

ABSTRACT
Suppression of interferon signaling is of paramount importance to a virus. Interferon signaling significantly reduces or halts the ability of a virus to replicate; therefore, viruses have evolved sophisticated mechanisms that suppress activation of the interferon pathway or responsiveness of the infected cell to interferon. Adenovirus has multiple modes of inhibiting the cellular response to interferon. Here, we report that E1A, previously shown to regulate interferon signaling in multiple ways, inhibits interferon-stimulated gene expression by modulating RuvBL1 function. RuvBL1 was previously shown to affect type I interferon signaling. E1A binds to RuvBL1 and is recruited to RuvBL1-regulated promoters in an interferon-dependent manner, preventing their activation. Depletion of RuvBL1 impairs adenovirus growth but does not appear to significantly affect viral protein expression. Although RuvBL1 has been shown to play a role in cell growth, its depletion had no effect on the ability of the virus to replicate its genome or to drive cells into S phase. E1A was found to bind to RuvBL1 via the C terminus of E1A, and this interaction was important for suppression of interferon-stimulated gene transcriptional activation and recruitment of E1A to interferon-regulated promoters. Here, we report the identification of RuvBL1 as a new target for adenovirus in its quest to suppress the interferon response.
IMPORTANCE For most viruses, suppression of the interferon signaling pathway is crucial to ensure a successful replicative cycle. Human adenovirus has evolved several different mechanisms that prevent activation of interferon or the ability of the cell to respond to interferon. The viral immediate-early gene E1A was previously shown to affect interferon signaling in several different ways. Here, we report a novel mechanism reliant on RuvBL1 that E1A uses to prevent activation of interferon-stimulated gene expression following infection or interferon treatment. This adds to the growing knowledge of how viruses are able to inhibit interferon and identifies a novel target used by adenovirus for modulation of the cellular interferon pathway.
KEYWORDS: E1A, Pontin, RuvBL1, adenovirus, interferon
INTRODUCTION
Human adenovirus (HAdV) infects and replicates in terminally differentiated cells, usually of the epithelium (1). In order to replicate within the infected cell, the virus needs to reprogram the intracellular environment to be more permissive to replication of the viral genome. This is mainly due to the type of cell that the virus infects, which is a terminally differentiated epithelial cell lacking in proteins and cofactors required for large-scale DNA replication (2). In addition to cellular reprogramming, the virus needs to be able to hide within the infected cell long enough to accomplish its replicative cycle and spread to neighboring cells. Adenoviruses have evolved several different strategies to suppress the innate and acquired immune systems and prevent the detection and killing of an infected cell. One of the major contributors to host immune evasion by HAdV is the viral E3 transcriptional unit (3). The HAdV5 E3 transcriptional unit encodes several proteins that are involved in host immune evasion, including E3-gp19k, responsible for the inhibition of transport of major histocompatibility complex class I (MHC-I) molecules to the cell surface (4); the E3-14.7K protein, which inhibits tumor necrosis factor (TNF)-induced cytolysis of HAdV-infected cells (5–7); and receptor internalization and degradation alpha (RIDα) and RIDβ, which drive internalization and degradation of cell surface receptors, together with the E3-6.7K protein, such as receptors for Fas, TRAIL receptor 1, and TNF receptor 1 (reviewed in reference 8). The other major players in inhibition of the antiviral response are the virus-associated (VA) RNAs expressed during infection, particularly VAI (9). VAI RNA targets protein kinase R, preventing its activation and stimulation of the interferon (IFN) response. VA RNA also targets other components of innate immunity, including the small interfering RNA (siRNA) machinery and other immune proteins (reviewed in reference 10).
Inhibition of host immune evasion by HAdV is not limited to the viral E3 transcriptional unit or the VA RNAs but also relies on the functions of the E1A and E4 orf3 proteins. E1A is the immediate-early gene first expressed after the viral genome has entered the cell nucleus. E1A drives cells into S phase, which enables viral genomes to be replicated (2). However, besides inducing S phase, E1A has a multitude of other functions, including suppression of the antiviral interferon response (reviewed in reference 11). Notably, E1A is able to suppress type I interferon-inducible gene expression via its N terminus/CR1 region (12). E1A also suppresses expression of HLA class II genes by type II IFN (IFN-γ) and IFN-β mRNA by blocking transcription initiation (13). Lastly, E1A inhibits histone H2B monoubiquitination by interfering with the RNF20 ubiquitin ligase (14). E1A also interacts with DREF, a component of promyelocytic leukemia protein (PML) bodies that appears to play a role in the innate antiviral response; interference with DREF function by E1A enhances virus growth (15). E4 orf3 is also involved in IFN suppression and inhibition of PML body function and in the immune response (16). Collectively, HAdV has evolved sophisticated mechanisms to block the immune response and prevent immune-mediated killing of infected cells.
The C terminus of E1A, encoded by the second exon of the gene, spans residues 186 to 289 in the largest isoform of HAdV5 (2), yet until recently only a few proteins that bind within the region had been identified (17). Our studies of new C terminus binding proteins have identified DREF (15) and Ku70 (18) as novel E1A interaction partners. Here, we report the identification of another novel E1A C terminus binding protein, RuvBL1 (also known as Pontin and TIP49a). Our study shows that E1A uses RuvBL1 to suppress activation of interferon-stimulated genes (ISGs) following viral infection. During infection, RuvBL1 and E1A are recruited to ISG promoters to drive transcriptional silencing. Depletion of RuvBL1 renders E1A unable to suppress ISG activation, and mutants of E1A unable to bind to RuvBL1 are deficient for growth and ISG suppression. Our results identify a novel interaction between the cellular protein RuvBL1 and HAdV5 E1A that is important for suppression of the interferon response.
RESULTS
RuvBL1 interacts with the C terminus of HAdV5 E1A.
Initial mass spectrometry analysis of proteins associated with the C terminus of E1A identified several peptides corresponding to the cellular protein RuvBL1 (data not shown). To verify that this interaction occurred during viral infection and with endogenous RuvBL1, HT1080 cells were infected with HAdV5 dl309 expressing wild-type (wt) E1A (Fig. 1B). Immunoprecipitation (IP) for E1A readily precipitated cellular RuvBL1. We did not detect any RuvBL1 immunoprecipitated from uninfected cells despite equal levels of RuvBL1 present. It is unclear why there are two bands of endogenous RuvBL1 present; it is possible that one is a modified form, as RuvBL1 has been shown to be posttranslationally modified (19).
FIG 1.

RuvBL1 binds to multiple regions of E1A. (A) Schematic representation of HAdV5 E1A289R and the locations of the deletion mutants used in this study. (B) HT1080 cells infected with HAdV5 dl309 or mock infected were lysed and immunoprecipitated for E1A using M73 and M58 antibodies cross-linked to protein A-Sepharose beads. The complexes were washed, eluted, and resolved by SDS-PAGE. RuvBL1 was detected using the polyclonal anti-RuvBL1 antibody, while E1A was detected using M73 monoclonal antibody. One milligram of total cell lysate was used per immunoprecipitation. (C) HT1080 cells mock infected or infected with dl309 or the mutants shown were immunoprecipitated for E1A using either M73 or M58 antibody (M58 was used for mutants dl1135 and dl1136, and M73 was used for all others; mock sample was immunoprecipitated with a mixture of M73 and M58), and the complexes were washed, eluted, and subsequently resolved by SDS-PAGE. RuvBL1 was detected using the rat monoclonal anti-HA (3F10) antibody, while E1A was detected using M73 monoclonal antibody. One milligram of total cell lysate was used per immunoprecipitation. (D) Bacterially expressed and purified GST-RuvBL1 and 6×His-E1A289R were mixed, incubated for 1 h, pulled down using either glutathione or Ni-nitrilotriacetic acid (NTA) beads, and washed. The complexes were eluted using SDS-PAGE sample buffer and resolved on a 10% acrylamide gel. Associated proteins were detected using either GST, RuvBL1, or M73 (for E1A) antibody as indicated. One milligram of each protein was used in the pulldown, and the input Coomassie-stained gel shows the input level of each protein.
To identify the region within the C terminus of E1A required for the interaction, we performed further co-IP experiments using transfected, hemagglutinin (HA)-tagged RuvBL1 and HAdV mutants dl1101 to dl1108 and dl1116 to dl1136 (Fig. 1C), which collectively have residues 2 to 127 and 205 to 289 of HAdV5 E1A deleted (20–22) (Fig. 1A and C). Wild-type E1A was most efficient in immunoprecipitating RuvBL1; however, mutants dl1132 and dl1133 were severely deficient for the interaction, indicating that E1A residues 224 to 254 are required for binding to RuvBL1. It is also worth noting that mutant dl1135 showed reduced binding, but it was consistently slightly above background in repeat experiments. Recently, RuvBL1 was shown to interact with the N terminus of E1A as part of the NuA4/TIP60 chromatin-remodeling complex (23). We therefore investigated whether any of our exon 1 E1A mutants (dl1101 through dl1108) (Fig. 1A) lost the ability to interact with RuvBL1 (Fig. 1B). Although none of our E1A N terminus mutants were completely defective for binding to RuvBL1, some showed reduced binding (Fig. 1). This suggests that E1A may bind to RuvBL1 via multiple regions on E1A.
Since E1A associates with multiple proteins (2), it was important to determine whether the association with RuvBL1 was direct or whether it was mediated by another protein. To test this, we performed a glutathione S-transferase (GST) pulldown assay using bacterially purified 6×His-tagged E1A and GST-tagged RuvBL1. When the assay was performed for E1A, RuvBL1 was efficiently pulled down by the nickel resin (Fig. 1D, left). Likewise, when the pulldown was performed using glutathione resin (binding GST-tagged RuvBL1), E1A was efficiently pulled down (Fig. 1D, right). These results demonstrate that the interaction between E1A and RuvBL1 is most likely direct.
Depletion of RuvBL1 reduces virus growth.
To determine how RuvBL1 affects virus growth, we assayed for viral replication in RuvBL1-depleted HT1080 cells (Fig. 2). Depletion of RuvBL1 via siRNA was efficient but not 100% complete, as some residual protein remained (Fig. 2, inset). Nevertheless, reduction of RuvBL1 protein levels had a pronounced effect on virus growth. Virus titers were reduced by more than 3-fold in RuvBL1-depleted cells compared to control siRNA-transfected cells. RuvBL1 depletion did not appear to have any effect on the viability or growth potential of the cells, as the cells continued to grow and divide normally (data not shown).
FIG 2.
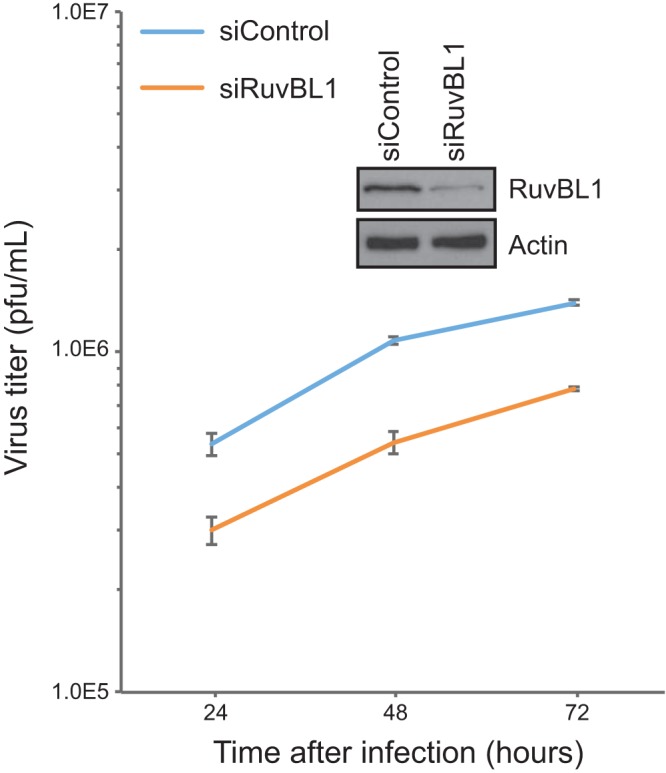
Depletion of RuvBL1 reduces HAdV growth. HT1080 cells were transfected with siRNA depleting RuvBL1 or a negative-control siRNA that depletes no human proteins. After depletion, the cells were infected with dl309 at an MOI of 10 and incubated for the indicated times, and virus was then harvested. The virus was quantified on 293 cells by plaque assay. The Western blot shows depletion of RuvBL1 at the time of infection. n = 3. The error bars represent standard deviations (SD).
RuvBL1 does not affect viral gene expression.
The reduced viral titers observed following siRNA-mediated knockdown of RuvBL1 could be attributed to a number of factors. Since RuvBL1 is a component of several chromatin-remodeling complexes, one possible explanation for the reduced virus growth could be that RuvBL1 directly participates in viral gene expression, and therefore, its depletion would result in reduced viral transcripts and fewer viruses. To investigate this possibility, we analyzed viral gene expression in infected HT1080 cells that were treated with control siRNA or siRNA targeting RuvBL1 (Fig. 3A and Table 1). Viral gene expression was reduced for E3, E4, and hexon 48 and 72 h after infection in RuvBL1-depleted cells compared to nondepleted cells. However, most genes were not significantly affected at 24 h and earlier in the infection (Fig. 3A and Table 1). To confirm the lack of a direct effect of RuvBL1 on viral gene expression during infection, we performed chromatin immunoprecipitation (ChIP) for RuvBL1 on viral promoters. We did not observe significant recruitment of RuvBL1 to any viral promoters despite repeated attempts using different RuvBL1 antibodies (data not shown), suggesting that RuvBL1 does not play a direct role in viral gene expression. To determine whether the reduced viral gene expression at later time points in the infection correlated with protein levels, we also investigated whether depletion of RuvBL1 affects viral proteins by Western blotting (Fig. 3B). We observed only a slight reduction in the levels of the E2 72,000-molecular-weight (72K) DNA binding protein (DBP), as well as some of the viral late proteins, 72 h after infection (Fig. 3B), but we saw no difference at earlier time points. We also did not observe reduced hexon protein levels in RuvBL1-depleted cells despite substantial reduction in hexon mRNA (Fig. 3). Lastly, we also investigated whether depletion of RuvBL1 affects the ability of the virus to replicate its genome or to drive cells into S phase (data not shown). We observed no significant differences in genome copies per cell or S phase induction in cells depleted of RuvBL1 versus control cells treated with nonspecific siRNA. Collectively, these results suggest that the reduced viral growth observed is unlikely to be caused by RuvBL1 having direct effects on viral gene expression and genome replication.
FIG 3.
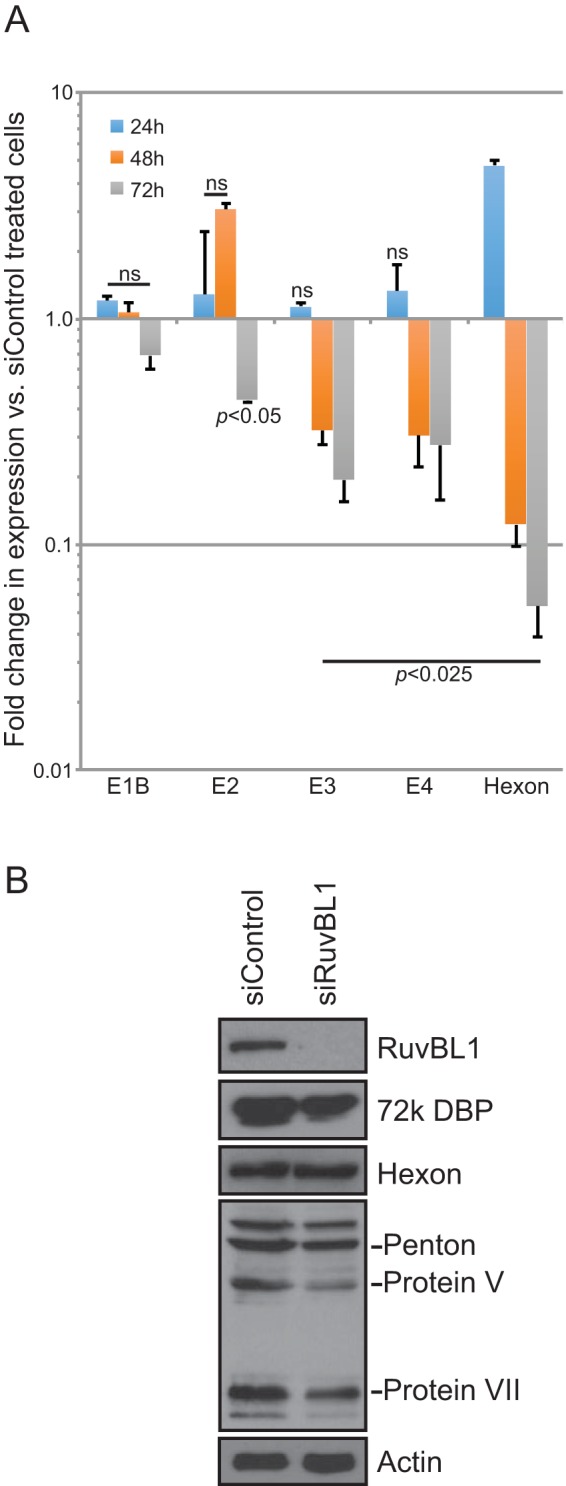
Effects of RuvBL1 depletion on viral gene and protein expression. (A) HT1080 cells were transfected with siRNA depleting RuvBL1 or a negative-control siRNA that depletes no human proteins. After depletion, the cells were infected with dl309 at an MOI of 10 and incubated for the indicated times, and total RNA was then extracted by the TRIzol method. cDNA was made from the total RNA using Vilo reverse transcriptase, and gene expression was quantified using the Bio-Rad CFX96 instrument and ABI SYBR Supermix for CFX reagent. n = 3. The error bars represent SD. The P values were determined using a t test. ns, not significant. (B) Western blot of dl309-infected (MOI, 10) HT1080 cells 72 h after infection that were depleted of RuvBL1 or treated with negative-control siRNA. Twenty micrograms of total cell lysate was loaded per lane, resolved by SDS-PAGE, and blotted for RuvBL1, E2 72K DBP, hexon and other viral late proteins, and actin.
TABLE 1.
Fold changes in expression of viral genes in RuvBL1-depleted cells versus cells treated with control siRNA
| Gene | Fold change ± SD (P value)a |
|||
|---|---|---|---|---|
| 6 h | 12 h | 16 h | 20 h | |
| E1A | 0.39 ± 0.14 (>0.05) | 0.63 ± 0.14 (>0.05) | 0.42 ± 0.014 (>0.05) | 1.17 ± 0.16 (>0.05) |
| E1B | 0.33 ± 0.10 (<0.01) | 0.50 ± 0.032 (>0.05) | 1.29 ± 0.27 (<0.025) | 0.88 ± 0.020 (>0.05) |
| E2A | ND | ND | 0.49 ± 0.25 (>0.05) | 0.36 ± 0.025 (<0.05) |
| E3A | ND | 1.75 ± 0.21 (>0.05) | 0.36 ± 0.043 (>0.05) | 0.55 ± 0.051 (>0.05) |
| E4 | ND | 1.57 ± 0.062 (>0.05) | 0.31 ± 0.069 (>0.05) | 0.87 ± 0.060 (= 0.05) |
| hexon | ND | ND | 0.29 ± 0.036 (>0.05) | 0.87 ± 0.17 (>0.05) |
ND, not detected. Values in boldface are significant.
RuvBL1 is required for E1A-mediated suppression of ISG expression.
Our investigation of the role of RuvBL1 in viral replication showed only minimal effects on various measures of viral fitness, yet we observed a modest, but consistent, reduction in the ability of the virus to grow following RuvBL1 depletion. Previous reports have implicated RuvBL1 in regulation of ISGs, specifically ISG56 and IFI6 (24). We therefore investigated whether depletion of RuvBL1 affects the ability of HAdV, and more specifically E1A, to suppress ISG56 and IFI6 following infection. To investigate this, we infected HT1080 cells with HAdV expressing wt E1A and assessed ISG56 and IFI6 levels 24 h after infection, comparing the results from RuvBL1-depleted cells to those from cells treated with a negative-control siRNA (Fig. 4). Infection with dl309 alone had a small effect on ISG expression, with ISG56 and IFI6 being induced 5-fold or less (Fig. 4B and C). We therefore treated the cells with IFN-α2a 24 h after infection and assayed for ISG56 and IFI6 expression 8 h later. IFN treatment potently induced expression of ISG56 in RuvBL1-depleted and infected cells, and we also observed a 10-fold induction of IFI6 in these cells after treatment. Interestingly, when RuvBL1 was present, as was the case in cells treated with the negative-control siRNA, the levels of ISG56 and IFI6 were much lower (Fig. 4B and C). In particular, the level of ISG56 was reduced from induction of over 60-fold versus untreated cells to less than 10-fold, while IFI6 was reduced by more than half. Infection of these cells with dl312, expressing no E1A (25), had no effect on ISG56 or IFI6 expression after IFN treatment compared to uninfected cells treated with IFN (data not shown). This observation suggests that IFN induction may rely, in part, on the ability of the virus to replicate, since dl312 does not grow in HT1080 cells (data not shown). We also observed similar results in IMR-90 normal lung fibroblasts (data not shown). Depletion of RuvBL1 had little effect on general transcription, as levels of the glucose-6-phosphate dehydrogenase gene (G6PD) (Fig. 4A) and others, including Nek9 and POLR2A (data not shown), were unchanged between depleted and control cells, although some, such as G6PD, were slightly elevated in infected cells as opposed to uninfected controls.
FIG 4.
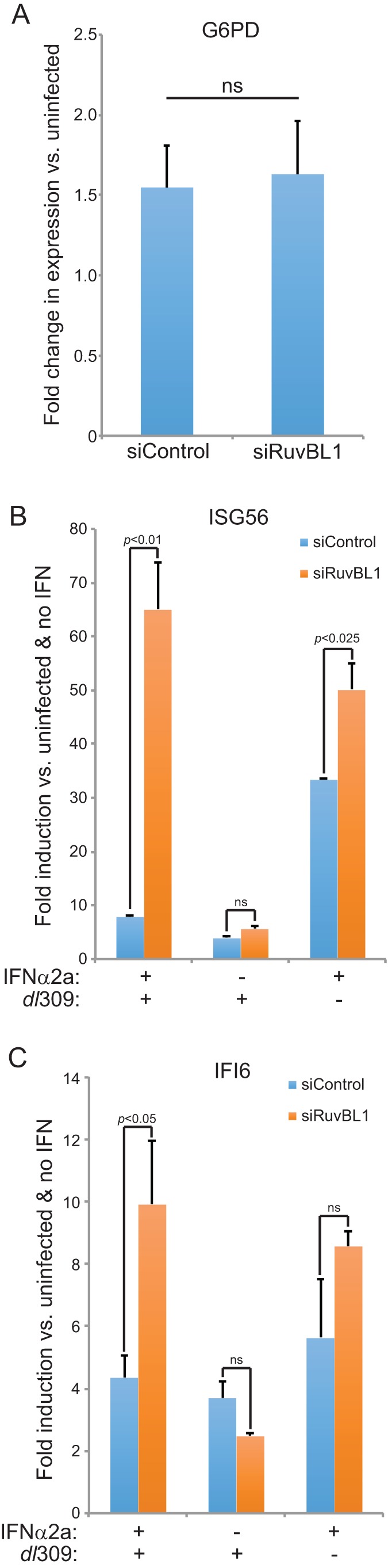
Depletion of RuvBL1 impairs the ability of E1A to suppress ISG activation. (A) HT1080 cells were treated with siRNA targeting RuvBL1 or a negative-control siRNA. The cells were then infected with dl309 at an MOI of 10, and 24 h later, total RNA was extracted with TRIzol reagent. Expression of G6PD was determined by qPCR using the Pfaffl method with SYBR green and a Bio-Rad CFX96 instrument and compared to uninfected cells. n = 3. The error bars represent SD. P values were determined using a t test. ns, not significant. (B) HT1080 cells were treated with siRNA targeting RuvBL1 or a negative-control siRNA. The cells were then infected with dl309 at an MOI of 10 and 24 h later treated with 1,000 U/ml of IFN-α2a (+) or vehicle (−) for 8 h prior to total RNA extraction with TRIzol reagent. Expression of ISG56 was determined by qPCR using the Pfaffl method with SYBR green and a Bio-Rad CFX96 instrument. n = 3. The error bars represent SD. P values were determined using a t test. ns, not significant. (C) HT1080 cells were treated with siRNA targeting RuvBL1 or a negative-control siRNA. The cells were then infected with dl309 at an MOI of 10 and 24 h later treated with 1,000 U/ml of IFN-α2a (+) or vehicle (−) for 8 h prior to total RNA extraction with TRIzol reagent. Expression of IFI6 was determined by qPCR using the Pfaffl method with SYBR green and a Bio-Rad CFX96 instrument. n = 3. The error bars represent SD. P values were determined using a t test. ns, not significant.
Unexpectedly, we observed that depletion of RuvBL1 had an enhancing effect on ISG expression after IFN treatment, contrary to a previously published result (24). Although this was not significant for IFI6, it was for ISG56, and based on the earlier study, we expected some reduction in ISG expression after RuvBL1 depletion. The enhanced expression of ISG56 and IFI6 in uninfected cells treated with IFN and depleted of RuvBL1 was observed in both HT1080 and IMR-90 cells (data not shown). Since the original report of RuvBL1 playing a role in ISG regulation used 293 cells (24) already expressing HAdV5 E1A, it is possible that this had unforeseen effects on ISG expression. Overall, our results suggest that during infection, HAdV uses RuvBL1 to suppress IFN-stimulated-gene expression in order to facilitate viral infection and that this effect is likely mediated by E1A.
Binding of RuvBL1 to E1A is required for ISG suppression.
Our results suggest that HAdV uses RuvBL1 to suppress IFN-stimulated-gene expression during infection. To further determine whether this effect is E1A dependent and whether it requires that RuvBL1 interact with E1A, we used E1A mutants dl1132 and dl1133, which are unable to bind to RuvBL1, to determine if they are able to suppress ISG56 and IFI6 expression after infection and IFN treatment. Infection followed by IFN treatment of HT1080 cells with dl1132 or dl1133 resulted in higher levels of expression of ISG56 and IFI6 than in cells infected with dl309 (Fig. 5B). Similarly, depletion of RuvBL1 did not affect the overall inability of E1A mutants dl1132 and dl1133 to suppress ISG expression (Fig. 5C). These effects were not due to differences in E1A expression, as the levels of the different E1A mutants were comparable (Fig. 5A). Interestingly, we observed a slight but consistent increase in RuvBL1 levels following viral infection (Fig. 5A). These observations suggest that suppression of ISG expression following infection relies not only on E1A expression, but also on an interaction of E1A, via its C terminus, with RuvBL1.
FIG 5.

Suppression of ISG activation relies on the ability of the E1A C terminus to interact with RuvBL1. (A) HT1080 cells were mock infected or infected with the indicated viruses at an MOI of 10, and 24 h later, the cells were lysed and Western blotting was performed for RuvBL1, E1A, and actin. (B) HT1080 cells were infected with the indicated viruses at an MOI of 10 and 24 h later treated with 1,000 U/ml of IFN-α2a for 8 h prior to total RNA extraction with TRIzol reagent. Expression of ISG56 was determined by qPCR using the Pfaffl method with SYBR green and a Bio-Rad CFX96 instrument. n = 3. The error bars represent SD. P values were determined using a t test. (C) HT1080 cells were transfected with siRNA targeting RuvBL1 or a negative-control siRNA, infected with the indicated viruses at an MOI of 10, and 24 h later treated with 1,000 U/ml of IFN-α2a for 8 h prior to total RNA extraction with TRIzol reagent. Expression of IFI6 was determined by qPCR using the Pfaffl method with SYBR green and a Bio-Rad CFX96 instrument and compared to that in RuvBL1-depleted and dl309-infected HT1080 cells. n = 3. The error bars represent SD. P values were determined using a t test.
RuvBL1 and E1A are recruited to ISG promoters following IFN treatment.
We wanted to investigate whether E1A is recruited to ISG promoters following infection as a potential mechanism of suppression. To investigate this, we performed ChIP of IFN-treated or untreated and infected HT1080 cells (Fig. 6). Unexpectedly, neither E1A nor RuvBL1 was present at a detectable level at the ISG56 or IFI6 promoter during infection prior to IFN treatment. Following IFN treatment, we observed considerable enrichment of E1A and RuvBL1 at the ISG56 and IFI6 promoters. Recruitment of RuvBL1 to these promoters was consistent with previous reports (24). Recruitment was specific to the promoter region, as we did not observe recruitment of either E1A or RuvBL1 to the 3′ end of the ISG56 gene under either untreated or IFN-treated conditions (Fig. 6). These observations suggest that E1A uses RuvBL1 for recruitment to ISG promoters in order to suppress IFN-mediated activation of ISG56 and IFI6.
FIG 6.
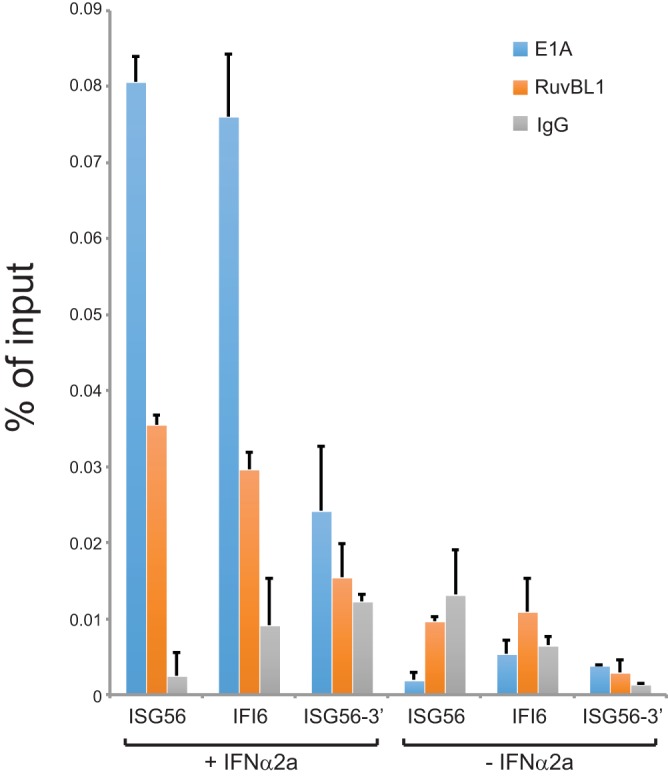
E1A is recruited to RuvBL1-regulated ISG promoters following IFN treatment. HT1080 cells were infected with dl309 at an MOI of 10 and 24 h later treated with 1,000 U/ml of IFN-α2a (+) or vehicle (−) for 8 h prior to fixation and ChIP. ChIP was performed for E1A using an M73 and M58 cocktail and for RuvBL1 using the rabbit anti-RuvBL1 antibody. Rabbit anti-rat antibody was used as a negative-control IgG. Promoter occupancy was determined by quantitative PCR as a percentage of input. n = 3. The error bars represent SD.
E1A binding to RuvBL1 is required for E1A recruitment to ISG promoters.
Our results show that following IFN treatment E1A is recruited to ISG56 and IFI6 promoters (Fig. 6). We also observed that E1A mutants unable to interact with RuvBL1 via the C terminus of E1A (dl1132 and dl1133) were not as efficient as wt E1A in suppressing ISG activation (Fig. 5). We therefore investigated whether these mutants are deficient for recruitment to ISG promoters after infection and IFN treatment. Wild-type E1A expressed in dl309-infected cells was efficiently recruited to ISG56 and IFI6 promoters; however, E1A mutant dl1132 was considerably reduced, while mutant dl1133 was below the background level of the IgG negative control (Fig. 7). Together, these results show that E1A requires interaction with RuvBL1 for efficient recruitment to ISG promoters during infection.
FIG 7.
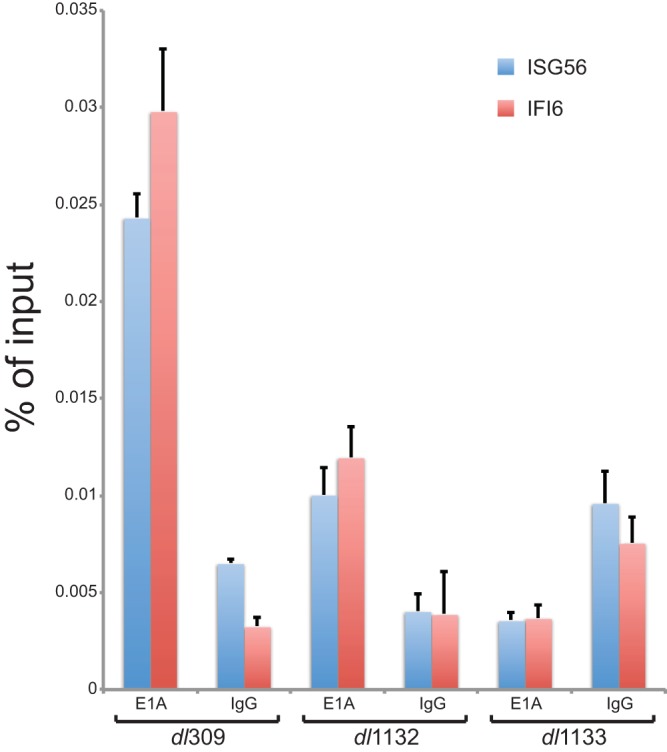
E1A recruitment to ISG promoters relies on E1A binding to RuvBL1. HT1080 cells were infected with the indicated viruses at an MOI of 10 and 24 h later treated with 1,000 U/ml of IFN-α2a for 8 h prior to fixation and ChIP. ChIP was performed for E1A using an M73 and M58 cocktail (E1A). Rabbit anti-rat antibody was used as a negative-control IgG (IgG). Promoter occupancy was determined by quantitative PCR as a percentage of input. n = 3. The error bars represent SD.
DISCUSSION
The present study reports the identification of a novel E1A C terminus binding protein, RuvBL1. We have mapped the interaction to residues 224 to 254, and possibly 271 to 284, in E1A289R of HAdV5 and have shown that this is a direct interaction via a GST pulldown assay (Fig. 1). Depletion of RuvBL1 was found to have a modest effect on virus growth, but unexpectedly, it had little direct effect on viral protein expression, viral genome replication, or the ability of the virus to drive arrested cells into S phase. Although we observed a significant reduction in several viral transcripts, particularly later in infection, it had minimal effect on protein levels (Fig. 3). This is likely caused by ribosomal saturation with already abundant viral mRNAs, and even a 10-fold reduction in these mRNAs maintains this saturation level, rendering ribosomes unable to translate all of the mRNAs available.
Our results show that RuvBL1 is required for efficient suppression of ISG expression after infection. Although we observed only a small degree of induction of ISGs after infection with dl309, we were able to assay the effects that RuvBL1 had on ISG expression after cells were treated with IFN-α2a following infection. Under these conditions, ISG56 and IFI6, previously shown to be regulated by RuvBL1 (24), were induced only when RuvBL1 was depleted by siRNA. When RuvBL1 levels were normal, ISG56 and IFI6 expression was considerably reduced in dl309-infected cells. There was also a slight decrease in IFI6 expression following infection in RuvBL1-depleted cells not treated with IFN (Fig. 4C). This was not statistically significant but nevertheless suggests an added layer of complexity in the regulation of ISG expression and deregulation by HAdV. Suppression of ISG56 and IFI6 relied, in part at least, on the ability of E1A to bind to RuvBL1 via its C terminus, since mutants unable to interact in this way were deficient in suppressing ISG levels (Fig. 5). Interestingly, we also observed that recruitment of E1A to RuvBL1-regulated promoters occurred only after IFN treatment (Fig. 6). Importantly, we observed that E1A mutants deficient for the interaction with RuvBL1 had a reduced ability to be recruited to ISG promoters (Fig. 7). Interestingly, the mutant dl1132 showed a reduced level of recruitment to ISG promoters compared to wt E1A (Fig. 7). This corresponds to its higher degree of suppression of ISG activation compared to dl1133 (Fig. 5A), which was not detected at ISG56 and IFI6 promoters (Fig. 7). It is possible that E1A dl1132 retains some residual binding to RuvBL1 that is not detected using standard coimmunoprecipitation. Unexpectedly, we observed that under RuvBL1 depletion conditions, IFN stimulation, and no infection, ISG expression was slightly elevated (Fig. 4). This is in contrast to a previously published report originally implicating RuvBL1 in ISG regulation (24). Since these authors used 293 cells expressing wt E1A, it is difficult to interpret their results in light of our current findings. Nevertheless, we observed elevation in ISG expression after RuvBL1 depletion consistently in HT1080 and IMR-90 cells, neither of which express any viral oncogenes.
During infection, E1A targets hub proteins, that is, proteins involved in multiple cellular processes, in order to remodel the intracellular environment to better support viral replication (2). In the present study, we explored how E1A affects ISG expression via RuvBL1; however, RuvBL1 is a multifunctional protein that plays roles in many cellular processes. It is therefore likely that by targeting RuvBL1, E1A is able to affect not only IFN signaling, but also other pathways regulated by RuvBL1. RuvBL1 belongs to the AAA+ (ATPase associated with diverse cellular activities) family of DNA helicases (26). Among many different activities and complexes, RuvBL1 has been found to participate in regulation of gene expression (as both an activator and a repressor) as part of several chromatin-remodeling complexes (including Ino80, SRCAP, and TIP60/NuA4), in mitosis, in DNA damage response, and in cell cycle and DNA replication checkpoint control (27). Interestingly, an ATPase-dead mutant of RuvBL1 almost completely abolished the ability of E1A to transform cells in cooperation with activated Ras (28). RuvBL1 was also shown to play a role in nonsense-mediated decay (29), a cellular surveillance mechanism that prevents translation of defective mRNAs. The many diverse roles of RuvBL1 in regulating a wide range of cellular processes point to its importance in the cell and suggest that RuvBL1 functions as a key molecular hub that links multiple signaling molecules. The hub-like nature of RuvBL1 makes it a prime target for E1A, and it would be of interest to explore whether E1A affects any of the other activities of RuvBL1.
A recent report showed that RuvBL1 binds to E1A via the N terminus as part of the NuA4/TIP60 chromatin-remodeling complex (23), and although the consequences of this interaction were not clear, they appear to be transcriptional. We therefore investigated whether there is a loss of binding by E1A to RuvBL1 with the use of N terminus deletion mutants (Fig. 1). Although we observed reduced interaction between RuvBL1 and some of the deletion mutants, we did not observe a total loss. Deletions within the C terminus of E1A, however, had a much more substantial effect on the ability of E1A to interact with RuvBL1 (Fig. 1). E1A mutants dl1132 and dl1133 showed binding below background level (Fig. 1), while mutant dl1135 showed binding near background level, suggesting that with the C terminus binding deleted, the contribution of the N-terminal binding is minimal. We also did not observe a loss of binding with E1A mutant dl1102, which has residues within E1A previously shown to be required for binding of RuvBL1 by the N terminus deleted (23). This is likely because only residues 1 to 80 were used in the mapping of RuvBL1 to the N terminus. Ultimately, binding of E1A via multiple regions is not novel and appears to be a common feature of how E1A targets cellular proteins. We have previously observed this with CtBP (30), p300/CBP (31), and pCAF (32), and one of the first proteins ever identified as binding to E1A, pRb, binds via the N terminus (CR1) and CR2, which is essential for E1A-mediated disruption of cell cycle regulation by pRb (33).
Interferon suppression is of paramount importance to a virus. Therefore, viruses have evolved multiple mechanisms that ensure that IFN signaling does not disrupt their growth. Treatment of infected cells with IFN has little effect on wt HAdV growth (9). The virus uses multiple mechanisms to suppress interferon signaling, including viral noncoding RNAs that target the host immune response and the ability of cells to respond to interferon (reviewed in reference 10). The viral E4 orf3 protein targets IFN and PML bodies (16, 34, 35), and E1A itself has been implicated in interferon response in several different ways (12–14). Since RuvBL1 was previously reported to be important in IFN signaling, we investigated the effects that E1A binding has on the ability of IFN to drive expression of two ISGs, ISG56 and IFI6, previously shown to be coregulated by RuvBL1 (24). During infection of HT1080 cells with dl309, we observed a very modest induction of ISGs, even in cells treated with IFN (Fig. 4 and data not shown), which is consistent with the ability of the virus to suppress the IFN pathway. However, upon depletion of RuvBL1 and IFN treatment, ISG56 and IFI6 were potently induced in dl309-infected cells (Fig. 4). Suppression of ISG expression was, in part, reliant on the ability of E1A to bind to RuvBL1 via the C terminus, since mutants dl1132 and dl1133, deficient for the interaction, were also less able to suppress activation of ISG56 and IFI6 after IFN treatment (Fig. 5). Our data suggest that E1A is selectively recruited to ISG promoters, in order to suppress them, only after they are activated by IFN treatment (Fig. 6), likely driven by recruitment of RuvBL1 to these promoters. This mode of action is similar to what was previously observed for E1A and CtBP (30), but with repression being the outcome rather than activation, as was the case with CtBP. Interestingly, the insect homologue of RuvBL1 was also shown to be a restriction factor for replication in diverse vector-borne viruses (including West Nile virus, Sindbis virus, dengue virus, Rift Valley fever virus, and vesicular stomatitis virus), suggesting that modulation of RuvBL1 activities may be a common strategy of viral pathogens in order to promote their replication (36).
RuvBL1 has a well-established role in cancer and cellular transformation (reviewed in reference 37). Importantly, RuvBL1 was previously shown to be required for E1A-mediated cellular transformation, in cooperation with activated H-Ras(G12V) (28), although that study did not report a physical interaction between E1A and RuvBL1. It is still likely that an interaction between E1A and RuvBL1 plays some role in the ability of E1A to deregulate the cell cycle and drive oncogenic transformation. However, we did not observe any significant effects on the ability of E1A to induce the S phase in infected cells following RuvBL1 depletion. This observation suggests that any transformation potential lost due to the reduction of RuvBL1 protein levels is not likely at the initial steps of inducing the cell cycle. Nevertheless, it would be of interest to investigate how RuvBL1 contributes to E1A-mediated transformation outside its role in IFN signaling.
In the present study, we have identified a novel interaction between the C terminus of E1A and the cellular protein RuvBL1. E1A was found to bind to RuvBL1 directly, and this interaction was important for recruitment of E1A to ISG promoters and suppression of ISG expression following infection and IFN stimulation. Viruses expressing E1A that was deficient for the interaction with RuvBL1 via the C terminus were also deficient for suppression of ISGs. Our study identifies yet another way in which HAdV, via the use of E1A, is able to suppress ISG activation in response to infection or IFN treatment. Several questions arise from our study. What is the mechanism of inhibition? Does the interaction of E1A with RuvBL1 via the N terminus of E1A contribute to ISG suppression? Does the C terminus of E1A interact with the related RuvBL2, and does this interaction affect ISG expression? Answering these questions will provide further insight into how HAdV, and E1A, modulate IFN signaling. In conclusion, we have identified RuvBL1 as a novel target of the E1A C terminus that is important for suppression of ISG expression.
MATERIALS AND METHODS
Antibodies.
Mouse monoclonal anti-E1A M73 and M58 antibodies were previously described (38) and were grown in house and used as the hybridoma supernatant. Mouse monoclonal anti-72K E2 DBP antibody was previously described (39) and was used at a dilution of 1:400 for Western blotting. Anti-RuvBL1 antibody was purchased from Cell Signaling Technologies (catalog number 74775) and was used at a dilution of 1:1,000 for Western blots, while 10 μl was used for ChIP. Anti-adenovirus type 5 antibody was purchased from Abcam (catalog number ab6982). Rat anti-HA antibody (Roche), clone 3F10, was used at a dilution of 1:5,000 for Western blotting. Secondary antibodies were acquired from Jackson ImmunoResearch and were used at a dilution of 1:200,000.
Cell and virus culture.
IMR-90 (ATCC CCL-186) and HT1080 (ATCC CCL-121) cells were grown in Dulbecco's modified Eagle's medium (DMEM) (HyClone) supplemented with 10% fetal bovine serum (Seradigm), streptomycin, and penicillin (HyClone). All virus infections were carried out in serum-free medium for 1 h, after which saved complete medium was added without removal of the infection medium. For interferon treatment of infected cells, 1,000 U/ml of IFN-α2a was added 24 h after infection for 8 h prior to analysis by quantitative PCR (qPCR) or ChIP.
Chromatin immunoprecipitation.
ChIP was carried out essentially as previously described (31). HT1080 cells were infected with the indicated adenoviruses at a multiplicity of infection (MOI) of 10 and harvested 24 h after infection for ChIP analysis. For immunoprecipitation of E1A, the monoclonal M73 and M58 antibodies were used. For immunoprecipitation of RuvBL1, the polyclonal anti-RuvBL1 antibody was used. Rabbit anti-mouse antibody was used as a negative-control IgG.
PCRs were carried out for HAdV5 early and major late promoters with SYBR Select master mix for CFX (Applied Biosystems) according to the manufacturer's directions, using 3% of the total ChIP DNA as the template on a CFX96 real-time PCR instrument (Bio-Rad). The annealing temperature was 60°C, and 40 cycles were run. Primers for viral promoters were previously described and are listed below (15).
EdU incorporation assay.
IMR-90 cells were grown until 100% confluent on Lab Tek II 4-chamber slides (Thermo-Fisher). After becoming fully confluent, the cells were incubated for a further 72 h to achieve growth arrest and treated with siRNA to deplete RuvBL1. Infections were carried out as described above at an MOI of 20 for dl309. One hour prior to fixation, the cells were pulsed with EdU for 1 h according to the manufacturer's specifications using the Click-It EdU-labeling kit for microscopy (Life Technologies). After EdU labeling, the cells were fixed in 3.7% formaldehyde, stained for EdU using the Click-It kit with Alexa Fluor 488, and labeled for E1A using M73 monoclonal antibody and Alexa Fluor 594-conjugated secondary antibody (Jackson ImmunoResearch). The cells were visualized using an LSM700 laser confocal microscope and the ZEN software suite.
Immunoprecipitation.
Transfected HT1080 cells were lysed in NP-40 lysis buffer (0.5% NP-40, 50 mM Tris [pH 7.8], 150 mM NaCl) supplemented with a protease inhibitor cocktail (Sigma). Cell lysate containing 1 mg of total protein was used for IP with the monoclonal M73 or M58 anti-E1A antibody. E1A was detected using the M73 or M58 monoclonal antibody, while RuvBL1 was detected using anti-HA rat antibody (clone 3F10). For immunoprecipitation of endogenous RuvBL1 with E1A, E1A beads were first prepared by direct cross-linking of M73 and M58 to protein A-Sepharose beads using dimethyl pimelimidate and then using the cross-linked beads in the immunoprecipitation to eliminate the antibody heavy chain and masking of RuvBL1.
PCR primers.
The primers used were as follows: IFI6, CTCGCTGATGAGCTGGTCT and TGCTGGCTACTCCTCATCCT; SG56, AAAAGCCCACATTTGAGGTG and GAAATTCCTGAAACCGACCA; ISG56 promoter, TTTCACTTTCCCCTTTCGGTTTCC and GGCTCCTCTGAGATCTGGCTATTC; IFI6 promoter, CTGGGCGGAGCTGGGAGAG and TGGGCACAGCAGCGAGTAAAC; ISG56 3′ end for ChIP, TCTGAACATTGAAAGGAACAAACTC and ACTCACTGCTTGGCGATAGG.
Any primers not listed were previously described (15, 18, 40, 41).
Plasmids.
The expression plasmid for pcDNA3.1-E1A was previously described (42), and it expresses all E1A isoforms. pcDNA-HA-RuvBL1 was generated by PCR amplification of RuvBL1 and cloning into the NheI/XbaI sites of pcDNA-HA.
Protein purification and GST pulldown assay.
Glutathione S-transferase fusions of RuvBL1 were made by subcloning the cDNA into pGEX-6P1 (GE Healthcare Life Sciences) in frame with the N-terminal GST tag. His-tagged E1A289R was made by subcloning the entire E1A289R cDNA into the pET42 vector (Novagen) in frame with a C-terminal 6×His tag. Proteins were expressed in Escherichia coli strain BL21(DE3) and purified on their respective resins according to the manufacturer's specifications. The GST pulldown assay was carried out as previously described (31).
Real-time gene expression analysis.
HT1080 or IMR-90 cells were infected with dl309 (43) at an MOI of 10. Total RNA was extracted using TRIzol reagent (Sigma) at the indicated time points according to the manufacturer's instructions. Total RNA (1.25 μg) was used in reverse transcriptase reactions using SuperScript Vilo reverse transcriptase (Invitrogen) according to the manufacturer's guidelines with random hexanucleotides for priming. The cDNA was subsequently used for real-time expression analysis using the Bio-Rad CFX96 real-time thermocycler. Analysis of expression data was carried out using the Pfaffl method (44) and was normalized to GAPDH mRNA levels and compared between siControl- and siRuvBL1-transfected cells. Total E1A was detected as previously described (41). Statistical analysis and the determination of the significance of real-time expression results were performed using Student's t test.
siRNA knockdown.
siRNA knockdown was carried out as previously described (31). Briefly, IMR-90 cells were transfected with RuvBL1-specific Silencer siRNA (Life Technologies number s16371) with SilentFect reagent (Bio-Rad) according to the manufacturer's specifications using a 10 nM final siRNA concentration. Silencer Select negative-control siRNA number 1 (Life Technologies) was used as the negative siRNA control.
Transfections.
Cells were plated in 10-cm plates at a density of 2.0 × 106 cells/plate 24 h prior to transfection. Transfection mixtures were prepared by mixing 1 ml of serum-free DMEM, 10 μg total plasmid DNA, and 20 μl of a linear 1-mg/ml solution of polyethylenimine 25-kDa reagent from Polysciences (number 23966-2). This was vortexed for 10 s and incubated at room temperature for 20 min. The complexes were then added to the cells and incubated for 24 to 48 h.
Viral genome quantification.
HT1080 cells depleted of RuvBL1 or treated with the control siRNA were lysed in lysis buffer (50 mM Tris, pH 8.1, 10 mM EDTA, and 1% SDS) on ice for 10 min. The lysates were sonicated briefly in a Covaris M220 focused ultrasonicator to break up the cellular chromatin and subjected to digestion using proteinase K (NEB) according to the manufacturer's specifications. Following digestion, viral DNA was purified using a GeneJet PCR purification kit (Thermo-Fisher). PCRs were carried out with SYBR Select master mix for CFX (Applied Biosystems) according to the manufacturer's directions using 2% total purified DNA as the template and a CFX96 real-time PCR instrument (Bio-Rad). A standard curve for absolute quantification was generated by serially diluting a pXC1 plasmid containing the left end of the HAdV5 genome starting with a concentration of 1.0 × 107 copies per reaction down to 1.0 copy per reaction. The primers used were the same as those used for expression analysis of the E1B region, the annealing temperature used was 60°C, and 40 cycles were run.
Viruses.
The viruses used in the study were HAdV5 mutant dl309 (43) expressing wt E1A but with much of the E3 region deleted, and HAdV5 E1A mutants dl1101, dl1102, dl1103, dl1104, dl1105, dl1106, dl1107, dl1108, dl1116, dl1132, dl1133, dl1134, dl1135, and dl1136, which were previously described (20, 21, 45) (the deletions are shown in Fig. 1A) and were generously donated by Joe Mymryk. All the viruses were amplified in low-passage-number 293 cells, and their titers were also determined on these 293 cells prior to performing the assays. All infections were carried out in serum-free medium for 1 h at an MOI of 10 unless otherwise specified in the figure legends.
Virus growth assay.
HT1080 cells were infected with HAdV5 dl309 at an MOI of 10 in serum-free medium. Virus was adsorbed for 1 h at 37°C under 5% CO2, after which the cells were bathed in conditioned medium and reincubated at 37°C under 5% CO2. Virus titers were determined 24, 48, and 72 h after infection, and plaque assays were performed on 293 cells by serial dilution.
ACKNOWLEDGMENTS
This work was supported by grants from the Natural Sciences and Engineering Research Council (grant number RGPIN/435375-2013) and Research Manitoba (MHRC Establishment Grant and Operating Grant). S.R. was supported by a University of Manitoba Graduate Fellowship award.
We are indebted to Joe Mymryk for countless reagents and invaluable discussions. We thank David E. Levy for providing us with primer sequences for ChIP of the ISG56 and IFI6 promoters. We also thank Andrea Soriano and Leandro Crisostomo for their assistance. P.P. also thanks Stanislawa Pelka for invaluable support and assistance.
REFERENCES
- 1.Berk AJ. 2013. Chapter 55. Adenoviridae. In Knipe DM, Howley PM (ed). Fields virology, 6th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Pelka P, Ablack JN, Fonseca GJ, Yousef AF, Mymryk JS. 2008. Intrinsic structural disorder in adenovirus E1A: a viral molecular hub linking multiple diverse processes. J Virol 82:7252–7263. doi: 10.1128/JVI.00104-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fessler SP, Delgado-Lopez F, Horwitz MS. 2004. Mechanisms of E3 modulation of immune and inflammatory responses. Curr Top Microbiol Immunol 273:113–135. [DOI] [PubMed] [Google Scholar]
- 4.Andersson M, Paabo S, Nilsson T, Peterson PA. 1985. Impaired intracellular transport of class I MHC antigens as a possible means for adenoviruses to evade immune surveillance. Cell 43:215–222. doi: 10.1016/0092-8674(85)90026-1. [DOI] [PubMed] [Google Scholar]
- 5.Gooding LR, Elmore LW, Tollefson AE, Brady HA, Wold WS. 1988. A 14,700 MW protein from the E3 region of adenovirus inhibits cytolysis by tumor necrosis factor. Cell 53:341–346. doi: 10.1016/0092-8674(88)90154-7. [DOI] [PubMed] [Google Scholar]
- 6.Gooding LR, Ranheim TS, Tollefson AE, Aquino L, Duerksen-Hughes P, Horton TM, Wold WS. 1991. The 10,400- and 14,500-dalton proteins encoded by region E3 of adenovirus function together to protect many but not all mouse cell lines against lysis by tumor necrosis factor. J Virol 65:4114–4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gooding LR, Sofola IO, Tollefson AE, Duerksen-Hughes P, Wold WS. 1990. The adenovirus E3-14.7K protein is a general inhibitor of tumor necrosis factor-mediated cytolysis. J Immunol 145:3080–3086. [PubMed] [Google Scholar]
- 8.Lichtenstein DL, Toth K, Doronin K, Tollefson AE, Wold WS. 2004. Functions and mechanisms of action of the adenovirus E3 proteins. Int Rev Immunol 23:75–111. doi: 10.1080/08830180490265556. [DOI] [PubMed] [Google Scholar]
- 9.Kitajewski J, Schneider RJ, Safer B, Munemitsu SM, Samuel CE, Thimmappaya B, Shenk T. 1986. Adenovirus VAI RNA antagonizes the antiviral action of interferon by preventing activation of the interferon-induced eIF-2 alpha kinase. Cell 45:195–200. doi: 10.1016/0092-8674(86)90383-1. [DOI] [PubMed] [Google Scholar]
- 10.Vachon VK, Conn GL. 2016. Adenovirus VA RNA: an essential pro-viral non-coding RNA. Virus Res 212:39–52. doi: 10.1016/j.virusres.2015.06.018. [DOI] [PubMed] [Google Scholar]
- 11.Hendrickx R, Stichling N, Koelen J, Kuryk L, Lipiec A, Greber UF. 2014. Innate immunity to adenovirus. Hum Gene Ther 25:265–284. doi: 10.1089/hum.2014.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ackrill AM, Foster GR, Laxton CD, Flavell DM, Stark GR, Kerr IM. 1991. Inhibition of the cellular response to interferons by products of the adenovirus type 5 E1A oncogene. Nucleic Acids Res 19:4387–4393. doi: 10.1093/nar/19.16.4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kalvakolanu DV, Bandyopadhyay SK, Harter ML, Sen GC. 1991. Inhibition of interferon-inducible gene expression by adenovirus E1A proteins: block in transcriptional complex formation. Proc Natl Acad Sci U S A 88:7459–7463. doi: 10.1073/pnas.88.17.7459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fonseca GJ, Thillainadesan G, Yousef AF, Ablack JN, Mossman KL, Torchia J, Mymryk JS. 2012. Adenovirus evasion of interferon-mediated innate immunity by direct antagonism of a cellular histone posttranslational modification. Cell Host Microbe 11:597–606. doi: 10.1016/j.chom.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 15.Radko S, Koleva M, James KM, Jung R, Mymryk JS, Pelka P. 2014. Adenovirus E1A targets the DREF nuclear factor to regulate virus gene expression, DNA replication, and growth. J Virol 88:13469–13481. doi: 10.1128/JVI.02538-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ullman AJ, Reich NC, Hearing P. 2007. Adenovirus E4 ORF3 protein inhibits the interferon-mediated antiviral response. J Virol 81:4744–4752. doi: 10.1128/JVI.02385-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cohen MJ, Yousef AF, Massimi P, Fonseca GJ, Todorovic B, Pelka P, Turnell AS, Banks L, Mymryk JS. 2013. Dissection of the C-terminal region of E1A re-defines the roles of CtBP and other cellular targets in oncogenic transformation. J Virol 87:10348–10355. doi: 10.1128/JVI.00786-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frost JR, Olanubi O, Cheng SK, Soriano A, Crisostomo L, Lopez A, Pelka P. 2017. The interaction of adenovirus E1A with the mammalian protein Ku70/XRCC6. Virology 500:11–21. doi: 10.1016/j.virol.2016.10.004. [DOI] [PubMed] [Google Scholar]
- 19.Kim JH, Lee JM, Nam HJ, Choi HJ, Yang JW, Lee JS, Kim MH, Kim SI, Chung CH, Kim KI, Baek SH. 2007. SUMOylation of Pontin chromatin-remodeling complex reveals a signal integration code in prostate cancer cells. Proc Natl Acad Sci U S A 104:20793–20798. doi: 10.1073/pnas.0710343105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jelsma TN, Howe JA, Evelegh CM, Cunniff NF, Skiadopoulos MH, Floroff MR, Denman JE, Bayley ST. 1988. Use of deletion and point mutants spanning the coding region of the adenovirus 5 E1A gene to define a domain that is essential for transcriptional activation. Virology 163:494–502. doi: 10.1016/0042-6822(88)90290-5. [DOI] [PubMed] [Google Scholar]
- 21.Mymryk JS, Bayley ST. 1993. Induction of gene expression by exon 2 of the major E1A proteins of adenovirus type 5. J Virol 67:6922–6928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boyd JM, Subramanian T, Schaeper U, La Regina M, Bayley S, Chinnadurai G. 1993. A region in the C-terminus of adenovirus 2/5 E1a protein is required for association with a cellular phosphoprotein and important for the negative modulation of T24-ras mediated transformation, tumorigenesis and metastasis. EMBO J 12:469–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao LJ, Loewenstein PM, Green M. 2016. Ad E1A 243R oncoprotein promotes association of proto-oncogene product MYC with the NuA4/Tip60 complex via the E1A N-terminal repression domain. Virology 499:178–184. doi: 10.1016/j.virol.2016.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gnatovskiy L, Mita P, Levy DE. 2013. The human RVB complex is required for efficient transcription of type I interferon-stimulated genes. Mol Cell Biol 33:3817–3825. doi: 10.1128/MCB.01562-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jones N, Shenk T. 1979. An adenovirus type 5 early gene function regulates expression of other early viral genes. Proc Natl Acad Sci U S A 76:3665–3669. doi: 10.1073/pnas.76.8.3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grigoletto A, Lestienne P, Rosenbaum J. 2011. The multifaceted proteins Reptin and Pontin as major players in cancer. Biochim Biophys Acta 1815:147–157. doi: 10.1016/j.bbcan.2010.11.002. [DOI] [PubMed] [Google Scholar]
- 27.Jha S, Dutta A. 2009. RVB1/RVB2: running rings around molecular biology. Mol Cell 34:521–533. doi: 10.1016/j.molcel.2009.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dugan KA, Wood MA, Cole MD. 2002. TIP49, but not TRRAP, modulates c-Myc and E2F1 dependent apoptosis. Oncogene 21:5835–5843. doi: 10.1038/sj.onc.1205763. [DOI] [PubMed] [Google Scholar]
- 29.Izumi N, Yamashita A, Iwamatsu A, Kurata R, Nakamura H, Saari B, Hirano H, Anderson P, Ohno S. 2010. AAA+ proteins RUVBL1 and RUVBL2 coordinate PIKK activity and function in nonsense-mediated mRNA decay. Sci Signal 3:ra27. doi: 10.1126/scisignal.2000468. [DOI] [PubMed] [Google Scholar]
- 30.Bruton RK, Pelka P, Mapp KL, Fonseca GJ, Torchia J, Turnell AS, Mymryk JS, Grand RJ. 2008. Identification of a second CtBP binding site in adenovirus type 5 E1A conserved region 3. J Virol 82:8476–8486. doi: 10.1128/JVI.00248-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pelka P, Ablack JN, Torchia J, Turnell AS, Grand RJ, Mymryk JS. 2009. Transcriptional control by adenovirus E1A conserved region 3 via p300/CBP. Nucleic Acids Res 37:1095–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pelka P, Ablack JN, Shuen M, Yousef AF, Rasti M, Grand RJ, Turnell AS, Mymryk JS. 2009. Identification of a second independent binding site for the pCAF acetyltransferase in adenovirus E1A. Virology 391:90–98. doi: 10.1016/j.virol.2009.05.024. [DOI] [PubMed] [Google Scholar]
- 33.Dyson N, Guida P, McCall C, Harlow E. 1992. Adenovirus E1A makes two distinct contacts with the retinoblastoma protein. J Virol 66:4606–4611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Soria C, Estermann FE, Espantman KC, O'Shea CC. 2010. Heterochromatin silencing of p53 target genes by a small viral protein. Nature 466:1076–1081. doi: 10.1038/nature09307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vink EI, Zheng Y, Yeasmin R, Stamminger T, Krug LT, Hearing P. 2015. Impact of adenovirus E4-ORF3 oligomerization and protein localization on cellular gene expression. Viruses 7:2428–2449. doi: 10.3390/v7052428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yasunaga A, Hanna SL, Li J, Cho H, Rose PP, Spiridigliozzi A, Gold B, Diamond MS, Cherry S. 2014. Genome-wide RNAi screen identifies broadly-acting host factors that inhibit arbovirus infection. PLoS Pathog 10:e1003914. doi: 10.1371/journal.ppat.1003914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rosenbaum J, Baek SH, Dutta A, Houry WA, Huber O, Hupp TR, Matias PM. 2013. The emergence of the conserved AAA+ ATPases Pontin and Reptin on the signaling landscape. Sci Signal 6:mr1. doi: 10.1126/scisignal.2003906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harlow E, Franza BR Jr, Schley C. 1985. Monoclonal antibodies specific for adenovirus early region 1A proteins: extensive heterogeneity in early region 1A products. J Virol 55:533–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reich NC, Sarnow P, Duprey E, Levine AJ. 1983. Monoclonal antibodies which recognize native and denatured forms of the adenovirus DNA-binding protein. Virology 128:480–484. doi: 10.1016/0042-6822(83)90274-X. [DOI] [PubMed] [Google Scholar]
- 40.Jung R, Radko S, Pelka P. 2015. The dual nature of Nek9 in adenovirus replication. J Virol 90:1931–1943. doi: 10.1128/JVI.02392-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Radko S, Jung R, Olanubi O, Pelka P. 2015. Effects of adenovirus type 5 E1A isoforms on viral replication in arrested human cells. PLoS One 10:e0140124. doi: 10.1371/journal.pone.0140124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pelka P, Scime A, Mandalfino C, Joch M, Abdulla P, Whyte P. 2007. Adenovirus E1A proteins direct subcellular redistribution of Nek9, a NimA-related kinase. J Cell Physiol 212:13–25. doi: 10.1002/jcp.20983. [DOI] [PubMed] [Google Scholar]
- 43.Jones N, Shenk T. 1979. Isolation of adenovirus type 5 host range deletion mutants defective for transformation of rat embryo cells. Cell 17:683–689. doi: 10.1016/0092-8674(79)90275-7. [DOI] [PubMed] [Google Scholar]
- 44.Pfaffl MW. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Egan C, Jelsma TN, Howe JA, Bayley ST, Ferguson B, Branton PE. 1988. Mapping of cellular protein-binding sites on the products of early-region 1A of human adenovirus type 5. Mol Cell Biol 8:3955–3959. doi: 10.1128/MCB.8.9.3955. [DOI] [PMC free article] [PubMed] [Google Scholar]