

Abstract
In this study, we described the identification of a large DNAJB2 (HSJ1) deletion in a family with recessive spinal muscular atrophy and Parkinsonism. After performing homozygosity mapping and whole genome sequencing, we identified a 3.8 kb deletion, spanning the entire DnaJ domain of the HSJ1 protein, as the disease-segregating mutation. By performing functional assays, we showed that HSJ1b-related DnaJ domain deletion leads to loss of HSJ1b mRNA and protein levels, increased HSJ1a mRNA and protein expressions, increased cell death, protein aggregation, and enhanced autophagy. Given the role of HSJ1 proteins in the degradation of misfolded proteins, we speculated that enhanced autophagy might be promoted by the elevated HSJ1a expression seen in HSJ1b-deficient cells. We also observed a significant reduction in both tau and brain-derived neurotrophic factor levels, which may explain the dopaminergic deficits seen in one of the affected siblings. We concluded that HSJ1b deficiency leads to a complex neurological phenotype, possibly due to the accumulation of misfolded proteins, caused by the lack of the DnaJ domain activity. We thus expand the phenotypic and genotypic spectrums associated with DNAJB2 disease and suggest relevant disease-associated mechanisms.
Keywords: DNAJB2, spinal muscular atrophy, Parkin-sonism, DnaJ domain deletion, WGS
Introduction
Juvenile Parkinsonism is a neurodegenerative condition characterized by the onset before age 20 of tremor, hypokinesia, muscular rigidity, and postural instability. This condition may have many etiologies, including metabolic or infectious diseases, pharmacotherapy, and genetic causes [Jankovic, 2008]. Atypical juvenile Parkinsonism (AJP) usually refers to a complex form of young-onset Parkinsonism that is inherited in a recessive manner and manifests with diverse neurological and psychiatric phenotypes along with classical Parkinsonism symptoms. In AJP, the following clinical manifestations may be seen: pyramidal signs, abnormalities of eye movement, psychiatric manifestations (depression, anxiety, psychosis, and impulse control disorders), intellectual disability, and other neurological symptoms such as ataxia and epilepsy. These complex forms of Parkinsonism do not always respond well to levodopa treatment and may become nonresponsive with disease progression [Paisan-Ruiz, et al., 2010].
In the case of AJP, pathogenic mutations in at least eight different genes have been reported to date. These include ATP13A2 [1p36; MIM#606693], DNAJC6 [1p31.3; MIM# 608375], FBXO7 [22q12.3; MIM# 260300], PLA2G6 [22q12.3; MIM# 612953], SPG11 [15q13-q15; MIM# 610844], ZFYVE26 [14q24.1; MIM# 270700], SYNJ1 [21q22.2; MIM# 615530], and VPS13C [15q22.2; MIM# 616840] genes [Edvardson, et al., 2012; Krebs, et al., 2013; Lesage, et al., 2016; Paisan-Ruiz, et al., 2009; Paisan-Ruiz, et al., 2010; Schicks, et al., 2011]. Mutations in most of these genes have also been implicated in other neurological phenotypes, suggesting that other genetic and/or environmental factors may be implicated in disease expression and presentation.
Hereditary motor and sensory neuropathies (HMSN) are the most common degenerative disorders of the peripheral nervous system and are associated with wide clinical and genetic heterogeneities. In addition to the classical clinical HMSN features, many other additional symptoms may occur in association with a large number of distinct genes [Tazir, et al., 2014; Werheid, et al., 2016]. The distal hereditary motor neuropathies (dHMN), also called distal spinal muscular atrophy (dSMA), are a rare group of hereditary neuropathies characterized by progressive muscle weakness and distal motor symptoms with lower motor neuron degeneration. At least 19 different dHMN genes have been reported in the past two decades [Gess, et al., 2014].
In this study, we aimed to elucidate the genetic causes of an autosomal recessive (AR) consanguineous family presenting with SMA and juvenile Parkinsonism. Parkinsonism was only observed in one patient. We performed a combination of homozygosity mapping (HM) and whole genome sequencing (WGS) to identify the disease-associated locus and candidate gene. Interestingly, a large deletion (3.8 kb) at locus 2q35 encompassing the first three coding exons of the DNAJB2 (MIM# 614881) gene was found to segregate with disease status in all affected members. DNAJB2 splice-site (c.229+1G>A; c.352+1G>A) and missense (p.Tyr5Cys) mutations were previously identified in rare forms of distal SMA-5 or HMSN [Blumen, et al., 2012; Gess, et al., 2014; Lupo, et al., 2016; Schabhuttl, et al., 2014]. None of the previously reported patients with DNAJB2 mutations presented with Parkinsonism. Functional analyses performed in HEK293 cells revealed that the mutation identified in this study resulted in the loss of DNAJB2 mRNA and protein expressions, increased cell death and autophagy, reduced MAPT mRNA levels, and altered brain-derived neurotrophic factor (BDNF) cellular levels.
In conclusion, we describe the identification of the first deletion reported at the DNAJB2 locus, further expanding its phenotypic and genotypic spectrums as well as its disease-associated mechanisms.
Materials and Methods
Subjects
A consanguineous family with early-onset AR SMA and Parkinsonism was clinically examined. The family consisted of healthy parents, who were first-degree relatives, as well as three affected and two unaffected siblings (Fig. 1A and B). The local ethics committee at Shahid Beheshti University of Medical Sciences approved this study, and we obtained informed consent according to the Declaration of Helsinki from all participants. DNA samples from all members were isolated from whole blood using standard procedures.
Figure 1.
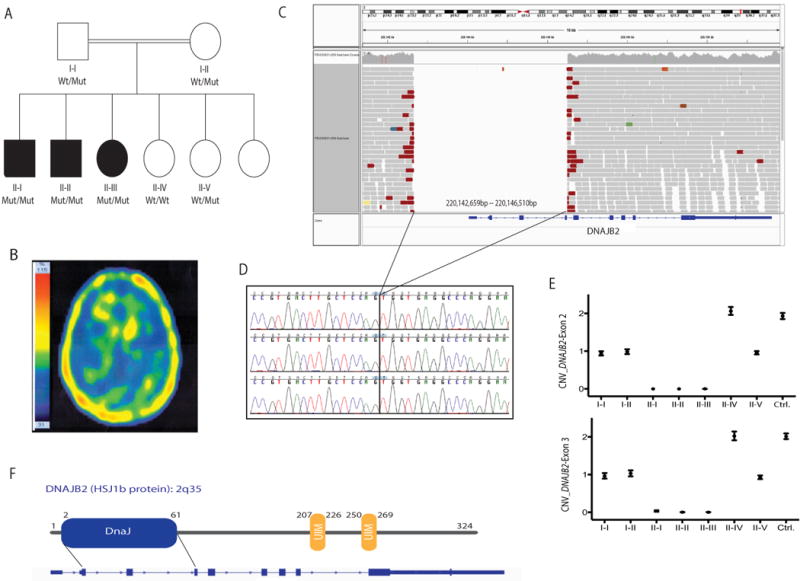
A 3.8 kb deletion at the DNAJB2 locus (2q35) was identified as the disease-segregating mutation in a recessive family with SMA and Parkinsonism. A: Pedigree structure of a family presenting with SMA and Parkinsonism (one patient; II–I). Homozygous mutation carriers are represented as Mut/Mut, heterozygous carriers as Wt/Mut, and non-carriers as Wt/Wt. Dark squares (males) and circles (females) indicate neurological disease. B: Brain TRODAT SPECT imaging of affected sibling II–I showing decreased presynaptic dopamine activity in the striata, more prominent on the right side. C: WGS reads of an affected sibling showing the 3.8 kb chromosome 2 deletion identified in all affected members. WGS reads were visualized by using the IGV tool. D: Sanger chromatogram sequences of the three affected siblings confirming the presence of a homozygous deletion on chromosome 2q35. E: CNV plots of DNAJB2exons 2 and 3 obtained through ddPCR (QX100 system, Bio-rad). Two exon copies (homozygous wt allele) are represented with a CNV score of 2, one exon copy (heterozygous mutant allele) with a CNV score of 1, and no copies with a CNV score of 0. F: HSJ1b protein structure located at the 2q35 locus. The functional DnaJ domain expands from exons 2 through 4.
Homozygosity Mapping
High-throughput SNP genotyping was carried out in all available family members (n = 7) (Fig. 1A) using the HumanOmniExpress Exome arrays v1.3 and HiScanSQ system (Illumina Inc., San Diego, CA). The GenomeStudio program (GS; Illumina) was used to undertake quality assessments and generate PLINK input reports [Purcell, et al., 2007] for HM. HM analyses were carried out as previously described [Krebs, et al., 2013].
Whole Genome Sequencing
WGS was carried out at the New York Genome Center (NYGC) in two affected family members (II–I, II–III). Detailed description of these analyses can be found in the Supp. Methods.
Validation of the Only Identified Disease-Segregating Mutation
First, direct Sanger sequencing using primers flanking the deletion breakpoints (a PCR product of 935 bp in mutation carriers) was used to validate the DNAJB2 deletion identified through WGS in all available family members. Primer sequences were designed by using a public primer design Website (http://ihg.gsf.de/ihg/ExonPrimer.html) (Supp. Table S1). All purified PCR products were sequenced in both forward and reverse directions with Applied Biosystems BigDye Terminator v3.1 sequencing chemistry as per the manufacturer’s instructions, and resolved and analyzed as described elsewhere [Krebs, et al., 2013]. Second, copy number variation (CNV) quantification of DNAJB2 exons 2 and 3 was performedusingtheDropletDigital PCR (ddPCR) QX100 system (Bio-Rad, Hercules, CA, USA) [Hindson, et al., 2011]. Taq-Man probes targeting DNAJB2 exons 2 and 3 as well as a reference gene (TERT) were acquired from Applied Biosystems (Life Technologies, Grand Island, NY, USA). A sample DNA control from a healthy individual was used as reference DNA, and a non-template control was included as negative control. CNV scores were calculated using the Quantasoft software as per the manufacturer’s instructions (Bio-Rad, Hercules, CA, USA).
HSJ1b Plasmid Validation and Site-Directed Mutagenesis
The pcDNA5/FRT/TO GFP DNAJB2b plasmid was a gift from Harm Kampinga (Addgene plasmid # 19496) (www.addgene.org) [Hageman and Kampinga, 2009]. The plasmid was expanded using a Qiagen Spin MiniPrep Kit (Qiagen, Hilden, Germany) (wildtype plasmid). Once validated, a plasmid deletion targeting coding exons 1 through 3 of the DNAJB2b insert was produced by using Agilent Technologies QuickChange II XL Site Directed Mutagenesis Kit (Agilent Technologies). Primers used for the deletion were generated using QuickChange Primer Design Program (Agilent Technologies Inc., Santa Clara, CA, USA) (Supp. Table S1). Cycling parameters were followed according to manufacturer’s recommendations. Primer sequences for both wild type and mutant DNAJB2b were designed using the Primer3 program (http://bioinfo.ut.ee/primer3-0.4.0/). All generated constructs were verified in both directions by Sanger sequencing, using the human sequence GRCh37.p13 as reference (Supp. Table S1).
Cell Transfection, Immunocytochemistry, Western Blotting, and Enzyme-Linked Immunosorbent Assay
Cell transfections were carried out as described in the Supp. Methods and by using human embryonic kidney 293 cells (HEK293T). Transfected cells with either wild type or mutant plasmid were then plated on poly-l-lysine (Sigma–Aldrich Co., St. Louis, MO, USA) treated in 18 mm cover glasses (Fisher Scientific, Hampton, NH, USA) contained in a six-well plate. For immunostaining, cells were fixed with 4% paraformaldehyde for 15 min, permeabilized with 0.1% Triton X-100 (Sigma–Aldrich Co., St. Louis, MO, USA) for 5 min, blocked 10% BSA, and incubated overnight at 4°C in a primary antibody that was appropriately diluted in 1% BSA. The primary antibodies used in this study included rabbit anti-DNAJB2 (1:250; Atlas Antibodies, Stockholm, Sweden), mouse anti-HSP70 (1:200; Pierce Antibody Products, Waltham, MA, USA), chicken anti-GFP (1:5,000; EMD Millipore, Billerica, MA, USA), and rabbit anti-LC3-I/II (1:200; EMD Millipore, Billerica, MA, USA). After washing, fluorophore-conjugated secondary antibodies were added for 1 hr at RT. The secondary antibodies included donkey anti-rabbit Alexa Fluor-647, donkey anti-mouse Rhodamine Red-X, and donkey anti-chicken Alexa Fluor-488 (all 1:200, from Jackson Immunoresearch Labs, West Grove, PA, USA). Cell nuclei were labeled using DAPI (1:400; Sigma-Aldrich Co., St. Louis, MO, USA). Coverslips were then mounted to the slides using Mowiol.
For Western blotting and ELISA, transfected cell lysates were collected using the radioactive immunoprecipitation assay buffer (Sigma-Aldrich Co., St. Louis, MO, USA) or IP Lysis Buffer (Thermo Scientific Inc, Waltham, MA, USA) with a phosphatase inhibitor (Roche, Basel, Switzerland). Protein concentrations were then determined by using the Pierce BCA Protein Assay kit (Thermo Fisher Scientific Inc, Waltham, MA, USA). Proteins (GAPDH, HSJ1a, HSJ1b) were detected by Western blotting; and human BDNF was detected by using the BDNF DuoSet ELISA Development System Kit (R&D Systems, Minneapolis, MN, USA; Supp. Methods).
Image Acquisition
Immunolabeling was visualized using a Leica SP5 DM microscope with a 63 × magnification objective. Confocal z-stack images were acquired in six random locations within coverslip per condition while blinded to genotype. Images were then analyzed using ImageJ (http://imagej.nih.gov/ij/) [Schneider, et al., 2012].
Quantitative PCR Analyses
Transfected cells were harvested and RNA was purified using the Qiagen RNeasy Mini Kit. RNA was then reverse transcribed into cDNA using the SuperScript IV Reverse Transcriptase System (Life Technologies, Grand Island, NY, USA). Gene expression levels of DNAJB2, Hsp70, MAPT, and GFP were determined by quantitative PCR (qPCR) and corresponding primers previously designed through the PRIMER3 program (http://bioinfo.ut.ee/primer3-0.4.0/; Supp. Table S1). All qPCR assays were performed on an Eco Real-Time PCR System (Illumina Inc, San Diego, CA, USA), as described elsewhere [Sanchez, et al., 2015].
Statistical Analyses
Statistical analyses were performed using the GraphPad Prism software version 6.0 (GraphPad, La Jolla, CA, USA). Data on graphs are presented as mean ± SEM. Statistical differences between wild type and mutant were calculated by using the Mann-Whitney non-parametric test. *P< 0.05, ***P< 0001.
Results
Clinical Report
The family consisted of unaffected parents as well as three affected and two unaffected siblings (Fig. 1A). All family members were examined. There was no family history of any neurological disorders or exposure to toxic agents.
Patient II–I is a 28-year-old man who at the age of 16 presented with a coarse resting tremor and rigidity, which gradually progressed. There was no history of any medication exposure, including antipsychotics. On neurological examination, mental status was normal, as were the cranial nerves apart from eye movements, which showed interrupted saccadic and smooth pursuit. There was mild distal atrophy of the muscles. Deep tendon reflexes were diminished and bilateral Babinski signs were present. Sensory examination was normal, as was coordination. He had a severe bilateral coarse resting tremor of the upper limbs, which worsened with action, and also tremor of the chin. There was asymmetric rigidity, more marked on the right, and severe bradykinesia, also more prominent on the right, which showed a slight response to levodopa therapy (1,000 mg per day in divided doses with carbidopa). Tremor was aggravated during walking. Brain MRI was normal. Dopamine transporter imaging (TRODAT SPECT scan) showed markedly decreased presynaptic dopamine activity in the striata, more severe on the right side (Fig. 1B). Electromyography (EMG) and nerve conduction testing revealed an active chronic motor neuron axonal disorder with huge motor unit action potentials (MUAPs) compatible with a SMA. No retinal abnormality was seen on ophthalmological examination. At 1-year follow-up, his tremor no longer responded to levodopa treatment.
Patients II–II (age 38) and II–III (age 40) had muscle atrophy of all four limbs, more marked distally, and diminished reflexes. The age at onset of symptoms in both patients was not clear, but both patients were symptomatic in the third decade of life. Position, vibration, light touch, and pin prick sensation was decreased. Abnormal smooth pursuits with saccadic intrusions were seen in patient II–II. EMG in both patients revealed an axonal sensorimotor polyneuropathy compatible with Charcot-Marie-Tooth hereditary neuropathy type 2 (CMT2). Cardiac problems were not seen, and ophthalmological examination revealed no retinal abnormality.
HM and WGS Analyses Identified a 3.8 kb Deletion at the DNAJB2 Locus as the Disease-Causing Mutation
Because HM is an efficient gene mapping method for recessive disorders in inbred populations, all available family members (n = 7) were subject to genome-wide SNP genotyping, and genotyping data was used to perform HM analyses. Four potential disease-associated loci, shared only by the three affected individuals, were identified (Table 1). WGS was then performed on two affected individuals (II–I and II–III), resulting in the identification of 396 genomic variations, including missense (n = 294), frameshift, splice-site, stop-gained, and start-lost (n = 102) mutations, common to both affected individuals. However, none of them was located in any of the four disease-associated loci previously identified through HM analyses. We also investigated whether identified genomic deletions to be shared exclusively by affected individuals were located in previously identified disease-associated loci. To our surprise, a large deletion (NM 001039550.1: c.−1618 229+17del) located in a disease-associated locus (2q35) was identified to be present in both affected members (http://databases.lovd.nl/shared/transcripts/00006537). Upon further inspecting this large deletion in the Integrative Genomics Viewer (IGV) tool [Thorvaldsdottir, et al., 2013], it was determined that the deletion spanned about 3.8 kb and encompassed the first four exons of the DNAJB2 gene (Fig. 1C) along with additional 5′ UTR intergenic regions. To ensure that the breakpoints of this deletion were correctly mapped, the candidate region on chromosome 2q35 was realigned by using SplazerS [Emde, et al., 2012], which confirmed the same breakpoints identified by GenomeSTRiP.
Table 1.
Potential Disease-Associated Loci Identified Through HM Analyses
| Flanking SNPs
|
SNP position (bp)
|
||||
|---|---|---|---|---|---|
| Chr | SNP1 | SNP2 | BP1 | BP2 | Size (kb) |
| 1 | Exm92700 | rs12084304 | 147380101 | 148937908 | 1,557.81 |
| 2 | rs6729330 | rs16860248 | 218694296 | 220609378 | 1,915.08 |
| 14 | rs7143698 | rs10134540 | 59804127 | 61623700 | 1,819.57 |
| 17 | rs4393605 | rs7209470 | 63446416 | 65630262 | 2,183.85 |
Bold indicates the disease-associated locus confirmed later by WGS.
No shared region of homozygosity was identified within the known Parkinsonism loci and no pathogenic mutation was identified in the known Parkinsonian genes in the patient (II–I) featuring Parkinsonism.
To validate the 3.8 kb DNAJB2 deletion and examine its segregation with the disease status, genomic primers flanking the deletion breakpoints identified through GenomeSTRiP and Splazer were designed and used to amplify a 935 bp PCR product, expanding the 3.8 kb DNAJB2 deletion in mutation carriers. The exact size of the DNAJB2 deletion encompassing exon 1 through exon 4 was confirmed to be of 3,851 bp in length, as previously determined by WGS analyses. We amplified the 935 bp PCR product in both unaffected parents (expected to be heterozygous carriers for the 3.8 kb deletion), three affected family members (expected to be homozygous carriers for the deletion), and one unaffected sibling (II–V) (Fig. 1D).
Consequently, by performing CNV analyses targeting DNAJB2 coding exons 1 and 2, we confirmed that both unaffected parents as well as one unaffected sibling II–V carried only one copy of each exon (CNV score of 1); all three affected members carried homozygous deletions for both exons (CNV score of 0); and unaffected family member II–IV carried two copies of each exon with 100% positive droplets and a CNV score of 2 for the both examined exons, indicative of a homozygous wt allele. These results confirmed the presence of a homozygous DNAJB2 deletion in all affected family members as well as its segregation with disease-status, further confirming its pathogenicity.
The DNAJB2 gene, also called heat shock protein J1 (HSJ1), encodes a molecular cochaperone member of the heat shock protein family Hsp40 with important cellular roles in correct protein folding, in response to protein misfolding, and in the degradation of misfolded proteins [Kampinga and Craig, 2010; Voisine, et al., 2010]. The HSJ1 proteins contain two functional domains, a DnaJ domain, a highly conserved ~70 amino-acid signature region, and two ubiquitin interacting motifs (UIMs) that promote the sorting of ubiquitinylated substrate proteins to the proteasome for degradation [Westhoff, et al., 2005]. The entire DnaJ domain, expanding exons 2 and 3 and the beginning of exon 4, was found to be deleted in our affected patients (Fig. 1F).
HSJ1b Mutant Cells Lacking the DnaJ Domain Showed Large GFP Aggregations and Reduced HSJ1b mRNA Expression
Because the DNAJB2 3.8 kb deletion encompassed the entire functional DnaJ domain of the HSJ1 protein, we then proceeded to determine its functional consequences in vitro by using HEK293 cells. HSJ1 encodes two alternatively spliced isoforms that differ in their C-termini. The HSJ1b (long isoform; NM_006736.5,) or canonical isoform undergoes post-translational modification that mediates its attachment to the cytoplasmic side of the endoplasmic reticulum membrane, whereas the HSJ1a isoform (short isoform, NM_001039550.1) is expressed in both the cytoplasm and nucleus. Both HSJ1 mRNAs are enriched in the brain and are indeed found in various nervous system regions, such as the cortex, cerebellum, striatum, and retina; however, HSJ1b was found to be more abundant in the frontal cortex and hippocampus of humans and mice than HSJ1a [Chapple and Cheetham, 2003; Cheetham, et al., 1992; Claeys, et al., 2010].
We therefore mutated the predominant HSJ1 isoform in the brain, HSJ1b. The first three coding exons (exons 2 through 4) containing the DnaJ domain were deleted in the pcDNA5/FRT/TO GFP DNAJB2b (HSJ1b) plasmid by site-directed mutagenesis. HEK293 cells were transfected and allowed to express either the non-mutated (wt) or mutated (mut) HSJ1b protein. After 48 hr, transfected cells were identified by the expression of green fluorescence protein (GFP), and transfection efficiency was assessed as previously described (Fig. 2A). QPCR analyses carried out in the transfected cells revealed that wt HSJ1b expressed significantly higher levels of GFP compared with mutant HSJ1b (Fig. 2B), confirming a decrease in HSJ1b mRNA expression in mutant cells. Furthermore, cells transfected with wt HSJ1b had small lysosome-like deposits (Fig. 2C, top panel, arrows), while mutant cells showed a higher number of chromatin or nuclei fragmentation or nuclear condensation revealed by DAPI staining as well as large GFP aggregations (Fig. 2C, bottom panel arrows; Fig. 2D).
Figure 2.
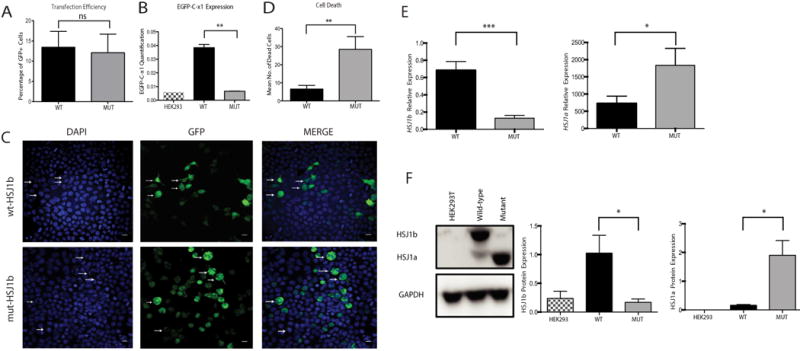
In vitro modeling of DNAJB2 exons 2 through 4 deletion leads to aggregations, cell death, and altered HSJ1 mRNA and protein expressions. A: Transfection efficiency. HEK293 cells were transfected in three independent experiments and only GFP+were considered for further analysis. B: Expression of HSJ1 b in non-transfected (black and white column), as well as wild type (black column) and mutant (solid gray column) transfected cells. Expression was evaluated by absolute quantification using the standard curve method. The y axis represents the mRNA expression (in number of copies) (P= 0.002, Mann-Whitney Test). C: HEK293 cells transfected with either wt HSJ1b-GFP (wt-HSJ1b) or mutant HSJ1b-GFP (mut-HSJ1b). Cells transfected with wt-HSJ1b do not show apoptotic nuclei and show small lysosomal deposits (white arrows). HEK293 cells transfected with Mut-DNAJB2 show apoptotic nuclei and aggregates (arrows). Scale bar: 50 μM. D: Quantification of HEK293 cells’ apoptotic nuclei in both wt and mutant HSJ1b cells. Cells transfected with mutant HSJ1b have a significantly higher number of apoptotic nuclei (Mean number of dead cells: WT = 3 and MUT = 24.50, P= 0.008, Mann-Whitney Test). E: HSJ1b (left) and HSJ1a (right) mRNA levels in wt (black column) and mutant (solid gray column) HSJ1b transfected cells. HSJ1b mRNA levels are highly decreased in mutant cells (left). F: Western blot analysis of HSJ1a, HSJ1b, and GAPDH proteins in cell lysatesfrom non-transfected cells (black and white columns), as well as wild type (black column) and mutant (solid gray column) HSJ1b transfected cells. HSJ1b or long isoform is of 36 kDa, HSJ1a or short isoform is of 31 kDa, and the GAPDH protein that was used as a loading control is of 37 kDa. Results are representative of four separate experiments. HSJ1b protein levels were significantly lower in mutant cells while HSJ1a were higher. Values represent the means ± SEM. *P< 0.05, **P< 0.01, ***P< 0.0001, ns = non-significant.
By using specific primers for each isoform, we observed that HSJ1b mRNA levels were significantly higher in wt than in mutant transfected cells (P < 0.0001), whereas HSJ1a mRNA levels were slightly higher in mutant carriers than in non-carriers (P = 0.03) (Fig. 2E). The mutant transfected cells also resulted in decreased protein levels of HSJ1b isoform (P = 0.03), and increased levels of HSJ1a protein (P = 0.03; Fig. 2F), indicating that the deletion of DNAJB2b exons 2 through 4 (coding exons 1–3) results in a highly significant decrease of HSJ1b expression at both mRNA and protein levels.
Enhanced Autophagy was Observed in HSJ1b-Deficient Mutant Cells
Because HSJ1 proteins are known to participate in preventing the accumulation of toxic protein aggregations via Hsp40-Hsp70 complexes [Chapple and Cheetham, 2003; Howarth, et al., 2007; Westhoff, et al., 2005], we also examined the autophagic protein degradation systems in both wt and mutant HSJ1b transfected cells. We investigated the expression and localization of the microtubule-associated protein1 light chain 3 (LC3), a well-known autophagy marker, in transfected cells. While LC3 was identified in both wt (Fig. 3A, upper panel) and mutant (Fig. 3A, lower panel) HSJ1b transfected cells, it was more abundant in mutant cells where, indeed, it localized in cells with aberrant nuclei and large aggregates (P = 0.002; Fig. 3A and B). Because the enhanced autophagy may be promoted by the increased HSJ1a expression seen in HSJ1b mutant cells, we also examined whether Hsp70 and LC3 proteins co-localized with HSJ1b transfected cells. Hsp70 and LC3 proteins were shown to co-localize with HSJ1 transfected cells (GFP+), being more predominant in mutant HSJ1b cells (Supp. Fig. S1A and B).
Figure 3.
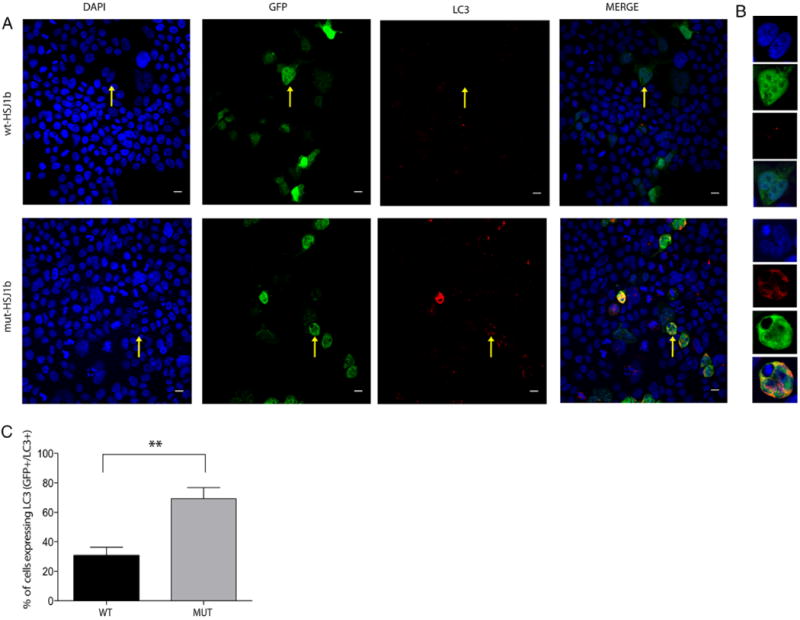
Increased co-localization of LC3 in mutant HSJ1b transfected cells versus wild type. A: LC3 localizes in both transfected (GFP+ cells) and non-transfected cells (GFP− cells), being highly localized in cells presenting aberrant nuclei and big aggregates (yellow arrow, lower panel, mut-HSJ1b). However, transfected HEK293 cells with wild type HSJ1b have small aggregates and low levels of LC3 (yellow arrow, upper panel, wt-HSJ1b). B: Zoomed images of a sample cell with low levels of LC3 (upper panel, wt-HSJ1b) and a sample cell with high levels of LC3 (lower panel, mut-HSJ1b). C: Quantification of HEK293 transfected cells expressing LC3 (GFP+/LC3+) in wild type (solid black column) and mutant (solid gray column) HSJ1b (percentage of GFP+/LC3+ cells, wt-HSJ1b = 34.5% and mut-HSJ1b-Mut = 64.6%, P = 0.002, Mann–Whitney non-parametric test. Values represent the mean SEM. **P < 0.01. Scale bar = 50 μm.
Tau Levels are Decreased in HSJ1b-Deficient Mutant Cells
It is well established that the DnaJ-domain-containing chaperones are required to regulate the ATPase activity of Hsp70 and subsequently modulate the substrate binding and ATPase cycle. It is thought that DnaJ domains bind the substrate (unfolded client protein) and then introduce it to Hsp70, in addition to stabilizing Hsp70-polypeptides complexes by driving the conversion from the Hsp70-ATP to Hsp70-ADP, which makes the protein substrate bind tightly [Mayer and Bukau, 2005; Turturici, et al., 2011]. It is also known that the vast majority of tau in the brain is controlled by the constitutively expressed form of heat shock protein 70 (Hsp70) and whether this Hsp70 complex degrades or preserves tau depends on its interaction with co-chaperones, such as Hsp40 [Jinwal, et al., 2013]. Therefore, whether tau levels were altered in our mutant cells versus wt was also examined. Although the expression of Hsp70 was not altered in mutant cells versus wild type (Fig. 4A), the examination of MAPT transcript levels in our wt and mutant transfected cells revealed a highly significant reduction of tau levels in mutant cells when compared with their wild type counterparts (Fig. 4B), suggesting that HSJ1b deficiency reduces tau levels possibly due to the lack of DnaJ-stimulated ATP turnover.
Figure 4.
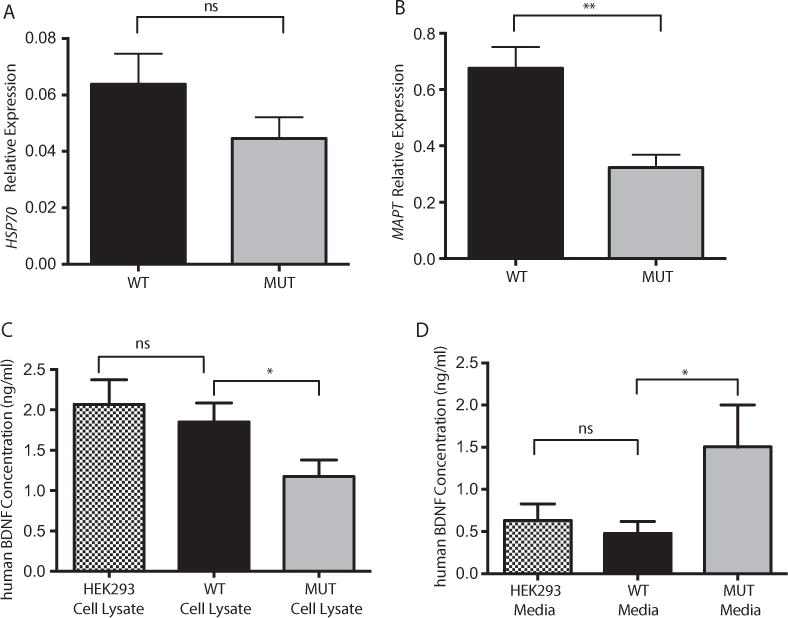
HSJ1b deficiency does not alter Hsp70 mRNA levels but alters both MAPT and BDNF levels. A: Hsp70 mRNA expression levels are not significantly altered in wt (black column) versus mutant (solid gray column) transfected HEK293 cells (P= ns). B: MAPT (TAU) mRNA levels are significantly reduced in HSJ1b mutant transfected HEK293 cells (solid gray column) when compared with their wild type counterparts (black column) (P= 0.001).C: BDNF levels are reduced in cell lysatesof HSJ1b mutant cells (P= 0.05) but not in wtHSJ1b transfected cells (black column) (ns), which showed similar BDNF levels than non-transfected cells (ns). D: Overexpression of wild type HSJ1b (black column) did not increase the release of BDNF into the cell culture media when compared with non-transfected cells (black and white column) (ns). BNDF media release is increased in mutant HSJ1b transfected cells (solid gray column) (P = 0.03). Results are representative of two (A, B) or four (C, D) separate experiments. Values represent means ± SEM. *P≤ 0.05, **P< 0.001, ns = non-significant.
BDNF Release is Altered in HSJ1b Mutant Cells Lacking the DnaJ Protein Domain
BDNF is a member of a family of proteins that is not only important for the normal development of the peripheral and central nervous system but also plays a key role in neuronal survival and synaptic plasticity in the adult brain [Arancio and Chao, 2007]. Along these lines, it has been shown that HSJ1b increases BDNF release in neuronal cells and consequently promotes neuronal survival. Specifically, high levels of both HSJ1 transcripts were observed when treating mouse neuronal cells overexpressing BDNF and HSJ1 with cystamine, but only a significantly increase in BDNF release was observed in neuronal cells overexpressing HSJ1b, suggesting a role of HSJ1b in neuronal survival [Borrell-Pages, et al., 2006]. We therefore analyzed whether BDNF levels were also altered in our mutant cells as compared with wt. While BDNF levels were not altered in the media or cell lysates of wt HSJ1b-overpressing cells (Fig. 4C and D), a significant decrease in BDNF levels were observed in cell lysates of mutant HSJ1b-overexpressing cells when compared with their wt counterparts (P = 0.05) (Fig. 4C). Likewise, BDNF levels were increased in the media of mutant HSJ1b transfected cells (P = 0.03) (Fig. 4D). Taken together, HSJ1b deficiency resulted in a significant decrease of BDNF in cell lysates, and a significant increase of BDNF levels in the media (Fig. 4C and D).
Discussion
Here, we describe the identification of a large DNAJB2 deletion (3.8 kb), spanning the entire DnaJ domain of the DNAJB2 gene, in a family with SMA and Parkinsonism. Three pathogenic mutations (two splice-site and one missense) in the DNAJB2 gene were recently associated with rare forms of dHMN in patients from Morocco, Turkey, Spain, and Austria [Blumen, et al., 2012; Gess, et al., 2014; Lupo, et al., 2016; Schabhuttl, et al., 2014]. None of the previously reported dHMN patients presented with Parkinsonism in addition to their motor symptoms, and no deletions or insertions were previously reported at the DNAJB2 locus [Blumen, et al., 2012; Gess, et al., 2014; Lupo, et al., 2016]. None of the two splice mutations previously reported affected the coding exons’ encoding for the DnaJ domain. Therefore, here we report for the first time a deletion involving the entire DnaJ domain of the HSJ1 protein as the disease-causing mutation in a complex family with SMA and Parkinsonism (one patient), which clearly highlights the relevance of the DnaJ function in HSJ1 disease-associated mechanisms. In vitro functional analysis of the HSJ1b protein lacking the entire DnaJ domain resulted in a significantly reduced HSJ1b expression at both mRNA and protein levels and increased apoptosis, clearly confirming the inactivity of the HSJ1b protein in mutant cells (Fig. 2). Interestingly, decreased HSJ1b levels have also been reported in Huntington disease (HD) brains [Borrell-Pages, et al., 2006], further implicating the role of HSJ1 proteins in other neurodegenerative diseases.
Our HSJ1b mutant cells also showed large GFP aggregates accompanied with increased cell death. As stated before, the DnaJ domains of the Hsp40 protein are known to regulate the ATP hydrolysis of Hsp70 proteins. Briefly, Hsp40 proteins bind to unfolded polypeptide chains and maintain them in a folding competent state, thereby reducing aggregation and cellular stress [Fan, et al., 2003]. Therefore, it is not surprising that the 3.8 kb DNAJB2 deletion identified in this study gives rise to protein aggregation and body inclusion formation, as the DnaJ-mediated Hsp70 activity is expected to be impaired due to the lack of DnaJ protein domain (Fig. 2).
We observed that LC3 levels were significantly elevated in mutant HSJ1b transfected cells, showing aberrant nuclei and large aggregates (Fig. 3), further confirming an enhanced autophagy in mutant HSJ1b transfected cells with increased cell death. Because we also showed that the increased LC3 levels co-localize with both Hsp70 and HSJ1 proteins (Supp. Fig. S1A and B), we hypothesize that autophagy might be induced by the increased HSJ1a expression seen in our HSJ1b mutant cells. HSJ1a isoform has been shown to promote autophagy and reduce protein aggregation and inclusion body formation in models of polyglutamine expansion diseases [Howarth, et al., 2007] and mutant p.C289G Parkin in a DnaJ-domain dependent manner, while overexpression of HSJ1a has no effect on a wild type Parkin protein [Rose, et al., 2011]. This suggests that the HSJ1a isoform may act as a compensatory mechanism by promoting autophagy only in cells with increased levels of aggregated proteins, which may justify the increased HSJ1a levels seen in our HSJ1b-deficient mutant cells. Although this clearly suggests a plausible role of HSJ1a in inducing autophagy in cells with the HSJ1b aberrant protein, further studies are required to confirm this hypothesis.
Tau levels, which are thought to be regulated by Hsp70–Hsp40 complexes, were also found to be downregulated in our mutant HSJ1b-overexpressing cells (Fig. 4B), suggesting that HSJ1b deficiency leads to the loss of tau. These results correlate with previous work, in which YM-01, an allosteric Hsp70 inhibitor was shown to inhibit DnaJ-stimulated ATP turnover rate and potentially reduce tau levels. Likewise, overexpression of HSJ1 in cells overexpressing tau was shown to suppress the efficacy of YM-01 and increase tau levels [Abisambra, et al., 2013]. Interestingly, tau-knockout mice showing decreased nigral tau levels have been reported to exhibit severe motor deficits, nigral neuronal loss, iron accumulation, cognitive deficits, and brain atrophy. Cognitive impairment was shown to occur in tandem with deficits in neurotrophic factors, such as BDNF, pro-BDNF, and tropomyosin receptor kinase B (TrkB), and motor deficits were rescued by levodopa administration [Lei, et al., 2012]. Taken together, one might hypothesize that the loss of tau seen in our deficient HSJ1b cells could lead to dopamine deficiency in the striatum, and thereby induce Parkinsonism. However, additional research is needed to confirm this hypothesis as two of our patients with the large HSJ1 deletion showed no signs of Parkinsonism (and did not have imaging of dopaminergic function performed).
BDNF provided by cortico-striatal projections is essential for the survival of striatal neurons in normal and pathological situations [Baquet, et al., 2004; Saudou, et al., 1998]. Along these lines, HSJ1b has been shown to regulate BDNF release in BDNF-overexpressing neuronal cells and consequently promotes neuronal survival [Borrell-Pages, et al., 2006]. We observed that BDNF levels were reduced in cell lysates of mutant HSJ1b-overexpressing cells but increased in the media. These results indicate that BDNF levels are also altered in HSJ1-associated neurodegeneration and this alteration may be compatible with what is observed in HD and PD patients. BDNF transport is specifically altered in HD patients, where BDNF levels are reduced in the striatum but not in the cortex [Gauthier, et al., 2004]. Low levels of BDNF concentrations are also observed in nigrostriatal dopamine regions of PD brains, while BDNF levels, which are thought to respond to signals released from the failing neurons, are increased in nigral glial cells [Knott, et al., 2002; Mogi, et al., 1999]. An increased BDNF expression in the serum of PD patients has also recently been reported [Ventriglia, et al., 2013]. Thus, while BDNF deficiency may result in cell toxicity, it may also activate a protective mechanism that increases BDNF release in adjacent cells and induces neuronal survival. Because BDNF levels were altered in our cells overexpressing mutant HSJ1b (Fig. 4C and D), one may hypothesize that HSJ1b may play an important role in the regulation of BDNF transport, as previously suggested.
Conclusions
We identified a large genomic deletion at the DNAJB2 locus, implicating the entire DnaJ domain of the HSJ1 protein, in patients with recessive SMA and Parkinsonism. Although Parkinsonism may not be triggered by this DNAJB2 deletion, as was only seen in one patient, we suggest that DNAJB2 genetic screening should be considered in rare forms of SMA with Parkinsonism. Additionally, CNV analysis at the DNAJB2 locus should be carried out in patients with SMA phenotypes. We showed that HSJ1 deficiency leads to the presentation of large cellular aggregates along with an increased cell death that may be due to the protein accumulation caused by the lack of the DnaJ domain. Enhanced autophagy, which may be promoted by an increased HSJ1a expression, was also observed in our HSJ1b-deficient mutant cells. Lastly, cells lacking the DnaJ domain of the HSJ1b protein showed low levels of both BDNF and tau proteins, suggesting a relevance role of these proteins in the HSJ1 disease-associated pathogenesis.
Supplementary Material
Acknowledgments
We thank the patients and relatives for their cooperation in this study. Microscopy and image analysis was performed at the Microscopy CORE at the Icahn School of Medicine at Mount Sinai.
Contract Grant Sponsor: Shahid Beheshti University of Medical Sciences; National Institute of Neurological Disorders and Stroke of the National Institutes of Health (NINDS; R01NS079388).
Footnotes
Additional Supporting Information may be found in the online version of this article.
Disclosure statement: The authors declare no conflict of interest.
References
- Abisambra J, Jinwal UK, Miyata Y, Rogers J, Blair L, Li X, Seguin SP, Wang L, Jin Y, Bacon J, et al. Allosteric heat shock protein 70 inhibitors rapidly rescue synaptic plasticity deficits by reducing aberrant tau. Biol Psychiatry. 2013;74:367–374. doi: 10.1016/j.biopsych.2013.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arancio O, Chao MV. Neurotrophins, synaptic plasticity and dementia. Curr Opin Neurobiol. 2007;17:325–330. doi: 10.1016/j.conb.2007.03.013. [DOI] [PubMed] [Google Scholar]
- Baquet ZC, Gorski JA, Jones KR. Early striatal dendrite deficits followed by neuron loss with advanced age in the absence of anterograde cortical brain-derived neurotrophic factor. J Neurosci. 2004;24:4250–4258. doi: 10.1523/JNEUROSCI.3920-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumen SC, Astord S, Robin V, Vignaud L, Toumi N, Cieslik A, Achiron A, Carasso RL, Gurevich M, Braverman I, et al. A rare recessive distal hereditary motor neuropathy with HSJ1 chaperone mutation. Ann Neurol. 2012;71:509–519. doi: 10.1002/ana.22684. [DOI] [PubMed] [Google Scholar]
- Borrell-Pages M, Canals JM, Cordelieres FP, Parker JA, Pineda JR, Grange G, Bryson EA, Guillermier M, Hirsch E, Hantraye P, et al. Cystamine and cysteamine increase brain levels of BDNF in Huntington disease via HSJ1b and transglutaminase. J Clin Invest. 2006;116:1410–1424. doi: 10.1172/JCI27607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapple JP, Cheetham ME. The chaperone environment at the cytoplasmic face of the endoplasmic reticulum can modulate rhodopsin processing and inclusion formation. J Biol Chem. 2003;278:19087–19094. doi: 10.1074/jbc.M212349200. [DOI] [PubMed] [Google Scholar]
- Cheetham ME, Brion JP, Anderton BH. Human homologues of the bacterial heat-shock protein DnaJ are preferentially expressed in neurons. Biochem J. 1992;284:469–476. doi: 10.1042/bj2840469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claeys KG, Sozanska M, Martin JJ, Lacene E, Vignaud L, Stockholm D, Laforet P, Eymard B, Kichler A, Scherman D, et al. DNAJB2 expression in normal and diseased human and mouse skeletal muscle. Am J Pathol. 2010;176:2901–2910. doi: 10.2353/ajpath.2010.090663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edvardson S, Cinnamon Y, Ta-Shma A, Shaag A, Yim YI, Zenvirt S, Jalas C, Lesage S, Brice A, Taraboulos A, et al. A deleterious mutation in DNAJC6 encoding the neuronal-specific clathrin-uncoating co-chaperone auxilin, is associated with juvenile parkinsonism. PLoS One. 2012;7:e36458. doi: 10.1371/journal.pone.0036458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emde AK, Schulz MH, Weese D, Sun R, Vingron M, Kalscheuer VM, Haas SA, Reinert K. Detecting genomic indel variants with exact breakpoints in single- and paired-end sequencing data using SplazerS. Bioinformatics. 2012;28:619–627. doi: 10.1093/bioinformatics/bts019. [DOI] [PubMed] [Google Scholar]
- Fan CY, Lee S, Cyr DM. Mechanisms for regulation of Hsp70 function by Hsp40. Cell Stress Chaperones. 2003;8:309–316. doi: 10.1379/1466-1268(2003)008<0309:mfrohf>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier LR, Charrin BC, Borrell-Pages M, Dompierre JP, Rangone H, Cordelieres FP, De Mey J, MacDonald ME, Lessmann V, Humbert S, et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118:127–138. doi: 10.1016/j.cell.2004.06.018. [DOI] [PubMed] [Google Scholar]
- Gess B, Auer-Grumbach M, Schirmacher A, Strom T, Zitzelsberger M, Rudnik-Schoneborn S, Rohr D, Halfter H, Young P, Senderek J. HSJ1-related hereditary neuropathies: novel mutations and extended clinical spectrum. Neurology. 2014;83:1726–1732. doi: 10.1212/WNL.0000000000000966. [DOI] [PubMed] [Google Scholar]
- Hageman J, Kampinga HH. Computational analysis of the human HSPH/HSPA/DNAJ family and cloning of a human HSPH/HSPA/DNAJ expression library. Cell Stress Chaperones. 2009;14:1–21. doi: 10.1007/s12192-008-0060-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindson BJ, Ness KD, Masquelier DA, Belgrader P, Heredia NJ, Makarewicz AJ, Bright IJ, Lucero MY, Hiddessen AL, Legler TC, et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem. 2011;83:8604–8610. doi: 10.1021/ac202028g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howarth JL, Kelly S, Keasey MP, Glover CP, Lee YB, Mitrophanous K, Chapple JP, Gallo JM, Cheetham ME, Uney JB. Hsp40 molecules that target to the ubiquitin-proteasome system decrease inclusion formation in models of polyglutamine disease. Mol Ther. 2007;15:1100–1105. doi: 10.1038/sj.mt.6300163. [DOI] [PubMed] [Google Scholar]
- Jankovic J. Parkinson’s disease: clinical features and diagnosis. J Neurol Neurosurg Psychiatry. 2008;79:368–376. doi: 10.1136/jnnp.2007.131045. [DOI] [PubMed] [Google Scholar]
- Jinwal UK, Akoury E, Abisambra JF, O’Leary JC, Thompson AD, 3rd, Blair LJ, Jin Y, Bacon J, Nordhues BA, Cockman M, et al. Imbalance of Hsp70 family variants fosters tau accumulation. FASEB J. 2013;27:1450–1459. doi: 10.1096/fj.12-220889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampinga HH, Craig EA. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat Rev Mol Cell Biol. 2010;11:579–592. doi: 10.1038/nrm2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott C, Stern G, Kingsbury A, Welcher AA, Wilkin GP. Elevated glial brain-derived neurotrophic factor in Parkinson’s diseased nigra. Parkinsonism Relat Disord. 2002;8:329–341. doi: 10.1016/s1353-8020(02)00008-1. [DOI] [PubMed] [Google Scholar]
- Krebs CE, Karkheiran S, Powell JC, Cao M, Makarov V, Darvish H, Di Paolo G, Walker RH, Shahidi GA, Buxbaum JD, et al. The Sac1 domain of SYNJ1 identified mutated in a family with early-onset progressive Parkinsonism with generalized seizures. Hum Mutat. 2013;34:1200–1207. doi: 10.1002/humu.22372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei P, Ayton S, Finkelstein DI, Spoerri L, Ciccotosto GD, Wright DK, Wong BX, Adlard PA, Cherny RA, Lam LQ, et al. Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat Med. 2012;18:291–295. doi: 10.1038/nm.2613. [DOI] [PubMed] [Google Scholar]
- Lesage S, Drouet V, Majounie E, Deramecourt V, Jacoupy M, Nicolas A, Cormier-Dequaire F, Hassoun SM, Pujol C, Ciura S, et al. Loss of VPS13C function in autosomal-recessive Parkinsonism causes mitochondrial dysfunction and increases PINK1/Parkin-dependent mitophagy. Am J Hum Genet. 2016;98:500–513. doi: 10.1016/j.ajhg.2016.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupo V, Garcia-Garcia F, Sancho P, Tello C, Garcia-Romero M, Villarreal L, Alberti A, Sivera R, Dopazo J, Pascual-Pascual SI, et al. Assessment of targeted next-generation sequencing as a tool for the diagnosis of Charcot-Marie-Tooth disease and hereditary motor neuropathy. J Mol Diagn. 2016;18:225–234. doi: 10.1016/j.jmoldx.2015.10.005. [DOI] [PubMed] [Google Scholar]
- Mayer MP, Bukau B. Hsp70 chaperones: cellular functions and molecular mechanism. Cell Mol Life Sci. 2005;62:670–684. doi: 10.1007/s00018-004-4464-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogi M, Togari A, Kondo T, Mizuno Y, Komure O, Kuno S, Ichinose H, Nagatsu T. Brain-derived growth factor and nerve growth factor concentrations are decreased in the substantia nigra in Parkinson’s disease. Neurosci Lett. 1999;270:45–48. doi: 10.1016/s0304-3940(99)00463-2. [DOI] [PubMed] [Google Scholar]
- Paisan-Ruiz C, Bhatia KP, Li A, Hernandez D, Davis M, Wood NW, Hardy J, Houlden H, Singleton A, Schneider SA. Characterization of PLA2G6 as a locus for dystonia-parkinsonism. Ann Neurol. 2009;65:19–23. doi: 10.1002/ana.21415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paisan-Ruiz C, Guevara R, Federoff M, Hanagasi H, Sina F, Elahi E, Schneider SA, Schwingenschuh P, Bajaj N, Emre M, et al. Early-onset L-dopa-responsive parkinsonism with pyramidal signs due to ATP13A2, PLA2G6, FBXO7 and spatacsin mutations. Mov Disord. 2010;25:1791–1800. doi: 10.1002/mds.23221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose JM, Novoselov SS, Robinson PA, Cheetham ME. Molecular chaperone-mediated rescue of mitophagy by a Parkin RING1 domain mutant. Hum Mol Genet. 2011;20:16–27. doi: 10.1093/hmg/ddq428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez E, Bergareche A, Krebs CE, Gorostidi A, Makarov V, Ruiz-Martinez J, Chorny A, Lopez de Munain A, Marti-Masso JF, Paisan-Ruiz C. SORT1 mutation resulting in sortilin deficiency and 75(NTR) upregulation in a family with essential tremor. ASN Neuro. 2015;7(4) doi: 10.1177/1759091415598290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saudou F, Finkbeiner S, Devys D, Greenberg ME. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell. 1998;95:55–66. doi: 10.1016/s0092-8674(00)81782-1. [DOI] [PubMed] [Google Scholar]
- Schabhuttl M, Wieland T, Senderek J, Baets J, Timmerman V, De Jonghe P, Reilly MM, Stieglbauer K, Laich E, Windhager R, et al. Whole-exome sequencing in patients with inherited neuropathies: outcome and challenges. J Neurol. 2014;261:970–982. doi: 10.1007/s00415-014-7289-8. [DOI] [PubMed] [Google Scholar]
- Schicks J, Synofzik M, Petursson H, Huttenlocher J, Reimold M, Schols L, Bauer P. Atypical juvenile parkinsonism in a consanguineous SPG15 family. Mov Disord. 2011;26:564–566. doi: 10.1002/mds.23472. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tazir M, Hamadouche T, Nouioua S, Mathis S, Vallat JM. Hereditary motor and sensory neuropathies or Charcot-Marie-Tooth diseases: an update. J Neurol Sci. 2014;347:14–22. doi: 10.1016/j.jns.2014.10.013. [DOI] [PubMed] [Google Scholar]
- Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turturici G, Sconzo G, Geraci F. Hsp70 and its molecular role in nervous system diseases. Biochem Res Int. 2011;2011:618127. doi: 10.1155/2011/618127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventriglia M, Zanardini R, Bonomini C, Zanetti O, Volpe D, Pasqualetti P, Gennarelli M, Bocchio-Chiavetto L. Serum brain-derived neurotrophic factor levels in different neurological diseases. Biomed Res Int. 2013;2013:901082. doi: 10.1155/2013/901082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voisine C, Pedersen JS, Morimoto RI. Chaperone networks: tipping the balance in protein folding diseases. Neurobiol Dis. 2010;40:12–20. doi: 10.1016/j.nbd.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werheid F, Azzedine H, Zwerenz E, Bozkurt A, Moeller MJ, Lin L, Mull M, Hausler M, Schulz JB, Weis J, et al. Underestimated associated features in CMT neuropathies: clinical indicators for the causative gene? Brain Behav. 2016:e00451. doi: 10.1002/brb3.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westhoff B, Chapple JP, van der Spuy J, Hohfeld J, Cheetham ME. HSJ1 is a neuronal shuttling factor for the sorting of chaperone clients to the proteasome. Curr Biol. 2005;15:1058–1064. doi: 10.1016/j.cub.2005.04.058. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.