of Streptomyces lividans xylanase B toward enhanced thermal and alkaline pH stability. World J Microbiol Biotechnol. 2009;25:93–100. [Google Scholar]
- 4.Gilkes NR, Henrissat B, Kilburn DG, Miller RC, Jr, Warren RA. Domains in microbial beta-1, 4-glycanases: sequence conservation, function, and enzyme families. Microbiol Rev. 1991;55:303–315. doi: 10.1128/mr.55.2.303-315.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Henrissat B, Davies G. Structural and sequence-based classification of glycoside hydrolases. Curr Opin Struct Biol. 1997;7:637–644. doi: 10.1016/s0959-440x(97)80072-3. [DOI] [PubMed] [Google Scholar]
- 6.Campbell RL, R DR, Wakarchuk WW, To R, Sung W, Yaguchi M. A comparison of the structures of the 20 kD axylanases from Trichoderma harzianum and Bacillus circulans. In: Suominen PRT, editor. Proceedings of the second TRICEL symposium on Trichoderma reesei cellulases and other hydrolases. 1993. pp. 63–72. [Google Scholar]
- 7.Törrönen A, Harkki A, Rouvinen J. Three-dimensional structure of endo-1,4-beta-xylanase II from Trichoderma reesei: two conformational states in the active site. EMBO J. 1994;13:2493–2501. doi: 10.1002/j.1460-2075.1994.tb06536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Törrönen A, Rouvinen J. Structural comparison of two major endo-1,4-xylanases from Trichoderma reesei. Biochemistry. 1995;34:847–856. doi: 10.1021/bi00003a019. [DOI] [PubMed] [Google Scholar]
- 9.McIntosh LP, Hand G, Johnson PE, Joshi MD, Korner M, Plesniak LA, Ziser L, Wakarchuk WW, Withers SG. The pKa of the general acid/base carboxyl group of a glycosidase cycles during catalysis: a 13C-NMR study of bacillus circulans xylanase. Biochemistry. 1996;35:9958–9966. doi: 10.1021/bi9613234. [DOI] [PubMed] [Google Scholar]
- 10.Connelly GP, McIntosh LP. Characterization of a buried neutral histidine in Bacillus circulans xylanase: internal dynamics and interaction with a bound water molecule. Biochemistry. 1998;37:1810–1818. doi: 10.1021/bi972085v. [DOI] [PubMed] [Google Scholar]
- 11.Plesniak LA, Connelly GP, Wakarchuk WW, McIntosh LP. Characterization of a buried neutral histidine residue in Bacillus circulans xylanase: NMR assignments, pH titration, and hydrogen exchange. Protein Sci. 1996;5:2319–2328. doi: 10.1002/pro.5560051118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wakarchuk WW, Campbell RL, Sung WL, Davoodi J, Yaguchi M. Mutational and crystallographic analysis of the active site residues of the Bacillus circulans xylanase. Protein Sci. 1994;3:467–475. doi: 10.1002/pro.5560030312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wakarchuk WW, Sung WL, Campbell RL, Cunningham A, Watson DC, Yaguchi M. Thermostabilization of the Bacillus subtilis xylanase by the introduction of disulfide bonds. Protein Eng, Des Sel. 1994;7:1379–1386. doi: 10.1093/protein/7.11.1379. [DOI] [PubMed] [Google Scholar]
- 14.Lawson SL, Wakarchuk WW, Withers SG. Positioning the acid/base catalyst in a glycosidase: studies with Bacillus circulans xylanase. Biochemistry. 1997;36:2257–2265. doi: 10.1021/bi9620215. [DOI] [PubMed] [Google Scholar]
- 15.Torronen A, Rouvinen J. Structural and functional properties of low molecular weight endo-1,4-beta-xylanases. J Biotechnol. 1997;57:137–149. doi: 10.1016/s0168-1656(97)00095-3. [DOI] [PubMed] [Google Scholar]
- 16.Kluepfel D, Vats-Mehta S, Aumont F, Shareck F, Morosoli R. Purification and characterization of a new xylanase (xylanase B) produced by Streptomyces lividans 66. Biochem J. 1990;267:45–50. doi: 10.1042/bj2670045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hurtubise Y, Shareck F, Kluepfel D, Morosoli R. A cellulase/xylanase-negative mutant of Streptomyces lividans 1326 defective in cellobiose and xylobiose uptake is mutated in a gene encoding a protein homologous to ATP-binding proteins. Mol Microbiol. 1995;17:367–377. doi: 10.1111/j.1365-2958.1995.mmi_17020367.x. [DOI] [PubMed] [Google Scholar]
- 18.Vieira DS, Degreve L, Ward RJ. Characterization of temperature dependent and substrate-binding cleft movements: Bacillus circulans family 11 xylanase: a molecular dynamics investigation. Biochim Biophys Acta, Gen Subj. 2009;1790:1301–1306. doi: 10.1016/j.bbagen.2009.04.017. [DOI] [PubMed] [Google Scholar]
- 19.Paës G, Tran V, Takahashi M, Boukari I, O'Donohue MJ. New insights into the role of the thumb-like loop in GH-11 xylanases. Protein Eng, Des Sel. 2007;20:15–23. doi: 10.1093/protein/gzl049. [DOI] [PubMed] [Google Scholar]
- 20.Cervera Tison M, Andre-Leroux G, Lafond M, Georis J, Juge N, Berrin JG. Molecular determinants of substrate and inhibitor specificities of the Penicillium griseofulvum family 11 xylanases. Biochim Biophys Acta, Proteins Proteomics. 2009;1794:438–445. doi: 10.1016/j.bbapap.2008.11.024. [DOI] [PubMed] [Google Scholar]
- 21.Murakami MT, Arni RK, Vieira DS, Degreve L, Ruller R, Ward RJ. Correlation of temperature induced conformation change with optimum catalytic activity in the recombinant G/11 xylanase A from Bacillus subtilis strain 168 (1A1) FEBS Lett. 2005;579:6505–6510. doi: 10.1016/j.febslet.2005.10.039. [DOI] [PubMed] [Google Scholar]
- 22.Pollet A, Vandermarliere E, Lammertyn J, Strelkov SV, Delcour JA, Courtin CM. Crystallographic and activity-based evidence for thumb flexibility and its relevance in glycoside hydrolase family 11 xylanases. Proteins: Struct, Funct, Genet. 2009;77:395–403. doi: 10.1002/prot.22445. [DOI] [PubMed] [Google Scholar]
- 23.Connelly GP, Withers SG, McIntosh LP. Analysis of the dynamic properties of Bacillus circulans xylanase upon formation of a covalent glycosyl-enzyme intermediate. Protein Sci. 2000;9:512–524. doi: 10.1110/ps.9.3.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Havukainen R, Torronen A, Laitinen T, Rouvinen J. Covalent binding of three epoxyalkyl xylosides to the active site of endo-1,4-xylanase II from Trichoderma reesei. Biochemistry. 1996;35:9617–9624. doi: 10.1021/bi953052n. [DOI] [PubMed] [Google Scholar]
- 25.Paës G, Cortes J, Simeon T, O'Donohue MJ, Tran V. Thumb-loops up for catalysis: a structure/function investigation of a functional loop movement in a GH11 xylanase. Comput Struct Biotechnol J. 2012;1:e201207001. doi: 10.5936/csbj.201207001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vandermarliere E, Bourgois TM, Rombouts S, van Campenhout S, Volckaert G, Strelkov SV, Delcour JA, Rabijns A, Courtin CM. Crystallographic analysis shows substrate binding at the − 3 to + 1 active-site subsites and at the surface of glycoside hydrolase family 11 endo-1,4-β-xylanases. Biochem J. 2008;410:71–79. doi: 10.1042/BJ20071128. [DOI] [PubMed] [Google Scholar]
- 27.Gauthier C, Li H, Morosoli R. Increase in sylanase production by Streptomyces lividans through simultaneous use of the Sec- and Tat-dependent protein export systems. Appl Environ Microbiol. 2005;71:3085–3092. doi: 10.1128/AEM.71.6.3085-3092.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 29.Wouters J, Georis J, Engher D, Vandenhaute J, Dusart J, Frere JM, Depiereux E, Charlier P. Crystallographic analysis of family 11 endo-beta-1,4-xylanase Xyl1 from Streptomyces sp. S38 Acta Crystallogr, Sect D: Biol Crystallogr. 2001;57:1813–1819. doi: 10.1107/s0907444901015153. [DOI] [PubMed] [Google Scholar]
- 30.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 31.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cavanagh J, Fairbrother W, Palmer AG, III, Skelton N, Rance M. Protein NMR Spectroscopy: Principles and Practice. 2nd. Elsevier Academic; San Diego: 2006. [Google Scholar]
- 34.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 35.Grzesiek S, Stahl SJ, Wingfield PT, Bax A. The CD4 determinant for downregulation by HIV-1 Nef directly binds to Nef. Mapping of the Nef binding surface by NMR Biochemistry. 1996;35:10256–10261. doi: 10.1021/bi9611164. [DOI] [PubMed] [Google Scholar]
- 36.Mittermaier A, Meneses E. Analyzing protein-ligand interactions by dynamic NMR spectroscopy. Methods Mol Biol. 2013;1008:243–266. doi: 10.1007/978-1-62703-398-5_9. [DOI] [PubMed] [Google Scholar]
- 37.Farrow NA, Muhandiram R, Singer AU, Pascal SM, Kay CM, Gish G, Shoelson SE, Pawson T, Forman-Kay JD, Kay LE. Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry. 1994;33:5984–6003. doi: 10.1021/bi00185a040. [DOI] [PubMed] [Google Scholar]
- 38.Loria JP, Rance M, Palmer AG., 3rd A Relaxation-Compensated Carr–Purcell–Meiboom–Gill Sequence for Characterizing Chemical Exchange by NMR Spectroscopy. J Am Chem Soc. 1999;121:2331–2332. [Google Scholar]
- 39.Carver JP, Richards RE. A general two-site solution for the chemical exchange produced dependence of T2 upon the Carr-Purcell pulse separation. J Magn Reson. 1972;6:89–105. [Google Scholar]
- 40.Palmer AG., 3rd NMR characterization of the dynamics of biomacromolecules. Chem Rev. 2004;104:3623–3640. doi: 10.1021/cr030413t. [DOI] [PubMed] [Google Scholar]
- 41.Axe JM, Boehr DD. Long-range interactions in the alpha subunit of tryptophan synthase help to coordinate ligand binding, catalysis, and substrate channeling. J Mol Biol. 2013;425:1527–1545. doi: 10.1016/j.jmb.2013.01.030. [DOI] [PubMed] [Google Scholar]
- 42.Selvaratnam R, VanSchouwen B, Fogolari F, Mazhab-Jafari MT, Das R, Melacini G. The projection analysis of NMR chemical shifts reveals extended EPAC autoinhibition determinants. Biophys J. 2012;102:630–639. doi: 10.1016/j.bpj.2011.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Case DA, Darden TA, Cheatham TE, III, Simmerling CL, Wang J, Duke RE, Luo R, Walker RC, Zhang W, Merz KM, Roberts B, Hayik S, Roitberg A, Seabra G, Swails J, Gotz AW, Kolossvary I, Wong KF, Paesani F, Vanicek J, Wolf RM, Liu J, Wu X, Brozell SR, Steinbrecher T, Gohlke H, Cai Q, Ye X, Wang J, Hsieh MJ, Cui G, Roe DR, Mathews DH, Seetin MG, Salomon-Ferrer R, Sagui C, Babin V, Luchko T, Gusarov S, Kovalenko A, Kollman PA. AMBER 12. University of California; San Francisco: 2012. [Google Scholar]
- 44.Ramanathan A, Agarwal PK. Evolutionarily Conserved Linkage between Enzyme Fold, Flexibility, and Catalysis. PLoS Biol. 2011;9:e1001193. doi: 10.1371/journal.pbio.1001193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Agarwal PK. Computational Studies of the mechanism of cis/trans isomerization in HIV-1 catalyzed by cyclophilin A. Proteins: Struct Funct, Genet. 2004;56:449–463. doi: 10.1002/prot.20135. [DOI] [PubMed] [Google Scholar]
- 46.Wakarchuk WW, Campbell RL, Sung WL, Davoodi J, Yaguchi M. Mutational and crystallographic analyses of the active site residues of the Bacillus circulans xylanase. Protein Sci. 1994;3:467–475. doi: 10.1002/pro.5560030312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ludwiczek ML, Heller M, Kantner T, McIntosh LP. A secondary xylan-binding site enhances the catalytic activity of a single-domain family 11 glycoside hydrolase. J Mol Biol. 2007;373:337–354. doi: 10.1016/j.jmb.2007.07.057. [DOI] [PubMed] [Google Scholar]
- 48.Cuyvers S, Dornez E, Delcour JA, Courtin CM. Occurrence and functional significance of secondary carbohydrate binding sites in glycoside hydrolases. Crit Rev Biotechnol. 2012;32:93–107. doi: 10.3109/07388551.2011.561537. [DOI] [PubMed] [Google Scholar]
- 49.Gagne D, Narayanan C, Doucet N. Network of long-range concerted chemical shift displacements upon ligand binding to human angiogenin. Protein Sci. 2015;24:525–533. doi: 10.1002/pro.2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ludwiczek ML, D'Angelo I, Yalloway GN, Brockerman JA, Okon M, Nielsen JE, Strynadka NC, Withers SG, McIntosh LP. Strategies for modulating the pH-dependent activity of a family 11 glycoside hydrolase. Biochemistry. 2013;52:3138–3156. doi: 10.1021/bi400034m. [DOI] [PubMed] [Google Scholar]
- 51.Hanoian P, Liu CT, Hammes-Schiffer S, Benkovic S. Perspectives on electrostatics and conformational motions in enzyme catalysis. Acc Chem Res. 2015;48:482–489. doi: 10.1021/ar500390e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Boehr DD, Liu X, Yang X. Targeting structural dynamics of the RNA-dependent RNA polymerase for anti-viral strategies. Curr Opin Virol. 2014;9:194–200. doi: 10.1016/j.coviro.2014.08.006. [DOI] [PubMed] [Google Scholar]
- 53.Gagne D, Doucet N. Structural and functional importance of local and global conformational fluctuations in the RNase A superfamily. FEBS J. 2013;280:5596–5607. doi: 10.1111/febs.12371. [DOI] [PubMed] [Google Scholar]
- 54.Kempf JG, Loria JP. Measurement of intermediate exchange phenomena. Methods Mol Biol. 2004;278:185–231. doi: 10.1385/1-59259-809-9:185. [DOI] [PubMed] [Google Scholar]
- 55.Muilu J, Torronen A, Perakyla M, Rouvinen J. Functional conformational changes of endo-1,4-xylanase II from Trichoderma reesei: a molecular dynamics study. Proteins: Struct, Funct, Genet. 1998;31:434–444. [PubMed] [Google Scholar]
- 56.Ramanathan A, Savol AJ, Agarwal PK, Chennubhotla CS. Event detection and sub-state discovery from biomolecular simulations using higher-order statistics: application to enzyme adenylate kinase. Proteins: Struct, Funct, Genet. 2012;80:2536–2551. doi: 10.1002/prot.24135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ramanathan A, Savol AJ, Langmead CJ, Agarwal PK, Chennubhotla CS. Discovering conformational substates relevant to protein function. PLoS One. 2011;6:e15827. doi: 10.1371/journal.pone.0015827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ramanathan A, Agarwal PK. Computational identification of slow conformational fluctuations in proteins. J Phys Chem B. 2009;113:16669–16680. doi: 10.1021/jp9077213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Palmer AG., 3rd Enzyme dynamics from NMR spectroscopy. Acc Chem Res. 2015;48:457–465. doi: 10.1021/ar500340a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kohen A. Role of dynamics in enzyme catalysis: substantial versus semantic controversies. Acc Chem Res. 2015;48:466–473. doi: 10.1021/ar500322s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hammes GG, Benkovic SJ, Hammes-Schiffer S. Flexibility, diversity, and cooperativity: pillars of enzyme catalysis. Biochemistry. 2011;50:10422–10430. doi: 10.1021/bi201486f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.De Vos D, Collins T, Nerinckx W, Savvides SN, Claeyssens M, Gerday C, Feller G, Van Beeumen J. Oligosaccharide binding in family 8 glycosidases: crystal structures of active-site mutants of the beta-1,4-xylanase pXyl from Pseudoaltermonas haloplanktis TAH3a in complex with substrate and product. Biochemistry. 2006;45:4797–4807. doi: 10.1021/bi052193e. [DOI] [PubMed] [Google Scholar]
- 63.Manley G, Loria JP. NMR insights into protein allostery. Arch Biochem Biophys. 2012;519:223–231. doi: 10.1016/j.abb.2011.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.