

ABSTRACT
The viral genotype has been shown to play an important role in HIV pathogenesis following transmission. However, the viral phenotypic properties that contribute to disease progression remain unclear. Most studies have been limited to the evaluation of Gag function in the context of a recombinant virus backbone. Using this approach, important biological information may be lost, making the evaluation of viruses obtained during acute infection, representing the transmitted virus, a more biologically relevant model. Here, we evaluate the roles of viral infectivity and the replication capacity of viruses from acute infection in disease progression in women who seroconverted in the CAPRISA 004 tenofovir microbicide trial. We show that viral replication capacity, but not viral infectivity, correlates with the set point viral load (Spearman r = 0.346; P = 0.045) and that replication capacity (hazard ratio [HR] = 4.52; P = 0.01) can predict CD4 decline independently of the viral load (HR = 2.9; P = 0.004) or protective HLA alleles (HR = 0.61; P = 0.36). We further demonstrate that Gag-Pro is not the main driver of this association, suggesting that additional properties of the transmitted virus play a role in disease progression. Finally, we find that although viruses from the tenofovir arm were 2-fold less infectious, they replicated at rates similar to those of viruses from the placebo arm. This indicates that the use of tenofovir gel did not select for viral variants with higher replication capacity. Overall, this study supports a strong influence of the replication capacity in acute infection on disease progression, potentially driven by interaction of multiple genes rather than a dominant role of the major structural gene gag.
IMPORTANCE HIV disease progression is known to differ between individuals, and defining which fraction of this variation can be attributed to the virus is important both clinically and epidemiologically. In this study, we show that the replication capacity of viruses isolated during acute infection predicts subsequent disease progression and drives CD4 decline independently of the viral load. This provides further support for the hypothesis that the replication capacity of the transmitted virus determines the initial damage to the immune system, setting the pace for later disease progression. However, we did not find evidence that the major structural gene gag drives this correlation, highlighting the importance of other genes in determining disease progression.
KEYWORDS: HIV, replication capacity, acute infection, disease progression, tenofovir, CAPRISA 004, infectivity, isolates, heterosexual transmission, pathogenesis
INTRODUCTION
HIV disease progression is well known to differ between individuals and populations (1). If left untreated, 70 to 80% of patients develop AIDS within 3 to 10 years. However, some succumb earlier (rapid progressors) while others progress more slowly or control their disease (1, 2). Defining which fraction of this variation can be attributed to the host as opposed to viral characteristics is important both clinically and epidemiologically.
Several host mechanisms have been identified that correlate with disease progression, the most important of which is the HLA background of individuals. Protective HLA I alleles, such as B*57, B*58:01, and B*81 (3, 4), are associated with strong CD8+ T-cell responses that target vulnerable regions of the HIV genome, thereby influencing the viral load (VL) and slowing down CD4+ T-cell decline. The stabilized VL reached after acute infection, referred to as the VL set point, is often used as a marker of disease progression, as it is associated with time to AIDS (5). However, genome-wide association studies estimate that host factors account for only 15 to 25% of the variation in set point VL (6, 7), and several studies have implied a role for viral factors. In transmission pairs, a strong correlation has been observed between the VLs of the donor and the recipient (8–12), demonstrating that the VL set point is at least partially heritable. Furthermore, there is evidence that transmission of viruses harboring cytotoxic T-lymphocyte escape or drug resistance mutations is associated with lower VL and higher CD4+ counts in the recipient (13–16). Overall, the viral genotype is estimated to account for 20 to 46% of the variation in the VL set point (17).
Exactly how viral factors contribute to disease progression remains unclear but is likely to involve the intrinsic replication capacity (RC) of a virus and/or its ability to induce persistent immune activation. In chronically infected individuals, it is well established that the in vitro HIV RC correlates with the concomitant plasma VL (18–21). However, less is known about how characteristics of acute and early viruses impact HIV pathogenesis, with some evidence suggesting that HIV transmission selects for viruses with high viral infectivity and RC (22, 23). It is plausible that during these early stages of infection, prior to adaptive immune responses, the infectivity and/or the RC of a virus contributes to the initial damage to the immune system, influencing later stages of infection. Recent studies on acutely subtype C-infected individuals from the Zambia Emory HIV Research Project (ZEHRP) revealed associations between the in vitro RC of Gag-MJ4 recombinant viruses and set point VL and CD4 decline (24, 25). However, using a similar approach, we and others did not find convincing associations between the in vitro RC of early Gag–Pro–NL4-3 recombinant viruses and disease progression (26, 27).
We therefore propose that additional properties of the transmitted virus drive clinical progression. Although in chronic infection Gag is a proven major determinant of viral fitness, other genes, such as env (28–30) and nef (31–33), are known to significantly impact HIV replication and pathogenesis. Moreover, the construction of recombinant viruses may artificially change the phenotype of interest. This was shown for env recombinants, whose neutralization sensitivity is impacted by the backbone subtype (34) or the cells used to generate the virus (35). Finally, the use of cloned viruses often implies the selection of one or a few variants as opposed to evaluating quasispecies (36). This can be confounding, especially in cases of multivariant or high-diversity transmission.
Therefore, in the current study, we took a different, more in vivo-relevant approach by isolating viruses from acutely infected individuals, which has been shown to preserve the phenotypic features of the circulating quasispecies in vivo (37). We then investigated whether viral infectivity and/or the RC impacted VL and CD4 decline in women who seroconverted in the CAPRISA 004 microbicide trial, which evaluated the use of 1% tenofovir (TFV) gel for the prevention of HIV infection. In this cohort, there was a high percentage (38.6%) of rapid progressors (38). We show that viral RC, but not viral infectivity, correlates with set point VL and CD4 decline and further demonstrate that the viral RC can drive CD4 decline independently of the VL or protective HLA alleles. Finally, we found that viruses from the TFV arm were less infectious and replicated at rates similar to those of viruses from the placebo (PLB) arm. This is important, as women assigned to the TFV gel arm had higher VL set points than those in the PLB arm, which raised concern that the drug might have selected for fitter viral variants (39).
RESULTS
Primary isolates.
To evaluate the roles of viral infectivity and the RC of acute infection viruses on disease progression, we isolated plasma virus from women who became HIV-1 infected while participating in the 1% TFV microbicide gel trial (40). Samples (n = 48) were obtained within 3 months of infection as a proxy for the transmitted virus. A total of 24 plasma samples from the TFV arm and 24 samples from the PLB arm were matched for time postinfection and the presence of protective HLA alleles (B*57, B*58:01, and B*81:01). Virus was successfully isolated from 39 plasma samples (20 PLB arm and 19 TFV arm samples) collected a median of 37 (range, 20 to 87) days postinfection (Table 1). All primary isolates were subtype C in their Gag and Nef sequences, with the exception of one A/D recombinant (CAP353) (27). Interestingly, one virus (CAP337) was phenotypically confirmed to be CXCR4 tropic (X4), while the remaining viruses were identified as CCR5 tropic (data not shown).
TABLE 1.
Characteristics of primary isolatesa
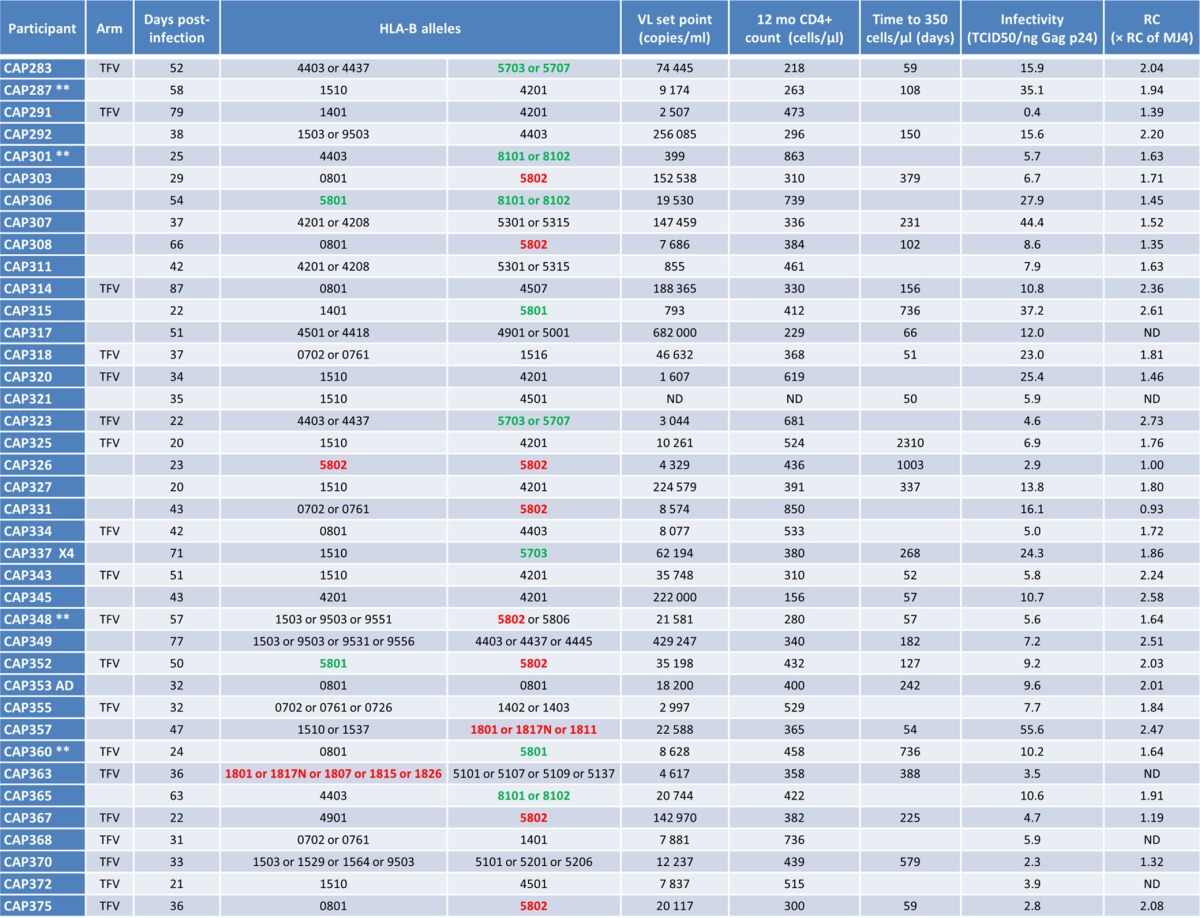
To evaluate whether the isolated viruses were representative of the in vivo plasma virus at the time of sampling, for 24 participants, we sequenced the Env V3V5 regions of multiple variants in the isolate and plasma samples and measured the genetic distance to the plasma consensus sequence (assumed to represent the transmitted/founder virus). The maximum DNA distance from the consensus (plasma, median = 0.2%, range = 0.0 to 1.7%; isolate, median = 0.2%, range = 0.0 to 1.5%) did not differ between plasma and isolate sequences (P = 0.1765; Wilcoxon signed-rank test). This was further confirmed by phylogenetic analysis showing that isolate sequences were dispersed within plasma-derived sequences (data not shown). Typical of early infection, most participants (21/24) exhibited a low-diversity quasispecies in plasma, with mean intrapatient DNA distances of 0.0 to 0.6% (median, 0.17%). For the three participants with higher diversity in vivo (CAP348, CAP360, and CAP375), virus isolation was able to capture multiple variants in at least two of the three participants (data not shown). Together, these data imply that the viral isolates are a good representation of the plasma virus in vivo.
Viral infectivity does not correlate with clinical markers of disease progression.
Particle infectivity was measured using the established cervical cell line TZM-bl and calculated as the infectious titer (50% tissue culture infectious dose [TCID50]) per nanogram normalized for the amount of input virus. This reporter cell line is Tat inducible and expresses high levels of both CD4 and CCR5/CXCR4 cellular receptors. Infectivity on TZM-bl cells did not correlate with the concomitant VL or with the VL or CD4+ count at 3 months postinfection (data not shown). At 12 months postinfection, a weak but insignificant correlation (Spearman r = 0.299; P = 0.068) was observed between viral infectivity and the VL set point, but no correlation was observed with the 12-month CD4+ count (Fig. 1A and B). Comparable results were obtained when infectivity was normalized per million viral RNA copies (with VL, Spearman r = 0.276, P = 0.094; with CD4, Spearman r = −0.228, P = 0.169). As expected, the X4 virus (Fig. 1A, red dot) had a higher-than-median infectivity (Fig. 1A and B and Table 1). Of note, a positive correlation (Spearman r = 0.436; P = 0.010) was observed between viral infectivity and the RC (data not shown), measured as described below.
FIG 1.

Associations between viral infectivity or replication capacity of viruses from acute infection and clinical markers of disease progression. Viral infectivity, calculated as the TCID50 per nanogram of Gag p24, did not correlate with the viral load set point (A) or the CD4+ T-cell count, calculated as the geometric mean of three consecutive visits over 12 months postinfection (B). (C and D) The same markers of disease progression were compared to viral replication capacity relative to the subtype C reference strain MJ4, revealing statistically significant correlations. Trend lines with 95% confidence intervals were generated using linear regression in order to visualize correlations. Blue dots, viruses derived from participants harboring protective HLA-B alleles (B*57, B*58:01, and B*81:01); red, CXCR4 tropic; green, subtype A/D recombinant in Gag and Nef.
The viral RC is associated with the 12-month VL set point and CD4+ count.
The viral RC was measured using the Tat-inducible green fluorescent protein (GFP) reporter T-cell line CEM-GXR (41) and ranged from 0.93 to 2.73 (median, 1.8) times the RC of the subtype C reference strain MJ4 (Table 1) (42). This is in line with previously reported MJ4-normalized RC data ranging from 0.01 to 3.5 times (24). The A/D recombinant CAP353 and the X4 CAP337 did not display a higher-than-median RC and were not associated with faster disease progression (Table 1).
As expected, due to the large fluctuations in the VL during acute infection, we found no correlations between the RC and the concomitant VL measured in the sample used for virus isolation. At 3 months postinfection, however, we observed a negative correlation of the RC with the CD4+ count (Spearman r = −0.452; P = 0.007) but no correlation with the VL (Spearman r = 0.267; P = 0.127). More importantly, at 12 months postinfection, an elevated RC was consistently associated with higher VLs and lower CD4+ counts (with VL, Spearman r = 0.346, P = 0.045; with CD4, Spearman r = −0.461, P = 0.006) (Fig. 1C and D).
As class I HLA-B alleles B*57, B*58:01, B*81:01, and B*39:10 were shown to be protective in this population (38, 43), we then excluded participants who harbored these alleles (Fig. 1, blue dots) and found an even stronger association between the RC and the VL or CD4+ count (Spearman r = 0.552, P = 0.003, and r = −0.562, P = 0.003, respectively), indicating that this association was not driven by the presence of these alleles. When only participants with protective HLA-B alleles were considered for analysis (n = 9), the correlation with the CD4+ count could still be observed, although it lost significance (Spearman r = −0.483; P = 0.194).
To evaluate the effect of the RC on disease progression rather than on a mean CD4+ count value over 12 months, a Kaplan-Meier survival analysis was performed with the endpoint defined as a CD4+ count below 350 cells/μl. In this analysis, the RC, expressed categorically as high (RC 2.04 to 2.73 times that of MJ4), middle (RC 1.64 to 2.01 times that of MJ4), or low (RC 0.93 to 1.64 times that of MJ4), was consistently associated with faster CD4 decline (Fig. 2).
FIG 2.
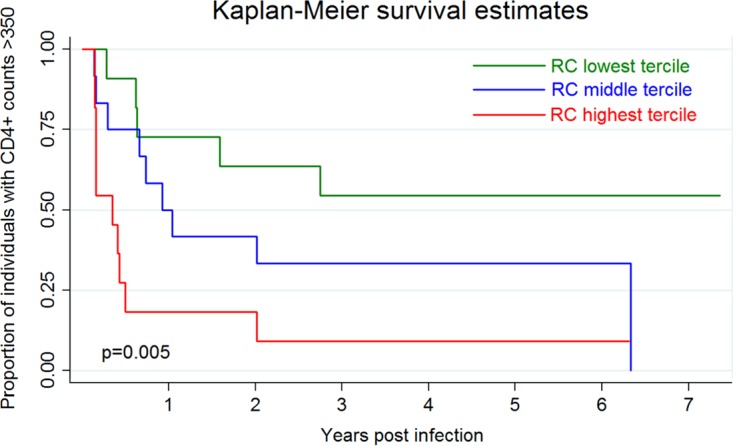
Viral RC drives CD4 decline. Kaplan-Meier survival analysis was performed to evaluate the effect of the viral RC on disease progression, defined as the time to a CD4+ T-cell count below 350 cells/μl. The curves represent the lowest, middle, and highest terciles of RCs and are compared with the log-rank test.
The viral RC drives CD4 decline independently of the VL.
As the VL is a well-known driver of CD4 decline, we evaluated the individual effects of the VL and the RC in a multivariable Cox proportional-hazard model (Table 2). To adjust for the presence of detrimental (B*58:02 and B*18) and protective (B*57, B*58:01, and B*81:01) HLA-B alleles, as well as potential differences between trial arms, we included these variables in our model. Although, as expected, the VL set point had a significant impact on CD4 decline (hazard ratio [HR] = 2.9), the viral RC remained a strong and independent predictor of CD4 decline (HR = 4.5) (Table 2). This independent effect is further illustrated by CAP287 and CAP308 who, despite early viral control (<10,000 copies/ml at set point), progressed to a CD4+ count below 350 cells/μl within just 4 months of infection (Table 1).
TABLE 2.
Host and viral characteristics independently predict CD4+ T cell decline
| Factor | Hazard ratioa | 95% CIb | P value |
|---|---|---|---|
| Tenofovir arm | 1.65 | 0.65–4.18 | 0.291 |
| Protective HLAs | 0.61 | 0.21–1.75 | 0.355 |
| Detrimental HLAs | 3.00 | 1.22–7.39 | 0.017 |
| Replication capacity | 4.52 | 1.43–14.3 | 0.010 |
| Viral load set point | 2.90 | 1.40–5.99 | 0.004 |
A multivariable Cox proportional-hazards model was used to evaluate the individual effects of the trial arm, host's HLA alleles, viral replication capacity (continuous variable), and viral load set point (log10 transformed) on disease progression using a single CD4+ T-cell count below 350 cells/μl as the endpoint.
CI, confidence interval.
When the RC was included as a categorical rather than a continuous variable, comparable hazard ratios were observed for both the lowest and middle RC terciles (HRs of 0.15 and 0.29, respectively, with overlapping confidence intervals), suggesting that the highest RC tercile largely drives CD4 decline (data not shown). This is illustrated by the fact that of the 11 participants infected with a high-RC virus, 9 progressed to a CD4+ count below 350 cells/μl within 1 year (Table 1). The remaining 2 individuals, CAP315 and CAP323, had protective HLAs and controlled VL, suggesting they mounted an effective immune response to the high-RC virus. CAP315, however, lost control of the VL after 12 months, possibly due to the presence of multiple B*57/58-linked polymorphisms associated with cytotoxic T-lymphocyte escape (T242N, A146P, and I147L) and compensation (I223V), which were not seen to revert by 12 months postinfection (data not shown). As this virus was isolated only 22 days after transmission, these mutations could have been transmitted (Table 1).
The presence of detrimental HLA-B alleles (B*58:02 and B*18) was significantly associated with increased risk (HR = 3.0) (Table 2). Interestingly, the presence of favorable alleles did not significantly impact disease progression in our sample set (25). Finally, as shown previously (39), being assigned to the TFV arm did not impact disease progression.
Gag-Pro does not drive the association between viral RC and disease progression.
Previously, we evaluated the RC of Gag–Pro–NL4-3 recombinant viruses derived from 75 participants (27). In the current study, we isolated HIV from 32 of these participants, allowing us to compare the RC of the full-genome virus to the Gag-Pro-mediated RC. Interestingly, the Gag-Pro-mediated RC did not correlate with the viral RC (Spearman r = −0.020; P = 0.91) (Fig. 3A), suggesting that the overall replicative fitness of the cognate viruses was driven by factors other than, or in addition to, the Gag-Pro function.
FIG 3.

Gag-Pro does not determine viral replication capacity. The replication capacity of Gag–Pro–NL4-3 recombinants (A), as well as the number of HLA-I-associated amino acid polymorphisms in Gag (B), did not correlate with the overall replication capacity of viruses isolated from the same participants. Both Gag-Pro-mediated and overall replication capacities were measured in the GFP reporter cell line CEM-GXR25 and were expressed relative to the subtype B strain NL4-3 and to the subtype C strain MJ4, respectively.
We then analyzed the contemporary Gag sequences generated from these participants, since the number of HLA-I-associated amino acid polymorphisms (occurring in or within five amino acids of well-defined, optimal epitopes [13]) was previously shown to negatively impact the RC (21, 26). While the number of HLA-I-associated polymorphisms did indeed correlate negatively with the Gag-Pro-mediated RC in our participant set (Spearman r = −0.253; P = 0.038; n = 68) (Fig. 4), the same was not seen with the virus RC (Spearman r = 0.196; P = 0.274; n = 33) (Fig. 3B). This suggests that although the accumulation of these polymorphisms reduced fitness, they did not impact the overall RC of the cognate viruses, further supporting a driving role for factors other than Gag-Pro influencing the viral RC.
FIG 4.

The total number of HLA-I-associated amino acid polymorphisms inversely correlates with Gag-Pro-mediated replication capacity. Polymorphisms were defined as occurring in or within 5 amino acids of well-defined, optimal epitopes in Gag (13). A trend line with 95% confidence interval was generated using linear regression to visualize the correlation.
Finally, in the Zambian ZEHRP cohort, Prince et al. and Claiborne et al. showed associations between the RCs of Gag-MJ4 recombinants and markers of disease progression (24, 25), as well as a negative association between the number of HLA-I-associated polymorphisms in Gag and the VL set point (13, 24). However, when we performed a similar analysis on our cohort for 71 to 73 participants (Fig. 5), we found that neither the Gag-Pro-mediated RC nor the number of HLA-I-associated polymorphisms correlated with the VL set point or 12-month CD4+ count (Fig. 5A, B, and D). We did, however, find a moderate but statistically insignificant correlation between Gag-Pro-mediated RC and the VL set point (Spearman r = 0.214; P = 0.073; n = 71) (Fig. 5C). When evaluated in a Cox proportional-hazard model, the Gag-Pro-mediated RC also did not impact disease progression (HR = 0.51; P = 0.651) (data not shown).
FIG 5.

Gag-Pro does not drive the association between viral replication capacity and disease progression. The number of HLA-I-associated amino acid polymorphisms in Gag (A and B), as well as the replication capacity of Gag–Pro–NL4-3 recombinants (C and D), did not correlate with the viral load set point or CD4+ T-cell count, calculated as the geometric mean of three consecutive visits over 12 months postinfection (reanalysis from reference 27).
Viruses isolated from TFV arm participants were less infectious but did not differ in RC.
Since viruses in this study were isolated from individuals who participated in the 1% TFV microbicide trial, we were interested in evaluating whether viral infectivity and/or the RC differed between trial arms. For this analysis, we considered either all viruses or the 16 TFV-PLB pairs that were matched for time postinfection and the presence of protective HLA-B alleles. Regardless of the sample set used, we found that the median viral RCs were similar between trial arms (Fig. 6). However, viruses from the TFV arm were found to be approximately 2-fold less infectious than viruses isolated from the PLB arm (Fig. 7A). Similar results were obtained when infectivity was normalized per million viral RNA copies (TCID50/million copies, 1.7-fold; Mann-Whitney P = 0.035) (data not shown) or when only the matched samples were considered (TCID50/nanogram p24, 2.26-fold; Mann-Whitney P = 0.0039) (data not shown). To evaluate whether the decreased infectivity in the TFV arm was due to lower viral entry efficiency, we mitigated this rate-limiting step by adding DEAE-dextran or by using spinoculation. As expected, viral infectivity increased by orders of magnitude, yet the difference between the trial arms disappeared (Fig. 7B and C), suggesting a role for viral entry.
FIG 6.

Viruses isolated from TFV arm participants did not differ in replication capacity. Viral replication capacity was expressed relative to the subtype C reference strain MJ4 and compared between the two arms of the 1% TFV microbicide trial. Medians are displayed both as lines and as numbers. Considered for analysis were either all 39 viruses (A) or only 16 TFV-PLB pairs that were matched for time postinfection and the presence of protective HLA-B alleles (B). Statistics were generated using the nonparametric Mann-Whitney U test.
FIG 7.

Viruses isolated from the TFV arm participants were 2-fold less infectious than those from the PLB arm participants. Viral infectivity, calculated as the TCID50 per nanogram of Gag p24, was measured using TZM-bl cells and compared between the two arms of the 1% TFV microbicide trial. Medians are displayed both as lines and as numbers. Infectivity was measured in standard cell culture medium (A), after spinoculation to enhance infectivity (B), or in the presence of the chemical enhancer DEAE-dextran (C). Statistics were generated using the nonparametric Mann-Whitney U test. W/O, without; W/, with.
DISCUSSION
To date, most research has focused on host factors to explain the variation in HIV pathogenesis between individuals. However, new data and analyses have shown that the VL set point is partially heritable (33%) (8, 11, 12), suggesting that transmitted viral factors also play an important role in disease progression and that they are potentially underestimated (17). As HIV rapidly evolves following transmission, it is crucial for pathogenesis studies to elucidate the properties of viruses collected soon after transmission, prior to extensive adaptation in the new host. Recent studies have suggested that HIV transmission selects for viruses with high infectivity and replication capacity (22). In this study, using viruses isolated from acutely infected women, we investigated how these early viral characteristics impact subsequent VL and CD4 decline.
Our results demonstrate that the presence of a virus with high RC during early infection contributes significantly to disease progression, both by increasing the VL set point and by accelerating CD4 decline. Interestingly, this effect on disease progression was strong (HR = 4.5) and independent of host protective HLA-B alleles and the set point VL. This supports the hypothesis that the RC of the transmitted virus determines the extent of damage to the gut-associated lymphoid tissue in the newly infected individual before the onset of an adaptive immune response and viral escape. Gut damage would then drive long-term immune activation and enhance CD4 decline (25).
Using viruses that are biologically representative of circulating virus in vivo, our results augment observations in a Zambian cohort study that used Gag-MJ4 recombinant viruses to investigate the relationship between the RC and disease progression (24, 25). Similarly, the Zambian studies showed that the RC correlates with the VL set point and is an independent predictor of CD4 decline. However, our study also identified some key differences. Although the presence of protective HLA-B alleles had a favorable effect on CD4 decline, as shown by Claiborne et al. (25), this was not significant in our sample set, potentially due to the much smaller sample size and/or the presence of transmitted escape and/or compensatory mutations. Second, in our cohort, the viral RC was more strongly associated with disease progression, explaining, respectively, up to 12% and 21% of the VL set point and the 12-month CD4+ count as opposed to the correlation coefficients reported by Prince et al. (24). This might be related to the fact that we evaluated full viruses as opposed to single-gene recombinant viruses.
Using comparisons with Gag–Pro–NL4-3 recombinant viruses, we found that Gag-Pro alone was not the main driver of this correlation with disease progression. Neither the RC of Gag–Pro–NL4-3 recombinant viruses nor the underlying number of HLA-I-associated polymorphisms in Gag correlated with the RC of viruses isolated from the same participants. Furthermore, in contrast to Prince et al., we did not find statistically significant correlations between Gag-Pro-mediated RC and the VL set point or 12-month CD4+ count. For the correlation with the VL set point, this might be due to a smaller sample size. However, for the association with the CD4+ count, sample sizes between the cohorts were very similar (CAPRISA, n = 71 versus ZEHRP, n = 63) (24). Since we observed strong correlations between the viral RC and disease progression, this implies that in our sample set, Gag-Pro-mediated RC cannot strongly correlate with viral RC. Hence, other genes are likely to be important contributors to the viral RC and as such might mask Gag-Pro influence on disease progression. This is further supported by the fact that, in contrast to the Zambian cohort, the number of HLA-I-associated polymorphisms in Gag did not correlate with the VL set point, indicating that the negative impact of these polymorphisms on the Gag-Pro-mediated RC was further compensated for outside of gag-pro.
The lack of a correlation between the Gag-Pro-mediated RC and the viral RC or disease progression could be due to a lack of compatibility of the subtype C Gag-Pro with the subtype B NL4-3 backbone, an important difference in methodology with the Zambian studies, which used a subtype C backbone (MJ4). However, observations from previous studies do not support this. Using the same NL4-3-based assay, the RC of subtype C/B recombinants from chronic infection was shown to correlate with viral loads and CD4 counts (20), as did the RC of subtype B/B recombinants (21). This indicates that our assay is clinically relevant despite the mismatched subtypes. Moreover, similar to what was reported for subtype B/B recombinants (21), we found a negative correlation between the number of HLA-I-associated polymorphisms in Gag and the Gag-Pro-mediated RC, suggesting that this number is meaningful and is not impacted by the subtype mismatch between the backbone and the Gag-Pro insert.
Since no association was found with disease progression, we did not expect the Gag-Pro-mediated RC to strongly correlate with the viral RC. However, the lack of even a weak association was surprising, given Gag-Pro's role in particle assembly and structure. Methodological differences in generating recombinant viruses as opposed to plasma isolates, including the introduction of genetic changes, might have increased variability, decreasing our power to detect weak correlations with our limited sample size (n = 32). Second, it remains possible that, despite being clinically and biologically relevant, the Gag–Pro–NL4-3 assay used in our cohort is less sensitive than the Gag-MJ4 assay used in the Zambian studies. This would further explain why we did not pick up strong correlations in the smaller studies on acute infection viruses between Gag-Pro function and markers of disease progression (21, 26). However, the latter might also be influenced by demographic differences between the two cohorts. Although certain Gag polymorphisms are known to carry substantial fitness costs, the effect of the transmitted Gag on disease progression ultimately depends on the degree of preadaptation to the recipient's HLA molecules (44–46). Hence, factors such as the cohort's HLA profile and the age of the epidemic can influence this correlation. Furthermore, since inflammation at the genital mucosa may affect the phenotype of the transmitted virus (47) and since both inflammatory profiles (48, 49) and the fitness of the transmitted virus (50) have been shown to differ between men and women, the fact that our cohort was exclusively female may have influenced differences in study findings.
Because previously we did not find a relationship between baseline Nef-mediated CD4+ HLA downregulation and the 12-month VL or CD4+ count (27), we are currently investigating the role of the viral envelope. Indeed, several studies have shown that Env can impact disease progression. Previous studies showed that viruses from elite controllers exhibit reduced entry efficiency (51) and that intraindividual envelope diversity correlates with in vitro RC (28). Although we did not evaluate entry efficiency directly, viral infectivity was measured in a single-cycle assay and hence could be considered an approximate measure of early Env characteristics. As expected, viral infectivity correlated with viral RC. This was previously shown for full-length infectious molecular clones generated from transmitted and nontransmitted viruses (52). However, the lack of a strong association between viral infectivity and disease progression suggests that the viral RC and its subsequent impact on disease progression are determined by the contributions of multiple structural viral genes rather than one major gene.
Finally, of the viruses in this study, 49% were derived from women infected while using a vaginal gel containing 1% TFV. We previously reported that TFV use did not alter the number of transmitted/founder variants identified in these women (53), nor did it select for transmitted drug resistance (54). However, higher VLs were observed in women who became infected in the TFV arm, raising concerns that TFV might have selected for the transmission of fitter variants (39), but our results provide no evidence to support this hypothesis, as viral infectivity was 2-fold lower in the TFV arm and the RCs did not differ between the trial arms. The reduced viral infectivity could be a consequence of genital inflammation. Although TFV gel itself does not cause inflammation (55), it has been reported to prolong preexisting inflammation (56). This would allow transmission of less infectious HIV (47). The difference in infectivity between the trial arms disappeared when viral entry was facilitated chemically or physically, suggesting that TFV arm viruses may have reduced entry efficiency. We are currently constructing pseudoviruses containing the viral isolates' env genes to evaluate entry efficiency in a more direct way.
Overall, this study supports a strong influence of acute viral RC on disease progression, potentially driven by the interaction of multiple genes rather than a dominant role of major structural genes, such as gag. This highlights the importance of evaluating more biologically relevant, full viruses as opposed to single-gene recombinants.
MATERIALS AND METHODS
Study subjects and samples.
Women participating in the CAPRISA 004 study, a randomized, controlled, double-blinded trial to assess the safety and effectiveness of the vaginal microbicide 1% TFV gel (40), underwent monthly HIV testing using 2 rapid HIV antibody tests (Abbott Determine and Unigold). The date of infection was estimated as the midpoint between the last negative and first positive antibody tests or as 14 days before the first positive PCR result if a woman had tested negative on rapid antibody tests but positive on retrospective PCR testing. After diagnosis, participants had VL (Roche Cobas AmpliPrep/TaqMan HIV-1 test, v2) and CD4+ T-cell (TruCount method; BD Biosciences, San Jose, CA) counts performed at each visit. Plasma samples collected within 3 months postinfection were selected for virus isolation from 24 TFV/PLB participant pairs and matched for time postinfection (maximum, 7 days difference) and the presence of protective HLA alleles (B*57, B*58:01, B*81:01, or B*39:10) (38, 43). The University of KwaZulu-Natal and University of Cape Town Research Ethics Committees approved this study, and all participants provided written informed consent.
Cells.
Peripheral blood mononuclear cells (PBMCs) were isolated from buffy coats of healthy blood donors (kindly provided by the Western Province Blood Transfusion Service, Cape Town, South Africa) through Lymphoprep density gradient centrifugation. CEM-GXR25 (41) and TZM-bl cells were maintained in R10 medium (RPMI 1640 medium or Dulbecco's modified Eagle's medium [DMEM] [Lonza, Belgium] containing 10% fetal bovine serum [FBS] [Biochrom], 2 mM glutamine [Lonza], 50 μg gentamicin/ml [Lonza], respectively).
Acute virus isolation.
Virus stocks were isolated from cryopreserved patient plasma using the μMCAS VitalVirus HIV isolation kit (Miltenyi Biotec, Germany) and 3 × 3 stimulation of PBMCs according to the manufacturer's protocol. Briefly, cryopreserved PBMCs from three HIV-seronegative donors were thawed, CD8 depleted using MACS CD8 MicroBeads (Miltenyi Biotec), pooled to 4 × 106/ml in R10 medium, and finally stimulated for 72 h with either 0.5 μg/ml phytohemagglutinin (PHA) (Remel, Thermo Fisher, USA), 5 μg/ml PHA, or the surface-immobilized anti-CD3 monoclonal antibody (MAb) OKT3 (Leaf; Biolegend, USA), i.e., 3 × 3 stimulation. Of note, the use of cryopreserved PBMCs allowed us to passage all the viruses through the same sets of donor PBMCs. Next, virus was purified from 1 ml patient plasma by incubation at room temperature for 30 min with 250 μl anti-CD44 magnetic beads and subsequent binding to μMACS columns (Miltenyi Biotec). Beads with bound virus were washed, eluted in R10 medium supplemented with 200 U/ml interleukin 2 (IL-2) (Gentaur, Belgium), and added to the 3 × 3-stimulated CD8-depleted PBMCs to a final concentration of 2 × 106/ml. The cells were infected by spinoculation for 1 h (1,200 × g; 25°C) and subsequently maintained at a concentration of 1 × 106 to 2 × 106/ml with expansion over 2 to 3 weeks to 30 to 40 ml by weekly addition of fresh 3 × 3-stimulated cells. The culture supernatants were monitored for p24 production using an Allianz HIV-1 p24 antigen enzyme-linked immunosorbent assay (ELISA) kit (PerkinElmer, USA) and were harvested and stored at −80°C on day 25.
Determination of coreceptor usage.
The coreceptor usage of the isolated viruses was determined by preincubating TZM-bl cells with a serial dilution of two CCR5 inhibitors, i.e., maraviroc (NIH Reagent Program) and PSC-RANTES (kindly provided by Oliver Hartley), and one CXCR4 inhibitor, i.e., JM-2987 (NIH Reagent Program). Subsequently, the cells were infected with 200 TCID50 of each virus in the presence of 20 μg/ml DEAE-dextran. After 48 h, luciferase activity was quantified, and nonlinear regression analysis was used to calculate the 50% inhibitory concentration. Viruses were considered R5 tropic if they were inhibited >98% by 5,000 nM maraviroc or 40 nM PSC-RANTES and <5% by 10,000 nM JM-2987.
Single-genome env amplification and sequencing.
Viral RNA was extracted from the isolate stocks and plasma samples using the Roche MagNA Pure Compact System and RNA isolation kit and reverse transcribed using Superscript III RT (Invitrogen, USA) with oligo(dT)20. Env amplicons were generated by limiting dilution of the cDNA template prior to nested PCR using primers that were previously described (57). PCR amplicons were confirmed on gel and sent for Sanger sequencing by the Central Analytical Facilities at the University of Stellenbosch. Sequences were excluded if they contained >3 double peaks in the chromatogram or deletions >30 nucleotides long. To include plasma-derived Env V3V5 sequences that were already available for comparison (53), all other sequences were truncated to the V3V5 region. Sequences were aligned in BioEdit to a subtype C acute (<3-month) consensus sequence, which was generated from available CAPRISA 002 plasma Env sequences (n = 592). MEGA6 (http://www.megasoftware.net/) was used to calculate pairwise DNA distances and to generate a maximum-likelihood tree.
Single-cycle infectivity.
Viral isolates were serially diluted and added in triplicate to 10,000 TZM-bl cells (NIH AIDS Reagent Program) in the presence or absence of 20 μg/ml DEAE-dextran (Sigma) or followed by spinoculation for 1 h (1,200 × g; 25°C). Luciferase activity was quantified after 48 h by adding Steadylite HTS (PerkinElmer) and measuring luminescence with a Promega Glomax 96 luminometer. The relative light units (RLU) generated per volume of virus stock were calculated using all virus dilutions in the linear range of the assay (2,000 to 600,000 RLU). The TCID50 was calculated using the method of Reed and Muench (58), and viral titers were expressed as infectious units per milliliter. Viral infectivity was calculated as TCID50 per nanogram of Gag p24 or per million viral RNA copies in each stock (Roche Cobas AmpliPrep/TaqMan HIV-1 test, v2).
Virus replication assay.
The in vitro RCs of the primary isolates were evaluated using the GFP reporter T-cell line CEM-GXR25. Briefly, 1 × 106 cells were infected in duplicate at a constant multiplicity of infection of 0.06% (on day 2) in a total volume of 1.5 ml. The percentage of GFP-positive (GFP+) cells was measured using flow cytometry (FACSCalibur; BD Biosciences) at days 2 through 10 postinfection by harvesting and refreshing 0.5 ml of the culture. For each virus, the background value measured in mock-treated controls was subtracted, and the natural-log slope of the percentage of GFP+ cells was calculated from day 3 during the exponential phase of viral spread (GFP+ cells < 11%). For ease of interpretation, replication capacities were normalized to the RC of the subtype C reference strain MJ4.
Gag-Pro-mediated RC and sequence analysis.
HIV-1 gag-protease regions were previously amplified from bulk plasma RNA by Chopera et al. (27) (GenBank accession numbers KF208740 to KF208816) and used to construct Gag–Pro–NL4-3 recombinant viruses whose RC was measured by flow cytometry using the CEM-GXR25 cell line (27). For the sequence analysis, HLA-I-associated polymorphisms were defined as occurring in or within 5 amino acids of well-defined, optimal epitopes in chronically clade C-infected individuals (Goepfert et al. [13]). The A/D recombinant CAP353 was excluded from this analysis.
Statistical methods.
Statistical analyses were conducted using GraphPad Prism 5 (GraphPad, USA) and STATA (StataCorp, USA). VL and CD4+ counts were determined as the geometric mean of 3 consecutive visits over 3 or 12 months postinfection, with the exception of CAP317 and CAP375, who started highly active antiretroviral therapy (HAART) at 25 and 34 weeks, respectively, and for whom the last known stable values were used. Correlations were performed using the Spearman rank test, and trend lines and 95% confidence intervals were generated using linear regression. Survival analysis was used to investigate the relationship between the RC and CD4 decline. The endpoint was defined as the time for a single CD4+ count to fall below 350 cells/μl. The first 7 weeks postinfection were excluded to allow CD4+ recovery after acute infection. A Cox proportional-hazards model was used to assess the risk associated with high viral RC with or without adjustment for potential confounding factors. The trial arm and the number of protective and detrimental HLA alleles (B*58:02 or B*18) were included in the model as explanatory variables.
ACKNOWLEDGMENTS
We acknowledge the CAPRISA 004 and the Tenofovir Gel Research for AIDS Prevention Science (TRAPS) study teams for coordinating the collection, processing, storage, and shipping of the samples. We also thank the Western Province Blood Transfusion Service in Cape Town for providing HIV-negative blood and Oliver Hartley for the gift of PSC-RANTES.
This work was supported by a grant from the Clinical Infectious Diseases Research Initiative (CIDRI), South Africa, to Philippe Selhorst, who holds a postdoctoral fellowship from the Claude Leon Foundation and CIDRI. The parent trial (CAPRISA 004) was supported by the United States Agency for International Development (USAID), FHI360 (USAID cooperative agreement GPO-A-00-05-00022-00, contract no. 132119), and the Technology Innovation Agency (LIFElab) of the South African government's Department of Science and Technology. Tenofovir was provided by Gilead Sciences, and the gel was manufactured and supplied for the CAPRISA 004 trial by CONRAD. The current study is part of the CAPRISA Program, which is funded by CONRAD, Eastern Virginia Medical School (USAID cooperative grant GP00-08-00005-00, subproject agreement PPA-09-046). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The views expressed do not necessarily reflect the views of USAID, Gilead Sciences, Eastern Virginia Medical School, or CONRAD.
REFERENCES
- 1.Pantaleo G, Fauci AS. 1996. Immunopathogenesis of HIV infection. Annu Rev Microbiol 50:825–854. doi: 10.1146/annurev.micro.50.1.825. [DOI] [PubMed] [Google Scholar]
- 2.Yue L, Pfafferott KJ, Baalwa J, Conrod K, Dong CC, Chui C, Rong R, Claiborne DT, Prince JL, Tang J, Ribeiro RM, Cormier E, Hahn BH, Perelson AS, Shaw GM, Karita E, Gilmour J, Goepfert P, Derdeyn CA, Allen SA, Borrow P, Hunter E. 2015. Transmitted virus fitness and host T cell responses collectively define divergent infection outcomes in two HIV-1 recipients. PLoS Pathog 11:e1004565. doi: 10.1371/journal.ppat.1004565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leslie A, Matthews PC, Listgarten J, Carlson JM, Kadie C, Ndung'u T, Brander C, Coovadia H, Walker BD, Heckerman D, Goulder PJR. 2010. Additive contribution of HLA class I alleles in the immune control of HIV-1 infection. J Virol 84:9879–9888. doi: 10.1128/JVI.00320-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tang J, Malhotra R, Song W, Brill I, Hu L, Farmer PK, Mulenga J, Allen S, Hunter E, Kaslow RA. 2010. Human leukocyte antigens and HIV type 1 viral load in early and chronic infection: predominance of evolving relationships. PLoS One 5:e9629. doi: 10.1371/journal.pone.0009629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lyles RH, Munoz A, Yamashita TE, Bazmi H, Detels R, Rinaldo CR, Margolick JB, Phair JP, Mellors JW. 2000. Natural history of human immunodeficiency virus type 1 viremia after seroconversion and proximal to AIDS in a large cohort of homosexual men. Multicenter AIDS Cohort Study. J Infect Dis 181:872–880. doi: 10.1086/315339. [DOI] [PubMed] [Google Scholar]
- 6.Fellay J, Shianna KV, Ge D, Colombo S, Ledergerber B, Weale M, Zhang K, Gumbs C, Castagna A, Cossarizza A, Cozzi-Lepri A, De Luca A, Easterbrook P, Francioli P, Mallal S, Martinez-Picado J, Miro JM, Obel N, Smith JP, Wyniger J, Descombes P, Antonarakis SE, Letvin NL, McMichael AJ, Haynes BF, Telenti A, Goldstein DB. 2007. A whole-genome association study of major determinants for host control of HIV-1. Science 317:944–947. doi: 10.1126/science.1143767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McLaren PJ, Coulonges C, Bartha I, Lenz TL, Deutsch AJ, Bashirova A, Buchbinder S, Carrington MN, Cossarizza A, Dalmau J, De Luca A, Goedert JJ, Gurdasani D, Haas DW, Herbeck JT, Johnson EO, Kirk GD, Lambotte O, Luo M, Mallal S, van Manen D, Martinez-Picado J, Meyer L, Miro JM, Mullins JI, Obel N, Poli G, Sandhu MS, Schuitemaker H, Shea PR, Theodorou I, Walker BD, Weintrob AC, Winkler CA, Wolinsky SM, Raychaudhuri S, Goldstein DB, Telenti A, de Bakker PIW, Zagury J-F, Fellay J. 2015. Polymorphisms of large effect explain the majority of the host genetic contribution to variation of HIV-1 virus load. Proc Natl Acad Sci U S A 112:14658–14663. doi: 10.1073/pnas.1514867112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hollingsworth TD, Laeyendecker O, Shirreff G, Donnelly CA, Serwadda D, Wawer MJ, Kiwanuka N, Nalugoda F, Collinson-Streng A, Ssempijja V, Hanage WP, Quinn TC, Gray RH, Fraser C. 2010. HIV-1 transmitting couples have similar viral load set-points in Rakai, Uganda. PLoS Pathog 6:e1000876. doi: 10.1371/journal.ppat.1000876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alizon S, von Wyl V, Stadler T, Kouyos RD, Yerly S, Hirschel B, Böni J, Shah C, Klimkait T, Furrer H, Rauch A, Vernazza PL, Bernasconi E, Battegay M, Bürgisser P, Telenti A, Günthard HF, Bonhoeffer S. 2010. Phylogenetic approach reveals that virus genotype largely determines HIV set-point viral load. PLoS Pathog 6:e1001123. doi: 10.1371/journal.ppat.1001123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hecht FM, Hartogensis W, Bragg L, Bacchetti P, Atchison R, Grant R, Barbour J, Deeks SG. 2010. HIV RNA level in early infection is predicted by viral load in the transmission source. AIDS 24:941–945. doi: 10.1097/QAD.0b013e328337b12e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lingappa JR, Thomas KK, Hughes JP, Baeten JM, Wald A, Farquhar C, de Bruyn G, Fife KH, Campbell MS, Kapiga S, Mullins JI, Celum C, Partners in Prevention HSV/HIV Transmission Study Team. 2013. Partner characteristics predicting HIV-1 set point in sexually acquired HIV-1 among African seroconverters. AIDS Res Hum Retroviruses 29:164–171. doi: 10.1089/aid.2012.0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yue L, Prentice HA, Farmer P, Song W, He D, Lakhi S, Goepfert P, Gilmour J, Allen S, Tang J, Kaslow RA, Hunter E. 2013. Cumulative impact of host and viral factors on HIV-1 viral-load control during early infection. J Virol 87:708–715. doi: 10.1128/JVI.02118-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goepfert PA, Lumm W, Farmer P, Matthews P, Prendergast A, Carlson JM, Derdeyn CA, Tang J, Kaslow RA, Bansal A, Yusim K, Heckerman D, Mulenga J, Allen S, Goulder PJR, Hunter E. 2008. Transmission of HIV-1 Gag immune escape mutations is associated with reduced viral load in linked recipients. J Exp Med 205:1009–1017. doi: 10.1084/jem.20072457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chopera DR, Woodman Z, Mlisana K, Mlotshwa M, Martin DP, Seoighe C, Treurnicht F, de Rosa DA, Hide W, Abdool Karim S, Gray CM, Williamson C. 2008. Transmission of HIV-1 CTL escape variants provides HLA-mismatched recipients with a survival advantage. PLoS Pathog 4:e1000033. doi: 10.1371/journal.ppat.1000033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miura T, Brockman MA, Brumme ZL, Brumme CJ, Pereyra F, Trocha A, Block BL, Schneidewind A, Allen TM, Heckerman D, Walker BD. 2009. HLA-associated alterations in replication capacity of chimeric NL4-3 viruses carrying gag-protease from elite controllers of human immunodeficiency virus type 1. J Virol 83:140–149. doi: 10.1128/JVI.01471-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miura T, Brumme ZL, Brockman MA, Rosato P, Sela J, Brumme CJ, Pereyra F, Kaufmann DE, Trocha A, Block BL, Daar ES, Connick E, Jessen H, Kelleher AD, Rosenberg E, Markowitz M, Schafer K, Vaida F, Iwamoto A, Little S, Walker BD. 2010. Impaired replication capacity of acute/early viruses in persons who become HIV controllers. J Virol 84:7581–7591. doi: 10.1128/JVI.00286-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fraser C, Lythgoe K, Leventhal GE, Shirreff G, Hollingsworth TD, Alizon S, Bonhoeffer S. 2014. Virulence and pathogenesis of HIV-1 infection: an evolutionary perspective. Science 343:1243727. doi: 10.1126/science.1243727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Campbell TB, Schneider K, Wrin T, Petropoulos CJ, Connick E. 2003. Relationship between in vitro human immunodeficiency virus type 1 replication rate and virus load in plasma. J Virol 77:12105–12112. doi: 10.1128/JVI.77.22.12105-12112.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Trkola A, Kuster H, Leemann C, Ruprecht C, Joos B, Telenti A, Hirschel B, Weber R, Bonhoeffer S, Günthard HF, The Swiss HIV Cohort Study. 2003. Human immunodeficiency virus type 1 fitness is a determining factor in viral rebound and set point in chronic infection. J Virol 77:13146–13155. doi: 10.1128/JVI.77.24.13146-13155.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wright JK, Brumme ZL, Carlson JM, Heckerman D, Kadie CM, Brumme CJ, Wang B, Losina E, Miura T, Chonco F, van der Stok M, Mncube Z, Bishop K, Goulder PJR, Walker BD, Brockman MA, Ndung'u T. 2010. Gag-protease-mediated replication capacity in HIV-1 subtype C chronic infection: associations with HLA type and clinical parameters. J Virol 84:10820–10831. doi: 10.1128/JVI.01084-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brockman MA, Brumme ZL, Brumme CJ, Miura T, Sela J, Rosato PC, Kadie CM, Carlson JM, Markle TJ, Streeck H, Kelleher AD, Markowitz M, Jessen H, Rosenberg E, Altfeld M, Harrigan PR, Heckerman D, Walker BD, Allen TM. 2010. Early selection in Gag by protective HLA alleles contributes to reduced HIV-1 replication capacity that may be largely compensated for in chronic infection. J Virol 84:11937–11949. doi: 10.1128/JVI.01086-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parrish NF, Gao F, Li H, Giorgi EE, Barbian HJ, Parrish EH, Zajic L, Iyer SS, Decker JM, Kumar A, Hora B, Berg A, Cai F, Hopper J, Denny TN, Ding H, Ochsenbauer C, Kappes JC, Galimidi RP, West AP, Bjorkman PJ, Wilen CB, Doms RW, O'Brien M, Bhardwaj N, Borrow P, Haynes BF, Muldoon M, Theiler JP, Korber B, Shaw GM, Hahn BH. 2013. Phenotypic properties of transmitted founder HIV-1. Proc Natl Acad Sci U S A 110:6626–6633. doi: 10.1073/pnas.1304288110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iyer SS, Bibollet-Ruche F, Sherrill-Mix S, Learn GH, Plenderleith L, Smith AG, Barbian HJ, Russel RM, Gondim MVP, Bahari CY, Shaw CM, Li Y, Decker T, Haynes BF, Shaw GM, Sharp PM, Borrow P, Hahn B. 2017. Resistance to type 1 interferons is a major determinant of HIV-1 transmission fitness. Proc Natl Acad Sci U S A 114:E590–E599. doi: 10.1073/pnas.1620144114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Prince JL, Claiborne DT, Carlson JM, Schaefer M, Yu T, Lahki S, Prentice HA, Yue L, Vishwanathan SA, Kilembe W, Goepfert P, Price MA, Gilmour J, Mulenga J, Farmer P, Derdeyn CA, Tang J, Heckerman D, Kaslow RA, Allen SA, Hunter E. 2012. Role of transmitted Gag CTL polymorphisms in defining replicative capacity and early HIV-1 pathogenesis. PLoS Pathog 8:e1003041. doi: 10.1371/journal.ppat.1003041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Y DT, Prince JL, Scully E, Macharia G, Micci L, Lawson B, Kopycinski J, Deymier MJ, Vanderford TH, Nganou-Makamdop K, Ende Z, Brooks K, Tang J, Yu T, Lakhi S, Kilembe W, Silvestri G, Douek D, Goepfert PA, Price MA, Allen SA, Paiardini M, Altfeld M, Gilmour J, Hunter E. 2015. Replicative fitness of transmitted HIV-1 drives acute immune activation, proviral load in memory CD4+ T cells, and disease progression. Proc Natl Acad Sci U S A 112:E1480–E1489. doi: 10.1073/pnas.1421607112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wright JK, Novitsky V, Brockman MA, Brumme ZL, Brumme CJ, Carlson JM, Heckerman D, Wang B, Losina E, Leshwedi M, van der Stok M, Maphumulo L, Mkhwanazi N, Chonco F, Goulder PJR, Essex M, Walker BD, Ndung'u T. 2011. Influence of Gag-protease-mediated replication capacity on disease progression in individuals recently infected with HIV-1 subtype C. J Virol 85:3996–4006. doi: 10.1128/JVI.02520-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chopera DR, Mann JK, Mwimanzi P, Omarjee S, Kuang XT, Ndabambi N, Goodier S, Martin E, Naranbhai V, Abdool Karim S, Abdool Karim Q, Brumme ZL, Ndung'u T, Williamson C, Brockman MA. 2013. No evidence for selection of HIV-1 with enhanced Gag-protease or Nef function among breakthrough infections in the CAPRISA 004 tenofovir microbicide trial. PLoS One 8:e71758. doi: 10.1371/journal.pone.0071758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Troyer RM, Collins KR, Abraha A, Fraundorf E, Moore DM, Krizan RW, Toossi Z, Colebunders RL, Jensen MA, Mullins JI, Vanham G, Arts EJ. 2005. Changes in human immunodeficiency virus type 1 fitness and genetic diversity during disease progression. J Virol 79:9006–9018. doi: 10.1128/JVI.79.14.9006-9018.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marozsan AJ, Moore DM, Lobritz MA, Fraundorf E, Abraha A, Reeves JD, Arts EJ. 2005. Differences in the fitness of two diverse wild-type human immunodeficiency virus type 1 isolates are related to the efficiency of cell binding and entry. J Virol 79:7121–7134. doi: 10.1128/JVI.79.11.7121-7134.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rangel HR, Weber J, Chakraborty B, Gutierrez A, Marotta ML, Mirza M, Kiser P, Martinez MA, Este JA, Quiñones-Mateu ME. 2003. Role of the human immunodeficiency virus type 1 envelope gene in viral fitness. J Virol 77:9069–9073. doi: 10.1128/JVI.77.16.9069-9073.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arien KK, Verhasselt B. 2008. HIV Nef: role in pathogenesis and viral fitness. Curr HIV Res 6:200–208. doi: 10.2174/157016208784325001. [DOI] [PubMed] [Google Scholar]
- 32.Lundquist CA, Tobiume M, Zhou J, Unutmaz D, Aiken C. 2002. Nef-mediated downregulation of CD4 enhances human immunodeficiency virus type 1 replication in primary T lymphocytes. J Virol 76:4625–4633. doi: 10.1128/JVI.76.9.4625-4633.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Watkins RL, Zou W, Denton PW, Krisko JF, Foster JL, Garcia JV. 2013. In vivo analysis of highly conserved Nef activities in HIV-1 replication and pathogenesis. Retrovirology 10:125. doi: 10.1186/1742-4690-10-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chenine AL, Wieczorek L, Sanders-Buell E, Wesberry M, Towle T, Pillis DM, Molnar S, McLinden R, Edmonds T, Hirsch I, O'Connell R, McCutchan FE, Montefiori DC, Ochsenbauer C, Kappes JC, Kim JH, Polonis VR, Tovanabutra S. 2013. Impact of HIV-1 backbone on neutralization sensitivity: neutralization profiles of heterologous envelope glycoproteins expressed in native subtype C and CRF01-AE backbone. PLoS One 8:e76104. doi: 10.1371/journal.pone.0076104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Louder MK, Sambor A, Chertova E, Hunte T, Barrett S, Ojong F, Sanders-Buell E, Zolla-Pazner S, McCutchan FE, Roser JD, Gabuzda D, Lifson JD, Mascola JR. 2005. HIV-1 envelope pseudotyped viral vectors and infectious molecular clones expressing the same envelope glycoprotein have a similar neutralization phenotype, but culture in PBMCs is associated with decreased neutralization sensitivity. Virology 339:226–238. doi: 10.1016/j.virol.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 36.Dykes C, Demeter LM. 2007. Clinical significance of human immunodeficiency virus type 1 replication fitness. Clin Microbiol Rev 20:550–578. doi: 10.1128/CMR.00017-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dalmau J, Codoñer FM, Erkizia I, Pino M, Pou C, Paredes R, Clotet B, Martinez-Picado J, Prado JG. 2012. In–depth characterization of viral isolates from plasma and cells compared with plasma circulating quasispecies in early HIV-1 infection. PLoS One 7:e32714. doi: 10.1371/journal.pone.0032714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mlisana K, Werner L, Garrett NJ, McKinnon LR, van Loggerenberg F, Passmore J-AS, Gray CM, Morris L, Williamson C, Abdool Karim SS. 2014. Rapid disease progression in HIV-1 subtype C-infected South African women. Clin Infect Dis 59:1322–1331. doi: 10.1093/cid/ciu573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Garrett NJ, Werner L, Naicker N, Naranbhai V, Sibeko S, Samsunder N, Gray C, Williamson C, Morris L, Abdool Karim QA, Abdool Karim SS. 2015. HIV disease progression in seroconvertors from the CAPRISA 004 tenofovir gel pre-exposure prophylaxis trial. J Acquir Immune Defic Syndr 68:55–61. doi: 10.1097/QAI.0000000000000367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abdool Karim Q, Abdool Karim SS, Frohlich JA, Grobler AC, Baxter C, Mansoor LE, Kharsany ABM, Sibeko S, Mlisana KP, Omar Z, Gengiah TN, Maarschalk S, Arulappan N, Mlotshwa M, Morris L, Taylor D. 2010. Effectiveness and safety of tenofovir gel, an antiretroviral microbicide, for the prevention of HIV infection in women. Science 329:1168–1174. doi: 10.1126/science.1193748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brockman MA, Tanzi GO, Walker BD, Allen TM. 2006. Use of a novel GFP reporter cell line to examine replication capacity of CXCR4- and CCR5-tropic HIV-1 by flow cytometry. J Virol Methods 131:134–142. doi: 10.1016/j.jviromet.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 42.Ndung'u T, Renjifo B, Essex M. 2001. Construction and analysis of an infectious human immunodeficiency virus type 1 subtype C molecular clone. J Virol 75:4964–4972. doi: 10.1128/JVI.75.11.4964-4972.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ntale RS, Chopera DR, Ngandu NK, Assis de Rosa D, Zembe L, Gamieldien H, Mlotshwa M, Werner L, Woodman Z, Mlisana K, Abdool Karim S, Gray CM, Williamson C. 2012. Temporal association of HLA-B*81:01- and HLA-B*39:10-mediated HIV-1 p24 sequence evolution with disease progression. J Virol 86:12013–12024. doi: 10.1128/JVI.00539-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Payne R, Muenchhoff M, Mann J, Roberts HE, Matthews P, Adland E, Hempenstall A, Huang K-H, Brockman M, Brumme Z, Sinclair M, Miura T, Frater J, Essex M, Shapiro R, Walker BD, Ndung'u T, McLean AR, Carlson JM, Goulder PJR. 2014. Impact of HLA-driven HIV adaptation on virulence in populations of high HIV seroprevalence. Proc Natl Acad Sci U S A 111:E5393–E5400. doi: 10.1073/pnas.1413339111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carlson JM, Du VY, Pfeifer N, Bansal A, Tan VYF, Power K, Brumme CJ, Kreimer A, DeZiel CE, Fusi N, Schaefer M, Brockman MA, Gilmour J, Price MA, Kilembe W, Haubrich R, John M, Mallal S, Shapiro R, Frater J, Harrigan PR, Ndung'u T, Allen S, Heckerman D, Sidney J, Allen TM, Goulder PJR, Brumme ZL, Hunter E, Goepfert PA. 2016. Impact of pre-adapted HIV transmission. Nat Med 22:606–613. doi: 10.1038/nm.4100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mónaco DC, Dilernia DA, Gartland AF, Yu T, Prince JL, Dennis KK, Qin K, Schaefer M, Claiborne DT, Kilembe W, Tang J, Price MA, Farmer P, Gilmour J, Bansal A, Allen S, Goepfert P, Hunter E. 2016. Balance between transmitted HLA preadapted and nonassociated polymorphisms is a major determinant of HIV-1 disease progression. J Exp Med 213:2049–2063. doi: 10.1084/jem.20151984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Selhorst P, Masson L, Ismail SD, Samsunder N, Garrett N, Mansoor LE, Abdool Karim Q, Abdool Karim SS, Passmore J-AS, Williamson C. 2017. Cervicovaginal inflammation facilitates acquisition of less infectious HIV variants. Clin Infect Dis 64:79–82. doi: 10.1093/cid/ciw663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Meier A, Chang JJ, Chan ES, Pollard RB, Sidhu HK, Kulkarni S, Wen TF, Lindsay RJ, Orellana L, Mildvan D, Bazner S, Streeck H, Alter G, Lifson JD, Carrington M, Bosch RJ, Robbins GK, Altfeld M. 2009. Sex differences in the Toll-like receptor-mediated response of plasmacytoid dendritic cells to HIV-1. Nat Med 15:955–959. doi: 10.1038/nm.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Griesbeck M, Ziegler S, Laffont S, Smith N, Chauveau L, Tomezsko P, Kourjian G, Porichis F, Hart M, Palmer CD, Sirignano M, Beisel C, Hildebrandt H, Cénac C, Villani A, Diefenbach TJ, Le Gall S, Schwartz O, Herbeuval J, Autran B, Chang JJ, Altfeld M, Chauveau L, Tomezsko P, Sharei A, Kourjian G, Porichis F, Hart M, Palmer CD, Sirignano M. 2015. Sex differences in plasmacytoid dendritic cell levels of IRF5 drive higher IFN-α production in women. J Immunol 195:5327–5336. doi: 10.4049/jimmunol.1501684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Carlson JM, Schaefer M, Monaco DC, Batorsky R, Claiborne DT, Prince J, Deymier MJ, Ende ZS, Klatt NR, DeZiel CE, Lin T-H, Peng J, Seese AM, Shapiro R, Frater J, Ndung'u T, Tang J, Goepfert P, Gilmour J, Price MA, Kilembe W, Heckerman D, Goulder PJR, Allen TM, Allen S, Hunter E. 2014. HIV transmission. Selection bias at the heterosexual HIV-1 transmission bottleneck. Science 345:1254031. doi: 10.1126/science.1254031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lassen KG, Lobritz MA, Bailey JR, Johnston S, Nguyen S, Lee B, Chou T, Siliciano RF, Markowitz M, Arts EJ. 2009. Elite suppressor-derived HIV-1 envelope glycoproteins exhibit reduced entry efficiency and kinetics. PLoS Pathog 5:e1000377. doi: 10.1371/journal.ppat.1000377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Deymier MJ, Ende Z, Fenton-May AE, Dilernia DA, Kilembe W, Allen SA, Borrow P, Hunter E. 2015. Heterosexual transmission of subtype C HIV-1 selects consensus-like variants without increased replicative capacity or interferon-α resistance. PLoS Pathog 11:e1005154. doi: 10.1371/journal.ppat.1005154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Valley-Omar Z, Sibeko S, Anderson J, Goodier S, Werner L, Arney L, Naranbhai V, Treurnicht F, Abrahams M-R, Bandawe G, Swanstrom R, Abdool Karim Q, Abdool Karim SS, Williamson C. 2012. CAPRISA 004 tenofovir microbicide trial: no impact of tenofovir gel on the HIV transmission bottleneck. J Infect Dis 206:35–40. doi: 10.1093/infdis/jis305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wei X, Hunt G, Karim SSA, Naranbhai V, Sibeko S, Karim QA, Li JF, Kashuba ADM, Werner L, Passmore J-AS, Morris L, Heneine W, Johnson JA. 2014. Sensitive tenofovir resistance screening of HIV-1 from the genital and blood compartments of women with breakthrough infections in the CAPRISA 004 tenofovir gel trial. J Infect Dis 209:1916–1920. doi: 10.1093/infdis/jiu026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Masson L, Passmore J-AS, Liebenberg LJ, Werner L, Baxter C, Arnold KB, Williamson C, Little F, Mansoor LE, Naranbhai V, Lauffenburger DA, Ronacher K, Walzl G, Garrett NJ, Williams BL, Couto-Rodriguez M, Hornig M, Lipkin WI, Grobler A, Abdool Karim Q, Abdool Karim SS. 2015. Genital inflammation and the risk of HIV acquisition in women. Clin Infect Dis 61:260–269. doi: 10.1093/cid/civ298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hladik F, Burgener A, Ballweber L, Gottardo R, Vojtech L, Fourati S, Dai JY, Cameron MJ, Strobl J, Hughes SM, Hoesley C, Andrew P, Johnson S, Piper J, Friend DR, Ball TB, Cranston RD, Mayer KH, McElrath MJ, McGowan I. 2015. Mucosal effects of tenofovir 1% gel. eLife 4:e04525. doi: 10.7554/eLife.04525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Salazar-Gonzalez JF, Bailes E, Kimmy T, Salazar MG, Guffey MB, Keele BF, Derdeyn CA, Farmer P, Hunter E, Allen S, Manigart O, Mulenga J, Anderson JA, Swanstrom R, Haynes BF, Athreya GS, Korber BTM, Sharp PM, Shaw GM, Hahn BH, Salazar-Gonzalez JF, Pham KT. 2008. Deciphering human immunodeficiency virus type 1 transmission and early envelope diversification by single-genome amplification and sequencing. J Virol 82:3952–3970. doi: 10.1128/JVI.02660-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Reed LJ, Muench H. 1938. A simple method of estimating fifty per cent endpoints. Am J Epidemiol 27:493–497. doi: 10.1093/oxfordjournals.aje.a118408. [DOI] [Google Scholar]