

Abstract
The blood-brain barrier (BBB) plays important roles in the recovery of traumatic brain injury (TBI) which is a major factor contributing to cerebral edema. Acid fibroblast growth factor (aFGF) contributes to maintain vascular integrity and restores nerve function. However, whether aFGF protects BBB following TBI remains unknown. The purpose of this study was to determine whether exogenous aFGF preserves BBB integrity by activating the PI3K-Akt-Rac1 pathway and inhibiting RhoA after TBI. BBB permeability was assessed using evans blue dye and fluorescein isothiocyanate dextran fluorescence. Neurofunctional tests, such as the garcia test, were conducted in a blinded fashion, and protein expression was evaluated via western blotting and immunofluorescence staining. Our results showed that aFGF improved neurofunctional deficits, preserved BBB integrity, and up-regulated tight junction proteins and adherens junction proteins 24 h after experimental TBI. However, the PI3K/Akt inhibitor LY294002 reversed the protective effects of aFGF on neurofunctional deficits and junction protein expression and significantly suppressed p-Akt and GTP-Rac1 activity. Furthermore, aFGF administration significantly decreased GTP-RhoA expression in the treated group compared with the vehicle group, while PI3K/Akt inhibition increased GTP-RhoA expression. Similar results were observed in vitro, as aFGF exerted protective effects on endothelial cell integrity by up-regulating junction proteins and PI3K-Akt-Rac1 pathway and down-regulating RhoA expression under oxygen-glucose deprivation/reoxygenation (OGD) conditions. These data suggest that exogenous aFGF reduces RhoA activity in part by activating the PI3K-Akt-Rac1 signaling pathway, thus improving neurofunctional deficits and preserving BBB integrity after TBI.
Keywords: Traumatic brain injury, blood-brain barrier, acid fibroblast growth factor, junction protein, RhoA
Introduction
Traumatic brain injury (TBI) occurs when a physical force causes brain dysfunction and pathology and it is a leading cause of death, disability, and stroke-related emergencies [1]. TBI comprises primary and secondary injuries. Secondary injuries occur over a period of time following an initial trauma and result from the activation of different pathways related to coagulation, inflammation, oxidation, and apoptosis [2]. Furthermore, brain edema severity is highly correlated with neurofunctional deficit and disability severity in TBI patients [3].
The blood-brain barrier (BBB) is maintained primarily by complex tight junctions (TJs) and specific solute carriers that regulate brain microenvironment homeostasis to ensure appropriate neuronal activity. TJs and adherens junctions (AJs) are the two main types of proteins that constitute BBB. TJs are composed of multiple protein components, such as occludins and claudins, and junction-related molecules linked to the actin cytoskeleton via zonula occludens (ZO) proteins, which play a critical role in maintaining BBB integrity and paracellular function [4]. The AJ components p120-catenin, β-catenin and VE-cadherin are endothelial cell molecules which are necessary for the maintenance of endothelial stability and BBB integrity [5]. AJs initiate brain endothelial cell-to-cell contact and promote cell maturation, maintenance, and plasticity and regulate tensile forces. Although TJs and AJs are essential and irreplaceable components of BBB in the central nervous system (CNS), the exact mechanism underlying their regulatory effects on BBB permeability remains unknown.
Previous studies [6] have shown that increases in BBB permeability are associated with junctional protein phosphorylation. In addition, increases in BBB permeability stimulate the recycling of junctional molecules, proteinase activated receptors and the subsequent activation of downstream effectors, such as ras homolog gene family member A (RhoA), phosphatidylinositol 3-kinase (PI3K), Rho-associated coiled-coil forming protein serine/threonine kinase (ROCK) of cytoskeletal dynamics and ras-related C3 botulinum toxin substrate 1 (Rac1) [7,8]. Studies on experimental TBI [9] have also shown that Rho family GTPase activation results in the disassembly of TJs and AJs, thus increasing BBB damage and ultimately causing vasogenic edema. In addition, the PI3K/Akt pathway is required for barrier function stability. According to a recent study [10], insulin stabilizes endothelial barrier function via Rac1 activation induced by PI3K/Akt pathway activity. Rac1 preserves and stabilizes endothelial cell barrier function, while RhoA antagonizes endothelial barrier function. However, the mechanism underlying the crosstalk among the PI3K/Akt pathway, Rac1 and RhoA in the setting of BBB disruption in mice after TBI remains unknown.
Human acidic fibroblast growth factor (aFGF) stimulates angiogenesis and proliferation of fibroblasts, which forms granulation tissue during the wound healing process. Interest in determining how aFGF affects the nervous system has inspired more clinical and animal studies. Some previous studies [11] have shown that FGF receptor (FGFR) signaling has an intimate relationship with the adhesion molecules of BBB and that aFGF activates Rac1 through the PI3K/Akt signaling pathway in human corneal endothelial cells and bone marrow stromal cells. However, no study has provided evidence regarding whether aFGF participates in BBB protection following TBI.
The purpose of the present study is to determine whether aFGF preserves BBB integrity and reduces neurofunctional deficits by activating the PI3K-Akt-Rac1 pathway and inhibiting RhoA after TBI. Evans blue (EB) dye, fluorescein isothiocyanate (FITC)-dextran fluorescence, garcia test, western blotting and immunofluorescence were used to evaluate BBB integrity and neurofunction and to quantify the levels of activated Akt, Rac1, RhoA, TJs and AJs expression on both an experimental mouse TBI model and an OGD-treated endothelial cell model. We found that exogenous aFGF administration attenuated experimental TBI in mice, specifically by preserving endothelial TJs and AJs, reducing vasogenic brain edema and attenuating neuronal dysfunction and BBB damage. We also found that aFGF induced PI3K-Akt-Rac1 signaling pathway activation and consequently inhibited RhoA GTPase activity, thus preserving endothelial TJs and AJs.
Materials and methods
Reagents and antibodies
aFGF was purchased from Zhejiang Grostre Biotechnology Co., Ltd. Flux of fluorescein isothiocyanate labeled dextran (FITC-dextran), PI3K inhibitor LY294002 and evans blue (EB)were purchased from Sigma (Sigma-Aldrich, St. Louis, MO). Anti-β-catenin and anti-p120-catenin were purchased from Abcam (Abcam, Cambridge, MA). Anti-claudin-5, anti-occludin, anti-Akt and anti-p-Akt (Ser473), anti-zonula occludens (ZO)-1, and the appropriate secondary antibodies were from Santa Cruz Biotechnology. GTP-Rac1, total-Rac1, GTP-RhoA and total-RhoA were detected using Rac1/Cdc42 and Rho Activation Assay Kits (Millipore, Temecula, CA). Mouse brain endothelial cells (bEnd.3) were purchased from Sciencell (Carlsbad, CA, USA).
Animal treatment
C57BL/6N male mice weighing 20-25 g were purchased from the Animal Center of the Chinese Academy of Sciences (Shanghai, China). All animal experiments were approved by the Animal Care and Use Committee of Wenzhou Medical University (Wenzhou, China) and conducted in accordance with the Temple University Guidelines. The mice were maintained in the animal care facility in a temperature-controlled room (25°C) for a minimum of 7 days under 12/12 h light/dark cycles and were allowed ad libitum access to food and water prior to the experiments. Mice were randomly distributed into four groups: (I) Sham group, (II) TBI group (mice were received same volume of saline), (III) TBI plus aFGF group (0.5 μg/g aFGF was intranasally administrated 1 h before induction of the TBI model), (IV) TBI plus aFGF and LY294002 group (0.5 μg/g aFGF and 50 nmol/kg LY294002 were intranasally administrated 1 h before induction of the TBI model, n = 6 in each group). To determine the effects of aFGF after TBI in mice, we subjected sham operation mice to surgery, but these mice were not subjected to a cortical impact. The animals were sacrificed 24 h after injury.
Surgical preparation for TBI
The TBI model of parasagittal fluid percussion (FP) brain injury was established as described previously. Briefly, the animals were anesthetized via intraperitoneal injections of 10 ml/kg 4% choral hydrate and mounted on a stereotaxic system (David Kopf Instruments, Tujunga, California). The surgeries and the subsequent steps were all performed under aseptic conditions. The skull was exposed by cutting the scalp at the midline. A 3-mm diameter manual trephine (Roboz Surgical Instrument Co., Gaithersburg, MD) was used to carefully penetrate the skull to remove the bone flap. The mice were subjected to TBI, and a craniotomy incision was made over the right parietal cortex between the lambda and the bregma, approximately 1 mm from the midline. The stereotaxic frame was tilted thus the plane of the cerebral cortex was perpendicular to an impactor tip, which impacted the cerebral cortex at a velocity of 4.0 m/sec and achieved a penetration depth of 1.5 mm and a dwell time of 600 ms. The mice were subsequently allowed to recover from anesthesia in a warm chamber for 24 h. The sham-operated control mice underwent only a parietal craniotomy and were not injured or treated.
BBB integrity and permeability assay
BBB integrity was studied in the experimental mice. We intravenously injected 0.25 ml of 2% EB dye into the mice. After 2 h, the mice were perfused with saline solution to wash away any dye remaining in the blood vessels. Brains tissue samples were harvested, and the right hemisphere was dissected and weighed. The samples were centrifuged at 12,000 g for 15 min to remove the supernatant. EB dye was detected via spectrophotometry (ex: 610 nm, em: 680 nm) and quantified according to a standard curve. To quantify low-molecular-weight molecular leakage, we injected the mice with 2% FITC-dextran (100 μg/ml, 0.2 ml). Two hours later, the mice were subjected to systemic intracardiac perfusion with 1 USP U/ml heparin in saline to flush the FITC-dextran out of the vasculature. Five minutes later, the mice were euthanized, and their perfused brains were subsequently harvested. Supernatant fluorescence was measured using an EnSpire Manager Multimode Plate Reader (ex: 485 nm, em: 535 nm) (PerkinElmer, Waltham, USA).
Neurofunctional assessment
Neurofunctional testing was conducted in a blinded fashion. The mice were evaluated via the garcia test, which is used to identify various sensorimotor deficits. This garcia test consists of 7 independent sub-tests, which examine spontaneous activity (I), axial sensation (II), vibrissae proprioception (III), limb symmetry (IV), lateral turning ability (V), forelimb outstretching (VI), and climbing (VII). Scores for each sub-test range from 0 (worst performance) to 3 (best performance), and the total garcia score (maximum score of 21 points) is calculated as the sum of all sub-test scores.
Cell culture and the in vitro oxygen glucose deprivation/reoxygenation (OGD) model
Endothelial cells bEnd.3 were maintained in an incubator at 37°C in a humidified atmosphere of 5% CO2/95% air in dulbecco’s modified eagle medium (DMEM) until they attached to their plates. After the endothelial cells reached 80%-90% confluence, the normal growth medium was replaced with medium without glucose. The cells were then incubated in an anaerobic chamber for 24 h. The oxygen level within the anaerobic chamber was maintained below 0.5%. After OGD, the glucose-free DMEM was replaced with normal growth medium, and the cells were once again incubated under normal culture conditions. The cells were pretreated with aFGF (100 ng/ml) before being subjected OGD. To evaluate the effects of PI3K/Akt activation on OGD further, we pretreated the cells with or without a specific inhibitor, LY294002 (20 μM), for 1 h. After 24 hours, the cells were detached and collected for further study. All experiments were performed in triplicate.
Epithelial barrier function assay
For the permeability experiments, FITC-dextran (70 kDa) flux across the endothelial cells was analyzed. Briefly, the endothelial cells were seeded onto polycarbonate 24-well transwell chambers with a 0.4 mm mean pore size and a 0.3 cm2 surface area (Millicell Hanging Cell Culture Inserts, USA) at a density of 2×104 cells/well in 100 μl of medium. The cells were incubated with FITC-dextran (1 mg/ml) in medium for 4 h at a constant temperature of 37°C. Then, the relative fluorescence passing through the chamber (in the lower chamber) was determined using an EnSpire Manager Multimode Plate Reader (PerkinElmer, Waltham, USA) at an excitation wavelength of 485 nm and an emission wavelength of 520 nm.
Western blot analysis
The levels of claudin-5, ZO-1, occludin, β-catenin, p120-catenin expression were determined by western blotting at 24 h after TBI. Brain tissue samples from the right peri-contusional cortex were harvested, and total proteins were purified using protein extraction reagents. The samples (100 μg per lane) were separated on a 12% gel and then transferred onto 0.22 mm PVDF membranes. The membranes were blocked in 5% non-fat milk for 1 h at room temperature before being incubated overnight with the following antibodies: claudin-5 (1:500), ZO-1 (1:200), occludin (1:500), p-Akt (1:500), p120-catenin (1:1000), β-catenin (1:1000), GTP-Rac1 (1:1000), total Rac1 (1:1000), GTP-RhoA (1:1000) and total RhoA (1:1000). The following day, the membranes were washed and treated with horseradish peroxidase-conjugated secondary antibodies for 2 h at room temperature before being scanned using a ChemiDicTM XRS+ Imaging System (Bio-Rad Laboratories, Hercules, CA, USA). Band densities were quantified with Multi Gauge Science Lab 2006 Software (FUJIFILM Corporation, Tokyo, Japan).
Immunofluorescence staining
The cells were fixed with 4% paraformaldehyde at 25°C for 15 min, permeabilized with 0.5% triton x-100 in PBS for 10 min, blocked with 10% BSA containing 0.05% tween-20 in PBS for 1 h, incubated with primary rabbit polyclonal antibodies against claudin-5 (1:100), occludin (1:100), p120-catenin (1:1000) and β-catenin (1:1000) in 1% BSA at 4°C overnight and finally incubated with Alexa-Fluor594/647 donkey anti-mouse/rabbit secondary antibodies (1:500; Invitrogen Corporation, Carlsbad, CA, USA) for 1 h at 37°C. Cellular nuclei were counterstained with DAPI (Sigma-Aldrich) for 5 min. After the cells were incubated with these secondary fluorescence antibodies and stained with DAPI, immunofluorescence images were captured under a fluorescence microscope (Keyence, Tokyo, Japan). The slides were mounted with Fluoromount-G® mounting media on glass and imaged under a fluorescence microscope at 400× magnifications using a Nikon ECLPSE 80i microscope.
Statistical analysis
Data were expressed as the mean ± SEM if normally distributed. Student’s t test was used between 2 groups if data were normally distributed. Otherwise, we used the Wilcoxon rank sum test instead. For analyzing 3 or more groups, statistical analysis was performed using one-way analysis of variance (ANOVA) if data were normally distributed and variance homogeneous; otherwise, the Kruskal-Wallis test was performed, followed by post-hoc adjustment using Bonferroni’s multiple comparisons test. Normality was checked using the Kolmogorov-Smirnov test and the homogeneity of variance was checked by the Levene test. A probability value P < 0.05 was considered to show statistical significance.
Results
aFGF attenuates TBI-induced neurofunctional deficits and BBB integrity disruption in mice
Given that TBI can disrupt cerebral autoregulation and cause BBB breakdown, we evaluated BBB integrity and neurofunction 24 h after modeling to determine the role of aFGF after TBI in mice. EB dye was intravenously injected into the brain after TBI (Figure 1A). Sham-operated mice received no EB dye. EB extravasation was markedly reduced in aFGF-treated mice compared with vehicle-treated mice 24 h after experimental TBI surgery (Figure 1A). Similar results were obtained regarding fluorescence intensity (Figure 1B). Furthermore, the garcia test was performed to evaluate neuronal dysfunction after TBI in mice. We found that garcia neuroscores were low in vehicle-treated mice but were significantly increased in aFGF-treated mice 24 h after modeling, suggesting that aFGF (0.5 μg/g) treatment significantly ameliorated neuronal dysfunction (Figure 1C). Given that FITC-dextran is commonly used to measure BBB permeability [12], FITC-dextran was used in the present study. As expected, the level of FITC-dextran passing through BBB increased significantly after TBI and decreased in mice receiving aFGF treatment (Figure 1D). These results indicate that aFGF treatment reduces neurological deficits by ameliorating BBB disruption following experimental TBI in mice.
Figure 1.
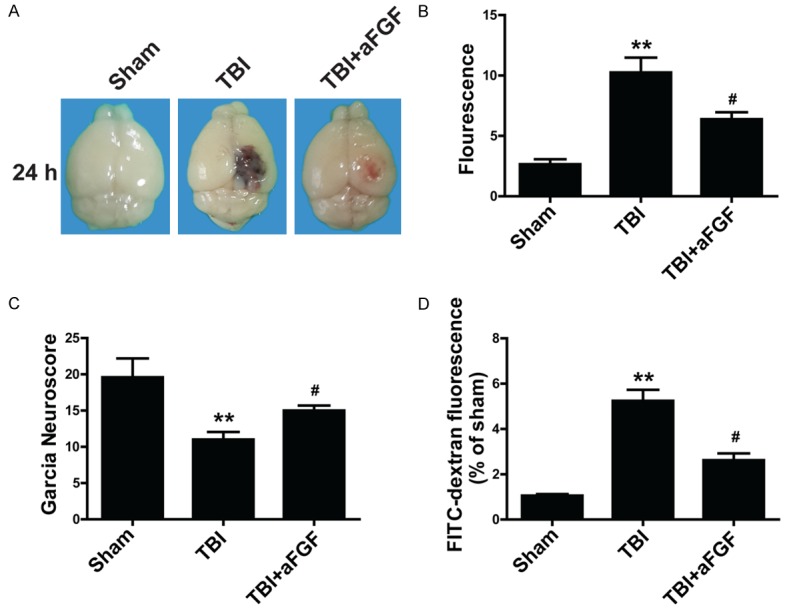
Exogenous aFGF (0.5 μg/g) administration improves neurofunctional deficits 24 h after experimental TBI in C57BL/6 mice. A. Representative effects of aFGF on EB extravasation. B. Summary data showing EB extravasation fluorescence. C. aFGF-treated mice performed differently on the garcia test than vehicle-treated mice. D. FITC-dextran fluorescence 24 h after TBI. *P < 0.05 versus sham group. **P < 0.01 versus sham group. #P < 0.05 versus TBI group. Data are presented as the mean ± standard error of the mean. n = 6.
To examine BBB integrity further, we examined the levels of TJs and AJs at 24 h after TBI. The western blotting and graphical quantification results indicated that the expression levels of each TJ (claudin-5, occludin and ZO-1) and AJ protein (p120-catenin and β-catenin) were increased in TBI tissue samples and were increased to greater extent when aFGF treatment preceded TBI, suggesting that aFGF preserved BBB integrity by regulating the expression of TJs and AJs (Figure 2 and Figure S1).
Figure 2.
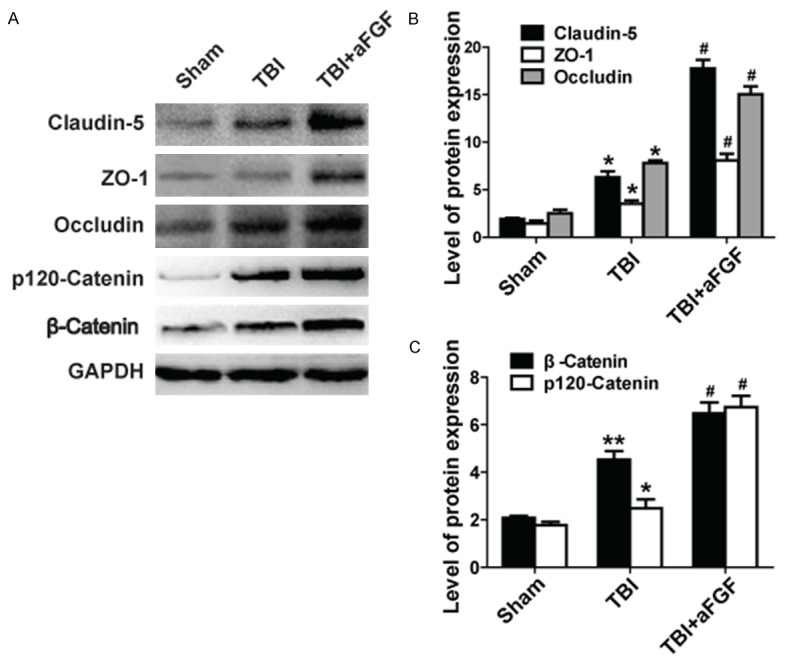
aFGF administration attenuates the loss of the TJ proteins claudin-5, occludin, and ZO-1. The AJ proteins p120-catenin and β-catenin improve BBB integrity following experimental TBI in mice. GAPDH was used as a loading control and for band density normalization. A. Representative western blots of the TJ proteins claudin-5, occludin, and ZO-1 and the AJ proteins p120-catenin and β-catenin. B. Quantified data regarding expression of the TJ proteins claudin-5, occludin, and ZO-1. C. Quantified data regarding expression of the AJ proteins p120-catenin and β-catenin. *P < 0.05 versus sham group. **P < 0.01 versus sham group. #P < 0.05 versus TBI group. Data are presented as the mean ± standard error of the mean. n = 6.
The protective effects exerted by aFGF against neurofunctional deficits and BBB integrity disruption after TBI are partially dependent on the PI3K/Akt pathway
We found in our previous study [13] that the PI3K/Akt pathway mediated the neuroprotective effects of bFGF. We therefore hypothesize that the PI3K/Akt pathway also mediates the effects of aFGF. To confirm our hypothesis, we intranasally injected the PI3K/Akt inhibitor LY294002 into the left striatum in each mouse. Neuronal dysfunction, EB extravasation and FITC-dextran fluorescence were evaluated at 24 h after experimental TBI. The results showed that aFGF plus LY294002 significantly increased EB extravasation compared with aFGF treatment alone (Figure 3A and 3B), indicating that aFGF and LY294002 co-administration can reversed the protective effects of aFGF. In addition, the garcia test results showed that animals treated with aFGF plus LY294002 exhibited significantly lower neuroscores than animals treated with aFGF alone (Figure 3C). Finally, FITC-dextran was used to determine the extent of BBB disruption and permeability alterations at 24 h after experimental TBI. The results showed that the amount of FITC-dextran passing through the basolateral side of the BBB was significantly increased in mice receiving aFGF plus LY294002 compared with that in mice receiving aFGF alone (Figure 3D). These data suggest that the PI3K/Akt inhibitor reversed the protective effects of aFGF on neurofunctional deficits after experimental TBI.
Figure 3.
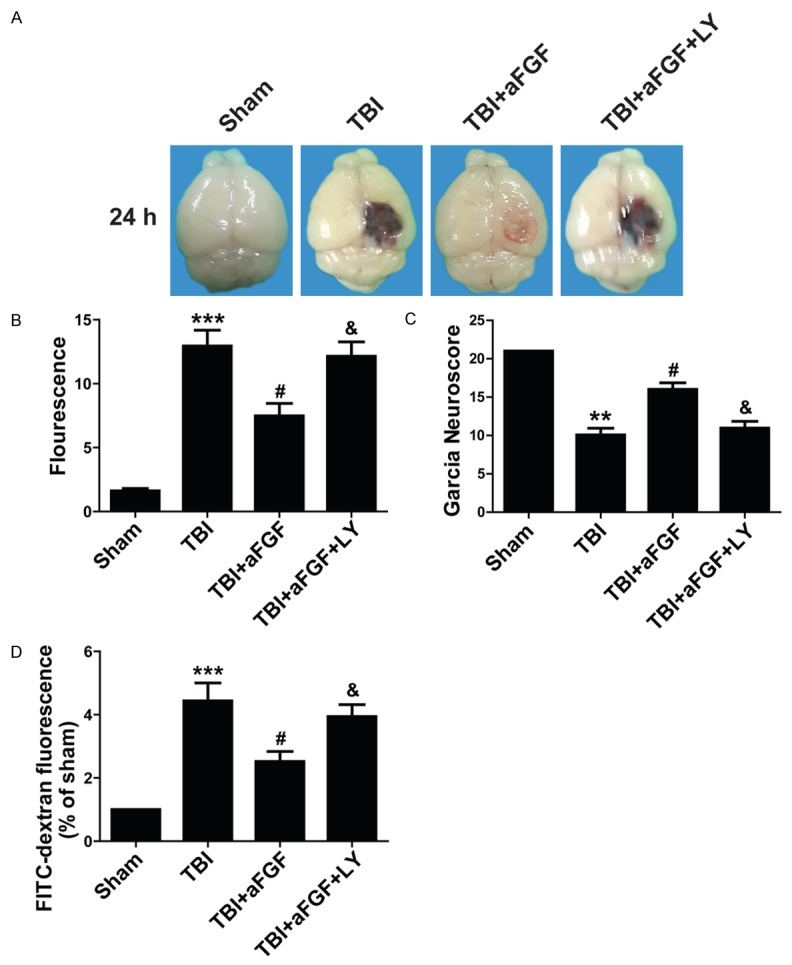
The PI3K/Akt inhibitor LY294002 (50 nmol/kg) reverses aFGF-induced BBB injury attenuation 24 h after experimental TBI in mice. A. EB extravasation in the mouse brain. B. Summary data showing EB extravasation fluorescence. C. Garcia test results. D. FITC-dextran fluorescence in mice subjected to TBI. *P < 0.05 versus sham group. ***P < 0.001 versus sham group. #P < 0.05 versus TBI group. &P < 0.05 versus TBI plus aFGF group. Data are presented as the mean ± standard error of the mean. n = 6.
After completion of these experiments, mice were perfused with paraformaldehyde, and their brains were processed for western blotting using antibodies against TJ and AJ proteins. The representative results pertaining to the ipsilateral cerebral cortex tissue samples from the sham, vehicle-treated, aFGF-treated, and aFGF plus LY294002-treated mice are shown in Figure 4 and Figure S2. Claudin-5 levels were significantly decreased in mice receiving aFGF plus LY294002 compared with mice receiving aFGF alone. Similar results were observed regarding occludin, ZO-1, p120-catenin and β-catenin levels (Figure 4 and Figure S2). These results indicate that LY294002 reverse the effects of aFGF on TJs and AJs and that the protective effects exerted by aFGF on neurofunctional deficits after experimental TBI are partially dependent on the PI3K/Akt pathway.
Figure 4.
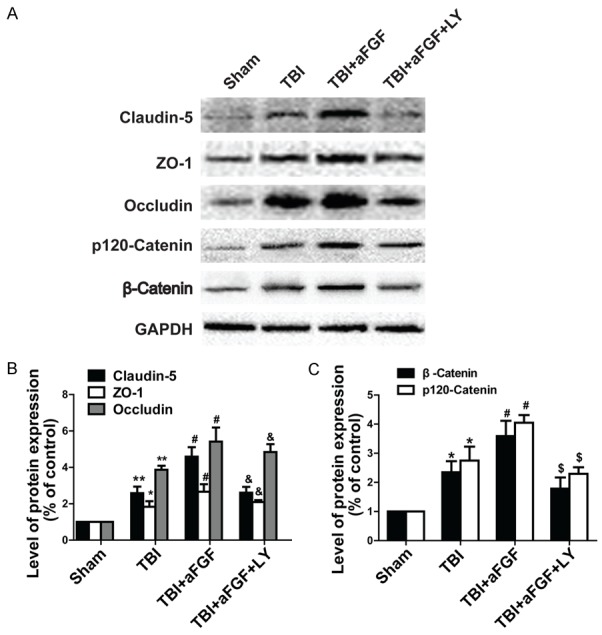
Addition of the Akt inhibitor LY294002 reverses the effects of aFGF on the expression of the TJ proteins claudin-5, occludin, and ZO-1 and the AJ proteins p120-catenin and β-catenin 24 h after TBI in mice. GAPDH was used as a loading control and for band density normalization. A. Representative western blots of the TJ proteins claudin-5, occludin, and ZO-1 and the AJ proteins p120-catenin and β-catenin. B. Quantified data regarding expression of the TJ proteins claudin-5, occludin, and ZO-1. C. Quantified data regarding expression of the AJ proteins p120-catenin and β-catenin. *P < 0.05 versus sham group. **P < 0.01 versus sham group. #P < 0.05 versus TBI group. &P < 0.05 versus TBI plus aFGF group regarding TJ expression. $P < 0.05 versus TBI plus aFGF group regarding AJ expression. Data are presented as the mean ± standard error of the mean. n = 6.
aFGF preserves BBB function by inhibiting RhoA activity via the PI3K-Akt-Rac1 signaling pathway following experimental TBI in mice
Given that the small GTPases RhoA and Rac1 play crucial roles in TJs and adhesion molecule expression in bEnd.3 cells and endothelial cells found in other organs [14], we performe western blot to confirm the hypothesis that aFGF protects BBB via PI3K-Akt-Rac1 pathway-dependent RhoA inhibition. The levels of phosphorylated Akt, GTP-Rac1, and GTP-RhoA expression were quantified after TBI. We found that the levels of p-Akt, GTP-RhoA and GTP-Rac1 expression were significantly enhanced following experimental TBI. In addition, aFGF administration significantly increased Akt phosphorylation and GTP-bound Rac1 activation in the treated group compared with the sham and vehicle groups (Figure 5). However, p-Akt and GTP-Rac1 activity was markedly diminished after administration of the Akt inhibitor LY294002. aFGF administration significantly decreased GTP-bound RhoA expression in the treated group compared with the sham and vehicle groups, but the Akt inhibitor LY294002 increased GTP-RhoA expression (Figure 5). Taken together, these findings suggest that aFGF protectes neurofunction and preserves BBB integrity 24 h after experimental TBI in part by inhibiting RhoA expression, up-regulating Rac1 expression and activating the PI3K-Akt-Rac1 signaling pathway.
Figure 5.
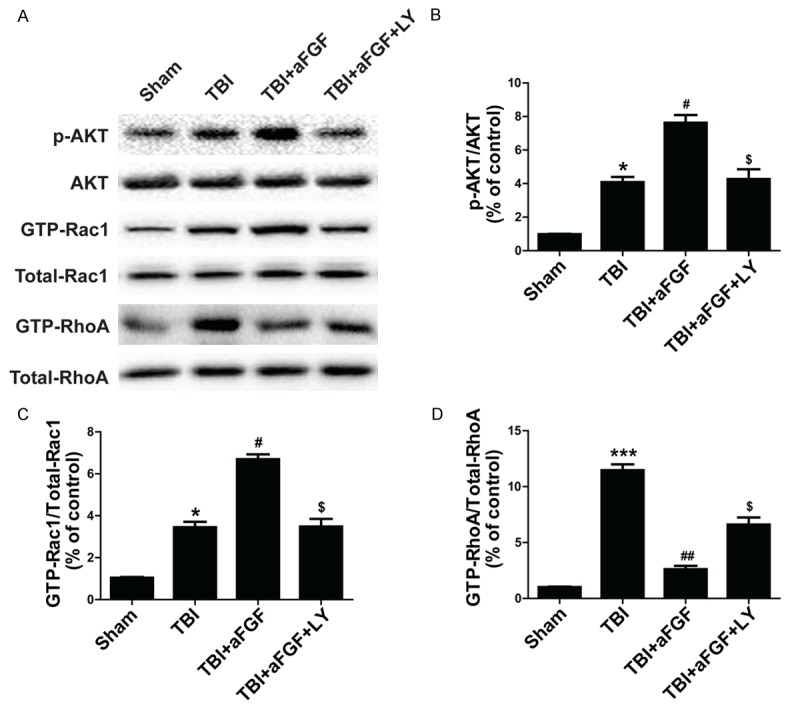
Addition of LY294002 reverses the effects of aFGF on p-Akt and GTP-Rac1 and aFGF-induced GTP-RhoA inhibition at 24 h after TBI-induced BBB destruction in mice. A. p-Akt, GTP-Rac1 and GTP-RhoA expression. B. Optical density analysis of p-Akt. C. Optical density analysis of GTP-Rac1. D. Optical density analysis of GTP-RhoA. *P < 0.05 versus sham group. ***P < 0.001 versus sham group. #P < 0.05 versus TBI group. ##P < 0.05 versus TBI group. $P < 0.05 versus TBI plus aFGF group. Data are presented as the mean ± standard error of the mean. n = 6.
The protective effects exerted by aFGF against OGD-induced BBB disruption in vitro are partially dependent on the PI3K/Akt pathway
To determine whether aFGF can ameliorate BBB injury, we studied endothelial cells in an OGD model. As was shown in Figure 6A, paracellular FITC-dextran permeability was dramatically increased in endothelial cells after OGD treatment, and FITC-dextran leakage was ameliorated in aFGF-treated cells. TJ and AJ proteins (claudin-5, occludin, ZO-1, p120-catenin, and β-catenin) expression was evaluated via western blotting after OGD treatment in vitro. As was shown in Figure 6B, OGD treatment markedly reduced the levels of TJs and AJs expression in the treated group compared with the control group. Meanwhile, aFGF significantly increased the levels of TJs and AJs in the aFGF-treated group compared with the OGD-treated group. Based on the western blot assay results, we performed claudin-5, occludin, β-catenin and p120-catenin immunostaining to confirm whether aFGF preserved BBB integrity in endothelial cells. The result showed that OGD treatment decreased the expression of TJ and AJ proteins, while aFGF administration preserved BBB integrity by significantly augmenting the expression of TJ and AJ proteins. However, the Akt inhibitor LY294002 reversed the protective effects exerted by aFGF on BBB integrity and reversed the TJs and AJs activation induced by aFGF (Figure 6B-D). Figure 7A showed that claudin-5 expression levels were significantly increased after aFGF treatment compared with the control group and that LY294002 partially reversed the effects of aFGF on the expression of TJ and AJ proteins. Similar results were observed regarding endothelial cell occludin, β-catenin and p120-catenin protein expression (Figure 7B-D). All these findings illustrate that aFGF increases BBB tightness in part by regulating the expression of TJ and AJ proteins via the PI3K/Akt pathway in OGD-treated endothelial cells.
Figure 6.
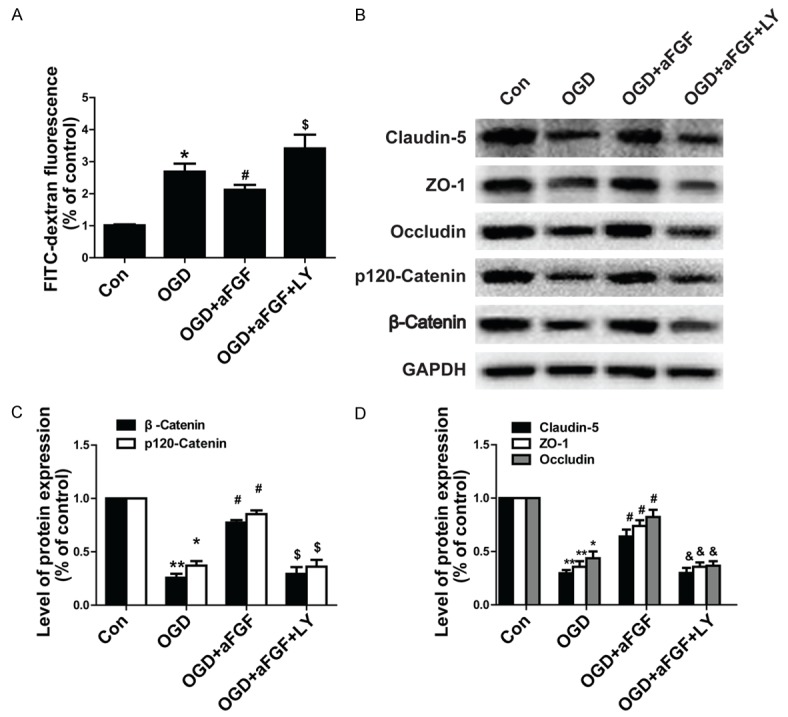
LY294002 reverses the protective effects exerted by aFGF against OGD-induced BBB destruction in endothelial cells. A. FITC-dextran fluorescence in endothelial cells under OGD conditions. B. Representative western blots of the TJ proteins claudin-5, occludin, and ZO-1 and the AJ proteins p120-catenin and β-catenin. GAPDH was used as a loading control and for band density normalization. C. Optical density analyses of claudin-5, occludin, and ZO-1. D. Optical density analyses of p120-catenin and β-catenin. *P < 0.05 versus control group. **P < 0.01 versus sham group. #P < 0.05 versus OGD group. $P < 0.05 versus OGD plus aFGF group regarding AJ expression. &P < 0.05 versus OGD plus aFGF group regarding TJ expression. Data are presented as the mean ± standard error of the mean.
Figure 7.
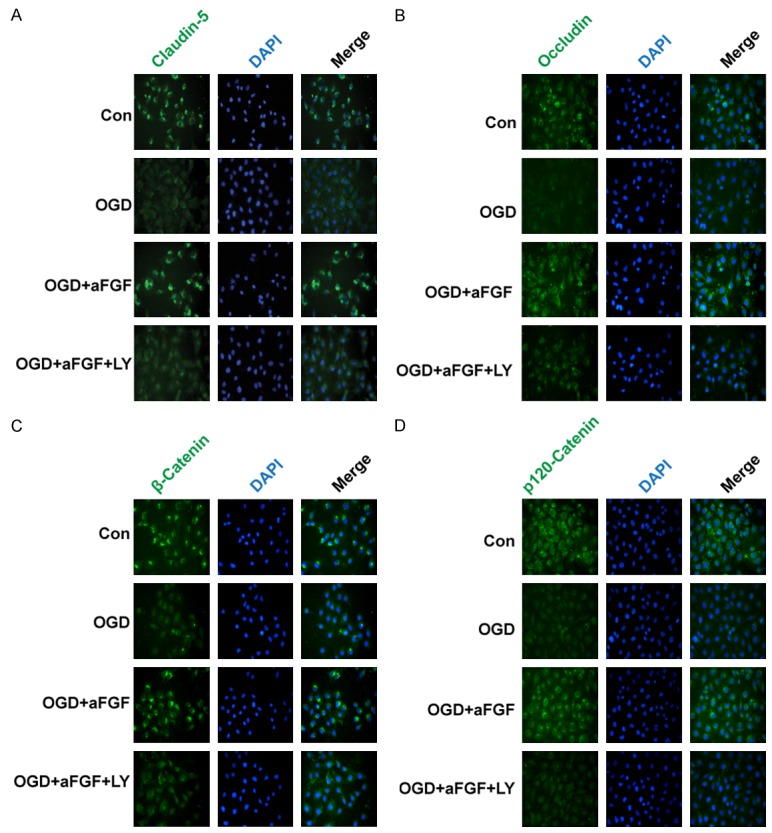
The Akt inhibitor LY294002 reverses the protective effects exerted by aFGF on OGD-treated TJ and AJ proteins in endothelial cells, as demonstrated using immunofluorescence staining. A-D. Immunofluorescence staining of a confluent endothelial cell monolayer for claudin-5, occludin, β-catenin and p120-catenin. Nuclei were labeled by DAPI. The proteins with clear bright signals are labeled. Magnification is 40×.
aFGF preserves BBB function by inhibiting RhoA activity via the PI3K-Akt-Rac1 signaling pathway under OGD conditions in vitro
Encouraged by the protective effects exerted by aFGF on RhoA activity via the PI3K-Akt-Rac1 signaling pathway after TBI in mice, we performed additional experiments to determine whether aFGF can inhibit RhoA function via the same mechanism in vitro. Endothelial cells subjected to OGD conditions were treated with aFGF or aFGF plus the Akt inhibitor LY294002. Western blot analyses of the endothelial cells were conducted, and the changes in the expression levels of phosphorylated Akt, GTP-Rac1, total Rac1, RhoA, GTP-RhoA and total RhoA were quantified after OGD treatment. The results showed that aFGF enhanced p-Akt and GTP-Rac1 expression and decreased GTP-RhoA expression under OGD conditions. The Akt inhibitor LY294002 was also administered to OGD-treated endothelial cells and significantly reversed the effects of aFGF on p-Akt, GTP-Rac1, and GTP-RhoA expression (Figure 8). These data demonstrate that the PI3K-Akt-Rac1 pathway mediated the protective effects exerted by aFGF by inhibiting RhoA. All these findings show that aFGF attenuates BBB injury by inhibiting RhoA expression and by activating the PI3K-Akt-Rac1 signaling pathway, both in vivo and in vitro.
Figure 8.

Addition of LY294002 reverses the effects of aFGF on p-Akt and GTP-Rac1 and aFGF-induced GTP-RhoA inhibition in OGD-treated endothelial cells. GAPDH was used as a loading control and for band density normalization. A. p-Akt, GTP-Rac1 and GTP-RhoA protein expression in OGD-treated endothelial cells treated with aFGF and LY294002. B-D. Optical density analysis of p-Akt, GTP-Rac1 and GTP-RhoA. *P < 0.05 versus control group. **P < 0.01 versus sham group. ***P < 0.001 versus sham group. #P < 0.05 versus OGD group. $P < 0.05 versus OGD plus aFGF group. Data are presented as the mean ± standard error of the mean.
Discussion
Traumatic brain injury is an unforeseen pervasive clinical disease caused by vehicle accidents, falls, football collisions, gunshot wounds and devastating earthquakes that result in debilitating neurological degeneration in survivors. TBI is a complex neurological injury that initiates many critical cellular and molecular pathways causing neurovascular dysfunction, BBB destruction, even cell apoptosis and death [15]. BBB is the interface between blood components and the neural tissues within the brain and spinal cord, whose disruption has been identified as an important hallmark of severe brain injury [16]. It was found in our previous study that bFGF treatment can attenuate BBB dysfunction and neurological deficits in a TBI model [13]. aFGF is not only similar to bFGF in terms of its chemical structure and biological function but also has some advantages over bFGF. The aim of the present study is to investigate the effects of aFGF on BBB function after experimental TBI. The result shows that aFGF decreases BBB permeability and increases the expression of TJ and AJ proteins. However, the PI3K/Akt inhibitor reversed these neuroprotective effects by regulating RhoA and Rac1 expression. These data suggest that aFGF increases BBB integrity via the PI3K-Akt-Rac1 signaling pathway by decreasing RhoA expression in a hydraulic pressure-induced TBI model in mice.
BBB dysfunction is an early event following TBI and plays a major role in the development of vasogenic edema and the exacerbation cytotoxic edema. One study [17] has reported that progressive cerebral edema increases BBB permeability as early as one day after TBI, and another study [18] has shown that BBB leakage can persist for as long as 5 weeks after brain injury in a rat model, indicating that brain injury definitely leads to BBB disruption. Several drugs have been reported to promote TBI repair by improving BBB integrity. For instance, poloxamer-188 attenuates TBI-induced BBB damage, thus decreasing cerebral edema in a mouse controlled cortical impact injury model [19]. Caffeic acid phenethyl ester protects BBB integrity and reduces contusion volumes following TBI in both rat and mouse models [20]. Moreover, studies [21] have also reported that growth factors, such as NGF, can attenuate TBI-induced cerebral edema, as determined via assessments of hemisphere water content at 12, 24 and 72 h after experimental TBI induction. It has also been previously reported that FGF administration preserves endothelial TJs and AJs, reduces perihematomal brain edema and improves neurofunctional deficits at 72 h after ICH induction [22]. Although some of these drugs are extracted from herbs, and others are endogenous or exogenous growth factors, all of them promote injury repair by preserving BBB integrity. The results of the present study indicate that experimental TBI increased EB extravasation and BBB permeability to FITC-dextran, while aFGF significantly decreased BBB permeability during the experimental period in mice. aFGF also increased garcia neuroscores (Figure 1), indicating that exogenous aFGF administration exerts protective effects on neural function and BBB permeability and promotes recovery from experimental TBI.
Cells such as astrocyte endfeet, pericytes, perivascular astrocytes and endothelial cells in the vasculature form BBB parenchyma, while cellular elements (TJs, AJs and the basal lamina) constitute its structure [23]. Structurally, endothelial cells and their junctions is the first barrier between the blood stream and nervous tissue [24]. In MDCK cells, claudin-5 and occludins have been shown to contribute to TJ localization and stability and to cooperate in regulating BBB permeability [25]. Moreover, the levels of ZO-1 and occludin expression were significantly decreased after TBI, and degrading ZO-1 and occludins in endothelial TJs facilitated capillary leakage, resulting in increased BBB permeability [26]. Moreover, studies [27] have reported that p120-catenin expression was correlated with increased BBB permeability in human brain microvascular endothelial cells (HBMEC) after LPS challenge. Tran et al [28] has suggested that β-catenin maintains BBB integrity and that its transcriptional expression plays a key role in BBB integrity. As was shown previously, TJs and AJs are vital to BBB permeability, and changes in their expression severely affect cell tightness. Our results indicate that aFGF markedly increases the expression of TJs and AJs both in vitro and in vivo (Figures 2 and 6) and attenuate brain damage. However, we have also noted differences between our in vivo and in vitro models. The levels of TJs and AJs are up-regulated after TBI in mice (Figure 2) but down-regulated after OGD treatment (Figure 6), as TJs and AJs take part in regulating BBB integrity and stress and play important compensatory roles under pathological TBI conditions. However, in cell OGD models, TJs and AJs are destroyed by the lack of oxygen and glucose and are thus unable to exert compensatory effects in mice.
Previous studies have reported that various molecular mechanisms regulate the expression of TJ and AJ proteins in BBB. For example, the Wnt/β-catenin signaling pathway [29] and sonic hedgehog (Shh) pathway [30] have been shown to regulate TJs and AJs maturation and gene transcription. The PI3K/Akt pathway is activated during BBB formation, leading to PI3K phosphorylation and phosphatidylinositol (3, 4, 5)-triphosphate generation [31]. This pathway plays a central role in 3-chloropropanediol-induced biphasic paracellular claudin-5 expression loss [32]. Li et al [33] have reported that LY294002 pretreatment significantly attenuates changes in BBB permeability, indicating that the RhoA/PI3K signaling pathway has an intimate relationship with BBB permeability. PI3K/Akt is a downstream signaling pathway activated by growth factors that exerts essential neuroprotective effects. Daniels et al [34] have reported that the small GTPase Rac1 regulates endothelial permeability and TJ formation in a BBB model in vitro. Another study [35] has shown that EGF treatment preserves blood-spinal cord barrier integrity and improves functional recovery after spinal cord injury by blocking Rac1 activation in endothelial cells. It’s been found in the present study that aFGF recovers TBI-induced BBB breakdown and that the PI3K/Akt inhibitor LY294002 abolishes the protective effects of aFGF on TJs and AJs, indicating that PI3K/Akt pathway activation is responsible for the protective effects of aFGF.
Despite the above findings, how some factors induce and maintain BBB function remains unclear. Several cytoplasmic signaling molecules, such as Rho kinase, Rac-1, and phospholipase C, have been localized to TJs and AJs and may regulate their assembly and disassembly [36]. One study [37] has reported that they contribute to TJs and AJs maintenance and endothelial cell polarity by regulating the small GTPase Rac1 and the Rho signaling pathway. Huang et al [22] have reported that exogenous FGF treatment reduces RhoA activity via FGFR-induced PI3K-Akt-Rac1 signaling pathway activation, thus preserving BBB integrity following ICH in mice. It’s been confirmed in the present study that aFGF increases GTP-Rac1 protein expression and decreases GTP-RhoA protein expression in experimental TBI mice and under OGD conditions in endothelial cells (Figures 5, 6, 7 and 8). PI3K-Akt inhibition significantly inhibits PI3K-Akt-Rac1 signaling pathway activity and increases RhoA expression compared with the aFGF-treated group. To the best of our knowledge, this is the first study to demonstrate that the PI3K-Akt-Rac1 pathway mediates the protective effects of aFGF by inhibiting RhoA.
Based on the findings of this study and those of our previous study, we conclude that both aFGF and bFGF can effectively protect BBB from damage following TBI by regulating junction protein expression via the PI3K-Akt-Rac1 pathway. Although bFGF and aFGF both play important roles in TBI patients, there are many differences between them. aFGF release is much slower than bFGF release in animal models, and the effects of aFGF last longer than those of bFGF [38]. aFGF also has stronger affinity for FGFRs [39]. Moreover, aFGF significantly reduces the occurrence of delayed neuronal death, while bFGF has only a slight effect on delayed neuronal death [40]. However, bFGF is more comprehensively accepted by researchers with respect to its role in healing brain injury. The neuroprotective effects of bFGF may be facilitated by its direct promotion of the up-regulation and secretion of neuronal trophic factors, such as NGF and BDNF [41]. Therefore, both aFGF and bFGF have specific advantages and should be used in different situations. However, aFGF and bFGF both have potential as therapies for TBI.
Acknowledgements
This work was supported by the Nature Science Foundation of Zhejiang Province (LY14H090013, LQ16H090007, LY14H150010), the National Natural Science Foundation of China (81501953) and Nature Science Foundation of Ningbo City (2015A610192, 2015A610213).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Hyder AA, Wunderlich CA, Puvanachandra P, Gururaj G, Kobusingye OC. The impact of traumatic brain injuries: a global perspective. NeuroRehabilitation. 2007;22:341–353. [PubMed] [Google Scholar]
- 2.Courtney A, Courtney M. The complexity of biomechanics causing primary blast-induced traumatic brain injury: a review of potential mechanisms. Front Neurol. 2015;6:221. doi: 10.3389/fneur.2015.00221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thurman DJ. The epidemiology of traumatic brain injury in children and youths: a review of research since 1990. J Child Neurol. 2016;31:20–27. doi: 10.1177/0883073814544363. [DOI] [PubMed] [Google Scholar]
- 4.Suzuki Y, Nagai N, Umemura K. A Review of the mechanisms of blood-brain barrier permeability by tissue-type plasminogen activator treatment for cerebral ischemia. Front Cell Neurosci. 2016;10:2. doi: 10.3389/fncel.2016.00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crosby CV, Fleming PA, Argraves WS, Corada M, Zanetta L, Dejana E, Drake CJ. VE-cadherin is not required for the formation of nascent blood vessels but acts to prevent their disassembly. Blood. 2005;105:2771–2776. doi: 10.1182/blood-2004-06-2244. [DOI] [PubMed] [Google Scholar]
- 6.Lee HS, Namkoong K, Kim DH, Kim KJ, Cheong YH, Kim SS, Lee WB, Kim KY. Hydrogen peroxide-induced alterations of tight junction proteins in bovine brain microvascular endothelial cells. Microvasc Res. 2004;68:231–238. doi: 10.1016/j.mvr.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 7.Citi S, Guerrera D, Spadaro D, Shah J. Epithelial junctions and Rho family GTPases: the zonular signalosome. Small GTPases. 2014;5:1–15. doi: 10.4161/21541248.2014.973760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goddard LM, Iruela-Arispe ML. Cellular and molecular regulation of vascular permeability. Thromb Haemost. 2013;109:407–415. doi: 10.1160/TH12-09-0678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yamamoto M, Ramirez SH, Sato S, Kiyota T, Cerny RL, Kaibuchi K, Persidsky Y, Ikezu T. Phosphorylation of claudin-5 and occludin by rho kinase in brain endothelial cells. Am J Pathol. 2008;172:521–533. doi: 10.2353/ajpath.2008.070076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gunduz D, Thom J, Hussain I, Lopez D, Hartel FV, Erdogan A, Grebe M, Sedding D, Piper HM, Tillmanns H, Noll T, Aslam M. Insulin stabilizes microvascular endothelial barrier function via phosphatidylinositol 3-kinase/Akt-mediated Rac1 activation. Arterioscler Thromb Vasc Biol. 2010;30:1237–1245. doi: 10.1161/ATVBAHA.110.203901. [DOI] [PubMed] [Google Scholar]
- 11.Hemmings BA, Restuccia DF. The PI3K-PKB/Akt pathway. Cold Spring Harb Perspect Biol. 2015:7. doi: 10.1101/cshperspect.a026609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reyes R, Guo M, Swann K, Shetgeri SU, Sprague SM, Jimenez DF, Barone CM, Ding Y. Role of tumor necrosis factor-alpha and matrix metalloproteinase-9 in blood-brain barrier disruption after peripheral thermal injury in rats. J Neurosurg. 2009;110:1218–1226. doi: 10.3171/2008.8.JNS08382. [DOI] [PubMed] [Google Scholar]
- 13.Wang ZG, Cheng Y, Yu XC, Ye LB, Xia QH, Johnson NR, Wei X, Chen DQ, Cao G, Fu XB, Li XK, Zhang HY, Xiao J. bFGF protects against blood-brain barrier damage through junction protein regulation via PI3K-Akt-Rac1 pathway following traumatic brain injury. Mol Neurobiol. 2016;53:7298–7311. doi: 10.1007/s12035-015-9583-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Spindler V, Schlegel N, Waschke J. Role of GTPases in control of microvascular permeability. Cardiovasc Res. 2010;87:243–253. doi: 10.1093/cvr/cvq086. [DOI] [PubMed] [Google Scholar]
- 15.Huang XT, Zhang YQ, Li SJ, Li SH, Tang Q, Wang ZT, Dong JF, Zhang JN. Intracerebroventricular transplantation of ex vivo expanded endothelial colony-forming cells restores blood-brain barrier integrity and promotes angiogenesis of mice with traumatic brain injury. J Neurotrauma. 2013;30:2080–2088. doi: 10.1089/neu.2013.2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Israeli D, Tanne D, Daniels D, Last D, Shneor R, Guez D, Landau E, Roth Y, Ocherashvilli A, Bakon M, Hoffman C, Weinberg A, Volk T, Mardor Y. The application of MRI for depiction of subtle blood brain barrier disruption in stroke. Int J Biol Sci. 2010;7:1–8. doi: 10.7150/ijbs.7.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beaumont A, Fatouros P, Gennarelli T, Corwin F, Marmarou A. Bolus tracer delivery measured by MRI confirms edema without blood-brain barrier permeability in diffuse traumatic brain injury. Acta Neurochir Suppl. 2006;96:171–174. doi: 10.1007/3-211-30714-1_38. [DOI] [PubMed] [Google Scholar]
- 18.Strbian D, Durukan A, Pitkonen M, Marinkovic I, Tatlisumak E, Pedrono E, Abo-Ramadan U, Tatlisumak T. The blood-brain barrier is continuously open for several weeks following transient focal cerebral ischemia. Neuroscience. 2008;153:175–181. doi: 10.1016/j.neuroscience.2008.02.012. [DOI] [PubMed] [Google Scholar]
- 19.Bao HJ, Wang T, Zhang MY, Liu R, Dai DK, Wang YQ, Wang L, Zhang L, Gao YZ, Qin ZH, Chen XP, Tao LY. Poloxamer-188 attenuates TBI-induced blood-brain barrier damage leading to decreased brain edema and reduced cellular death. Neurochem Res. 2012;37:2856–2867. doi: 10.1007/s11064-012-0880-4. [DOI] [PubMed] [Google Scholar]
- 20.Zhao J, Pati S, Redell JB, Zhang M, Moore AN, Dash PK. Caffeic Acid phenethyl ester protects blood-brain barrier integrity and reduces contusion volume in rodent models of traumatic brain injury. J Neurotrauma. 2012;29:1209–1218. doi: 10.1089/neu.2011.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lv Q, Fan X, Xu G, Liu Q, Tian L, Cai X, Sun W, Wang X, Cai Q, Bao Y, Zhou L, Zhang Y, Ge L, Guo R, Liu X. Intranasal delivery of nerve growth factor attenuates aquaporins-4-induced edema following traumatic brain injury in rats. Brain Res. 2013;1493:80–89. doi: 10.1016/j.brainres.2012.11.028. [DOI] [PubMed] [Google Scholar]
- 22.Huang B, Krafft PR, Ma Q, Rolland WB, Caner B, Lekic T, Manaenko A, Le M, Tang J, Zhang JH. Fibroblast growth factors preserve blood-brain barrier integrity through RhoA inhibition after intracerebral hemorrhage in mice. Neurobiol Dis. 2012;46:204–214. doi: 10.1016/j.nbd.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xue Q, Liu Y, Qi H, Ma Q, Xu L, Chen W, Chen G, Xu X. A novel brain neurovascular unit model with neurons, astrocytes and microvascular endothelial cells of rat. Int J Biol Sci. 2013;9:174–189. doi: 10.7150/ijbs.5115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haseloff RF, Dithmer S, Winkler L, Wolburg H, Blasig IE. Transmembrane proteins of the tight junctions at the blood-brain barrier: structural and functional aspects. Semin Cell Dev Biol. 2015;38:16–25. doi: 10.1016/j.semcdb.2014.11.004. [DOI] [PubMed] [Google Scholar]
- 25.Balda MS, Flores-Maldonado C, Cereijido M, Matter K. Multiple domains of occludin are involved in the regulation of paracellular permeability. J Cell Biochem. 2000;78:85–96. [PubMed] [Google Scholar]
- 26.Lucke-Wold BP, Logsdon AF, Smith KE, Turner RC, Alkon DL, Tan Z, Naser ZJ, Knotts CM, Huber JD, Rosen CL. Bryostatin-1 Restores Blood Brain Barrier Integrity following Blast-Induced Traumatic Brain Injury. Mol Neurobiol. 2015;52:1119–1134. doi: 10.1007/s12035-014-8902-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu N, Li AL, Zhou XP, Chen Q, Cao W. P120 catenin attenuates lipopolysaccharide-induced blood-brain barrier dysfunction and inflammatory responses in human brain microvascular endothelial cells. Int J Clin Exp Pathol. 2015;8:4204–4212. [PMC free article] [PubMed] [Google Scholar]
- 28.Tran KA, Zhang X, Predescu D, Huang X, Machado RF, Gothert JR, Malik AB, Valyi-Nagy T, Zhao YY. Endothelial beta-catenin signaling is required for maintaining adult blood-brain barrier integrity and central nervous system homeostasis. Circulation. 2016;133:177–186. doi: 10.1161/CIRCULATIONAHA.115.015982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stenman JM, Rajagopal J, Carroll TJ, Ishibashi M, McMahon J, McMahon AP. Canonical Wnt signaling regulates organ-specific assembly and differentiation of CNS vasculature. Science. 2008;322:1247–1250. doi: 10.1126/science.1164594. [DOI] [PubMed] [Google Scholar]
- 30.Alvarez JI, Dodelet-Devillers A, Kebir H, Ifergan I, Fabre PJ, Terouz S, Sabbagh M, Wosik K, Bourbonniere L, Bernard M, van Horssen J, de Vries HE, Charron F, Prat A. The Hedgehog pathway promotes blood-brain barrier integrity and CNS immune quiescence. Science. 2011;334:1727–1731. doi: 10.1126/science.1206936. [DOI] [PubMed] [Google Scholar]
- 31.Murillo CA, Rychahou PG, Evers BM. Inhibition of alpha5 integrin decreases PI3K activation and cell adhesion of human colon cancers. Surgery. 2004;136:143–149. doi: 10.1016/j.surg.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 32.Camire RB, Beaulac HJ, Brule SA, McGregor AI, Lauria EE, Willis CL. Biphasic modulation of paracellular claudin-5 expression in mouse brain endothelial cells is mediated through the phosphoinositide-3-kinase/AKT pathway. J Pharmacol Exp Ther. 2014;351:654–662. doi: 10.1124/jpet.114.218339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li Z, Liu XB, Liu YH, Xue YX, Liu J, Teng H, Xi Z, Yao YL. Low-dose endothelial monocyte-activating polypeptide-II increases blood-tumor barrier permeability by activating the RhoA/ROCK/PI3K signaling pathway. J Mol Neurosci. 2016;59:193–202. doi: 10.1007/s12031-015-0668-5. [DOI] [PubMed] [Google Scholar]
- 34.Daniels BP, Holman DW, Cruz-Orengo L, Jujjavarapu H, Durrant DM, Klein RS. Viral pathogen-associated molecular patterns regulate blood-brain barrier integrity via competing innate cytokine signals. MBio. 2014;5:e01476–01414. doi: 10.1128/mBio.01476-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zheng B, Ye L, Zhou Y, Zhu S, Wang Q, Shi H, Chen D, Wei X, Wang Z, Li X, Xiao J, Xu H, Zhang H. Epidermal growth factor attenuates blood-spinal cord barrier disruption via PI3K/Akt/Rac1 pathway after acute spinal cord injury. J Cell Mol Med. 2016;20:1062–1075. doi: 10.1111/jcmm.12761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gonzalez-Mariscal L, Tapia R, Chamorro D. Crosstalk of tight junction components with signaling pathways. Biochim Biophys Acta. 2008;1778:729–756. doi: 10.1016/j.bbamem.2007.08.018. [DOI] [PubMed] [Google Scholar]
- 37.Dejana E, Orsenigo F. Endothelial adherens junctions at a glance. J Cell Sci. 2013;126:2545–2549. doi: 10.1242/jcs.124529. [DOI] [PubMed] [Google Scholar]
- 38.Wu S, Wu X, Zhu W, Cai WJ, Schaper J, Schaper W. Immunohistochemical study of the growth factors, aFGF, bFGF, PDGF-AB, VEGF-A and its receptor (Flk-1) during arteriogenesis. Mol Cell Biochem. 2010;343:223–229. doi: 10.1007/s11010-010-0517-3. [DOI] [PubMed] [Google Scholar]
- 39.Herbert C, Lassalle G, Alcouffe C, Bono F. Approaches targeting the FGF-FGFR system: a review of the recent patent literature and associated advanced therapeutic agents. Pharm Pat Anal. 2014;3:585–612. doi: 10.4155/ppa.14.45. [DOI] [PubMed] [Google Scholar]
- 40.Cuevas P, Carceller F, Munoz-Willery I, Gimenez-Gallego G. Intravenous fibroblast growth factor penetrates the blood-brain barrier and protects hippocampal neurons against ischemia-reperfusion injury. Surg Neurol. 1998;49:77–83. doi: 10.1016/s0090-3019(97)00193-6. discussion 83-74. [DOI] [PubMed] [Google Scholar]
- 41.Shimizu F, Sano Y, Abe MA, Maeda T, Ohtsuki S, Terasaki T, Kanda T. Peripheral nerve pericytes modify the blood-nerve barrier function and tight junctional molecules through the secretion of various soluble factors. J Cell Physiol. 2011;226:255–266. doi: 10.1002/jcp.22337. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.