

ABSTRACT
The human protein DDX3X is a DEAD box ATP-dependent RNA helicase that regulates transcription, mRNA maturation, and mRNA export and translation. DDX3X concomitantly modulates the replication of several RNA viruses and promotes innate immunity. We previously showed that herpes simplex virus 1 (HSV-1), a human DNA virus, incorporates DDX3X into its mature particles and that DDX3X is required for optimal HSV-1 infectivity. Here, we show that viral gene expression, replication, and propagation depend on optimal DDX3X protein levels. Surprisingly, DDX3X from incoming viral particles was not required for the early stages of the HSV-1 infection, but, rather, the protein controlled the assembly of new viral particles. This was independent of the previously reported ability of DDX3X to stimulate interferon type I production. Instead, both the lack and overexpression of DDX3X disturbed viral gene transcription and thus subsequent genome replication. This suggests that in addition to its effect on RNA viruses, DDX3X impacts DNA viruses such as HSV-1 by an interferon-independent pathway.
IMPORTANCE Viruses interact with a variety of cellular proteins to complete their life cycle. Among them is DDX3X, an RNA helicase that participates in most aspects of RNA biology, including transcription, splicing, nuclear export, and translation. Several RNA viruses and a limited number of DNA viruses are known to manipulate DDX3X for their own benefit. In contrast, DDX3X is also known to promote interferon production to limit viral propagation. Here, we show that DDX3X, which we previously identified in mature HSV-1 virions, stimulates HSV-1 gene expression and, consequently, virion assembly by a process that is independent of its ability to promote the interferon pathway.
KEYWORDS: DDX3X, helicase, herpes, host-pathogen interaction, DNA virus, RNA virus, interferon, herpes simplex virus, host-pathogen interactions, transcriptional regulation, translational control
INTRODUCTION
The human DDX3 protein is a member of a large family of DEAD box ATP-dependent RNA helicases. In humans, it is encoded by the X (DDX3X) and Y (DDX3Y) chromosomes, albeit the latter is restricted to testes (1). It participates in different stages of cellular gene expression, such as transcription, mRNA maturation, and mRNA export and translation (2). Given these crucial roles in RNA biology, several RNA viruses interact with DDX3X, often with important consequences for viral replication. This includes hepatitis C virus (HCV), norovirus, West Nile virus, and Japanese encephalitis virus (3–7). This is also the case for HIV and hepatitis B virus (HBV), a peculiar DNA virus that relies on an RNA template and reversed transcription to replicate its genome (8, 9). Furthermore, DDX3X also contributes to innate immunity against these viruses. For instance, DDX3X stimulates interferon (IFN) type I production by binding IKKε (IκB kinase epsilon) and TBK1 (tank-binding kinase 1), leading to IRF3 phosphorylation and activation (10, 11). These findings position DDX3X as a critical player for the replication and immunity against RNA-based viruses. However, its role is not restricted to these viruses since it also impacts innate immunity against DNA viruses. For instance, the HBV viral polymerase binds IKKε and blocks its ability to interact with DDX3X, thereby hampering interferon production to the benefit of the virus (12). Similarly, the vaccinia virus (VACV) K7 protein binds and sequesters DDX3X and prevents its interaction with the above kinases, once again blocking interferon activation in favor of viral propagation (13). DDX3X is therefore an important mediator of host-pathogen interactions that acts via multiple and likely parallel routes on many RNA and at least two distinct DNA viruses.
Herpes simplex virus 1 (HSV-1) is a ubiquitous human pathogen that is dormant in 80 to 90% of the population but causes clinical symptoms in roughly a third of humans. It is primarily associated with cold sores but is also responsible for severe infections in both immunocompetent and immunodeficient individuals (14). Replication of herpesviruses occurs in the cell nucleus, where the virus takes over the host gene expression machinery during an active infection. Three classes of HSV-1 genes, namely, the immediate early (IE), early (E), and late (L) genes, are expressed in a sequential manner by the cellular RNA polymerase II and a collection of transcriptional and translational cellular factors (15). This expression cascade is stimulated by ICP0, ICP4, and VP16, three tegument components present in mature viral particles that promptly act as transactivators following viral entry in the cell (16–20). As for other viruses, HSV-1 gene expression is clearly dependent on both viral and host proteins.
Past studies from our laboratory revealed that HSV-1 incorporates DDX3X into its mature particles (16). Most interestingly, depleting either the viral or the cellular pool of DDX3X significantly impairs HSV-1 infectivity (21). As the exact function of DDX3X in the HSV-1 replication cycle is not clear, we examined more closely the nature of this host-pathogen interaction. The present data from multiple assays show that DDX3X impacts the propagation of HSV-1. They also reveal that the portion of DDX3X present in the incoming virions is not required for the early stages of HSV-1 entry. In contrast, depletion of DDX3X from the cell impacted viral particle assembly and intracellular and extracellular viral yields. Interestingly, either reducing or overexpressing the cellular pool of DDX3X downregulated HSV-1 gene expression. This dependency on DDX3X protein levels was corroborated in rescue experiments using cells that were depleted by a small interfering RNA (siRNA) targeting DDX3X (siDDX3X) and that concomitantly expressed an siRNA-resistant DDX3X construct. At the mechanistic level, DDX3X modulated the transcription of immediate early, early, and late viral genes, including the aforementioned viral transactivating proteins ICP0, ICP4, and VP16. This appeared independent of the ability of DDX3X to promote IFN-β production. We conclude that DDX3X modulates HSV-1 yields by a novel mechanism implicating viral gene transcription, irrespective of the interferon pathway.
RESULTS
DDX3X is required for optimal viral yields.
We previously reported by proteomics that HSV-1 incorporates the host protein DDX3X in mature virions (16) and that both the cellular and virion-incorporated pools of DDX3X influence viral yields (21). To independently validate the role of the cellular DDX3X in viral replication, we resorted to the BHK21-derived tsET24 cell line, which harbors a temperature-sensitive (ts) DDX3X inactive at 39.5°C but functional at 34°C (22). Infection of the tsET24 and parental BHK21 cell lines with wild-type HSV-1 showed that viral yields were strongly reduced at the nonpermissive temperature in the tsET24 cell line but not affected in the parental cell line (Fig. 1). This confirmed that DDX3X is indeed a modulator of the HSV-1 life cycle. It also ruled out that nonspecific off-target effects were responsible for our past findings (21).
FIG 1.

DDX3X is necessary for HSV-1 replication. The DDX3X thermosensitive tsET24 and parental BHK21 cell lines were incubated for 24 h prior to infection at the permissive (34°C) or nonpermissive (39.5°C) temperature and infected with HSV-1 K26GFP (MOI of 5) for an additional 24 h at that same temperature. Afterwards, supernatants were collected and titrated on Vero cells. Titers were normalized to the mean value obtained with samples infected at 34°C (arbitrarily set to 100%). Data represent the averages of two independent experiments, each titrated in duplicates. The error bars represent the standard deviations of the means.
HSV-1 yields are sensitive to DDX3X levels.
Given that viral yields were lowered upon depletion of the host protein DDX3X, we probed whether overexpression of the protein would have the opposite effect on HSV-1. An initial examination by immunofluorescence microscopy indicated that overexpression did not influence the subcellular localization of DDX3X and that the level of exogenous DDX3X, based on quantitation with the DDX3X antibody, was similar to that of its endogenous counterpart (Fig. 2A). Thus, cells were first transfected with wild-type DDX3X and then infected, and extracellular virus production was measured by plaque assay. Unexpectedly, an excess of DDX3X also reduced viral infectivity (Fig. 2B). This suggested that optimal viral replication required a carefully controlled level of DDX3X.
FIG 2.

(A) Overexpressed DDX3X colocalizes with endogenous DDX3X. HeLa cells grown on coverslips were mock treated or transfected with pSG-N-4xHA-TEV-N-term DDX3X for 24 h. Cells were fixed and reacted with antibodies against total DDX3X (red) or HA-specific antibodies to detect exogenous DDX3X (green). Nuclei were labeled with Hoechst (blue). Samples were analyzed by confocal laser scanning microscopy. Scale bar, 10 μm. These results are representative of three individual experiments. (B) Effect of DDX3X overexpression on HSV-1 infectivity. HeLa cells grown in six-well plates were either mock treated or transfected with plasmids that express only the tag (GS-STOP) or the exogenous DDX3X fused to the tag (GS-DDX3X) for 24 h and then infected with wild-type HSV-1 at an MOI of 5 for 18 h. Supernatants, containing extracellular viruses, were then collected and titrated on Vero cells. Data represents the pool of five individual experiments. Bilateral Student's t tests (with standard deviations shown) were performed to detect significant hits compared to results with the transfection agent-only control. (*, P < 0.05; **, P < 0.01).
To better define the relationship between DDX3X and the infection, we opted to rescue DDX3X in siRNA-depleted cells. To this end, we first determined which of the four siRNAs targeting DDX3X worked best (Fig. 3A) and generated a translationally silent DDX3X mutation resistant to this specific siRNA. We then treated cells with this unique siRNA to reduce endogenous DDX3X levels and rescued them with the above-described siRNA-resistant DDX3X construct. Cells were subsequently infected with HSV-1 K26GFP, a recombinant virus expressing a green fluorescent protein (GFP)-tagged VP26 minor capsid component (23), and the presence of DDX3X and the virus was monitored by fluorescence microscopy. Note that the exogenous DDX3X was expressed at levels similar to those of its endogenous counterpart and double the total DDX3X in the absence of siRNA (Fig. 3B and C, compare GS-DDX3X and transfection agent only). The results also revealed that, as expected, silencing DDX3X significantly reduced DDX3X. Interestingly, overexpression of DDX3X nearly abolished HSV-1 production (Fig. 3B and C). Rescuing the depleted cells with exogenous DDX3X restored the infection up to 63% compared to the level in untransfected cells (Fig. 3B and C, bottom). The virus was clearly sensitive to the DDX3X protein levels.
FIG 3.

(A) RNA interference efficiency. HeLa cells seeded in 24 wells were individually transfected for 48 h with each of the different siRNAs against DDX3X (a, b, c, and d) that are present in the SMARTpool. Cell lysates were collected, and Western blotting was done as described previously. Values represent the amounts with respect to the untransfected control. The data are representative of two individual experiments. Normal levels of DDX3X best support HSV-1 propagation. (B) HeLa cells grown on coverslips were sequentially mock transfected or treated with siDDX3Xa in the presence or absence of a plasmid coding for the siRNA-resistant DDX3X mutant (pGS-TAP-tagged DDX3X, here called GS-DDX3X) for 48 h. All but the mock-infected samples were subsequently infected at an MOI of 5 with HSV-1 K26GFP (green signal) for 18 h. The cells were finally fixed and reacted with primary antibodies against DDX3X and appropriate secondary antibodies, a method which detects both endogenous and exogenous DDX3X (red signal), while nuclei were labeled with Hoechst (blue signal). Samples were analyzed by fluorescence microscopy. (C) Fluorescence intensities (relative units [RU]) were quantified for 100 cells using ImageJ. Scale bar, 10 μm. The data are representative of three independent experiments. Bars indicate the means and error bars indicate standard deviations of the mean (bilateral Student t tests; **, P < 0.01 and ***, P < 0.001; NS, not significant).
The virus functionally interacts with endogenous DDX3X.
To understand the role of DDX3X in the viral life cycle, we next examined whether the virus influenced DDX3X levels. To this end, we probed DDX3X expression by Western blotting in three cell lines commonly used to study HSV-1, namely, HeLa, 143B, and Vero cells. HSV-1 infection had no impact on DDX3X protein levels in HeLa and Vero cells but reduced them by half in 143B cells (Fig. 4). We also examined whether the virus altered the subcellular localization of DDX3X. As shown in Fig. 5 (left panels), the endogenous DDX3X was primarily localized in cytoplasmic granules in uninfected cells. Attempts to identify these granules with a variety of markers unfortunately failed to unambiguously identify them. In agreement with previous studies, some DDX3X could also be detected in the nucleus (Fig. 5, insets), consistent with its shuttling across the nuclear envelopes (24, 25). Upon infection, DDX3X levels were once again unaffected in HeLa and Vero cells but reduced in 143B cells, as reported above. However, DDX3X was somewhat aggregated in all three cell lines (Fig. 5A, compare left and right panels). Furthermore, the GFP-tagged viral particles often, but not always, colocalized with DDX3X in the cytoplasm (Fig. 5B), in agreement with the incorporation of this cellular protein in mature virions (21). A time course revealed that DDX3X started to aggregate between 3 and 6 h postinfection (hpi) and that this intensified with time (Fig. 6). As noted above, DDX3X also partially colocalized with the ICP4 viral marker within the nucleus. Albeit the virus can alter the localization of DDX3X and although viral proteins partially colocalize with it, we cannot at this point formally infer that viral particles themselves functionally interact with DDX3X. However, given that the cellular protein is incorporated in mature virions, this seems plausible, and it would be interesting to find whether this occurs in the nucleus, cytoplasm, or elsewhere.
FIG 4.

Impact of HSV-1 on endogenous DDX3X cellular levels. HeLa, Vero, and 143B cells were seeded in 10-cm plates 24 h prior to infection. Cells were then either mock treated (−) or infected (+) with wild-type HSV-1 at an MOI of 5 for 18 h. Total cell lysates were then collected, and DDX3X cellular levels were probed by Western blotting. γ-Tubulin was used as a loading control. Numbers below the blots indicate the average levels of DDX3X, normalized for γ-tubulin, from five independent experiments.
FIG 5.

Endogenous DDX3X subcellular localization. (A) HeLa, Vero, and 143B cells were seeded on coverslips 24 h prior to infection. Cells were then either mock infected or inoculated with HSV-1 K26GFP (green) at an MOI of 5 for 18 h. Cells were then fixed, permeabilized, and immunostained with an antibody specific for DDX3X (red), and the nuclei were stained with Hoechst (blue). Cells were finally analyzed by confocal laser scanning microscopy. (B) Close-up view in the cytoplasm of infected HeLa cells. Scale bar, 10 μm. Results are representative of three independent experiments.
FIG 6.

Time course of DDX3X aggregation. HeLa cells grown on coverslips were infected with wild-type HSV-1 at MOI of 5 for 3, 6, 9, and 24 hpi. Cells were fixed at the indicated time points and reacted with antibodies against endogenous DDX3X (red) or ICP4 (green; to delineate infected cells), while nuclei were labeled with Hoechst (blue). Samples were analyzed by fluorescence microscopy. Scale bar, 10 μm. These results are representative of two individual experiments. Arrows indicate the colocalization of DDX3X with ICP4.
DDX3X downregulation alters novel viral particle assembly.
A number of scenarios may justify the presence of a host protein in mature virions. One explanation might be that it is needed immediately after cell entry to initiate an infection. A second scenario would be that the protein boosts viral replication postentry. A third one is that DDX3X virion incorporation may be a bystander effect of a previously occurring interaction taking place during viral particle assembly or transport toward the cell periphery. Note that these options are not mutually exclusive. To address the first of these scenarios, we directly probed whether the virion-associated pool of DDX3X is required for viral entry. To this end, we used a viral strain that encodes luciferase under a constitutive cytomegalovirus (CMV) immediate early promoter (26). We concomitantly infected cells with this virus, a wild-type virus (i.e., without the luciferase cassette), or no virus at all (mock infection). To specifically probe the role of DDX3X during entry, we additionally infected the cells with the luciferase-coding virus depleted of DDX3X or, as a control, depleted of VP16, a transactivating viral protein incorporated in virions and known to jump-start viral gene expression (21). To synchronize the infection, viral adsorption was performed at 4°C for 1 h, and the cells were subsequently transferred to 37°C for an additional hour. Other control cells were maintained at 4°C throughout the experiment to prevent viral entry. Entry of the virus into the cells was then monitored via luciferase expression. Viruses devoid of the luciferase gene, mock-infected cells, or the luciferase-encoding virus incubated at 4°C all gave signals at the background level (Fig. 7). In contrast, incubation of the untreated luciferase-positive virus at 37°C gave a strong signal, which was normalized to 100%. Infection by VP16- or DDX3X-depleted luciferase-positive virions resulted in similar signals, indicating that their presence in the mature virus was not essential for this early phase of the infection. This also suggested that the CMV immediate early promoter driving the luciferase is insensitive to VP16, as expected. Together, these observations suggested that DDX3X acted downstream of viral entry.
FIG 7.

Virion-associated DDX3X has no effect on HSV-1 entry. 143B cells preseeded in 24-well plates were mock infected or infected at an MOI of 30 for 1 h at 4°C with various HSV-1 viruses, as indicated below each bar. These included wild-type HSV-1 (luciferase-negative) and untreated and DDX3X- or VP16-depleted HSV-1(17+)Lox-Luc viruses. To enable viral entry, the cells were then incubated at 37°C for another hour and subsequently lysed at room temperature for 30 min in the presence of luciferin and energy. As a control, one sample was left at 4°C throughout the experiment to prevent viral entry. Samples were then transferred to 96-well plates and analyzed with a luminometer. Values represent the mean relative light units (RLU) from two independent experiments, and error bars indicate the standard deviations of the means. Asterisks indicate the results of bilateral Student's t tests (***, P < 0.001). ScrVP16, scrambled siRNA targeting VP16.
Thus far, our findings pointed to a requirement for DDX3X in the HSV-1 life cycle at some time after viral entry. To define whether this phenomenon is due to a block in viral egress from the intracellular to the extracellular environment or to a reduction in the assembly of viral particles, we next examined intracellular viral yields. The results indicated that DDX3X depletion from cells significantly reduced the amount of intracellular viral particles, as did the knockdown of the transactivator and structural VP16 protein (Fig. 8). Consistently, less GFP signal was observed by immunofluorescence following an HSV-1 K26 GFP viral infection after DDX3X or VP16 depletion (Fig. 9). Thus, DDX3X was required for the efficient assembly of nascent viral particles. To ascertain whether this truly translated into fewer viral particles, we performed an electron microscopy analysis under depleted or DDX3X overexpression conditions. As shown in Fig. 10A and quantified in panel B, both scenarios significantly reduced the total number of viral particles associated with the cells, as did the control siVP16 depletion.
FIG 8.

Impact of DDX3X knockdown on intracellular virions. 143B cells were transfected with siRNA pools targeting DDX3X or VP16 and infected with K26GFP at an MOI of 5. Cells were collected and lysed, and viruses were titrated on Vero cells. The error bars show the standard deviations of the means of two independent experiments. Bilateral Student's t tests were performed to detect significant hits compared to results with the transfection agent-only control (***, P < 0.001).
FIG 9.

Inhibition of DDX3X-reduced viral particle assembly. 143B cells were mock infected, mock transfected, or treated with a nontargeting siRNA or with pooled siRNAs against DDX3X or VP16 as indicated. All but the mock-infected samples were incubated with HSV-1 K26GFP (green signal) at an MOI of 5 for 18 h. The cells were fixed, and the nuclei were stained with Hoechst 33342 (blue). Right panels are enlargements of the boxed sections present in the left panels. Scale bar, 10 μm.
FIG 10.

DDX3X plays a role in viral assembly. HeLa cells were either transfected with siDDX3X, siVP16, GS-DDX3X or just treated with a nontargeting siRNA (scVP16) or transfection reagent (Pepmute). All but the mock-infected samples were infected with HSV-1 strain 17+ at an MOI of 5 for 18 h. The cells were fixed and processed for Epon embedding (see Materials and Methods) to be observed by electron microscopy. (A) Representative cells for each condition. Enlargements (boxed higher-magnification images) were added to help visualize the mature virions more clearly. (B) The data represent the quantification of total cell-associated viral particles, both intracellular and those bound to the extracellular surface of the cells, for 12 individual cells of each condition from two independent experiments. Error bars indicate the standard deviations of the means Asterisks show the results of bilateral Student's t tests (***, P < 0.001). Scale bars are as indicated on each figure.
DDX3X modulates viral gene expression.
Given the reduced production of viral particles upon DDX3X depletion and given that DDX3X is a known modulator of host gene transcription and translation, we wondered if DDX3X could also regulate viral gene expression. To address this, we probed representative candidates of the three viral kinetics classes, namely, ICP0 and ICP4 (immediate early proteins), ICP8 and pUL23 (early proteins), and VP16 and pUL31 (late proteins) under normal, reduced (siDDX3X), or enhanced (DDX3X overexpression) conditions. We infected cells for 9 h since it is an intermediate time point when both early and late viral proteins are detectable. Both DDX3X conditions negatively affected the expression of all classes of viral proteins, including once again VP16 (Fig. 11A). Note that these Western blots were not performed with film but, rather, with a ChemiDoc digital instrument, which gives very good linear detection signals (Fig. 11B). Quantification of several independent experiments, which were normalized against a γ-tubulin loading control, confirmed these findings (Fig. 11C). To evaluate if the reduced protein levels were the consequence of reduced transcription, the samples were further analyzed by quantitative reverse transcription-PCR (qRT-PCR). The data showed that most of the viral gene transcripts were negatively impacted by both reduced and overabundant DDX3X (Fig. 12), albeit the changes were slightly less than those in the corresponding protein levels (Table 1).
FIG 11.

Effect of depletion or overexpression of DDX3X on HSV-1 gene expression. (A) HeLa cells seeded in six-well plates were transfected for 48 h with pooled siRNA against DDX3X and/or a plasmid coding for siRNA-resistant DDX3X. Cells were subsequently infected at an MOI of 5 with wild-type HSV-1 and harvested at 9 hpi and lysed. Twenty micrograms of the lysates was directly loaded onto SDS-PAGE gels and analyzed by Western blotting. γ-Tubulin was used as the loading control. (B) HeLa cells were seeded in a 10-cm petri dish for 24 h; cell lysates were then collected using three freeze-thaw cycles. Different amounts of cell lysate were loaded on an SDS-PAGE gel in duplicates to exclude potential loading errors. Western blotting was done using DDX3X or γ-tubulin antibodies. Linearity of the results was assayed by measuring the signal intensity of each point (mean of the duplicates). R2 values were measured using GraphPad Prism, version 6. (C) Protein expression was evaluated, normalized to the level of γ-tubulin, and compared to the values obtained for infected but nontransfected cells. The reported values represent the average of two experiments. The error bars indicate standard deviations of the means. Bilateral Student's t tests were performed (*, P < 0.05; **, P < 0.01, ***, P < 0.001). Note that throughout this study, Western blots were quantified on a ChemiDoc MP system with a 4 orders of magnitude dynamic range, not film which has a poor linearity. In panels A and C, proteins are classified as immediate early (α), early (β), and late (γ).
FIG 12.

Impact of DDX3X on viral gene transcription. HeLa cells were seeded in six-well plates and transfected for 48 h either with siDDX3Xa or an siRNA-resistant DDX3X plasmid. Following 8 h of infection with wild-type virus at an MOI of 5, total RNA was collected and reverse transcribed into cDNAs. Expression levels of immediate early (α), early (β), or late (γ) viral genes were analyzed by qRT-PCR. Values represent the averages of two experiments. All the values were normalized to the level of GAPDH. Bilateral Student's t tests were performed to detect significant hits compared to results with the transfection agent-only control, arbitrarily set at 100% (*, P < 0.05; **, P < 0.01).
TABLE 1.
Effect of DDX3X depletion or overexpression on HSV-1 gene expression
| Gene | Protein | Value under the indicated DDX3X condition (%)a |
|||
|---|---|---|---|---|---|
| Depletion |
Overexpression |
||||
| mRNA level | Protein level | mRNA level | Protein level | ||
| RL2 | ICP0 | 72 ± 10 | 36 ± 2* | 47 ± 29 | 12 ± 9 |
| RS1 | ICP4 | 71 ± 7 | 18 ± 0.6† | 48 ± 14 | 8 ± 4* |
| UL29 | ICP8 | 65 ± 4 | 52 ± 6 | 43 ± 15 | 28 ± 5 |
| UL23 | TK | 69 ± 10 | 44 ± 14 | 58 ± 12 | 9 ± 2* |
| UL31 | pUL31 | 71 ± 7 | 39 ± 13 | 48 ± 14 | 11 ± 0.04 |
| UL48 | VP16 | 58 ± 7 | 22 ± 1* | 41 ± 10 | 1 ± 1* |
Significant differences between mRNA and protein levels (means ± standard deviations) for each condition in two independent experiments based on bilateral Student's t tests are indicated. *, P < 0.05; †, P < 0.001.
DDX3X's effect on the virus is independent of IFN-β production.
Given the role of DDX3X in innate immunity (see introduction), we probed by qRT-PCR the impact of DDX3X modulation on interferon type I production using IFN-β as a gauge. As reported elsewhere (10), depleting endogenous DDX3X had no effect on the already low level of IFN-β mRNA in uninfected cells, while DDX3X overexpression strongly stimulated its expression (Fig. 13). Meanwhile, IFN-β production was slightly increased by the virus compared to the level in mock-treated cells, but this was not statistically significant, which is perhaps consistent with the ability of the virus to counteract this innate response. Not surprisingly in this context, depleting or overproducing DDX3X in infected cells did not have any major impact on IFN-β levels (Fig. 13). Thus, as previously documented by other investigators, DDX3X positively promoted IFN-β levels in uninfected cells but had a limited effect in HSV-1-infected cells. Therefore, the well-known ability of DDX3X to modulate the interferon type I pathway did not appear to play a critical role here since both DDX3X knockdown and overexpression reduced viral gene transcription. This suggested that DDX3X may act on viral propagation by an interferon-independent pathway.
FIG 13.

IFN transcription levels. HeLa cells grown in six-well plates were transfected for 48 h with siDDX3Xa and/or an siRNA-resistant DDX3X plasmid. Cells were either mock treated or subsequently infected with wild-type virus for 8 h at an MOI of 5. Total RNA was collected, and IFN-β mRNA was measured by qRT-PCR normalized to GAPDH levels. Data represent the average of two independent experiments. Bilateral Student's t tests were performed to detect significant hits compared to results with the transfection agent-only control (NS, not significant; ***, P < 0.001). RU, relative units.
To independently evaluate if DDX3X significantly impacted HSV-1 propagation via innate immunity, we measured viral genome copies by quantitative PCR (qPCR) under normal, depleted, or overexpressed DDX3X conditions. The data illustrated that viral genome replication was strongly inhibited under conditions of both depletion and overexpression (Fig. 14), in agreement with the scenario whereby DDX3X acted on the virus independently of its ability to modulate IFN-β production. To confirm this hypothesis, we resorted to a well-characterized DDX3X point mutant (K230A) that abolishes its ATPase—and consequently helicase—activities without perturbing its ability to modulate the interferon pathway (8, 10, 13). Our rationale was that if DDX3X primarily acted via the interferon type I pathway, the mutant should behave like wild-type DDX3X and block viral propagation, but if the ATPase activity is the main driver, the mutant would not have any impact and would allow the virus to replicate normally. The data showed that the K230A mutant was normally expressed and completely inactive (Fig. 15B). This indicated that DDX3X acted on the virus via its ATPase/helicase activities independently of the interferon pathway.
FIG 14.
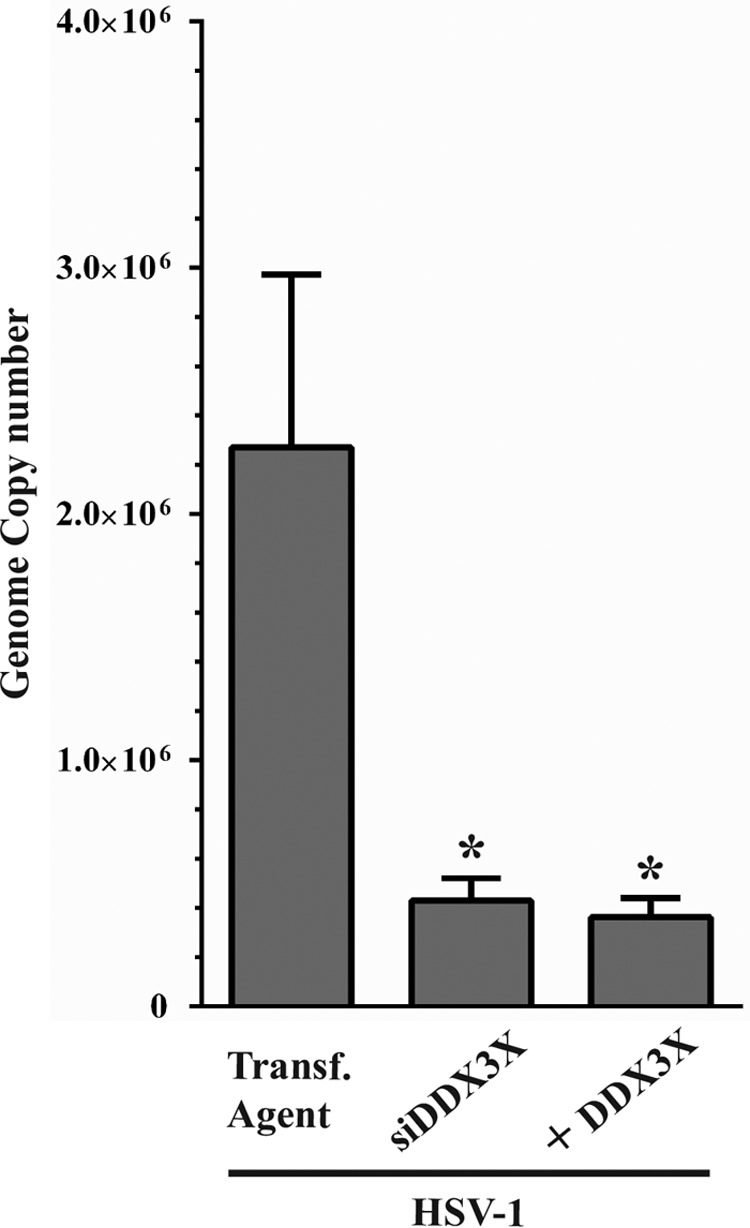
Viral genome copies upon siDDX3X depletion or overexpression. HeLa cells seeded in six-well plates were transfected for 48 h with siDDX3Xa and/or an siRNA-resistant DDX3X plasmid. Cells were then infected with wild-type virus for 8 h at an MOI of 5. Total DNA was then collected, and viral genome copy numbers were measured by qPCR. Data represent the average of two experiments. Bilateral Student's t tests were performed to detect significant hits compared to results with the transfection agent-only control (*, P < 0.05).
FIG 15.

(A) The K230A DDX3X mutant is expressed at levels similar to those of the wild type. HeLa cells grown on coverslips were mock treated or transfected with pSG-N-4xHA-TEV-N-term K230A for 24 h. Cells were fixed and labeled with antibodies against total DDX3X (red) or with HA-specific antibodies to detect mutant DDX3X (green). Nuclei were labeled with Hoechst (blue). Samples were analyzed using confocal laser scanning microscopy. Images are representative of three individual experiments. Scale bar, 10 μm. (B) Overexpression of an ATPase/helicase-inactive but interferon-competent DDX3X mutant has no impact on viral propagation. HeLa cells grown in six-well plates were either mock treated or transfected for 24 h with exogenous wild-type (wt) DDX3X or the ATPase/helicase-deficient but interferon-competent K230A mutant. They were then infected with wild-type HSV-1 for 18 h at an MOI of 5. Extracellular viruses in the supernatant were then collected and titrated on Vero cells. Data represents the pool of five individual experiments. Statistical significance was assessed based on bilateral Student's t tests (**, P < 0.01; ***, P < 0.001).
DISCUSSION
The DDX3X RNA helicase modulates HSV-1 propagation.
The present data, along with our past findings (21), indicate that depletion of DDX3X either in cells or in HSV-1 mature viral particles led to a significant reduction of infectious HSV-1 particles. Two independent data sets confirmed that this was specific and not the result of off-target effects associated with RNA interference. First, viral yields were significantly rescued by an siRNA-resistant DDX3X construct (Fig. 3). Second, orthogonal validation of the results with the tsET24 cell line, where DDX3X is nonfunctional at the restrictive temperature, further proved that the phenotypes were directly linked to DDX3X (Fig. 1). We conclude that DDX3X is required for optimal HSV-1 propagation.
Despite the contribution of DDX3X to viral yields, overexpression of DDX3X unexpectedly also reduced virus production (Fig. 2 and 3). This was corroborated by the fact that we detected HSV-1 K26GFP expression in only 0.9% of the cells overexpressing DDX3X (Fig. 3). This inhibition notably required the ATPase and helicase activities of DDX3X (Fig. 15). Though initially counterintuitive, this apparent contradiction has also been reported for hepatitis B and C viruses (9, 27). We presume that reduced or overabundant DDX3X levels exert their effects by distinct mechanisms and postulate that DDX3X stimulates viral production when it is present in rate-limiting or normal amounts. In contrast, overabundant functional DDX3X may compete with one of its molecular partners and repress the endogenous DDX3X machinery. This seemed in line with our inability to generate cell lines stably overexpressing DDX3X (data not shown). In this context, it is worth noting that HSV-1 did not alter the DDX3X protein levels in HeLa and Vero cells (Fig. 4 and 5). Why the virus reduced DDX3X protein levels to a significant extent in 143B cells remains to be elucidated. Nonetheless, our working hypothesis is supported by our rescue experiments whereby transfection of siDDX3X-resistant exogenous DDX3X in siDDX3X-treated cells partially rescued both normal levels of DDX3X and viral output (Fig. 3). Hence, DDX3X is an important host partner for HSV-1.
DDX3X acts on HSV-1 gene expression.
Distinct nonexclusive scenarios could explain the presence of DDX3X in mature HSV-1 particles. First, the virion-associated DDX3X may be required to initiate an efficient infection. However, this was mandatory as viral entry was unperturbed by the depletion of DDX3X within the viral particles prior to the entry assay (Fig. 7). It remains possible, though, that the cellular pool of DDX3X takes over this task. Second, DDX3X incorporation in virions could be due to an interaction of the host protein and structural components of the virus and merely reflect a past role(s) or simply a sticky protein. Although we cannot formally rule this out, it is clear that DDX3X plays an active role in the assembly of novel viral particles by regulating the transcription of viral genes (Fig. 11). Given the stronger impact of DDX3X on viral protein levels than on transcription (Table 1), it is conceivable that DDX3X also acts on other steps of viral gene expression, namely, mRNA transport, stability, or translation or even viral protein stability. On the other hand, the more pronounced effect of DDX3X on protein levels may simply be the consequence of the relative abundance of viral transcripts, albeit this is hard to correlate (28). We conclude that DDX3X clearly modulates HSV-1 gene transcription with potential impacts on other steps of gene expression. The presence of DDX3X in various mature extracellular herpesviruses, including HSV-1, pseudorabies virus (PRV), and human CMV (HCMV) (29), suggests that the present findings may apply to several viruses. One major outstanding question is why these viruses incorporate DDX3X in their viral particles.
Mechanism of DDX3X action.
Whether DDX3X acts directly on various viral genes or indirectly via a few select molecules is unclear. Thus far, DDX3X was shown to bind to the IFN-β and the tumor suppressor p21 promoters, thus suggesting a direct role as a transcription cofactor (10, 30). However, precise binding motifs have yet to be identified.
An intriguing scenario is the stimulatory role that DDX3X plays in innate immunity upon TBK1 and IKKε binding and IRF3 activation (10, 13, 31). As expected, modulating DDX3X levels did alter interferon production in uninfected cells but had minimal impacts in infected cells (Fig. 12), presumably because of the well-documented ability of the virus to circumvent this pathway (32). Since either increasing or decreasing DDX3X levels reduced viral yields (Fig. 1 to 3 and 8 to 10), gene expression (Fig. 11 and 12 and Table 1), and genome copy numbers (Fig. 14), this suggested that DDX3X acted on HSV-1 independently of the interferon pathway. This model is supported by the observation that a helicase-deficient DDX3X mutant, which is still able to stimulate interferon production (10, 13), was completely dead in our hands and failed to limit HSV-1 propagation (Fig. 15). So, we conclude that DDX3X most likely modulates HSV-1 gene expression in an interferon-independent manner.
DDX3X is a key player for both RNA and DNA viruses.
DDX3X is a multifunctional cellular protein that interacts with viruses at several levels. Much of the current literature focuses on RNA viruses, given that DDX3X has RNA helicase activity. Not surprisingly, DDX3X was found to promote genome replication of many RNA viruses (33). However, DDX3X binds TBK1/IKKε, which stimulate both IRF3 activation and interferon type I production, thereby promoting an antiviral state (10, 11). This implies that DDX3X has dual functions that can either facilitate or hamper viruses. Interestingly, the latter property is not limited to RNA viruses but also operates on vaccinia virus and hepatitis B virus, two DNA viruses (12, 13). Accordingly, DDX3X influences viral outcomes both via innate immunity and RNA viral gene duplication. The present study further suggests that DDX3X also acts on DNA viruses by a third mechanism, namely, by modulating viral gene expression.
MATERIALS AND METHODS
Cells, viruses, and plasmids.
HeLa (ATCC CCL-2), Vero (ATCC CCL-81), 143B thymidine kinase-negative (TK−) (ATCC CRL-8303), tsET24, and BHK21 cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 2 mM l-glutamine and either 10% fetal bovine serum (FBS) or 5% bovine growth serum (BGS). 143B cells were also supplemented with 15 μg/ml 5-bromo-2 deoxyuridine (BrdU; Sigma) except prior to transfection and infection. The tsET24 thermosensitive and parental BHK21 cell lines (a kind gift from Takeshi Sekiguchi, Kyushu University [22]) were passaged at the permissive temperature of 34°C.
HSV-1 K26GFP (strain KOS; provided by Prashant Desai, Johns Hopkins University [23]) is a fluorescent virus tagging the minor VP26 capsid component. Wild-type HSV-1 strain F was obtained from the American Type Culture Collection (ATCC VR-735). The HSV-1(17+)Lox-Luc viral strain is derived from strain 17+ and encodes a luciferase gene under the constitutive immediate early CMV promoter positioned between the UL55 and UL56 HSV-1 genes (26). All viruses were propagated on BHK cells and titrated on Vero cells as previously described (34).
The pGS-TAP-tagged DDX3X (DDX3X fused to protein G and streptavidin binding peptide with a tandem affinity purification [TAP] tag), pSG-N-4xHA-TEV-N-term DDX3X (hemagglutinin [HA]-tagged DDX3X; TEV is tobacco etch virus) and pSG-N-4xHA-TEV-N-term K230A (defective mutant) eukaryotic expression plasmids were a kind gift from G. Superti-Furga (Research Center for Molecular Medicine) (10). An siRNA-resistant plasmid was derived from pGS-TAP-tagged DDX3X using a QuikChange II XL site-directed mutagenesis kit (Agilent Technologies) and forward primer 5′-GGAAAGAGGAAAGATTGATTAGACTTTTGTAAGTATCTGGTCCTGGATGAAGCTGATCGGATGTTGGATATGGG-3′ and reverse primer 5′-CCCCATATCCAACATCCGATCAGCTTCATCCAGGACCAGATACTACAAAAGTCTAATCCAATCTTTCCTCTTTCC-3′, as per the manufacturer's instructions (bold sequences indicate the silent point mutations). We also generated a plasmid that expresses only the GS tag by changing the first methionine in the DDX3X sequence to a stop codon. In addition, three other amino acids at the beginning of the DDX3X sequence were also changed to stop codons to make sure that protein was not expressed, using the following primers: forward primer, 5′-CTG GTC CAG CCC GAG CGC ATT TTA CAC TCA CAC TTA ACT CTA GGA GCC TGC TTT TTT GTA CAA AC-3′; reverse primer, 5′-GTT TGT ACA AAA AAG CAG GCT CC TAG AGT TAA GTG TGA GTG TAA AAT GCG CTC GGG CTG GAC CAG-3′ (mutations are in boldface). This plasmid served as a negative control in our experiments and is referred to as GS-STOP in the manuscript.
Antibodies.
Primary antibodies were as follows: the anti-human DDX3 rabbit R648 polyclonal serum (immunofluorescence, 1:200; Western blot, 1:2,000) was a kind gift from A. Patel (University of Glasgow Centre for Virus Research) (35). Viral antibodies were generously provided by several laboratories and used at the indicated dilutions for Western blotting: VP16 (1:1,000; Helena Browne), TK (1:2,500; James R. Smiley), and pUL31 (1:1,000; Joel D. Baines). All other antibodies were purchased from commercial vendors, including anti-VP5 (1:5,000; Cedarlane), γ-tubulin (1:5,000; Sigma-Aldrich), and ICP4 (immunofluorescence, 1:500; Western blot, 1:5,000), ICP8 (1:1,000), and ICP0 (1:1,000) were all purchased from Abcam. All secondary antibodies were purchased from Molecular Probes.
Fluorescence microscopy.
HeLa, Vero, or 143B cells were seeded overnight on coverslips in 24-well plates at a concentration of 1.5 × 104 cells/well. Cells were mock treated or infected with HSV-1 K26GFP at a multiplicity of infection (MOI) of 5. After a 1-h adsorption at 37°C, cells were washed twice in phosphate-buffered saline (PBS), fresh medium was added, and the cells were incubated for another 18 h prior to fixation and permeabilization with 3% paraformaldehyde and 0.1% Triton X-100. The cells were then incubated with a rabbit anti-DDX3X antibody for 1 h and further incubated for 45 min with a goat anti-rabbit antibody coupled to Alexa Fluor 568 (Molecular Probes). The samples were finally stained with Hoechst 33342 (Sigma-Aldrich) and examined on an Axiophot epifluorescence or LSM700 confocal microscope (Zeiss). Fluorescence intensities were calculated by ImageJ by subtracting the background signal then dividing this net signal by the cell area.
Western blotting.
HeLa, Vero, or 143B cells were mock treated or infected with wild-type HSV-1 at an MOI of 5 and collected 18 h later by scraping them in lysis buffer (10 mM Tris, pH 7.4, 150 mM NaCl, 2 mM MgCl2, 5 mM dithiothreitol [DTT], 1 mM EDTA, 1% Igepal, and a cocktail of protease inhibitors). After centrifugation for 5 min at 4°C and 500 × g, cell pellets were resuspended in lysis buffer and incubated on ice for 1 h. Then they were passed through 27-gauge 1/2-in. needles three times and treated with DNase for 30 min at 37°C (500 U/ml; Roche). Cell debris was removed by spinning at 2,500 × g for 10 min, and cell lysates were collected. Samples (typically 20 μg) were loaded on 8% acrylamide SDS-PAGE gels in protein sample buffer (50 mM Tris-HCl, pH 6.8, 2% SDS, 0.1% bromophenol blue, 10% glycerol, and 2% β-mercaptoethanol). Proteins were then transferred to polyvinylidene difluoride (PVDF) membranes, which were then incubated for 1 h in blocking buffer (5% nonfat dry milk, 13.7 mM NaCl, 0.27 mM KCl, 0.2 mM KH2PO4, 1 mM Na2HPO4, and 0.1% Tween 20). The membranes were ultimately reacted with antibodies as indicated in each figure legend. When indicated (Fig. 3A, 4, and 11), protein expression levels were quantified with a ChemiDoc MP System (Bio-Rad), which has a dynamic range of 4 orders of magnitude, and Image lab, version 5.0, software (Bio-Rad).
Thermosensitive DDX3X.
Thermosensitive tsET24 and parental BHK21 cells were plated in six-well plates and incubated at the permissive temperature of 34°C or the nonpermissive temperature of 39.5°C for 24 h. The cells were then mock treated or infected with HSV-1 K26GFP at an MOI of 5 at 34°C or 39.5°C. Cells and supernatants were collected at 24 h postinfection (hpi), and HSV-1 production was titrated on Vero cells by plaque assays.
DDX3X Overexpression.
HeLa cells were seeded on coverslips in 24-well plates at a concentration of 2.0 × 104 cells/well. The next day, cells were transfected for 24 h with 0.5 μg/well of pGS-TAP-tagged DDX3X using LipoD293 reagent (SignaGen). Cells were subsequently infected for a further 18 h with wild-type HSV-1 K26GFP at an MOI of 5. The samples were finally fixed and permeabilized as described above and observed on an Axiophot epifluorescence microscope (Zeiss). To measure the impact of DDX3X overexpression on viral yields, HeLa cells were seeded at a concentration of 5.5 × 105 cells/well in six-well plates and transfected with pSG-N-4xHA-TEV-N-term DDX3X or pSG-N-4xHA-TEV-N-term K230A for 24 h with 4 μg/well of DDX3X constructs. Cells were then infected for a further 18 h with wild-type HSV-1 at an MOI of 5. Supernatants containing extracellular viruses were collected and titrated on Vero cells.
RNA interference.
siRNAs against human DDX3X were either used as a SMARTpool of four siRNAs (Dharmacon) or individually tested as indicated in the respective figure legends. These siRNA were the following: siDDX3Xa, GCAAATACTTGGTGTTAGA; siDDX3Xb, ACATTGAGCTTACTCGTTA; siDDX3Xc, CTATATTCCTCCTCATTTA; and siDDX3Xd, GGTATTAGCACCAACGAGA. siRNAs (25-nm) were transfected into 143B or HeLa cells using either Pepmute (Signagen) or Lipofectamine 2000 (Thermo Fisher) for 48 h.
Generation of depleted virions.
To produce depleted virions, 1 × 106 143B cells were seeded in 10-cm dishes for 24 h. These cells were then either transfected or mock treated for 48 h with 25 nM siRNA against DDX3X, VP16, or scVP16 (scrambled siRNA targeting VP16) (21) using Lipofectamine 2000 (Invitrogen). Cells were then infected for another 24 h with HSV-1(17+)Lox-Luc or wild-type virus at an MOI of 5. In order to separate intracellular and extracellular virions, the tissue culture medium was first removed, and cells were scraped, centrifuged at 250 × g for 5 min at 4°C, and resuspended in MNT (30 mM morpholinoethanesulfonic acid, 100 mM NaCl, and 20 mM Tris, pH 7.4). Meanwhile, the extracellular medium was concentrated at 40,000 × g for 40 min at 4°C, and viral pellets were resuspended in the MNT buffer. All viruses were then titrated on Vero cells.
DDX3X rescue.
HeLa cells were seeded on coverslips at a concentration of 2.0 × 104 cells/well. Cells were transfected for 24 h with 25 nM siRNA against DDX3X (siDDX3Xa) or with transfection agent only as control. Cells were then transfected a second time with an siRNA-resistant DDX3X construct using LipoD293 reagent for a further 24 h. They were finally infected with HSV-1 K26GFP at an MOI of 5 for 18 h. The cells were then fixed and permeabilized as described above and examined with an Axiophot epifluorescence microscope. Quantification of fluorescence signals was done with ImageJ (version 1.48), and values were normalized per cell areas.
Entry assay.
143B cells were seeded in 24-well plates 24 h before infection at a concentration of 1 ×105 cells/well. Cells were inoculated with wild-type HSV-1, untreated HSV-1(17+)Lox-Luc, depleted viruses (see above), or, as a control, no virus at all under conditions that enable the virus to bind the cells but not penetrate them (MOI of 30; 4°C for 1 h). Samples were then shifted to 37°C for another 1 h and lysed for 30 min at room temperature with 100 μl/well of lysis buffer from a firefly luciferase assay kit (Biotium). Samples were then transferred to 96-well plates and analyzed by a LUMIstar Galaxy luminometer (BMG Labtech). Luminescence was quantified using LUMIstar Galaxy software, version 4.30-0.
qRT-PCR.
Following DDX3X depletion or overexpression (see above), HeLa cells were infected with wild-type HSV-1 for 8 h. Total RNA was extracted using an SV Total RNA Isolation System (Promega). The RNA was then reverse transcribed with a high-capacity cDNA reverse transcription kit (Applied Biosystems) according to the manufacturer's instructions. The cDNA was then analyzed by quantitative PCR using a LightCycler 480 (Roche). Viral (ICP0, ICP4, UL23, ICP8, VP16, and UL31) or cellular (DDX3X and IFN-β) genes were quantified using the standard curve method and normalized to an endogenous control (glyceraldehyde-3-phosphate dehydrogenase [GAPDH]). All PCRs were performed using SYBR Green (Molecular Probes), and primers are as indicated in Table 2.
TABLE 2.
PCR primers
| Gene | Protein | Forward primer (5′ → 3′) | Reverse primer (5′ → 3′) |
|---|---|---|---|
| RL2 | ICP0 | CTGTCGCCTTACGTGAACAA | CATCCAGAGGCTGTTCCACT |
| RS1 | ICP4 | CGACACGGATCCACGACCC | GATCCCCCTCCCGCGCTTCGTCCG |
| UL23 | TK | GTAATGACAAGCGCCCAGAT | ATGCTGCCCATAAGGTATCG |
| UL29 | ICP8 | ACATTACGTTCACGGCCTTC | GGCCATCGACACGATAGACT |
| UL48 | VP16 | GGACGAGCTCCACTTAGACG | AGGGCATCGGTAAACATCTG |
| UL31 | pUL31 | GTGAAGACCACTCCCGTCTC | ATCGTGTTGATCTGCTGCAC |
| UL55 | gB | TTTGTGTACATGTCCCCGTTTTAC | AGAAGCCGTCGACCTGCTT |
| DDX3X | DDX3X | TGCTGGCCTAGACCTGAACT | TTGATCCACTTCCACGATCA |
| GAPDH | GAPDH | GAGTCAACGGATTTGGTCGT | TTGATTTTGGAGGGATCTCG |
| IFN-β | IFN-β | AAACTCATGAGCAGTCTGCA | AGGAGATCTTCAGTTTCGGAGG |
Viral genome copies.
DDX3X was either depleted or overexpressed in HeLa cells as detailed above. Cells were then infected with wild-type HSV-1 for 8 h. Total DNA was purified from each condition using GenElute Mammalian Genomic DNA Miniprep kits (Sigma) as per the manufacturer's instructions. For the qPCR analysis, gB-specific primers (Table 2) were used with the above SYBR Green assay using GAPDH as the internal control.
Electron microscopy.
HeLa cells seeded in 10-cm dishes were either depleted of DDX3X or VP16 with an siRNA or transfected to overexpress DDX3X. Forty-eight hours later, the cells were infected with HSV-1 strain 17+ for 18 h. Samples were subsequently fixed (2.5% glutaraldehyde, 2% paraformaldehyde, 0.1 M cacodylate buffer, pH 7.2). Fixed cells were washed twice in 0.1 M cacodylate buffer, pH 7.2, and spun at 3,300 × g. The cells were resuspended in postfixation buffer (1% osmium tetroxide, 0.1 M cacodylate buffer) for 1 h at 4°C. Next, samples were gradually dehydrated using ethanol at 30%, 50%, 70%, 95%, and 100%. Cells were permeabilized with propylene oxide and then embedded in Epon [Epon 812; dodecenyl succinic anhydride (DDSA), nadic methyl anhydride (NMA) plus tri(dimethyl amino methyl) phenol (DMP-30)]. Embedded samples were cut with a Leica (MZ6) Ultracut UCT ultramicrotome (80- to 90-nm thickness). A Phillips 300 transmission electron microscope was used to analyze the sections.
Statistical analysis.
Virus titers, protein abundance, and fluorescence were normalized to the values obtained for the controls as mentioned in each figure legend and analyzed with bilateral Student's t tests using GraphPad Prism, version 5 (GraphPad Software).
ACKNOWLEDGMENTS
We are indebted to Arvind Patel, Helena Browne, James R. Smiley, Joel D. Baines, Giulio Superti-Furga, Takeshi Sekiguchi, and Prashant Desai for antibodies, viruses, plasmids, and cell lines. We also thank Diana Matheoud for her kind guidance to determine interferon levels and for her critical readings of the manuscript.
This study was funded by the Canadian Institutes of Health Research to R.L. (grant MOP258030). B.S. has been funded by the Deutsche Forschungsgemeinschaft (German Research Council; Program Project Grant SFB900, TP C2; Excellence Cluster EXC 62/1, Rebirth—From Regenerative Biology to Reconstructive Therapy).
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
We declare that we have no conflicts of interest.
REFERENCES
- 1.Ditton HJ, Zimmer J, Kamp C, Rajpert-De Meyts E, Vogt PH. 2004. The AZFa gene DBY (DDX3Y) is widely transcribed but the protein is limited to the male germ cells by translation control. Hum Mol Genet 13:2333–2341. doi: 10.1093/hmg/ddh240. [DOI] [PubMed] [Google Scholar]
- 2.Sharma D, Jankowsky E. 2014. The Ded1/DDX3 subfamily of DEAD-box RNA helicases. Crit Rev Biochem Mol Biol 49:343–360. doi: 10.3109/10409238.2014.931339. [DOI] [PubMed] [Google Scholar]
- 3.Ariumi Y, Kuroki M, Abe K, Dansako H, Ikeda M, Wakita T, Kato N. 2007. DDX3 DEAD-box RNA helicase is required for hepatitis C virus RNA replication. J Virol 81:13922–13926. doi: 10.1128/JVI.01517-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Randall G, Panis M, Cooper JD, Tellinghuisen TL, Sukhodolets KE, Pfeffer S, Landthaler M, Landgraf P, Kan S, Lindenbach BD, Chien M, Weir DB, Russo JJ, Ju J, Brownstein MJ, Sheridan R, Sander C, Zavolan M, Tuschl T, Rice CM. 2007. Cellular cofactors affecting hepatitis C virus infection and replication. Proc Natl Acad Sci U S A 104:12884–12889. doi: 10.1073/pnas.0704894104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ko C, Lee S, Windisch MP, Ryu WS. 2014. DDX3 DEAD-box RNA helicase is a host factor that restricts hepatitis B virus replication at the transcriptional level. J Virol 88:13689–13698. doi: 10.1128/JVI.02035-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vashist S, Urena L, Chaudhry Y, Goodfellow I. 2012. Identification of RNA-protein interaction networks involved in the norovirus life cycle. J Virol 86:11977–11990. doi: 10.1128/JVI.00432-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chahar HS, Chen S, Manjunath N. 2013. P-body components LSM1, GW182, DDX3, DDX6 and XRN1 are recruited to WNV replication sites and positively regulate viral replication. Virology 436:1–7. doi: 10.1016/j.virol.2012.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yedavalli VS, Neuveut C, Chi YH, Kleiman L, Jeang KT. 2004. Requirement of DDX3 DEAD box RNA helicase for HIV-1 Rev-RRE export function. Cell 119:381–392. doi: 10.1016/j.cell.2004.09.029. [DOI] [PubMed] [Google Scholar]
- 9.Wang H, Kim S, Ryu WS. 2009. DDX3 DEAD-Box RNA helicase inhibits hepatitis B virus reverse transcription by incorporation into nucleocapsids. J Virol 83:5815–5824. doi: 10.1128/JVI.00011-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Soulat D, Burckstummer T, Westermayer S, Goncalves A, Bauch A, Stefanovic A, Hantschel O, Bennett KL, Decker T, Superti-Furga G. 2008. The DEAD-box helicase DDX3X is a critical component of the TANK-binding kinase 1-dependent innate immune response. EMBO J 27:2135–2146. doi: 10.1038/emboj.2008.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gu L, Fullam A, Brennan R, Schroder M. 2013. Human DEAD box helicase 3 couples IκB kinase epsilon to interferon regulatory factor 3 activation. Mol Cell Biol 33:2004–2015. doi: 10.1128/MCB.01603-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang H, Ryu WS. 2010. Hepatitis B virus polymerase blocks pattern recognition receptor signaling via interaction with DDX3: implications for immune evasion. PLoS Pathog 6:e1000986. doi: 10.1371/journal.ppat.1000986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schroder M, Baran M, Bowie AG. 2008. Viral targeting of DEAD box protein 3 reveals its role in TBK1/IKKepsilon-mediated IRF activation. EMBO J 27:2147–2157. doi: 10.1038/emboj.2008.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pellett PE, Roizman B. 2007. The family Herpesviridae: a brief introduction, p 2479–2500. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 15.Weir JP. 2001. Regulation of herpes simplex virus gene expression. Gene 271:117–130. doi: 10.1016/S0378-1119(01)00512-1. [DOI] [PubMed] [Google Scholar]
- 16.Loret S, Guay G, Lippé R. 2008. Comprehensive characterization of extracellular herpes simplex virus type 1 virions. J Virol 82:8605–8618. doi: 10.1128/JVI.00904-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Loret S, Lippé R. 2012. Biochemical analysis of infected cell polypeptide (ICP)0, ICP4, UL7 and UL23 incorporated into extracellular herpes simplex virus type 1 virions. J Gen Virol 93:624–634. doi: 10.1099/vir.0.039776-0. [DOI] [PubMed] [Google Scholar]
- 18.Everett RD. 2000. ICP0, a regulator of herpes simplex virus during lytic and latent infection. Bioessays 22:761–770. doi:. [DOI] [PubMed] [Google Scholar]
- 19.Watson RJ, Clements JB. 1980. A herpes simplex virus type 1 function continuously required for early and late virus RNA synthesis. Nature 285:329–330. doi: 10.1038/285329a0. [DOI] [PubMed] [Google Scholar]
- 20.Yang WC, Devi-Rao GV, Ghazal P, Wagner EK, Triezenberg SJ. 2002. General and specific alterations in programming of global viral gene expression during infection by VP16 activation-deficient mutants of herpes simplex virus type 1. J Virol 76:12758–12774. doi: 10.1128/JVI.76.24.12758-12774.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stegen C, Yakova Y, Henaff D, Nadjar J, Duron J, Lippé R. 2013. Analysis of virion-incorporated host proteins required for herpes simplex virus type 1 infection through a RNA interference screen. PLoS One 8:e53276. doi: 10.1371/journal.pone.0053276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sekiguchi T, Iida H, Fukumura J, Nishimoto T. 2004. Human DDX3Y, the Y-encoded isoform of RNA helicase DDX3, rescues a hamster temperature-sensitive ET24 mutant cell line with a DDX3X mutation. Exp Cell Res 300:213–222. doi: 10.1016/j.yexcr.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 23.Desai P, Person S. 1998. Incorporation of the green fluorescent protein into the herpes simplex virus type 1 capsid. J Virol 72:7563–7568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Soto-Rifo R, Ohlmann T. 2013. The role of the DEAD-box RNA helicase DDX3 in mRNA metabolism. Wiley Interdiscip Rev RNA 4:369–385. doi: 10.1002/wrna.1165. [DOI] [PubMed] [Google Scholar]
- 25.Pene V, Li Q, Sodroski C, Hsu CS, Liang TJ. 2015. Dynamic interaction of stress granules, DDX3X, and IKK-α mediates multiple functions in hepatitis C virus infection. J Virol 89:5462–5477. doi: 10.1128/JVI.03197-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nagel CH, Pohlmann A, Sodeik B. 2014. Construction and characterization of bacterial artificial chromosomes (BACs) containing herpes simplex virus full-length genomes. Methods Mol Biol 1144:43–62. doi: 10.1007/978-1-4939-0428-0_4. [DOI] [PubMed] [Google Scholar]
- 27.Shih JW, Tsai TY, Chao CH, Wu Lee YH. 2008. Candidate tumor suppressor DDX3 RNA helicase specifically represses cap-dependent translation by acting as an eIF4E inhibitory protein. Oncogene 27:700–714. doi: 10.1038/sj.onc.1210687. [DOI] [PubMed] [Google Scholar]
- 28.Wren JD, Conway T. 2006. Meta-analysis of published transcriptional and translational fold changes reveals a preference for low-fold inductions. OMICS 10:15–27. doi: 10.1089/omi.2006.10.15. [DOI] [PubMed] [Google Scholar]
- 29.Lippé R. 2012. Deciphering novel host-herpesvirus interactions by virion proteomics. Front Microbiol 3:181. doi: 10.3389/fmicb.2012.00181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chao CH, Chen CM, Cheng PL, Shih JW, Tsou AP, Lee YH. 2006. DDX3, a DEAD box RNA helicase with tumor growth-suppressive property and transcriptional regulation activity of the p21waf1/cip1 promoter, is a candidate tumor suppressor. Cancer Res 66:6579–6588. doi: 10.1158/0008-5472.CAN-05-2415. [DOI] [PubMed] [Google Scholar]
- 31.Pomerantz JL, Baltimore D. 1999. NF-kappaB activation by a signaling complex containing TRAF2, TANK and TBK1, a novel IKK-related kinase. EMBO J 18:6694–6704. doi: 10.1093/emboj/18.23.6694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Paladino P, Mossman KL. 2009. Mechanisms employed by herpes simplex virus 1 to inhibit the interferon response. J Interferon Cytokine Res 29:599–607. doi: 10.1089/jir.2009.0074. [DOI] [PubMed] [Google Scholar]
- 33.Schroder M. 2011. Viruses and the human DEAD-box helicase DDX3: inhibition or exploitation? Biochem Soc Trans 39:679–683. doi: 10.1042/BST0390679. [DOI] [PubMed] [Google Scholar]
- 34.Turcotte S, Letellier J, Lippé R. 2005. Herpes simplex virus type 1 capsids transit by the trans-Golgi network, where viral glycoproteins accumulate independently of capsid egress. J Virol 79:8847–8860. doi: 10.1128/JVI.79.14.8847-8860.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Angus AG, Dalrymple D, Boulant S, McGivern DR, Clayton RF, Scott MJ, Adair R, Graham S, Owsianka AM, Targett-Adams P, Li K, Wakita T, McLauchlan J, Lemon SM, Patel AH. 2010. Requirement of cellular DDX3 for hepatitis C virus replication is unrelated to its interaction with the viral core protein. J Gen Virol 91:122–132. doi: 10.1099/vir.0.015909-0. [DOI] [PMC free article] [PubMed] [Google Scholar]