

Abstract
Endoplasmic reticulum (ER) stress play important roles in the spinal cord injury (SCI), which including blood-spinal cord barrier (BSCB) disruption. Lithium chloride (LiCl) is a clinical drug for bipolar mood disorders and contributes to neuroprotection. This study aims to investigate the effects of LiCl on BSCB disruption and the ER stress pathway induced by spinal cord injury. We examined the integrity of the BSCB with Evans Blue dye and macrophages extravasation, measured the microvessels loss, the junction proteins degeneration, the activation ER stress, and the locomotor function recovery. Our data indicated that LiCl treatment could attenuates BSCB disruption and improved the recovery of functional locomotion in rats SCI model, reduced the structure damage and number loss of microvessels, increased the expressions of junction proteins, including p120, β-catenin, occludin, and claudin-5, via reversed the upregulated ER stress associated proteins. In addition, LiCl significantly inhibited the increase of ER stress markers and prevents loss of junction proteins in thapsigargin (TG)-treated human brain microvascular endothelial cells (HBMEC). These findings suggest that LiCl treatment alleviates BSCB disruption and promote the neurological function recovery after SCI, partly through inhibiting the activation of ER stress.
Keywords: Blood-spinal cord barrier, spinal cord injury, endoplasmic reticulum stress, lithium chloride
Introduction
The blood-spinal cord barrier (BSCB) is composed of a tightly sealed monolayer of endothelial cells and adjacent perivascular cells, including astrocytes (astrocytic endfeet) and pericytes. These surrounding perivascular cells wrap the abluminal capillary surface providing physical support and stability the BSCB [1]. Junctional complexes between the endothelial cells, comprised of tight junction (TJ), adherens junction (AJ), and gap junction, also play a critical role in maintaining the integrity of the BSCB [2]. Traumatic spinal cord injury (SCI) is initially caused by the initial mechanical insult, but SCI pathology is largely determined by prolonged secondary processes associated with damage of the BSCB, including blood infiltration, loss of microvasculature, and degeneration of junctional complexes. The disruption of the BSCB after SCI elicits a robust release of neurotoxic products that can compromise synaptic and neuronal functions, which eventually induces extensive neuronal programmed cell death and permanent neurological deficits as a result [3,4]. Thus, by targeting the BSCB disruption should be considered as a feasible approach for treating neurological deficits after SCI.
Lithium has been the mainstay of treatment for bipolar disorder for more than 60 years. Lithium mediates its neuroprotective effects in a variety of neurodegenerative conditions including stroke, Alzheimer disease, Parkinson disease, and Huntington disease [5-7]. A few studies have proposed that lithium chloride (LiCl) promotes barrier tightness by increasing the expression and distribution of TJ proteins via inhibiting Glycogen Synthase Kinase-3β in the brain [8]. Although there have been encouraging reposts claiming that LiCl improves locomotor functions after SCI [9,10], as yet the mechanism by which treatment of LiCl prevents disruption of BSCB after SCI is not fully understood.
The endoplasmic reticulum (ER) is an important subcellular organelle that facilitates proper folding of newly synthesized secretory and membranous proteins and also serves as an intracellular Ca2+ store [11]. Activation of ER stress following SCI leads to an accumulation of unfolded proteins in the ER lumen triggering the unfolded protein response (UPR). Prolong ER stress results in inhibition of protein synthesis and depletion of Ca2+ from ER stores, which eventually increases CHOP expression and activates caspase-dependent neuronal cell death [12,13]. Deletion of CHOP protects loss of microvasculature following SCI [14]. It has been shown that treatment of lithium reduced ER stress-mediated apoptosis via inhibition of caspase-12 and cleavage of caspase-3 in the ER [15]. Protracted lithium pretreatment is cytoprotective against thapsigargin (TG) induced cytotoxicity resulting from ER stress in PC12 cells [16]. Recently, a few studies have shown that, in rat model of SCI, inhibition of ER stress upregulated levels of TJ and AJ protein, and thus improving the BSCB integrity [17,18]. However, much less is known about the effect of LiCl on ER stress in the BSCB. In the present study, we sought to investigate the potential therapeutic effect of LiCl on the BSCB disruption from both in vivo and in vitro approaches, and out findings demonstrated that treatment of LICl attenuated ER stress-induced loss of TJ and AJ proteins, thus improving the BSCG integrity after SCI.
Materials and methods
Animals
Adult female Sprague-Dawley (SD) rats (220-250 g) were purchased from the Chinese Academy of Sciences, Shanghai, China. All experimental procedures were approved by the ethics committee of Wenzhou Medical University and performed in accordance with the Guide for the Care and Use of Laboratory Animals.
Animal model of SCI
SD rats were maintained in a temperature-controlled environment (23-25°C) with 12 h light/dark cycles and free access to food and water. Aniamls were randomly divided into three groups: (1) control group, (2) SCI group, and (3) SCI + LiCl group. Rats were anaesthetized with 2% pentobarbital sodium (40 mg/kg) intraperitoneally with all fur and muscle adjacent to the spinous processes dislodged to expose the vertebral column. A laminectomy was performed at the T9 vertebral level to expose the spinal cord, where a moderate contusion was done using a vascular clip (15 g forces, Oscar, China) for 1 min. Then, the incision sites were closed in layers. In the SCI + LiCl group, LiCl dissolved in PBS was administered intraperitoneally (20 mg/kg/d) 2 h before injury and after once daily until animals were sacrificed. An equal amount of PBS was administered to vehicle-treated rats. Rats in the sham-operated control group were subjected to the same surgical procedure without compression injury. Manual urinary bladder emptying was peformed twice daily until bladder function returned in these animals.
Cell culture and viability assay
Primary cultures of human brain microvascular endothelial cells (HBMEC) were purchased from ScienCell Research Laboratories (ScienCell Research Laboratories, San Diego, CA, USA), and maintained in endothelial cell medium in a humidified atmosphere containing 5% CO2 at 37°C. HBMEC were expanded and treated with LiCl alone (10 mM), TG alone (10 μM) or LiCl plus TG for 24 h. At the end of experiments, cells were harvested for immunofluorescence staining, and western blot analyses. For a viability assay, cells were seeded on a 96-well plate at 7 × 103 cells/well. After treating different drugs in each group for 24 h, HBMEC were incubated with MTT at 37°C for 4 h. Medium was removed subsequently and 100 µl of DMSO was added to each well before measurement. The absorbance was measured at 550 nm with a spectrophotometer (Thermo Electron Corporation, France), and cell viability was expressed as a percentage relative to the control group treated with LiCl alone.
Evans Blue-BSCB disruption
The integrity of the BSCB was examined with use of Evans Blue dye extravasation, as previously described [19]. In brief, animals received a tail injection of 2% Evans Blue dye (4 ml/kg, Sigma-Aldrich) 1 day after SCI before sacrifice. For qualitative measurement of Evans Blue extravasation, one centimeter of the T9 spinal cord surrounding the injury site was extracted, weighed, and snap-frozen in -80°C. Samples (400 mg) were then homogenized in 400 μL of N, N’-dimethylformamide (DMF) and incubated at 70°C for 72 h. Samples were centrifuged at 18,000 rpm for 20 min twice and the supernatant was collected, Collected samples were aliquoted (200 μL) into a 96-well glass plate, and the fluorescence signal was quantified at an excitation wavelength of 620 nm and an emission wavelength of 680 nm using a spectrophotometer. Data were normalized to the original sample weight, and Evans Blue concentration was calculated based on a standard curve of Evans Blue in DMF (data reported as Evans Blue per spinal cord weight: μg/g). On the other hand, animals were perfused with PBS and subsequently with 4% formaldehyde (PFA). Spinal cords were sectioned into 20-μm thick and the fluorescence signal of Evans Blue was captured using a fluorescence microscope and the relative fluorescence intensity was determined by Image ProPlus (Media Cybernetics, Rockville, MD, USA).
Locomotion recovery assessment
Functional deficits induced by SCI in rats were evaluated using the 21-point Basso-Beattie-Bresnahan (BBB) locomotion scale and footprint analysis. The BBB locomotion scale was conducted at 1, 3, 7, 14, 21, and 28 d after SCI in rats. Animals were placed in open experimental field and were allowed to move freely for 5 min. Crawling ability was evaluated according to the BBB scale, and scores ranging from 0 (no limb movement or weight support) to 21 (normal locomotion) were awarded. For footprint analysis, both animal’s forepaws and hindpaws were dipped in red and blue ink (nontoxic) and were allowed to leave a trail of footprints as they walk across a narrow box (1 m long and 7 cm wide). The footprints were scanned, and digitized images were analyzed.
Transmission electron microscopy (TEM) analysis
Spinal cord tissues were fixed in 2.5% glutaraldehyde overnight, and then were post-fixed in 2% osmium tetroxide before blocking with 2% uranyl acetate. Following dehydration in a series of acetone wash, tissues were embedded in araldite and sectioned. Toluidine blue staining was performed and observed using a Hitachi TEM.
Immunofluorescence
Animals were anesthetized and transcardially perfused with 0.9% NaCl, followed by 4% PFA at 1 d after injury, T7-T9 spinal cord segments near the lesion epicenter were fixed in 4% PFA for 24 h and embedded in paraffin for transverse sectioning (5 μm). Sections were incubated with 5% bovine serum albumin (BSA) in a 37°C oven for 30 min following by an overnight incubation of appropriate primary antibodies at 4°C. Primary antibodies are listed as follows: rabbit anti-CD68 (1:400, Abcam, Cambridge, UK), mouse anti-CD31 (1:400, Abcam), rabbit anti-claudin-5 (1:100; Santa Cruz, CA, USA), rabbit anti-OCC (1:100, Proteintech), rabbit anti-PDI (1:400, Abcam), and rabbit anti-CHOP (1:100; Santa Cruz). After that, sections were washed at room temperature, and were incubated with goat anti-rabbit Alexa Fluor 488 (1:1000, Abcam), or donkey anti-mouse TR (1:1000, Abcam) for 1 h [20,21].
For in vitro study, cells were fixed with 4% FPA for 30 min at room temperature followed by blocking with 5% BSA in a 37°C oven for 30 min. After blocking, cells were incubated with primary rabbit antibody anti-β-catenin (1:400, Abcam) at 4°C overnight, and secondary goat antibody anti-rabbit Alexa Fluor 488 (1:1000, Abcam). The nuclei were stained with DAPI for 5 min at room temperature [22]. Images were captured by a confocal microscopy. The CD68 positive cells were counted at eight randomly selected fields from per sample by using the Image ProPlus.
Western blot analysis
Proteins from the spinal cord tissues and HBMEC were purified in an NP-40 lysis buffer. An equal amount of protein was separated by 11.5% gel, transferred onto a PVDF membrane (Bio-Rad Laboratories) [23]. Membranes were blocked with 5% milk-TBST for 90 min at room temperature, and were subsequently incubated with the primary antibodies: rabbit anti-CD31 (1:1000, Abcam), rabbit anti-p120 (1:1000, Abcam), rabbit anti-β-caternin (1:1000, Abcam), rabbit anti-occludin (1:1000, Cell Signal Technology), rabbit anti-claudin-5 (1:300; Santa Cruz), mouse anti-ATF-6 (1:1000, Abcam), rabbit anti-PDI (1:1000, Abcam), rabbit anti-GRP78 (1:1000, Abcam), and rabbit anti-CHOP (1:300; Santa Cruz). After washing, membranes were incubated with appropriate secondary antibodies for 1 h at room temperature. Proteins were detected using an enhanced chemiluminescence (ECL) kit and imaged by the VersaDoc Imaging System (Bio-Rad Laboratories). Signal intensities were densitometrically quantified using Image Lab 3.0 software (Bio-Rad). Data were normalized to total or loading controls [24,25].
Statistical analysis
Statistical analyses were carried out using the SPSS 2.0 statistical software. Data were expressed as the means ± SEM. Statistical significance was determined by Student t test if comparing only two experimental groups or one-way ANOVA followed by Dunnett’spost hoc test if analyzing more than two groups. Differences were considered to be statistically significant at P < 0.05.
Results
LiCl attenuates BSCB disruption antor recoved promotes locomory after SCI
The integrity of BSCB after injury was determined based on permeability of Evans Blue dye. Evans blue is a dye that binds albumin and has been used extensively as a marker of extravascular protein leakage. As shown in Figure 1A-C, increased Evans Blue dye was detected in the SCI group as compared with the sham-operated group, suggesting that SCI elicits BSCB disruption in rats. Interestingly, BSCB influx of Evans Blue dyes was significantly reduced in the SCI group that received treatment of LiCl (Figure 1A-C). These results were further corroborated by rats received LiCl showing a concentration-dependent decrease in Evans Blue dye fluorescence in the injured spinal cords at 1 d after SCI when compared to the no-treatment group after SCI (Figure 1D, 1E). During SCI, BSCB disruption causes influx of pro-inflammatory factors and other normally impermeable toxic molecules. Significantly increased inflammation as determined by enhanced CD68, a macrophage-specific marker, was observed in animals 1 d after SCI. Our results demonstrated that reduced staining for CD68 + cells in LiCl-treated rats after SCI compared to the non-treated group (Figure 1F, 1G), suggesting LiCl attenuated influx of inflammatory cells into the BSCB induced by SCI in rats.
Figure 1.
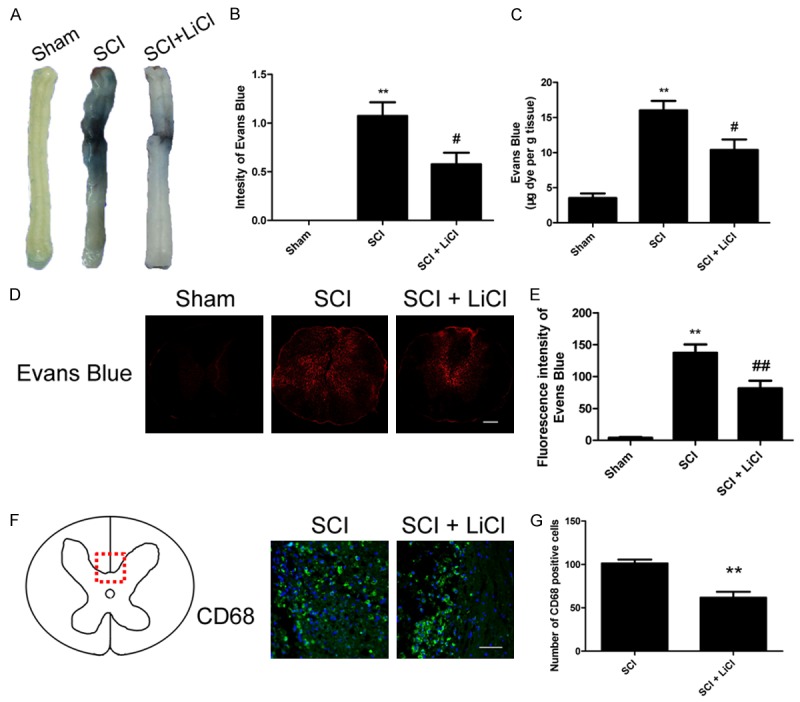
LiCl attenuates BSCB disruption after SCI. After SCI, rat were administered with LiCl intraperitoneally (20 mg/kg/d), and BSCB permeability was measured at 1 d after SCI by using Evans Blue dye (n = 4 per group). A, B. Representative whole spinal cords and quantification of BSCB permeability data in the sham, SCI, SCI + LiCl group showing Evans Blue dye permeabilized into the spinal cord. **P < 0.01 vs sham group, #P < 0.05 vs SCI group. C. Quantification data of EB content of spinal cord (μg/g) in each group. **P < 0.01 vs sham group, #P < 0.05 vs SCI group. D, E. Representative confocal images of an EB extravasation and quantification of the fluorescence intensity of EB in each group. Scale bar = 1 mm, **P < 0.01 vs sham group, ##P < 0.01 vs SCI group. F, G. Immunofluorescence staining of CD68 and DAPI and quantification of the number of CD68 positive cells in selected area of spinal cords in each group. Scale bar = 50 μm, **P < 0.01 vs SCI group.
Hindlimb locomotor function was evaluated 4 weeks after injury in rats using the Basso, Beattie and Bresnahan (BBB) scale and footprint analysis. Sham rats showed no altered locomotor function while both SCI experimental groups showed reduced BBB scores indicating there locomoter functions were significantly impaired. BBB scores were not different between animals received LiCl and with no treatment 1, 3, and 7 d after injury. However, rats received treatment of LiCl showed improved BBB scores as compared to their non-treated controls at 14, 21 and 28 d after injury (Figure 2A). Furthermore, footprint analyses also demonstrated that LiCl improved forelimb-hindlimb coordination in rats 28 d after injury, which was contrast to non-treated SCI group showing inconsistent coordination and extensive drags (Figure 2B). Taken together, these findings suggested that LiCl improved BSCB disruption accelerating locomotor recovery in rats after SCI.
Figure 2.
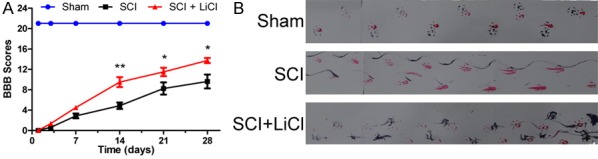
LiCl facilitates improved locomotor recovery from SCI. After SCI, rat were administered with LiCl intraperitoneally (20 mg/kg/d) once per day for 28 d, and locomotor recovery was assessed via the BBB scale (n = 8 per group), and footprint analysis. A. The BBB scores in the sham, SCI, and SCI + NBP groups. *P < 0.05, **P < 0.01 versus the SCI group. B. Footprint analysis results of each group. Both animal’s forepaws and hindpaws were respectively dipped in red and blue dye.
LiCl prevents endothelial cells damage after SCI
The disruption of BSCB is associated with endothelial cell (EC) death and vascular remodeling. We performed immunostaining for CD31, an EC-specific marker, in animals. Our results demonstrated reduced CD31 staining in the non-treated SCI group as compared with the sham group. Nevertheless, a significantly increased CD31 expression was found in LiCl-treated rats 1 d after injury (Figure 3A-C). ECs and the surrounding parenchyma in spinal cord were further examined using electron microscopy, (Figure 3D). Capillary ECs showed organelle swelling and formation of numerous large vacuoles in their cytoplasm, and intracellular edema of the ECs was also observed in the SCI group with no treatment. Furthermore, TJs between the ECs were hardly found in the non-treated SCI group. In contrast, LiCl treatment effectively protected capillary ECs from SCI, intact TJs were noted in these animals after injury.
Figure 3.
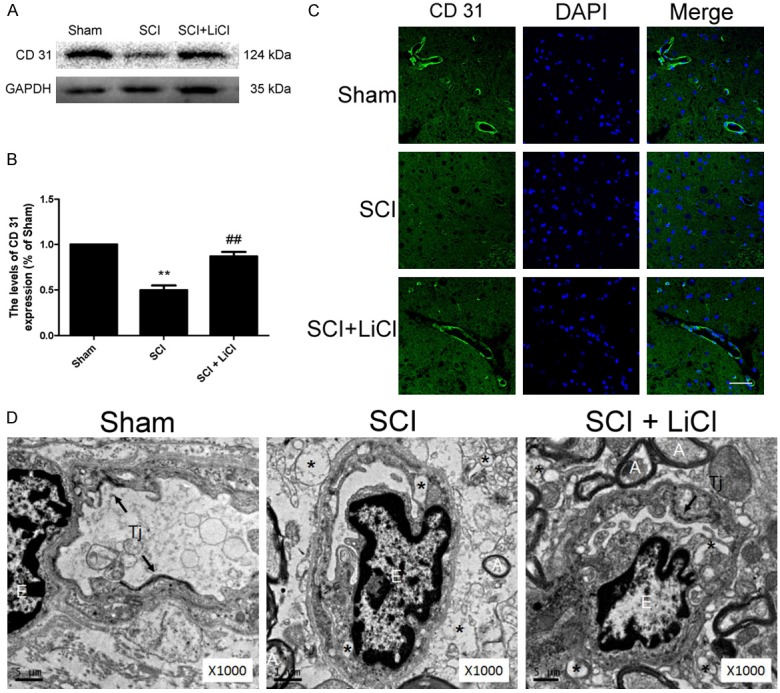
LiCl preventes endothelial cells damage after SCI. A, B. Western blot and quantification of CD31 at 1 d after injury in the sham, SCI, and SCI + NBP groups. n = 4 per group, **P < 0.01 vs sham group, ##P < 0.01 vs SCI group. C. Immunofluorescence staining of CD31 and DAPI at 1 d after injury in each group. Scale bar = 10 μm, n = 4 per group. D. Representative transmission electron microscope photographs at 1 d after injury in each group. n = 4 per group. E-endothelial cell, A-axon, Tj-tight junction, Asterisks indicate extracellular and intracellular edema.
LiCl prevents loss of AJ and TJ proteins after SCI
Both TJ and AJ proteins play an important role in sealing the gap between ECs, which is essential for the integrity of BSCB [26]. One day after injury, the expression of p120 and β-catenin were significantly decreased in the SCI group compared with the sham-operated rats. Decreased levels of oocludin (OCC) and claudin-5 were found in the SCI group 1 d after injury (Figure 4C, 4D). Interestingly, treatment of LiCl prevented reduced p120, β-catenin, OCC, and claudin-5 in rats after injury (Figure 4A-D). Reduced TJ and AJ protein expression was in agreement with immunostaining analyses showing that attenuated OCC and claudin-5 signals were detected in rats after injury, whereas LiCl treatment reversed these findings in animals with SCI (Figure 4E, 4F). Taken together, these observations suggest that LiCl prevented loss of TJ and AJ protein expression associated with BSCB disruption during SCI.
Figure 4.

LiCl prevents loss of tight junction (TJ) and adheren junction (AJ) proteins after SCI. A, B. Western blot and quantification of p120 and β-catenin at 1 d after injury in the sham, SCI, and SCI + NBP groups. n = 4 per group, *P < 0.05 vs sham group, ##P < 0.01 vs SCI group. C, D. Western blot and quantification of Occludin (OCC) and Claudin-5 at 1 d after injury in each group. n = 4 per group. *P < 0.05 vs sham group, #P < 0.05, ##P < 0.01 vs SCI group. E, F. Immunofluorescence staining of OCC and Claudin-5 at 1 d after injury in each group, nuclei are labeled with DAPI. Scale bar = 10 μm, n = 4 per group, arrows indicate microvessels.
LiCl Inhibits the activation of ER stress after SCI
Previous work demonstrated that ER stress markers were elevated due to an acute SCI [17]. We found that significant increases of ER stress associated protein, such as GRP78, PDI, ATF-6, and CHOP in the SCI group 1 d after injury. Treatment of LiCl markedly reduced levels of these proteins in animals during SCI (Figure 5A-D). An observed increase of PDI and CHOP immunostaining in the SCI group was reversed by treatment of LiCI after injury (Figure 5E, 5F). Furthermore, the level of cleaved caspase-12, a downstream molecule of CHOP, was markedly increased in the injured spinal cord of animals, and this increase was attenuated by treatment of LiCl (Figure 5G, 5H). These results indicated that LiCl effectively inhibited cell death mediated by increased ER stress after SCI in rats.
Figure 5.
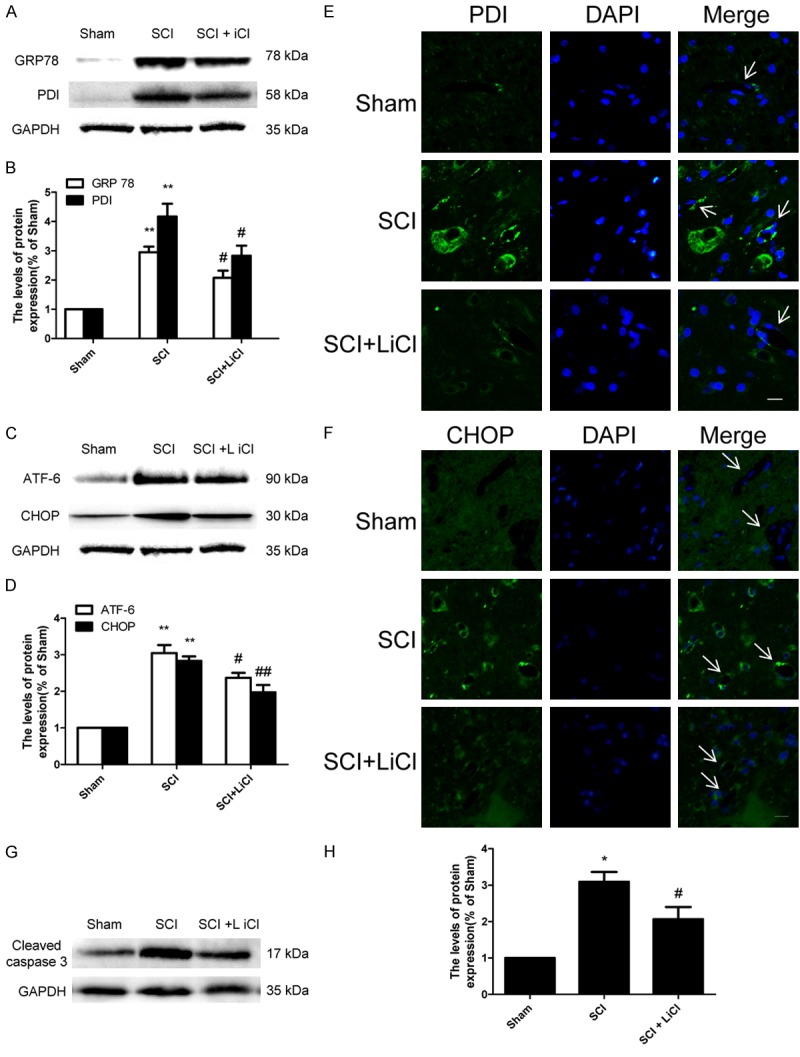
LiCl Inhibits the activation of ER stress after SCI. A-D. Western blot and quantification of GRP 78, PDI, ATF-6 and CHOP at 1 d after injury in the sham, SCI, and SCI + NBP groups. n = 4 per group, **P < 0.01 vs sham group, #P < 0.05, ##P < 0.01 vs SCI group. E, F. Immunofluorescence staining of PDI and CHOP at 1 d after injury in each group, nuclei are labeled with DAPI. Scale bar = 10 μm, n = 4 per group. G, H. Western blot and quantification of cleaved caspase 3 at 1 d after injury in each group. n = 4 per group, *P < 0.05 vs sham group, #P < 0.05 vs SCI group.
LiCl prevents loss of AJ and TJ proteins in TG-treated HBMEC
In parallel to our in vivo study, we examined whether inhibition of ER stress was involved in the protective effect mediated by treatment of LiCl in vitro. HBMEC treated with TG exhibited a reduce in p120, β-catenin, OCC, and claudin-5 protein levels when compared with controls (Figure 6A-D). However, LiCl incubation reversely up-regulated p120, β-catenin, OCC, and Claudin-5 protein expression in HBMEC treated TG (Figure 6A-D). These results were also in agreement with subsequent immunofluorescence staining demonstrating that HBMEC treated with TG showed that β-catenin immunostaining was reduced, yet a higher level of β-catenin immunostaining was observed the LiCl-treated group (Figure 6E).
Figure 6.
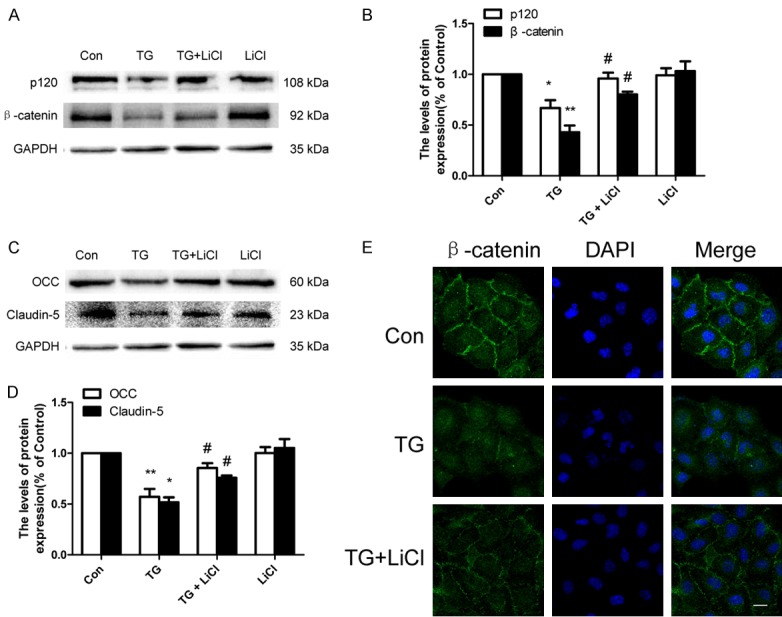
LiCl prevents loss of TJ and AJ proteins in TG-treated HBMEC. HBMEC were treated with TG (10 μM) or together with LiCl (10 mM) or LiCl alone for 24 h. A-D. Western blot and quantification of p120, β-catenin, OCC and Claudin-5 in the control, TG, and TG + LiCl and LiCl groups. *P < 0.05, **P < 0.01 vs control group, #P < 0.05 vs TG group. E. Immunofluorescence staining of β-catenin in each group, nuclei are labeled with DAPI. Scale bar = 10 μm.
LiCl inhibits ER stress in TG-treated HBMEC
Our in vitro study also showed that levels of ER stress associated proteins, including GRP78, PDI, ATF-6, and CHOP were significantly increased in HBMECs when treated with TG, which were reversed by treatment of LiCl (Figure 7A-D). However, no effect of PBA alone on these proteins was observed. We also studied the effect of TG on HBMEC viability. In MTT assays, TG inhibited cell viability and this inhibition was partly reversed by LiCl treatment (Figure 7E). These results suggested that LiCl effectively inhibited ER stress-associated apoptosis in HBMEC in vitro.
Figure 7.
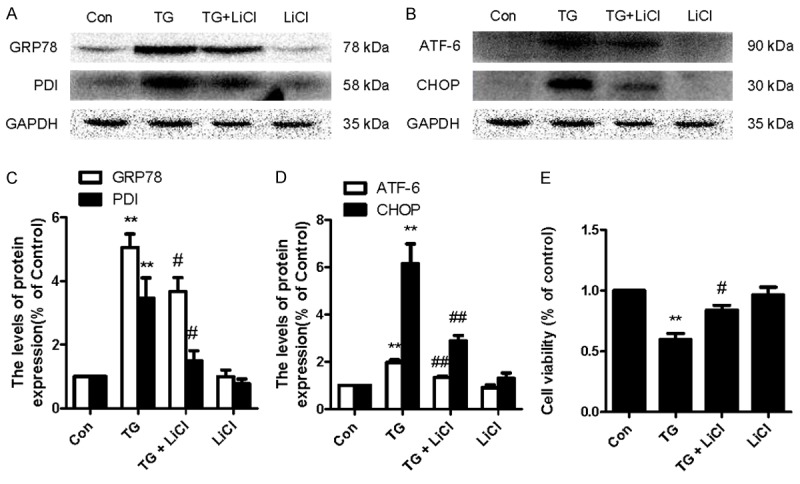
LiCl inhibits ER stress in TG-treated HBMEC. HBMEC were treated with TG (10 μM) or together with LiCl (10 mM) or LiCl alone for 24 h. A-D, Western blot and quantification of GRP78, PDI, ATF-6, and CHOP in the control, TG, and TG + LiCl and LiCl groups. *P < 0.05, **P < 0.01 vs control group, #P < 0.05, ##P < 0.01 vs TG group. E. MTT results of LiCl -treated HBMEC induced by TG. **P < 0.01 vs control group, #P < 0.05 versus TG group.
Discussion
Under normal physiological conditions, the BSCB represents a tight barrier between the circulating blood and central nervous system (CNS). The dense junction proteins, which seal the space between adjacent endothelial cells, play an important role in maintenance of integrity of the BSCB. SCI induced by initial mechanical insult resulted in rapid, permanent damages in microvasculature leading to endothelial dysfunction and increased BSCB leakage [27]. In our study, the BSCB was disrupted and permeability was significantly increased at 1 d after SCI, which was effectively prevented by LiCl treatment. Moreover, treatment of LiCl reduced the infiltration of macrophages following the disruption of BSCB. ECs and their TJs and AJs are the primary components of the BSCB, thus, loss of ECs and their junction proteins may directly result in microcirculation disruption, and eventually induced extensive cell death in spinal cord neurons, glial cells, and axons [28]. In line with previous studies, our results demonstrated that severely reduced ECs, altered microvasculature, and damaged surrounding parenchyma were observed in rats after SCI, whereas LiCl significantly attenuated these damages. Furthermore, our previous reports showed that the levels of AJ and TJ proteins are dramatically decreased at 1 or 3 d after SCI, supporting the notion that changes in the expression and distribution of AJ and TJ proteins are closely related to the permeability of BSCB during [18,29]. Our data showed that p120, β-catenin, OCC, and claudin-5 were markedly degraded at 1 d after SCI, but LiCl reversed the degradation of these molecules. Our in vitro study also showed that treatment of LiCl enhanced the expression of AJ and TJ proteins in HBMEC, which was consistent with previous study supporting our in vivo study. As a result, treatment LiCl could effectively improve the locomotor recovery after SCI [8]. Taken together, our result suggests that treatment of LiCl has a therapeutic effect in maintenance of the BSCB integrity, and therefore improves SCI in animals.
The mechanisms by which BSCB breakdown occurs, progress and the consequences of a compromised barrier are manifold [30]. Previous works show that ER stress-targeted therapeutic strategies effectively protects the spinal cord against protracted damage that occurs in the sub-acute phase by alleviating inflammatory and ischemic injury [14]. Our previous studies have shown that prolonged ER stress might activate an apoptotic pathway by activating CHOP and caspase-12 causing PC12 cells apoptosis and secondary injury after SCI [31,32]. Furthermore, in a model of SCI, we previously showed that inhibition of ER stress by PBA, a specific ER stress inhibitor, prevented loss of TJ and AJ proteins, and eventually resulted in improved BSCB integrity [17]. Other studies also showed that protracted lithium pretreatment of PC12 cells inhibited an intracellular calcium increase, up-regulated the antiapoptotic protein Bcl-2, and blocked Bcl-2 down-regulation against TG-induced ER stress [16]. In this study, we first reported that LiCl treatment effectively inhibited the increase of the up-regulated ER stress associated proteins at 1 day after SCI, which was concurrent with LiCl conferring protection against BSCB disruption. Furthermore, the activation of ER stress induced by TG was attenuated by LiCl, and TG-mediated degradation of AJ and TJ proteins was prevented as a result in HBMEC. In sum, these results indicated that ER stress plays an important role in BSCB disruption, LiCI prevented BSCB disruption via inhibiting ER stress in rats after SCI. Previous findings showed that inhibition of GSK-3β by LiCl also increased the half-life of occludin and claudin-5, and therefore produced a gradual and sustained increase in transendothelial electrical resistance in HBMEC [10]. The signaling mechanisms by which LiCl prevents the loss of ECs and the integrity of the BSCB remain unclear. Several molecular mechanisms possibly underlying this phenomena are as follows: 1) treatment of LiCl might attenuate accumulation of misfolded and unfolded proteins in the ER lumen through inhibition of GSK-3β activity, thus preserve both AJ and TJ proteins expression; 2) LiCl might inhibit GSK-3β-mediated mitochondria dependent apoptotic pathway and increased ER stress, and therefore reduce loss of ECs and neuronal death under pathological conditions of SCI.
Today lithium is licensed for clinical use for treating bipolar mood disorders. Over the past years, LiCl has generated considerable excitement for its potential therapeutic effects for a wide variety of neurological disorders [5-7]. In this study, we first demonstrate that LiCl played a critical role in maintaining the BSCB integrity, which significant improved functional recovery in animals, and this protective effective was mediated by via down-regulation of ER under pathological conditions such as SCI. Taken together, the present study has paved the pathway for new treatment of SCI.
In conclusion, our research demonstrated that the increased permeability of Evans Blue dye, enhanced filtration of macrophage, loss of microvasculature, and degradation of junctional complexes such as p120, β-catenin, OCC and claudin-5 under pathological conditions of SCI. Strikingly, treatment of LiCl inhibited activation of ER stress, which preserved the integrity of the BSCB, and as a result improved locomoter deficits were observed in rats after SCI. In addition, treatment of LiCl significantly inhibited ER stress-mediated loss of TJ and AJ proteins and cell death in HBVECs. In sum, these data demonstrate that LiCl is considered to be a potential therapeutic agent for treating BSCB disruption induced by SCI.
Acknowledgements
This study was partially supported by a research grant from National Natural Science Foundation of China (81572237, 81572227, 81302775, 81501953, 81601980), Zhejiang Provincial Natural Science Foundation (Y14H170002, LY17H090017, Y14H170008, Q16H090023), and Technologies Science and technology Program of Zhejiang Provincial (2016C33107).
Disclosure of conflict of interest
None.
Authors’ contribution
J.X., H.-Z.X. and Z.-L.H. conceived and designed the experiments. Z.-L.H., Q.-Q.W., J.-W.L., Z.-M.Z., J.C., and H.-Y.Z. performed the experiments. Z.-L.H., Q.-Q.W., Y.-L.Z., H.-Z.X. and J.X. analyzed the data. Z.-L.H. Y.-L.Z., and Z.-G.W. wrote the paper.
References
- 1.Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiol Dis. 2010;37:13–25. doi: 10.1016/j.nbd.2009.07.030. [DOI] [PubMed] [Google Scholar]
- 2.Stamatovic SM, Johnson AM, Keep RF, Andjelkovic AV. Junctional proteins of the blood-brain barrier: new insights into function and dysfunction. Tissue Barriers. 2016;4:e1154641. doi: 10.1080/21688370.2016.1154641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev. 2005;57:173–185. doi: 10.1124/pr.57.2.4. [DOI] [PubMed] [Google Scholar]
- 4.Gerzanich V, Woo SK, Vennekens R, Tsymbalyuk O, Ivanova S, Ivanov A, Geng Z, Chen Z, Nilius B, Flockerzi V, Freichel M, Simard JM. De novo expression of Trpm4 initiates secondary hemorrhage in spinal cord injury. Nat Med. 2009;15:185–191. doi: 10.1038/nm.1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chiu CT, Chuang DM. Molecular actions and therapeutic potential of lithium in preclinical and clinical studies of CNS disorders. Pharmacol Ther. 2010;128:281–304. doi: 10.1016/j.pharmthera.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chiu CT, Wang Z, Hunsberger JG, Chuang DM. Therapeutic potential of mood stabilizers lithium and valproic acid: beyond bipolar disorder. Pharmacol Rev. 2013;65:105–142. doi: 10.1124/pr.111.005512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang ZF, Fessler EB, Chuang DM. Beneficial effects of mood stabilizers lithium, valproate and lamotrigine in experimental stroke models. Acta Pharmacol Sin. 2011;32:1433–1445. doi: 10.1038/aps.2011.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ramirez SH, Fan S, Dykstra H, Rom S, Mercer A, Reichenbach NL, Gofman L, Persidsky Y. Inhibition of glycogen synthase kinase 3beta promotes tight junction stability in brain endothelial cells by half-life extension of occludin and claudin-5. PLoS One. 2013;8:e55972. doi: 10.1371/journal.pone.0055972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zakeri M, Afshari K, Gharedaghi MH, Shahsiah R, Rahimian R, Maleki F, Dehpour AR, Javidan AN. Lithium protects against spinal cord injury in rats: role of nitric oxide. J Neurol Surg A Cent Eur Neurosurg. 2014;75:427–433. doi: 10.1055/s-0033-1345098. [DOI] [PubMed] [Google Scholar]
- 10.Yang ML, Li JJ, So KF, Chen JY, Cheng WS, Wu J, Wang ZM, Gao F, Young W. Efficacy and safety of lithium carbonate treatment of chronic spinal cord injuries: a double-blind, randomized, placebo-controlled clinical trial. Spinal Cord. 2012;50:141–146. doi: 10.1038/sc.2011.126. [DOI] [PubMed] [Google Scholar]
- 11.Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2008;7:1013–1030. doi: 10.1038/nrd2755. [DOI] [PubMed] [Google Scholar]
- 12.Zhang HY, Zhang X, Wang ZG, Shi HX, Wu FZ, Lin BB, Xu XL, Wang XJ, Fu XB, Li ZY, Shen CJ, Li XK, Xiao J. Exogenous basic fibroblast growth factor inhibits ER stress-induced apoptosis and improves recovery from spinal cord injury. CNS Neurosci Ther. 2013;19:20–29. doi: 10.1111/cns.12013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee JY, Maeng S, Kang SR, Choi HY, Oh TH, Ju BG, Yune TY. Valproic acid protects motor neuron death by inhibiting oxidative stress and endoplasmic reticulum stress-mediated cytochrome C release after spinal cord injury. J Neurotrauma. 2014;31:582–594. doi: 10.1089/neu.2013.3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fassbender JM, Saraswat-Ohri S, Myers SA, Gruenthal MJ, Benton RL, Whittemore SR. Deletion of endoplasmic reticulum stress-induced CHOP protects microvasculature post-spinal cord injury. Curr Neurovasc Res. 2012;9:274–281. doi: 10.2174/156720212803530627. [DOI] [PubMed] [Google Scholar]
- 15.Ghribi O, Herman MM, Savory J. Lithium inhibits Abeta-induced stress in endoplasmic reticulum of rabbit hippocampus but does not prevent oxidative damage and tau phosphorylation. J Neurosci Res. 2003;71:853–862. doi: 10.1002/jnr.10511. [DOI] [PubMed] [Google Scholar]
- 16.Hiroi T, Wei H, Hough C, Leeds P, Chuang DM. Protracted lithium treatment protects against the ER stress elicited by thapsigargin in rat PC12 cells: roles of intracellular calcium, GRP78 and Bcl-2. Pharmacogenomics J. 2005;5:102–111. doi: 10.1038/sj.tpj.6500296. [DOI] [PubMed] [Google Scholar]
- 17.Zhou Y, Ye L, Zheng B, Zhu S, Shi H, Zhang H, Wang Z, Wei X, Chen D, Li X, Xu H, Xiao J. Phenylbutyrate prevents disruption of blood-spinal cord barrier by inhibiting endoplasmic reticulum stress after spinal cord injury. Am J Transl Res. 2016;8:1864–1875. [PMC free article] [PubMed] [Google Scholar]
- 18.Ye LB, Yu XC, Xia QH, Yang Y, Chen DQ, Wu F, Wei XJ, Zhang X, Zheng BB, Fu XB, Xu HZ, Li XK, Xiao J, Zhang HY. Regulation of Caveolin-1 and junction proteins by bFGF contributes to the integrity of blood-spinal cord barrier and functional recovery. Neurotherapeutics. 2016;13:844–858. doi: 10.1007/s13311-016-0437-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xanthos DN, Pungel I, Wunderbaldinger G, Sandkuhler J. Effects of peripheral inflammation on the blood-spinal cord barrier. Mol Pain. 2012;8:44. doi: 10.1186/1744-8069-8-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Feng ZC, Donnelly L, Li J, Krishnamurthy M, Riopel M, Wang R. Inhibition of Gsk3beta activity improves beta-cell function in c-KitWv/+ male mice. Lab Invest. 2012;92:543–555. doi: 10.1038/labinvest.2011.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feng ZC, Popell A, Li J, Silverstein J, Oakie A, Yee SP, Wang R. c-Kit receptor signaling regulates islet vasculature, beta-cell survival, and function in vivo. Diabetes. 2015;64:3852–3866. doi: 10.2337/db15-0054. [DOI] [PubMed] [Google Scholar]
- 22.Feng ZC, Riopel M, Li J, Donnelly L, Wang R. Downregulation of Fas activity rescues early onset of diabetes in c-Kit(Wv/+) mice. Am J Physiol Endocrinol Metab. 2013;304:E557–565. doi: 10.1152/ajpendo.00453.2012. [DOI] [PubMed] [Google Scholar]
- 23.Choi J, Diao H, Feng ZC, Lau A, Wang R, Jevnikar AM, Ma S. A fusion protein derived from plants holds promising potential as a new oral therapy for type 2 diabetes. Plant Biotechnol J. 2014;12:425–435. doi: 10.1111/pbi.12149. [DOI] [PubMed] [Google Scholar]
- 24.Feng ZC, Li J, Turco BA, Riopel M, Yee SP, Wang R. Critical role of c-Kit in beta cell function: increased insulin secretion and protection against diabetes in a mouse model. Diabetologia. 2012;55:2214–2225. doi: 10.1007/s00125-012-2566-5. [DOI] [PubMed] [Google Scholar]
- 25.Li J, Feng ZC, Yeung FS, Wong MR, Oakie A, Fellows GF, Goodyer CG, Hess DA, Wang R. Aldehyde dehydrogenase 1 activity in the developing human pancreas modulates retinoic acid signalling in mediating islet differentiation and survival. Diabetologia. 2014;57:754–764. doi: 10.1007/s00125-013-3147-y. [DOI] [PubMed] [Google Scholar]
- 26.Zhang HY, Wang ZG, Wu FZ, Kong XX, Yang J, Lin BB, Zhu SP, Lin L, Gan CS, Fu XB, Li XK, Xu HZ, Xiao J. Regulation of autophagy and ubiquitinated protein accumulation by bFGF promotes functional recovery and neural protection in a rat model of spinal cord injury. Mol Neurobiol. 2013;48:452–464. doi: 10.1007/s12035-013-8432-8. [DOI] [PubMed] [Google Scholar]
- 27.Benton RL, Maddie MA, Minnillo DR, Hagg T, Whittemore SR. Griffonia simplicifolia isolectin B4 identifies a specific subpopulation of angiogenic blood vessels following contusive spinal cord injury in the adult mouse. J Comp Neurol. 2008;507:1031–1052. doi: 10.1002/cne.21570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fan ZK, Lv G, Wang YF, Li G, Yu DS, Wang YS, Zhang YQ, Mei XF, Cao Y. The protective effect of salvianolic acid B on blood-spinal cord barrier after compression spinal cord injury in rats. J Mol Neurosci. 2013;51:986–993. doi: 10.1007/s12031-013-0083-8. [DOI] [PubMed] [Google Scholar]
- 29.Liebner S, Czupalla CJ, Wolburg H. Current concepts of blood-brain barrier development. Int J Dev Biol. 2011;55:467–476. doi: 10.1387/ijdb.103224sl. [DOI] [PubMed] [Google Scholar]
- 30.Obermeier B, Daneman R, Ransohoff RM. Development, maintenance and disruption of the blood-brain barrier. Nat Med. 2013;19:1584–1596. doi: 10.1038/nm.3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ohri SS, Maddie MA, Zhang Y, Shields CB, Hetman M, Whittemore SR. Deletion of the pro-apoptotic endoplasmic reticulum stress response effector CHOP does not result in improved locomotor function after severe contusive spinal cord injury. J Neurotrauma. 2012;29:579–588. doi: 10.1089/neu.2011.1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ohri SS, Hetman M, Whittemore SR. Restoring endoplasmic reticulum homeostasis improves functional recovery after spinal cord injury. Neurobiol Dis. 2013;58:29–37. doi: 10.1016/j.nbd.2013.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]