

ABSTRACT
The coronavirus (CoV) S protein requires cleavage by host cell proteases to mediate virus-cell and cell-cell fusion. Many strains of the murine coronavirus mouse hepatitis virus (MHV) have distinct, S-dependent organ and tissue tropisms despite using a common receptor, suggesting that they employ different cellular proteases for fusion. In support of this hypothesis, we found that inhibition of endosomal acidification only modestly decreased entry, and overexpression of the cell surface protease TMPRSS2 greatly enhanced entry, of the highly neurovirulent MHV strain JHM.SD relative to their effects on the reference strain, A59. However, TMPRSS2 overexpression decreased MHV structural protein expression, release of infectious particles, and syncytium formation, and endogenous serine protease activity did not contribute greatly to infection. We therefore investigated the importance of other classes of cellular proteases and found that inhibition of matrix metalloproteinase (MMP)- and a disintegrin and metalloprotease (ADAM)-family zinc metalloproteases markedly decreased both entry and cell-cell fusion. Suppression of virus by metalloprotease inhibition varied among tested cell lines and MHV S proteins, suggesting a role for metalloprotease use in strain-dependent tropism. We conclude that zinc metalloproteases must be considered potential contributors to coronavirus fusion.
IMPORTANCE The family Coronaviridae includes viruses that cause two emerging diseases of humans, severe acute respiratory syndrome (SARS) and Middle East respiratory syndrome (MERS), as well as a number of important animal pathogens. Because coronaviruses depend on host protease-mediated cleavage of their S proteins for entry, a number of protease inhibitors have been proposed as antiviral agents. However, it is unclear which proteases mediate in vivo infection. For example, SARS-CoV infection of cultured cells depends on endosomal acid pH-dependent proteases rather than on the cell surface acid pH-independent serine protease TMPRSS2, but Zhou et al. (Antiviral Res 116:76–84, 2015, doi:10.1016/j.antiviral.2015.01.011) found that a serine protease inhibitor was more protective than a cathepsin inhibitor in SARS-CoV-infected mice. This paper explores the contributions of endosomal acidification and various proteases to coronavirus infection and identifies an unexpected class of proteases, the matrix metalloproteinase and ADAM families, as potential targets for anticoronavirus therapy.
KEYWORDS: TMPRSS2, coronavirus, membrane fusion, metalloprotease, virus entry
INTRODUCTION
Entry of enveloped viruses requires viral surface proteins to attach to the cell surface and to undergo conformational changes that drive fusion of the viral and cellular membranes. Both steps can also involve host cell factors: attachment requires a cellular receptor recognized by the viral attachment protein, and fusion may require cellular processes, such as endocytosis and endosomal acidification and/or cleavage of viral surface proteins by host cell proteases. While receptor availability is a major determinant of viral species and tissue tropism, other host fusion factors can also contribute. For example, low-pathogenicity strains of avian influenza virus require cleavage of the fusion protein HA by trypsin-like proteases, confining the virus to the digestive and respiratory tracts where such enzymes are available, whereas high-pathogenicity strains have HA sequences that can be cleaved by ubiquitously expressed proteases and thus cause systemic infection (reviewed in reference 1). Host cell fusion factors are therefore potential targets for antiviral therapy.
The emergence of severe acute respiratory syndrome (SARS) and Middle East respiratory syndrome (MERS) coronaviruses (CoV) as causes of human diseases has prompted interest in anticoronavirus strategies, including inhibition of host cell proteases involved in coronavirus fusion (2). Coronaviruses rely on a single spike (S) protein for attachment and fusion, and fusion requires proteolytic cleavage of S by host proteases during the viral replication cycle (reviewed in reference 3). S comprises an N-terminal S1 portion, containing the receptor-binding domain (RBD), and a C-terminal S2 portion, containing the fusion machinery. In some coronavirus species, S is cleaved at the S1/S2 boundary, typically by a furin-like protease in the producing cell during virus assembly and/or egress; S1 and S2 remain associated via noncovalent interactions (3). In murine coronavirus, furin cleavage at S1/S2 is not required for infection but appears to be necessary for cell-cell fusion, as mutation of the site (4–6) or pharmacologic inhibition of furin-like proteases (7) affects syncytium formation; insertion of a furin cleavage site at S1/S2 also enhances cell-cell fusion by SARS coronavirus (8). Fusion is also thought to require an additional proteolytic cleavage within S2 at the N terminus of the fusion peptide, resulting in a new C-terminal fragment sometimes called S2′ (reviewed in references 3 and 9). Members of at least four groups of proteases have been implicated in the S2′ cleavage: cathepsins B and L, which are acid-dependent endosomal cysteine proteases; transmembrane protease, serine (TMPRSS) family proteases, especially TMPRSS2, which are acid-independent serine proteases generally found at the cell surface; elastases, which are common serine family proteases in lung tissue; and furin-like proprotein convertases. Inhibitors of cathepsins B and L block entry by many coronaviruses, including SARS-CoV (10, 11) and MERS-CoV (12, 13), feline coronavirus (14), and the mouse hepatitis virus (MHV) strain MHV-2 (4). The role of TMPRSS2 is less clear. It appears to promote infection by SARS-CoV (15–17), MERS-CoV (12, 13), the human respiratory coronavirus NL63 (18), and clinical isolates of the human respiratory coronavirus 229E (19), especially when endosomal acidification or cathepsin activity is inhibited, and also increases infection and virus release by otherwise trypsin-dependent strains of porcine epidemic diarrhea virus (PEDV) (20). TMPRSS2 is thought to cleave and thus activate some influenza HA proteins (21, 22), and it may increase SARS-CoV entry by cleaving the S protein or by enhancing virus particle uptake via specific cleavage of the SARS-CoV receptor ACE2 (16, 23). A number of extracellular proteases, including elastase, enhance SARS-CoV infection (24), and a putative elastase site has been identified within S2 (25). The fourth protease, furin, has a cleavage substrate motif that is found at the N terminus of the putative fusion peptide in MERS-CoV (26, 27) and the embryo-adapted Beaudette strain of the avian coronavirus infectious bronchitis virus (IBV) (8). Furin does appear to cleave at this site, and furin activity enhances MERS-CoV infection (27). Finally, a fifth category of proteases plays a controversial role in SARS-CoV entry: the metalloprotease ADAM17/TACE has been reported to enhance SARS-CoV uptake by cleaving ACE2 (28, 29), although other authors have disagreed (23, 30). Inhibition of proteases implicated in viral fusion is now under investigation as an anticoronavirus strategy, with promising results: camostat, an inhibitor of serine proteases, including TMPRSS2, was recently shown to reduce mortality in a mouse model of SARS-CoV infection, whereas a cathepsin inhibitor that decreased SARS-CoV entry in vitro had minimal effect in the infected mice (2). The effect of TMPRSS2 seems particularly context specific: clinical but not culture-adapted strains of 229E are TMPRSS2 dependent (19), and MERS-CoV requires TMPRSS2 for infection of some respiratory cells but not other cell lines (31). The diversity of proteases involved in coronavirus entry may thus complicate the search for effective treatments, as the protease dependence of a particular coronavirus may vary among target cells.
If the specific protease dependence of coronavirus fusion depends on the cell type being infected, as the in vivo data suggest, then coronaviruses may have evolved to use different proteases to infect different sites. This would make protease use a potential determinant of coronavirus organ and tissue tropism, as is the case for avian influenza. We sought to explore this possibility using the murine coronavirus MHV as a model. MHV is useful for studying the contribution of host fusion factors to coronavirus tropism because infection of the laboratory mouse, a natural host, has identified a number of strains that appear to use the same receptor, CEACAM1a, but exhibit diverse cell, tissue, and organ specificities. We chose to focus on the brain-adapted strain JHM.SD (formerly named MHV4; GenBank accession number FJ647219.1) because its extreme neurovirulence is largely S protein dependent (32, 33) and because the JHM.SD spike also displays an unusual cell-to-cell spread phenotype that indicates exceptional susceptibility to S2′ cleavage: JHM.SD forms syncytia when infected cells are overlaid on nonpermissive (i.e., receptor-lacking) cells, a process known as receptor-independent spread (34). Furthermore, CEACAM1a is poorly expressed in the brain and almost absent from neurons, yet viruses bearing the JHM.SD spike spread extensively in infected brains and in neurons from wild-type or Ceacam1a−/− mice (35, 36). These properties led us to speculate that the JHM.SD spike adapted to the low level of receptor in the brain by becoming more sensitive to cleavage by an available protease. The CEACAM1a-independent cell-to-cell spread phenotype and the insensitivity of JHM.SD infection to endosomotropic weak bases (37) strongly suggested the involvement of a cell surface protease, such as TMPRSS2. We therefore hypothesized that JHM.SD infection is more sensitive than that of other MHV strains to a cell surface protease and that this difference is responsible for the neurotropism of the JHM.SD spike.
In this study, we examined the dependence of JHM.SD infection on endosomal acidification (and thereby acid-dependent endosomal proteases) and the neutral cell surface protease TMPRSS2, and we found that JHM.SD was less sensitive to inhibition of endosomal acidification but more sensitive to TMPRSS2 expression than the moderately neurovirulent reference strain A59. However, inhibitor studies revealed at best a minor role for surface serine proteases in MHV virus-cell and cell-cell fusion; instead, an unidentified cell surface metalloprotease appears to mediate these activities in the cell lines examined. These results suggest that sensitivity to a metalloprotease available in the brain underlies the tropism of JHM.SD.
RESULTS
JHM.SD is relatively resistant to bafilomycin A.
We first used luciferase reporter viruses to determine whether JHM.SD and A59 differed in sensitivity to inhibitors of endosomal proteases. Bafilomycin A inhibits the endosomal H+-ATPase and thereby indirectly inhibits acid-activated endosomal proteases, such as cathepsins (38). The assay was performed in L2 cells, an MHV-susceptible cell line previously used to compare the effect of bafilomycin A between A59 and MHV-2 (4). As entry of cell-associated MHV has been observed >1 h after washing (39), the treatment was maintained throughout the infection (Fig. 1, top), and the effects of bafilomycin on late infection events were assessed separately by infecting untreated cells in parallel and beginning treatment at 1 h postinfection (hpi), after which bafilomycin A was previously shown to have minimal effect on MHV-2 infection of L2 cells (4) (Fig. 1, middle). As postentry treatment significantly affected A59 infection, the effect of the preinfection treatment was divided by the effect of the postinfection treatment to correct for any postentry effects (Fig. 1, bottom). After correction, pretreatment with bafilomycin A significantly decreased both JHM.SD and A59 infection at both doses. The effect did not differ significantly between the 10 nM and 100 nM doses for either virus but was significantly smaller for JHM.SD than for A59 at both doses. These results suggest that JHM.SD can use acid-dependent endosomal entry for infection of L2 cells but also exploits an acidification-independent entry route that is less available to A59.
FIG 1.

JHM.SD is less sensitive than A59 to bafilomycin A. Pretreated L2 cells were infected with rJHM.SD-fluc or rA59-fluc at an approximate multiplicity of infection (MOI) of 0.5 and then assayed for luciferase activity 7 hpi as described in Materials and Methods. In parallel, cells were infected and then treated with DMSO-bafilomycin A beginning 1 h postinfection, and the pretreatment effect (relative to DMSO alone for each virus) was divided by the corresponding posttreatment effect to correct for posttreatment effects. (Top and middle) An asterisk indicates significant difference between the 0 and 10 nM or 100 nM treatment within each virus (2-way ANOVA with Dunnett's multiple comparisons of simple effects within columns). (Bottom) After correction, the effect of bafilomycin A was significantly smaller for JHM.SD than for A59 (n = 5; P < 0.0001 for the bafilomycin A effect, P < 0.0001 for the virus strain effect, and P < 0.0008 for the interaction, all by 2-way ANOVA). Symbols: *, significant difference (Tukey's multiple comparisons between all cell means) within each MHV strain between the bafilomycin A treatment and the 0 nM bafilomycin A control; #, significant difference between JHM.SD and A59 at the indicated bafilomycin A concentration (Tukey's multiple comparisons between all cell means). Data shown are representative of 3 independent experiments with n = 5 technical replicates.
TMPRSS2 expression increases JHM.SD infection.
We next considered whether acidification-independent JHM.SD infection involves the cell surface serine protease TMPRSS2, as has been shown for other coronaviruses. To address this possibility, we cotransfected HEK-293T cells with an MHV receptor (murine Ceacam1a-4L [mCeacam1a-4L]) and increasing amounts of human TMPRSS2 (hTMPRSS2) to generate receptor-bearing cells that also expressed TMRPSS2 protein. We found that TMPRSS2 expression increased JHM.SD infection but not A59 infection (Fig. 2A). Both the amount of TMPRSS2 transfected and the virus strain had statistically significant and interactive effects on luciferase activity, whereas transfection with a catalytically inactive mutant of TMPRSS2 had minimal effect (data not shown). Treatment with the surface serine protease inhibitor camostat beginning 3 h before infection abolished the effect of TMPRSS2 on JHM.SD infection (Fig. 2B), demonstrating that camostat inhibits TMPRSS2 as expected and also suggesting that TMPRSS2 activity is required near the time of infection. TMPRSS2 also increased JHM.SD infection in the presence of bafilomycin A (Fig. 2C), indicating that the effect of TMPRSS2 does not depend on endosomal acidification. Finally, TMPRSS2 transfection did increase A59 infection in the presence of bafilomycin A (Fig. 2D), indicating that TMPRSS2-dependent infection is available to A59 when the acidification-dependent pathway is blocked. Taken together, these results show that TMPRSS2 activity can promote MHV infection; specifically, TMPRSS2 increases JHM.SD infection of untreated cells and both JHM.SD and A59 infection of bafilomycin A-treated cells.
FIG 2.

TMPRSS2 activity directly mediates bafilomycin A-independent MHV infection. (A) JHM.SD is more sensitive than A59 to TMPRSS2 transfection. HEK-293T cells cotransfected with pCAGGS-mCeacam1a-4L and pCAGGS-hTMPRSS2-FLAG were infected with the indicated virus. Using two-way ANOVA (n = 5), P values were <0.0001 for the effects of TMPRSS2 and the virus strain and their interaction. Asterisks indicate the TMPRSS2 transfection levels at which the 2 viruses were significantly different from each other by Tukey's multiple comparison. (B) Camostat abrogates the effect of TMPRSS2 on JHM.SD infection. Transfected HEK-293T cells were treated with DMSO or camostat (final DMSO concentration, 1.5%) prior to infection. Using two-way ANOVA, P values were <0.0001 for TMPRSS2 transfection, camostat treatment, and their interaction. Number signs indicate significant differences from the no-TMPRSS2 control within the DMSO group using Dunnett's multiple comparisons (no significant difference from the no-TMPRSS2 control at any level of TMPRSS2 transfection within the camostat group). (C) TMPRSS2 enhances JHM.SD infection in the presence of bafilomycin A. Transfected HEK-293T cells were treated with DMSO or bafilomycin A (final DMSO concentration, 0.5%) prior to infection with JHM.SD-fluc. Using two-way ANOVA, P values were <0.0001 for the effects of TMPRSS2 and bafilomycin A and their interaction. Symbols: #, significant difference from the no-TMPRSS2 control within the DMSO group; †, significant difference from the no-TMPRSS2 control within the bafilomycin A group (determined by Dunnett's multiple-comparison test). (D) TMPRSS2 overcomes bafilomycin A inhibition of A59 infection. Transfected HEK-293T cells were treated with DMSO or bafilomycin A (final DMSO concentration, 0.5%) prior to infection with rA59-fluc. Using two-way ANOVA, P values were <0.0001 for the effects of TMPRSS2 and bafilomycin A and their interaction. Symbols: #, the TMPRSS2 transfection levels at which A59 infection differed from the no-TMPRSS2 control within the DMSO group; †, the TMPRSS2 transfection levels at which A59 infection differed from the no-TMPRSS2 control within the bafilomycin A group (determined by Dunnett's multiple-comparison test). All data are representative of at least 2 independent experiments with n = 5 technical replicates.
TMPRSS2 overexpression decreases productive MHV infection and syncytium formation.
We next investigated whether TMPRSS2 cleaves the JHM S spike and whether it cleaves the viral receptor CEACAM1a, as reported for the SARS-CoV receptor ACE2. We first cotransfected HEK-293T cells with mCeacam1a and hTMPRSS2 and examined protein size and expression by immunoblotting. CEACAM1a appeared as a doublet, with a major band consistent with the 110-kDa size of the full-length glycoprotein and a slower-migrating minor band sometimes visible in highly concentrated CEACAM1 preparations, especially protein purified from mouse intestinal brush border membranes (40, 41). Increasing expression of TMPRSS2 eliminated the upper band and ultimately decreased the strength of the major CEACAM1a band; however, no new species consistent with cell-associated TMPRSS2-cleaved CEACAM1a was seen in either uninfected or A59-infected cells (Fig. 3A, top). These changes seemed most consistent with either shedding of CEACAM1a from the cell surface or suppression of CEACAM1a expression. As a soluble receptor generally neutralizes MHV (42) and has been shown to induce S1/S2 dissociation of JHM S and, thus, to compromise infectivity (43), we did not attribute the effect of TMPRSS2 expression on infection to shedding of soluble CEACAM1a by TMPRSS2. Wild-type TMPRSS2 was difficult to detect in cell lysates, although the active-site mutant TMPRSS2-S441A was well expressed (Fig. 3A, bottom), consistent with reports of autocatalysis and subsequent shedding of the peripheral (C-terminal) TMPRSS2 fragment (44). We next infected cotransfected HEK-293T cells but found that expression of mCeacam1a from the high-level expression vector pCAGGS severely limited expression of viral proteins and release of infectious virus (data not shown); therefore, mCeacam1a was expressed from a constitutive low-level expression vector using the thymidine kinase promoter (pTK) in this and all further experiments in which late postinfection events (>8 hpi) were examined in human cell lines. Under these conditions, transfection with pCAGGS-hTMPRSS2-FLAG, which had promoted JHM.SD entry (Fig. 2), actually decreased productive MHV infection. Increasing expression of active TMPRSS2 decreased the levels of the viral structural proteins S and N in cells infected with JHM.SD, A59, or a chimeric virus bearing the MHV-2 spike (rA59/SMHV-2) that is entirely cathepsin dependent for entry (Fig. 3B). However, the polyclonal anti-S antibody AO4 recognized a possible TMPRSS2 S2′ product (expected size of 60 to 80 kDa [8, 26, 27]) only for MHV-2 S. Immunoblotting with the monoclonal anti-S antibody 5B19.2, which recognizes JHM.SD and A59 S and has been mapped to the MHV fusion peptide, revealed an approximately 150-kDa species of S that increased in density with increasing TMPRSS2 expression (Fig. 3C). This size is inconsistent with S2′ cleavage and indicates cleavage C terminal to the fusion peptide, which would be expected to inactivate S for fusion. Furthermore, increasing expression of TMPRSS2 significantly decreased release of infectious virus for JHM.SD, rA59, and rA59/SMHV-2 (Fig. 3D). Finally, expression of active TMPRSS2 actually decreased syncytium formation by a chimeric virus bearing the JHM.SD spike (rA59/SJHM.SD-EGFP); the effect was blocked by the serine protease inhibitor camostat, confirming that the decrease in cell-cell fusion was due to TMPRSS2 activity (Fig. 3E). Taken together, these results indicate that the level of TMPRSS2 overexpression that promoted JHM.SD entry in Fig. 3 is detrimental to productive infection; the loss of both cell-associated CEACAM1a and viral protein expression indicates that this level of TMPRSS2 activity is cytotoxic. Coexpression of MHV S and TMPRSS2 did result in new S cleavage products, but these cleavages may not represent S2′ cleavage and did not promote cell-cell fusion. We concluded that we could not further elucidate the role of TMPRSS2 in MHV infection using this overexpression system.
FIG 3.

TMPRSS2 overexpression decreases productive MHV infection. (A) TMPRSS2 decreases CEACAM1a protein levels. HEK-293T cells cotransfected with pCAGGS-mCeacam1a-4L and pCAGGS-hTMPRSS2-FLAG or pCAGGS-hTMPRSS2-S441A-FLAG were infected with rA59/SJHM.SD-EGFP and lysed for immunoblotting at 18 hpi. (B) TMPRSS2 decreases cell-associated MHV protein levels. HEK-293T cells cotransfected with pTK-mCeacam1a-4L and pCAGGS-hTMPRSS2-FLAG or pCAGGS-hTMPRSS2-S441A-FLAG were infected as indicated and lysed for immunoblotting 18 hpi. Goat polyclonal anti-S antibody AO4 was used to detect the S protein and mouse anti-N monoclonal antibody 1-16-1 to detect N protein. The vertical lines indicate boundaries between nonadjacent lanes (rA59/SJHM.SD-EGFP and rA59-EGFP were run on the same gel, but their positions were exchanged for consistency with other panels; rA59/SMHV-2-EGFP and the mock-infected cells were run in parallel on a separate gel). (C) TMPRSS2 cleavage of S may be nonproductive. Probing the lysates shown in panel C with anti-S2 MAb 5B19.2, previously mapped to the fusion peptide, detected an ∼150-kDa fragment (black box) inconsistent with S2′ cleavage. (D) TMPRSS2 decreases MHV titer. HEK-293T cells were cotransfected with pTK-mCeacam1a-4L and 200 ng of pCAGGS-hTMPRSS2-FLAG or pCAGGS-hTMPRSS2-S441A-FLAG and infected with the indicated viruses; cell supernatants were collected at 18 hpi and titers determined. Both active and inactive TMPRSS2 significantly decreased the MHV titer (using 2-way ANOVA with Dunnett's multiple comparisons of each TMPRSS2 level with the 0-ng control within each virus, P < 0.0001 for the effect of the virus, P < 0.0001 for the effect of TMPRSS2 transfection, and P < 0.0045 for the interaction; *, P < 0.05; **, P < 0.01; ***, P < 0.001; and ****, P < 0.0001 for the multiple comparisons). Data are representative of 2 independent experiments performed in triplicate. (E) TMPRSS2 activity decreases syncytium size. HEK-293β5 cells were cotransfected with pTK-mCeacam1a-4L and pCAGGS-hTMPRSS2-FLAG or pCAGGS-hTMPRSS2-S441A-FLAG and infected as described for panels B to D, treated at 2 hpi with DMSO or camostat as indicated (final DMSO concentration of 0.1% for all treatments), and fixed for microscopy at 18 hpi.
MHV infection is sensitive to metalloprotease inhibition.
Given the difficulty of interpreting results from cells overexpressing TMPRSS2, we sought to augment our findings in TMPRSS2-expressing HEK-293T cells by examining the role of endogenous mouse TMPRSS-family proteases in MHV-permissive cell lines. We first examined murine Tmprss2 (mTmprss2) mRNA expression by reverse transcription-quantitative PCR (RT-qPCR) but found that the mTmprss2 mRNA levels in MHV target cells were at or below the limit of detection; Table 1 shows the results from the only experiment in three attempts in which we consistently detected mTmprss2 in L2 cells. A single preliminary experiment using primary mouse cells yielded similar results, with very low levels of mTmprss2 mRNA in neurons and astrocytes and none detected in microglia or macrophages (data not shown). We concluded that endogenous TMPRSS2 is unlikely to contribute to MHV infection in these target cells. We next investigated whether another member of the TMPRSS family facilitates JHM.SD infection of L2 cells by treating L2 cells with the surface serine protease inhibitor camostat, which is expected to inhibit all 20 TMPRSS family members. As described for Fig. 1, cells were pretreated with protease inhibitors with and without bafilomycin (Fig. 4, top), and the results were normalized to those from cells treated postentry (Fig. 4, middle and bottom). Camostat only slightly decreased JHM.SD infection: the significant 31% reduction shown in Fig. 4 was the strongest effect observed in 3 independent experiments, and the effect did not reach significance in one of those experiments. We therefore tested a variety of other protease inhibitors: the non-cell-penetrating cysteine protease inhibitor E64, the non-cell-penetrating aspartyl protease inhibitor pepstatin A, and the metalloprotease inhibitor batimastat. Batimastat strongly decreased infection by both JHM.SD and A59 even after correction for the substantial postentry effect (Fig. 4); most strikingly, combined treatment with batimastat and bafilomycin A completely inhibited infection by both viruses. Therefore, MHV infection of L2 cells appears to involve a batimastat-sensitive metalloprotease.
TABLE 1.
Tmprss2 mRNA expression in mouse cell linesa
| Sample | CT (Actb) | CT (Tmprss2) | No. of reactions with CT (Tmprss2) ≤ 40b | ΔCT |
|---|---|---|---|---|
| Reference RNA | 17.9 | 24.3 | NA | 6.3 |
| Kidney | 22.3 | 27.4 | NA | 5.1 |
| Prostate | 20.1 | 25.9 | NA | 5.8 |
| DBT | 16.5 (0.3) | 39.1 (1.1) | 4/9 | 22.6 (1.0) |
| 17Cl1 | 16.0 (0.2) | 38.3 (1.6) | 6/9 | 22.3 (1.5) |
| L2 | 16.3 (0.1) | 33.5 (0.3) | 9/9 | 17.2 (0.3) |
Threshold cycle (CT) values for cell lines are the means (standard deviations) from n = 3 cDNA samples prepared from separate wells of each cell type; each cDNA sample was then assayed in triplicate (9 reactions total).
The final cycle number (40) was used for the Tmprss2 CT value where no signal was otherwise detected. Data are representative of two independent experiments. NA, not applicable.
FIG 4.

Metalloprotease inhibitor batimastat reduces JHM.SD and A59 infection of L2 cells. L2 cells were treated with DMSO or the indicated inhibitors as described in Materials and Methods (1.5% DMSO final concentration) and infected with rJHM.SD-fluc (left) or rA59-fluc (right), and luciferase activity was measured at 8 hpi. For each treatment, the effect of pretreatment relative to DMSO alone was divided by the effect of posttreatment relative to DMSO alone to correct for postentry effects, and the results were analyzed using 2-way ANOVA with Dunnett's multiple comparisons tests, comparing each protease inhibitor alone or with bafilomycin A to DMSO alone or with bafilomycin A, respectively. Both bafilomycin A and protease inhibitor treatment had significant and interactive effects on both JHM.SD and A59 infection (P < 0.0001 for protease inhibition, bafilomycin A, and the interaction for both JHM.SD and A59). Asterisks indicate the level of significance of the results of Dunnett's multiple-comparison tests of simple effects within columns (protease inhibitor versus DMSO control within each bafilomycin A treatment group; *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001). Data shown are representative of 3 independent experiments with n = 5 technical replicates.
To ensure that the effect of batimastat was due to metalloprotease inhibition, we next employed another hydroxamate metalloprotease inhibitor, TAPI-1. Batimastat and TAPI-1 were applied to three MHV-susceptible cell lines and their effects on cell viability and JHM.SD infection assessed. In all three cell lines, both drugs were essentially nontoxic (Fig. 5A) and significantly decreased JHM.SD infection; in L2 and DBT cells, both drugs also further decreased infection when combined with bafilomycin A (Fig. 5B). The effects of the inhibitors varied in magnitude among the cell types (Fig. 5C): infection of 17Cl1 cells decreased greatly in response to bafilomycin A, accounting for 85% of the total variation (2-way analysis of variance [ANOVA]), but varied only slightly in response to metalloprotease inhibitors (7.7% of total variation), whereas infection of DBT cells decreased markedly in response to metalloprotease inhibitors (83% of total variation) but actually increased slightly in response to bafilomycin A alone. We did not observe the complete inhibition of infection by combined bafilomycin A/metalloprotease inhibitor treatment seen in Fig. 4, which we attribute to differences in the infection procedure (synchronization was omitted because 17Cl1 cells did not tolerate washing after incubation at 4°C). Together, these results indicate that metalloprotease inhibition decreases MHV infection in multiple cell types and that the decrease in reporter gene expression is not due to cytotoxicity. As MHV does not encode a viral metalloprotease, the most logical conclusion is that a cell surface metalloprotease contributes to MHV entry.
FIG 5.

Cell-penetrating and extracellular metalloprotease inhibitors are nontoxic and decrease JHM.SD infection in multiple cell lines. Cells were pretreated with batimastat or TAPI-1 and/or bafilomycin A (final DMSO concentration, 1.5% for all treatments in all cells), infected with JHM.SD-fluc (MOI of 0.5) (A) or mock infected (B), washed, and incubated for an additional 6 h at 37°C and 30 min at room temperature in the presence of inhibitor before cell viability (A) and viral luciferase activity (B) were assessed. Representative data from two independent experiments with n = 5 technical replicates are shown. (A) Both batimastat and TAPI-1 were essentially nontoxic under the tested conditions. (B) Both batimastat and TAPI-1 decreased infection in all cell types. Asterisks show the results of two-way ANOVA with Dunnett's multiple-comparison test of simple effects within columns (metalloprotease inhibitor versus DMSO) for L2 and 17Cl1 cells and Tukey's multiple-comparison test between all cell means (not all results are shown) for DBT cells (*, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001). (C) Tabular results of two-way ANOVA on the data from panel B.
Metalloprotease inhibitors reduce MHV-mediated syncytium formation without blocking S1/S2 cleavage.
Addition of batimastat 1 h postinfection strongly decreased luciferase reporter activity (Fig. 4) with minimal evidence of cytotoxicity (Fig. 5). As MHV-induced cell-cell fusion begins only shortly after detectable viral protein expression, we hypothesized that batimastat inhibits MHV S-induced cell-cell fusion. We therefore examined the effects of protease inhibitors on syncytium formation and MHV spike cleavage in cells infected with isogenic enhanced green fluorescent protein (EGFP)-expressing MHV strains bearing the syncytium-forming JHM.SD or A59 spike or the non-syncytium-forming MHV-2 spike. In parallel, we treated cells with camostat to determine whether TMPRSS family proteases contributed to cell-cell fusion. As S1/S2 cleavage is a prerequisite for MHV-mediated cell-cell fusion, we also employed furin inhibitor I as a positive control for inhibition of S1/S2 cleavage and syncytium formation. Addition of batimastat 5 h postinfection substantially decreased syncytium formation by rA59/SJHM.SD-EGFP and rA59-EGFP (Fig. 6A); the effect of batimastat was even greater than that of furin inhibitor I, while camostat had no apparent effect. Immunoblotting of infected cell lysates showed that both batimastat and furin inhibitor I reduced the levels of JHM.SD and A59 S but not MHV-2 S protein, consistent with a loss of JHM.SD and A59 expansion through syncytium formation.
FIG 6.

Batimastat inhibits syncytium formation but not S1/S2 cleavage. (A) L2 cells were infected (MOI of 0.01) as indicated. Five hours postinfection, the medium was replaced with fresh medium containing the indicated inhibitor (final DMSO concentration of 1%). Fifteen hours postinfection, the cells were fixed and infection analyzed by bright-field and fluorescence microscopy. (B) L2 cells were infected (MOI of 0.1) as indicated. At 5 hpi, the medium was replaced with fresh medium containing the indicated inhibitor. At 16 hpi, the supernatant was removed and the cells lysed and subjected to immunoblotting with a polyclonal anti-S antibody (AO4). β-Tubulin was detected as a loading control.
S1/S2 cleavage is associated with MHV syncytium formation (4, 45), but the 110-kDa S1/S2 cleavage products were still detected in batimastat-treated cells, indicating that batimastat does not inhibit cell-cell fusion by blocking S1/S2 cleavage. Unfortunately, inhibition of S2′ cleavage could not be assessed because no bands consistent with S2′ were visible in the control samples, as was the case for Fig. 3. In contrast, furin inhibitor I greatly decreased S1/S2 cleavage of the A59 spike (Fig. 6B) but did not completely inhibit cell-cell fusion (Fig. 6A); surprisingly, it did not completely inhibit cleavage of the JHM.SD spike even at the relatively high concentration of 100 μM (Fig. 6B), suggesting that the JHM.SD S1/S2 site is either exceptionally susceptible to furin cleavage or cleavable by other cellular proteases. These results indicate that MHV-mediated cell-cell fusion relies on a batimastat-sensitive metalloprotease that is not required for cleavage at S1/S2.
TMPRSS2 restores infection but not cell-cell fusion in the presence of metalloprotease inhibition.
We next investigated whether metalloprotease and TMPRSS2 are redundant for MHV infection by treating hTMPRSS2-transfected cells with batimastat, bafilomycin A, or both inhibitors. Both batimastat and bafilomycin A decreased JHM.SD infection in HEK-293 cells, as in mouse cells. TMPRSS2 expression significantly increased infection in a dose-dependent manner under all conditions (Fig. 7A). The ability of TMPRSS2 to replace metalloprotease activity in cell-to-cell fusion was difficult to assess because TMPRSS2 overexpression itself decreases syncytium size (Fig. 3E). The overwhelming majority of infection foci in TMPRSS2-transfected cells were unicellular regardless of treatment (Fig. 7B). We therefore conclude that TMPRSS2 can replace metalloprotease activity for MHV entry but not for cell-cell fusion.
FIG 7.
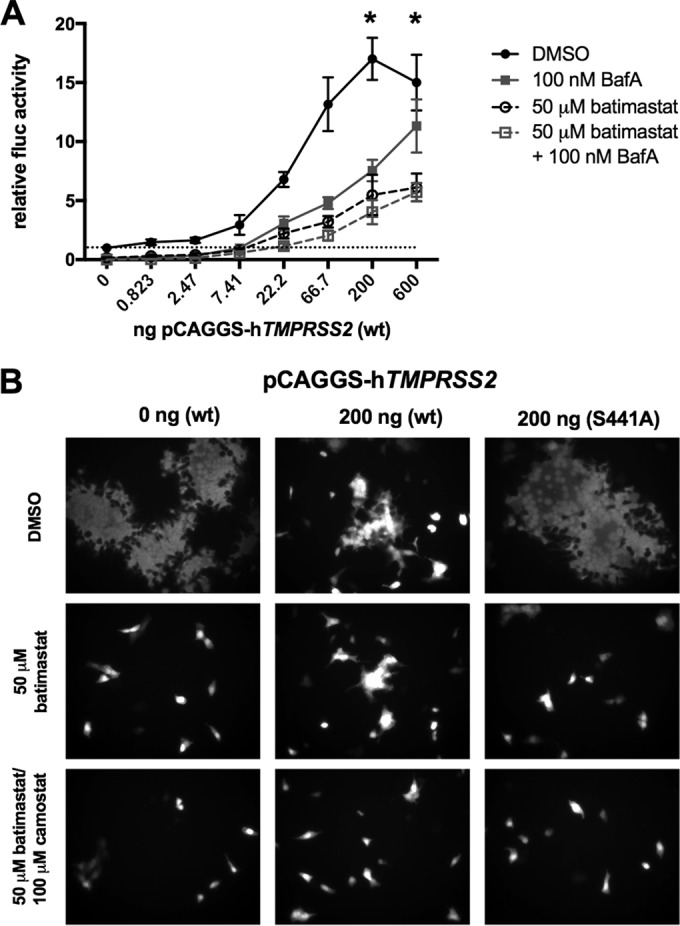
TMPRSS2 is an alternative to metalloprotease for JHM.SD entry. (A) TMPRSS2 rescues blockade of JHM.SD entry by bafilomycin A, batimastat, or both. HEK-293T cells cotransfected with pTK-mCeacam1a-4L and pCAGGS-hTMPRSS2-FLAG were pretreated with batimastat and/or bafilomycin A (final DMSO concentration, 1.5%) and infected with JHM.SD-fluc (MOI, 0.05 PFU/cell), and the treatment was maintained until the luciferase activity was assayed at 7.5 hpi. Using two-way ANOVA (n = 5), P values were <0.0001 for drug treatment, TMPRSS2 level, and the interaction between them; *, transfection levels at which Dunnett's multiple-comparisons tests showed significant differences from the baseline for all treatments. Data are representative of two independent experiments with n = 5 technical replicates. (B) TMPRSS2 inefficiently rescues MHV cell-to-cell spread in the presence of batimastat. HEK-293β5 cells cotransfected with pTK-mCeacam1a-4L and pCAGGS-hTMPRSS2-FLAG or pCAGGS-hTMPRSS2-S441A-FLAG were infected with rA59/SJHM.SD-EGFP (MOI, 0.1); at 2 hpi, the medium was replaced with medium containing DMSO, batimastat, camostat, or both, as indicated (final DMSO concentration, 0.1% for all treatments), and syncytium formation was assessed at 18 h.
The MHV-2 spike protein is resistant to metalloprotease inhibition during entry.
Finally, we sought to link the effect of metalloprotease inhibition to cleavage of MHV S during entry. We reasoned that if the metalloprotease cleaves MHV S to activate it for fusion, then infection by the MHV-2 spike, being entirely dependent on cathepsin B and/or L activity, should be resistant to metalloprotease inhibitors. To test this hypothesis, L2 cells were pretreated with batimastat and/or bafilomycin A and infected at 4°C (as described for Fig. 1 and 4) with the isogenic chimeric viruses rA59-EGFP, rA59/SJHM.SD-EGFP, and rA59/SMHV-2-EGFP. To separate the effects of metalloprotease inhibition on entry and cell-cell fusion, both inhibitors were removed at 1 hpi and replaced with medium containing dimethyl sulfoxide (DMSO) only (termed pretreatments); in parallel, infected cells pretreated with DMSO alone were treated with inhibitors beginning at 1 hpi (posttreatments); the DMSO control cells were treated with fresh medium containing DMSO at each time point. Consistent with previous results (4) and our hypothesis, rA59/SMHV-2-EGFP entry was highly sensitive to bafilomycin A but resistant to batimastat treatment (Fig. 8). All three viruses were essentially blocked by combined treatment with bafilomycin A and batimastat, as seen in Fig. 4, and the effects of postinfection treatment were also S dependent, as seen in Fig. 6. These strain-dependent effects strongly suggest that batimastat inhibits a metalloprotease that interacts with the viral S protein, and the pronounced suppression of JHM.SD and A59 by batimastat treatment from −3 to 1 hpi demonstrates that the drug inhibits MHV entry.
FIG 8.

Effect of metalloprotease inhibition on MHV infection is S strain specific. L2 cells were treated with batimastat (50 μM) and/or bafilomycin A (100 nM) or DMSO as described in Materials and Methods (1.5% DMSO final concentration for all conditions) and infected with isogenic chimeric viruses bearing the S protein from JHM.SD, A59, or MHV-2 (MOI, 0.5). Two-way ANOVA showed significant effects of treatment, strain, and interaction (P < 0.0001 for all); the asterisks represent the results of Tukey's multiple-comparison tests between each pair of viruses under each condition (level of significance for at least 2 of the 3 comparisons; *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001). Data are representative of 2 independent experiments with n = 5 technical replicates.
DISCUSSION
Coronaviruses use diverse proteases for entry at different cellular sites, including acid-dependent endosomal/lysosomal proteases, such as cathepsins, to enter cells via the late endosome (4, 8, 10–14, 18, 46), furin, to enter via the early endosome (39, 47), and TMPRSS2, to enter presumably at or near the cell surface (13, 15–18, 48). Among MHV strains, MHV-2 depends on the endosomal cysteine proteases cathepsin B and L, while A59 can be blocked by combined inhibition of cysteine and aspartyl lysosomal proteases (4, 39). This study extends the analysis of MHV protease use to include the brain-adapted JHM.SD strain of MHV, which mediates cell-cell fusion independent of the receptor protein, and cell surface serine proteases, such as TMPRSS2. We found that bafilomycin A (an inhibitor of endosomal acidification and therefore indirectly of acid-dependent endosomal proteases) only modestly decreased entry, and overexpression of TMPRSS2 greatly enhanced entry of JHM.SD relative to their effects on strain A59. However, inhibition of endogenous surface serine protease activity had only a modest effect on JHM.SD infection; instead, an unidentified batimastat-sensitive metalloprotease appeared to be most important for both viral entry and virus-mediated cell-cell fusion. The list of proteases that may mediate coronavirus fusion must therefore be expanded to include metalloproteases. The contribution of metalloprotease activity to JHM.SD infection varied widely among the tested cell lines, suggesting that either the level of MHV-promoting metalloprotease activity or the efficiency of the endosomal entry pathway differs between cell types. We also observed MHV strain-dependent effects of bafilomycin A and batimastat on viruses bearing the MHV-2, A59, and JHM.SD spike proteins. Together, these results suggest that metalloprotease use underlies the spike-dependent neurovirulence of JHM.SD (32, 33).
Batimastat and TAPI-1, the metalloprotease inhibitors used in the present study, are broad-spectrum inhibitors of two metalloprotease families: the matrix metalloproteases (MEROPS family M10; abbreviated as MMP) and the a disintegrin and metalloprotease group (MEROPS family M12; abbreviated as ADAM or ADAMTS) (49). One member of these families, ADAM17, was previously shown to mediate uptake of SARS-CoV S-pseudotyped particles but not productive infection (23, 28–30); otherwise, these proteases have not, to the best of our knowledge, been implicated in viral entry. In the mouse, these families comprise some 69 catalytically active proteases (49, 50, and http://merops.sanger.ac.uk); we are currently working to identify the specific metalloprotease(s) involved in MHV infection.
Many studies of protease use in coronavirus fusion have employed exogenous expression of cellular proteases or addition of recombinant soluble proteases. The results we obtained from overexpression of TMRPSS2 suggest that such experiments should be interpreted with caution. Although TMPRSS2 increased JHM.SD entry, the level of overexpression used was detrimental to virus production and cell-cell fusion. We judged the potential for confounding effects to be significant and did not attempt to determine whether the loss of CEACAM1a and MHV protein in cell lysates was due to shedding, loss of MHV amplification by cell-to-cell spread, or general cytotoxicity. It remains unclear whether endogenous expression of any TMPRSS protease in any cell type promotes JHM.SD entry without decreasing overall virus yield, as would be necessary for JHM.SD to have evolved to use these proteases for central nervous system (CNS) infection. We therefore conclude that enhancement of JHM.SD infection by TMPRSS2 suggests that this virus is susceptible to fusion activation by nonendosomal proteases, but we cannot infer a role for TMPRSS2 in MHV infection of the CNS from the results of the present study.
Our findings pose three apparent discrepancies with published results that must be addressed. First, A59 was previously shown to be largely insensitive to inhibition of endosomal acidification (4). However, in that case the inhibition was removed shortly after infection, and the infection was measured as the viral titer at 16 hpi. A59 can enter asynchronously (39), and it seems likely that some A59 infection occurred after the removal of the inhibitor or that robust second-round infection compensated for entry inhibition during inoculation. More recently, the presence of a postfusion A59 S2 cleavage fragment was not affected by the metalloprotease inhibitor phosphoramidon (26); however, phosphoramidon does not inhibit batimastat-sensitive MMPs and ADAM proteases (49 and http://merops.sanger.ac.uk). Finally, JHM.SD infection was previously reported to resist inhibition by endosomotropic weak bases (37), which indirectly inhibit cathepsins similarly to bafilomycin A; however, that study was performed in DBT cells, in which we observed increased JHM.SD infection after bafilomycin A treatment (Fig. 5). Therefore, we consider the results of the current study to be consistent with the published literature.
Given that the coronavirus S2 domain requires proteolysis for fusion activation (8), that the metalloprotease inhibitor acts early in infection and also blocks cell-cell fusion, and that the effect varies among MHV S proteins, the simplest interpretation of our findings is that a metalloprotease cleaves some MHV S proteins to activate S2 for fusion. Collectively, the results of the present study and other studies of coronavirus entry suggest that there are parallel acid-dependent (i.e., late endosomal/lysosomal) and acid-independent (i.e., surface or early endosomal metalloprotease or TMPRSS2) pathways for MHV entry (Fig. 9A). In this model, JHM.SD cannot efficiently access the acid-dependent pathway and relies on the acid-independent pathway, making it less sensitive to bafilomycin A and more sensitive to metalloprotease inhibition and TMPRSS2 overexpression. In contrast, A59 enters efficiently via the acid-dependent pathway but is less efficiently cleaved by acid-independent proteases, making it more susceptible to bafilomycin A and less susceptible to TMPRSS2 expression or metalloprotease inhibition, while MHV-2 is blocked by bafilomycin A alone and is relatively resistant to metalloprotease inhibition. Simultaneous blockade of both pathways by combined bafilomycin A/batimastat treatment therefore abrogates infection by all strains. The mechanism underlying these interstrain differences is less clear; we suspect that the instability of JHM.SD S inactivates many potentially infectious particles due to S1/S2 dissociation and/or premature fusion triggering during endosomal uptake and/or acidification, whereas the more stable A59 and MHV-2 spike proteins survive until appropriately activated by endosomal proteases. Metalloprotease or TMPRSS2 cleavage would thus rescue JHM.SD infection by allowing virions to enter the acid-independent pathway (up to 90% or more of the total inoculum, judging by the >10-fold enhancement of infection by TMPRSS2 in HEK-293T cells shown in Fig. 2). Such parallel fusion protein processing could greatly expand the cell and organ tropism of MHV.
FIG 9.

Independent and sequential cleavage models of MHV entry. (A) In the independent pathway model, JHM.SD is more efficiently cleaved by cell surface acid-independent proteases (vertical stripes) such as metalloprotease or TMPRSS2 and fuses at the plasma membrane, whereas A59 better survives endocytosis and/or is more efficiently cleaved by acid-dependent endosomal proteases (horizontal stripes). In the sequential cleavage model (B), cleavage by acid-independent proteases produces a metastable intermediate that is more readily cleaved by endosomal proteases, and JHM.SD S is more efficiently cleaved by acid-independent proteases but less efficiently cleaved by endosomal proteases.
Although our findings seem most consistent with parallel entry pathways (Fig. 9A), we cannot rule out a sequential cleavage model in which all MHV strains ultimately enter via endosomal protease cleavage, but prior cleavage by surface proteases greatly increases the efficiency of endosomal protease cleavage (Fig. 9B). Assuming that endosomal proteases retain some activity in the face of high-concentration bafilomycin A treatment, we can hypothesize that the native JHM.SD spike is poorly cleaved by acid-dependent proteases (creating dependence on acid-independent proteases) but is readily cleaved and, thus, primed by acid-independent proteases (decreasing the effect of bafilomycin A by making the residual endosomal protease activity more effective), while the A59 spike is a better substrate for endosomal proteases in the absence of prior cleavage (making it less dependent on acid-independent protease activity) but also less efficiently primed (rendering it more susceptible to bafilomycin A). In this case, MHV-2 might be unaffected by metalloprotease cleavage because S1/S2 cleavage is required to expose the metalloprotease site. This model could even be invoked to explain the effect of batimastat on cell-cell fusion, as some paramyxovirus fusion proteins have been shown to require secretion, internalization, cathepsin cleavage, and recycling to the plasma membrane for activation (reviewed in reference 51). The idea of multiple proteolytic cleavage sites in S2 is not unprecedented. At least two groups have now reported heterogeneous coronavirus S postentry cleavage products suggestive of cleavage at multiple sites within S2 (26, 27), consistent with the idea that S2 is cleaved by more than one protease during virus-cell fusion. However, which of these cleavages are fusion activating, which prime the protein for a definitive fusion-activating cleavage, and which are simply destructive remains to be clarified.
In conclusion, MHV strains JHM.SD and A59 exhibit strain-specific dependence on endosomal acidification and acid-independent proteases, including a previously unsuspected batimastat-sensitive metalloprotease, for infection. These results hint that cleavage of MHV spike by different proteases either provides alternative entry pathways or greatly facilitates a definitive fusion-activating event. MHV strains differ markedly in organ and tissue tropism in a spike-dependent manner despite the use of a common receptor; the current study joins other recent work in suggesting that the availability of specific fusion-activating proteases greatly influences coronavirus virulence.
MATERIALS AND METHODS
Viruses and cells.
Recombinant MHV strains rJHM.SD-fluc and rA59-fluc were generated by targeted RNA recombination according to the methods developed by Kuo et al. (52) and Masters and Rottier (53). The firefly luciferase gene was included between the viral E and M genes, as originally done by de Haan et al. (54). The luciferase-expressing viruses were grown in DBT cells, and stocks were expanded from single luciferase-positive plaques. Recombinant MHV strains icJHM.SD (55), rA59 (32), and rA59/SMHV-2 (56) and enhanced green fluorescent protein (EGFP)-expressing strains rA59/SJHM.SD-EGFP, rA59-EGFP (57), and rA59/SMHV-2-EGFP (4) were grown in 17Cl1 cells. The titers of all viruses were determined on L2 cells as previously described (58), and all multiplicity of infection (MOI) calculations were based on L2 cell titers. Viruses were diluted in Dulbecco's modified Eagle's medium (DMEM) with 2% fetal bovine serum (FBS) for inoculation unless otherwise indicated.
Human embryonic kidney (HEK)-293T cells were maintained in DMEM (11965-084; Gibco) with 100 U/ml penicillin, 100 μg/ml streptomycin, and 10% fetal bovine serum (FBS); HEK-293β5 cells (human embryonic kidney cells overexpressing the human β5 integrin subunit) were maintained in the same medium with 100 μg/ml G418. DBT cells were maintained in DMEM with 100 U/ml penicillin, 100 μg/ml streptomycin, 5% FBS, and 10% tryptose phosphate broth. L2 and 17Cl1 cells were maintained in reconstituted DMEM (12100-061; Gibco) with 0.37% sodium bicarbonate, 10 mM HEPES, 2 mM glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 10% FBS.
Inhibitors.
Bafilomycin A1 (96000; Calbiochem), camostat (SML0057; Sigma), E64 (E3132; Sigma), pepstatin A (11359053001; Roche Boehringer Mannheim), batimastat (SML0041; Sigma), and furin inhibitor I (344930; Calbiochem) were dissolved in dimethyl sulfoxide (DMSO) at 200× (bafilomycin A) or 100× (protease inhibitors) the highest final concentration indicated for the entry and L2 cell spread assays and at 2,000× the final concentration for the HEK-293β5 cell spread assays. The inhibitors were stored in working aliquots at −20°C and added to the medium immediately before use.
Plasmids and transfection.
pCAGGS-hTMPRSS2-FLAG (wild type) and pCAGGS-hTMPRSS2-S441A-FLAG are described in reference 16. pCAGGS-mCeacam1a-4L was made by inserting a PCR product amplified from BgpD (GenBank accession number X67279.1) into the EcoRI and KpnI sites of pCAGGS-MCS. pTK-mCeacam1a-4L was made by inserting a PCR product amplified from BgpD into the NotI backbone of pTKbeta (ATCC 77178). The nucleotide sequences of all PCR-derived segments were confirmed by sequencing. The day before transfection, HEK-293T cells were seeded in 6-well plates at 1 × 106 cells/well and HEK-293β5 cells were seeded in 24-well plates at 1.3 × 105 cells/well. For transfection, 3 μg plasmid (comprising 1,200 ng of pCAGGS-mCeacam1a-4L or pTK-mCeacam1a-4L, up to 600 ng pCAGGS-hTMPRSS2-FLAG, and the balance as empty vector pCAGGS-MCS) was diluted into 200 μl of Opti-MEM, and then 9 μg of polyethylenimine (at 1 μg/μl, pH 7.0; 3:1, wt/wt; 23966; Polysciences, Inc.) was added. The reaction mixtures were incubated at room temperature for 15 min and then added to the cells (150 μl/well for 6-well plates and 30 μl/well for 24-well plates); for consistency, the amounts of pCAGGS-hTMPRSS2-FLAG are always shown as the equivalent amount for a 6-well plate well. Transfected HEK-293T cells were reseeded into smaller wells 1 day posttransfection (dpt) as described for the individual experiments.
Entry and viability assays.
L2 cells (1 × 105 cells/well), 17Cl1 cells (5 × 104 cells/well), and DBT cells (5 × 104 cells/well) were seeded in tissue culture-treated white 96-well plates 1 day before infection. In the luciferase reporter experiments (Fig. 1, 2, 4, and 5), the cells were pretreated for 3 h with protease inhibitors or DMSO and/or for 1 h with bafilomycin A or additional DMSO (preinfection treatment); the postinfection treatment cells (Fig. 1 and 4) were not disturbed. Virus was diluted to achieve the indicated MOI at 50 μl/well, and inhibitors were added to the inoculum at the indicated concentrations (preinfection treatment only); virus-free diluent with inhibitors was prepared in parallel. In experiments using only L2 cells (Fig. 1 and 4), the medium was aspirated and cold inoculum was added on ice; in parallel, one well per treatment was inoculated with virus-free diluent containing inhibitors to provide a background value (none of the drugs affected this value throughout the experiments). The inoculated cells were incubated at 4°C for 1 h, washed once with cold phosphate-buffered saline (PBS), and refed with cold medium containing the inhibitors; the plates were then moved to a 37°C incubator (t = 0). For postinfection treatment, the medium was aspirated and replaced with warm medium containing DMSO or inhibitor(s) at 1 h (Fig. 1 and 4). At 7 to 8 h, the medium was aspirated, the cells were washed once with PBS, Glo Lysis buffer (E2661; Promega) was added at 100 μl/well, and the plates were stored at −80°C prior to analysis. For analysis, the plates were warmed to room temperature, 100 μl/well of Steady-Glo reagent (E2510; Promega) was added to the lysates, and the total luminescence was measured using a BioTek Synergy HT instrument. The experiments shown in Fig. 5 comparing L2, 17Cl1, and DBT cells were performed similarly with the following modifications. Postinoculation, all cells were immediately incubated at 37°C (t = 0). Cells were washed with warm PBS and refed with warm medium containing inhibitors at 1 h, removed from the incubator, allowed to equilibrate to room temperature at 7 h, and analyzed for luciferase activity at 7.5 h by adding 100 μl/well of Steady-Glo reagent directly to the culture medium and measuring the activity within 30 min. In parallel, separate plates were treated, mock infected, and refed with medium containing inhibitors and assessed for cell viability by adding 100 μl/well of reconstituted CellTiter-Glo reagent (G7571; Promega) directly to the culture medium and proceeding according to the manufacturer's instructions. No postinfection treatment-only condition was included in the viability assays. The HEK-293T experiments shown in Fig. 2 and 7 were performed similarly to the L2/DBT/17Cl1 cell experiments, with the following modifications. HEK-293T cells were reseeded into 96-well plates at 5 × 104 cells/well on day 1 posttransfection and infected on day 2 posttransfection. All cells were infected at an MOI of 0.05 PFU/cell, and luciferase activity was assessed at 7.5 hpi. The EGFP reporter experiments (Fig. 8) were performed similarly to experiments shown in Fig. 1 and 4, with the following modifications. The preinfection treatment cells were washed and refed with medium containing DMSO alone at 1 h. The postinfection treatment condition cells were treated with DMSO alone during pretreatment and infection and washed and refed with medium containing inhibitors beginning at 1 h. At 10 h, the cells were washed once with PBS, lysed with reporter lysis buffer (E3971; Promega), and frozen at −80°C to complete lysis. After thawing, the EGFP fluorescence endpoint was read with a BioTek Synergy HT plate reader (485-nm excitation filter, 528-nm emission filter; band pass for each, 20 nm; gain, 120). In all experiments, the final DMSO concentration was the same across all treatments and is indicated in the figure legend for each experiment.
Tmprss2 mRNA measurement.
Confluent wells of cells in 6-well plates were harvested and mRNA purified using TRIzol reagent (Thermo Fisher Scientific) according to the manufacturer's instructions, with the following modifications: a second chloroform extraction (1:1, vol/vol) was performed on the aqueous phase, and an ethanol precipitation step was added after the isopropanol precipitation. Kidney and prostate from 7- to 8-week-old male C57BL/6 mice were homogenized in TRIzol and RNA purified similarly. RNA was diluted to 200 ng/μl and treated using the Turbo DNA-free kit (Thermo Fisher Scientific) to remove residual genomic DNA. Mouse universal reference total RNA (S3296; BD Biosciences) was used as an additional control. cDNA was prepared from 350 ng of DNA-free RNA per 20-μl reaction mixture using Superscript III reverse transcriptase (Thermo Fisher Scientific) according to the manufacturer's instructions. The amounts of Actb and Tmprss2 cDNA were assessed using quantitative PCR with iQ SYBR green supermix (Bio-Rad) with 2 μl cDNA per 25-μl reaction mixture; the organ cDNA was diluted 1:5 prior to qPCR. The Tmprss2 primers spanned the junction of exons 12 and 13 (forward, 5′-ACAACAACCTAATCACACCAGCCAT-3′; reverse, 5′-AGCCACCAGATCCCATTCTTCAAAG-3′).
Immunoblotting.
L2 cells were seeded at 2.5 × 105 cells/well, and HEK-293T cells were seeded 1 dpt at 3.5 × 105 cells/well in 24-well plates and infected the next day (2 dpt for the HEK-293T cells) with the indicated virus diluted in DMEM with 2% FBS (t = 0 hpi). The cells were incubated at 37°C for 1 h and rocked by hand every 15 min, washed 3× (L2 cells) or 1× (HEK-293T cells) with warm PBS, and refed with fresh medium. For the L2 cells, the medium was removed at 5 hpi and replaced with medium containing the indicated treatment with a final DMSO concentration of 1%. At 16 hpi (L2 cells) or 18 hpi (HEK-293T cells), the cells were washed once with PBS and lysed with 1% NP-40 alternative (492018; EMD Millipore) in PBS with EDTA-free protease inhibitor cocktail (11836170001; Roche) plus 2 mM EDTA. Lysates were centrifuged at 700 × g at 4°C for 10 min to pellet the nuclei, and the postnuclear supernatants were used immediately or stored at −80°C. The total protein concentration in the lysates was checked using a bicinchoninic acid protein assay kit (23225; Pierce) and differed minimally among samples in all cases, so loading of equal amounts of total protein or equal volumes of lysate was used interchangeably. Lysates were run on NuPAGE 3 to 8% Tris-acetate gels (Invitrogen) using the manufacturer-recommended reagents, and the proteins were transferred to polyvinylidene difluoride (PVDF) membranes. Membranes were blocked for 30 min at room temperature or overnight at 4°C in Tris-buffered saline with 0.1% Tween 20 (TBST) with 10% nonfat dry milk (here termed block), incubated with primary antibody (polyclonal goat anti-S AO4, a kind gift from K. V. Holmes, 1:1,000 in block; monoclonal mouse anti-S2 5B19.2 [59], 1:1,000 in block; monoclonal mouse anti-N clone 1-16-1, from J. L. Leibowitz, 1:1,000 in block; monoclonal mouse anti-CEACAM1 CC1 [60], 1:1,000 in block; or polyclonal rabbit anti-FLAG [F7425; Sigma], 1:1,000 in TBST) for 1 h at room temperature or overnight at 4°C. They were then washed for 20 min with block and twice for 10 min each with TBST, incubated 30 min with species-specific horseradish peroxidase-conjugated secondary antibody in block (rabbit anti-goat IgG [1:5,000; 61-1620; Invitrogen], goat anti-mouse IgG, [1:5,000; 31430; Pierce], or donkey anti-rabbit IgG [1:10,000; NA934V; GE]), developed with Western Lightning or Western Lightning plus enhanced chemiluminescence reagent (PerkinElmer), and imaged using an Amersham Imager 600 (GE). For detection of additional substrates, membranes were stripped twice for 10 min each with mild stripping buffer (1.5% glycine, 0.1% sodium dodecyl sulfate, 1% Tween 20, pH 2.2) and washed twice with PBS and twice with TBST before blocking and reprobing. Horseradish peroxidase-conjugated anti-β-tubulin (21058; Abcam) was used as a loading control, diluted 1:1,000 in TBST, and hybridized for 1 h at room temperature, followed by washing and detection as described for conjugated secondary antibodies.
Syncytium formation assay.
L2 cells (100% confluent) or HEK-293β5 cells on dpt 2 (∼80% confluent) in 24-well plates were infected with the indicated EGFP-expressing virus at an MOI of 0.01 (L2) or 0.1 (HEK-293β5) PFU/cell. The cells were incubated for 1 h at 37°C, rocked at 15-min intervals, and then washed with PBS and refed with L2 medium or HEK-293T medium (t = 0). Five hours postinfection (L2) or 1 h postinfection (HEK-293β5), the medium was replaced by medium containing DMSO or the indicated inhibitor(s) at the indicated concentration(s) (final DMSO concentration of 1% for L2 cells and 0.1% for HEK-293β5 cells). At 16 hpi (L2) or 18 hpi (HEK-293β5), the cells were washed once with PBS, fixed for 20 min with 4% paraformaldehyde in PBS, and washed three times for 5 min each with PBS. Fluorescence was detected using a Nikon Eclipse TE2000-U fluorescence microscope with a 488-nm excitation filter.
ACKNOWLEDGMENTS
This work was supported by NIH grants R01AI600210 to S.R.W. and K08AI098503 to J.M.P.
REFERENCES
- 1.Bertram S, Glowacka I, Steffen I, Kuhl A, Pohlmann S. 2010. Novel insights into proteolytic cleavage of influenza virus hemagglutinin. Rev Med Virol 20:298–310. doi: 10.1002/rmv.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhou Y, Vedantham P, Lu K, Agudelo J, Carrion R Jr, Nunneley JW, Barnard D, Pohlmann S, McKerrow JH, Renslo AR, Simmons G. 2015. Protease inhibitors targeting coronavirus and filovirus entry. Antiviral Res 116:76–84. doi: 10.1016/j.antiviral.2015.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heald-Sargent T, Gallagher T. 2012. Ready, set, fuse! The coronavirus spike protein and acquisition of fusion competence. Viruses 4:557–580. doi: 10.3390/v4040557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Qiu Z, Hingley ST, Simmons G, Yu C, Das Sarma J, Bates P, Weiss SR. 2006. Endosomal proteolysis by cathepsins is necessary for murine coronavirus mouse hepatitis virus type 2 spike-mediated entry. J Virol 80:5768–5776. doi: 10.1128/JVI.00442-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yamada YK, Takimoto K, Yabe M, Taguchi F. 1997. Acquired fusion activity of a murine coronavirus MHV-2 variant with mutations in the proteolytic cleavage site and the signal sequence of the S protein. Virology 227:215–219. doi: 10.1006/viro.1996.8313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gombold JL, Hingley ST, Weiss SR. 1993. Fusion-defective mutants of mouse hepatitis virus A59 contain a mutation in the spike protein cleavage signal. J Virol 67:4504–4512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Haan CA, Stadler K, Godeke GJ, Bosch BJ, Rottier PJ. 2004. Cleavage inhibition of the murine coronavirus spike protein by a furin-like enzyme affects cell-cell but not virus-cell fusion. J Virol 78:6048–6054. doi: 10.1128/JVI.78.11.6048-6054.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Belouzard S, Chu VC, Whittaker GR. 2009. Activation of the SARS coronavirus spike protein via sequential proteolytic cleavage at two distinct sites. Proc Natl Acad Sci USA 106:5871–5876. doi: 10.1073/pnas.0809524106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Belouzard S, Millet JK, Licitra BN, Whittaker GR. 2012. Mechanisms of coronavirus cell entry mediated by the viral spike protein. Viruses 4:1011–1033. doi: 10.3390/v4061011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Simmons G, Gosalia DN, Rennekamp AJ, Reeves JD, Diamond SL, Bates P. 2005. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc Natl Acad Sci USA 102:11876–11881. doi: 10.1073/pnas.0505577102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang IC, Bosch BJ, Li F, Li W, Lee KH, Ghiran S, Vasilieva N, Dermody TS, Harrison SC, Dormitzer PR, Farzan M, Rottier PJ, Choe H. 2006. SARS coronavirus, but not human coronavirus NL63, utilizes cathepsin L to infect ACE2-expressing cells. J Biol Chem 281:3198–3203. doi: 10.1074/jbc.M508381200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qian Z, Dominguez SR, Holmes KV. 2013. Role of the spike glycoprotein of human Middle East respiratory syndrome coronavirus (MERS-CoV) in virus entry and syncytia formation. PLoS One 8:e76469. doi: 10.1371/journal.pone.0076469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gierer S, Bertram S, Kaup F, Wrensch F, Heurich A, Kramer-Kuhl A, Welsch K, Winkler M, Meyer B, Drosten C, Dittmer U, von Hahn T, Simmons G, Hofmann H, Pohlmann S. 2013. The spike protein of the emerging betacoronavirus EMC uses a novel coronavirus receptor for entry, can be activated by TMPRSS2, and is targeted by neutralizing antibodies. J Virol 87:5502–5511. doi: 10.1128/JVI.00128-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Regan AD, Shraybman R, Cohen RD, Whittaker GR. 2008. Differential role for low pH and cathepsin-mediated cleavage of the viral spike protein during entry of serotype II feline coronaviruses. Vet Microbiol 132:235–248. doi: 10.1016/j.vetmic.2008.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matsuyama S, Nagata N, Shirato K, Kawase M, Takeda M, Taguchi F. 2010. Efficient activation of the severe acute respiratory syndrome coronavirus spike protein by the transmembrane protease TMPRSS2. J Virol 84:12658–12664. doi: 10.1128/JVI.01542-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shulla A, Heald-Sargent T, Subramanya G, Zhao J, Perlman S, Gallagher T. 2011. A transmembrane serine protease is linked to the severe acute respiratory syndrome coronavirus receptor and activates virus entry. J Virol 85:873–882. doi: 10.1128/JVI.02062-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Glowacka I, Bertram S, Muller MA, Allen P, Soilleux E, Pfefferle S, Steffen I, Tsegaye TS, He Y, Gnirss K, Niemeyer D, Schneider H, Drosten C, Pohlmann S. 2011. Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response. J Virol 85:4122–4134. doi: 10.1128/JVI.02232-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kawase M, Shirato K, van der Hoek L, Taguchi F, Matsuyama S. 2012. Simultaneous treatment of human bronchial epithelial cells with serine and cysteine protease inhibitors prevents severe acute respiratory syndrome coronavirus entry. J Virol 86:6537–6545. doi: 10.1128/JVI.00094-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shirato K, Kanou K, Kawase M, Matsuyama S. 2017. Clinical isolates of human coronavirus 229E bypass the endosome for cell entry. J Virol 91:e01387-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shirato K, Matsuyama S, Ujike M, Taguchi F. 2011. Role of proteases in the release of porcine epidemic diarrhea virus from infected cells. J Virol 85:7872–7880. doi: 10.1128/JVI.00464-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chaipan C, Kobasa D, Bertram S, Glowacka I, Steffen I, Tsegaye TS, Takeda M, Bugge TH, Kim S, Park Y, Marzi A, Pohlmann S. 2009. Proteolytic activation of the 1918 influenza virus hemagglutinin. J Virol 83:3200–3211. doi: 10.1128/JVI.02205-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bottcher E, Matrosovich T, Beyerle M, Klenk HD, Garten W, Matrosovich M. 2006. Proteolytic activation of influenza viruses by serine proteases TMPRSS2 and HAT from human airway epithelium. J Virol 80:9896–9898. doi: 10.1128/JVI.01118-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heurich A, Hofmann-Winkler H, Gierer S, Liepold T, Jahn O, Pohlmann S. 2014. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J Virol 88:1293–1307. doi: 10.1128/JVI.02202-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matsuyama S, Ujike M, Morikawa S, Tashiro M, Taguchi F. 2005. Protease-mediated enhancement of severe acute respiratory syndrome coronavirus infection. Proc Natl Acad Sci U S A 102:12543–12547. doi: 10.1073/pnas.0503203102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Belouzard S, Madu I, Whittaker GR. 2010. Elastase-mediated activation of the severe acute respiratory syndrome coronavirus spike protein at discrete sites within the S2 domain. J Biol Chem 285:22758–22763. doi: 10.1074/jbc.M110.103275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wicht O, Burkard C, de Haan CA, van Kuppeveld FJ, Rottier PJ, Bosch BJ. 2014. Identification and characterization of a proteolytically primed form of the murine coronavirus spike proteins after fusion with the target cell. J Virol 88:4943–4952. doi: 10.1128/JVI.03451-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Millet JK, Whittaker GR. 2014. Host cell entry of Middle East respiratory syndrome coronavirus after two-step, furin-mediated activation of the spike protein. Proc Natl Acad Sci USA 111:15214–15219. doi: 10.1073/pnas.1407087111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haga S, Nagata N, Okamura T, Yamamoto N, Sata T, Yamamoto N, Sasazuki T, Ishizaka Y. 2010. TACE antagonists blocking ACE2 shedding caused by the spike protein of SARS-CoV are candidate antiviral compounds. Antiviral Res 85:551–555. doi: 10.1016/j.antiviral.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haga S, Yamamoto N, Nakai-Murakami C, Osawa Y, Tokunaga K, Sata T, Yamamoto N, Sasazuki T, Ishizaka Y. 2008. Modulation of TNF-alpha-converting enzyme by the spike protein of SARS-CoV and ACE2 induces TNF-alpha production and facilitates viral entry. Proc Natl Acad Sci USA 105:7809–7814. doi: 10.1073/pnas.0711241105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Glowacka I, Bertram S, Herzog P, Pfefferle S, Steffen I, Muench MO, Simmons G, Hofmann H, Kuri T, Weber F, Eichler J, Drosten C, Pohlmann S. 2010. Differential downregulation of ACE2 by the spike proteins of severe acute respiratory syndrome coronavirus and human coronavirus NL63. J Virol 84:1198–1205. doi: 10.1128/JVI.01248-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Park JE, Li K, Barlan A, Fehr AR, Perlman S, McCray PB Jr, Gallagher T. 2016. Proteolytic processing of Middle East respiratory syndrome coronavirus spikes expands virus tropism. Proc Natl Acad Sci USA 113:12262–12267. doi: 10.1073/pnas.1608147113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Phillips JJ, Chua MM, Lavi E, Weiss SR. 1999. Pathogenesis of chimeric MHV4/MHV-A59 recombinant viruses: the murine coronavirus spike protein is a major determinant of neurovirulence. J Virol 73:7752–7760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Phillips JJ, Chua MM, Rall GF, Weiss SR. 2002. Murine coronavirus spike glycoprotein mediates degree of viral spread, inflammation, and virus-induced immunopathology in the central nervous system. Virology 301:109–120. doi: 10.1006/viro.2002.1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gallagher TM, Buchmeier MJ, Perlman S. 1992. Cell receptor-independent infection by a neurotropic murine coronavirus. Virology 191:517–522. doi: 10.1016/0042-6822(92)90223-C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bender SJ, Phillips JM, Scott EP, Weiss SR. 2010. Murine coronavirus receptors are differentially expressed in the central nervous system and play virus strain-dependent roles in neuronal spread. J Virol 84:11030–11044. doi: 10.1128/JVI.02688-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miura TA, Travanty EA, Oko L, Bielefeldt-Ohmann H, Weiss SR, Beauchemin N, Holmes KV. 2008. The spike glycoprotein of murine coronavirus MHV-JHM mediates receptor-independent infection and spread in the central nervous systems of Ceacam1a−/− mice. J Virol 82:755–763. doi: 10.1128/JVI.01851-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gallagher TM, Escarmis C, Buchmeier MJ. 1991. Alteration of the pH dependence of coronavirus-induced cell fusion: effect of mutations in the spike glycoprotein. J Virol 65:1916–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oda K, Nishimura Y, Ikehara Y, Kato K. 1991. Bafilomycin A1 inhibits the targeting of lysosomal acid hydrolases in cultured hepatocytes. Biochem Biophys Res Commun 178:369–377. doi: 10.1016/0006-291X(91)91823-U. [DOI] [PubMed] [Google Scholar]
- 39.Burkard C, Verheije MH, Wicht O, van Kasteren SI, van Kuppeveld FJ, Haagmans BL, Pelkmans L, Rottier PJ, Bosch BJ, de Haan CA. 2014. Coronavirus cell entry occurs through the endo-/lysosomal pathway in a proteolysis-dependent manner. PLoS Pathog 10:e1004502. doi: 10.1371/journal.ppat.1004502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Williams RK, Jiang GS, Snyder SW, Frana MF, Holmes KV. 1990. Purification of the 110-kilodalton glycoprotein receptor for mouse hepatitis virus (MHV)-A59 from mouse liver and identification of a nonfunctional, homologous protein in MHV-resistant SJL/J mice. J Virol 64:3817–3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pensiero MN, Dveksler GS, Cardellichio CB, Jiang GS, Elia PE, Dieffenbach CW, Holmes KV. 1992. Binding of the coronavirus mouse hepatitis virus A59 to its receptor expressed from a recombinant vaccinia virus depends on posttranslational processing of the receptor glycoprotein. J Virol 66:4028–4039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zelus BD, Wessner DR, Dveksler GS, Holmes KV. 1998. Neutralization of MHV-A59 by soluble recombinant receptor glycoproteins. Adv Exp Med Biol 440:3–9. [DOI] [PubMed] [Google Scholar]
- 43.Gallagher TM. 1997. A role for naturally occurring variation of the murine coronavirus spike protein in stabilizing association with the cellular receptor. J Virol 71:3129–3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Afar DE, Vivanco I, Hubert RS, Kuo J, Chen E, Saffran DC, Raitano AB, Jakobovits A. 2001. Catalytic cleavage of the androgen-regulated TMPRSS2 protease results in its secretion by prostate and prostate cancer epithelia. Cancer Res 61:1686–1692. [PubMed] [Google Scholar]
- 45.Hingley ST, Leparc-Goffart I, Seo SH, Tsai JC, Weiss SR. 2002. The virulence of mouse hepatitis virus strain A59 is not dependent on efficient spike protein cleavage and cell-to-cell fusion. J Neurovirol 8:400–410. doi: 10.1080/13550280260422703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Simmons G, Bertram S, Glowacka I, Steffen I, Chaipan C, Agudelo J, Lu K, Rennekamp AJ, Hofmann H, Bates P, Pohlmann S. 2011. Different host cell proteases activate the SARS-coronavirus spike-protein for cell-cell and virus-cell fusion. Virology 413:265–274. doi: 10.1016/j.virol.2011.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yamada Y, Liu DX. 2009. Proteolytic activation of the spike protein at a novel RRRR/S motif is implicated in furin-dependent entry, syncytium formation, and infectivity of coronavirus infectious bronchitis virus in cultured cells. J Virol 83:8744–8758. doi: 10.1128/JVI.00613-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bertram S, Dijkman R, Habjan M, Heurich A, Gierer S, Glowacka I, Welsch K, Winkler M, Schneider H, Hofmann-Winkler H, Thiel V, Pohlmann S. 2013. TMPRSS2 activates the human coronavirus 229E for cathepsin-independent host cell entry and is expressed in viral target cells in the respiratory epithelium. J Virol 87:6150–6160. doi: 10.1128/JVI.03372-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rawlings ND, Walter M, Barrett AJ, Bateman A. 2014. MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res 42:D503–D509. doi: 10.1093/nar/gkt953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.UniProt Consortium. 2014. Activities at the Universal Protein Resource (UniProt). Nucleic Acids Res 42:D191–D198. doi: 10.1093/nar/gkt1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chang A, Dutch RE. 2012. Paramyxovirus fusion and entry: multiple paths to a common end. Viruses 4:613–636. doi: 10.3390/v4040613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kuo L, Godeke GJ, Raamsman MJ, Masters PS, Rottier PJ. 2000. Retargeting of coronavirus by substitution of the spike glycoprotein ectodomain: crossing the host cell species barrier. J Virol 74:1393–1406. doi: 10.1128/JVI.74.3.1393-1406.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Masters PS, Rottier PJ. 2005. Coronavirus reverse genetics by targeted RNA recombination. Curr Top Microbiol Immunol 287:133–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.de Haan CA, van Genne L, Stoop JN, Volders H, Rottier PJ. 2003. Coronaviruses as vectors: position dependence of foreign gene expression. J Virol 77:11312–11323. doi: 10.1128/JVI.77.21.11312-11323.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang R, Li Y, Cowley TJ, Steinbrenner AD, Phillips JM, Yount BL, Baric RS, Weiss SR. 2015. The nsp1, nsp13, and M proteins contribute to the hepatotropism of murine coronavirus JHM.WU. J Virol 89:3598–3609. doi: 10.1128/JVI.03535-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Das Sarma J, Fu L, Tsai JC, Weiss SR, Lavi E. 2000. Demyelination determinants map to the spike glycoprotein gene of coronavirus mouse hepatitis virus. J Virol 74:9206–9213. doi: 10.1128/JVI.74.19.9206-9213.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Das Sarma J, Scheen E, Seo SH, Koval M, Weiss SR. 2002. Enhanced green fluorescent protein expression may be used to monitor murine coronavirus spread in vitro and in the mouse central nervous system. J Neurovirol 8:381–391. doi: 10.1080/13550280260422686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hingley ST, Gombold JL, Lavi E, Weiss SR. 1994. MHV-A59 fusion mutants are attenuated and display altered hepatotropism. Virology 200:1–10. doi: 10.1006/viro.1994.1156. [DOI] [PubMed] [Google Scholar]
- 59.Collins AR, Knobler RL, Powell H, Buchmeier MJ. 1982. Monoclonal antibodies to murine hepatitis virus-4 (strain JHM) define the viral glycoprotein responsible for attachment and cell-cell fusion. Virology 119:358–371. doi: 10.1016/0042-6822(82)90095-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Holmes KV, Williams RK, Cardellichio CB, Compton SR, Stephensen CB, Snyder SW, Frana MF, Jiang GS, Smith A, Knobler RL. 1990. Is the 110K glycoprotein the only receptor for MHV and does its expression determine species specificity? Adv Exp Med Biol 276:37–44. doi: 10.1007/978-1-4684-5823-7_6. [DOI] [PubMed] [Google Scholar]