

ABSTRACT
Hepatitis C virus (HCV) envelope glycoprotein complex is composed of E1 and E2 subunits. E2 is the receptor-binding protein as well as the major target of neutralizing antibodies, whereas the functions of E1 remain poorly defined. Here, we took advantage of the recently published structure of the N-terminal region of the E1 ectodomain to interrogate the functions of this glycoprotein by mutating residues within this 79-amino-acid region in the context of an infectious clone. The phenotypes of the mutants were characterized to determine the effects of the mutations on virus entry, replication, and assembly. Furthermore, biochemical approaches were also used to characterize the folding and assembly of E1E2 heterodimers. Thirteen out of 19 mutations led to viral attenuation or inactivation. Interestingly, two attenuated mutants, T213A and I262A, were less dependent on claudin-1 for cellular entry in Huh-7 cells. Instead, these viruses relied on claudin-6, indicating a shift in receptor dependence for these two mutants in the target cell line. An unexpected phenotype was also observed for mutant D263A which was no longer infectious but still showed a good level of core protein secretion. Furthermore, genomic RNA was absent from these noninfectious viral particles, indicating that the D263A mutation leads to the assembly and release of viral particles devoid of genomic RNA. Finally, a change in subcellular colocalization between HCV RNA and E1 was observed for the D263A mutant. This unique observation highlights for the first time cross talk between HCV glycoprotein E1 and the genomic RNA during HCV morphogenesis.
IMPORTANCE Hepatitis C virus (HCV) infection is a major public health problem worldwide. It encodes two envelope proteins, E1 and E2, which play a major role in the life cycle of this virus. E2 has been extensively characterized, whereas E1 remains poorly understood. Here, we investigated E1 functions by using site-directed mutagenesis in the context of the viral life cycle. Our results identify unique phenotypes. Unexpectedly, two mutants clearly showed a shift in receptor dependence for cell entry, highlighting a role for E1 in modulating HCV particle interaction with a cellular receptor(s). More importantly, another mutant led to the assembly and release of viral particles devoid of genomic RNA. This unique phenotype was further characterized, and we observed a change in subcellular colocalization between HCV RNA and E1. This unique observation highlights for the first time cross talk between a viral envelope protein and genomic RNA during morphogenesis.
KEYWORDS: hepatitis C virus, glycoprotein, envelope proteins, viral entry, viral assembly, glycoproteins, viral envelope
INTRODUCTION
Hepatitis C virus (HCV) infection is a major public health problem, with around 170 million people infected worldwide (1). HCV infection has a high propensity for establishing a chronic infection, and, in the long term, this can lead to cirrhosis and hepatocellular carcinoma. Although recent improvements in the standard-of-care therapy have been achieved, the available treatments remain very expensive and far from being accessible to the majority of HCV-infected patients (2).
HCV is a plus-stranded RNA virus which belongs to the Hepacivirus genus in the Flaviviridae family. The viral genome contains a single open reading frame generating a polyprotein which is sequentially processed by both cellular and virally encoded proteases into 10 mature viral proteins. Among these polypeptides, the structural proteins (core, E1, and E2) are the components of the viral particle (reviewed in reference 3).
The E1 and E2 envelope glycoproteins are two highly glycosylated type I transmembrane proteins, each with an N-terminal ectodomain and a well-conserved C-terminal transmembrane domain. By being part of the viral particle, HCV envelope glycoproteins E1 and E2 play an essential role in virion morphogenesis as well as in HCV entry into liver cells. These two steps necessitate timely and coordinated control of HCV glycoprotein functions. Furthermore, HCV entry is a complex multistep process involving at least four major entry factors. They include scavenger receptor BI (SR-BI) (4), tetraspanin CD81 (5), and tight junction proteins claudin-1 (CLDN1) (6), and occludin (OCLN) (7).
Until recently, research on HCV glycoproteins has been focused mainly on E2 because it is the receptor-binding protein interacting with CD81 and SR-BI. E2 is also the major target of neutralizing antibodies, and it was postulated to be the fusion protein (reviewed in reference 8). However, the structure of E2 does not fit with what one would expect for a fusion protein (9, 10), suggesting that E1 alone or in association with E2 might be responsible for the fusion step. Interestingly, several studies characterizing novel inhibitors of late steps of HCV entry have shown that some resistant mutations can be found in E1 (11–13), reinforcing the hypothesis that this protein plays a major role during the fusion process. Furthermore, E1 also plays a role in modulating the exposure of the CD81-binding region on E2 (14). Together, these observations indicate that E1 plays a more important role than previously thought in the HCV life cycle. It is therefore essential to better understand how E1 plays an active role in HCV entry and assembly.
Recently, the crystal structure of the N-terminal half of the E1 ectodomain was reported (15). This partial structure reveals a complex network of covalently linked, intertwined homodimers. We took advantage of this report to investigate the functional role of E1 by alanine replacement of residues in the context of an infectious clone. Among 19 mutants, eight showed reduced viral infectivity, and five were no longer infectious. Interestingly, two attenuated mutants, T213A and I262A, showed a shift of dependence in virus entry factor from CLDN1 to CLDN6. Importantly, another mutation, D263A, which abolished virus infectivity, led to the secretion of viral particles devoid of genomic RNA but containing core protein and HCV glycoproteins, highlighting the cross talk between HCV glycoprotein E1 and genomic RNA during HCV morphogenesis.
RESULTS
Amino acid conservation in the N-terminal region of the E1 ectodomain and mutagenesis rationale.
The secondary structures present in the N-terminal 79 amino acid residues of E1 are presented in Fig. 1A (15). The analysis of E1 amino acid sequence conservation among all HCV genotypes shows that the most conserved residues do not necessarily match the secondary structure elements that have been identified in the crystallographic structure of the N-terminal domain of the E1 ectodomain (Fig. 1B). This highlights that both the secondary structures and peculiar features of the loops, which contain crucial cysteine residues as well as glycosylation sites, are crucial for the biological function(s) of E1. It is worth noting that the less conserved region in the N-terminal region of the E1 ectodomain corresponds to the disordered loop between β4 and β5 that was not seen in the electron density of the crystallographic structure.
FIG 1.

E1 N-terminal region sequence analyses. (A) The sequence of the 79-amino-acid region comprising positions 192 to 270 of E1 of the HCV JFH1 strain (AB047639; genotype 2a) is indicated (polyprotein numbering). The A4 epitope (specific for genotype 1a) is comprised of N-terminal amino acids 197 to 207. The secondary structures corresponding to the α-helix and β strands previously identified are highlighted in blue and yellow, respectively. Amino acids mutated in this study are indicated by a red dot. (B) Amino acid repertoire of the N-terminal region of the E1 ectodomain. The amino acid repertoire was deduced from ClustalW multiple alignment of 19 reference sequences from all confirmed genotypes and subtypes (https://euhcvdb.ibcp.fr/euHCVdb/). Amino acids observed at a given position in fewer than two distinct sequences were not included. Amino acids observed at a given position in more than 17 distinct sequences are shown in uppercase letters. The degree of amino acid conservation at each position can be inferred from the extent of variability (with the observed amino acid listed in decreasing order of frequency from top to bottom), together with the similarity index according to ClustalW convention (asterisk, invariant; colon, highly similar; dot, similar).
Here, we mutated residues in the context of an infectious clone. The effects of mutation of cysteine residues and glycosylation sites have already been reported previously (14, 16), and these residues were therefore not included in this study. We produced a series of 19 mutants in which residues were individually replaced by alanine residues, and we concentrated our study on structured segments (β1 to β5 strands and α-helix) or conserved amino acids located close to these secondary structures (Fig. 1A). Mutations were introduced in a modified version of the plasmid harboring the full-length JFH1 genome in which the N-terminal E1 sequence has been modified to reconstitute the A4 epitope, which is present in the E1 of genotype 1a (17), and therefore allows the identification of the E1 of genotype 2a for which there is no antibody easily available. We did not introduce any mutation in β2 since it contains the monoclonal antibody (MAb) A4 epitope sequence. It is worth noting that the introduction of the A4 epitope had no effect on HCV infectivity (data not shown), indicating that this modification is not interfering with the phenotype of E1 mutants characterized in our study.
Effect of E1 mutations on HCV infectivity.
We first determined whether our mutants are functional for replication. For this, we analyzed the expression of several HCV proteins at 48 h postelectroporation. For all the mutants, the levels of expression of E2 and NS5A were similar to those of the wild-type virus (Fig. 2A). However, we observed a weaker signal for the E1 glycoprotein in the case of the Q193A and V194A mutants, which is likely due to a weaker recognition of E1 by MAb A4, whose minimum epitope has been mapped immediately downstream of these two residues. Together, our data indicate that our mutations do not affect HCV genome replication.
FIG 2.

Effects of mutations on viral protein expression and infectivity. (A) Huh-7 cells were electroporated with viral RNA transcribed from JFH1-derived mutants, and they were lysed at 48 h postelectroporation. Viral proteins were separated by SDS-PAGE and detected by Western blotting with MAbs A4 (anti-E1), 3/11 (anti-E2), and anti-NS5A. Western blotting with an anti-beta-tubulin antibody was performed in parallel to verify that equal amounts of cell lysates had been loaded. (B) Infectivity of E1 mutants. Huh-7 cells were electroporated with viral RNA transcribed from JFH1-derived mutants. At 48, 72, and 96 h postelectroporation, infectivities of the supernatants and intracellular viruses were determined by titration. Error bars indicate standard errors of the means from at least three independent experiments. Results were compared to those for the wild type, and a P value of <0.05 was determined for intracellular mutants Q193A, V194A, I212A, T213A, W214A, Q215A, V220A, L221A, H222A, R231A, G233A, W239A, V240A, P244A, I262A, D263A and M264A. A P value of <0.05 was determined for extracellular mutants I212A, T213A, W214A, Q215A, H222A, W239A, and D263A. WT, wild type; FFU, focus-forming units.
We then measured the effects of the mutations on the production of infectious virus by determining intra- and extracellular infectivities. As shown in Fig. 2B, we observed three phenotypes for virus infectivity: (i) complete loss of infectivity, (ii) no effect on infectivity, or (iii) reduced infectivity. In the β1 strand we observed a slight decrease in extracellular infectivity for the Q193A and V194A mutants, indicating that these mutations only slightly affect HCV infectivity. Four mutations (S208A, T213A, V220A, and L221A) in the α-helix had only a slight effect or no effect at all on HCV infectivity, whereas others produced a drastic reduction in HCV infectivity (I212A and Q215A) or totally abolished it (W214A and H222A). Most mutants within the β-sheet (β3 to β5) showed no change in infectivity (G233A, M264A, and V265A) or only a slight decrease (R231A, V240A, P244A, and I262A). However, with two of them (W239A and D263A) infectivity was totally abolished. Overall, the intracellular infectivity profiles were similar to those observed for extracellular viruses, excluding any effect of the mutations on infectious particle release. This first analysis indicates that the α-helix and the β-sheet contain essential residues for HCV infectivity.
Determination of virion release.
To determine the effect of mutations on viral secretion in the case of reduced or abolished infectivity, we measured the expression of core protein in cell lysates and supernatants. As shown in Fig. 3A, the levels of the intracellular core proteins of the mutants were comparable to the wild-type level, excluding any effect of the mutations on HCV genomic replication. In contrast, the levels of extracellular core proteins were reduced for several mutants (Fig. 3B). For most mutants, the levels of extracellular core proteins paralleled those of extracellular infectivity (compare Fig. 2 and 3). However, mutant D263A showed a good level of core protein release despite the absence of infectivity in the supernatant (Fig. 3B), which was confirmed in an additional experiment in the presence of a mutant virus defective in virus assembly (Fig. 3C), suggesting that this mutation leads to the release of noninfectious viral particles.
FIG 3.

Analysis of core protein release for E1 mutants. Huh-7 cells were electroporated with viral RNA transcribed from JFH1-derived mutants. At 48 h postelectroporation, the amount of intracellular core antigen was determined in cell lysates (A) and supernatants (B). A control experiment with a genome defective in virus assembly, ΔE1E2, is shown in panel C. HCV core protein was quantified by a fully automated chemiluminescent microparticle immunoassay. Error bars indicate standard deviations. Results were compared to those for the wild type, and a P value of <0.05 was determined for extracellular mutants I212A, T213A, W214A, Q215A, H222A, and W239A.
Effect of E1 mutations on HCV glycoprotein folding and E1E2 heterodimerization.
Given the cooperativity between E1 and E2 in their respective folding, we analyzed the effect of E1 mutations on the formation of E1E2 heterodimers. To study the effect of mutations on the folding of E1 and E2, we used a pulldown assay using the CD81 large extracellular loop (CD81-LEL) that recognizes correctly folded E2. As shown in Fig. 4A, E2 proteins from all the mutants were recognized by CD81-LEL. However, lower signals were observed for several of them: I212A, T213A, W214A, Q215A, V220A, H222A, R231, W239A, and D263A. Importantly, for some mutants, the presence of E1 was not detected or only barely detected (Q193A, V194A, I212A, H222A, W239A, I262A, and D263A) or was reduced (T213A). As discussed above, the weaker E1 signals for mutants Q193A and V194A are likely due to the involvement of these two residues in the A4 epitope. For the other mutants, the absence or drastic decrease of E1 coprecipitation suggests that these mutations affect the interaction between E1 and E2, at least in the context of properly folded E2. Since these mutants (I212A, H222A, W239A, I262A, and D263A) are also affected in their infectivity (Fig. 2B), our data suggest that the assembly defect of the E1E2 heterodimer could be responsible for the decrease in infectivity. However, in the case of the I262A mutant, infectivity was only 1 log10 lower than that of the wild type (Fig. 2B), suggesting that the residual interaction between E1 and E2 is sufficient to maintain a certain level of infectivity. In contrast, E1 and E2 from mutants W214A and Q215A were precipitated by CD81 despite a loss of infectivity, suggesting that the functional defect for these mutants is not due to a global effect on E1E2 folding but, rather, on virion assembly itself.
FIG 4.

Effect of E1 mutations on HCV glycoprotein folding and E1E2 heterodimerization. (A) Interaction of viral glycoproteins with HCV entry factor CD81. At 48 h postelectroporation, lysates were analyzed by GST pulldown using a CD81-LEL-GST fusion protein. Pulled-down E1 and E2 were analyzed by SDS-PAGE and detected by Western blotting with MAbs A4 and 3/11. (B and C) Interaction of HCV glycoproteins with the conformation-sensitive anti-E1E2 MAb AR5A and anti-E1 MAb IGH526, as indicated. At 48 h postelectroporation, E1 and E2 from cell lysates were analyzed by immunoprecipitation with MAbs AR5A and IGH526. Precipitated E1 and E2 were then separated by SDS-PAGE and detected by Western blotting with MAbs A4 and 3/11. The weaker E1 signals observed with two of our mutants (Q193A and V194A) are likely due to the involvement of these two residues in the A4 epitope.
To further characterize the folding of E1E2 complex, we also performed an immunoprecipitation experiment with conformation-sensitive MAbs. We first used MAb AR5A that recognizes a conformational epitope shared between E1 and E2 (18). For five of our mutants (I212A, H222A, W239A, I262A, and D263A), HCV glycoproteins were not detected or only weakly recognized by MAb AR5A (Fig. 4B), which correlated with the data obtained with CD81 pulldown of E1E2. Since these mutants were also altered in their infectivity, one can expect that alteration in protein folding is responsible for the phenotype of these viruses.
Finally, we also used the conformation-sensitive anti-E1 MAb IGH526, whose core epitope is located at amino acid positions 314 to 324 (19). For this analysis, we focused mainly on mutants showing alterations in infectivity. As shown in Fig. 4C, a decrease in the recognition of E1 was observed for the I212A, T213A, H222A, W239A, and D263A mutants, which correlated with an alteration in recognition of E1E2 complex by CD81 and AR5A (Fig. 4A and B). It is noteworthy, that the I262A mutant was relatively well recognized by MAb IGH526 (Fig. 4C), which contrasts with the altered recognition by CD81 and AR5A, suggesting that E1 might have achieved an advanced state of folding for this mutant despite alterations in E1E2 interactions. A decrease in E1 recognition by MAb IGH526, which correlated with a slight reduction of recognition by CD81, was also observed for the R231A mutant (Fig. 4C). This could again explain the slight decrease in infectivity observed for this mutant (Fig. 4A).
Effect of E1 mutations on sensitivity to antibody neutralization and inhibition by CD81 coreceptor.
The above-described analyses of the effects of E1 mutations on the folding of the envelope proteins were performed in the context of intracellular proteins. However, incorporation of the envelope glycoproteins on the surface of the viral particle during the assembly process leads to conformational changes that occur in the quaternary structure of these proteins (15, 20). The biochemical analyses of the glycoproteins associated with the viral particle are not easily performed because they require large amounts of viral particles. A more sensitive alternative approach is to determine the sensitivity of the mutant viruses to inhibition mediated by the presence of CD81-LEL or neutralizing MAbs. We therefore used CD81-LEL as well as MAbs AR5A and IGH526 for these experiments. In these analyses, we focused on infectious mutants showing a decrease in infectivity of approximately 1 log10 (T213A, R231A, and I262A), and the S208A mutant was used as a control. Interestingly, we observed a strong decrease in sensitivity to inhibition by CD81-LEL, AR5A, and IGH526 for the T213A and I262A mutants (Fig. 5). These results indicate conformational changes in the envelope proteins present on the surfaces of these two mutants, which are in line with the alterations observed in our biochemical approach (Fig. 4), suggesting a similar effect of the mutations on the conformation of HCV glycoproteins present on the surface of the viral particle. However, for some mutants, discrepancies were observed between biochemical analyses and neutralization results. Indeed, the R231A mutant showed some alteration according to the biochemical analysis but was neutralized at the wild-type level. In contrast, the I262A mutant was well recognized by IGH526 in the immunoprecipitation experiment but was much less sensitive to neutralization by this antibody. This indicates that the biochemical results do not necessarily parallel the functional phenotype.
FIG 5.
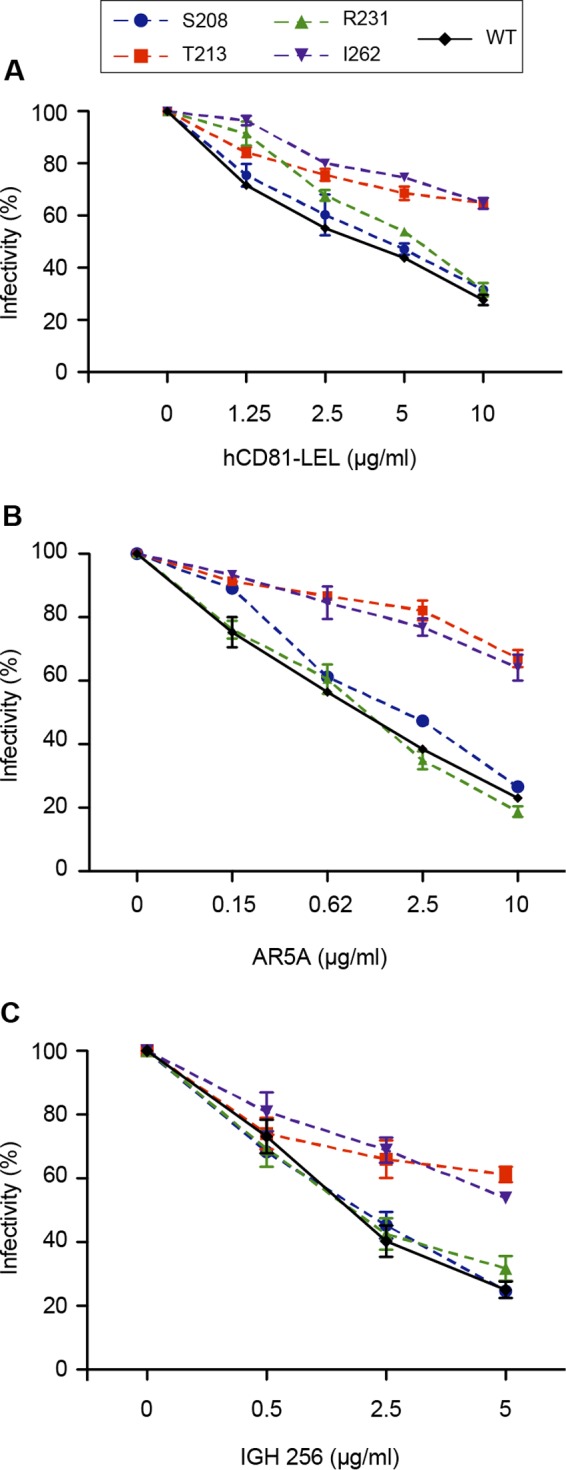
Effect of E1 mutations on sensitivity to antibody neutralization and inhibition by CD81. CD81 inhibition assays (A), AR5A neutralization experiments (B), and IGH526 neutralization experiments (C) were performed by incubating E1 mutants or wild-type virus with increasing concentrations of human CD81-LEL, MAb AR5A, or MAb IGH526. After 2 h of incubation at 37°C, the mixtures were put into contact with target cells, and the inoculum was replaced by fresh medium at 6 h postinfection. At 72 h postinfection, residual infectivity was measured by immunofluorescence. The values are the combined data from three independent experiments. The error bars represent standard errors of the means. Results were compared to those for the wild type, and a P value of <0.05 was determined for mutants T213A and I262A under all inhibitory conditions.
Effect of E1 mutations on the recognition of HCV receptors.
To further characterize the phenotype of the T213A, R231A, and I262A mutants, we analyzed their dependence on known receptors. For this, we analyzed the sensitivity of our mutants to inhibition by antireceptor MAbs previously reported to affect HCV entry. Similar dose-dependent decreases in infectivity were observed for mutant (S208A, T213A, R231A, and I262A) and wild-type viruses in the presence of anti-CD81 MAb JS81 or anti-SR-BI MAb Cla-I (Fig. 6A and B). In contrast, the T213A and I262A mutants were less inhibited by anti-CLDN1 MAb OM8A9-A3 (Fig. 6C), suggesting that these viruses were less dependent on CLDN1 to infect Huh-7 cells. Since these cells also express CLDN6, another tight junction protein that can be used by some viruses in the absence of CLDN1 (21), we also tested the sensitivity of the T213A and I262A mutants to inhibition by anti-CLDN6 MAb 342927. As shown in Fig. 6D, the T213A and I262A mutants were more sensitive than the wild-type virus to inhibition by anti-CLDN6 MAb, whereas the S208A and R231A mutants showed the same profile as the wild-type virus (data not shown). Furthermore, when we coincubated these mutants with both anti-CLDN1 and anti-CLDN6 MAbs, the T213A and I262A mutants were inhibited to a similar extent as the wild-type virus (Fig. 6E). In the absence of antibodies against OCLN, we used a cell line knocked out for this receptor (22) to determine the dependence of our mutants to OCLN for virus entry. None of our mutants were able to infect this cell line (data not shown), indicating a similar dependence on OCLN. Altogether, our data indicate that the T213A and I262A mutations induce a shift in receptor usage from CLDN1 toward CLDN6.
FIG 6.
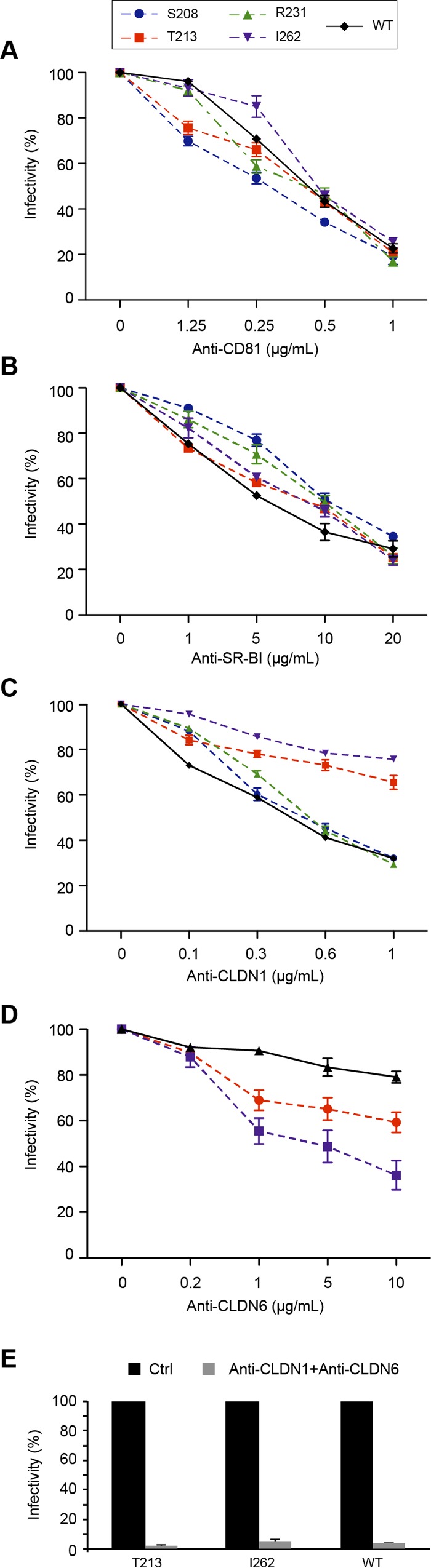
Effect of E1 mutations on the recognition of HCV receptors. (A to D) Huh-7 cells were preincubated for 2 h at 37°C with increasing concentrations of antibodies targeting HCV receptors: anti-CD81 MAb JS81, anti-SR-BI MAb Cla-I, anti-CLDN1 MAb OM8A9-A3, and anti-CLDN6 MAb 342927, as indicated. Cells were then incubated with E1 mutants or wild-type virus, and the inoculum was replaced by fresh medium at 6 h postinfection. At 72 h postinfection, cells were processed for immunofluorescence to quantify residual infectivity. The values are the combined data from three independent experiments. The error bars represent standard errors of the means. Results were compared to those for the wild type, and a P value of <0.05 was determined for mutants T213A and I262A in the presence of anti-CLDN1 and anti-CLDN6 antibodies. (E) Effect of E1 mutations on the recognition of CLDN1 and CLDN6. Huh-7 cells were preincubated for 2 h at 37°C with a combination of anti-CLDN1 (5 μg/ml) and anti-CLDN6 (10 μg/ml) MAbs. Cells were then incubated with E1 mutants or wild-type virus, and the inoculum was replaced by fresh medium at 6 h postinfection. At 72 h postinfection, cells were processed for immunofluorescence to quantify the residual infectivity. The values are the combined data from three independent experiments. The error bars represent standard errors of the means.
Characterization of the D263A mutant.
Since the D263A mutant had lost infectivity but showed a good level of core protein secretion, it was further characterized. For this, we analyzed the released viral material on an iodixanol density gradient. As a negative control, we used a viral genome carrying a large in-frame deletion in the E1E2 coding region known to affect the release of viral particles (ΔE1E2). The different fractions obtained after gradient sedimentation were analyzed to determine infectivity as well as the content of core protein and genomic RNA. As shown in Fig. 7B, core protein of the wild-type virus showed two peaks, one in fractions 1 and 2 and the other one in fractions 5 to 7. Fractions 1 and 2 corresponded to the peak of infectivity and the first peak of genomic RNA, whereas fractions 5 to 7 corresponded to the second peak of genomic RNA which was noninfectious (Fig. 7A). Although core protein of the D263A mutant was detected in fractions 1 to 9, the majority peaked in fractions 5 to 7, together with the noninfectious peak of the wild type-virus (Fig. 7A). Surprisingly, HCV RNA was at a background level for the D263A mutant, very similar to the level of the control ΔE1E2 virus, which is defective in virus assembly (Fig. 7B). This was not due to the absence of viral replication since intracellular RNA levels were similar for both the D263A mutant and the wild-type virus (Fig. 7C). Together, our data indicate that the D263A mutation leads to the assembly and release of particulate material devoid of genomic RNA.
FIG 7.
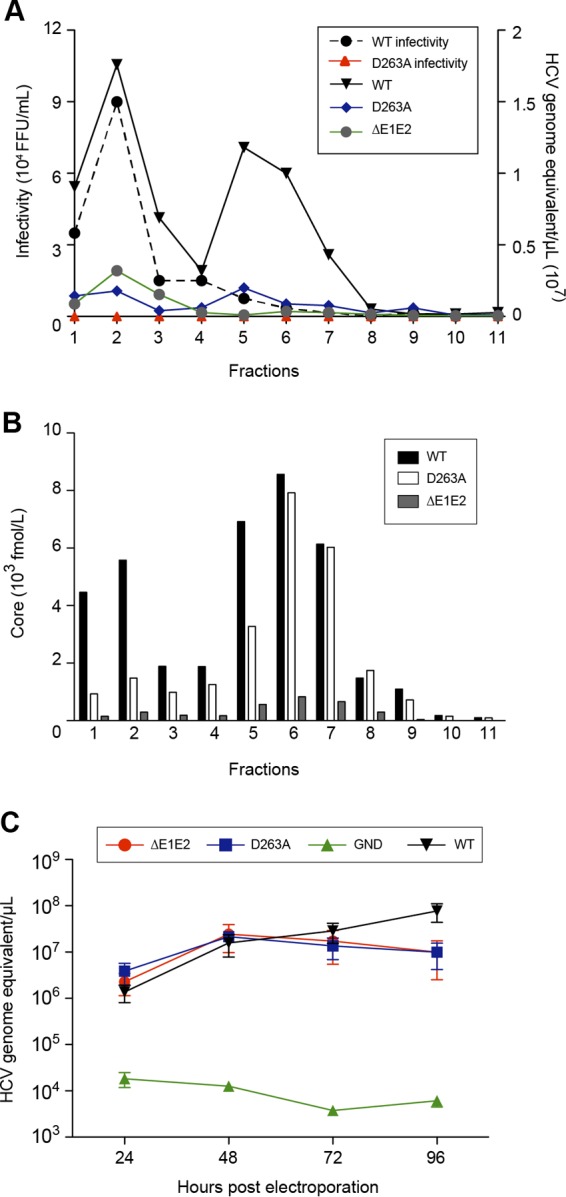
Characterization of the secreted form of the D263A mutant. Concentrated supernatant of cells electroporated with HCV was separated by sedimentation through a 10 to 50% iodixanol gradient. Fractions were collected from the top and analyzed for their infectivity as well as their viral RNA (A) and core protein content (B). (C) Analyses of intracellular genomic replication for the D263A mutant. Intracellular HCV genome copies in electroporated cells were quantified at different times postelectroporation. Control experiments with the wild type and ΔE1E2 and GND mutants were performed. The error bars represent standard deviations.
To further understand the molecular basis of the absence of genomic RNA in secreted virus particles in the context of the D263A mutation, we investigated whether the core protein was able to oligomerize into capsid-like structures. Oligomerization of core proteins expressed by this mutant was analyzed on an iodixanol gradient by ultracentrifugation, as described in Materials and Methods. The core protein complexes of the wild-type virus were detected in fractions 6 to 8 (Fig. 8). These fractions correspond to highly ordered multimeric complexes (23). A similar profile of sedimentation was observed with the D263A mutant as well as with the assembly-defective control ΔE1E2 virus (Fig. 8). However, there was a slight shift toward lower density with the D263A mutant. Furthermore, a small proportion of the core protein was found in fractions 2 and 3 of both the D263A and ΔE1E2 mutants. This likely corresponds to monomeric and/or dimeric forms of the core protein, as previously suggested (24). When the wild-type core protein was treated with 1% SDS before ultracentrifugation, only the monomeric form of the core protein was detected in fractions 1 and 2 at the top of the gradient. This profile is due to disruption of core protein complexes by SDS, as shown previously (23) (Fig. 8). These results therefore suggest that the D263A mutation does not drastically affect core protein multimerization.
FIG 8.

Analysis of core protein oligomerization by velocity sedimentation. Cells electroporated with the D263A mutant, ΔE1E2, or wild-type JFH1 RNA genome were lysed at 48 h postelectroporation. Lysates were subjected to velocity sedimentation on a 10 to 50% iodixanol density gradient, followed by Western blot analysis of core protein. Fractions were collected from the top. A control gradient was performed in parallel with extracts of cells electroporated with the wild-type genome that had been treated with 1% SDS. The input represents 8% of lysates.
Finally, we recently showed that E1 forms homotrimers during the assembly process (20). We therefore also tested the capacity of the D263A mutant to form such trimers, but we did not detect any defect in E1 trimerization (data not shown).
Subcellular localization of HCV proteins during the assembly process of the D263A mutant.
Since the D263A mutant leads to the production of viral particles devoid of genomic RNA, we further investigated the subcellular colocalization of HCV proteins to determine whether this mutation would induce a mislocalization of the E1 glycoprotein. For this, Huh-7 cells were electroporated with D263A mutant RNA, and the cells were fixed with paraformaldehyde at 48 h postelectroporation before being processed for immunofluorescence. E1 mutated at position D263 showed a colocalization with E2 similar to what was observed for the wild-type virus (Fig. 9). Furthermore, there was no difference in the core and NS5A protein colocalization with lipid droplets, the site where HCV assembly is supposed to take place (25). Finally, we also analyzed whether the D263A mutation affects the colocalization of core protein or E1 with the viral RNA. For this, we analyzed the localization of HCV RNA by fluorescence in situ hybridization (FISH). Although the D263A mutation did not change the colocalization of core protein with HCV RNA (Fig. 10A), a significant decrease in subcellular colocalization between HCV RNA and E1 was observed for this mutant (Fig. 10B and C), supporting the hypothesis that E1 could play a role in the recruitment of HCV RNA during virus assembly.
FIG 9.

Subcellular localization of HCV proteins in the context of the D263A mutant. Electroporated cells grown on coverslips were fixed at 48 h postelectroporation and processed for immunofluorescence with antibodies against viral proteins (E1, E2, core protein [C], and NS5A). Lipid droplets were stained with BODIPY 493/503 (green). Rat anti-E2 MAb 3/11 was used for the colocalization with E1 (mouse anti-E1 MAb A4). Anti-core MAb ACAP-27 was used for the colocalization with lipid droplets (LD). The NS5A protein was labeled with anti-NS5A (9E10). Nuclei were stained with DAPI (blue). Representative confocal images of individual cells are shown with the merged images in the right column. Bar, 25 μm.
FIG 10.
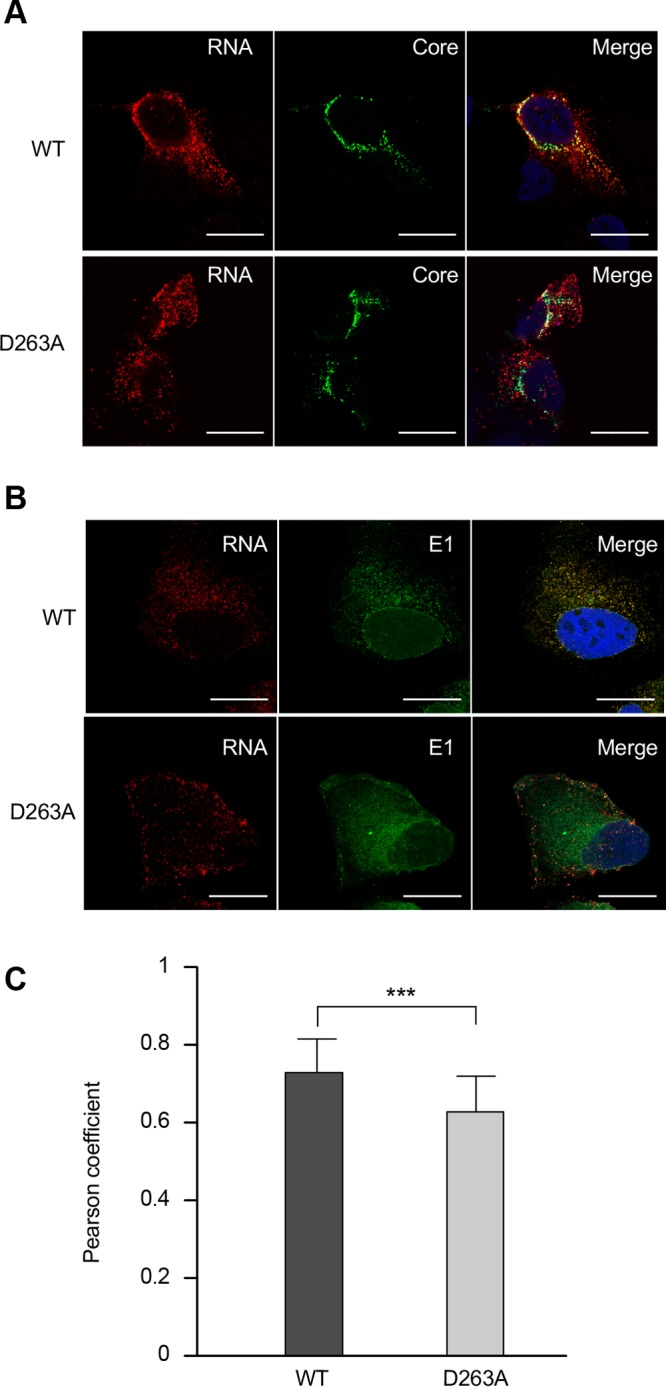
Subcellular localization of the D263A mutant and wild-type RNA by fluorescence in situ hybridization (FISH). Huh-7 cells were electroporated with the D263A and wild-type (WT) RNA genomes, fixed at 48 h postelectroporation, and processed for HCV positive-strand-specific RNA detection, followed by immunofluorescence staining for core protein with MAb ACAP-27 (A) or for E1 with MAb A4 (B). Scale bar, 20 μm. (C) Pearson's correlation coefficient. Error bars represent standard deviations from 21 different images (***, P = 0.0004).
DISCUSSION
For a long time, E1 remained poorly studied. However, recent structural studies on E2 suggested that E1 might play an active role in the fusion process (26), prompting us to initiate a functional study of this protein based on the recently published structure of its N terminus (15). Our data identify residues in the α-helix and the β-sheet that are important for the assembly and release of infectious viral particles. Characterization of our mutants also highlights cross talk between E1 and E2 during HCV morphogenesis. Furthermore, neutralization experiments indicate that mutations in two of our mutants induce a shift in receptor usage from CLDN1 toward CLDN6. Finally, characterization of the mutant D263A shows that this virus leads to assembly and release of viral particles devoid of genomic RNA, indicating that E1 plays a role in the incorporation of HCV RNA into the nucleocapsid.
Several mutations in E1 affect the folding of E2. This is the case for the I212A, T213A, H222A, and W239A mutants, as shown by CD81 pulldown. CD81 is often used as a probe to determine the folding of E2 since its binding region is located in E2 and since it is conformation dependent (27). HCV glycoproteins have been shown to assemble as noncovalent E1E2 heterodimers (28), and these proteins are known to cooperate for the formation of a functional complex (29). The folding of E1 has indeed initially been shown to be dependent on the coexpression of E2 (30, 31). Later on, it was reported that E1 can also affect the folding of E2 (32, 33). Our observation that mutations in E1 can affect the recognition of E2 by CD81 is therefore in line with the fact that E1 can play an active role in the folding of E2 in the context of E1E2. It is to be noted, however, that E2 expressed alone is well recognized by CD81 (5), suggesting that mutations in E1 can push E2 toward a conformation poorly recognized by CD81, which is in line with the cross talk observed between these two proteins (34, 35). I212, T213, and H222 correspond to highly conserved residues located in the α-helix of E1 (Fig. 11), whereas W239 belongs to the β4 strand.
FIG 11.

Positions of identified amino acid residues on the 3D structure of the E1 N-terminal region. The residues identified in this study are highlighted on the structural model of the N-terminal region of E1 from the JFH1 isolate. This model was built using the Swiss-Model web server (53) with the crystallographic structure of E1 (H77 strain) (PDB accession number 4UOI) as the template. (A) Residues for which the mutation toward an alanine showed an interesting or lethal phenotype are shown in green or orange, respectively, and their side chains are shown in stick rendering. (B) Hydrogen bond network established by the D263 residue with others residues from the β4 and β5 strands. The figure was generated using the PyMOL Molecular Graphics System, version 1.8 (Schrödinger, LLC). Ct, C terminus; Nt, N terminus.
Several mutations in E1 affect E1E2 interaction. This is particularly the case for mutants W239A, I262A, and D263A, as shown by the lack of E1 signal after CD81 pulldown. Residues involved in E1E2 interactions have been identified in the transmembrane domains of these two proteins (36). Moreover, a study based on chimeric E1E2 heterodimers derived from different genotypes has also recently identified residues in the ectodomain of E1, at positions 308, 330, and 345, as being involved in functional interactions between E1 and E2 (35). However, in that study, by Douam and coworkers, no biochemical analysis was performed to determine the physical interaction between these two proteins. Amino acid residues W239A, I262A, and D263A identified in our study as affecting E1E2 interaction are located in a β-sheet structure identified in the N-terminal region of E1 (Fig. 11) (15). I262 and D263, which belong to the β5 strand, and W239, which belongs to the β4 strand, are close in space in the structure of E1. Indeed the D263 residue is directly facing the W239 residue. Therefore, our data suggest that this β-sheet is involved in interactions with E2. Interestingly, D263 and W239, two highly conserved residues, are part of a rather hydrophobic surface that has been described by El Omari et al. as likely being involved in interaction with another protein partner (15). It has to be noted that one of our mutations, I262A, is not lethal for the virus, suggesting that alteration in E1E2 interactions does not necessarily abolish viral infectivity. However, for this particular mutant, E1E2 interaction was not totally abolished, and the remaining infectivity might be explained by this residual interaction. If I262 is close to the D263 and W239 residues, its side chain does not point in the same direction. Indeed, the side chains of D263 and W239 are exposed to the E1 surface, directly available to potentially interact with another protein, whereas the side chain of I262 is buried in the E1 structure and makes hydrophobic contacts with the α-helix (Fig. 11). Thus, the side chain of I262 is unlikely to be directly involved in the interaction with E2, and this may explain the peculiar phenotype observed for the I262 mutant.
Our data indicate that mutations in E1 can affect the tropism of HCV for CLDN1. Indeed, the T213A and I262A mutants preferentially use CLDN6 instead of CLDN1 in Huh-7 cells. A similar shift in receptor dependence has recently been reported for another E1 mutant that has been selected by long-term culturing and passage of the HCV Jc1 isolate through CLDN1 knockout (KO) Huh-7.5 cells (37). Together, these data point to a role for E1 in the HCV cell entry process as a modulator of entry factor usage. It has been previously reported that many HCV isolates can naturally use both CDLN1 and CLDN6 for host cell entry (21, 38–40), so a change in receptor dependence from CLDN1 to CLDN6 is not entirely surprising. However, it is not clearly known whether E1 interacts directly with CLDN1. It has been reported that mutations in E1 that affect the infectivity of pseudoparticles bearing HCV glycoproteins can modulate the binding of these particles to CLDN1-expressing cells, suggesting a role for E1 in HCV glycoprotein interaction with CLDN1 (35). However, one cannot exclude the possibility that these E1 mutations may be functioning indirectly by influencing how E2 interacts with CLDN1. We have indeed already observed that mutations in E1 can affect E2-CD81 interaction, indicating that E1 plays a role in modulating the receptor binding capacity of E2 (14). The most surprising observation is that mutations located in different regions of the E1 primary sequence can affect the dependence on CLDN1. Indeed, the mutations identified in our work are at positions 213 and 262, which belong to the α-helix and the β5 strand, respectively (Fig. 1), whereas the mutation identified by Hopcraft and Evans is located at position 316 (37), which is located in the epitope of neutralizing MAb IGH526 (19). However, T213 in the α-helix and I262 in the β5 strand are proximate in space in the E1 structure. Moreover, I262 establishes hydrophobic interactions with the α-helix that contribute to the folding back of this helix on the β-sheet of E1 (Fig. 11). Interestingly, our biochemical and neutralization data indicate that both the T213A and I262A mutations affect E1 recognition by IGH526, suggesting that amino acid positions 213, 262, and 316 might be in close proximity in the three-dimensional (3D) structure of E1. Whether the alteration in CLDN1 dependence is clinically relevant remains to be determined.
The most surprising observation of our study is the production of HCV particles devoid of genomic RNA in the case of the D263A mutation. The D263 amino acid is highly conserved. Indeed, an aspartate residue is present at this position in 18 out of 19 reference sequences from all confirmed genotypes and subtypes (Fig. 1B) as well as in 96% of the full E1 sequences from the European HCV Database (https://euhcvdb.ibcp.fr/euHCVdb/) (41). It has been shown that, in a cell-free assay, HCV core proteins produced in bacteria self-assemble into nucleocapsids (42). However, in this case viral or nonviral RNA molecules were associated with the particles. Intracellular assembly of HCV capsids has also been described in the past (43). However, these particles were not secreted, and they were shown to form abortive budding events. In our case, we detected secretion of viral particles that contain at least the core protein but no genomic RNA. Due to the low production of particles and the fact that we cannot amplify them by reinfection, we could not determine whether HCV envelope glycoproteins are associated with these particles. We presume that they should be present in the envelope. E1 has been shown to interact with the capsid protein in the context of a heterologous expression system, at least after oligomerization of the capsid protein (44). Together with our data, this suggests that E1 plays a major role in HCV particle assembly. In the context of our D263A mutant, one can speculate that, due to loss of interaction with E2, E1 might be able to directly interact with the capsid protein in the absence of interaction with the genomic RNA. Interestingly, we also observed a decrease in subcellular colocalization between HCV RNA and E1 in the D263A mutant, which is in line with the hypothesis that, through its interaction with core protein, E1 might play a role in recruiting the genomic RNA. As already mentioned above, in the E1 structure, the D263 residue that is located in the β-sheet has its side chain exposed at the molecular surface (Fig. 11). Interestingly, the D263 carboxylic function of its side chain is at the center of a hydrogen bond network, which comprises the R237, W239, and H261 side chains (Fig. 11B). This polar interaction network might be involved in the stabilization of D263, R237, W239, and H261 side chains toward an E1 conformation suitable to interact with an essential biological partner.
To conclude, our mutagenesis study highlights cross talk between E1 and E2 during HCV morphogenesis. Our data also indicate the role of E1 in modulating functional interactions between E1E2 complex and CLDN1. Finally, this study describes for the first time cross talk between E1 and the genomic RNA during HCV morphogenesis.
MATERIALS AND METHODS
Cell culture.
Huh-7 hepatoma cells (45) were grown in Dulbecco's modified essential medium (DMEM; ThermoFisher) supplemented with GlutaMAX, 10% fetal calf serum, and nonessential amino acids (NEAA).
Antibodies.
Anti-HCV monoclonal antibodies (MAbs) A4 (anti-E1) (46) and 3/11 (anti-E2; kindly provided by J. A. McKeating, University of Birmingham, Birmingham, United Kingdom) (47) were produced in vitro by using a MiniPerm apparatus (Heraeus) as recommended by the manufacturer. Anti-E1E2 MAb AR5A (18) and anti-E1 MAb IGH526 (19) were kindly provided by M. Law (Scripps Research Institute, La Jolla, CA, USA). The anti-NS5A MAb 9E10 (48) and a polyclonal antibody (49) were a gift from C. M. Rice (Rockefeller University, New York, NY) and M. Harris (University of Leeds, United Kingdom), respectively. Anti-core MAb ACAP-27 (50) was kindly provided by J. F. Delagneau (Bio-Rad, France). Anti-SR-BI MAb C167 was a gift from A. Nicosia (Okairos, Rome, Italy) (51). Anti-CLDN1 MAb OM8A9-A3 has been previously described (52). Commercially available anti-CD81 MAb JS81 (BD Pharmingen), anti-SR-BI polyclonal antibody (Abcam), anti-SR-BI Mab Cla-I (BD Biosciences), and anti-CLDN6 MAb clone 342927 (R&D Systems) were used in this work. Secondary antibodies used for immunofluorescence were purchased from Jackson ImmunoResearch. Control anti-tubulin antibody was from Sigma.
Structural model of E1.
The structural model of the N-terminal region of the E1 ectodomain was constructed using the JFH1 amino acid sequence and the crystallographic structure of E1 from the H77 strain (Protein Data Bank [PDB] accession number 4UOI, chain F) as the template with the Swiss-Model server (53).
Mutagenesis and virus production.
The virus used in this study is a modified version of the JFH1 isolate (genotype 2a; GenBank accession number AB237837) (54), kindly provided by T. Wakita (National Institute of Infectious Diseases, Tokyo, Japan) (17). Mutants were generated by site-directed mutagenesis. Selected residues were replaced by alanines. The restriction enzyme XbaI was used to linearize plasmids encoding viral RNAs. The linearized plasmids were then treated with mung bean nuclease (New England BioLabs) with the aim of obtaining blunt-ended DNA. For in vitro transcription, 2 μg of linearized DNA was transcribed using a MEGAscript kit according to the manufacturer's protocol (Ambion). The in vitro transcription reaction mixture was set up and incubated at 37°C for 4 h, and transcripts were precipitated by the addition of equal volumes of LiCl and nuclease-free water. The mixture was chilled at −20°C for 30 min and then centrifuged at 4°C for 15 min at 14,000 × g. The supernatants were then removed, and the RNA pellets were washed with 70% ethanol and resuspended in RNase-free water. Infectivity analyses were performed as previously described (14). Briefly, supernatants containing extracellular virus were removed at different times after electroporation, and cell debris was removed by centrifugation for 5 min at 10,000 × g. Infected cells were washed with phosphate-buffered saline (PBS) and harvested by treatment with trypsin, and intracellular viral particles were obtained after four freeze-thaw cycles. Cell lysates were clarified by centrifugation at 10,000 × g for 7 min. Clarified supernatants containing extracellular virus and intracellular virus were used for infection of naive Huh-7 cells. Infected cells were then fixed with ice-cold methanol (100%) and immunostained with anti-E1 or anti-NS5A antibodies. The nonreplicative control HCV genome contained a GND mutation (GND HCV) in the NS5B active site, as previously reported (55). The assembly-deficient HCV control (ΔE1E2) containing an in-frame deletion introduced into the E1E2 regions has been previously described (54).
Immunofluorescence.
Immunofluorescence analyses were performed as previously described (24). Briefly, Huh-7 cells electroporated with 10 μg of wild-type or mutant RNA were grown on 12-mm coverslips or in 96-well plates. After 48 h, the cells were washed twice with PBS and then fixed with cold methanol (100%) for 5 min. The methanol was removed by washing the cells twice with PBS. The cells were then blocked with 10% goat or horse serum for at least 10 min, followed by washing with PBS. The primary anti-E1, anti-E2, and anti-NS5A antibodies were diluted in 10% goat serum/horse serum, and the coverslips were incubated with antibodies at room temperature for 25 min. The cells were then washed three times in PBS. The secondary antibody was diluted in goat serum/horse serum (1/500), and coverslips were incubated with a Cy3-conjugated antibody for 20 min. The cells were washed again with PBS. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). The coverslips were mounted on glass slides using 7 μl of mounting medium (Mowiol 4-88; Calbiochem). Confocal microscopy was performed with an LSM 880 confocal laser scanning microscope (Zeiss) using a ×63 (1.4 numerical aperture) oil immersion lens. Double-label immunofluorescence signals were sequentially collected by using single fluorescence excitation and acquisition settings to avoid crossover. Images were processed by using ImageJ software.
Equilibrium density gradient analysis.
Equilibrium density gradient analyses were performed as previously described (14) after concentration of the viral preparation by polyethylene glycol (PEG) precipitation as described previously (56). Briefly, viruses were harvested at 48 h following electroporation. Approximately 80 ml of virus supernatants was precipitated using PEG 6000 to a final concentration of 8%. The mixture was shaken for 1 h on ice, centrifuged at 8,000 rpm (Beckman JLA-10.5 rotor) for 25 min, and then resuspended in 1 ml of sterile PBS. The virus was then loaded on a 10 to 50% iodixanol gradient. The gradients were spun for 16 h at 36,000 rpm in an SW41 rotor (Beckman) and fractionated from the top.
Core protein oligomerization.
Huh-7 cells electroporated with mutant and wild-type RNA genomes were lysed at 48 h postelectroporation in lysis buffer (PBS–0.3% NP-40 and a protease inhibitor cocktail [Roche]) for 15 min at room temperature. Cell lysates were precleared by centrifugation at 14,000 rpm for 5 min at 4°C. Each sample was layered on top of an 11-ml, 10 to 50% iodixanol gradient and centrifuged in a Beckman SW41 Ti rotor (Beckman) at 36,000 rpm for 16 h at 4°C. Fractions of 1 ml were collected from the top of each tube and analyzed by SDS-PAGE and immunoblotting.
HCV core protein quantification.
HCV core protein was quantified by a fully automated chemiluminescent microparticle immunoassay according to the manufacturer's instructions (Architect HCVAg; Abbott, Germany) as previously described (57, 58).
Western blotting.
Western blotting experiments were performed as previously described (13). Cells were lysed in PBS lysis buffer (1% Triton X-100, 20 mM N-ethylmaleimide [NEM], 2 mM EDTA, protease inhibitor cocktail [Roche]). Cell lysates were then precleared by centrifugation at 14,000 × g for 5 min at 4°C. Protein samples were heated for 7 min at 70°C in Laemmli sample buffer. Following separation by SDS-PAGE, the proteins were transferred onto nitrocellulose membranes (Hybond-ECL; Amersham) and detected with specific antibodies. Following incubation with primary antibodies, the membranes were incubated with the corresponding peroxidase-conjugated anti-rat (Jackson), anti-rabbit (Amersham), anti-sheep (Amersham), and anti-mouse (Dako) antibodies. The proteins were then detected by enhanced chemiluminescence (ECL) (Amersham) as recommended by the manufacturer.
CD81 interaction and immunoprecipitation assays.
CD81 pulldown and immunoprecipitation experiments were performed as previously described (59). Cells were lysed in PBS lysis buffer (1% Triton X-100, 20 mM NEM, 2 mM EDTA, protease inhibitor cocktail [Roche]). Cell lysates were then cleared by centrifugation at 14,000 × g for 15 min at 4°C. For CD81 pulldown, glutathione-Sepharose beads (glutathione-Sepharose 4B; Amersham Bioscience) were washed twice with cold PBS to remove the storage buffer. For each cell lysate sample, 50 μl of glutathione beads was incubated with 10 μg of human CD81 (hCD81) large extracellular loop (LEL) glutathione S-transferase (GST) recombinant protein in 1 ml of cold PBS containing 1% Triton X-100 for 2 h at 4°C. Following incubation, the glutathione-Sepharose beads were washed with cold PBS. Cell lysate samples containing E1E2 proteins were then incubated with CD81-LEL complexed with glutathione beads overnight at 4°C. The following day, the beads were washed five times with cold PBS and 1% Triton X-100. Finally, the beads were resuspended in 30 μl of Laemmli buffer. Samples were heated at 70°C and loaded onto 10% SDS-PAGE gels, followed by Western blotting to reveal the proteins of interest. For immunoprecipitation, 70 μl of protein A-Sepharose beads was incubated with 10 μg of rabbit anti-human IgG (Dako) in 1 ml of cold PBS and 1% Triton X-100 for 2 h at 4°C. In parallel, 100 μl of cell lysates was incubated with 2 μg of MAb AR5A (anti-E1E2) or MAb IGH526 (anti-E1) in 400 μl of cold PBS and 1% Triton X-100 for 2 h at 4°C. Next, protein A-Sepharose beads were washed twice with cold PBS and 1% Triton X-100 and added to cell lysates. The mixture was then incubated for 90 min at 4°C. After incubation, the beads were washed five times with cold PBS and 1% Triton X-100. Finally, the beads were resuspended in 30 μl of Laemmli buffer. The presence of HCV envelope glycoproteins was then detected by Western blotting.
Entry inhibition assays and neutralization assays.
Viruses or cells were preincubated with human CD81-LEL, MAb AR5A, MAb IGH526, or antireceptor antibody for 2 h at 37°C. The viruses were then put in contact with Huh-7 cells. At 6 h postinfection, the inoculum was removed, and the cells were further incubated for 72 h with complete medium. The cells were then processed for immunofluorescence to measure residual infectivity.
FISH and colocalization with viral proteins.
In situ hybridization was performed as previously described (60). Briefly, cells were washed once with PBS and fixed with 500 μl of 4% paraformaldehyde for 20 min at room temperature, followed by three washes with PBS. Fixed cells were processed for FISH analysis, using a QuantiGene ViewRNA in situ hybridization cell assay (Affymetrix) as recommended by the manufacturer.
Graphs and statistics.
Prism, version 5.0c (GraphPad Software, Inc., La Jolla, CA), software was used to prepare graphs and to determine statistical significance of differences between data sets using a Mann-Whitney test.
ACKNOWLEDGMENTS
We thank J. F. Delagneau, M. Harris, M. Law, J. McKeating, A. Nicosia, C. Rice, and T. Wakita for providing essential reagents. We also thank Sophana Ung for his help in preparing the figures. The immunofluorescence analyses were performed with the help of the imaging core facility of the BioImaging Center Lille Nord-de-France.
This work was supported by the French National Agency for Research on AIDS and Viral Hepatitis (ANRS) and the ANR through the ERA-NET Infect-ERA program (ANR-13-IFEC-0002-01). J.G.H. was successively supported by fellowships from the Lebanese Development Association and the ANRS.
J.G.H., Y.R., M.H., F.D., T.F.B., G.D., M.L., and J.D. conceived and designed the experiments; J.G.H., Y.R., V.D., and M.L. performed the experiments; T.F.B. provided reagents and edited the manuscript; J.G.H., Y.R., X.H., M.L., and J.D. analyzed the data; J.G.H., M.L., and J.D. wrote the paper.
REFERENCES
- 1.Mohd Hanafiah K, Groeger J, Flaxman AD, Wiersma ST. 2013. Global epidemiology of hepatitis C virus infection: new estimates of age-specific antibody to HCV seroprevalence. Hepatology 57:1333–1342. doi: 10.1002/hep.26141. [DOI] [PubMed] [Google Scholar]
- 2.Webster DP, Klenerman P, Dusheiko GM. 2015. Hepatitis C. Lancet 385:1124–1135. doi: 10.1016/S0140-6736(14)62401-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Popescu CI, Riva L, Vlaicu O, Farhat R, Rouillé Y, Dubuisson J. 2014. Hepatitis C virus life cycle and lipid metabolism. Biology 3:892–921. doi: 10.3390/biology3040892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scarselli E, Ansuini H, Cerino R, Roccasecca RM, Acali S, Filocamo G, Traboni C, Nicosia A, Cortese R, Vitelli A. 2002. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J 21:5017–5025. doi: 10.1093/emboj/cdf529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pileri P, Uematsu Y, Campagnoli S, Galli G, Falugi F, Petracca R, Weiner AJ, Houghton M, Rosa D, Grandi G, Abrignani S. 1998. Binding of hepatitis C virus to CD81. Science 282:938–941. doi: 10.1126/science.282.5390.938. [DOI] [PubMed] [Google Scholar]
- 6.Evans MJ, von Hahn T, Tscherne DM, Syder AJ, Panis M, Wölk B, Hatziioannou T, McKeating JA, Bieniasz PD, Rice CM. 2007. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 446:801–805. doi: 10.1038/nature05654. [DOI] [PubMed] [Google Scholar]
- 7.Ploss A, Evans MJ, Gaysinskaya VA, Panis M, You H, de Jong YP, Rice CM. 2009. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 457:882–886. doi: 10.1038/nature07684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lavie M, Penin F, Dubuisson J. 2015. HCV envelope glycoproteins in virion assembly and entry. Future Virol 10:297–312. doi: 10.2217/fvl.14.114. [DOI] [Google Scholar]
- 9.Khan AG, Whidby J, Miller MT, Scarborough H, Zatorski AV, Cygan A, Price AA, Yost SA, Bohannon CD, Jacob J, Grakoui A, Marcotrigiano J. 2014. Structure of the core ectodomain of the hepatitis C virus envelope glycoprotein 2. Nature 509:381–384. doi: 10.1038/nature13117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kong L, Giang E, Nieusma T, Kadam RU, Cogburn KE, Hua Y, Dai X, Stanfield RL, Burton DR, Ward AB, Wilson IA, Law M. 2013. Hepatitis C virus E2 envelope glycoprotein core structure. Science 342:1090–1094. doi: 10.1126/science.1243876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perin PM, Haid S, Brown RJP, Doerrbecker J, Schulze K, Zeilinger C, von Schaewen M, Heller B, Vercauteren K, Luxenburger E, Baktash YM, Vondran FW, Speerstra S, Awadh A, Mukhtarov F, Schang LM, Kirschning A, Müller R, Guzman CA, Kaderali L, Randall G, Meuleman P, Ploss A, Pietschmann T. 2016. Flunarizine prevents hepatitis C virus membrane fusion in a genotype-dependent manner by targeting the potential fusion peptide within E1. Hepatology 63:49–62. doi: 10.1002/hep.28111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vausselin T, Calland N, Belouzard S, Descamps V, Douam F, Helle F, François C, Lavillette D, Duverlie G, Wahid A, Fénéant L, Cocquerel L, Guérardel Y, Wychowski C, Biot C, Dubuisson J. 2013. The antimalarial ferroquine is an inhibitor of hepatitis C virus. Hepatology 58:86–97. doi: 10.1002/hep.26273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vausselin T, Séron K, Lavie M, Mesalam AA, Lemasson M, Belouzard S, Fénéant L, Danneels A, Rouillé Y, Cocquerel L, Foquet L, Rosenberg AR, Wychowski C, Meuleman P, Melnyk P, Dubuisson J. 2016. Identification of a new benzimidazole derivative as an antiviral against hepatitis C virus. J Virol 90:8422–8434. doi: 10.1128/JVI.00404-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wahid A, Helle F, Descamps V, Duverlie G, Penin F, Dubuisson J. 2013. Disulfide bonds in hepatitis C virus glycoprotein E1 control the assembly and entry functions of E2 glycoprotein. J Virol 87:1605–1617. doi: 10.1128/JVI.02659-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.El Omari K, Iourin O, Kadlec J, Sutton G, Harlos K, Grimes JM, Stuart DI. 2014. Unexpected structure for the N-terminal domain of hepatitis C virus envelope glycoprotein E1. Nat Commun 5:4874. doi: 10.1038/ncomms5874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Helle F, Vieyres G, Elkrief L, Popescu CI, Wychowski C, Descamps V, Castelain S, Roingeard P, Duverlie G, Dubuisson J. 2010. Role of N-linked glycans in the functions of hepatitis C virus envelope proteins incorporated into infectious virions. J Virol 84:11905–11915. doi: 10.1128/JVI.01548-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goueslain L, Alsaleh K, Horellou P, Roingeard P, Descamps V, Duverlie G, Ciczora Y, Wychowski C, Dubuisson J, Rouille Y. 2010. Identification of GBF1 as a cellular factor required for hepatitis C virus RNA replication. J Virol 84:773–787. doi: 10.1128/JVI.01190-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Giang E, Dorner M, Prentoe JC, Dreux M, Evans MJ, Bukh J, Rice CM, Ploss A, Burton DR, Law M. 2012. Human broadly neutralizing antibodies to the envelope glycoprotein complex of hepatitis C virus. Proc Natl Acad Sci U S A 109:6205–6210. doi: 10.1073/pnas.1114927109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kong L, Kadam RU, Giang E, Ruwona TB, Nieusma T, Culhane JC, Stanfield RL, Dawson PE, Wilson IA, Law M. 2015. Structure of hepatitis C virus envelope glycoprotein E1 antigenic site 314–324 in complex with antibody IGH526. J Mol Biol 427:2617–2628. doi: 10.1016/j.jmb.2015.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Falson P, Bartosch B, Alsaleh K, Tews BA, Loquet A, Ciczora Y, Riva L, Montigny C, Montpellier C, Duverlie G, Pécheur E-I, le Maire M, Cosset FL, Dubuisson J, Penin F. 2015. Hepatitis C virus envelope glycoprotein E1 forms trimers at the surface of the virion. J Virol 89:10333–10346. doi: 10.1128/JVI.00991-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haid S, Grethe C, Dill MT, Heim M, Kaderali L, Pietschmann T. 2014. Isolate-dependent use of claudins for cell entry by hepatitis C virus. Hepatology 59:24–34. doi: 10.1002/hep.26567. [DOI] [PubMed] [Google Scholar]
- 22.Shirasago Y, Shimizu Y, Tanida I, Suzuki T, Suzuki R, Sugiyama K, Wakita T, Hanada K, Yagi K, Kondoh M, Fukasawa M. 2016. Occludin-knockout human hepatic Huh7.5.1-8-derived cells are completely resistant to hepatitis C virus infection. Biol Pharm Bull 39:839–848. doi: 10.1248/bpb.b15-01023. [DOI] [PubMed] [Google Scholar]
- 23.Ai LS, Lee YW, Chen SS. 2009. Characterization of hepatitis C virus core protein multimerization and membrane envelopment: revelation of a cascade of core-membrane interactions. J Virol 83:9923–9939. doi: 10.1128/JVI.00066-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alsaleh K, Delavalle PY, Pillez A, Duverlie G, Descamps V, Rouille Y, Dubuisson J, Wychowski C. 2010. Identification of basic amino acids at the N-terminal end of the core protein that are crucial for hepatitis C virus infectivity. J Virol 84:12515–12528. doi: 10.1128/JVI.01393-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miyanari Y, Atsuzawa K, Usuda N, Watashi K, Hishiki T, Zayas M, Bartenschlager R, Wakita T, Hijikata M, Shimotohno K. 2007. The lipid droplet is an important organelle for hepatitis C virus production. Nat Cell Biol 9:1089–1097. doi: 10.1038/ncb1631. [DOI] [PubMed] [Google Scholar]
- 26.Li Y, Modis Y. 2014. A novel membrane fusion protein family in Flaviviridae? Trends Microbiol 22:176–182. doi: 10.1016/j.tim.2014.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cocquerel L, Voisset C, Dubuisson J. 2006. Hepatitis C virus entry: potential receptors and their biological functions. J Gen Virol 87:1075–1084. doi: 10.1099/vir.0.81646-0. [DOI] [PubMed] [Google Scholar]
- 28.Deleersnyder V, Pillez A, Wychowski C, Blight K, Xu J, Hahn YS, Rice CM, Dubuisson J. 1997. Formation of native hepatitis C virus glycoprotein complexes. J Virol 71:697–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lavie M, Goffard A, Dubuisson J. 2007. Assembly of a functional HCV glycoprotein heterodimer. Curr Issues Mol Biol 9:71–86. [PubMed] [Google Scholar]
- 30.Michalak JP, Wychowski C, Choukhi A, Meunier JC, Ung S, Rice CM, Dubuisson J. 1997. Characterization of truncated forms of hepatitis C virus glycoproteins. J Gen Virol 78:2299–2306. doi: 10.1099/0022-1317-78-9-2299. [DOI] [PubMed] [Google Scholar]
- 31.Patel J, Patel AH, McLauchlan J. 2001. The transmembrane domain of the hepatitis C virus E2 glycoprotein is required for correct folding of the E1 glycoprotein and native complex formation. Virology 279:58–68. doi: 10.1006/viro.2000.0693. [DOI] [PubMed] [Google Scholar]
- 32.Brazzoli M, Helenius A, Foung SK, Houghton M, Abrignani S, Merola M. 2005. Folding and dimerization of hepatitis C virus E1 and E2 glycoproteins in stably transfected CHO cells. Virology 332:438–453. doi: 10.1016/j.virol.2004.11.034. [DOI] [PubMed] [Google Scholar]
- 33.Cocquerel L, Quinn ER, Flint M, Hadlock KG, Foung SK, Levy S. 2003. Recognition of native hepatitis C virus E1E2 heterodimers by a human monoclonal antibody. J Virol 77:1604–1609. doi: 10.1128/JVI.77.2.1604-1609.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Albecka A, Montserret R, Krey T, Tarr AW, Diesis E, Ball JK, Descamps V, Duverlie G, Rey F, Penin F, Dubuisson J. 2011. Identification of new functional regions in hepatitis C virus envelope glycoprotein E2. J Virol 85:1777–1792. doi: 10.1128/JVI.02170-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Douam F, Dao Thi VL, Maurin G, Fresquet J, Mompelat D, Zeisel MB, Baumert TF, Cosset FL, Lavillette D. 2014. Critical interaction between E1 and E2 glycoproteins determines binding and fusion properties of hepatitis C virus during cell entry. Hepatology 59:776–788. doi: 10.1002/hep.26733. [DOI] [PubMed] [Google Scholar]
- 36.Op De Beeck A, Montserret R, Duvet S, Cocquerel L, Cacan R, Barberot B, Le Maire M, Penin F, Dubuisson J. 2000. The transmembrane domains of hepatitis C virus envelope glycoproteins E1 and E2 play a major role in heterodimerization. J Biol Chem 275:31428–31437. doi: 10.1074/jbc.M003003200. [DOI] [PubMed] [Google Scholar]
- 37.Hopcraft SE, Evans MJ. 2015. Selection of a hepatitis C virus with altered entry factor requirements reveals a genetic interaction between the E1 glycoprotein and claudins. Hepatology 62:1059–1069. doi: 10.1002/hep.27815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zheng A, Yuan F, Li Y, Zhu F, Hou P, Li J, Song X, Ding M, Deng H. 2007. Claudin-6 and claudin-9 function as additional coreceptors for hepatitis C virus. J Virol 81:12465–12471. doi: 10.1128/JVI.01457-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meertens L, Bertaux C, Cukierman L, Cormier E, Lavillette D, Cosset FL, Dragic T. 2008. The tight junction proteins claudin-1, -6, and -9 are entry cofactors for hepatitis C virus. J Virol 82:3555–3560. doi: 10.1128/JVI.01977-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fofana I, Zona L, Thumann C, Heydmann L, Durand SC, Lupberger J, Blum HE, Pessaux P, Gondeau C, Reynolds GM, McKeating JA, Grunert F, Thompson J, Zeisel MB, Baumert TF. 2013. Functional analysis of claudin-6 and claudin-9 as entry factors for hepatitis C virus infection of human hepatocytes by using monoclonal antibodies. J Virol 87:10405–10410. doi: 10.1128/JVI.01691-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Combet C, Garnier N, Charavay C, Grando D, Crisan D, Lopez J, Dehne-Garcia A, Geourjon C, Bettler E, Hulo C, Le Mercier P, Bartenschlager R, Diepolder H, Moradpour D, Pawlotsky JM, Rice CM, Trépo C, Penin F, Deléage G. 2007. euHCVdb: the European hepatitis C virus database. Nucleic Acids Res 35:D363–D366. doi: 10.1093/nar/gkl970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kunkel M, Lorinczi M, Rijnbrand R, Lemon SM, Watowich SJ. 2001. Self-assembly of nucleocapsid-like particles from recombinant hepatitis C virus core protein. J Virol 75:2119–2129. doi: 10.1128/JVI.75.5.2119-2129.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Blanchard E, Brand D, Trassard S, Goudeau A, Roingeard P. 2002. Hepatitis C virus-like particle morphogenesis. J Virol 76:4073–4079. doi: 10.1128/JVI.76.8.4073-4079.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nakai K, Okamoto T, Kimura-Someya T, Ishii K, Lim CK, Tani H, Matsuo E, Abe T, Mori Y, Suzuki T, Miyamura T, Nunberg JH, Moriishi K, Matsuura Y. 2006. Oligomerization of hepatitis C virus core protein is crucial for interaction with the cytoplasmic domain of E1 envelope protein. J Virol 80:11265–11273. doi: 10.1128/JVI.01203-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nakabayashi H, Taketa K, Miyano K, Yamane T, Sato J. 1982. Growth of human hepatoma cells lines with differentiated functions in chemically defined medium. Cancer Res 42:3858–3863. [PubMed] [Google Scholar]
- 46.Dubuisson J, Hsu HH, Cheung RC, Greenberg HB, Russell DG, Rice CM. 1994. Formation and intracellular localization of hepatitis C virus envelope glycoprotein complexes expressed by recombinant vaccinia and Sindbis viruses. J Virol 68:6147–6160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Flint M, Maidens C, Loomis-Price LD, Shotton C, Dubuisson J, Monk P, Higginbottom A, Levy S, McKeating JA. 1999. Characterization of hepatitis C virus E2 glycoprotein interaction with a putative cellular receptor, CD81. J Virol 73:6235–6244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lindenbach BD, Evans MJ, Syder AJ, Wölk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, Rice CM. 2005. Complete replication of hepatitis C virus in cell culture. Science 309:623–626. doi: 10.1126/science.1114016. [DOI] [PubMed] [Google Scholar]
- 49.Macdonald A, Crowder K, Street A, McCormick C, Saksela K, Harris M. 2003. The hepatitis C virus non-structural NS5A protein inhibits activating protein-1 function by perturbing Ras-ERK pathway signaling. J Biol Chem 278:17775–17784. doi: 10.1074/jbc.M210900200. [DOI] [PubMed] [Google Scholar]
- 50.Maillard P, Krawczynski K, Nitkiewicz J, Bronnert C, Sidorkiewicz M, Gounon P, Dubuisson J, Faure G, Crainic R, Budkowska A. 2001. Nonenveloped nucleocapsids of hepatitis C virus in the serum of infected patients. J Virol 75:8240–8250. doi: 10.1128/JVI.75.17.8240-8250.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Catanese MT, Loureiro J, Jones CT, Dorner M, von Hahn T, Rice CM. 2013. Different requirements for scavenger receptor class B type I in hepatitis C virus cell-free versus cell-to-cell transmission. J Virol 87:8282–8293. doi: 10.1128/JVI.01102-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fofana I, Krieger SE, Grunert F, Glauben S, Xiao F, Kremer SF, Soulier E, Royer C, Thumann C, Mee CJ, McKeating JA, Dragic T, Pessaux P, Stoll-Keller F, Schuster C, Thompson J, Baumert TF. 2010. Monoclonal anti-claudin 1 antibodies prevent hepatitis C virus infection of primary human hepatocytes. Gastroenterology 139:953–964, 964.e1–4. doi: 10.1053/j.gastro.2010.05.073. [DOI] [PubMed] [Google Scholar]
- 53.Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, Kiefer F, Gallo Cassarino T, Bertoni M, Bordoli L, Schwede T. 2014. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res 42:W252–W258. doi: 10.1093/nar/gku340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Kräusslich H-G, Mizokami M, Bartenschlager R, Liang TJ. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med 11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Delgrange D, Pillez A, Castelain S, Cocquerel L, Rouille Y, Dubuisson J, Wakita T, Duverlie G, Wychowski C. 2007. Robust production of infectious viral particles in Huh-7 cells by introducing mutations in hepatitis C virus structural proteins. J Gen Virol 88:2495–2503. doi: 10.1099/vir.0.82872-0. [DOI] [PubMed] [Google Scholar]
- 56.Calland N, Albecka A, Belouzard S, Wychowski C, Duverlie G, Descamps V, Hober D, Dubuisson J, Rouillé Y, Séron K. 2012. (−)-Epigallocatechin-3-gallate is a new inhibitor of hepatitis C virus entry. Hepatology 55:720–729. doi: 10.1002/hep.24803. [DOI] [PubMed] [Google Scholar]
- 57.Mederacke I, Wedemeyer H, Ciesek S, Steinmann E, Raupach R, Wursthorn K, Manns MP, Tillmann HL. 2009. Performance and clinical utility of a novel fully automated quantitative HCV-core antigen assay. J Clin Virol 46:210–215. doi: 10.1016/j.jcv.2009.08.014. [DOI] [PubMed] [Google Scholar]
- 58.Morota K, Fujinami R, Kinukawa H, Machida T, Ohno K, Saegusa H, Takeda K. 2009. A new sensitive and automated chemiluminescent microparticle immunoassay for quantitative determination of hepatitis C virus core antigen. J Virol Methods 157:8–14. doi: 10.1016/j.jviromet.2008.12.009. [DOI] [PubMed] [Google Scholar]
- 59.Lavie M, Sarrazin S, Montserret R, Descamps V, Baumert TF, Duverlie G, Séron K, Penin F, Dubuisson J. 2014. Identification of conserved residues in hepatitis C virus envelope glycoprotein E2 that modulate virus dependence on CD81 and SRB1 entry factors. J Virol 88:10584–10597. doi: 10.1128/JVI.01402-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Poenisch M, Metz P, Blankenburg H, Ruggieri A, Lee JY, Rupp D, Rebhan I, Diederich K, Kaderali L, Domingues FS, Albrecht M, Lohmann V, Erfle H, Bartenschlager R. 2015. Identification of HNRNPK as regulator of hepatitis C virus particle production. PLoS Pathog 11:e1004573. doi: 10.1371/journal.ppat.1004573. [DOI] [PMC free article] [PubMed] [Google Scholar]