

Abstract
Tetrahydrocurcumin (THC) has been identified as a multi-functional neuroprotective agent in numerous neurological disorders. Oxidative stress as a result of injury may induce neuronal apoptosis after traumatic brain injury (TBI). Treatment with THC may improve neurological function following TBI by attenuating oxidative stress and apoptosis and by enhancing autophagy. The purpose of this study was to investigate the mechanism of neuroprotection by THC against oxidative stress-induced neuronal apoptosis after TBI. We hypothesized that neuroprotection by THC may involve modulation of autophagy and the mitochondria apoptotic pathway. We used western blot analysis to evaluate the effect of THC on proteins involved in mitochondrial autophagy and apoptosis after TBI. The terminal deoxynucleotidyl transferase dUTP nick end-labeling (TUNEL) assay and immunofluorescence staining were used to confirm the role of THC in apoptosis and autophagy, respectively. THC-induced neuroprotection was assessed by neurological severity scoring (NSS) and by measuring the brain water content. We demonstrated that treatment with THC increased expression of autophagy-associated proteins LC3-II and Beclin-1 at 24 h post-TBI. Treatment with THC also reduced expression of malondialdehyde (MDA) and increased glutathione peroxidase (GPx) activity. Further, treatment with THC attenuated apoptosis by modulating mitochondrial apoptosis and reducing oxidative stress. Treatment with 3-methyladenine (3-MA) mitigated autophagy activation and reversed the inhibitory effect of THC on the translocation of Bax to the mitochondrial membrane. Moreover, treatment with THC improved neurological function and reduced the brain water content in rats after TBI. We concluded that the neuroprotective effects of THC are mediated by enhancing autophagy activation and by attenuation of oxidative stress and apoptosis after TBI, probably by modulating the mitochondrial apoptotic pathway. We suggest that THC may be an effective therapeutic agent to treat TBI.
Keywords: Traumatic brain injury, tetrahydrocurcumin, oxidative stress, autophagy, mitochondrial apoptosis pathway
Introduction
Traumatic brain injury (TBI) is defined as a mechanical injury accompanied by serious complications. TBI causes numerous deaths, acute and chronic deficits in survivors, and has been found to have a devastating impact on families and society [1,2].
The mechanism of TBI involves both primary and secondary brain damage. Although the primary brain injury is the major factor, the secondary injury, which involves oxidative stress [4], provides the opportunity for clinical intervention [3]. Damaged mitochondria at the center of the injury produce excess reactive oxygen species (ROS) [5], and the dysfunctional mitochondria may be degraded by autophagy to partially protect neurons and to reduce the oxidative burden [6,7]. During autophagy, the membrane permeability of damaged mitochondria increases and mitochondrial apoptosis-associated proteins promote neuronal cell death [8], thus, enhancing autophagy may be an effective therapeutic strategy for TBI.
Curcumin (Cur) is abundant in the roots of the Curcuma longa Linn plant. An active metabolite of Cur, Tetrahydrocurcumin (THC), has been shown to have greater antioxidant activity than Cur and other curcuminoids in vitro and in vivo [9,10]. A previous study suggested that THC could prevent human leukemia by enhancing autophagy [11]. In cisplatin-induced oxidative renal damage, THC exhibited antioxidant and anti-apoptotic effects to protect the kidney from nephrotoxicity [12]. Moreover, intraperitoneal injection of THC was shown to improve cerebral ischemia through the modulation of autophagy [13]. However, the role of THC in the mitochondrial autophagy pathway in TBI remains unclear.
In this study, we investigated the effects of THC on mitochondrial apoptosis and autophagy in a TBI model to determine whether THC provides neuroprotection after TBI by reducing apoptosis and autophagy caused by damaged mitochondria.
Material and methods
Animal preparation
Adult male Sprague-Dawley (SD) rats weighing 250-300 g were purchased from the Experimental Animal Center of Jinling Hospital. All rats were allowed ad libitum access to food and water under conditions of controlled temperature (24 ± 0.5°C) in a 12 h light/dark cycle room. The experimental protocols were approved by the Institutional Animal Care and Use Committee at Jinling hospital and conformed to the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals.
TBI model in rats
The rat model of TBI used in this study was a modified version of the weight-drop model according to previous study [2]. Briefly, rats were placed in a stereotaxic frame after intraperitoneal anesthesia with sodium pentobarbital (50 mg/kg). After disinfection, a 2.0 cm midline scalp incision was made and the skull was exposed. On the left parietal cortex, a 6-mm craniotomy was performed through the skull; the center of the hole was 2.5 mm lateral to the midline on the mid-coronal plane. The dura was left intact during the operation. Impact was performed by releasing a 40 g weight onto the dura from a height of 25 cm along a steel shaft, which translated to 1000 × g/cm. By this time, the scalp wound was thoroughly disinfected and closed with suture. Sham animals were subjected to the same procedures without injury. Then rats were returned to the cages and maintained at a temperature of 24.0 ± 0.5°C.
Experimental design and drug administration
Rats were randomly assigned to four groups: The sham group, the TBI group, the TBI + vehicle group and the TBI + THC group (25 mg/kg). THC was purchased from Sigma-Aldrich (Saint Louis, MO, USA) and dissolved in saline containing 1% dimethyl sulfoxide. THC (5 mg/kg, 25 mg/kg, 50 mg/kg and 100 mg/kg) or equivalent volumes of vehicle (1% dimethyl sulfoxide in saline) were administered by intraperitoneal injection 30 min after TBI according to previous study [13]. To further confirm the effect of autophagy after TBI, rats were randomly assigned to the 3-MA group: TBI + THC (25 mg/kg) + 3-MA (200 nM). The 3-methyladenine (3-MA) (Sigma-Aldrich, M9281), used as an autophagy inhibitor, was dissolved in the same vehicle liquid and injected into the intracerebral ventricle according to a previous report [14].
Western blot analysis
Western blot analysis was performed as previously described [15]. The left cerebral cortical tissue was lysed in ice cold RIPA buffer containing 1% of a protease inhibitor cocktail. The protein content of the lysate was determined by the Bradford method. Equal amounts of proteins were resolved on a 10%-12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gel and transferred onto polyvinylidene fluoride (PVDF) membrane (Immobilon-P, Millipore Billerica MA, USA). The membrane was blocked with 5% non-fat dry milk in TBST (Tris-buffered saline with 0.05% Tween 20) for 2 h at room temperature, and then incubated overnight at 4°C, separately with the appropriate primary antibodies against the specific proteins. Specifically, LC3 at 1:1000 (#NB600-1384, Novus Biologicals, Minneapolis, MN, USA), Beclin-1 at 1:1000 (#NB500-249, Novus Biologicals), caspase-3 at 1:1000 (#9662, Cell Signaling Technology, Danvers, MA, USA), Bcl-2 at 1:1000 (#4223, Cell Signaling Technology), COX IV at 1:1000 (#11967, Cell Signaling Technology), Bax at 1:200 (#ab32503, Abcam, Cambridge, UK), cytochrome c at 1:10000 (#ab133504, Abcam), and β-actin at 1:5000 (Bioworld Technology, Saint Louis Park, MN, USA) in a blocking buffer. Afterwards, the membrane was washed three times with TBST for 15 min, and then incubated with the secondary antibodies, namely HRP conjugated secondary antibodies at 1:5000 (goat; Bioworld Technology) for 2 h at room temperature. Finally, following a 20-min wash with TBST, the protein bands were visualized via enhanced chemiluminescence (ECL) (Millipore, Billerica, MA, USA) and exposure to X-ray film. The western blot results were analyzed using Un-Scan-It 6.1 software (Silk Scientific Inc., Orem, UT, USA).
Determination of malondialdehyde (MDA) content and glutathione peroxidase (GPx) activity
The MDA content and GPx activity were evaluated according to previous study [4]. Tissue samples were homogenized in 2 ml phosphate buffer saline (PBS, pH 7.4) and centrifuged at 12,000 rpm for 15 min/4°C. First, we determined the concentrations of total protein by the Bradford method. Then, according to the manufacturer’s instructions of the commercial kits for MDA and GPx (Nanjing Jiancheng Biochemistry Co., Nanjing, China) detection, we measured the content of MDA and the activity of GPx using a spectrophotometer. The content of MDA was expressed as nmol/mg protein, and the activity of GPx was expressed as U/mg protein.
Terminal deoxynucleotidyl transferase dUTP nick end-labeling (TUNEL)
The brain tissue sections were examined for apoptotic cells using a terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) detection kit (Roche, South San Francisco, CA, USA) following the manufacturer’s instructions. Briefly, the sections were incubated with the TUNEL reagent for 1 h at 37°C. The sections were then washed three times in PBS, counterstained with DAPI for 2 min, and rinsed with PBS. Cover slips were applied with mounting media. Fluorescence was imaged on an Olympus IX71 inverted microscope system. The number of apoptotic cells to DAPI stained-cells was regarded as apoptotic index (apoptotic cells/DAPI). Six random vision fields (40 ×) surrounding contusion in each coronal section were chosen, and the mean number of apoptotic index in the six views was regarded as the data of each section. A total of four sections from each animal were used for quantification. The final average number of the four sections was regarded as the data for each sample. Data are presented as the mean number of apoptotic index per 40 × magnification field. All the processes were conducted by two investigators who were blinded to the grouping.
Immunofluorescence staining
The level of autophagy was assessed according to a previous immunostaining protocol as follows: the slides of each coronal sections were incubated in blocking buffer for 2 h, then washed with PBS three times for 10 min. Next, the slides were incubated with anti-LC3 antibody (1:200), in a dark place overnight at 4°C. Afterwards, the slides were washed three times with PBS and incubated with another antibody, namely anti-NeuN (1:100), under similar conditions. The following day, the slides were thoroughly washed with PBS and incubated with the corresponding secondary antibodies for 1 h. After the wash with PBS, the slides were stained with DAPI for 2 min to show the location of nucleus. Coverslips were applied with mounting media. The fluorescently-stained cells were imaged on an Olympus IX71 inverted microscope system and analyzed using the Image-Pro Plus 6.0 software (Media Cybernetics, Silver Spring, MD, USA).
Brain water content
Brain water content was measured as previously described [4]. Animals were anesthetized with sodium pentobarbital (50 mg/kg; i.p) and the brains were quickly dissected at 24 h after TBI. The cerebellum and brainstem were discarded, while the left cortical tissue was harvested. The wet weight of each cortical tissue was measured, then dried for 72 h at 80°C and the dry weight determined. The percentage of brain water content was calculated using the following formula = [(wet weight-dry weight)/wet weight] × 100%.
Assessment of neurological function
The neurological function of all rats before TBI and at 24 h after TBI were evaluated, according to previous studies [16,17], by three investigators who were blinded to the experimental groups. The grading of neurological severity scoring (NSS) was as follows: severe injury (score: 13-18); moderate injury (score: 7-12); mild injury (score: 1-6); no injury (score: 0) (Table 1).
Table 1.
Neurological severity scores (NSS)
| Points | |
|---|---|
| Motor tests | |
| Raising rat by the tail (normal = 0; maximum = 3) | |
| Flexion of forelimb | 1 |
| Flexion of hindlimb | 1 |
| Head moved > 10° to vertical axis within 30 s | 1 |
| Placing rat on the floor (normal = 0; maximum = 3) | |
| Normal walk | 0 |
| Inability to walk straight | 1 |
| Circling toward the paretic side | 2 |
| Fall down to the paretic side | 3 |
| Sensory tests (normal = 0; maximum = 2) | |
| Placing test (visual and tactile test) | 1 |
| Proprioceptive test (deep sensation, pushing the paw against the table edge to stimulate limb muscles) | 2 |
| Beam balance tests (normal = 0; maximum = 6) | |
| Balances with steady posture | 0 |
| Grasps side of beam | 1 |
| Hugs the beam and one limb falls down from the beam | 2 |
| Hugs the beam and two limbs fall down from the beam, or spins on beam (> 60 s) | 3 |
| Attempts to balance on the beam but falls off (> 40 s) | 4 |
| Attempts to balance on the beam but falls off (> 20 s) | 5 |
| Falls off: No attempt to balance or hang on to the beam (< 20 s) | 6 |
| Reflexes absent and abnormal movements (normal = 0; maximum = 4) | |
| Pinna reflex (head shake when touching the auditory meatus) | 1 |
| Corneal reflex (eye blink when lightly touching the cornea with cotton) | 1 |
| Startle reflex (motor response to a brief noise from snapping a clipboard paper) | 1 |
| Seizures, myoclonus, myodystony | 1 |
| Maximum points | 18 |
Statistical analysis
Data were analyzed with SPSS 17.0 software package (SPSS, Inc., Chicago, IL, USA) and the statistically significant difference was set at P < 0.05. Data are expressed as the mean ± Standard Deviation (SD). Comparisons between two groups were performed using Student’s t test and Multiple comparisons were performed using a one-way ANOVA followed by Tukey’s test.
Results
THC-induced autophagy in the injured cortex after TBI
We evaluated the effect of THC on autophagy with the established autophagy activation biological markers LC3 and Beclin-1. Previous studies [4,13] indicated that 24 h post-TBI is the most appropriate timepoint to evaluate autophagy in rats. Four doses (5, 25, 50, and 100 mg/kg) of THC were tested to evaluate the dose effect of THC on LC3 and Beclin-1 expression at 24 h post-TBI. LC3-II expression increased 30% in the TBI group when compared with the sham group (P < 0.01) (Figure 1A, 1B). Administration of 5 mg/kg of THC did not affect autophagy, but administration of 25 mg/kg of THC increased LC3-II expression (P < 0.001) (Figure 1A, 1B). As the THC concentration administered increased to 50 mg/kg and 100 mg/kg, LC3-II expression gradually declined relative to the 25 mg/kg group (P < 0.01) (Figure 1A, 1B), but remained higher than in the TBI group. Similarly, the highest level of Beclin-1 expression was reached with 25 mg/kg of THC at 24 h post-TBI (P < 0.01) (Figure 1A, 1C). Because the 25 mg/kg dose of THC showed the strongest effect on autophagy, we used this dose in the forthcoming experiments.
Figure 1.
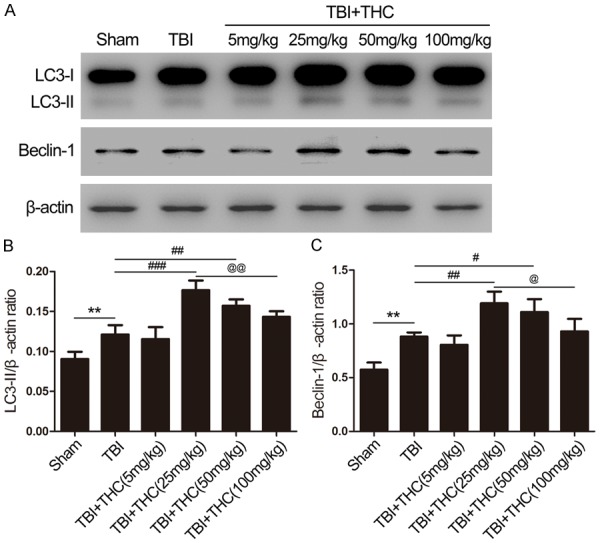
The effect of THC on autophagy in different groups. (A) The expression of LC3-II and Beclin-1 was assessed by western blot analysis in rats at 24 h after TBI. Quantitative analysis the ratios of LC3-II (B) and Beclin-1 (C), normalized against β-actin. Data are presented as mean ± SD (n = 5 rats per group). **P < 0.01 vs. Sham group; #P < 0.05, ##P < 0.01, ###P < 0.001 vs. TBI group; @P < 0.05, @@P < 0.01 vs. TBI + THC (25 mg/kg) group.
To confirm the optimal time at which THC-induced autophagy, we evaluated treatment with 25 mg/kg of THC at three timepoints (6, 24, and 72 h) post-TBI. LC3-II expression increased at 24 h post-TBI relative to the sham group (P < 0.01) (Figure 2A, 2B), whereas only minor alterations were seen at 6 h post-TBI relative to the TBI + vehicle group (P < 0.05) (Figure 2A, 2B). LC3-II expression was enhanced at the 24 h timepoint relative to the 6 h time point (P < 0.001), LC3-II expression decreased by the 72 h timepoint relative to the 24 h timepoint (P < 0.01) (Figure 2A, 2B). Beclin-1 expression exhibited a similar pattern; Beclin-1 expression began increasing 6 h post-TBI (P < 0.05) (Figure 2A, 2C), reached its highest level at 24 h post-TBI when compared with the 6 h timepoint (P < 0.05), and then gradually decreased (P < 0.01) (Figure 2A, 2C). These data confirmed utilization of the 24 h post-TBI timepoint in the forthcoming experiments.
Figure 2.
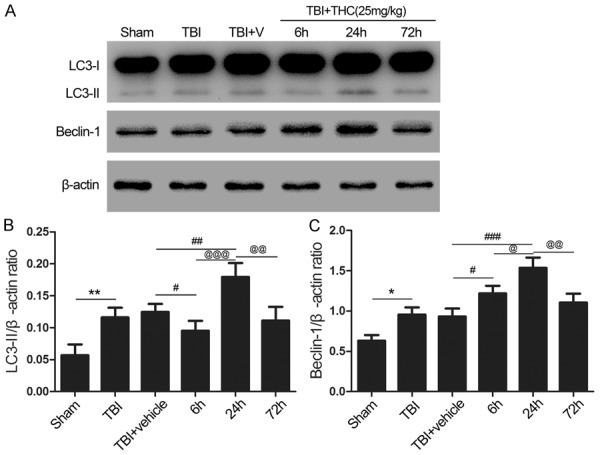
The appropriate time of THC on activation of autophagy after TBI in rats. (A) Western blot ssay of LC3-II and Beclin-1 expression in different groups. High expression of LC3-II and Beclin-1 were observed in the group of 24 h time point. Quantitative analysis the ratios of LC3-II (B) and Beclin-1 (C), normalized against β-actin. Data are presented as mean ± SD (n = 5 rats per group). *P < 0.05, **P < 0.01 vs. Sham group; #P < 0.05, ##P < 0.01, ###P < 0.001 vs. TBI + vehicle group; @P < 0.05, @@@P < 0.001 vs. 6 h group; @@P < 0.01 vs. 24 h group.
To further confirm the optimal dose and time for THC-induced autophagy post-TBI, four groups were evaluated: a sham group, a TBI group, a TBI + vehicle group, and a TBI + THC group. Evaluation of LC3 puncta upon treatment with 25 mg/kg of THC at 24 h post-TBI was performed by immunofluorescence. Only a few LC3-positive cells were present in the sham group, and the number of LC3-positive cells increased with TBI (Figure 3). Upon treatment with 25 mg/kg of THC post-TBI, the number of LC3-positive cells increased further. These data suggest that treatment with 25 mg/kg of THC enhances autophagy at 24 h post-TBI.
Figure 3.

Representing immunofluorescence images of LC3 and NeuN co-expression surrounding the injured cortex after THC treatment at 24 h with TBI in rats. In the cytoplasm, LC3 punctate dots was labeled with red. Neurons are labeled with NeuN (green) and nuclei of all cells are labeled with DAPI (blue). THC treatment significantly increased the proportion of LC3 positive neurons (red) after TBI. Bar = 20 µm.
THC treatment reduced oxidative stress in the injured cortex after TBI
To investigate the effect of THC on oxidative stress after TBI, we measured malondialdehyde (MDA) and glutathione peroxidase (GPx) levels in four groups of rats: a sham group, a TBI group, a TBI + vehicle group, and a TBI + THC group. MDA and GPx levels reflect lipid peroxidation and antioxidant levels, respectively. MDA levels were higher in the TBI and TBI + vehicle groups relative to the sham group (P < 0.01, P < 0.001) (Figure 4A). Treatment with 25 mg/kg of THC reduced MDA levels (P < 0.05) (Figure 4A). GPx activities were reduced in the TBI and TBI + vehicle groups relative to the sham group (P < 0.01) (Figure 4B), but treatment with THC rescued GPx activity after TBI (P < 0.01) (Figure 4B). These data suggest that THC alleviates oxidative stress and enhances antioxidant enzyme activity after TBI.
Figure 4.
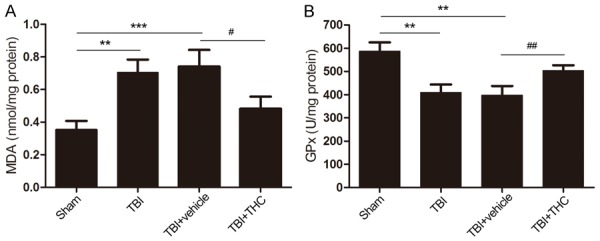
The effect of THC on anti-oxidative stress after TBI in rats. Measurements the level of malondialdehyde (MDA) (A) and the activity of glutathione peroxidase (GPx) (B) in different groups. Data are presented as mean ± SD (n = 5 rats per group). **P < 0.01, ***P < 0.001 vs. Sham group; #P < 0.05, ##P < 0.01 vs. TBI + vehicle group.
THC reduced apoptosis in the injured cortex after TBI
To determine whether treatment with 25 mg/kg of THC could reduce apoptosis in the injured cortex 24 h post-TBI, we used the terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end-labeling (TUNEL) immunofluorescence staining assay to detect apoptotic cells. Although only a few TUNEL-positive cells were found in the cortex of the sham group, the apoptotic index of the cortex increased after TBI (P < 0.001) (Figure 5A, 5B). THC treatment reversed this post-TBI apoptotic index; the number of TUNEL-positive cells decreased in the TBI + THC group relative to the TBI + vehicle group (P < 0.05) (Figure 5B). No difference in the number of apoptotic cells was found between the TBI group and the TBI + vehicle group. These data suggest that THC treatment could reduce TBI-induced apoptosis.
Figure 5.
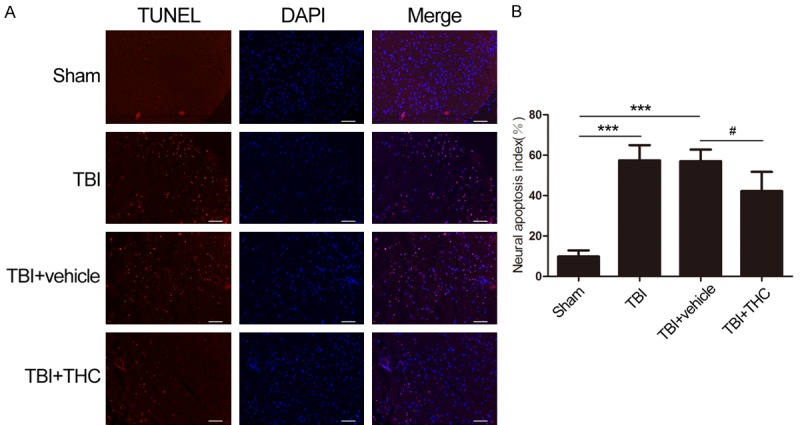
The effect of THC on anti-apoptosis was evaluated by TUNEL staining after TBI in rats. A. Representing immunofluorescence images that showed similar results demonstrating the effect of THC on the anti-apoptosis at 24 h induced by TBI. Apoptotic neurons are labeled with red, surrounding the injured site and nuclei are labeled with DAPI (blue). B. The ratio of positive cells against neural cells. Data are presented as mean ± SD (n = 3 rats per group). Bar = 20 µm. ***P < 0.001 vs. Sham group; #P < 0.05 vs. TBI + vehicle group.
We also used western blot analysis to examine expression of cleaved caspase-3, an indicator of apoptosis. Cleaved caspase-3 expression was higher at 24 h post-TBI relative to the sham group (P < 0.001) (Figure 6A, 6B). Additionally, treatment with THC reduced the cleaved caspase-3 level relative to the TBI + vehicle group (P < 0.05) (Figure 6A, 6B). These results indicate that 25 mg/kg of THC could reduce the level of apoptosis at 24 h post-TBI.
Figure 6.
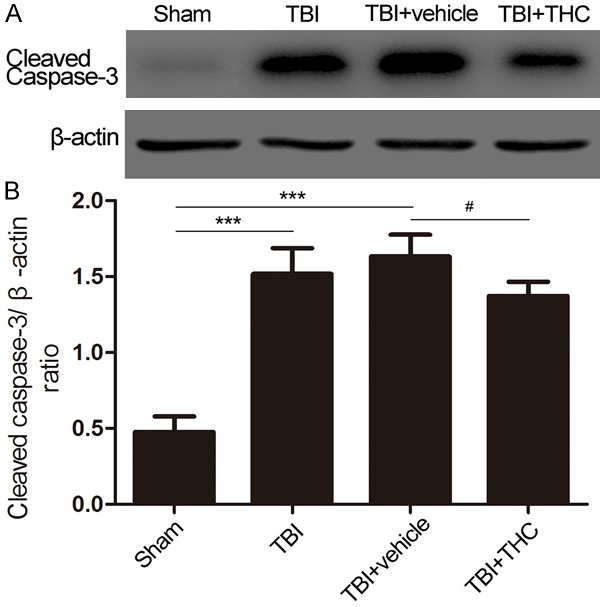
The effect of THC on anti-apoptosis was assessed at 24 h after TBI in rats. A. The expression of cleaved caspase-3 was evaluated in different groups by western blot analysis. B. Quantitative analysis the ratio of cleaved caspase-3, normalized against β-actin. Data are presented as mean ± SD (n = 5 rats per group). ***P < 0.001 vs. Sham group; #P < 0.05 vs. TBI + vehicle group.
THC altered expression of apoptotic factors in the injured cortex after TBI
To determine whether the anti-apoptotic effect of the THC treatment is connected to the mitochondrial apoptotic pathway, we examined expression of biological markers of the mitochondrial apoptotic pathway, namely Bax, Bcl-2, and cytochrome c upon TBI. When mitochondria were injured by TBI, expression of the pro-apoptotic factor Bax increased when compared with the sham group (P < 0.001) (Figure 7A-E), expression of the anti-apoptotic factor Bcl2 decreased when compared with the sham group (P < 0.001), and cytochrome c was released from the mitochondria into the cytoplasm. However, treatment with 25 mg/kg of THC at 24 h post-TBI reversed the expression levels of mitochondrial Bax and Bcl-2 relative to the TBI + vehicle group (P < 0.05, P < 0.01) (Figure 7A-E). Expression of mitochondrial cytochrome c was lower in the TBI and TBI + vehicle groups when compared with the sham group (P < 0.001) (Figure 7A, 7G). Administration of 25 mg/kg of THC increased mitochondrial cytochrome c expression post-TBI (P < 0.01).
Figure 7.
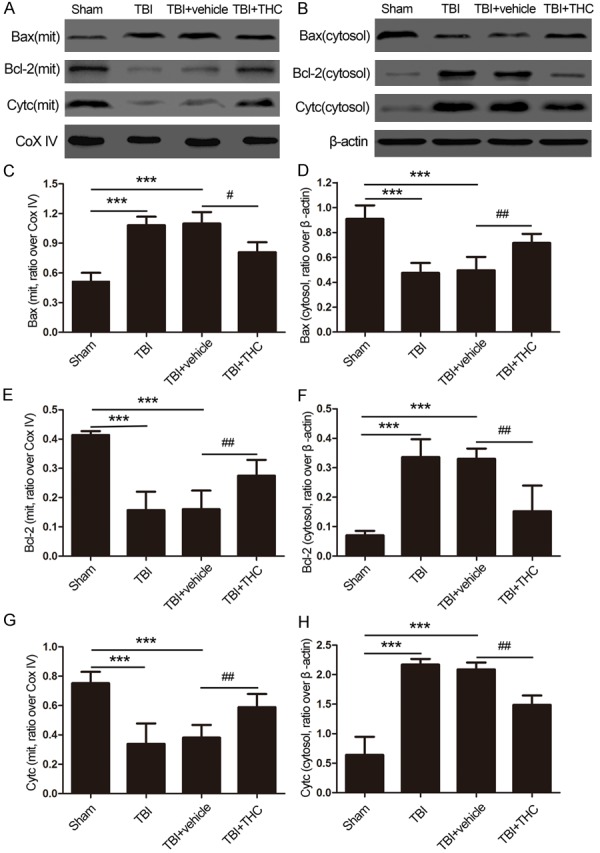
The effect of THC on mitochondrial apoptotic associated proteins at 24 h after TBI in rats. The expression of Bax, Bcl-2 and cytochrome c in mitochondria (A) and in cytoplasm (B) was evaluated by western blot analysis. Quantitative analysis the ratios of Bax (C), Bcl-2 (E) and cytochrome c (G) in mitochondria and in cytoplasm (D, F, H), normalized against Cox IV or β-actin. Data are presented as mean ± SD (n = 5 rats per group). ***P < 0.001 vs. Sham group; #P < 0.05, ##P < 0.01 vs. TBI + vehicle group.
We also evaluated cytoplasmic protein levels. The level of cytoplasmic Bax protein decreased, whereas the levels of cytoplasmic Bcl-2 and cytochrome c proteins increased at 24 h post-TBI relative to the sham group (P < 0.001) (Figure 7B, 7D, 7F, 7H). After treatment with 25 mg/kg of THC, the level of cytoplasmic Bax protein increased, whereas the levels of cytoplasmic Bcl-2 and cytochrome c proteins decreased when compared with the TBI + vehicle group (P < 0.01) (Figure 7B, 7D, 7F, 7H). These results indicate that 25 mg/kg of THC partially inhibits the release of cytochrome c and the expression of pro-apoptotic factors, and increases the expression of anti-apoptotic factors in the mitochondria.
THC suppressed apoptosis through modulation of autophagy in the injured cortex after TBI
We found that autophagy was activated upon treatment with 25 mg/kg of THC at 24 h post-TBI. To determine whether this autophagy activation is associated with the mitochondrial apoptotic pathway, we used the autophagy inhibitor 3-MA and examined the expression of mitochondrial apoptosis-associated proteins by western blot analysis. Expression of two autophagy markers, Beclin-1 and LC3-II, were reduced in the TBI + THC + 3-MA group when compared with the TBI + THC group (P < 0.001) (Figure 8A-C). Similarly, treatment with THC + 3-MA reduced expression of Bax, a protein associated with the mitochondrial apoptotic pathway, relative to the group treated with THC alone (P < 0.01) (Figure 8D, 8F). In addition, treatment with THC + 3-MA increased the level of cleaved caspase-3 relative to treatment with THC alone (P < 0.05) (Figure 8D, 8E). These data indicate that THC suppresses the mitochondrial apoptotic pathway by activating autophagy after TBI.
Figure 8.

The effect of THC on mitochondrial apoptotic associated proteins after autophagy inhibitor 3-MA treatment at 24 h following TBI. (A) Western blot assay of Beclin-1 and LC3-II expression in different groups. Quantitative analysis the ratios of Beclin-1 (B) and LC3-II (C) in, normalized against β-actin. (D) The expression of cleaved caspase-3 and Bax was evaluated by western blot analysis. Quantitative analysis the ratios of cleaved caspase-3 (E) and Bax (F), normalized against β-actin. Data are presented as mean ± SD (n = 5 rats per group). **P < 0.01, ***P < 0.001 vs. Sham group; ##P < 0.01, ###P < 0.001 vs. TBI group; @P < 0.05, @@P < 0.01, @@@P < 0.001 vs. TBI + THC group.
THC improved neurological function and ameliorated cerebral edema in the injured hemisphere after TBI
Neurological function and brain water content were analyzed to evaluate the neuroprotective effect of 25 mg/kg of THC at 24 h post-TBI. We used the neurological severity scoring (NSS) system to evaluate neural impairment, and we measured the brain water content to determine the degree of brain edema. All of the rats were scored as 0 before the operation. We found that the NSS and brain water content increased after TBI treatment relative to the sham group (P < 0.001) (Figure 9A, 9B). Treatment with 25 mg/kg of THC improved neurological function and reduced the brain water content when compared with the TBI + vehicle group (P < 0.01) (Figure 9A, 9B). These data demonstrate that THC provides neuroprotection after TBI in rats.
Figure 9.
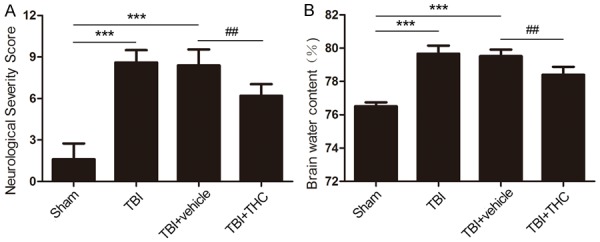
Evaluation the effect of THC on neuroprotection at 24 h with TBI in rats. Neurological assessment by neurological severity score (A) and measurements the level of brain water content (B) in different groups. Data are presented as mean ± SD (n = 7 rats per group). ***P < 0.001 vs. Sham group; ##P < 0.01 vs. TBI + vehicle group.
Discussion
In this study, we investigated the neuroprotective effects of THC against TBI-induced secondary brain injury in a rat model of TBI. The major findings of this include: 1) In the injured cortex of the rat, autophagy increased at 24 h post-TBI and treatment with 25 mg/kg of THC treatment further enhanced autophagy. 2) Treatment with THC reduced oxidative stress and apoptosis of neuronal cells, probably via modulation of autophagy and mitochondrial apoptotic pathways. 3) Treatment with THC attenuated the brain water content and improved neurological function at 24 h post-TBI.
Previous studies demonstrated that THC penetrated the blood brain barrier after peripheral injection [18], exhibited potent antioxidant properties, and modulated autophagy [18,19]. In cerebral ischemia [13] and neurodegenerative diseases [19], THC showed neuroprotective effects by regulating autophagy and attenuating oxidative stress. In addition, THC-induced autophagy in human leukemia HL-60 cells, thereby exerting an anticancer effect [11]. Furthermore, we found that intraperitoneal injection with 25 mg/kg of THC 30 min post-TBI activated autophagy. We also found that THC treatment was optimal at 24 h post-TBI, which is consistent with a previous study [20].
Oxidative stress, a pathology resulting from secondary brain injury, is caused by accumulation of ROS after TBI [4]. During normal respiration, mitochondria produce ROS to oxidize proteins and DNA [5]. Damaged mitochondria at the center of an injury produces excess ROS, which obstructs mitochondrial function and upsets the normal balance of endogenous oxidant and antioxidant mechanisms [21]. To evaluate the level of oxidative stress after TBI, MDA and GPx, markers of lipid peroxidation and antioxidation, were measured [22]. We showed that treatment with 25 mg/kg of THC reduced the MDA level and induced GPx activity indicating that THC functions as an antioxidant after TBI.
In the pathophysiology of TBI, dysfunctional mitochondria can promote cell death through the mitochondrial apoptotic pathway [23-25]. Briefly, an increase in expression of pro-apoptotic factors relative to expression of anti-apoptotic BCL2 family proteins leads to BAX-BAK dependent pore formation and increases mitochondrial permeability. Afterwards, killer proteins, such as cytochrome c, are released into the cytoplasm from the mitochondrial intermembrane space and trigger a cascade of molecular interactions, including caspases-3 activation, leading to degradation of DNA and essential proteins, which ultimately results in cell death [22,26,27]. Our data indicate that treatment with 25 mg/kg of THC inhibits the translocation of Bax to the mitochondrial membrane, increases the expression of the anti-apoptotic protein Bcl-2 in the mitochondria, and reduces the release of cytochrome c from the mitochondria to the cytoplasm. We also showed that THC protects neurons from the mitochondrial apoptotic pathway following TBI.
Autophagy has been found to be involved in various brain injury diseases, including neurodegenerative diseases [28-30] and cerebral ischemia [31-33]. It is still unclear whether autophagy functions as a cell death or a cell survival pathway [34]. Clark found that autophagy increased in human brain tissue after TBI and that oxidative stress exacerbated neuropathological damage in mice after TBI by influencing autophagy [35]. Viscomi found that activation of autophagy by rapamycin provided neuroprotection by inhibiting the release of cytochrome c after CNS focal damage [36]. We showed that treatment with THC enhanced activation of autophagy and protected the brain from mitochondrial apoptosis in a rat model of TBI.
Our study had several limitations. First, when examining the relationship between autophagy and apoptosis, we only used the autophagy inhibitor 3-MA, but to be more convincing, we should have used additional autophagy inhibitors, such as Wortmannin or Becn+/- rats. Second, we only showed the anti-apoptotic effect of THC by inhibiting the mitochondrial apoptotic pathway via activation of autophagy. Additional regulatory mechanisms of autophagy and apoptosis should be investigated. Third, we did not examine the effects of THC in vitro, thus further studies are required to verify our findings.
In conclusion, we demonstrated that treatment with THC provides neuroprotection by improving neurological function and ameliorating cerebral edema in a rat model of TBI. Moreover, administration of THC reduced oxidative stress and reduced the number of apoptotic neurons following TBI. THC also activated autophagy and inhibited the mitochondrial apoptotic pathway after TBI. Treatment with the autophagy inhibitor 3-MA confirmed the relationship between autophagy and the mitochondrial apoptosis pathway. We suggest that modulation of autophagy may be an effective therapeutic strategy for TBI.
Acknowledgements
This work was supported by grants from Jinling Hospital of Nanjing, School of Medicine, Nanjing University (No. 2010 M023) and the Major Innovative Project of Medical Science and Technology of Nanjing Military Command (No. 14ZX16) and the Project of Nanjing Science and Technology Development (No. 201605078) and the National Natural Science Foundation, China (No. 81400980).
Disclosure of conflict of interest
None.
References
- 1.Xu J, Wang H, Ding K, Zhang L, Wang C, Li T, Wei W, Lu X. Luteolin provides neuroprotection in models of traumatic brain injury via the Nrf2-ARE pathway. Free Radic Biol Med. 2014;71:186–195. doi: 10.1016/j.freeradbiomed.2014.03.009. [DOI] [PubMed] [Google Scholar]
- 2.Xiong Y, Mahmood A, Chopp M. Animal models of traumatic brain injury. Nat Rev Neurosci. 2013;14:128–142. doi: 10.1038/nrn3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ding K, Wang H, Xu J, Li T, Zhang L, Ding Y, Zhu L, He J, Zhou M. Melatonin stimulates antioxidant enzymes and reduces oxidative stress in experimental traumatic brain injury: the Nrf2-ARE signaling pathway as a potential mechanism. Free Radic Biol Med. 2014;73:1–11. doi: 10.1016/j.freeradbiomed.2014.04.031. [DOI] [PubMed] [Google Scholar]
- 4.Wei W, Wang H, Wu Y, Ding K, Li T, Cong Z, Xu J, Zhou M, Huang L, Ding H, Wu H. Alpha lipoic acid inhibits neural apoptosis via a mitochondrial pathway in rats following traumatic brain injury. Neurochem Int. 2015;87:85–91. doi: 10.1016/j.neuint.2015.06.003. [DOI] [PubMed] [Google Scholar]
- 5.Hagberg H, Mallard C, Rousset CI, Thornton C. Mitochondria: hub of injury responses in the developing brain. Lancet Neurology. 2014;13:217–232. doi: 10.1016/S1474-4422(13)70261-8. [DOI] [PubMed] [Google Scholar]
- 6.Maiuri MC, Criollo A, Kroemer G. Crosstalk between apoptosis and autophagy within the Beclin 1 interactome. EMBO J. 2010;29:515–516. doi: 10.1038/emboj.2009.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen J, Wang Q, Yin FQ, Zhang W, Yan LH, Li L. MTRR silencing inhibits growth and cisplatin resistance of ovarian carcinoma via inducing apoptosis and reducing autophagy. Am J Transl Res. 2015;9:1510–1527. [PMC free article] [PubMed] [Google Scholar]
- 8.Galluzzi L, Blomgren K, Kroemer G. Mitochondrial membrane permeabilization in neuronal injury. Nat Rev Neurosci. 2009;10:481–494. doi: 10.1038/nrn2665. [DOI] [PubMed] [Google Scholar]
- 9.Blommaart EF, Luiken JJ, Meijer AJ. Autophagic proteolysis: control and specificity. Histochem J. 1997:365–385. doi: 10.1023/a:1026486801018. [DOI] [PubMed] [Google Scholar]
- 10.Fengsrud M, Roos N, Berg T, Liou W, Slot JW, Seglen PO. Ultrastructural and immunocytochemical characterization of autophagic vacuoles in isolated hepatocytes: effects of vinblastine and asparagine on vacuole distributions. Exp Cell Res. 1995;221:504–519. doi: 10.1006/excr.1995.1402. [DOI] [PubMed] [Google Scholar]
- 11.Wu JC, Lai CS, Badmaev V, Nagabhushanam K, Ho CT, Pan MH. Tetrahydrocurcumin, a major metabolite of curcumin, induced autophagic cell death through coordinative modulation of PI3K/Akt-mTOR and MAPK signaling pathways in human leukemia HL-60 cells. Mol Nutr Food Res. 2011;55:1646–1654. doi: 10.1002/mnfr.201100454. [DOI] [PubMed] [Google Scholar]
- 12.Song KI, Park JY, Lee S, Lee D, Jang HJ, Kim SN, Ko H, Kim HY, Lee JW, Hwang GS, Kang KS, Yamabe N. Protective effect of tetrahydrocurcumin against cisplatin-induced renal damage: in vitro and in vivo studies. Planta Med. 2015;81:286–291. doi: 10.1055/s-0035-1545696. [DOI] [PubMed] [Google Scholar]
- 13.Tyagi N, Qipshidze N, Munjal C, Vacek JC, Metreveli N, Givvimani S, Tyagi SC. Tetrahydrocurcumin ameliorates homocysteinylated cytochrome-c mediated autophagy in hyperhomocysteinemia mice after cerebral ischemia. J Mol Neurosci. 2012;47:128–138. doi: 10.1007/s12031-011-9695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang L, Ding K, Wang H, Wu Y, Xu J. Traumatic brain injury-induced neuronal apoptosis is reduced through modulation of pi3k and autophagy pathways in mouse by FTY720. Cell Mol Neurobiol. 2016;36:131–142. doi: 10.1007/s10571-015-0227-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ulbrich F, Lerach T, Biermann J, Kaufmann KB, Lagreze WA, Buerkle H, Loop T, Goebel U. Argon mediates protection by Interleukin-8 suppression via a TLR2/TLR4/STAT3/NF-kappaB pathway in a model of apoptosis in neuroblastoma cells in-vitro and following ischemia-reperfusion injury in rat retina in-vivo. J Neurochem. 2016;138:859–73. doi: 10.1111/jnc.13662. [DOI] [PubMed] [Google Scholar]
- 16.Chen J, Li Y, Wang L, Zhang Z, Lu D, Lu M, Chopp M. Therapeutic benefit of intravenous administration of bone marrow stromal cells after cerebral ischemia in rats. Stroke. 2001;32:1005–1011. doi: 10.1161/01.str.32.4.1005. [DOI] [PubMed] [Google Scholar]
- 17.Liu F, Chen MR, Liu J, Zou Y, Wang TY, Zuo YX, Wang TH. Propofol administration improves neurological function associated with inhibition of pro-inflammatory cytokines in adult rats after traumatic brain injury. Neuropeptides. 2016;58:1–6. doi: 10.1016/j.npep.2016.03.004. [DOI] [PubMed] [Google Scholar]
- 18.Cheng KK, Yeung CF, Ho SW, Chow SF, Chow AH, Baum L. Highly stabilized curcumin nanoparticles tested in an in vitro blood-brain barrier model and in Alzheimer’s disease Tg2576 mice. AAPS J. 2013;15:324–336. doi: 10.1208/s12248-012-9444-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mishra S, Mishra M, Seth P, Sharma SK. Tetrahydrocurcumin confers protection against amyloid beta-induced toxicity. Neuroreport. 2011;22:23–27. doi: 10.1097/WNR.0b013e328341e141. [DOI] [PubMed] [Google Scholar]
- 20.Ding K, Wang H, Wu Y, Zhang L, Xu J, Li T, Ding Y, Zhu L, He J. Rapamycin protects against apoptotic neuronal death and improves neurologic function after traumatic brain injury in mice via modulation of the mTOR-p53-Bax axis. J Surg Res. 2015;194:239–247. doi: 10.1016/j.jss.2014.09.026. [DOI] [PubMed] [Google Scholar]
- 21.Grivennikova VG, Kareyeva AV, Vinogradov AD. What are the sources of hydrogen peroxide production by heart mitochondria? Biochim Biophys Acta. 2010;1797:939–944. doi: 10.1016/j.bbabio.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robertson CL, Scafidi S, McKenna MC, Fiskum G. Mitochondrial mechanisms of cell death and neuroprotection in pediatric ischemic and traumatic brain injury. Exp Neurol. 2009;218:371–380. doi: 10.1016/j.expneurol.2009.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blomgren K, Hagberg H. Free radicals, mitochondria, and hypoxia-ischemia in the developing brain. Free Radic Biol Med. 2006;40:388–397. doi: 10.1016/j.freeradbiomed.2005.08.040. [DOI] [PubMed] [Google Scholar]
- 24.Ma J, Shui S, Han X, Guo D, Li T, Yan L. microRNA 22 attenuates neuronal cell apoptosis in a cell model of traumatic brain injury. Am J Transl Res. 2016;4:1895–1902. [PMC free article] [PubMed] [Google Scholar]
- 25.Zhen Y, Ding C, Sun J, Wang Y, Li S, Dong L. Activation of the calcium-sensing receptor promotes apoptosis by modulating the JNK/p38 MAPK pathway in focal cerebral ischemia-reperfusion in mice. Am J Transl Res. 2016;2:911–921. [PMC free article] [PubMed] [Google Scholar]
- 26.Rasola A, Sciacovelli M, Pantic B, Bernardi P. Signal transduction to the permeability transition pore. FEBS Lett. 2010;584:1989–1996. doi: 10.1016/j.febslet.2010.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Q, Bian H, Guo L, Zhu H. Pharmacologic preconditioning with berberine attenuating ischemia-induced apoptosis and promoting autophagy in neuron. Am J Transl Res. 2016;2:1197–1207. [PMC free article] [PubMed] [Google Scholar]
- 28.Rubinsztein DC, DiFiglia M, Heintz N, Nixon RA, Qin ZH, Ravikumar B, Stefanis L, Tolkovsky A. Autophagy and its possible roles in nervous system diseases, damage and repair. Autophagy. 2005;1:11–22. doi: 10.4161/auto.1.1.1513. [DOI] [PubMed] [Google Scholar]
- 29.Qin ZH, Wang Y, Kegel KB, Kazantsev A, Apostol BL, Thompson LM, Yoder J, Aronin N, DiFiglia M. Autophagy regulates the processing of amino terminal huntingtin fragments. Hum Mol Genet. 2003;12:3231–3244. doi: 10.1093/hmg/ddg346. [DOI] [PubMed] [Google Scholar]
- 30.Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–1295. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- 31.Adhami F, Liao G, Morozov YM, Schloemer A, Schmithorst VJ, Lorenz JN, Dunn RS, Vorhees CV, Wills-Karp M, Degen JL, Davis RJ, Mizushima N, Rakic P, Dardzinski BJ, Holland SK, Sharp FR, Kuan CY. Cerebral ischemia-hypoxia induces intravascular coagulation and autophagy. Am J Pathol. 2006;169:566–583. doi: 10.2353/ajpath.2006.051066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koike M, Shibata M, Tadakoshi M, Gotoh K, Komatsu M, Waguri S, Kawahara N, Kuida K, Nagata S, Kominami E, Tanaka K, Uchiyama Y. Inhibition of autophagy prevents hippocampal pyramidal neuron death after hypoxic-ischemic injury. Am J Pathol. 2008;172:454–469. doi: 10.2353/ajpath.2008.070876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Balduini W, Carloni S, Buonocore G. Autophagy in hypoxia-ischemia induced brain injury. Autophagy. 2009;5:221–223. doi: 10.4161/auto.5.2.7363. [DOI] [PubMed] [Google Scholar]
- 34.Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science. 2004;306:990–995. doi: 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Clark RS, Bayir H, Chu CT, Alber SM, Kochanek PM, Watkins SC. Autophagy is increased in mice after traumatic brain injury and is detectable in human brain after trauma and critical illness. Autophagy. 2014;4:88–90. doi: 10.4161/auto.5173. [DOI] [PubMed] [Google Scholar]
- 36.Viscomi MT, D’Amelio M, Cavallucci V, Latini L, Bisicchia E, Nazio F, Fanelli F, Maccarrone M, Moreno S, Cecconi F, Molinari M. Stimulation of autophagy by rapamycin protects neurons from remote degeneration after acute focal brain damage. Autophagy. 2012;8:222–235. doi: 10.4161/auto.8.2.18599. [DOI] [PubMed] [Google Scholar]