

Abstract
Glioblastoma (GBM) is the most common brain tumor with poor response to current therapeutics. Alisertib (ALS), a second-generation selective Aurora kinase A (AURKA) inhibitor, has shown potent anticancer effects on solid tumors in animal studies. This study aimed to investigate the killing effect of ALS on GBM cell line DAOY and the possible underlying mechanisms using both bioinformatic and cell-based approaches. Our molecular docking showed that ALS preferentially bound AURKA over AURKB via hydrogen bond formation, charge interaction, and π-π stacking. ALS also bound key regulating proteins of cell cycle, apoptosis and autophagy, such as cyclin-dependent kinase 1 (CDK1/CDC2), CDK2, cyclin B1, p27 Kip1, p53, cytochrome C, cleaved caspase 3, Bax, Bcl-2, Bcl-xl, phosphatidylinositol 3-kinase (PI3K), protein kinase B (Akt), mammalian target of rapamycin (mTOR), 5’-adenosine monophosphate-activated protein kinase (AMPK), p38 mitogen-activated protein kinase (MAPK), beclin 1, phosphatase and tensin homolog (PTEN), and microtubule-associated protein light chain 3 (LC3). ALS exhibited potent growth-inhibitory, pro-apoptotic, and pro-autophagic effects on DAOY cells in a concentration-dependent manner. Notably, ALS remarkably induced G2/M arrest mainlyvia regulating the expression of CDK1/CDC2, CDK2, cyclin B1, p27 Kip1, and p53 in DAOY cells. ALS significantly induced the expression of mitochondria-mediated pro-apoptotic proteins such as Baxbut inhibited the expression of anti-apoptotic proteins such as Bcl-2 and Bcl-xl, with a significant increase in the release of cytochrome C and the activation of caspases 3 and 9. ALS also induced PI3K/Akt/mTOR and p38 MAPK signaling pathways while activating the AMPK signaling pathway. Taken together, these findings indicate that ALS exerts a potent inhibitory effect on cell proliferation and induces mitochondria-dependent apoptosis and autophagy with the involvement of PI3K/Akt/mTOR- and p38 MAPK-mediated signaling pathways in DAOY cells. ALS is a promising anticancer agent for GBM treatment.
Keywords: Glioblastoma, alisertib, aurora kinase A, aurora kinase B, molecular docking, hydrogen bond, cell cycle, apoptosis, autophagy, PI3K/Akt/mTOR pathway, DAOY cell
Introduction
Glioblastoma (GBM) arising from astrocytes is the most common and aggressive brain tumor, with a worldwide occurrence rate of 3.4 out of 100,000 people in 2012, age-adjusted using the world standard population and accounting for 70% of all brain tumors [1]. This represented an estimated 139,608 males and 116,605 females diagnosed worldwide with a primary malignant brain tumor in 2012, an overall total of 256,213 individuals. Diffuse intrinsic pontine GBM is 15% of all childhood brain tumors and is the primary cause of brain tumor-related deaths in children. In 2013, the estimated incidence of primary brain cancers in the United States (US) was 23,130, with 14,080 Americans dying from GBM [2]. An estimated 26,070 new cases of primary malignant brain tumors are expected to be diagnosed and an estimated 16,947 deaths will be attributed to primary malignant brain tumors in the US in 2017 [3]. Multifocal GBM has been defined as GBM being found synchronously in multiple foci and there is a presumed microscopic connection. It has a poor survival rate when compared with other brain tumors [4,5]. The multiple lesions made the treatment very complicated since one operation was not enough. Current treatment of GBM utilizes a multidisciplinary coordinated approach usually involving neurosurgery, radiation therapy and chemotherapy [6]. To date, there have only been five drugs and one device approved by the Food and Drug Administration to treat brain tumors. These include everolimus, bevacizumab, carmustine, lomustine, temozolomide, and carmustine implant. The average survival rate for all primary pediatric (0-19 years of age) malignant brain tumors is 73.6%. Although in recent years the diagnosis and treatment have made great progress, the prognosis of patients with high-grade gliomas is still poor, with a median survival of 13-15 months post-diagnosis [7]. Low-grade gliomas are slower growing than their high-grade counterparts, with a median survival of 4.7 to 9.8 years [8]. Additionally, GBM has innate abilities to proliferate extensively, recurrence often occurs approximately 32-36 weeks after initial treatment. Overall survival for GBM patients remains poor with only 15-20% of patients surviving longer than three years [9]. For adults with more aggressive GBM when treated with concurrent temozolamide and radiation therapy, the median survival is about 14.6 months and two-year survival is 30% [5,9]. As such, there is an urgent need for the discovery of potent and safe therapeutics for GBM.
Accumulating evidence has demonstrated that abnormalities in mitosis can provide a source of the genomic instability that is often associated with tumorigenesis, thus targeting cell cycle, mitosis, and its key regulators is a promising strategy to treat cancer [10-12]. Aurora kinases are a family of serine/threonine kinases and consist of three members, Aurora kinase A/B/C (AURKA/B/C) that play a key role in mitosis, with specific functions in cell signaling and mitotic division [13-15]. Among these, AURKA and AURKB play central roles in mitosis, whereas AURKC has a unique role in meiosis. AURKA is expressed in late S phase, peaking at G2/M phase, and declining at G1 phase, which plays an important role in centrosome maturation and separation, bipolar spindle assembly, and mitotic entry [16]. AURKA phosphorylates a variety of proteins, including targeting protein for Xklp2, transforming acidic coiled-coil containing protein 3, Bora, breast cancer 1, M-phase inducer phosphatase 2 (CDC25B), Par-3 family cell polarity regulator, DLG associated protein 5, histone deacetylase 6, the mitotic kinesin Eg5, kinesin family member 2A, and p53, etc. Protein phosphatase type 1 binds AURKA and inhibits its activity by dephosphorylating Thr288 during mitosis, while phosphorylation at Ser342 decreases the kinase activity. AURKB localizes to kinetochores in mitosis and to the midbody during cytokinesis, where it phosphorylates several proteins, including inner centromere protein, histone H3, G-protein subunit-α6, kinesin family member 2C, Sun RNA methyltransferase family member 2, and RacGTPase-activating protein 1. The three members of the Aurora kinases of varying peptide lengths share similar catalytic domains located in the C-terminus, but their N-terminal extensions are of variable length and display little or no similarity. They have a conserved adenosine triphosphate binding pocket but different amino acid sequences at the N-terminal domain. AURKA localized to chromosome 20q13.2, the most characterized member of Aurora kinase family, plays a critical role in chromosome maturation and separation as well as bipolar spindle assembly during G2/M phase of mitosis [17]. High-level expression of AURKA is associated with centrosome amplification, mitotic abnormalities, chromosomal instability, and malignant transformation in cancer development, progression, and metastasis [15,18]. AURKA is a potent oncogene resulting in genetic instability and dedifferentiated morphology. Aberrant expression and activity of AURKA often occur in various malignancies, including the bladder, breast, colon, liver, ovaries, pancreas, stomach, esophagus, and melanoma tumors [18]. One study has demonstrated that in WHO grade II diffuse astrocytomas, grade III anaplastic astrocytomas and grade IV glioblastomas, AURKA protein is upregulated with increased tumor grade [19]. Aurora kinases therefore represent promising targets for anticancer therapeutics and a number of Aurora kinase inhibitors have been developed with some of which currently undergoing clinical evaluation.
Alisertib (ALS, previously known as MLN8237, see Figure 1) is an investigational, orally available, and selective small-molecule AURKA inhibitor [20]. ALS has the ability to selectively inhibit AURKA and thereby induces cell-cycle arrest, aneuploidy, polyploidy, mitotic catastrophe, and cell death. In preclinical studies, ALS exhibited potent AURKA inhibition and high antitumor activity in a wide range of tumor cells [20]. The studies by our laboratory have shown that ALS induces cancer cell apoptosis and autophagy and inhibits epithelial to mesenchymal transition via activation of mitochondrial pathway, up-regulation of p53 and inhibition of phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt)/mammalian target of rapamycin (mTOR) signaling pathway in breast, pancreatic, ovarian, and gastric cancer and leukemia [21-27]. However, there is a lack of evidence for the anticancer effect of ALS in GBM therapy. In this study, we explored the anticancer effect of ALS in GBM via running molecular docking and cell-based assays. The docking was performed to check how ALS bound AURKA and AURKB and to compare the differences in the binding mode with other Aurora kinase inhibitors, including the selective inhibitor of AURKB barasertib (AZD1152-HQPA), the selective AURKA inhibitor MLN8054, the potent inhibitor of AURKA and AURKB and vascular endothelial growth factor receptor 2 CYC116 (4-methyl-5-[2-(4-morpholin-4-ylanilino)pyrimidin-4-yl]-1,3-thiazol-2-amine), and the potent and highly selective pan-AURKA, AURKB and AURKC inhibitors AMG-900 (N-(4-((3-(2-aminopyrimidin-4-yl)pyridin-2-yl)oxy)phenyl)-4-(4-methylthiophen-2-yl)phthalazin-1-amine), danusertib, and tozasertib (VX-680 & MK-0457) (for their chemical structures, see Figure 1). ALS was also docked into selected key regulators of cell cycle, apoptosis and autophagy. To validate the docking findings, we also examined the effects of ALS on cell cycle distribution, apoptosis, and autophagy on DAOY cells and the potential mechanisms.
Figure 1.
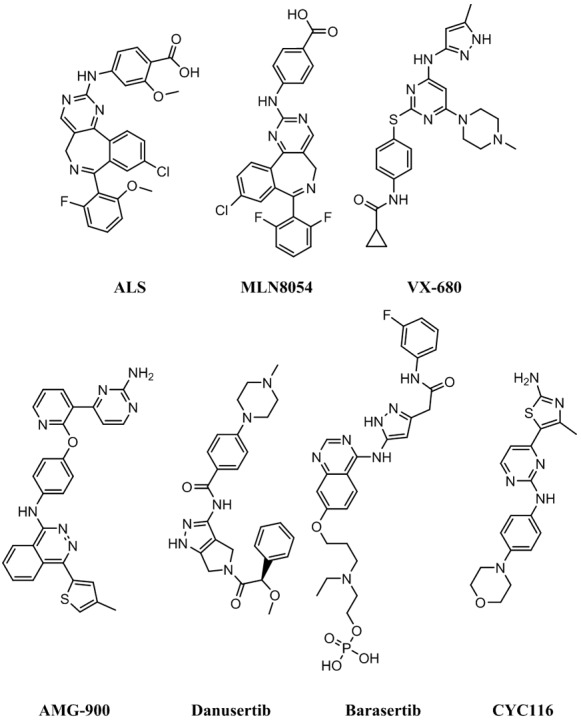
Chemical structures of ALS, MLN8054, tozasertib (VX-680), AMG-900, danusertib, barasertib, and CYC116.
Materials and methods
Chemicals and reagents
ALS (also known as MLN8237) was purchased from Selleckchem Inc. (Houston, TX, USA). The autophagy inhibitors SB202190(4-[4-fluorophenyl]-2-[4-hydroxyphenyl]-5-[4-pyridyl]-1H-imidazole) and wortmannin (WM) were purchased from Invivogen Inc. (San Diego, CA, USA). Dulbecco’s phosphate-buffered saline (PBS), fetal bovine serum (FBS), thiazolyl blue tetrazolium bromide (MTT), dimethyl sulfoxide (DMSO), 7-amino-actinomycin D (7-AAD), propidium iodide (PI), and RNase A were purchased from Sigma-Aldrich Inc. (St Louis, MO, USA). Dulbecco’s Modified Eagle’s Medium (DMEM) was purchased from Corning Cellgro Inc. (Herndon, VA, USA). The AnnexinV: phycoerythrinv (PE) apoptosis detection kit was purchased from BD Bio-sciences Inc. (San Jose, CA, USA). A Cyto-ID® autophagy detection kit was obtained from Enzo Life Sciences Inc. (Farmingdale, NY, USA). The polyvinylidene difluoride membrane was sourced from Millipore Inc. (Bedford, MA, USA). The pierce bicinchoninic acid (BCA) protein assay kit, skim milk, and Western blotting substrate were all obtained from Thermo Scientific Inc. (Hudson, NH, USA).
Primary antibodies against human AURKA, phosphorylated (p)-AURKA at Thr288, cyclin-dependent kinase 1 (CDK1/CDC2), CDK2, cyclin B1, p27 Kip1, p53, phosphorylated (p)-p53 at Lys379, cytochrome C, cleaved caspase 3, cleaved caspase 9, Bcl-2-like protein4/Bcl-2-associated X protein (Bax), B-cell lymphoma 2 (Bcl-2), B-cell lymphoma-extra-large (Bcl-xl), the p53 upregulated modulator of apoptosis (PUMA), phosphatidylinositide 3-kinase (PI3K), phosphorylated (p)-PI3K at Tyr458, protein kinase A (Akt), phosphorylated (p)-Akt at Ser473, mammalian target of rapamycin (mTOR), phosphorylated (p)-mTOR at Ser2448, the AMP-activated protein kinase (AMPK), phosphorylated (p)-AMPK at Thr172, p38 MAPK, phosphorylated (p)-38 MAPK at Thr180/Tyr182, beclin 1, phosphatase and tensin homolog (PTEN), microtubule-associated protein light chain 3 (LC3)-I and LC3-II were all purchased from Cell Signaling Technology Inc. (Beverly, MA, USA). The antibody against human β-actin was obtained from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA).
Molecular docking
In order to determine the molecular interactions between the target protein and Aurora kinase inhibitors, we employed the Discovery Studio® program 3.1 designed by Accelrys Inc. (San Diego, CA, USA). The 3D crystal protein structures of AURKA and AURKB were obtained from the Research Collaborator for Structural Bioinformatics Protein Data Bank (PDB; http://www.rcsb.org/pdb/). ALS, barasertib, CYC116, AMG-900, danusertib, and VX-680 were docked into the active sites of AURKA (PDB ID: 4UTD) and AURKB (PDB ID: 4AF3) as previously described by us [28-36]. The 2D structures of the ligands were retrieved from the PubChem database (Figure 1). The CDOCKER interaction energy (CIE) was calculated. A lowCIE may reflect a high-affinity binding.
There were five key regulators of cell cycle for the molecular docking experiments, including human CDK1/CDC2 (PDB ID: 4YC6), CDK2 (PDB ID: 3L2O), cyclin B1 (PDB ID: 2JGZ), cyclin dependent kinase inhibitor 1B/p27 Kip1 (PDB ID: 2AST), and p53 (PDB ID: 5A7B); seven key regulators of apoptosis for the molecular docking experiments, including human cytochrome C (PDB ID: 4Y6I), cleaved caspase 3 (PDB ID: 4PRY), cleaved caspase 9 (PDB ID: 4RHW), Bax (PDB ID: 4ZII), Bcl-2 (PDB ID: 3BL2), Bcl-xl (PDB ID: 3MXO), and PUMA (PDB ID: 4BPI); eight key regulators of autophagy for the molecular docking experiments, including human PI3K (PDB ID: 4WAF), Akt (PDB ID: 4GV1), mTOR (PDB ID: 4DRH), AMPK (PDB ID: 4YEF), p38 MAPK (PDB ID: 3D83), beclin 1 (PDB ID: 3Q8T), PTEN (PDB ID: 5BZZ), and LC3 (PDB ID: 4ZDV).
The proteins and ligands were prepared prior to docking. The preparation of proteins were cleaned, modified, and prepared for defining and editing the binding site. During preparation for ALS, MLN8054, tozasertib (VX-680), AMG-900, danusertib, barasertib, and CYC116, duplicate structures were deleted; and ionization change, tautomer or isomer generation, Lipinski filter and 3D generator were all set “true”. A harmonic potential with the force constant of 300 kcal/mol was applied outside the grid boundary. Following the preparation of proteins, ligands and grid setting, ALS, MLN8054, tozasertib (VX-680), AMG-900, danusertib, barasertib, and CYC116 were docked into binding sites of AURKA and AURKB and ALS were docked into binding sites of the key regulators involved in cell cycle, apoptosis, and autophagy. Electrostatic energy and van der Waals forces were considered during the docking process. For each defined van der Waals force or electrostatic probe, the interactions with all protein atoms were stored at predetermined grid points. For ligand atoms located between grid points, a trilinear interpolation was used to approximate the energies.
Cell line and cell culture
The DAOY cell line (ATCC® HTB186™) was obtained from the American Type Culture Collection (Manassas, VA, USA) and cultured in DMEM medium supplemented with 10% heat-inactivated FBS and 1% penicillin/streptomycin. The cells were maintained in a 5% CO2/95% air humidified incubator at 37°C. ALS was dissolved in DMSO at a stock concentration of 50 mM and stored at -20°C. It was freshly diluted to the desired concentration with culture medium. The final concentration of DMSO was at 0.05% (v/v). The control cells received the vehicle only.
Cell viability assay
The MTT assay was performed to examine the effect of ALS on cell viability. Briefly, DAOY cells were seeded into a 96-well culture plate at a density of 8 × 103 cells/well. After 24 h incubation, the cells were treated with ALS at concentrations ranging from 0.01-50 μM for 24 or 48 h. Following the drug treatment, 10 μL of MTT stock solution (5 mg/mL) was added to each well and incubated for 2-4 h. Then, the medium was carefully removed and 100 μL DMSO was added into each well. The plate was placed on a rocker to mix the solution thoroughly for 30 seconds and incubated at 37°C for 10 min. The absorbance was measured using a Synergy™ H4 Hybrid micro plate reader (BioTek Inc., Winooski, VT, USA) at wavelengths of 560 nm (MTT formazan) and 670 nm (background). The concentration required for IC50 value was determined using the relative viability over ALS concentration curve and nonlinear regression analysis using GraphPad Prism 5.0 software (San Diego, CA, USA). The experiment was performed at least three times.
Flow cytometry
The effect of ALS on cell cycle distribution of DAOY cells was examined using flow cytometry. Briefly, DAOY cells were treated with ALS at 0.1, 1, and 5 μM for 24 h. In separate experiments, DAOY cells were treated with 1 μM ALS for 4, 8, 12, 24, 48, and 72 h. After treated with ALS, cells were trypsinized and fixed in 70% ethanol at -20°C overnight. Following the fixation, cells were collected and resuspended in 1 mL of PBS containing 1 mg/mL ribonuclease A and 50 µg/mL PI. Thereafter, cells were incubated in the dark for 30 min at room temperature. A total number of 1 × 104 cells were subject to cell cycle analysis using a flow cytometer (BD LSR II Analyzer, Becton Dickinson Immunocytometry Systems, San Jose, CA, USA).
Quantification of cellular apoptosis
The effect of ALS on the apoptosis of DAOY cells was quantified using the annexin V: PE apoptosis detection kit according to the manufacturer’s instructions. Annexin V is a 35-36 kDa Ca2+-dependent phospholipid binding protein that has a high affinity for negatively charged phospholipid phosphatidylserine and binds to cells that are actively undergoing apoptosis with exposed phospholipid phosphatidylserine. DAOY cells were plated in six-well cell culture plates at a density of 1 × 105 cells per well. After incubation overnight, the cells were incubated with ALS at 0.1, 1, and 5 μM for 24 h. Then cells were trypsinized and washed twice with 4°C PBS. A quota of cell suspension (100 μL) was transferred into a 5 mL clean test tube and incubated with 5 μL PE annexin V and 5 μL 7-AAD (a vital nucleic acid dye) in the dark for 15 min at room temperature. A quota of 1 × binding buffer (250 μL) was then added to each test tube and the number of apoptotic cells was counted using a flow cytometer within 1 h.
Determination of cellular autophagy
To determine the effect of ALS on autophagy in DAOY cells, the intracellular autophagy level was examined using flow cytometry as previous described [37,38]. Briefly, the cells were treated with fresh medium alone, control vehicle alone (0.05% DMSO, v/v), or ALS at 0.1, 1, and 5 μM. After 24 h of incubation, the cells were trypsinized and collected with 1 × assay buffer containing 5% FBS. To investigate the mechanisms for ALS-induced autophagy, cells were pre-treated with 10 μM SB202190 or 10 mM WM for 1 h in DAOY cells, then co-treated with 1 μM ALS for a further 24 h. Groups of cells treated with each of these compounds alone were also included. All inhibitors were dissolved in DMSO at a final concentration of 0.05% (v/v). Cells were resuspended in 250 µL of phenol red free culture medium (Thermo Fisher Scientific Inc.; No 1294895) containing 5% FBS and 250 µL of the diluted Cyto-ID® green stain solution (Enzo Life Sciences Inc.; No: ENZ-51031-K200) was added to each sample and mixed well. Cells were incubated for 30 min at 37°C in the dark. Following the incubation, cells were collected by centrifugation at 250 × g for 3 min, washed with 1 × assay buffer, resuspended in 500 µL fresh 1 × assay buffer containing 5% FBS and subject to flow cytometric analysis.
Confocal fluorescence microscopy for autophagy detection
To further detect the autophagy-inducing effect of ALS on DAOY cells, the confocal microscopic examination was performed. The assay provides a rapid and quantitative approach to monitor autophagy in live cells without the need for cell transfection and allows the measurement and differentiation between autophagic flux and autophagolysosome accumulation [39]. DAOY cells were seeded into 8-well chamber slides. The cells were treated with ALS at 0.1, 1, and 5 μM for 24 h. In separate experiments, to investigate the mechanisms for ALS-induced autophagy, cells were pretreated with 10 μM SB202190 or 10 μM WM, then co-treated with 1 μM ALS for a further 24 h. When cells reached ~70% confluence, they were washed with 1 × assay buffer provided in the Cyto-ID® autophagy detection kit. The kit was used to measure cellular autophagic vacuoles. Following the incubation with 100 μL microscopy dual detection reagent for 30 min at 37°C in the dark, the cells were washed with 200 μL fresh 1 × assay buffer containing 5% FBS to remove unbound detection reagent. The slides were covered with glass cover slips and sealed with polish oil. Samples were examined using a Leica TCS SP2 laser scanning confocal microscope (Leica Microsystems, Wetzlar, Germany) using a standard fluorescein isothiocyanate filter set for imaging the autophagic signal at wavelengths of 405/488 nm.
Western blot analysis
The expression level of various cellular proteins involved in cell cycle, apoptosis, and autophagy was determined using Western blot assays. DAOY cells was washed with pre-cold PBS after 24 h treatment with ALS at 0.1, 1, and 5 μM, lysed on ice with radioimmunoprecipitation assay (RIPA) buffer containing the phosphatase inhibitor and protease inhibitor cocktail and centrifuged at 3000 × g for 15 min at 4°C. The supernatant was collected and the protein concentrations were measured using a Pierce™ BCA protein assay kit. An equal amount of protein sample (30 µg) was resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) sample loading buffer and electrophoresed on 7% or 12% SDS-PAGE mini-gel after thermal denaturation at 95°C for 5 min. The proteins were transferred onto polyvinylidene difluoride membrane at 400 mA for 2 h at 4°C. The membranes were probed with indicated primary antibody overnight at 4°C and then blotted with appropriate horseradish peroxidase-conjugated secondary anti-mouse or anti-rabbit antibody. Visualization was performed using a ChemiDoc™ XRS system (Bio-Rad, Hercules, CA, USA) with an enhanced chemiluminescence kit (Thermal Scientific) and the blots were analyzed using Image Lab 3.0 (Bio-Rad). The protein level was normalized to the matching densitometric value of the internal control β-actin.
Statistical analysis
Data are presented as the mean ± standard deviation (SD). Comparisons of multiple groups were evaluated by one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison procedure. Values of P < 0.05 were considered statistically different. Assays were performed at least three times independently.
Results
Molecular interactions
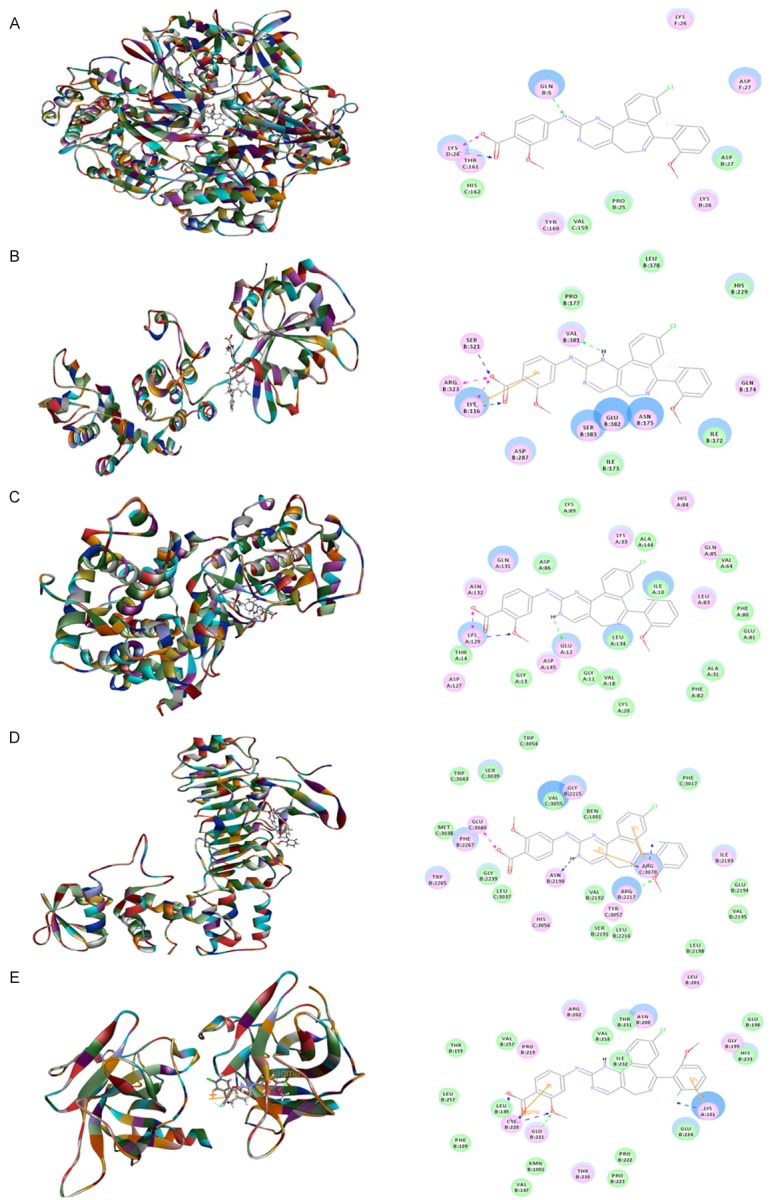
To explore how ALS interacts with AURKA and AURKB and to compare the differences in molecular interactions between ALS and other Aurora kinase inhibitors, we first carried out docking experiments using the Discovery Studio program 3.1. Each compound-enzyme complex with the highest CIE was selected and the 2D and 3D pictures of these complexes collected.
After docking ALS into the active sites of AURKA (PDB: 4UTD, residues 122-403) and AURKB (PDB: 4AF3, residues 55-344), there were 50 and 16 conformations generated for ALS-AURKA and ALS-AURKB interactions, respectively. There was a remarkable difference in CIEs between ALS-AURKA (-64.6 kcal/mol) and ALS-AURKB (-25.3 kcal/mol) binding. The docking data are shown in Figure 2 and Table 1. ALS bound AURKA through formation of hydrogen bond at Trp277, charge interaction at Lys162 and Asp274, and π-π stacking at Glu260 (Figure 2A and Table 1). ALS also bound AURKB via hydrogen bond at Pro226 and charge interaction at Arg230 (Figure 2F and Table 1).
Figure 2.

Molecular docking of ALS, MLN8054, tozasertib (VX-680), AMG-900, danusertib, barasertib, and CYC116 into the active sites of human AURKA (PDB ID: 4UTD) and AURKB (PDB ID: 4AF3). ALS (A), MLN8054 (B), tozasertib (VX-680) (C), AMG-900 (D), and danusertib (E) were readily docked into the active sites of AURKA. ALS (F), MLN8054 (G), tozasertib (VX-680) (H), barasertib (I), and CYC116 (J) were readily docked into the active sites of AURKB.
Table 1.
Molecular interactions between ALS, MLN8054, tozasertib (VX-680), AMG-900, danusertib, barasertib, and CYC116 with human AURKA and AURKB
| Ligand | CIE (kcal/mol) | H-bond number | Residues involved in H-bond formation | Charge interaction | Residues involved in charge interaction | π-π stacking | Residues involved in π-π stacking | Conformation number |
|---|---|---|---|---|---|---|---|---|
| AURKA (PDB ID: 4UTD) | ||||||||
| Alisertib | -64.625 | 1 | N-Trp277 | 3 | O-Lys162 | 1 | Glu260 | 50 |
| O-Asp274 | ||||||||
| MLN8054 | -37.9176 | 1 | O-Lys162 | 3 | O-Lys162 | 1 | Lys143 | 51 |
| O-Glu181 | ||||||||
| O-Asp274 | ||||||||
| Tozasertib (VX-680) | -56.9129 | 6 | O-Lys162 | 0 | - | 4 | Phe144 | 367 |
| H-Glu181 | Lys162 | |||||||
| H-Asp256 | Lys258 | |||||||
| N-Asn261 | Trp277 | |||||||
| H-Asp274 | ||||||||
| AMG-900 | -45.4247 | 3 | N-Lys258 | 0 | - | 2 | Leu139 | 90 |
| O-Lys258 | Lys162 | |||||||
| Danusertib | -51.3649 | 3 | N-Lys162 | 0 | - | 1 | Trp277 | 93 |
| O-Lys258 (2) | ||||||||
| Barasertib | Failed to dock | |||||||
| CYC116 | Failed to dock | |||||||
| AURKB (PDB ID: 4AF3) | ||||||||
| Alisertib | -25.2975 | 1 | H-Pro226 | 1 | O-Arg230 | 0 | - | 16 |
| MLN8054 | -38.4163 | 1 | H-Ser275 | 1 | O-Lys202 | 0 | - | 50 |
| Tozasertib (VX-680) | -43.7821 | 0 | - | 1 | N-Glu274 | 0 | - | 183 |
| Barasertib | 49.5692 | 1 | H-Glu265 | 1 | N-Lys202 | 0 | - | 6 |
| CYC116 | -50.3493 | 1 | H-Glu265 | 1 | N-Glu265 | 0 | - | 360 |
| AMG-900 | Failed to dock | |||||||
| Danusertib | Failed to dock | |||||||
Five key regulators of cell cycle for the molecular docking experiments, including human CDK1/CDC2 (PDB ID: 4YC6), CDK2 (PDB ID: 3L2O), cyclin B1 (PDB ID: 2JGZ), p27 Kip1 (PDB ID: 2AST), and p53 (PDB ID: 5A7B). Abbreviation: -, none; AURKA, Aurora kinase A; AURKB, Aurora kinase B; CIE, CDOCKER Interaction Energy; PDB, Protein Data Bank.
Following prediction and validation of the molecular targets, we examined the possible interactions between MLN8054, tozasertib (VX-680), AMG-900, danusertib, barasertib, and CYC116 into the active sites of AURKA and AURKB (Figure 2 and Table 1). There were 51 and 50 conformations generated for MLN8054-AURKA and MLN8054-AURKB binding, respectively. MLN8054 bound AURKA by hydrogen bond formation at Lys162, charge interaction at Lys162, Glu181, and Asp274, and π-π stacking with Lys143 (Figure 2B). MLN8054 also formed a hydrogen bond with Ser275 and charge interaction with Lys202 at the active site of AURKB (Figure 2G). The CIE was -37.9 and -38.4 kcal/mol for MLN8054-AURKA and MLN8054-AURKB binding, respectively. Tozasertib (VX-680) bound AURKA via charge interaction at Lys162, Asn261, Glu181, Asp274, and Asp256, and π-π stacking at Phe144, Lys162, Lys258, and Trp277 with a CIE of -56.9 kcal/mol and bound AURKB through charge interaction at Glu274 with a CIE of -43.8 kcal/mol (Figure 2C and 2H). There were 367 and 183 conformations for tozasertib-AURKA and tozasertib-AURKB binding, respectively.
AMG-900 bound AURKA with 90 conformations involving hydrogen bond formation at Lys258, and π-π stacking at Leu139 and Lys162 with a CIE of 45.4 kcal/mol (Figure 2D). Danusertib had 93 conformations at the active site of AURKA involving hydrogen bond formation at Lys162 and Lys258 and π-π stacking at Trp277 with a CIE of 51.4 kcal/mol (Figure 2E). There were 6 conformations for barasertib-AURKB binding. Barasertib bound AURKB via hydrogen bond formation at Glu265 and charge interaction at Lys202 (Figure 2I). The CIE of barasertib-AURKB binding was 49.6 kcal/mol. CYC116 bound AURKB through hydrogen bond formation and charge interaction at Glu265 (Figure 2J). The CIE of CYC116-AURKB was -50.3 kcal/mol. There were 360 conformations produced for CYC116-AURKB binding. In addition, the docking results showed that there was no position generated for barasertib and CYC116 with AURKA, while AMG-900 and danusertib did not bind to AURKB.
Molecular interactions of ALS with key cell cycle regulators
To explore the mechanism for the ALS inducing effect on cell cycle distribution in DAOY cells, we examined the possible binding modes of ALS with human CDK1/CDC2 (PDB ID: 4YC6, residues 1-297), CDK2 (PDB ID: 3L2O, residues 55-387), cyclin B1 (PDB ID: 2JGZ, residues 167-426), p27 Kip1 (PDB ID: 2AST, residues 89-424), and p53 (PDB ID: 5A7B, residues 94-312). ALS was easily docked into the active sites of human CDK1/CDC2, CDK2, cyclin B1, p27 Kip1, and p53 (Figure 3). As shown in Table 2, the CIE ranged from -43.3 to -178.6 kcal/mol between ALS and human CDK1/CDC2, CDK2, cyclin B1, p27 Kip1, and p53. ALS bound CDK1/CDC2 via formation of a hydrogen bond with Gln5, three hydrogen bonds with Lys26 and two charge interactions with Lys26 at the active site (Figure 3A). ALS bound CDK2 via formation of three hydrogen bonds at Lys116, Ser321, and Val381, two charge interactions at Lys116 and Arg323, and one π-π interaction at Lys116 (Figure 3). ALS bound cyclin B1 via a hydrogen bond with Glu12 and two hydrogen bonds with Lys129 (Figure 3C). ALS bound p27 Kip1 (Figure 3D). In addition, ALS bound p53 via formation of two π-π interactions at Lys101 and Cys220 and five hydrogen bonds at Lys101, Cys220, and Glu221 (Figure 3E).
Figure 3.
Molecular interactions between ALS and human CDK1/CDC2, CDK2, cyclin B1, p27 Kip1, and p53. A. CDK1/CDC2 (PDB ID: 4YC6); B. CDK2 (PDB ID: 3L2O); C. cyclin B1 (PDB ID: 2JGZ); D. p27 Kip1 (PDB ID: 2AST); and E. p53 (PDB ID: 5A7B).
Table 2.
Molecular interactions between ALS with human CDK1/CDC2, CDK2, cyclin B1, p27 Kip1, and p53
| Protein | (CIE) kcal/mol) | H-bond number | Residues involved in H-bond formation | Charge interaction | Residues involved in charge interaction | π-π stacking | Residues involved in π-π stacking | Conformation number |
|---|---|---|---|---|---|---|---|---|
| CDK1/CDC2 (4YC6) | -43.322 | 4 | N-Gln5 | 2 | O-Lys26 (2) | 0 | - | 50 |
| O-Lys26 (3) | ||||||||
| CDK2 (3L2O) | -55.0356 | 3 | O-Lys116 | 2 | O-Lys116 | 1 | Lys116 | 50 |
| O-Ser321 | O-Arg323 | |||||||
| H-Val381 | ||||||||
| Cyclin B1 (2JGZ) | -63.3496 | 3 | H-Glu12 | 0 | - | 0 | - | 50 |
| O-Lys129 (2) | ||||||||
| p27 Kip1 (2AST) | -52.5836 | - | - | - | - | - | 50 | |
| p53 (5A7B) | -178.584 | 5 | F-Lys101 | 0 | - | 2 | Lys101 | 1 |
| O-Cys220 (3) | Cys220 | |||||||
| O-Glu221 |
ALS was docked into the active sites of CDK1/CDC2 (PDB ID: 4YC6), CDK2 (PDB ID: 3L2O), cyclin B1 (PDB ID: 2JGZ), p27 Kip1 (PDB ID: 2AST), and p53 (PDB ID: 5A7B). Abbreviation: -, none; CIE, CDOCKER Interaction Energy.
Molecular interactions of ALS with key apoptotic pathway regulators
Next, we evaluated the molecular interactions between ALS and several key regulating proteins of apoptosis including cytochrome C (PDB ID: 4Y6I, residues 5-112), cleaved caspase 3 (PDB ID: 4PRY, residues 1-277), cleaved caspase 9 (PDB ID: 4RHW, residues 1-100), Bax (PDB ID: 4ZII, residues 1-170), Bcl-2 (PDB ID: 3BL2, residues 5-135), Bcl-xl (PDB ID: 3MXO, residues 90-289), and PUMA (PDB ID: 4BPI, residues 132-152) using a computational approach. ALS was easily docked into the active sites of all these proteins (Figure 4). As shown in Table 3, the CIE ranged from -29.9 to 139.0 kcal/mol. ALS bound cytochrome C via formation of five hydrogen bonds at Gln87, Thr17, Ser48, Lys67, and Ser48, charge interaction at Lys67, and three π-π interactions at His84, Tyr50, and Trp106 (Figure 4A). ALS bound cleaved caspase 3 via formation of π-π interaction at Tyr274, hydrogen bond at Met39, and charge interaction at Tyr274 (Figure 4B). ALS bound cleaved caspase 9 via formation of four π-π interactions at Arg44, Arg52, Arg45, and Arg45, two hydrogen bonds at Arg56 and Arg44, and three charge interactions at Arg56, Arg44, and Asp53 (Figure 4C). ALS interacted with Bax via formation of hydrogen bond with Glu44 and charge interaction with Asp33 at the active site (Figure 4D). ALS bound Bcl-2 via formation of hydrogen bond at Lys19 (Figure 4E). In addition, ALS bound Bcl-xl via two hydrogen bonds at Arg118 and Glu177, one charge interaction at Arg235, and two π-π interactions at Arg118 and Arg152 (Figure 4F). ALS bound PUMA via formation of four hydrogen bonds at LYS87, Arg91, Lys87, and Arg91, two charge interactions at Arg91, and four π-π interactions at Arg91, Lys87, Lys87 and Lys87 (Figure 4G).
Figure 4.

Molecular interactions between ALS and human cytochrome C, cleaved caspase 3, cleaved caspase 9, Bax, Bcl-2, Bcl-xl, and PUMA. A. Cytochrome C (PDB ID: 4Y6I); B. cleaved caspase 3 (PDB ID: 4PRY); C. Cleaved caspase 9 (PDB ID: 4RHW); D. BAX (PDB ID: 4ZII); E. Bcl-2 (PDB ID: 3BL2); F. Bcl-xl (PDB ID: 3MXO); G. PUMA (PDB ID: 4BPI).
Table 3.
Molecular interactions between ALS with human cytochrome C, cleaved caspase 3, cleaved caspase 9, Bax, Bcl-2, Bcl-xl, and PUMA
| Protein | CIE (kcal/mol) | H-bond number | Residues involved in H-bond formation | Charge interaction | Residues involved in charge interaction | π-π stacking | Residues involved in π-π stacking | Conformation number |
|---|---|---|---|---|---|---|---|---|
| Cytochrome C (4Y6I) | -38.8443 | 5 | O-Thr17 | 1 | O-Lys67 | 3 | Tyr50 | 49 |
| O-Ser48 (2) | His84 | |||||||
| O-Lys67 | Trp106 | |||||||
| N-Gln87 | ||||||||
| Cleaved caspase 3 (4PRY) | 139.022 | 1 | H-Met39 | 1 | O-Lys82 | 1 | Tyr274 | 2 |
| Cleaved caspase 9 (4RHW) | -54.0237 | 2 | H-Agr44 | 3 | O-Arg44 | 4 | Arg44 | 50 |
| O-Arg56 | O-Asp53 | Arg45 (2) | ||||||
| O-Arg56 | Arg52 | |||||||
| Bax (4ZII) | -33.2721 | 1 | H-Glu44 | 1 | O-Asp33 | 0 | - | 51 |
| Bcl-2 (3BL2) | -45.5841 | 1 | O-Lys19 | 0 | - | 0 | - | 50 |
| Bcl-xl (3MXO) | --48.6738 | 2 | O-Arg118 | 1 | O-Arg235 | 2 | Arg118 | 32 |
| H-Glu177 | Arg152 | |||||||
| PUMA (4BPI) | -29.8631 | 4 | O-Lys87 | 2 | O-Arg91 (2) | 4 | Lys87 (3) | 15 |
| O-Arg91 (2) | Arg91 |
Seven key regulators of apoptosis for the molecular docking experiments, including human cytochrome C (PDB ID: 4Y6I), cleaved caspase 3 (PDB ID: 4PRY), cleaved caspase 9 (PDB ID: 4RHW), Bax (PDB ID: 4ZII), Bcl-2 (PDB ID: 3BL2), Bcl-xl (PDB ID: 3MXO), and PUMA (PDB ID: 4BPI). Abbreviation: -, none; CIE, CDOCKER Interaction Energy.
Molecular interactions of ALS with key autophagic pathway regulators
Following the computational prediction of the molecular targets of ALS, we further explored the possible binding modes of ALS with key regulating proteins involved in autophagy. ALS was easily docked into the active sites of human PI3K (PDB ID: 4WAF, residues 2-1068), Akt (PDB ID: 4GV1, residues 144-480), mTOR (PDB ID: 4DRH, residues 2025-2114), AMPK (PDB ID: 4YEF, residues 76-156), p38 MAPK (PDB ID: 3D83, residues 1-360), beclin 1 (PDB ID: 3Q8T, residues 174-264), PTEN (PDB ID: 5BZZ, residues 14-351), and LC3 (PDB ID: 4ZDV, residues 2-120) (Figure 5). As shown in Table 4, the CIE ranged from -65.0 to -26.5 kcal/mol. ALS bound PI3K via formation of four hydrogen bonds with Arg348, Arg373, Lys942, and Gln1014, three charge interactions with Arg348, Arg349, and Arg373, and three π-π interactions at Arg348, Lys942, and Lys943 (Figure 5A). ALS bound Akt via two charge interactions at Lys179 and Asp292 and two π-π interactions at Lys276 and His354 (Figure 5B). ALS bound mTOR via formation of five hydrogen bonds at Arg2042, Lys2066, and Glu2067, one charge interaction at Lys2066, and one π-π interaction at Arg2060 (Figure 5C). ALS was docked into AMPK via formation of two hydrogen bonds at Lys102, one charge interaction with Lys102, and one π-π interaction at Ala114 (Figure 5D). ALS bound p38 MAPK via formation of one hydrogen bond at Lys152, one charge interaction at Lys152, and two π-π stacking interactions at Arg67 and Arg149 (Figure 5E). ALS bound beclin 1 via formation of four hydrogen bonds at Gly172, Ser173, and Asp174, and one charge interaction at Gly172 (Figure 5F). ALS bound PTEN via formation of charge interaction at Asp252 (Figure 5G). In addition, ALS bound LC3 via formation of one hydrogen bond at Lys42 and two charge interactions at Arg10 and Lys49 (Figure 5H).
Figure 5.

Molecular interactions between ALS and human PI3K, Akt, mTOR, AMPK, p38 MAPK, Beclin 1, PTEN, and LC3. A. PI3K (PDB ID: 4WAF); B. Akt (PDB ID: 4GV1); C. mTOR (PDB ID: 4DRH); D. AMPK (PDB ID: 4YEF); E. p38 MAPK (PDB ID: 3D83); F. Beclin 1 (PDB ID: 3Q8T); G. PTEN (PDB ID: 5BZZ); H. LC3 (PDB ID: 4ZDV).
Table 4.
Molecular interactions between ALS with human PI3K, Akt, mTOR, AMPK, p38 MAPK, beclin 1, PTEN, and LC3
| Protein | CIE (kcal/mol) | H-bond number | Residues involved in H-bond formation | Charge interaction | Residues involved in charge interaction | π-π stacking | Residues involved in π-π stacking | Conformation number |
|---|---|---|---|---|---|---|---|---|
| PI3K (4WAF) | -61.6782 | 5 | N-Arg348 (2) | 3 | O-Arg348 | 3 | Arg348 | 50 |
| O-Arg373 | O-Asp349 | Lys942 | ||||||
| O-Lys942 | O-Arg373 | Lys943 | ||||||
| H-Gln1014 | ||||||||
| Akt (4GV1) | -33.1251 | 2 | - | 2 | O-Lys179 | 2 | Lys276 | 1 |
| O-Asp292 | His354 | |||||||
| mTOR (4DRH) | -55.8744 | 5 | O-Arg2042 | 1 | O-Lys2066 | 1 | Arg2060 | 50 |
| O-Lys2066 (3) | ||||||||
| H-Glu2067 | ||||||||
| AMPK (4YEF) | -65.0290 | 2 | O-Lys102 (2) | 1 | O-Lys102 | 1 | Ala114 | 50 |
| p38 MAPK (3D83) | -57.4358 | 1 | O-Lys152 | 1 | O-Lys152 | 2 | Arg67 | 50 |
| Arg149 | ||||||||
| Beclin 1 (3Q8T) | -26.4804 | 4 | O-Gly172 (2) | 1 | O-Gly172 | 0 | - | 50 |
| O-Ser173 | ||||||||
| O-Asp174 | ||||||||
| PTEN (5BZZ) | -51.7995 | 0 | - | 1 | O-Asp252 | 0 | - | 1 |
| LC3 (4ZDV) | -52.3843 | 1 | O-Lys42 | 2 | O-Arg10 | 0 | - | 15 |
| O-Lys49 |
Eight key regulators of autophagy for the molecular docking experiments, including human PI3K (PDB ID: 4WAF), Akt (PDB ID: 4GV1), mTOR (PDB ID: 4DRH), AMPK (PDB ID: 4YEF), p38 MAPK (PDB ID: 3D83), beclin 1 (PDB ID: 3Q8T), PTEN (PDB ID: 5BZZ), and LC3 (PDB ID: 4ZDV). Abbreviation: -, none; CIE, CDOCKER Interaction Energy.
ALS inhibits the proliferation of in DAOY cells
To examine the effect of ALS on the viability of DAOY cells, we first tested the cell viability using the MTT assay. Incubation of cells with ALS at concentrations ranging from 0.01 to 50 μM for 24 and 48 h significantly decreased the cell viability. The concentration-dependent inhibitory effects of ALS on the growth of DAOY cells are shown in Figure 6A. In comparison to the control cells (100%), the viability was 97.5%, 93.0%, 85.2%, 78.8%, 61.4%, 57.6%, and 4.2% when DAOY cells were treated with ALS at 0.01, 0.1, 0.5, 1, 5, 10, and 50 μM, respectively, for 24 h. The viability was 94.8%, 90.8%, 57.6%, 19.0%, 8.8%, 1.7%, and 1.1% when DAOY cells were treated with ALS at 0.01, 0.1, 0.5, 1, 5, 10, and 50 μM, respectively, for 48 h. The 50% inhibition of growth (IC50) values were 8.3 μM for DAOY cells after 24 h and 4.8 μM for DAOY cells after 48 h incubation with ALS.
Figure 6.

Effects of ALS on the proliferation, expression of p-AURKA and AURKA, and cell cycle in DAOY cells. Cells were treated with ALS at concentrations of 0.1, 1, and 5 µM for 24 h or 1 µ Mover 72 h, then the protein samples were subject to Western blotting assay and cells were subject to flow cytometry analysis. A. Cell viability was determined by the MTT assay. B. Representative blots for p-AURKA and AURKA in DAOY cells. C. Bar graphs showing the relative expression level of p-AURKA, AURKA, and p-AURKA/AURKA in DAOY cells. D. Representative fluorescence histograms showing the effect on cell cycle distribution of DAOY cells treated with ALS at 0.1, 1, and 5 µM for 24 h. E. Bar graphs showing the percentage of DAOY cells in G1, S, and G2/M phases when treated with ALS at 0.1, 1, and 5 µM for 24 h. F. Representative fluorescence histograms showing the effect on cell cycle distribution of DAOY cells when treated with 1 µM ALS at 4, 8, 12, 24, 48, and 72 h. G. Bar graphs showing the percentage of DAOY cells in G1, S, and G2/M phases when treated with 1 µM ALS over 72 h. *P < 0.05 and ***P < 0.001.
ALS downregulates the phosphorylation of AURKA in DAOY cells
ALS is a selective inhibitor of AURKA that plays a critical role cell mitosis. We examined the effect of ALS on the p-AURKA and AURKA in DAOY cells (Figure 6B). As shown in Figure 6C, in comparison to the control, there was a 7.7%, 27.8%, and 33.8% reduction in the level of p-AURKA at Thr288 when DAOY cells were treated with ALS at 0.1, 1, and 5 μM for 24 h, respectively (P < 0.05 or 0.001). Similarly, the ratio of p-AURKA/AURKA was decreased 42.4%, 51.7%, and 40.5% (P < 0.001). However, there was a 61.5.0%, 54.7%, and 14.0% increase in the level of AURKA when treated with ALS at 0.1, 1, and 5 μM for 24 h (P < 0.05 or 0.001). It is unknown why ALS up-regulated AURKA.
ALS induces G2/M arrest via regulation of the expression of CDK1/CDC2, CDK2, cyclin B1, p27 Kip1, and p53 in DAOY cells
We next examined the effect of ALS on cell cycle distribution in DAOY cells using a flow cytometer. Treatment with ALS led to a marked G2/M arrest in concentration- and time-dependent manners in DAOY cells (Figure 6D and 6F). The percentage of G2/M, G1, and S phase was 13.1%, 51.4%, and 35.5% at the basal level and 11.6%, 51.6%, and 36.9% at the non-treated control cells, respectively. There was no significant difference observed in the number of cells between the non-treated control cells and the 0.05% DMSO treated cells. When DAOY cells were incubated with ALS at 0.1, 1, and 5 μM for 24 h, compared with the control cells, the percentage of cells in G2/M phase was 45.8%, 72.4%, and 68.4%, respectively; the percentage of cells in G1 phase was 21.0%, 8.7%, and 8.4%, respectively; and the percentage of cells in S phase was 33.3%, 18.9%, and 23.1%, respectively (Figure 6E).
We conducted further experiments to evaluate the effect of ALS at 1 μM on cell cycle distribution in DAOY cells over a 72 h period. The percentage of cells at G2/M phase was increased from 21.9% at the basal level to 25.9%, 48.0%, 61.1%, 72.4%, 88.0%, and 87.9% after 4, 8, 12, 24, 48, and 72 h, respectively. On the contrary, the percentage of DAOY cells in G1 phase was significantly decreased from 49.3% at the basal level to 25.9%, 20.7%, 11.1%, 8.7%, 5.2%, and 3.7% after 4, 8, 12, 24, 48, and 72 h, respectively; and the percentage of DAOY cells in S phase from 28.9% at the basal level to 44.5%, 31.3%, 27.8%, 18.9%, 6.8%, and 8.4% after 4, 8, 12, 24, 48, and 72 h, respectively (Figure 6G).
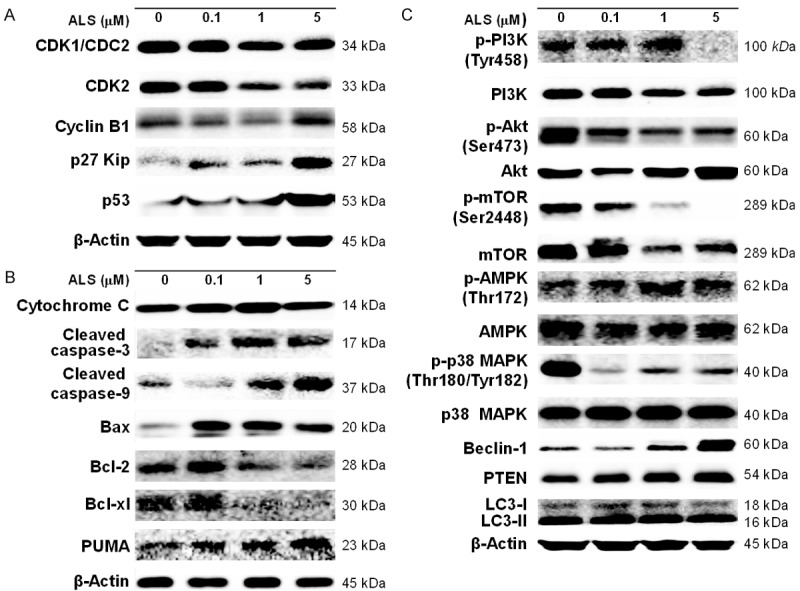
We used the Western blotting assay to examine the expression levels of key regulators responsible for the G2 checkpoint including CDK1/CDC2, CDK2, cyclin B1, p27 Kip1, and p53 (Figures 7A and 8A). As shown in Figure 7A, when DAOY cells treated with ALS at 0.1, 1, and 5 μM for 24 h, the expression level of CDK1/CDC2 and CDK2 were decreased 10.5%, 28.8%, 25.0% and 4.6%, 24.5%, 25.8%, respectively, in comparison with control cells. The expression level of cyclin B1 was concentration dependently decreased 7.7% and 9.5% when DAOY cells were treated with ALS at 0.1 and 1 μM and there was an ~50% decrease when DAOY cells were treated with 5 μM ALS, compared with control cells (Figure 7A). When DAOY cells were treated with ALS at 0.1 and 5 μM for 24 h, the expression of p27 Kip1 was significant increased 1.2- and 1.7-fold, respectively, in comparison with control cells. Moreover, the expression level of p53 was significantly increased in DAOY cells after ALS treatment (Figure 7A). In comparison to the control cells, there was a 1.2-, 1.3-, and 2.3-fold increase when treated with ALS at 0.1, 1, and 5 μM for 24 h, respectively.
Figure 7.
DAOY cells were incubated with ALS at 0.1, 1, and 5 µM for 24 h and then subject to Western blot assay. A. Representative blots of CDK1/CDC2, CDK2, cyclin B1, p27 Kip1, and p53 in DAOY cells. B. Representative blots of cytochrome C, cleaved caspase 3, cleaved caspase 9, Bax, Bcl-2, Bcl-xl, and PUMA in DAOY cells. C. Representative blots of PI3K, Akt, mTOR, AMPK, p38 MAPK, beclin 1, PTEN, and LC3 in DAOY cells.
Figure 8.

ALS modulates the expression level of key regulators involved in cell cycle, apoptosis, and autophagy in DAOY cells. A. Bar graphs showing the expression level of CDK1/CDC2, CDK2, cyclin B1, p27 Kip1, and p53 in DAOY cells. B. Bar graphs showing the expression level of cytochrome C, cleaved caspase 3, cleaved caspase 9, Bax, Bcl-2, Bcl-xl, and PUMA in DAOY cells. C. Bar graphs showing the expression level of PI3K, Akt, mTOR, AMPK, p38 MAPK, beclin 1, PTEN, and LC3 in DAOY cells. *P < 0.05, **P < 0.01, and ***P < 0.001.
ALS induces apoptosis via activation of the mitochondria-dependent pathway in DAOY cells
To further examine the cancer cell killing effect of ALS in DAOY cells, cellular apoptosis was quantified by flow cytometric analysis. The number of apoptotic cells was first quantified and the results are shown in Figure 9A. The number of apoptotic cells at the basal level was 2.87% in DAOY cells when treated with the control vehicle only (0.05% DMSO, v/v). When DAOY cells were treated with ALS 0.1, 1, and 5 μM for 24 h, the total percentage of apoptotic cells (early and late apoptosis) were 4.7%, 7.4%, and 11.1%, respectively, with a 1.6-, 2.6-, and 3.9-fold increase, respectively, compared with control cells (P < 0.05 or 0.001, Figure 9B).
Figure 9.

ALS induces apoptotic and autophagic cell death in DAOY cells determined by flow cytometry and confocal microscopy. DAOY cells were incubated with ALS at 0.1, 1, and 5 µM for 24 h and then subject to flow cytometry analysis and evaluated using a lysosome-specific fluorescence dye and the images were analyzed. A. Flow cytometry plots showing specific cell populations (live, early apoptosis, and late apoptosis) in DAOY cells. B. Bar graphs showing the percentage of apoptotic DAOY cells. C. Flow cytometric plots showing autophagy of cells treated with ALS at 0.1, 1, and 5 µM for 24 h. D. Bar graphs showing percentage of autophagic cells in DAOY cells. E. Representative images show autophagy of DAOY cells (stained in green). The level of autophagy was Magnification, × 60; scale bar, 5 µM. F. Bar graphs showing the autophagy of cells in DAOY cells. *P < 0.05 and ***P < 0.001.
Next, the effects of ALS on the mitochondria-related apoptotic pathway were examined in DAOY cells (Figures 7B and 8B). We first examined the expression level of cytochrome C, cleaved caspase 3, and cleaved caspase 9 in DAOY cells when treated with ALS. Compared with control cells, treatment of cells with ALS at 0.1 and 1 μM for 24 h significantly increased the level of cytosolic cytochrome C 1.3- and 1.4-fold, respectively (P < 0.001; Figures 7B and 8B). Compared with control cells, the level of cleaved caspase 3 and cleaved caspase 9 was increased. Treatment of DAOY cells with 1 μM ALS significantly increased the level of cleaved caspase 3 1.2-fold (P < 0.01) and treatment of cells with ALS at 1 and 5 mM increased the expression level of cleaved caspase 9 1.4- and 1.6-fold, respectively (P < 0.001; Figures 7B and 8B).
In addition, we investigated the expression of pro-apoptotic protein Bax and anti-apoptotic proteins Bcl-2 and Bcl-xl in DAOY cells. When DAOY cells were treated with ALS at 0.1, 1, and 5 μM for 24 h, there was a 1.5-, 1.4-, and 1.1-fold increase in the expression level of Bax, respectively, compared with control cells (P < 0.05 or 0.001; Figures 7B and 8B). In contrast, the expression level of Bcl-2 was decreased 23.6% and 31.8%, respectively, after 1 and 5 μM ALS treatment (P < 0.001). The expression level of Bcl-xl was reduced 8.5% and 11.6%, respectively, in DAOY cells when treated with ALS at 1 and 5 μM (P < 0.05 or 0.01). In addition, treatment of DAOY cells with ALS at 0.1, 1, and 5 μM for 24 h significantly increased the expression level of PUMA 1.2-, 1.2-, and 1.1-fold, respectively (P < 0.05, 0.01, or 0.001; Figures 7B and 8B).
ALS induces autophagy via inhibition of the PI3K/Akt/mTOR signaling pathway in DAOY cells
After observing binding of ALS with key autophagy regulators, we next examined the effect of ALS on autophagy of DAOY cells using flow cytometric analysis, confocal microscopy, and Western blotting analysis. The percent of autophagic cells at the basal level was 5.6% for DAOY cells. Incubation of DAOY cells with ALS for 24 h increased the percentage of autophagic cells in a concentration-dependent manner. When DAOY cells were treated with 0.1 μM ALS for 24 h, the percentage of autophagic cells was only slightly increased compared with control cells. However, there was a 6.5- and 8.0-fold increase in the percentage of autophagic cells when treated with ALS at 1 and 5 μM for 24 h, respectively, compared with control cells (P < 0.001; Figure 9C and 9D). We further tested the autophagy-inducing effect of ALS on DAOY cells using confocal microscopic examination (Figure 9E). In comparison with control cells, ALS increased autophagy in a concentration-dependent manner in DAOY cells. There was a 1.8- and 2.0-fold increase in autophagic death of DAOY cells when treated with ALS at 1 and 5 μM for 24 h (P < 0.001), respectively, but a lower concentration of ALS (0.1 μM) did not significantly affect autophagy of DAOY cells (Figure 9F).
Next, we examined the effects of ALS on the level of p-PI3K at Tyr458, PI3K, p-Akt at Ser473, Akt, p-mTOR at Ser2448, mTOR, p-AMPK at Thr172, AMPK, p38 MAPK at Thr180/Tyr182, p38 MAPK, beclin1, PTEN, and LC3-I/LC3-II using Western blotting analysis (Figures 7C and 8C). Exposure of DAOY cells to ALS led to a decrease in the level of p-PI3K at Tyr458 and PI3K, compared with control cells. In DAOY cells, the p-PI3K/PI3K ratio was decreased 34.5% when DAOY cells were treated with 5 μM ALS (P < 0.001). Compared with control cells, the level of p-Akt at Ser473 was decreased in a concentration-dependent manner, whilst there was no significant alteration in the level of Akt (Figures 7C and 8C). The p-Akt/Akt ratio was decreased 32.4%, 52.5%, and 48.5% in DAOY cells upon the exposure to ALS at 0.1, 1, and 5 μM for 24 h, respectively (P < 0.001). Treatment of DAOY cells with ALS at 0.1, 1, and 5 μM for 24 h significantly reduced mTOR phosphorylation at Ser2448 and the level of mTOR. The p-mTOR/mTOR ratio was significantly decreased by 62.6%, 74.0%, and 79.2%, respectively, compared with control cells (P < 0.001). In comparison with control cells, there was a concentration-dependent increase in the AMPK phosphorylation level at Thr172 in DAOY cells treated with ALS (Figures 7C and 8C). However, the p-AMPK/AMPK ratio was only significant increased by 1.1-fold when DAOY cells were treated with ALS at 1 μM (P < 0.01).
Furthermore, the involvement of p38 MAPK signaling pathway in ALS-induced autophagy was tested using Western blotting assay. As shown in Figure 10, exposure of DAOY cells to ALS at 0.1, 1, and 5 μM ALS for 24 h, ALS markedly induced the phosphorylation of p38 MAPK at Thr180/Tyr182. However, incubation of cells with ALS did not significantly affect the expression of total p38 MAPK. The ratio of p-p38 MAPK over p38 MAPK was concentration-dependently decreased when DAOY cells were treated with ALS at 0.1, 1, and 5 μM (P < 0.001). We also examined the effect of ALS on the expression level of Beclin 1 and LC3-I/LC3-II. There was a 2.0-fold increase in the level of beclin 1 in DAOY cells when treated with ALS at 5 μM for 24 h (P < 0.001). However, treatment of DAOY cells with ALS at 0.1 and 1 μM for 24 h increased beclin 1 1.1- and 1.3-fold, respectively; but none of these increases was statistically significant. After treatment of cells with ALS, our Western blotting analysis revealed two clear bands: LC3-I with an apparent mobility of about 18 kDa and LC3-II with an apparent mobility of 16 kDa in DAOY cells. In DAOY cells, there was a concentration-dependent increase in level of LC3-II and no significant alteration in the level of LC3-I. Compared with control cells, the ratio of LC3-II over LC3-I was remarkably increased 1.2- and 1.3-fold in DAOY cells when treated with ALS at 0.1 and 1 μM for 24 h, respectively (P < 0.001). In addition, the expression level of PTEN was significantly increased. There was a 1.2-, 1.3-, and 1.5-fold increase in the level of PTEN when DAOY cells were treated with ALS at 0.1, 1, and 5 μM for 24 h, respectively (P < 0.01 or 0.001).
Figure 10.

ALS induces autophagy via PI3K- and p38 MAPK-mediated signaling pathways in DAOY cells. A. Flow cytometric plots showing autophagy of cells treated with ALS in presence and absence of inhibitors. B. Bar graphs showing percentage of autophagic cells treated with ALS in presence and absence of inhibitors. C. Representative images show autophagy of DAOY cells (stained in green). The level of autophagy was evaluated using a lysosome-specific fluorescence dye and the images were analyzed. Magnification, × 60; scale bar, 5 µM. D. Bar graphs showing the autophagy of cells treated with ALS ± the inhibiting compound. *P < 0.05, **P < 0.01, and ***P < 0.001.
SB202190 and wortmannin (WM) enhance ALS-induced autophagy in DAOY cells
Since we observed that ALS induced significant autophagy in DAOY cells involving PI3K/Akt/mTOR pathway, the flow cytometry and confocal fluorescence microscopy were used to evaluate the effect of inhibitors of autophagy on basal and ALS-induced autophagy in DAOY cells. SB202190, is a selective p38 MAPK inhibitor that can induce autophagic vacuole formation through cross-inhibition of the PI3K/mTOR pathway. WM is a well-known PI3K inhibitor. We applied 10 μM SB202190 and 10 μM WM to examine the effect of ALS on the cellular autophagy level. Compared with control cells, treatment of cells with 10 μM SB 202190 increased 8.3-fold in autophagy (P < 0.01) and 10 μM WM only slightly increased (P > 0.05). However, the results showed that the autophagy level was increased 4.7- and 3.9-fold in cells co-treated with SB202190 and ALS or WM and ALS, respectively, compared with ALS alone (Figure 10A and 10B). In addition, we determined the autophagy-inducing effects of ALS in DAOY cells using confocal microscopic examination. In DAOY cells, co-incubation of SB202190 or WM at 10 μM significantly enhanced ALS-induced autophagy, compared to the control cells receiving ALS alone. In DAOY cells, SB202190 and WM at 10 μM significantly increased ALS-induced autophagy 3.6- and 2.4-flod (P < 0.001), respectively, compared to the control cells receiving ALS alone (Figure 10C and 10D).
Discussion
GBM is the most aggressive brain cancer with poor prognosis, and treatment of advanced GBM remains a major challenge in current clinical practice. There is a strong and urgent need to seek new effective chemotherapeutic drugs for GBM. The Aurora kinases are potentially good therapeutic targets for the therapy of GBM and other cancers. AURKA is often overexpressed in cancer cells, promoting cancer cell proliferation and survival, cell migration and invasion, and metastasis and inducing resistance to chemotherapeutic agents [40]. This gene may play a role in tumor development and progression. Presently, over a dozen of Aurora kinase inhibitors are in various phases of clinical development. The majority of the inhibitors (tozasertib/VX-680/, danusertib/PHA-739358, CYC116, SNS-314, AMG-900, AT-9283, SCH-1473759, ABT-348, PF-03814735, R-763/AS-703569, KW-2449 and TAK-901) are pan-selective (isoform non-selective) and few are AURKA (alisertib, MLN8237, VX-689/MK5108 and ENMD 2076) and AURKB (AZD1152 and GSK1070916) sub-type selective. In present study, we have demonstrated the cancer cell killing effect of ALS on DAOY cells and the underlying mechanisms.
Molecular docking has become a valuable tool of investigating the binding sites, interactome and protein targets of drugs [41]. We first performed docking to examine how ALS, MLN8054, tozasertib (VX-680), AMG-900, danusertib, barasertib, and CYC116 bound differently to AURKA and AURKB. These compounds are all known inhibitors of Aurora kinases with distinct binding affinity, mode, and preference. Our docking studies showed that ALS could bind to both AURKA and AURKB but with different binding energy. ALS formed hydrogen bond formation, charge interactions, and π-π stacking interactions with AURKA with a lower binding energy compared with binding to AURKB. This implied that ALS had a binding preference toward AURKA. Indeed, ALS has a selectivity of more than 200-fold for AURKA over AURKB in a cell-free assay [42]. Similarly, MLN8054, a selective AURKA inhibitor [43], bound AURKA preferentially over AURKB. Barasertib is a highly selective AURKB inhibitor with IC50 of 0.37 nM in a cell-free assay, ~3700 fold more selective for AURKB over AURKA [44]. CYC116 potently bound AURKB, but failed to dock into AURKA. CYC116 is a potent inhibitor of AURKA and AURKB with K i of 8.0 and 9.2 nM, respectively, and is less potent to vascular endothelial growth factor receptor 2 with a K i of 44 nM. CYC116 kills cancer cells via inhibition of Aurora kinase autophosphorylation, reduction of histone H3 phosphorylation, polyploidy, followed by cell death, resulting from a failure in cytokinesis [45]. Our docking has also demonstrated the potent binding of barasertib to AURKB but failed to dock into AURKA. In contrast, both AMG-900 and danusertib bound AURKA but failed to dock into AURKB. Danusertib is a pan-inhibitor for Aurora A/B/C with IC50 of 13, 79, and 61 nM, respectively, in cell-free assays. AMG 900 is a potent and highly selective pan-Aurora kinases inhibitor for Aurora A/B/C with IC50 of 5, 4, and 1 nM, respectively [46]. The pan-inhibitor of Aurora kinases, tozasertib (VX-680), bound both AURKA and AURKB with different CIE values. Tozasertib (VX-680) is a pan-Aurora inhibitor, mostly against AURKA with K i of 0.6 nM in a cell-free assay, less potent towards AURKB and AURKC [47].
It is important to realize the role of critical residues in the active sites of Aurora kinases that bind to Aurora kinase inhibitors and such knowledge can assist in the design of more potent sub-type selective Aurora kinase inhibitors [48]. Aurora kinases share a common sequence and structure consisting of a highly conserved C-terminal cataytic domain and a short N-terminal domain that varies in size [49]. The catalytic domain of AURKA has the typical bilobal kinase fold, comprised of an N-terminal β-strand domain (residues 127-215) and a C-terminal α-helical domain (residues 216-385). These domains are linked together by a hinge region (residues 210-216) that plays an important role in forming the catalytic active site and residues 273-292 are located at the N-terminal end of the activation loop of AURKA which adopt a unique conformation [50]. Residues Lys162 and Asp274 are essential for AURKA activity but do not form hydrogen bond to each other as seen in crystal structures of several other protein kinases [50]. Residue Trp277 in the activation loop is stabilized in the flexible glycine rich loop [51]. In our docking study, we have observed that Lys162, Asp274, and Trp277 all bound ALS and tozasertib (VX-680). Not all Lys162, Asp274, and Trp277 bound MLN8054, AMG-900, and danusertib.
Cell cycle consists of four distinct sequential phases, namely, G0/G1, S, G2 and M, which is closely regulated by a complex series of signaling pathways, checkpoint control, kinases and other proteins. In cancer, as a result of genetic mutations, this regulatory process malfunctions, resulting in uncontrolled cell proliferation and growth, invasion, and metastasis [52]. In present study, treatment of DAOY cells with ALS resulted in a significant decrease in the ratio of p-AURKA/AURKA, suggesting that the inhibition of phosphorylation of AURKA contributes, at least in part, to the inhibitory effect of ALS on cell proliferation and the inducing effect on cell-cycle arrest in G2/M phase. AURKA can enable cancer cells to become chemo- and radio-resistant through dysregulation of cell cycle progression and the DNA damage response. We found that ALS arrested DAOY cells in G2/M phase in concentration- and time-dependent manners. We further explored the effect of ALS on key regulators of cell cycle checkpoints, including CDK1/CDC2, CDK2, cyclin B1, p27 Kip1, and p53. AURKA acts as a key regulatory component of the p53 pathway, and particularly the checkpoint-response pathways critical for oncogenic transformation of cells, by phosphorylating and stabilizing p53. The molecular docking study showed that ALS could be easily docked into the active sites of CDK1/CDC2, CDK2, cyclin B1, p27 Kip1, and p53 via hydrogen bond formation, charge interaction, and/or π-π interaction. Particularly, ALS can be docked to p53 with the lowest CIE and more hydrogen bond formations. p53 is a target of AURKA and ALS may kill cancer cells via direct binding with p53.
The CDK1/CDC2-cyclin B1 complex is pivotal in regulating the G2/M phase transition and mitosis. During a normal cell cycle, the progression of cells in G2 phase to M phase is triggered by the activation of the cyclin B1-dependent CDK1/CDC2 kinase, which is regulated by a series of phosphorylation and dephosphorylation events and protein-protein interactions involving a network of signaling pathways [53]. In general, a cell with suppressed CDK1/CDC2-cyclin B1 activity would tend to be arrested in the G2/M phase, whereas a cell with elevated CDK1/CDC2-cyclin B1 activity would be favored to enter mitosis [54]. Activation of CDK1 appears to be an upstream event of AURKA activation. Furthermore, p27 Kip1 and p53 are another two important factors that contribute to the regulation of cell cycle. p27 Kip1 is a member of the Cip/Kip family of cyclin-dependent kinase inhibitors regulated by p53 and enforces the G1 restriction point via its inhibitory binding to CDK2/cyclin E complex, thereby inducing cell-cycle arrest at the G2/M phase [55]. p53 is a transcription factors that plays a major role in the cellular response to DNA damage and other genomic aberrations [56]. Activation of p53 can lead to either cell cycle arrest and DNA repair or apoptosis. Aurora kinase inhibitors such as ALS activate p53 signaling, contributing to the anti-proliferative and anticancer effects [57]. We have observed the expression level of CDK1/CDC2, CDK2, and cyclin B1 was remarkably decreased in DAOY cells when treated with ALS, however, the expression level of p27 Kip1 and p53 was markedly increased, which provides an explanation for the inducing effect of ALS on G2/M phase arrest in DAOY cells.
Programmed cell death, referring to apoptosis, autophagy, and programmed necrosis, is proposed to be death of a cell in any pathological format when mediated by an intracellular program. These three forms of programmed cell death may jointly decide the fate of malignant neoplastic cells; apoptosis and programmed necrosis invariably contribute to cell death, whereas autophagy can have either pro-survival or pro-death roles [58]. Apoptosis, or type I programmed cell death, is a fundamental biological phenomenon, with an essential role in removing unwanted or abnormal cells in multicellular organisms. It contributes to distinct biochemical and genetic pathways that play a critical role in the development and homeostasis in normal tissues. Apoptosis is not only a special type of cell death, but also has great biological significance and involves complex molecular mechanisms [59]. Studies have indicated that AURKA plays a crucial role in growth by inhibiting cell apoptosis and propelling cell cycle [40]. The pro-apoptotic effect of ALS has been reported in various cancer cell lines, including lymphoma, breast cancer, colorectal, lung, melanoma, gastric, and leukemia [20]. In our study, we found that ALS induced apoptotic cell death of DAOY cells in a concentration-dependent manner. Apoptosis is executed by members of the caspase family of cysteine proteases, which can be activated by two main pathways, namely the extrinsic death receptor pathway and the intrinsic mitochondria/cytochrome C mediated pathway [59]. The two pathways are linked and both triggered by the activation of caspases 3, 6, and 7. Caspases are a class of cysteine proteases that contain abundant cysteine at the active site and specifically catalyze hydrolysis of the aspartate portion in the substrate [60].
In the mitochondria/cytochrome C pathway, the release of cytochrome C from the mitochondrial inter-membrane space occurs during the early stages of apoptotic cell death [61]. Various stimuli converge on mitochondria and activate one or more members of the BH3-only protein family, which are tightly regulated by Bcl-2 family members. The Bcl-2 family is another group of proteins involved in apoptosis, members of which have anti-apoptotic effects as well as pro-apoptotic effects [62]. Tumor cells can acquire resistance to apoptosis by the overexpression of anti-apoptotic proteins such as Bcl-2 or by the downregulation or mutation of pro-apoptotic proteins such as Bax. The promotes the assembly of Bax oligomers within mitochondrial outer membranes, leading to cytochrome C release to cytosol, which in turn induces the apoptotic protease-activating factor-1 to generate apoptosome and activates caspas-9, eventually triggering caspase 3 activation and apoptosis. Furthermore, Bcl-2 can be inhibited by posttranslational modification and/or increased expression of PUMA, which is an essential regulator of p53-mediated cell apoptosis [63]. In present study, the molecular docking study showed that ALS was readily docked into the active sites of cytochrome C, cleaved caspase 3, cleaved caspase 9, Bax, Bcl-2, Bcl-xl, and PUMA via hydrogen bond formation, charge interaction, and/or π-π interaction. ALS bound cleaved caspase 3 with the lowest CIE. These results suggest that caspase 3 is a key protease involved in apoptosis, acting as an executor and terminator of apoptosis. In our Western blotting assay, we observed a significantly increased cytosolic level of cytochrome C after ALS treatment, subsequently activating cleaved caspase 9. Activated caspase 9 in turn activates cleaved caspase 3 and activated caspase 3 ultimately induces apoptosis with a marked increase in Bax but a decrease in Bcl-2 and Bcl-cl levels. As a consequence, the rise in the ratio of proapoptotic protein/antiapoptotic protein shifts the balance to apoptotic status. Moreover, we noted a concentration-dependent increase in the expression of PUMA in DAOY cells. These results indicate that ALS effectively induces mitochondria-dependent apoptosis in DAOY cells.
Autophagy, also known as a type II programed cell death, is a complicated process executed through multiple steps from intracellular membrane/vesicle reorganization to form double-membraned autophagosomes, which eventually degrade the contents via acidic lysosomal hydrolases [64]. Targeting autophagy for cancer treatment is promising but controversial, due to its multifaceted roles in the regulation of cell survival and cell death. Under certain circumstances, autophagy has been known to promote cellular survival during nutrient depletion and is essential for maintaining cellular hemostasis by degrading damaged organelles and proteins [65]. However, increasing evidence shows that autophagy is a promising and emerging target for GBM treatment via regulation of autophagy-related key functional proteins and signaling pathways [66-68]. As shown by our flow cytometric analysis and confocal microscopic examination, ALS clearly induced autophagy in DAOY cells. We also observed that the autophagy-inducing effect of ALS outweighed its apoptosis-inducing effect. Moreover, we explored the binding modes of ALS to several key autophagy-regulating proteins using Discovery Studio® program 3.1. The results demonstrated that ALS was easily docked into the active sites of PI3K, Akt, mTOR, AMPK, p38 MAPK, beclin 1, PTEN, and LC3 via hydrogen bond formation, charge interaction, and/or π-π interaction. These findings suggest that ALS may induce autophagy in DAOY cells via inhibition of AURKA and direct interactions with autophagy-regulating macromolecules.
Of note, treatment of DAOY cells with ALS resulted in differential effects on the phosphorylation and expression levels of PI3K/Akt/mTOR axis, AMPK, and p38 MAPK. These findings demonstrated that ALS inhibited the p-PI3K at Tyr458 and p-p38 MAPK at Thr180/Tyr182 but enhanced the p-AMPK at Thr172. ALS treatment caused significant alterations in the expression and phosphorylation levels of key functional proteins regulating autophagy signaling pathway. The PI3K/Akt/mTOR signaling pathway is a central pathway involved in autophagy through the regulation of cell growth, motility, protein synthesis, cell metabolism, cell survival, and cell death in response to various stimuli [63,69]. This critical pathway acts as a convergence point for a multitude of upstream signals and in turn stimulates the activity of numerous downstream effectors. Recently, the PI3K/AKT/mTOR signaling pathway has come to be regarded as the key regulator of a series of cell processes, as it can be deregulated by various genetic and epigenetic mechanisms in a wide range of cancer cells [58]. PI3K catalyzes the formation of phosphatidylinositol-3,4,5-triphosphate via phosphorylation of phosphatidylinositol, phosphatidylinositol-4-phosphate and phosphatidylinositol-4,5 bisphosphate [70]. In this study, ALS significantly inhibited the phosphorylation of PI3K at Tyr458 and the ratio of p-PI3K/PI3K in DAOY cells.
Akt is involved in the regulation of various signaling downstream pathways, including metabolism, cell proliferation, survival, growth, and angiogenesis. It has been reported that AURKA promotes cancer cell survival through the activation of Akt [51,71]. Thus, inhibition of AURKA may result in cancer cell death via inactivation of Akt. Our findings showed that the p-Akt/Akt ratio was significantly decreased by ALS in a concentration-dependent manner in DAOY cells. ALS treatment inhibited the p-AURKA, which may contribute, at least in part, to the inactivation of Akt in DAOY cells. mTOR plays a key role in cell growth, autophagic cell death and homeostasis. mTOR is phosphorylated at Ser2448 via the PI3K/Akt signaling pathway and is auto phosphorylated at Ser2481 [72]. Our results suggest that accumulation of ALS led to a significant decrease in p-Akt level and that low levels of p-Akt produced by the ALS correlated with low levels of p-mTOR. These results are consistent with the role of PI3K/Akt/mTOR axis in the regulation of autophagy. These findings indicate that ALS has a significant autophagy-inducing effect on DAOY cells via inhibition of the PI3K/Akt/mTOR pathway.
AMPK and p38 MAPK are upstream signaling molecules in the Akt/mTOR pathway and play important role in regulation of cell proliferation and death [73]. AMPK is a heterotrimeric complex composed of a catalytic α subunit, regulatory β and γ subunits and an important regulator of cell death under various conditions, through activation of c-Jun N-terminal kinase and p53 and inhibition of mTOR [74]. It is highly conserved from yeast to plants and animals and plays a key role in the regulation of energy homeostasis, cell survival and cell death. Incubation of DAOY cells with ALS for 24 h showed a promoting effect on the phosphorylation of AMPK at Thr172. There was no significant change in the expression of total AMPK compared with control cells, but the ratio of p-AMPK/AMPK was increased at 1 µM ALS in DAOY cells compared with control cells, which may contribute to the inhibition of mTOR. p38 MAPK regulates cellular responses to cytokines and thus controls cell differentiation, cell death, cell migration, and invasion [75]. It is responsive to stress stimuli, such as inflammatory cytokines, growth factors, lipopolysaccharide, ultraviolet irradiation, heat shock and osmotic shock [74]. Activated p38 MAPK can p-MAPK-activated protein kinase 2, several transcription factors including activating transcription factor, Mycassociated factor X and myocyte enhancer factor-2 and induce the expression of a number of target genes [76]. However, the molecular mechanisms that link p38 MAPK to autophagy are not yet fully understood. Our findings show that ALS significantly inactivates p-p38 MAPK signaling, which is responsive to a variety of stress stimuli and regulates autophagy. It can enhance the ALS-induced autophagy in DAOY cells.
Beclin 1, PTEN and LC3 are three specific markers of cellular autophagy and they are all involved in autophagic process, especially in its early stage. Beclin 1 promotes autophagy and inhibits proliferation of cancer cells and the suppression of beclin 1 expression impairs autophagy, which as a platform for recruitment of other Atgs that is critical for autophagosome formation [77]. In this study, treatment of DAOY with ALS significantly increased the expression of beclin 1. PTEN inhibits Akt/mTOR and MAPK signaling, leading to cell death and growth regulation [78]. We found that ALS induced remarkable increased the expression of PTEN in a concentration-dependent manner. LC3, associated with the formation of the autophagic vacuole, is currently considered as a specific molecular marker for autophagosomes in mammals. LC3 proteins can be divided into two forms: LC3-I and LC3-II. LI48 LC3-I is consequently proteolytically cleaved and lipidated by Atg3 and Atg7 to form LC3-II, which localizes to the autophagosome membrane. LC3-II migrated more rapidly than LC3-I on SDS-PAGE, leading to the appearance of two bands after immunoblotting, LC3-I with an apparent mobility of about 18 kDa and LC3-II with an apparent mobility of 16 kDa. There was a striking increase in the level of LC3-II/ LC3-I in DAOY cells treated with ALS using Western blotting assay.
We further explored the autophagy-inducing effect of ALS in DAOY cells using chemical inhibitors of autophagy. We employed SB202190, a p38 MAPK inhibitor and WM, which is an irreversible and selective phosphatidylinositol PI3K inhibitor to examine the levels of ALS-induced autophagy. The results showed that modulation of p38 MAPK by SB202190 and PI3K by WM altered ALS-induced autophagy in DAOY cells. SB202190 or WM completely blocked ALS-induced PI3K or p38 MAPK activation and both remarkably increased autophagic cell death in DAOY cells. These suggest that p38 MAPK is involved in the regulation of PI3K activation and autophagy process induced by ALS. AMPK and p38 MAPK play an important role in ALS-induced autophagy of DAOY cells. Taken together, ALS-induced inhibition of PI3K/Akt/mTOR and p38 MAPK pathways and activation of AMPK contribute to the autophagy-inducing effect of ALS, which may contribute to its anticancer activity. Moreover, we also provided evidence about the existence of crosstalk between the p38 MAPK and Akt/mTOR signaling pathways in DAOY cells treated with ALS.
Interestingly, we found that although ALS increased AURKA phosphorylation, it induced the expression of ALS via unknown mechanism. This effect might negate the cancer cell killing effect of ALS. In May 2015, Takeda Pharmaceutical Company Limited discontinued the Phase III trial of alisertib for patients with relapsed or refractory peripheral T-cell lymphoma due to lack of satisfactory efficacy. The reasons for the lack of efficacy of ALS may be complicated, probably related to compensated role of AURKB, improper regimen, off-target effects, etc. Combined use of ALS with cytotoxic agent may be a strategy to enhance its efficacy.
Conclusions
ALS preferentially bound AURKA over AURKB via hydrogen bond formation, charge interactions, and π-π stacking interactions. ALS potently inhibited the proliferation, induced cell cycle arrest in G2/M phase, activated the mitochondria-dependent apoptotic pathway, and promoted autophagy in DAOY cells. Our present study suggests that ALS is a promising new potential agent for the treatment of GBM and AURKA may represents a useful target for GBM treatment. However, due to the complexity of cancer as a systemic disease, the fate of tumor cells is not determined by one signaling pathway. In the future, further studies are needed to determine the precise mechanism and to reveal other potential targets of ALS in the treatment of GBM. A recent study has shown that ALS has antitumor activity in bevacizumab resistant, patient derived orthotopic models of glioblastoma in nude mice [79]. Another study has demonstrated that the simultaneous inhibition of phosphoinositide-dependent kinase 1 and AURKA by ALS induces the differentiation and apoptosis of GBM stem cells [80]. In order to verify the efficacy and safety, more studies of ALS efficacy with and without other co-therapies in animal models of GBM are warranted before it enters Phase I study in GBM.
Acknowledgements
The authors appreciate the financial support from the Startup Fund of the College of Pharmacy, University of South Florida, Tampa, Florida 33612, USA and from the Startup Grant of Huaqiao University, Xiamen, Fujian 361021, China.
Disclosure of conflict of interest
None.
Authors’ contribution
Participated in research design: Zheng Liu, Feng Wang, Zhi-Wei Zhou, He-Chun Xia, Yin-Xue Yang, Zhi-Xu He, Tao Sun, & Shu-Feng Zhou; Conducted experiments: Zheng Liu, Feng Wang, Zhi-Wei Zhou, & Xin-Yu Wang; Performed data analysis: Zheng Liu, Feng Wang, Zhi-Wei Zhou, & Xin-Yu Wang; Wrote or contributed to the writing of the manuscript: Zheng Liu, Zhi-Wei Zhou, Yin-Xue Yang, Zhi-Xu He, Tao Sun, & Shu-Feng Zhou.
References
- 1.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 2.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 3.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 4.Omuro A, DeAngelis LM. Glioblastoma and other malignant gliomas: a clinical review. JAMA. 2013;310:1842–1850. doi: 10.1001/jama.2013.280319. [DOI] [PubMed] [Google Scholar]
- 5.Wirsching HG, Galanis E, Weller M. Glioblastoma. Handb Clin Neurol. 2016;134:381–397. doi: 10.1016/B978-0-12-802997-8.00023-2. [DOI] [PubMed] [Google Scholar]
- 6.Mesti T, Ocvirk J. Malignant gliomas: old and new systemic treatment approaches. Radiol Oncol. 2016;50:129–138. doi: 10.1515/raon-2015-0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Groot JF. High-grade gliomas. Continuum (Minneap Minn) 2015;21:332–344. doi: 10.1212/01.CON.0000464173.58262.d9. [DOI] [PubMed] [Google Scholar]
- 8.Kumthekar P, Raizer J, Singh S. Low-grade glioma. Cancer Treat Res. 2015;163:75–87. doi: 10.1007/978-3-319-12048-5_5. [DOI] [PubMed] [Google Scholar]
- 9.Woehrer A, Bauchet L, Barnholtz-Sloan JS. Glioblastoma survival: has it improved? Evidence from population-based studies. Curr Opin Neurol. 2014;27:666–674. doi: 10.1097/WCO.0000000000000144. [DOI] [PubMed] [Google Scholar]
- 10.Lapenna S, Giordano A. Cell cycle kinases as therapeutic targets for cancer. Nat Rev Drug Discov. 2009;8:547–566. doi: 10.1038/nrd2907. [DOI] [PubMed] [Google Scholar]
- 11.Pitts TM, Davis SL, Eckhardt SG, Bradshaw-Pierce EL. Targeting nuclear kinases in cancer: development of cell cycle kinase inhibitors. Pharmacol Ther. 2014;142:258–269. doi: 10.1016/j.pharmthera.2013.12.010. [DOI] [PubMed] [Google Scholar]
- 12.Dominguez-Brauer C, Thu KL, Mason JM, Blaser H, Bray MR, Mak TW. Targeting mitosis in cancer: emerging strategies. Mol Cell. 2015;60:524–536. doi: 10.1016/j.molcel.2015.11.006. [DOI] [PubMed] [Google Scholar]
- 13.Sampath SC, Ohi R, Leismann O, Salic A, Pozniakovski A, Funabiki H. The chromosomal passenger complex is required for chromatin-induced microtubule stabilization and spindle assembly. Cell. 2004;118:187–202. doi: 10.1016/j.cell.2004.06.026. [DOI] [PubMed] [Google Scholar]
- 14.Krenn V, Musacchio A. The Aurora B kinase in chromosome bi-orientation and spindle checkpoint signaling. Front Oncol. 2015;5:225. doi: 10.3389/fonc.2015.00225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cicenas J, Cicenas E. Multi-kinase inhibitors, AURKs and cancer. Med Oncol. 2016;33:43. doi: 10.1007/s12032-016-0758-4. [DOI] [PubMed] [Google Scholar]
- 16.Dar AA, Goff LW, Majid S, Berlin J, El-Rifai W. Aurora kinase inhibitors-rising stars in cancer therapeutics? Mol Cancer Ther. 2010;9:268–278. doi: 10.1158/1535-7163.MCT-09-0765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hirota T, Kunitoku N, Sasayama T, Marumoto T, Zhang D, Nitta M, Hatakeyama K, Saya H. Aurora-A and an interacting activator, the LIM protein Ajuba, are required for mitotic commitment in human cells. Cell. 2003;114:585–598. doi: 10.1016/s0092-8674(03)00642-1. [DOI] [PubMed] [Google Scholar]
- 18.Yan M, Wang C, He B, Yang M, Tong M, Long Z, Liu B, Peng F, Xu L, Zhang Y, Liang D, Lei H, Subrata S, Kelley KW, Lam EW, Jin B, Liu Q. Aurora-A kinase: a potent oncogene and target for cancer therapy. Med Res Rev. 2016;36:1036–1079. doi: 10.1002/med.21399. [DOI] [PubMed] [Google Scholar]
- 19.Lehman NL, O’Donnell JP, Whiteley LJ, Stapp RT, Lehman TD, Roszka KM, Schultz LR, Williams CJ, Mikkelsen T, Brown SL, Ecsedy JA, Poisson LM. Aurora A is differentially expressed in gliomas, is associated with patient survival in glioblastoma and is a potential chemotherapeutic target in gliomas. Cell Cycle. 2012;11:489–502. doi: 10.4161/cc.11.3.18996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Durlacher CT, Li ZL, Chen XW, He ZX, Zhou SF. An update on the pharmacokinetics and pharmacodynamics of alisertib, a selective Aurora kinase A inhibitor. Clin Exp Pharmacol Physiol. 2016;43:585–601. doi: 10.1111/1440-1681.12571. [DOI] [PubMed] [Google Scholar]
- 21.Ding YH, Zhou ZW, Ha CF, Zhang XY, Pan ST, He ZX, Edelman JL, Wang D, Yang YX, Zhang X, Duan W, Yang T, Qiu JX, Zhou SF. Alisertib, an Aurora kinase A inhibitor, induces apoptosis and autophagy but inhibits epithelial to mesenchymal transition in human epithelial ovarian cancer cells. Drug Des Devel Ther. 2015;9:425–464. doi: 10.2147/DDDT.S74062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yuan CX, Zhou ZW, Yang YX, He ZX, Zhang X, Wang D, Yang T, Wang NJ, Zhao RJ, Zhou SF. Inhibition of mitotic Aurora kinase A by alisertib induces apoptosis and autophagy of human gastric cancer AGS and NCI-N78 cells. Drug Des Devel Ther. 2015;9:487–508. doi: 10.2147/DDDT.S74127. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 23.Wang F, Li H, Yan XG, Zhou ZW, Yi ZG, He ZX, Pan ST, Yang YX, Wang ZZ, Zhang X, Yang T, Qiu JX, Zhou SF. Alisertib induces cell cycle arrest and autophagy and suppresses epithelial-to-mesenchymal transition involving PI3K/Akt/mTOR and sirtuin 1-mediated signaling pathways in human pancreatic cancer cells. Drug Des Devel Ther. 2015;9:575–601. doi: 10.2147/DDDT.S75221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li JP, Yang YX, Liu QL, Pan ST, He ZX, Zhang X, Yang T, Chen XW, Wang D, Qiu JX, Zhou SF. The investigational Aurora kinase A inhibitor alisertib (MLN8237) induces cell cycle G2/M arrest, apoptosis, and autophagy via p38 MAPK and Akt/mTOR signaling pathways in human breast cancer cells. Drug Des Devel Ther. 2015;9:1627–1652. doi: 10.2147/DDDT.S75378. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 25.Niu NK, Wang ZL, Pan ST, Ding HQ, Au GH, He ZX, Zhou ZW, Xiao G, Yang YX, Zhang X, Yang T, Chen XW, Qiu JX, Zhou SF. Pro-apoptotic and pro-autophagic effects of the Aurora kinase A inhibitor alisertib (MLN8237) on human osteosarcoma U-2 OS and MG-63 cells through the activation of mitochondria-mediated pathway and inhibition of p38 MAPK/PI3K/Akt/mTOR signaling pathway. Drug Des Devel Ther. 2015;9:1555–1584. doi: 10.2147/DDDT.S74197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shu LP, Zhou ZW, Zi D, He ZX, Zhou SF. A SILAC-based proteomics elicits the molecular interactome of alisertib (MLN8237) in human erythroleukemia K562 cells. Am J Transl Res. 2015;7:2442–2461. [PMC free article] [PubMed] [Google Scholar]
- 27.Ren BJ, Zhou ZW, Zhu DJ, Ju YL, Wu JH, Ouyang MZ, Chen XW, Zhou SF. Alisertib induces cell cycle arrest, apoptosis, autophagy and suppresses EMT in HT29 and Caco-2 cells. Int J Mol Sci. 2015;17:41. doi: 10.3390/ijms17010041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang ZX, Sun J, Howell CE, Zhou QY, He ZX, Yang T, Chew H, Duan W, Zhou ZW, Kanwar JR, Zhou SF. Prediction of the likelihood of drug interactions with kinase inhibitors based on in vitro and computational studies. Fundam Clin Pharmacol. 2014;28:551–582. doi: 10.1111/fcp.12069. [DOI] [PubMed] [Google Scholar]
- 29.Yin JJ, Sharma S, Shumyak SP, Wang ZX, Zhou ZW, Zhang Y, Guo P, Li CZ, Kanwar JR, Yang T, Mohapatra SS, Liu W, Duan W, Wang JC, Li Q, Zhang X, Tan J, Jia L, Liang J, Wei MQ, Li X, Zhou SF. Synthesis and biological evaluation of novel folic acid receptor-targeted, beta-cyclodextrin-based drug complexes for cancer treatment. PLoS One. 2013;8:e62289. doi: 10.1371/journal.pone.0062289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu YH, Mo SL, Bi HC, Hu BF, Li CG, Wang YT, Huang L, Huang M, Duan W, Liu JP, Wei MQ, Zhou SF. Regulation of human pregnane X receptor and its target gene cytochrome P450 3A4 by Chinese herbal compounds and a molecular docking study. Xenobiotica. 2011;41:259–280. doi: 10.3109/00498254.2010.537395. [DOI] [PubMed] [Google Scholar]
- 31.Mo SL, Liu WF, Chen Y, Luo HB, Sun LB, Chen XW, Zhou ZW, Sneed KB, Li CG, Du YM, Liang J, Zhou SF. Ligand- and protein-based modeling studies of the inhibitors of human cytochrome P450 2D6 and a virtual screening for potential inhibitors from the Chinese herbal medicine, Scutellaria baicalensis (Huangqin, Baikal Skullcap) Comb Chem High Throughput Screen. 2012;15:36–80. doi: 10.2174/138620712798280826. [DOI] [PubMed] [Google Scholar]
- 32.Yang LP, Zhou ZW, Chen XW, Li CG, Sneed KB, Liang J, Zhou SF. Computational and in vitro studies on the inhibitory effects of herbal compounds on human cytochrome P450 1A2. Xenobiotica. 2012;42:238–255. doi: 10.3109/00498254.2011.610833. [DOI] [PubMed] [Google Scholar]
- 33.Mo SL, Liu WF, Li CG, Zhou ZW, Luo HB, Chew H, Liang J, Zhou SF. Pharmacophore, QSAR, and binding mode studies of substrates of human cytochrome P450 2D6 (CYP2D6) using molecular docking and virtual mutations and an application to chinese herbal medicine screening. Curr Pharm Biotechnol. 2012;13:1640–1704. doi: 10.2174/138920112800958779. [DOI] [PubMed] [Google Scholar]
- 34.Su J, Chang C, Xiang Q, Zhou ZW, Luo R, Yang L, He ZX, Yang H, Li J, Bei Y, Xu J, Zhang M, Zhang Q, Su Z, Huang Y, Pang J, Zhou SF. Xyloketal B, a marine compound, acts on a network of molecular proteins and regulates the activity and expression of rat cytochrome P450 3a: a bioinformatic and animal study. Drug Des Devel Ther. 2014;8:2555–2602. doi: 10.2147/DDDT.S73476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Qiu JX, Zhou ZW, He ZX, Zhang X, Zhou SF, Zhu S. Estimation of the binding modes with important human cytochrome P450 enzymes, drug interaction potential, pharmacokinetics, and hepatotoxicity of ginger components using molecular docking, computational, and pharmacokinetic modeling studies. Drug Des Devel Ther. 2015;9:841–866. doi: 10.2147/DDDT.S74669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Han XD, Zhou ZW, Yang W, Ye HC, Xu YZ, Huang YF, Zhang T, Zhou SF. A computational and functional study elicits the ameliorating effect of the Chinese herbal formula Huo Luo Xiao Ling Dan on experimental ischemia-induced myocardial injury in rats via inhibition of apoptosis. Drug Des Devel Ther. 2015;9:1063–1102. doi: 10.2147/DDDT.S76336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Qiu JX, Zhou ZW, He ZX, Zhao RJ, Zhang X, Yang L, Zhou SF, Mao ZF. Plumbagin elicits differential proteomic responses mainly involving cell cycle, apoptosis, autophagy, and epithelial-to-mesenchymal transition pathways in human prostate cancer PC-3 and DU145 cells. Drug Des Devel Ther. 2015;9:349–417. doi: 10.2147/DDDT.S71677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li YC, He SM, He ZX, Li M, Yang Y, Pang JX, Zhang X, Chow K, Zhou Q, Duan W, Zhou ZW, Yang T, Huang GH, Liu A, Qiu JX, Liu JP, Zhou SF. Plumbagin induces apoptotic and autophagic cell death through inhibition of the PI3K/Akt/mTOR pathway in human non-small cell lung cancer cells. Cancer Lett. 2014;344:239–259. doi: 10.1016/j.canlet.2013.11.001. [DOI] [PubMed] [Google Scholar]
- 39.Balan M, Pal S. A novel CXCR3-B chemokine receptor-induced growth-inhibitory signal in cancer cells is mediated through the regulation of Bach-1 protein and Nrf2 protein nuclear translocation. J Biol Chem. 2014;289:3126–3137. doi: 10.1074/jbc.M113.508044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Do TV, Xiao F, Bickel LE, Klein-Szanto AJ, Pathak HB, Hua X, Howe C, O’Brien SW, Maglaty M, Ecsedy JA, Litwin S, Golemis EA, Schilder RJ, Godwin AK, Connolly DC. Aurora kinase A mediates epithelial ovarian cancer cell migration and adhesion. Oncogene. 2014;33:539–549. doi: 10.1038/onc.2012.632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baig MH, Ahmad K, Roy S, Ashraf JM, Adil M, Siddiqui MH, Khan S, Kamal MA, Provaznik I, Choi I. Computer aided drug design: success and limitations. Curr Pharm Des. 2016;22:572–581. doi: 10.2174/1381612822666151125000550. [DOI] [PubMed] [Google Scholar]
- 42.Manfredi MG, Ecsedy JA, Chakravarty A, Silverman L, Zhang M, Hoar KM, Stroud SG, Chen W, Shinde V, Huck JJ, Wysong DR, Janowick DA, Hyer ML, Leroy PJ, Gershman RE, Silva MD, Germanos MS, Bolen JB, Claiborne CF, Sells TB. Characterization of Alisertib (MLN8237), an investigational small-molecule inhibitor of aurora A kinase using novel in vivo pharmacodynamic assays. Clin Cancer Res. 2011;17:7614–7624. doi: 10.1158/1078-0432.CCR-11-1536. [DOI] [PubMed] [Google Scholar]
- 43.Manfredi MG, Ecsedy JA, Meetze KA, Balani SK, Burenkova O, Chen W, Galvin KM, Hoar KM, Huck JJ, LeRoy PJ, Ray ET, Sells TB, Stringer B, Stroud SG, Vos TJ, Weatherhead GS, Wysong DR, Zhang M, Bolen JB, Claiborne CF. Antitumor activity of MLN8054, an orally active small-molecule inhibitor of Aurora A kinase. Proc Natl Acad Sci U S A. 2007;104:4106–4111. doi: 10.1073/pnas.0608798104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang J, Ikezoe T, Nishioka C, Tasaka T, Taniguchi A, Kuwayama Y, Komatsu N, Bandobashi K, Togitani K, Koeffler HP, Taguchi H, Yokoyama A. AZD1152, a novel and selective aurora B kinase inhibitor, induces growth arrest, apoptosis, and sensitization for tubulin depolymerizing agent or topoisomerase II inhibitor in human acute leukemia cells in vitro and in vivo . Blood. 2007;110:2034–2040. doi: 10.1182/blood-2007-02-073700. [DOI] [PubMed] [Google Scholar]
- 45.Wang S, Midgley CA, Scaerou F, Grabarek JB, Griffiths G, Jackson W, Kontopidis G, McClue SJ, McInnes C, Meades C, Mezna M, Plater A, Stuart I, Thomas MP, Wood G, Clarke RG, Blake DG, Zheleva DI, Lane DP, Jackson RC, Glover DM, Fischer PM. Discovery of N-phenyl-4-(thiazol-5-yl)pyrimidin-2-amine aurora kinase inhibitors. J Med Chem. 2010;53:4367–4378. doi: 10.1021/jm901913s. [DOI] [PubMed] [Google Scholar]
- 46.Payton M, Bush TL, Chung G, Ziegler B, Eden P, McElroy P, Ross S, Cee VJ, Deak HL, Hodous BL, Nguyen HN, Olivieri PR, Romero K, Schenkel LB, Bak A, Stanton M, Dussault I, Patel VF, Geuns-Meyer S, Radinsky R, Kendall RL. Preclinical evaluation of AMG 900, a novel potent and highly selective pan-aurora kinase inhibitor with activity in taxane-resistant tumor cell lines. Cancer Res. 2010;70:9846–9854. doi: 10.1158/0008-5472.CAN-10-3001. [DOI] [PubMed] [Google Scholar]
- 47.Harrington EA, Bebbington D, Moore J, Rasmussen RK, Ajose-Adeogun AO, Nakayama T, Graham JA, Demur C, Hercend T, Diu-Hercend A, Su M, Golec JM, Miller KM. VX-680, a potent and selective small-molecule inhibitor of the Aurora kinases, suppresses tumor growth in vivo. Nat Med. 2004;10:262–267. doi: 10.1038/nm1003. [DOI] [PubMed] [Google Scholar]
- 48.Sarvagalla S, Coumar MS. Structural biology insight for the design of sub-type selective Aurora kinase inhibitors. Curr Cancer Drug Targets. 2015;15:375–393. doi: 10.2174/1568009615666150421110401. [DOI] [PubMed] [Google Scholar]
- 49.Elkins JM, Santaguida S, Musacchio A, Knapp S. Crystal structure of human aurora B in complex with INCENP and VX-680. J Med Chem. 2012;55:7841–7848. doi: 10.1021/jm3008954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Littlepage LE, Wu H, Andresson T, Deanehan JK, Amundadottir LT, Ruderman JV. Identification of phosphorylated residues that affect the activity of the mitotic kinase Aurora-A. Proc Natl Acad Sci U S A. 2002;99:15440–15445. doi: 10.1073/pnas.202606599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cheetham GM, Knegtel RM, Coll JT, Renwick SB, Swenson L, Weber P, Lippke JA, Austen DA. Crystal structure of aurora-2, an oncogenic serine/threonine kinase. J Biol Chem. 2002;277:42419–42422. doi: 10.1074/jbc.C200426200. [DOI] [PubMed] [Google Scholar]
- 52.Kohrman AQ, Matus DQ. Divide or conquer: cell cycle regulation of invasive behavior. Trends Cell Biol. 2017;27:12–25. doi: 10.1016/j.tcb.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Peng CY, Graves PR, Thoma RS, Wu Z, Shaw AS, Piwnica-Worms H. Mitotic and G2 checkpoint control: regulation of 14-3-3 protein binding by phosphorylation of Cdc25C on serine-216. Science. 1997;277:1501–1505. doi: 10.1126/science.277.5331.1501. [DOI] [PubMed] [Google Scholar]
- 54.Wang TH, Wang HS, Soong YK. Paclitaxel-induced cell death: where the cell cycle and apoptosis come together. Cancer. 2000;88:2619–2628. doi: 10.1002/1097-0142(20000601)88:11<2619::aid-cncr26>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 55.Yoon MK, Mitrea DM, Ou L, Kriwacki RW. Cell cycle regulation by the intrinsically disordered proteins p21 and p27. Biochem Soc Trans. 2012;40:981–988. doi: 10.1042/BST20120092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Carvajal LA, Manfredi JJ. Another fork in the road-life or death decisions by the tumour suppressor p53. EMBO Rep. 2013;14:414–421. doi: 10.1038/embor.2013.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pitts TM, Bradshaw-Pierce EL, Bagby SM, Hyatt SL, Selby HM, Spreafico A, Tentler JJ, McPhillips K, Klauck PJ, Capasso A, Diamond JR, Davis SL, Tan AC, Arcaroli JJ, Purkey A, Messersmith WA, Ecsedy JA, Eckhardt SG. Antitumor activity of the aurora a selective kinase inhibitor, alisertib, against preclinical models of colorectal cancer. Oncotarget. 2016;7:50290–50301. doi: 10.18632/oncotarget.10366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ouyang L, Shi Z, Zhao S, Wang FT, Zhou TT, Liu B, Bao JK. Programmed cell death pathways in cancer: a review of apoptosis, autophagy and programmed necrosis. Cell Prolif. 2012;45:487–498. doi: 10.1111/j.1365-2184.2012.00845.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Estaquier J, Vallette F, Vayssiere JL, Mignotte B. The mitochondrial pathways of apoptosis. Adv Exp Med Biol. 2012;942:157–183. doi: 10.1007/978-94-007-2869-1_7. [DOI] [PubMed] [Google Scholar]
- 60.Logue SE, Martin SJ. Caspase activation cascades in apoptosis. Biochem Soc Trans. 2008;36:1–9. doi: 10.1042/BST0360001. [DOI] [PubMed] [Google Scholar]
- 61.Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 62.Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014;15:49–63. doi: 10.1038/nrm3722. [DOI] [PubMed] [Google Scholar]
- 63.Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol. 2008;9:231–241. doi: 10.1038/nrm2312. [DOI] [PubMed] [Google Scholar]
- 64.Grasso D, Garcia MN, Iovanna JL. Autophagy in pancreatic cancer. Int J Cell Biol. 2012;2012:760498. doi: 10.1155/2012/760498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yang S, Kimmelman AC. A critical role for autophagy in pancreatic cancer. Autophagy. 2011;7:912–913. doi: 10.4161/auto.7.8.15762. [DOI] [PubMed] [Google Scholar]
- 66.Yang MC, Loh JK, Li YY, Huang WS, Chou CH, Cheng JT, Wang YT, Lieu AS, Howng SL, Hong YR, Chou AK. Bcl2L12 with a BH3-like domain in regulating apoptosis and TMZ-induced autophagy: a prospective combination of ABT-737 and TMZ for treating glioma. Int J Oncol. 2015;46:1304–1316. doi: 10.3892/ijo.2015.2838. [DOI] [PubMed] [Google Scholar]
- 67.Catalano M, D’Alessandro G, Lepore F, Corazzari M, Caldarola S, Valacca C, Faienza F, Esposito V, Limatola C, Cecconi F, Di Bartolomeo S. Autophagy induction impairs migration and invasion by reversing EMT in glioblastoma cells. Mol Oncol. 2015;9:1612–1625. doi: 10.1016/j.molonc.2015.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rajagopalan V, Vaidyanathan M, Janardhanam VA, Bradner JE. Pre-clinical analysis of changes in intra-cellular biochemistry of glioblastoma multiforme (GBM) cells due to c-Myc silencing. Cell Mol Neurobiol. 2014;34:1059–1069. doi: 10.1007/s10571-014-0083-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rodon J, Dienstmann R, Serra V, Tabernero J. Development of PI3K inhibitors: lessons learned from early clinical trials. Nat Rev Clin Oncol. 2013;10:143–153. doi: 10.1038/nrclinonc.2013.10. [DOI] [PubMed] [Google Scholar]
- 70.Porta C, Paglino C, Mosca A. Targeting PI3K/Akt/mTOR signaling in cancer. Front Oncol. 2014;4:64. doi: 10.3389/fonc.2014.00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Melaiu O, Cristaudo A, Melissari E, Di Russo M, Bonotti A, Bruno R, Foddis R, Gemignani F, Pellegrini S, Landi S. A review of transcriptome studies combined with data mining reveals novel potential markers of malignant pleural mesothelioma. Mutat Res. 2012;750:132–140. doi: 10.1016/j.mrrev.2011.12.003. [DOI] [PubMed] [Google Scholar]
- 72.Rabinowitz JD, White E. Autophagy and metabolism. Science. 2010;330:1344–1348. doi: 10.1126/science.1193497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dunlop EA, Tee AR. The kinase triad, AMPK, mTORC1 and ULK1, maintains energy and nutrient homoeostasis. Biochem Soc Trans. 2013;41:939–943. doi: 10.1042/BST20130030. [DOI] [PubMed] [Google Scholar]
- 74.Denton D, Nicolson S, Kumar S. Cell death by autophagy: facts and apparent artefacts. Cell Death Differ. 2012;19:87–95. doi: 10.1038/cdd.2011.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Arthur JS, Ley SC. Mitogen-activated protein kinases in innate immunity. Nat Rev Immunol. 2013;13:679–692. doi: 10.1038/nri3495. [DOI] [PubMed] [Google Scholar]
- 76.Koul HK, Pal M, Koul S. Role of p38 MAP kinase signal transduction in solid tumors. Genes Cancer. 2013;4:342–359. doi: 10.1177/1947601913507951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang W, Fan H, Zhou Y, Duan P, Zhao G, Wu G. Knockdown of autophagy-related gene BECLIN1 promotes cell growth and inhibits apoptosis in the A549 human lung cancer cell line. Mol Med Rep. 2013;7:1501–1505. doi: 10.3892/mmr.2013.1379. [DOI] [PubMed] [Google Scholar]
- 78.Mester J, Eng C. When overgrowth bumps into cancer: the PTEN-opathies. Am J Med Genet C Semin Med Genet. 2013;163C:114–121. doi: 10.1002/ajmg.c.31364. [DOI] [PubMed] [Google Scholar]
- 79.Kurokawa C, Geekiyanage H, Allen C, Iankov I, Schroeder M, Carlson B, Bakken K, Sarkaria J, Ecsedy JA, D’Assoro A, Friday B, Galanis E. Alisertib demonstrates significant antitumor activity in bevacizumab resistant, patient derived orthotopic models of glioblastoma. J Neurooncol. 2017;131:41–48. doi: 10.1007/s11060-016-2285-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Daniele S, Sestito S, Pietrobono D, Giacomelli C, Chiellini G, Di Maio D, Marinelli L, Novellino E, Martini C, Rapposelli S. Dual inhibition of PDK1 and Aurora kinase A: an effective strategy to induce differentiation and apoptosis of human glioblastoma multiforme stem cells. ACS Chem Neurosci. 2017;8:100–114. doi: 10.1021/acschemneuro.6b00251. [DOI] [PubMed] [Google Scholar]