

ABSTRACT
Epstein-Barr virus (EBV)-associated diseases of epithelial cells, including tumors that have latent infection, such as nasopharyngeal carcinoma (NPC), and oral hairy leukoplakia (OHL) lesions that have lytic infection, frequently express the viral latent membrane protein 1 (LMP1). In lytically infected cells, LMP1 expression is activated by the BRLF1 (R) immediate early (IE) protein. However, the mechanisms by which LMP1 expression is normally regulated in epithelial cells remain poorly understood, and its potential roles in regulating lytic reactivation in epithelial cells are as yet unexplored. We previously showed that the differentiation-dependent cellular transcription factors KLF4 and BLIMP1 induce lytic EBV reactivation in epithelial cells by synergistically activating the two EBV immediate early promoters (Zp and Rp). Here we show that epithelial cell differentiation also induces LMP1 expression. We demonstrate that KLF4 and BLIMP1 cooperatively induce the expression of LMP1, even in the absence of the EBV IE proteins BZLF1 (Z) and R, via activation of the two LMP1 promoters. Furthermore, we found that differentiation of NOKs-Akata cells by either methylcellulose suspension or organotypic culture induces LMP1 expression prior to Z and R expression. We show that LMP1 enhances the lytic infection-inducing effects of epithelial cell differentiation, as well as 12-O-tetradecanoylphorbol-13-acetate (TPA) and sodium butyrate treatment, in EBV-infected epithelial cells by increasing expression of the Z and R proteins. Our results suggest that differentiation of epithelial cells activates a feed-forward loop in which KLF4 and BLIMP1 first activate LMP1 expression and then cooperate with LMP1 to activate Z and R expression.
IMPORTANCE The EBV protein LMP1 is expressed in EBV-associated epithelial cell diseases, regardless of whether these diseases are due to lytic infection (such as oral hairy leukoplakia) or latent infection (such as nasopharyngeal carcinoma). However, surprisingly little is known about how LMP1 expression is regulated in epithelial cells, and there are conflicting reports about whether it plays any role in regulating viral lytic reactivation. In this study, we show that epithelial cell differentiation induces LMP1 expression by increasing expression of two cellular transcription factors (KLF4 and BLIMP1) which cooperatively activate the two LMP1 promoters. We also demonstrate that LMP1 promotes efficient lytic reactivation in EBV-infected epithelial cells by enhancing expression of the Z and R proteins. Thus, in EBV-infected epithelial cells, LMP1 expression is promoted by differentiation and positively regulates lytic viral reactivation.
KEYWORDS: Epstein-Barr virus
INTRODUCTION
Epstein-Barr virus (EBV) is a ubiquitous human gammaherpesvirus that infects the majority of the human population worldwide (1). It causes infectious mononucleosis and is associated with both lymphoid and epithelial cell malignancies, including posttransplant lymphoproliferative disease, diffuse large B cell lymphomas, African Burkitt lymphoma, Hodgkin's lymphoma, undifferentiated nasopharyngeal carcinoma (NPC), and gastric carcinoma (1, 2). Like other herpesviruses, EBV undergoes latent and lytic modes of infection during its life cycle. The latent form of infection allows the virus to persist for the lifetime of the host, while the lytic form of infection enables infectious virion production and transmission from cell to cell and from host to host. Thus, both forms of infection are essential for the long-term success of the virus.
EBV infection in tumors is largely latent, since this form of infection does not kill the tumor cell and allows expression of virally carried oncogenes. There are at least three different types of EBV latency, two of which (types I and II) are found in EBV-infected epithelial cell tumors (1). In type I latency (the most restricted form), only one viral protein, EBNA1, is expressed, in addition to the virally encoded small nuclear RNAs (EBERs), long noncoding RNAs, and microRNAs. In type II latency, the virally encoded membrane proteins (latent membrane protein 1 [LMP1], LMP2A, and LMP2B) are also expressed, in addition to EBNA1. EBV often exhibits type II latency in NPC tumors, but it generally has type I latency in EBV-positive gastric carcinomas (3–7).
In contrast to the latent EBV infection that occurs in undifferentiated epithelial cell tumors, EBV infection of normal oropharyngeal epithelial cells is largely restricted to the more differentiated cells and is lytic (1, 8–13). Lytic EBV infection is initiated by the transcriptional effects of the two EBV immediate early (IE) proteins, BZLF1 (Z) and BRLF1 (R) (14–21). Both Z and R are required for lytic viral DNA replication. Activation of the Z and R IE promoters by cellular transcription factors leads to the initial production of the immediate early proteins, Z and R, respectively (reviewed in references 20 to 22); the Z and R transcription factors then autoregulate their own promoters as well as each other's promoters, leading to further upregulation of Z and R expression (23–26). We recently showed that epithelial cell differentiation activates the latent-to-lytic switch in EBV-infected epithelial cells by increasing expression of two cellular transcription factors, KLF4 and BLIMP1, that synergistically activate the Z and R promoters (11).
LMP1 is required for EBV transformation of B cells in vitro (27, 28) and is expressed during both latent and lytic infections. LMP1 mimics the effects of CD40 signaling and activates multiple cellular pathways, including NF-κB, mitogen-activated protein kinase (MAPK), phosphatidylinositol 3-kinase (PI3K), and c-Jun N-terminal kinase (JNK) pathways (reviewed in references 29 and 30). In addition to B cell lymphomas that have type II or type III latency, LMP1 expression also commonly occurs in EBV-infected NPCs and is thought to contribute to the formation of these tumors (31–38).
LMP1 is also expressed during lytic EBV infection (39–42), and oral hairy leukoplakia lesions in patients (caused by lytic EBV infection of differentiated tongue epithelium) have high-level LMP1 expression (43, 44). Furthermore, transfection of R into latently infected 293 cells activates LMP1 expression, and both of the two known LMP1 promoters (the proximal EDL1 promoter and the distal TR promoter) can be activated by R in reporter gene assays (39). However, whether LMP1 plays any role in regulating the latent-to-lytic switch of EBV or is required for efficient virion production remains controversial. One study found that Z-transfected 293 cells infected with an LMP1-deleted (B95.8 strain) EBV mutant produced as many infectious virions as 293 cells infected with wild-type (WT) EBV (45), suggesting that LMP1 is not required for the later parts of lytic EBV replication in this cell type. In contrast, another study examining the phenotype of an LMP1-deleted (Akata strain) EBV mutant reported that LMP1 is required for efficient viral egress in lytically infected Burkitt lymphoma cells (46). Conversely, other studies performed in B cells found that LMP1 negatively regulates the initial steps of lytic EBV reactivation through its effects on the NF-κB, protein kinase Cδ (PKCδ), and sumoylation pathways (47–49). Thus, the role(s) of LMP1 in regulating the EBV latent-to-lytic switch may be influenced by the cellular environment and/or the type of EBV latency.
Most previous studies examining the regulation of LMP1 expression were performed in B cells. LMP1 transcripts can be initiated from two different promoters in the EBV genome. The more proximal promoter, LMP1-EDL1, is the predominant promoter used in EBV-infected B cells with type III latency and is activated by the viral EBNA2 protein (50–52). The more distal promoter, LMP1-TR (located near the terminal repeats), is used in B cells with type II latency and in NPCs (50–52). Multiple layers of regulation act to ensure tight control of LMP1 transcription from both of these promoters. In addition to viral factors, such as EBNA2, EBNA-LP, EBNA3C, and microRNAs, cellular transcription factors, including RBPJ, PU.1, SpiB, IRF4, EBF1, IRF7, Pax5, Notch-1, ATF-1, CREB-1, AP-2, NF-κB family members, and STAT family members, also regulate the activity of the LMP1 promoters (53–66). Epigenetic modifications of the viral genome, such as promoter DNA methylation, histone acetylation, and histone methylation, also regulate LMP1 expression (67).
In the present study, we examined whether epithelial cell differentiation regulates LMP1 expression in EBV-infected epithelial cells and dissected the mechanisms for this effect. We also investigated the role of LMP1 expression during differentiation-induced lytic EBV reactivation. We found that differentiation of EBV-infected epithelial cells activates LMP1 expression prior to activating either BZLF1 or BRLF1 expression. Furthermore, we show that two different cellular transcription factors, KLF4 and BLIMP1, contribute to this effect. In addition, we demonstrate that LMP1 is required for efficient Z and R expression in EBV-infected epithelial cells. These unexpected and somewhat surprising results suggest that cellular differentiation initially activates LMP1 transcription in EBV-infected epithelial cells and that the LMP1 protein then cooperates with differentiation-induced cellular transcription factors to enhance Z and R expression.
RESULTS
Cellular differentiation induces LMP1 expression in epithelial cells.
We previously showed that epithelial cell differentiation induces lytic EBV reactivation in the NOKs-Akata cell line (a telomerase-immortalized normal oral keratinocyte cell line that is stably infected with the Akata strain of EBV) (11). To determine whether differentiation of NOKs-Akata cells affects LMP1 expression, cells were differentiated by suspension in methylcellulose for 48 h, and the levels of various EBV proteins and involucrin (an epithelial cell differentiation marker) were examined by immunoblot analysis. As expected, cells suspended in methylcellulose had greatly increased involucrin expression, confirming that the cells had undergone at least partial differentiation (Fig. 1A, left panel). Consistent with our previous results (11), differentiated NOKs-Akata cells also had increased expression of the viral immediate early lytic proteins, Z and R, as well as the early lytic protein, BMRF1 (Fig. 1A, left panel). Note that differentiation-mediated reactivation in NOKs-Akata cells also led to a large increase in the level of LMP1 expression (Fig. 1A, left panel). Although LMP1 expression (derived from the LMP1-EDL1 promoter) in B cells with type III latency is driven by the EBV-encoded EBNA2 protein, methylcellulose-treated NOKs-Akata cells had no detectable EBNA2 expression (Fig. 1A, right panel). Other methods of differentiating epithelial cells in vitro, such as growing them in organotypic raft cultures or treating them with high concentrations of calcium and serum, also induced LMP1 expression in NOKs-Akata cells (Fig. 1B). These results confirm that lytic EBV reactivation in and/or differentiation per se of NOKs-Akata cells leads to enhanced LMP1 expression.
FIG 1.

Cellular differentiation induces LMP1 expression in epithelial cells. (A) NOKs-Akata cells were either grown in a monolayer or suspended in methylcellulose (1.6% in K-SFM) for 48 h. For the left panel, immunoblot analysis was performed to compare the levels of viral proteins (Z, R, BMRF1, and LMP1), the differentiation-dependent cellular protein involucrin, and the cellular protein tubulin (as a loading control). For the right panel, immunoblot analysis was performed to compare the levels of viral proteins (EBNA2 and LMP1) and the cellular protein tubulin (as a loading control). Kem I and Kem III are isogenic Burkitt lymphoma cell lines that have type I and type III latency, respectively, and they served as negative and positive controls for LMP1 and EBNA2 expression. (B) Immunoblot analysis of NOKs and NOKs-Akata cells grown in untreated monolayer cultures (mono; left panels), organotypic cultures (rafts; middle panels) (2 independent rafts per cell type were analyzed), or monolayer cultures treated with high calcium (final concentration, 1.2 mM) and high serum (20% final concentration) for 0, 24, 48, or 72 h (right panels). (Top) LMP1-specific immunoblotting. (Bottom) Actin-specific immunoblotting. The left and middle panels were taken from the same immunoblot. (C) HONE-Akata cells were treated with nothing, TPA (20 ng/μl), or sodium butyrate (3 mM) for 48 h. Immunoblot analysis was performed to compare the levels of viral proteins (Z, R, BMRF1, and LMP1) and the cellular protein tubulin (as a loading control). Molecular mass markers are also indicated for the LMP1 blot.
To determine if lytic infection-inducing agents, such as 12-O-tetradecanoylphorbol-13-acetate (TPA) and sodium butyrate, can enhance LMP1 expression in EBV-infected epithelial cells that cannot undergo differentiation, we treated HONE-Akata cells (now thought to be an EBV-infected HeLa cell line [68]) with either TPA or sodium butyrate and examined the levels of Z, R, BMRF1, and LMP1. Treatment with either of these chemicals reactivated expression of the lytic EBV proteins Z, R, and BMRF1 and also increased the expression of LMP1 (Fig. 1C). These results confirm that LMP1 expression is strongly induced during lytic reactivation in epithelial cells by both differentiation-dependent and differentiation-independent stimuli.
R (but not Z) activates LMP1 expression in EBV-infected epithelial cells.
To examine whether expression of Z and/or R is sufficient to activate LMP1 expression in the context of the intact viral genome in epithelial cells, an Akata bacmid mutant that is deleted for the BZLF1, BRLF1, and BRRF1 (“Na”) genes was constructed and used to create a stably infected HeLa cell line. As shown in Fig. 2, transfection of these cells with a Z expression vector did not activate LMP1 expression from the endogenous viral genome, while transfection of an R expression vector activated LMP1 expression. Interestingly, in contrast to their effect on LMP1 expression (where R alone was sufficient to induce LMP1), the combination of both Z and R was required to induce expression of the early lytic BMRF1 protein. This result confirms that R, not Z, primarily drives LMP1 transcription during the lytic form of EBV infection.
FIG 2.
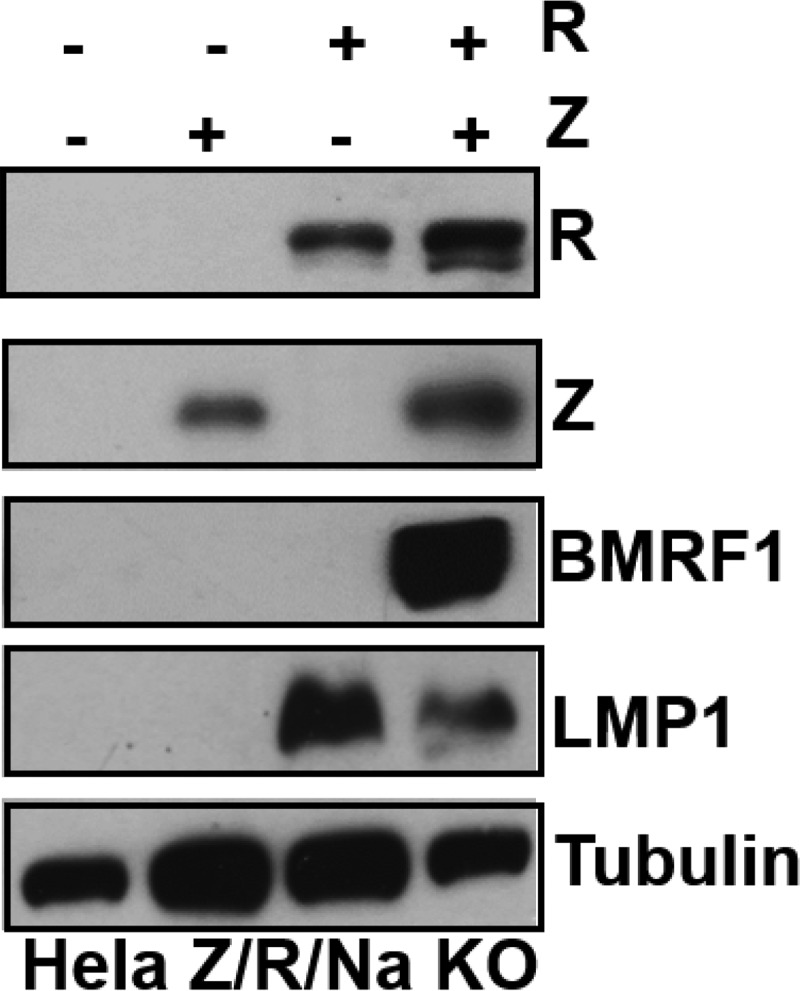
R activates LMP1 expression in EBV-infected epithelial cells. HeLa cells infected with a Z/R/BRRF1-deleted EBV mutant (Hela Z/R/Na KO) were transfected with either control vector or Z and R expression vectors (either alone or in combination), as indicated. Immunoblot analysis was performed to compare the levels of transfected Z and R and induction of the EBV proteins LMP1 and BMRF1 from the endogenous viral genome. The cellular protein tubulin was used as a loading control.
LMP1 is required for the ability of cellular differentiation to efficiently induce lytic EBV reactivation in NOKs-Akata cells.
To determine whether LMP1 induced during cellular differentiation has any role in regulating the latent-to-lytic switch of EBV, we performed LMP1 knockdown studies. NOKs-Akata cells were transfected with two different control small interfering RNAs (siRNAs) or two different siRNAs targeting LMP1 and then treated with or without methylcellulose suspension for 48 h to induce differentiation. Suspension in methylcellulose robustly increased expression of LMP1, as well as expression of the lytic proteins Z, R, and BMRF1, in cells transfected with a control siRNA (Fig. 3A). However, preventing methylcellulose-induced LMP1 expression by use of an siRNA targeting LMP1 decreased the ability of methylcellulose to induce the expression of the Z, R, and BMRF1 lytic proteins while not affecting induction of the cellular involucrin protein. Similar results were obtained when the experiment was repeated using a different control siRNA and another LMP1-directed siRNA (targeting a different region of the LMP1 transcript) (Fig. 3B).
FIG 3.

LMP1 is required for the ability of cellular differentiation to induce lytic EBV reactivation efficiently in NOKs-Akata cells. (A and B) NOKs-Akata cells were transfected with either of two different control siRNAs or two different LMP1 siRNAs and then were either left in monolayer culture or suspended in methylcellulose for 48 h. Immunoblot analysis was performed using 5 μg of protein lysate to detect the expression of viral proteins (LMP1, Z, R, and BMRF1) and the cellular proteins involucrin and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (as a loading control). (C) NOKs-Akata cells were transfected with either two different control siRNAs or two different LMP1 siRNAs for 48 h, and immunoblot analysis was performed using 25 μg of protein lysate to detect the endogenous expression levels of viral proteins (LMP1, Z, R, and BMRF1) and the cellular protein tubulin (as a loading control).
NOKs-Akata cells grown in monolayer cultures (in the absence of differentiating agents) have low-level lytic viral protein expression that can be detected when immunoblots are loaded with a large amount of protein. To determine whether LMP1 also plays a role in this “constitutive” lytic protein expression (which is likely due to a small number of spontaneously differentiating cells), NOKs-Akata cells were transfected with two different control siRNAs or two different LMP1 siRNAs, followed by immunoblot analysis of a gel loaded with 25 μg of protein lysate (versus 5 μg of protein loaded for the blots shown in Fig. 3A and B). Knockdown of LMP1 by use of two different siRNAs led to decreases in the constitutive expression of Z, R, and BMRF1 in NOKs-Akata cells (Fig. 3C).
LMP1 is also required for efficient chemically induced lytic reactivation in HONE-Akata cells.
To determine if the role of LMP1 in lytic reactivation is unique to NOKs-Akata cells or specific to the differentiation stimulus, HONE-Akata cells were treated with either control siRNAs or LMP1-directed siRNAs and then exposed to TPA or sodium butyrate (Fig. 4). Knockdown of LMP1 expression decreased the amounts of IE and early lytic EBV protein expression observed after either TPA treatment (Fig. 4A) or sodium butyrate treatment (Fig. 4B) of HONE-Akata cells. Knocking down LMP1 also inhibited the ability of TPA to induce the expression of the lytic EBV proteins Z, R, and BMRF1 in AGS gastric carcinoma cells superinfected with EBV (Fig. 4C). Overall, these results suggest that LMP1 is required for efficient lytic EBV reactivation in epithelial cells in response to a variety of different types of lytic infection-inducing agents and indicate that LMP1 expression is specifically required for efficient activation of the two EBV IE proteins, Z and R.
FIG 4.

LMP1 is required for efficient chemically induced lytic reactivation in epithelial cells. HONE-Akata cells were transfected with either two different control siRNAs or two different LMP1 siRNAs, followed by treatment with or without 20 ng/μl TPA (A) or 3 mM sodium butyrate (B) for 48 h. Immunoblot analysis was performed to detect the expression of viral proteins (LMP1, R, Z, and BMRF1) and the cellular protein tubulin (as a loading control), as indicated. (C) AGS-Akata cells were transfected with either a control siRNA or an LMP1 siRNA, followed by treatment with or without 20 ng/μl TPA for 48 h. Immunoblot analysis was performed to detect the expression of viral proteins (LMP1, Z, R, and BMRF1) and the cellular protein tubulin (as a loading control).
LMP1 expression is detected before Z expression during raft culture-mediated differentiation of NOKs-Akata cells.
The unexpected finding that LMP1 increases the ability of lytic infection-inducing agents to enhance Z and R expression in EBV-infected epithelial cells suggested the possibility that LMP1 expression may be activated by these agents prior to Z and R expression. To test this hypothesis, we differentiated NOKs-Akata cells in organotypic air-interface (raft) cultures. This technique, which we previously used to show that epithelial cell differentiation induces the latent-to-lytic switch of EBV (11), leads to the formation of a stratified epithelium that can be formalin fixed and cross-sectioned for immunofluorescence staining at the single-cell level. Moreover, the cross sections can be used to study the kinetics of viral gene expression during epithelial cell differentiation-mediated lytic reactivation. As shown in Fig. 5, similar to our previous findings (11), we observed Z-expressing cells only in the upper (most differentiated) layers of the NOKs-Akata raft cultures (Fig. 5A). In contrast, LMP1-expressing cells were observed in all cell layers except for the basal layer itself. Furthermore, many more LMP1-expressing cells than Z-expressing cells were observed. Uninfected NOKs cells that were grown in raft culture stained negative for Z and LMP1, as expected. Similar results were obtained with an independently generated NOKs-Akata clone (Fig. 5B). However, we could not determine if LMP1 expression was induced before or after R expression in these experiments, since we have not yet identified an antibody that detects the R protein by immunohistochemistry or immunofluorescence assay.
FIG 5.

LMP1 expression is detected before Z expression during raft culture-mediated differentiation of NOKs-Akata cells. (A and B) Uninfected NOKs cells (right panels) and two independent clones of EBV-infected NOKs-Akata cells (left panels) were grown in organotypic raft cultures, and immunofluorescence analysis was performed to detect the expression of EBV proteins (Z and LMP1, both stained red), as indicated. Examples of Z-stained cells are indicated by red arrows. DAPI staining is shown in blue.
LMP1 is expressed before the Z and R proteins during methylcellulose-mediated differentiation of NOKs-Akata cells.
To further examine if differentiation of NOKs-Akata cells activates LMP1 expression prior to Z and R expression, NOKs-Akata cells were treated by methylcellulose suspension for various amounts of time, and protein extracts were obtained at each time point for immunoblot analysis. As shown in Fig. 6, enhanced expression of the epithelial cell differentiation marker involucrin was first observed after 6 h of methylcellulose treatment, and it became even more marked at later time points. Expression of BLIMP1, which is required for differentiation of epithelial cells, was observed at the 2-h time point, while expression of KLF4 (which we previously showed to be expressed in both differentiated and undifferentiated NOKs cells [11]) was not significantly affected by methylcellulose treatment. Most importantly, methylcellulose treatment increased LMP1 protein expression at a much earlier time point (6 h) than that for increased expression of the Z and R proteins (12 to 24 h). Expression of the early lytic BMRF1 protein was not significantly increased until the 48-h time point. These results suggest that LMP1 expression precedes Z and R expression during methylcellulose-induced differentiation of NOKs-Akata cells, and thus LMP1 may be activated by a differentiation-dependent mechanism that is independent of Z and R function.
FIG 6.

The LMP1 protein is expressed before the Z and R proteins during methylcellulose-mediated differentiation of NOKs-Akata cells. NOKs-Akata cells were suspended in 1.6% methylcellulose (in K-SFM) and harvested at the indicated time points posttreatment. Immunoblot analysis was performed to detect the expression of viral proteins (Z, R, BMRF1, and LMP1) and cellular proteins (involucrin, KLF4, BLIMP1, and tubulin), as indicated. Tubulin was used as a loading control.
LMP1 transcripts are expressed before Z/R transcripts in methylcellulose-differentiated NOKs-Akata cells.
To confirm that methylcellulose treatment activates LMP1 transcription prior to Z/R transcription, quantitative PCR (qPCR) analysis was performed using RNAs harvested at various time points (Fig. 7A and B). As shown in Fig. 7C and D, the LMP1 primer set 1 used in this analysis can amplify transcripts derived from either of the two LMP1 promoters, and the Z/R primer set amplifies transcripts derived from either the Z or R IE promoter. The LMP1 transcript level was clearly increased (16-fold) after 2 h of methylcellulose suspension; in contrast, the level of the Z/R transcripts was not highly increased until 24 h after methylcellulose suspension (Fig. 7A). Similar results were obtained in a second independent experiment (Fig. 7B). Interestingly, LMP1 primer set 2 (Fig. 7C), which can amplify only the message derived from the distal promoter, did not show a detectable transcript in the presence or absence of methylcellulose (although this primer set amplified transcripts in Mutu III Burkitt lymphoma cells) (data not shown). These results indicate that the LMP1 message level is increased prior to any increase in Z/R transcripts during methylcellulose-mediated lytic reactivation. Thus, LMP1 expression may initially be activated by differentiation-associated cellular transcription factors. Our results also suggest that the methylcellulose-induced LMP1 transcript is primarily derived from the proximal EDL1 promoter.
FIG 7.

LMP1 transcripts are expressed before Z/R transcripts in methylcellulose-differentiated NOKs-Akata cells. (A and B) RNAs were isolated from NOKs-Akata cells suspended in 1.6% methylcellulose for the indicated times. qPCR analysis was performed on reverse-transcribed RNAs to detect the transcript levels for total LMP1 or Z plus R, using the LMP1 primer set 1 and Z/R primers shown in panels C and D. An LMP1 primer set (set 2) that detects only the LMP1-TRp-initiated LMP1 transcript did not detect the LMP1 message in methylcellulose-treated NOKs-Akata cells, although it detected the transcript in Mutu III Burkitt lymphoma cells (data not shown). Beta-globin was used as a housekeeping gene for normalization. Data are plotted as fold changes (increases) relative to the 0-h sample, whose level was set to 1.
KLF4 and BLIMP1 can activate the LMP1 promoters in reporter gene assays.
Differentiation of normal epithelial cells induces expression of both the KLF4 and BLIMP1 cellular transcription factors, and both of these transcription factors are required for normal epithelial cell differentiation (69–73). We previously showed that KLF4 and BLIMP1 synergistically activate both the Z and R EBV immediate early promoters (11). Furthermore, KLF4 and BLIMP1 were recently found to promote differentiation-dependent expression of HPV late structural proteins in HPV-infected epithelial cells (74). Given our finding that LMP1 expression is increased by differentiating agents in EBV-infected NOKs cells, we performed luciferase reporter gene assays to determine if KLF4 or BLIMP1 activates one or both of the two LMP1 promoters in uninfected NOKs cells.
As shown in Fig. 8A, either KLF4 alone or BLIMP1 alone produced low-level activation of both the proximal (LMP1-EDL1) and distal (LMP1-TR) promoters. Furthermore, the combination of both KLF4 and BLIMP1 together synergistically activated both promoters, although the effect on the proximal promoter was stronger. In contrast, the promoterless control luciferase vector did not respond to either KLF4 or BLIMP1. These results indicate that both LMP1 promoters are synergistically activated by the BLIMP1-KLF4 combination in EBV-negative epithelial cells.
FIG 8.

KLF4 and BLIMP1 synergistically activate the LMP1 promoters. (A) Reporter gene constructs containing either the proximal LMP1 promoter (LMP1-EDL1), the distal LMP1 promoter (LMP1-TR), or no promoter sequences upstream of the luciferase gene were cotransfected into EBV-negative NOKs cells with either the control vector, the KLF4 vector alone, the BLIMP1 vector alone, or the combination of the KLF4 and BLIMP1 vectors. Luciferase assays were performed 2 days after transfection. Total luciferase activity for each of the conditions from a representative experiment is shown (data are averages ± standard deviations of results from three replicates), as well as the fold increase in activity induced by KLF4 or BLIMP1. Similar results were obtained in three separate experiments. (B) NOKs-Akata cells (left) or CNE-Akata cells (right) were transfected with either the control vector or the KLF4 and BLIMP1 expression vectors, and immunoblotting was performed to detect LMP1, EBNA2, Z, R, and tubulin. Kem III cells served as the positive control for EBNA2 and LMP1. (C) HeLa cells infected with a mutant EBV strain with deletions of the Z/R/BRRF1 genes were transfected with either the control vector or the KLF4 and BLIMP1 expression vectors (either alone or in combination). Immunoblot analysis was performed to compare the levels of transfected KLF4 and BLIMP1 and the induction of EBV LMP1 from the EBV genome. The cellular protein tubulin was used as a loading control.
We next asked whether KLF4 and/or BLIMP1 can also turn on expression of LMP1 from the endogenous viral genome in the absence of Z and R expression. First, we confirmed that KLF4 and BLIMP1 can activate LMP1 expression without concomitant EBNA2 expression in NOKs-Akata cells and CNE-Akata cells (Fig. 8B). However, since KLF4/BLIMP1 also activated Z and R expression in these EBV-positive epithelial cell lines, LMP1 expression may potentially have been driven by R rather than by KLF4/BLIMP1. We next examined whether HeLa cells stably infected with a Z/R/BRRF1-deleted EBV mutant can express LMP1 when transfected with either a KLF4 expression vector alone, a BLIMP1 expression vector alone, or the combination of both vectors. As shown in Fig. 8C, overexpression of KLF4 alone was sufficient to induce expression of LMP1 from the Z/R/BRRF1-deleted EBV genome, while BLIMP1 alone had no effect; furthermore, the combination of KLF4 and BLIMP1 induced more LMP1 expression than that with KLF4 alone. These results confirm that KLF4 and BLIMP1 can activate expression of LMP1 in the context of the intact EBV genome even in the absence of Z or R expression. Thus, KLF4 and BLIMP1 likely contribute to differentiation-dependent LMP1 expression in EBV-infected epithelial cells.
DISCUSSION
Repression of lytic EBV infection is essential for the development of EBV-infected epithelial cell tumors. However, the cellular and viral factors that determine whether EBV remains latent or lytic in epithelial cells are not currently well understood. We recently showed that epithelial cell differentiation promotes lytic EBV reactivation through differentiation-dependent expression of two different cellular transcription factors, KLF4 and BLIMP1, and demonstrated that KLF4 and BLIMP1 synergistically activate both the Zp and Rp EBV IE promoters (11). Here we have discovered that epithelial cell differentiation also activates expression of the EBV LMP1 protein (even in the absence of Z and R expression), and we show that this effect is at least partially mediated by the ability of KLF4 and BLIMP1 to activate the two LMP1 promoters. Furthermore, although LMP1 has been shown to inhibit lytic EBV reactivation in B cells, we demonstrate that epithelial cell differentiation induces LMP1 expression prior to Z and R expression, and we show that LMP1 enhances the efficiency of lytic EBV reactivation in epithelial cells. These results suggest that differentiation-induced LMP1 expression promotes lytic EBV reactivation in epithelial cells, in sharp contrast to the effect of LMP1 in EBV-infected B cells.
LMP1 acts as a constitutively active CD40-like receptor and has been reported to negatively regulate lytic reactivation in EBV-infected B cells by various mechanisms, including activation of the NF-κB pathway (47), induction of interleukin-32 (IL-32) via the PKCδ pathway (49), and sumoylation of the cellular transcriptional repressor, KAP1 (48). In contrast, one study, using an LMP1-deleted EBV mutant, found that LMP1 positively regulates virion production in Burkitt lymphoma cells by increasing viral egress (46). The conflicting results obtained from previous studies of B cell lines suggest that the effects of LMP1 on lytic EBV reactivation may differ depending upon the particular part of the lytic viral life cycle examined, with LMP1 inhibiting lytic reactivation of latent infection but promoting the final stages of viral production during lytic infection.
In contrast to the previous studies of B cell lines, we show here that LMP1 enhances lytic EBV reactivation in EBV-infected epithelial cell lines. This effect was observed using a variety of different lytic infection-inducing agents (including differentiation stimuli, TPA, and sodium butyrate) and occurred in three different EBV-infected epithelial cell lines (NOKs-Akata, HONE-Akata, and AGS-Akata). Unexpectedly, knockdown of LMP1 decreased the ability of lytic infection-inducing stimuli to activate expression of the two immediate early proteins, Z and R. Furthermore, we showed that LMP1 expression occurs before Z and R expression in both raft-cultured NOKS-Akata cells and methylcellulose-differentiated NOKs-Akata cells. These surprising results suggest that LMP1 may function as an “immediate early” protein during lytic reactivation in epithelial cells, since its expression level is increased at time points prior to the increase in Z and R gene expression and it appears to be required for efficient Z and R expression in these cells.
We found that LMP1 was not detectably expressed in the undifferentiated basal layer cells of raft-cultured NOKs-Akata cells but was expressed in even the lowest layers of suprabasal (differentiated) cells. Although this result may simply reflect the increased Z and R expression that occurs in the suprabasal layers (allowing the LMP1 promoter to be activated by these IE transcription factors), we obtained a variety of additional results suggesting that LMP1 expression can be activated by epithelial cell differentiation independently of any Z and/or R effect. First, in raft-cultured NOKS-Akata cells, Z expression was restricted to the most differentiated cell layers, while (in comparison to Z) LMP1 was expressed in less differentiated cell layers. Second, in methylcellulose-differentiated NOKs-Akata cells, LMP1 activation occurred before Z and R activation at both the protein and transcript levels. Third, we demonstrated that the differentiation-dependent cellular transcription factors KLF4 and BLIMP1 can activate both LMP1 promoters in EBV-negative NOKs cells. Finally, we showed that KLF4 and BLIMP1 overexpression can also activate LMP1 expression in the context of the intact viral genome in HeLa cells infected with a Z/R/BRRF1-deleted Akata strain mutant. Interestingly, the combination of KLF4 and BLIMP1 did not activate LMP1 expression in 293 cells (thought to be derived from fetal neuronal cells [75]) infected with the same EBV mutant (data not shown), suggesting that the ability of these factors to activate LMP1 may be cell type dependent.
The cellular transcription factors KLF4 and BLIMP1 are required for the regulation of numerous cellular genes that are induced by terminal differentiation of epithelial cells, and we previously reported that KLF4 and BLIMP1 synergize with each other to cooperatively activate both the Z and R immediate early promoters of EBV (11). Furthermore, KLF4 was recently demonstrated to directly interact with and cooperate with BLIMP1 to activate transcription of late viral genes of human papillomaviruses (HPVs) during epithelial cell differentiation (74). Together, these findings suggest that DNA viruses may commonly use the KLF4/BLIMP1 transcription factors to regulate their life cycles during epithelial cell differentiation.
Here we found that overexpression of BLIMP1 alone does not activate either of the two LMP1 promoters in the presence of endogenous KLF4 expression in NOKs cells, although it synergistically activates both of these promoters with cotransfected KLF4 (Fig. 8). We (11) and others (74) have shown that in contrast to that in normal undifferentiated epithelial cells, KLF4 is expressed even in undifferentiated NOKs cells, although the transcriptional activity of this constitutively expressed KLF4 may be somewhat impaired, since it does not by itself lead to epithelial cell differentiation or activation of lytic EBV promoters. Since BLIMP1 expression remains differentiation dependent in NOKs cells, and since both KLF4 and BLIMP1 are required to activate lytic EBV expression, EBV presumably stays latent in undifferentiated NOKs cells due to the lack of BLIMP1 expression. In addition, if KLF4 and BLIMP1 must interact directly to activate certain EBV promoters (including the two LMP1 promoters), it is possible that constitutively expressed KLF4 in NOKs-Akata cells is already tied up in other protein complexes and therefore less available for interactions with BLIMP1 on these viral promoters. The Laimins lab reported that posttranslational modifications of KLF4 also regulate its functions in HPV-infected NOKs cells (74), which may potentially similarly contribute to the differentiation-dependent increase in lytic EBV protein expression.
Although the exact mechanism(s) by which LMP1 enhances Z and R expression in EBV-infected epithelial cells has not yet been determined, it is noteworthy that NF-κB activation (a major downstream signal of LMP1) has previously been shown to be required for transforming growth factor beta (TGF-β)-induced activation of these proteins in Burkitt lymphoma cells (76). Since we have not found that overexpression of LMP1 is sufficient by itself to induce lytic EBV reactivation in EBV-infected epithelial cells (data not shown), the activating effect of LMP1 on lytic EBV reactivation may occur only when the protein is expressed at physiologic levels and may require that other inducing stimuli (such as epithelial cell differentiation factors) are also present. Although our results obtained using two different control siRNAs and two different LMP1-directed siRNAs strongly suggest that lytic LMP1 expression contributes to differentiation-mediated viral reactivation, in the future it will be important to confirm this finding by using NOKs-Akata cells infected with an LMP1-deleted EBV mutant. Unfortunately, since the NOKs parental cells are already hygromycin resistant (due to their immortalization by use of a hygromycin-resistant TERT vector), we have been unable to stably infect them with two different LMP1-deleted EBV mutants (in B95.8 and M81 strain bacmids) that use the hygromycin resistance gene to stably select for EBV-infected cell lines.
Together our results suggest the model of differentiation-induced lytic EBV reactivation in epithelial cells that is shown in Fig. 9. First, differentiation leads to increased expression of the cellular transcription factors KLF4 and BLIMP1. KLF4 and BLIMP1 then activate expression of the LMP1 promoter, leading to LMP1 expression. LMP1 in combination with KLF4 and BLIMP1 helps to induce expression of the Z and R proteins. The R protein then further activates expression of the LMP1, Z, and R promoters, and Z induces further activation of the R and Z promoters, resulting in a positive feed-forward loop. Finally, our results also suggest that loss of LMP1 expression in EBV-infected NPCs (which is reported to occur in up to 20 to 60% of tumors) may reflect a less differentiated state and may be selected for in some tumors as a mechanism to promote viral latency.
FIG 9.

Proposed model depicting the effects of epithelial cell differentiation on the EBV lytic reactivation cascade. Epithelial cell differentiation activates expression of the differentiation-dependent cellular transcription factors KLF4 and BLIMP1. KLF4 and BLIMP1 then activate the promoters of the LMP1, Z, and R EBV proteins. The LMP1 protein (which is produced earlier than the Z and R proteins) enhances activation of the Z and R promoters in differentiating cells, through an as yet unknown mechanism. The R protein then furthers activates the LMP1, Z, and R promoters, while the Z protein further activates the Z and R promoters.
MATERIALS AND METHODS
Cell lines and culture.
The NOKs cell line (a gift from Karl Munger, Tufts University) is a telomerase-immortalized normal oral keratinocyte cell line that was established as previously described (77). This cell line was maintained in an undifferentiated state by growth in keratinocyte serum-free medium (K-SFM) (Life Technologies Inc.) supplemented with epidermal growth factor, pituitary extract, and 0.2% penicillin-streptomycin (pen-strep). The NOKs-Akata cell line was derived by coculturing uninfected NOKs cells with the Akata-GFP Burkitt lymphoma cell line (a gift from Kenzo Takada, Hokkaido University, Japan, via Bill Sugden, University of Wisconsin-Madison) for 24 h, followed by selection with 50 μg/ml of G418 as previously described (78). The Akata-GFP Burkitt lymphoma cell line is a type I latency Burkitt lymphoma line that lost its endogenous EBV genome and was then superinfected with an Akata EBV strain containing inserted green fluorescent protein (GFP) and G418 resistance genes as previously described (79). It also supports a low level of constitutive lytic gene expression and was maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), 1% pen-strep, and 500 μg/ml G418. Kem I and Kem III are EBV-infected Burkitt lymphoma cell lines that exhibit type I and type III latency, respectively (received from Jeff Sample). They were maintained in RPMI 1640 medium supplemented with 10% FBS and 1% pen-strep. HONE-Akata cells (a gift from Lawrence Young, University of Birmingham) and CNE-Akata cells (a gift from Dolly Huang, University of Hong Kong, via Diane Hayward at Johns Hopkins University) were established by superinfecting the respective epithelial carcinoma cell lines with the Akata strain of EBV. HONE cells and CNE cells were previously thought to have originated from nasopharyngeal carcinomas but were recently also shown to have derived at least partially from HeLa cells (68). HONE-Akata cells and CNE-Akata cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS, 1% pen-strep, and 400 μg/ml G418. AGS-Akata cells (a gift from Lindsey Hutt-Fletcher, Louisiana State University) are AGS gastric carcinoma cells superinfected with the Akata strain of EBV as previously described (80). They were grown in F-12 medium supplemented with 10% FBS, 1% pen-strep, and 400 μg/ml G418. The HeLa cell line is an HPV-infected cervical carcinoma cell line and was grown in DMEM supplemented with 10% FBS and 1% pen-strep.
Construction of EBV mutant genomes.
EBV AK-BAC-GFP, which was described previously (79), is the parent to the EBV Akata RZNa KO bacmid, which was constructed using the previously described Escherichia coli GS1783-based en passant method (81). Briefly, coordinates 89,674 to 93,036 (GenBank accession no. KC207813.1), spanning the BZLF1, BRFL1, and BRRF1 genes, were replaced with an ampicillin resistance marker by homologous recombination. A PCR product containing the ampicillin gene and homology arms for EBV Akata was generated using pEP-Kan-S2 (81) and primers listed in Table 1. Subsequently, this bacmid was transferred into E. coli BM2710 (82), which was used for infection of NOKs and HeLa cells. The integrity of the bacmid was confirmed by restriction enzyme digestion with at least 2 enzymes (BamHI and EcoRI). Furthermore, the mutation was confirmed by high-fidelity PCR amplification and sequencing of the mutated junction by use of primers listed in Table 1.
TABLE 1.
Primers used for construction of the EBV Akata RZNa KO bacmid
| Primer name | Sequence | Purpose |
|---|---|---|
| Akata RZNa KO primer 1 | AGTCAACATCCAGGCTTGGGCACATCTGCTTCAACAGGAGTACTACGACGGCATCTCCATTAGGGATAACAGGGTAATTTACCAATGCTTAATCAGTGAG | EBV Akata RZNa KO bacmid construction |
| Akata RZNa KO primer 2 | CGATTACCTTCCTCATACCTATGGAGATGCCGTCGTAGTACTCCTGTTGAAGCAGATGTGCGGGGAAATGTGCGCGGAAC | EBV Akata RZNa KO bacmid construction |
| RZNa Span-Fwd | GTGTTGCAGTATGTACAGTTAGC | Sequencing of the mutated junction |
| RZNa Span-Rev | GTAATGGCATCCGTGACCTC | Sequencing of the mutated junction |
Plasmids and siRNAs.
Plasmid DNAs were purified using Qiagen plasmid maxiprep kits as described by the manufacturer. pCDNA3.1-HA-KLF4 (Addgene plasmid 34593; a gift from Michael Ruppert) expresses a cytomegalovirus (CMV) promoter-driven human KLF4 protein that is hemagglutinin (HA) tagged at the amino terminus (83). pCDNA3.1-BLIMP1 (a gift from Kenneth Wright, Lee Moffitt Cancer Center) expresses a CMV promoter-driven human BLIMP1 protein that is FLAG tagged at the amino terminus (84). pSG5-Z and pSG5-R (a gift from S. D. Hayward, Johns Hopkins University) express simian virus 40 (SV40) promoter-driven Z and R immediately early viral lytic proteins, respectively, and were constructed as previously described (85, 86). pCpGL-basic is a promoterless vector driving the luciferase gene and has been engineered to lack any CpG in the vector background, as previously described (87). The LMP1-EDL1 promoter (positions 169483 to 1700643 in the B95.8 EBV genome) and the LMP1-TR promoter (positions 169986 to 170595 in the B95.8 EBV genome) were PCR amplified from the EBV B95.8 genome and cloned upstream of the luciferase gene in pCpGL-basic by use of SpeI and BglII restriction sites. RNA duplexes were synthesized by Dharmacon Research (Lafayette, CO). The sequences of the two different custom-ordered LMP1 siRNAs are AAGAGACCUUCUCUGUCCACU and GGUCAAAGAACAAGGCCAAUU. Two different nontargeting siGENOME siRNAs were also obtained from Dharmacon.
Organotypic raft cultures.
Uninfected NOKs cells and NOKs-Akata cells were grown in organotypic raft cultures as previously described (11). Briefly, the dermal equivalent was prepared in transwell inserts (24-mm diameter and 0.4-μm pore size; Costar) by coating with a layer of 1 ml collagen (3 mg/ml; Wako Chemicals) premix containing F-12 medium, 10% FBS, and 1% pen-strep. This was followed by coating with another layer of 2.5 ml collagen premix containing F-12 medium, 10% FBS, 1% pen-strep, and 4.5 × 105 early-passage human fibroblasts (EF-1-F). This dermal equivalent was allowed to incubate in F-12 medium supplemented with 10% FBS and 1% pen-strep at 37°C and 5% carbon dioxide. After 4 days, 2.1 × 105 uninfected NOKs or NOKs-Akata cells were plated on the dermal equivalent in keratinocyte plating medium (F medium [1.88 mM Ca2+]) containing 0.5% FBS, adenine (24 μg/ml), cholera toxin (8.4 ng/ml), hydrocortisone (2.4 μg/ml), and insulin (5 μg/ml). After growth of the NOKs cells for 4 days, until confluence, the raft cultures were raised to the air-liquid interface and fed through the pores of transwell inserts with cornification medium (keratinocyte plating medium containing 5% FBS and 10 μM C8:0). Raft cultures were grown at the air-liquid interface for 11 days, with the cornification medium replaced every other day. Finally, the rafts were harvested and embedded in 2% agar-1% formalin, followed by fixation in 10% neutral buffered formalin overnight. The raft tissues were then paraffin embedded and cut into 4-μm cross sections.
Immunofluorescence studies.
Formalin-fixed, paraffin-embedded raft tissues were deparaffinized and then subjected to immunofluorescence analysis as described previously (88). The primary antibodies used were an anti-Z (BZ.1) monoclonal antibody (1:200) (SC-53904; Santa Cruz Biotechnology) and a prediluted anti-LMP1 (CS1-4) monoclonal antibody (1:5) (ab7502; Abcam). The secondary antibody used was an Alexa 647-conjugated goat anti-mouse antibody (1:5,000) (A-31571; Life Technologies). The stained sections were mounted using ProLong Gold antifade reagent with DAPI (4′,6-diamidino-2-phenylindole; Life Technologies).
Chemical reagents.
Cells were treated for 48 h (unless noted otherwise in Results and the figure legends) with the following chemical reagents: TPA (20 ng/ml; Sigma), sodium butyrate (3 mM; Sigma), methylcellulose (1.6% in K-SFM; Sigma), calcium chloride (1.2 mM), and serum (20% by volume).
Transient transfection.
Lipofectamine 2000 reagent (Thermo Fisher Scientific) and RNAiMax reagent (Invitrogen) were used to transfect epithelial cells with plasmid DNA and siRNA, respectively. In general, 500 ng of total plasmid DNA was used to transfect all epithelial cells in a well of a 12-well plate. A total of 12.5 pmol siRNA was used to transfect HONE-Akata and AGS-Akata cells in a well of a 12-well plate, and 4 pmol of siRNA was used to transfect NOKs-Akata cells. The cells were transfected with siRNA 24 h prior to treatment with TPA, sodium butyrate, or methylcellulose.
Immunoblot analysis.
Cell lysates were harvested in Sumo lysis buffer including protease inhibitors (Roche) as described previously (89). Protein concentrations were determined using the Sumo protein assay (Bio-Rad), and proteins were separated in SDS-10% polyacrylamide gels and then transferred to a nitrocellulose membrane. Membranes were blocked in phosphate-buffered saline (PBS) containing 5% milk and 0.1% Tween 20 solution. Membranes were then incubated with the following primary antibodies: anti-Z (sc-53904; Santa Cruz) (1:250), anti-BMRF1 (MAB8186; Millipore) (1:3,000), anti-R rabbit polyclonal antibody directed against the R peptide (peptide sequence EDPDEETSQAVKALREMAD) (1:2,500), anti-KLF4 (4038; Cell Signaling) (1:1,000), anti-BLIMP1 (9115; Cell Signaling) (1:1,000), anti-LMP1 (ab78113; Abcam) (1:1,000), anti-β-actin (A5441; Sigma) (1:5,000), anti-tubulin (T5168; Sigma) (1:2,000), anti-actin (Abcam) (1:1,000), and anti-involucrin (I9018; Sigma) (1:3,000). The secondary antibodies used were horseradish peroxidase (HRP)-labeled goat anti-mouse antibody (Fisher Scientific) (1:5,000) and HRP-labeled anti-rabbit antibody (Fisher Scientific) (1:5,000).
Reporter gene assays.
Uninfected NOKs cells were harvested in 1× reporter lysis buffer (Promega) 48 h after transfection with the reporter gene constructs, with or without the transcription factors. The harvested cells were lysed by one freeze-thaw cycle, followed by quantification of relative luciferase units by use of a BD Monolight 3010 luminometer (BD Biosciences) and a luciferase assay system (Promega).
RT-qPCR analysis.
Total RNA was extracted by use of RNA-Bee reagent (Tel-Test Inc.) from NOKs-Akata cells that were resuspended in methylcellulose for the indicated time points. The extracted RNA was then treated with DNase, followed by reverse transcription using random primers and GoScript reverse transcriptase (RT) (Promega). Real-time PCR was performed on the reverse-transcribed cDNA by using iTaq Universal SYBR green mix (Bio-Rad) in a Bio-Rad CFX96 machine. cDNA (1.5 μl) was used for 40 cycles of 15 s at 95°C and 30 s at 60°C, using primers that will detect (i) LMP1 transcripts originating from both the TR and EDL1 promoters (LMP1-TR+EDL1; forward primer 5′-TGAGTAGGAGGGTGA-3′ and reverse primer 5′-CTATTCCTTTGCTCTCATGC-3′), (ii) LMP1 transcripts originating only from the TR promoter (LMP1-TR; forward primer 5′-GGCAGTACGGGTACAGATTTC-3′ and reverse primer 5′-TTCTAACACAAACACACGCTTTC-3′), (iii) a region that overlaps both the Z and R transcripts (Z+R; forward primer 5′-ACCTCAACCTGGAGACAATTC-3′ and reverse primer 5′-TGTCTGCTAGCTGTTGTTCTT-3′), (iv) a region that specifically detects R transcripts (R; forward primer 5′-GACTTTCTGAGGCTAACTCCTG-3′ and reverse primer 5′-CACACTCCCGGCTGTAAAT-3′), and (v) beta-actin transcripts (forward primer 5′-GCCGGGACCTGACTGACTAC-3′ and reverse primer 5′-TTCTCCTTAATGTCACGCACGAT-3′). Beta-actin was used as a housekeeping gene, and transcripts were quantified using the ΔΔCq method for each time point.
ACKNOWLEDGMENTS
We thank Bill Sugden, Karl Munger, Kenzo Takada, Lawrence Young, Diane Hayward, Dolly Huang, Alan Rickinson, and Jeffrey Sample for cell lines, Kenneth Wright for the BLIMP1 expression vector, Harlene Edwards for histochemical assistance, and members of the Johannsen, Kenney, Lambert, and Mertz laboratories for suggestions and discussions.
REFERENCES
- 1.Longnecker R, Kieff E, Cohen J. 2013. Epstein-Barr virus, p 1898–1959. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Kutok JL, Wang F. 2006. Spectrum of Epstein-Barr virus-associated diseases. Annu Rev Pathol 1:375–404. doi: 10.1146/annurev.pathol.1.110304.100209. [DOI] [PubMed] [Google Scholar]
- 3.Oh ST, Seo JS, Moon UY, Kang KH, Shin D-J, Yoon SK, Kim WH, Park J-G, Lee SK. 2004. A naturally derived gastric cancer cell line shows latency I Epstein-Barr virus infection closely resembling EBV-associated gastric cancer. Virology 320:330–336. doi: 10.1016/j.virol.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 4.Iizasa H, Nanbo A, Nishikawa J, Jinushi M, Yoshiyama H. 2012. Epstein-Barr virus (EBV)-associated gastric carcinoma. Viruses 4:3420–3439. doi: 10.3390/v4123420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Young LS, Dawson CW, Clark D, Rupani H, Busson P, Tursz T, Johnson A, Rickinson AB. 1988. Epstein-Barr virus gene expression in nasopharyngeal carcinoma. J Gen Virol 69:1051–1065. doi: 10.1099/0022-1317-69-5-1051. [DOI] [PubMed] [Google Scholar]
- 6.Brooks L, Yao QY, Rickinson AB, Young LS. 1992. Epstein-Barr virus latent gene transcription in nasopharyngeal carcinoma cells: coexpression of EBNA1, LMP1, and LMP2 transcripts. J Virol 66:2689–2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Young LS, Dawson CW. 2014. Epstein-Barr virus and nasopharyngeal carcinoma. Chin J Cancer 33:581–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Souza YG, Greenspan D, Felton JR, Hartzog GA, Hammer M, Greenspan JS. 1989. Localization of Epstein-Barr virus DNA in the epithelial cells of oral hairy leukoplakia by in situ hybridization of tissue sections. N Engl J Med 320:1559–1560. doi: 10.1056/NEJM198906083202315. [DOI] [PubMed] [Google Scholar]
- 9.Greenspan JS, Greenspan D, Lennette ET, Abrams DI, Conant MA, Petersen V, Freese UK. 1985. Replication of Epstein-Barr virus within the epithelial cells of oral “hairy” leukoplakia, an AIDS-associated lesion. N Engl J Med 313:1564–1571. doi: 10.1056/NEJM198512193132502. [DOI] [PubMed] [Google Scholar]
- 10.Webster-Cyriaque J, Raab-Traub N. 1998. Transcription of Epstein-Barr virus latent cycle genes in oral hairy leukoplakia. Virology 248:53–65. doi: 10.1006/viro.1998.9268. [DOI] [PubMed] [Google Scholar]
- 11.Nawandar DM, Wang A, Makielski K, Lee D, Ma S, Barlow E, Reusch J, Jiang R, Wille CK, Greenspan D, Greenspan JS, Mertz JE, Hutt-Fletcher L, Johannsen EC, Lambert PF, Kenney SC. 2015. Differentiation-dependent KLF4 expression promotes lytic Epstein-Barr virus infection in epithelial cells. PLoS Pathog 11:e1005195. doi: 10.1371/journal.ppat.1005195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Young LS, Lau R, Rowe M, Niedobitek G, Packham G, Shanahan F, Rowe DT, Greenspan D, Greenspan JS, Rickinson AB. 1991. Differentiation-associated expression of the Epstein-Barr virus BZLF1 transactivator protein in oral hairy leukoplakia. J Virol 65:2868–2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Temple RM, Zhu J, Budgeon L, Christensen ND, Meyers C, Sample CE. 2014. Efficient replication of Epstein-Barr virus in stratified epithelium in vitro. Proc Natl Acad Sci U S A 111:16544–16549. doi: 10.1073/pnas.1400818111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Countryman J, Jenson H, Seibl R, Wolf H, Miller G. 1987. Polymorphic proteins encoded within BZLF1 of defective and standard Epstein-Barr viruses disrupt latency. J Virol 61:3672–3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Countryman J, Miller G. 1985. Activation of expression of latent Epstein-Barr herpesvirus after gene transfer with a small cloned subfragment of heterogeneous viral DNA. Proc Natl Acad Sci U S A 82:4085–4089. doi: 10.1073/pnas.82.12.4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ragoczy T, Heston L, Miller G. 1998. The Epstein-Barr virus Rta protein activates lytic cycle genes and can disrupt latency in B lymphocytes. J Virol 72:7978–7984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takada K, Shimizu N, Sakuma S, Ono Y. 1986. trans activation of the latent Epstein-Barr virus (EBV) genome after transfection of the EBV DNA fragment. J Virol 57:1016–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zalani S, Holley-Guthrie E, Kenney S. 1996. Epstein-Barr viral latency is disrupted by the immediate-early BRLF1 protein through a cell-specific mechanism. Proc Natl Acad Sci U S A 93:9194–9199. doi: 10.1073/pnas.93.17.9194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rooney CM, Rowe DT, Ragot T, Farrell PJ. 1989. The spliced BZLF1 gene of Epstein-Barr virus (EBV) transactivates an early EBV promoter and induces the virus productive cycle. J Virol 63:3109–3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Speck SH, Chatila T, Flemington E. 1997. Reactivation of Epstein-Barr virus: regulation and function of the BZLF1 gene. Trends Microbiol 5:399–405. doi: 10.1016/S0966-842X(97)01129-3. [DOI] [PubMed] [Google Scholar]
- 21.Kenney SC, Mertz JE. 2014. Regulation of the latent-lytic switch in Epstein-Barr virus. Semin Cancer Biol 26:60–68. doi: 10.1016/j.semcancer.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Murata T, Tsurumi T. 2014. Switching of EBV cycles between latent and lytic states. Rev Med Virol 24:142–153. doi: 10.1002/rmv.1780. [DOI] [PubMed] [Google Scholar]
- 23.Flemington E, Speck SH. 1990. Autoregulation of Epstein-Barr virus putative lytic switch gene BZLF1. J Virol 64:1227–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Urier G, Buisson M, Chambard P, Sergeant A. 1989. The Epstein-Barr virus early protein EB1 activates transcription from different responsive elements including AP-1 binding sites. EMBO J 8:1447–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ragoczy T, Miller G. 2001. Autostimulation of the Epstein-Barr virus BRLF1 promoter is mediated through consensus Sp1 and Sp3 binding sites. J Virol 75:5240–5251. doi: 10.1128/JVI.75.11.5240-5251.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu P, Speck SH. 2003. Synergistic autoactivation of the Epstein-Barr virus immediate-early BRLF1 promoter by Rta and Zta. Virology 310:199–206. doi: 10.1016/S0042-6822(03)00145-4. [DOI] [PubMed] [Google Scholar]
- 27.Kaye KM, Izumi KM, Kieff E. 1993. Epstein-Barr virus latent membrane protein 1 is essential for B-lymphocyte growth transformation. Proc Natl Acad Sci U S A 90:9150–9154. doi: 10.1073/pnas.90.19.9150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dirmeier U, Neuhierl B, Kilger E, Reisbach G, Sandberg ML, Hammerschmidt W. 2003. Latent membrane protein 1 is critical for efficient growth transformation of human B cells by Epstein-Barr virus. Cancer Res 63:2982–2989. [PubMed] [Google Scholar]
- 29.Kieser A, Sterz KR. 2015. The latent membrane protein 1 (LMP1). Curr Top Microbiol Immunol 391:119–149. doi: 10.1007/978-3-319-22834-1_4. [DOI] [PubMed] [Google Scholar]
- 30.Lam N, Sugden B. 2003. CD40 and its viral mimic, LMP1: similar means to different ends. Cell Signal 15:9–16. doi: 10.1016/S0898-6568(02)00083-9. [DOI] [PubMed] [Google Scholar]
- 31.Zhao Y, Wang Y, Zeng S, Hu X. 2012. LMP1 expression is positively associated with metastasis of nasopharyngeal carcinoma: evidence from a meta-analysis. J Clin Pathol 65:41–45. doi: 10.1136/jclinpath-2011-200198. [DOI] [PubMed] [Google Scholar]
- 32.Tsao SW, Tramoutanis G, Dawson CW, Lo AKF, Huang DP. 2002. The significance of LMP1 expression in nasopharyngeal carcinoma. Semin Cancer Biol 12:473–487. doi: 10.1016/S1044579X02000901. [DOI] [PubMed] [Google Scholar]
- 33.Horikawa T, Yoshizaki T, Kondo S, Furukawa M, Kaizaki Y, Pagano JS. 2011. Epstein-Barr virus latent membrane protein 1 induces Snail and epithelial-mesenchymal transition in metastatic nasopharyngeal carcinoma. Br J Cancer 104:1160–1167. doi: 10.1038/bjc.2011.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pathmanathan R, Prasad U, Sadler R, Flynn K, Raab-Traub N. 1995. Clonal proliferations of cells infected with Epstein-Barr virus in preinvasive lesions related to nasopharyngeal carcinoma. N Engl J Med 333:693–698. doi: 10.1056/NEJM199509143331103. [DOI] [PubMed] [Google Scholar]
- 35.Horikawa T, Sheen TS, Takeshita H, Sato H, Furukawa M, Yoshizaki T. 2001. Induction of c-Met proto-oncogene by Epstein-Barr virus latent membrane protein-1 and the correlation with cervical lymph node metastasis of nasopharyngeal carcinoma. Am J Pathol 159:27–33. doi: 10.1016/S0002-9440(10)61669-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Horikawa T, Yang J, Kondo S, Yoshizaki T, Joab I, Furukawa M, Pagano JS. 2007. Twist and epithelial-mesenchymal transition are induced by the EBV oncoprotein latent membrane protein 1 and are associated with metastatic nasopharyngeal carcinoma. Cancer Res 67:1970–1978. doi: 10.1158/0008-5472.CAN-06-3933. [DOI] [PubMed] [Google Scholar]
- 37.Lin S, Zong Y, Lin H, Zhong B, Li Z, Liang Y. 2003. Effects of eb virus encoded LMP1 on differentiation of nasopharyngeal carcinoma cells. Chin J Cancer Res 15:257–261. doi: 10.1007/BF02974888. [DOI] [Google Scholar]
- 38.Pathmanathan R, Prasad U, Chandrika G, Sadler R, Flynn K, Raab-Traub N. 1995. Undifferentiated, nonkeratinizing, and squamous cell carcinoma of the nasopharynx. Variants of Epstein-Barr virus-infected neoplasia. Am J Pathol 146:1355–1367. [PMC free article] [PubMed] [Google Scholar]
- 39.Chang Y, Lee H-H, Chang S-S, Hsu T-Y, Wang P-W, Chang Y-S, Takada K, Tsai C-H. 2004. Induction of Epstein-Barr virus latent membrane protein 1 by a lytic transactivator Rta. J Virol 78:13028–13036. doi: 10.1128/JVI.78.23.13028-13036.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boos H, Berger R, Kuklik-Roos C, Iftner T, Mueller-Lantzsch N. 1987. Enhancement of Epstein-Barr virus membrane protein (LMP) expression by serum, TPA, or n-butyrate in latently infected Raji cells. Virology 159:161–165. doi: 10.1016/0042-6822(87)90360-6. [DOI] [PubMed] [Google Scholar]
- 41.Lu C-C, Jeng Y-Y, Tsai C-H, Liu M-Y, Yeh S-W, Hsu T-Y, Chen M-R. 2006. Genome-wide transcription program and expression of the Rta responsive gene of Epstein-Barr virus. Virology 345:358–372. doi: 10.1016/j.virol.2005.09.064. [DOI] [PubMed] [Google Scholar]
- 42.Yuan J, Cahir-McFarland E, Zhao B, Kieff E. 2006. Virus and cell RNAs expressed during Epstein-Barr virus replication. J Virol 80:2548–2565. doi: 10.1128/JVI.80.5.2548-2565.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Webster-Cyriaque J, Middeldorp J, Raab-Traub N. 2000. Hairy leukoplakia: an unusual combination of transforming and permissive Epstein-Barr virus infections. J Virol 74:7610–7618. doi: 10.1128/JVI.74.16.7610-7618.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Walling DM, Ling PD, Gordadze AV, Montes-Walters M, Flaitz CM, Nichols CM. 2004. Expression of Epstein-Barr virus latent genes in oral epithelium: determinants of the pathogenesis of oral hairy leukoplakia. J Infect Dis 190:396–399. doi: 10.1086/422039. [DOI] [PubMed] [Google Scholar]
- 45.Geiser V, Cahir-McFarland E, Kieff E. 2011. Latent membrane protein 1 is dispensable for Epstein-Barr virus replication in human embryonic kidney 293 cells. PLoS One 6:e22929. doi: 10.1371/journal.pone.0022929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ahsan N, Kanda T, Nagashima K, Takada K. 2005. Epstein-Barr virus transforming protein LMP1 plays a critical role in virus production. J Virol 79:4415–4424. doi: 10.1128/JVI.79.7.4415-4424.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Prince S, Keating S, Fielding C, Brennan P, Floettmann E, Rowe M. 2003. Latent membrane protein 1 inhibits Epstein-Barr virus lytic cycle induction and progress via different mechanisms. J Virol 77:5000–5007. doi: 10.1128/JVI.77.8.5000-5007.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bentz GL, Moss CR, Whitehurst CB, Moody CA, Pagano JS. 2015. LMP1-induced sumoylation influences the maintenance of Epstein-Barr virus latency through KAP1. J Virol 89:7465–7477. doi: 10.1128/JVI.00711-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lai K-Y, Chou Y-C, Lin J-H, Liu Y, Lin K-M, Doong S-L, Chen M-R, Yeh T-H, Lin S-J, Tsai C-H. 2015. Maintenance of Epstein-Barr virus latent status by a novel mechanism, latent membrane protein 1-induced interleukin-32, via the protein kinase Cδ pathway. J Virol 89:5968–5980. doi: 10.1128/JVI.00168-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sadler RH, Raab-Traub N. 1995. The Epstein-Barr virus 3.5-kilobase latent membrane protein 1 mRNA initiates from a TATA-less promoter within the first terminal repeat. J Virol 69:4577–4581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hudson GS, Farrell PJ, Barrell BG. 1985. Two related but differentially expressed potential membrane proteins encoded by the EcoRI Dhet region of Epstein-Barr virus B95-8. J Virol 53:528–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chang MH, Ng CK, Lin YJ, Liang CL, Chung PJ, Chen ML, Tyan YS, Hsu CY, Shu CH, Chang YS. 1997. Identification of a promoter for the latent membrane protein 1 gene of Epstein-Barr virus that is specifically activated in human epithelial cells. DNA Cell Biol 16:829–837. doi: 10.1089/dna.1997.16.829. [DOI] [PubMed] [Google Scholar]
- 53.Fåhraeus R, Jansson A, Ricksten A, Sjöblom A, Rymo L. 1990. Epstein-Barr virus-encoded nuclear antigen 2 activates the viral latent membrane protein promoter by modulating the activity of a negative regulatory element. Proc Natl Acad Sci U S A 87:7390–7394. doi: 10.1073/pnas.87.19.7390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lo AKF, To KF, Lo KW, Lung RWM, Hui JWY, Liao G, Hayward SD. 2007. Modulation of LMP1 protein expression by EBV-encoded microRNAs. Proc Natl Acad Sci U S A 104:16164–16169. doi: 10.1073/pnas.0702896104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Le Roux A, Kerdiles B, Walls D, Dedieu JF, Perricaudet M. 1994. The Epstein-Barr virus determined nuclear antigens EBNA-3A, -3B, and -3C repress EBNA-2-mediated transactivation of the viral terminal protein 1 gene promoter. Virology 205:596–602. doi: 10.1006/viro.1994.1687. [DOI] [PubMed] [Google Scholar]
- 56.Ning S, Hahn AM, Huye LE, Pagano JS. 2003. Interferon regulatory factor 7 regulates expression of Epstein-Barr virus latent membrane protein 1: a regulatory circuit. J Virol 77:9359–9368. doi: 10.1128/JVI.77.17.9359-9368.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Demetriades C, Mosialos G. 2009. The LMP1 promoter can be transactivated directly by NF-kappaB. J Virol 83:5269–5277. doi: 10.1128/JVI.00097-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Johansson P, Jansson A, Rüetschi U, Rymo L. 2009. Nuclear factor-kappaB binds to the Epstein-Barr virus LMP1 promoter and upregulates its expression. J Virol 83:1393–1401. doi: 10.1128/JVI.01637-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Johansson P, Jansson A, Rüetschi U, Rymo L. 2010. The p38 signaling pathway upregulates expression of the Epstein-Barr virus LMP1 oncogene. J Virol 84:2787–2797. doi: 10.1128/JVI.01052-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen H, Lee JM, Zong Y, Borowitz M, Ng MH, Ambinder RF, Hayward SD. 2001. Linkage between STAT regulation and Epstein-Barr virus gene expression in tumors. J Virol 75:2929–2937. doi: 10.1128/JVI.75.6.2929-2937.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Murata T, Noda C, Narita Y, Watanabe T, Yoshida M, Ashio K, Sato Y, Goshima F, Kanda T, Yoshiyama H, Tsurumi T, Kimura H. 2016. Induction of Epstein-Barr virus oncoprotein LMP1 by transcription factors AP-2 and early B cell factor. J Virol 90:3873–3889. doi: 10.1128/JVI.03227-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sjöblom A, Yang W, Palmqvist L, Jansson A, Rymo L. 1998. An ATF/CRE element mediates both EBNA2-dependent and EBNA2-independent activation of the Epstein-Barr virus LMP1 gene promoter. J Virol 72:1365–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Höfelmayr H, Strobl LJ, Stein C, Laux G, Marschall G, Bornkamm GW, Zimber-Strobl U. 1999. Activated mouse Notch1 transactivates Epstein-Barr virus nuclear antigen 2-regulated viral promoters. J Virol 73:2770–2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Arvey A, Tempera I, Tsai K, Chen H-S, Tikhmyanova N, Klichinsky M, Leslie C, Lieberman PM. 2012. An atlas of the Epstein-Barr virus transcriptome and epigenome reveals host-virus regulatory interactions. Cell Host Microbe 12:233–245. doi: 10.1016/j.chom.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Johannsen E, Koh E, Mosialos G, Tong X, Kieff E, Grossman SR. 1995. Epstein-Barr virus nuclear protein 2 transactivation of the latent membrane protein 1 promoter is mediated by J kappa and PU.1. J Virol 69:253–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhao B, Zou J, Wang H, Johannsen E, Peng C, Quackenbush J, Mar JC, Morton CC, Freedman ML, Blacklow SC, Aster JC, Bernstein BE, Kieff E. 2011. Epstein-Barr virus exploits intrinsic B-lymphocyte transcription programs to achieve immortal cell growth. Proc Natl Acad Sci U S A 108:14902–14907. doi: 10.1073/pnas.1108892108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen H-S, Martin KA, Lu F, Lupey LN, Mueller JM, Lieberman PM, Tempera I. 2014. Epigenetic deregulation of the LMP1/LMP2 locus of Epstein-Barr virus by mutation of a single CTCF-cohesin binding site. J Virol 88:1703–1713. doi: 10.1128/JVI.02209-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Strong MJ, Baddoo M, Nanbo A, Xu M, Puetter A, Lin Z. 2014. Comprehensive high-throughput RNA sequencing analysis reveals contamination of multiple nasopharyngeal carcinoma cell lines with HeLa cell genomes. J Virol 88:10696–10704. doi: 10.1128/JVI.01457-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Segre JA, Bauer C, Fuchs E. 1999. Klf4 is a transcription factor required for establishing the barrier function of the skin. Nat Genet 22:356–360. doi: 10.1038/11926. [DOI] [PubMed] [Google Scholar]
- 70.Cordani N, Pozzi S, Martynova E, Fanoni D, Borrelli S, Alotto D, Castagnoli C, Berti E, Viganò MA, Mantovani R. 2011. Mutant p53 subverts p63 control over KLF4 expression in keratinocytes. Oncogene 30:922–932. doi: 10.1038/onc.2010.474. [DOI] [PubMed] [Google Scholar]
- 71.Chew YC, Adhikary G, Xu W, Wilson GM, Eckert RL. 2013. Protein kinase C δ increases Kruppel-like factor 4 protein, which drives involucrin gene transcription in differentiating keratinocytes. J Biol Chem 288:17759–17768. doi: 10.1074/jbc.M113.477133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Magnúsdóttir E, Kalachikov S, Mizukoshi K, Savitsky D, Ishida-Yamamoto A, Panteleyev AA, Calame K. 2007. Epidermal terminal differentiation depends on B lymphocyte-induced maturation protein-1. Proc Natl Acad Sci U S A 104:14988–14993. doi: 10.1073/pnas.0707323104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chiang M-F, Yang S-Y, Lin I-Y, Hong J-B, Lin S-J, Ying H-Y, Chen C-M, Wu S-Y, Liu F-T, Lin K-I. 2013. Inducible deletion of the Blimp-1 gene in adult epidermis causes granulocyte-dominated chronic skin inflammation in mice. Proc Natl Acad Sci U S A 110:6476–6481. doi: 10.1073/pnas.1219462110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gunasekharan VK, Li Y, Andrade J, Laimins LA. 2016. Post-transcriptional regulation of KLF4 by high-risk human papillomaviruses is necessary for the differentiation-dependent viral life cycle. PLoS Pathog 12:e1005747. doi: 10.1371/journal.ppat.1005747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shaw G, Morse S, Ararat M, Graham FL. 2002. Preferential transformation of human neuronal cells by human adenoviruses and the origin of HEK 293 cells. FASEB J 16:869–871. [DOI] [PubMed] [Google Scholar]
- 76.Oussaief L, Ramírez V, Hippocrate A, Arbach H, Cochet C, Proust A, Raphaël M, Khelifa R, Joab I. 2011. NF-κB-mediated modulation of inducible nitric oxide synthase activity controls induction of the Epstein-Barr virus productive cycle by transforming growth factor beta 1. J Virol 85:6502–6512. doi: 10.1128/JVI.02560-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Piboonniyom S, Duensing S, Swilling NW, Hasskarl J, Hinds PW, Münger K. 2003. Abrogation of the retinoblastoma tumor suppressor checkpoint during keratinocyte immortalization is not sufficient for induction of centrosome-mediated genomic instability. Cancer Res 63:476–483. [PubMed] [Google Scholar]
- 78.Wille CK, Nawandar DM, Panfil AR, Ko MM, Hagemeier SR, Kenney SC. 2013. Viral genome methylation differentially affects the ability of BZLF1 versus BRLF1 to activate Epstein-Barr virus lytic gene expression and viral replication. J Virol 87:935–950. doi: 10.1128/JVI.01790-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kanda T, Yajima M, Ahsan N, Tanaka M, Takada K. 2004. Production of high-titer Epstein-Barr virus recombinants derived from Akata cells by using a bacterial artificial chromosome system. J Virol 78:7004–7015. doi: 10.1128/JVI.78.13.7004-7015.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Molesworth SJ, Lake CM, Borza CM, Turk SM, Hutt-Fletcher LM. 2000. Epstein-Barr virus gH is essential for penetration of B cells but also plays a role in attachment of virus to epithelial cells. J Virol 74:6324–6332. doi: 10.1128/JVI.74.14.6324-6332.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tischer BK, Smith GA, Osterrieder N. 2010. En passant mutagenesis: a two step markerless red recombination system. Methods Mol Biol 634:421–430. doi: 10.1007/978-1-60761-652-8_30. [DOI] [PubMed] [Google Scholar]
- 82.Grillot-Courvalin C, Goussard S, Huetz F, Ojcius DM, Courvalin P. 1998. Functional gene transfer from intracellular bacteria to mammalian cells. Nat Biotechnol 16:862–866. doi: 10.1038/nbt0998-862. [DOI] [PubMed] [Google Scholar]
- 83.Lin C-C, Liu L-Z, Addison JB, Wonderlin WF, Ivanov AV, Ruppert JM. 2011. A KLF4-miRNA-206 autoregulatory feedback loop can promote or inhibit protein translation depending upon cell context. Mol Cell Biol 31:2513–2527. doi: 10.1128/MCB.01189-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Györy I, Fejér G, Ghosh N, Seto E, Wright KL. 2003. Identification of a functionally impaired positive regulatory domain I binding factor 1 transcription repressor in myeloma cell lines. J Immunol 170:3125–3133. doi: 10.4049/jimmunol.170.6.3125. [DOI] [PubMed] [Google Scholar]
- 85.Hardwick JM, Lieberman PM, Hayward SD. 1988. A new Epstein-Barr virus transactivator, R, induces expression of a cytoplasmic early antigen. J Virol 62:2274–2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sarisky RT, Gao Z, Lieberman PM, Fixman ED, Hayward GS, Hayward SD. 1996. A replication function associated with the activation domain of the Epstein-Barr virus Zta transactivator. J Virol 70:8340–8347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Klug M, Rehli M. 2006. Functional analysis of promoter CpG methylation using a CpG-free luciferase reporter vector. Epigenetics 1:127–130. doi: 10.4161/epi.1.3.3327. [DOI] [PubMed] [Google Scholar]
- 88.Balsitis SJ, Sage J, Duensing S, Münger K, Jacks T, Lambert PF. 2003. Recapitulation of the effects of the human papillomavirus type 16 E7 oncogene on mouse epithelium by somatic Rb deletion and detection of pRb-independent effects of E7 in vivo. Mol Cell Biol 23:9094–9103. doi: 10.1128/MCB.23.24.9094-9103.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Adamson AL, Kenney S. 2001. Epstein-Barr virus immediate-early protein BZLF1 is SUMO-1 modified and disrupts promyelocytic leukemia bodies. J Virol 75:2388–2399. doi: 10.1128/JVI.75.5.2388-2399.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]