

Abstract
Increasing evidence suggests that EPCs improve neovascularization and endothelial regeneration via the production of paracrine factors. VEGF and IL-8 are major cytokines involved in EPC-mediated angiogenesis and re-endothelialization. In our previous studies, Hcy impaired EPC migratory and adhesive activities. We devised this study to determine whether Hcy could affect the expression and secretion of VEGF and IL-8 from EPCs. We found that high levels of Hcy (100-500 μM) decreased the EPC-mediated protein secretion and mRNA expression of VEGF and IL-8. Moreover, PIO, a PPARγ agonist, has been suggested to regulate EPC adhesion, migration, survival. In this study, PIO normalized the production of these cytokines by EPCs stimulated with Hcy. These effects of Hcy and PIO were primarily mediated by PKC and ROS via NADPH oxidase. We further confirmed this mechanism via knockdown of the NADPH oxidase subunits p67phox and Nox2. Furthermore, the PPARγ inhibitor GW9662 was not observed to abrogate the beneficial effect of PIO, indicating that PIO protected EPC paracrine function against Hcy in a PPARγ-independent manner.
Keywords: Paracrine cytokines, reactive oxygen species, p67phox, siRNA
Introduction
EPCs play a protective role in the cardiovascular system by enhancing the process of new vessel formation and the maintenance of endothelial homeostasis. Accumulating evidence demonstrates that EPCs may induce neovascularization and vascular regeneration primarily via paracrine signaling, that is, through the secretion of growth factors and pro-angiogenic cytokines [1]. Among these cytokines, VEGF and IL-8 constitute the most notable factors detected in EPC-CM and have been proven to improve angiogenesis and endothelial integrity by promoting the proliferation, migration and survival of endothelial cells [2-4]. However, the regulation of the EPC secretome remains a great challenge for investigators, and few studies have been performed to investigate how EPCs regulate their secretome to mediate beneficial paracrine effects under pathological stimulation.
Our previous studies revealed that hyperhomocysteine (HHcy) reduced the number of EPCs and impaired their functional activity, including their proliferative, migratory, adhesive and in vitro vasculogenesis capacities [5,6]. We also demonstrated one possible mechanism by which Hcy reduced EPC quantity and activity. Hcy accelerated the onset of EPC senescence, which involved the inhibition of telomerase activity and Akt phosphorylation in EPCs [7]. Furthermore, Hcy promoted the formation of ROS, primarily by NADPH oxidase, within EPCs and increased EPC apoptosis [8]. However, it remains unclear whether Hcy could affect the paracrine function of EPCs.
Hcy may disrupt the development of the extra-embryonic vasculature by reducing the expression of VEGF [9]. Moreover, the Hcy-induced decrease in VEGF production by podocytes was abolished by treatment with Nox2 siRNA or diphenyleneiodonium (DPI) [10]. Short-term hyperhomocysteinemia-induced oxidative stress increased VEGF expression in the retina [11]. PKC activation represents a common signaling pathway by which Hcy exerts its pathogenic actions in the vasculature. Severe hyperhomocysteinemia was demonstrated to cause endothelial dysfunction via PKC activation [12]. Hcy also activated NADPH oxidase in monocytes via PKCβ [13]. Several signaling pathways, such as PKC and NADPH oxidase, may be involved in the Hcy-induced alteration of the EPC secretome.
PIO, a PPAR agonist, has been used to treat type 2 diabetes for a decade. Despite the well-known ability of thiazolidinediones to modulate the transcription of the insulin-sensitive genes involved in the control of glucose and lipid metabolism, PIO exerts beneficial effects that are substantially higher than expected. PIO was proven to improve the migratory response and the adhesive capacity of EPCs in patients suffering from diabetes mellitus and coronary artery disease [14,15]. PIO also prevented apoptosis of EPCs and increased in vivo neoangiogenesis in mice [16]. In addition, PIO ameliorated Ang II-induced senescence of EPCs [17]. However, none of these previous studies have investigated the protective effect of PIO on EPC cytokine production and its related mechanisms, especially with respect to EPCs subjected to a high Hcy level.
Based on these considerations, the hypothesis of this study is that Hcy alters cytokine secretion by EPCs. The Hcy-induced effect on EPC paracrine function may be initiated by the activation of PKC and NADPH oxidase. The underlying protective effects of PIO on cytokine production by EPCs may involve reducing oxidative stress via Nox and modulating PKC activation in EPCs.
Therefore, these experiments were performed to determine whether Hcy induced a decrease in VEGF and IL-8 production in EPCs. PKC and ROS produced by NADPH oxidase may play an important role in the Hcy-induced reduction in VEGF and IL-8 production. PIO attenuated the disruption of cytokine production in Hcy-stimulated EPCs.
Materials and methods
Materials
Peripheral blood mononuclear cells were obtained from healthy volunteers. Informed consent was obtained from all volunteers. Male Sprague-Dawley (SD) rats (250-300 g) were purchased from the Experimental Animal Center of Zhejiang Province. Endothelial Cell Growth Medium-2 (EGM-2) was purchased from LONZA (Walkersville, MD, USA). Lympholyte-H cell separation media was purchased from CEDARLANE (Burlington, USA). Fetal bovine serum (FBS) was purchased from GIBCO (Los Angeles, USA). Hcy and Diphenyleneiodonium chloride (DPI) were purchased from Sigma Chemical (St. Louis, MO, USA). PIO was generously provided by Huadong Medicine Co. Ltd (Hangzhou, Zhejiang, China). 5-(and-6)-chloromethyl-2’,7’-dichlorodihydrofluorescein diacetate, acetyl ester (CM-H2DCFDA) was purchased from Invitrogen (Carlsbad, CA, USA). PIO was purchased from CAYMAN (Ann Arbor, Michigan, USA). Lucigenin and NADPH tetrasodium salt were purchased from Enzo Life Sciences (Plymouth Meeting, PA, USA). Bisindolylmaleimide I (GF 109203×) was purchased from Calbiochem (Darmstadt, Germany). The anti-gp91 (NOX2) antibody was purchased from Millipore (Bedford, Massachusetts, USA). The anti-p67phox, anti-PKC α, and anti-PKC β antibodies were purchased from Santa Cruz Bio- technology, Inc. (Santa Cruz, CA, USA) The anti-phospho-PKC α/βII antibody was obtained from Cell Signaling Technology (Cambridge, Massachusetts, USA). The HRP-conjugated mouse monoclonal anti-GAPDH antibody was purchased from Kangchen (Shanghai, China). PIO, DPI, GF109203× and GW9662 were dissolved in 0.1% dimethyl sulfoxide (DMSO), and the vehicle (0.1% DMSO) was added to the control samples.
Isolation and cultivation of EPCs
The EPCs were isolated, cultured and characterized according to previously described techniques. Briefly, peripheral blood mononuclear cells (PBMNCs) were isolated from healthy volunteers via Ficoll density gradient centrifugation and then cultured on human FN-coated dishes in EGM-2 containing 20% FBS, VEGF, fibroblast growth factor-2 (FGF-2), epidermal growth factor (EGF), insulin-like growth factor (IGF), and ascorbic acid. After 3 days in culture, the non-adherent cells were removed by washing with phosphate-buffered saline (PBS), and the adherent cells were maintained in fresh medium for another 4 days.
Animals and in vivo re-endothelialization assay
Male SD rats weighing 250-300 g were used for injection of human EPC-CM. All animal experimental procedures were approved by the Zhejiang University Institutional Animal Care and Use Committee. The animals were anesthetized using ketamine (100 mg/kg IP). A cannula was introduced into the common carotid artery via the external carotid artery. Vascular injury of the common carotid artery was induced via the passage and inflation of a balloon catheter (Medtronic, 1.25×15 mm) through an arteriotomy in the external carotid artery three times. The injured segment was transiently isolated using temporary ligatures. Endothelial Basal Medium-2 (EBM-2), human EPC-CM, or Hcy-pretreated EPC-CM was infused into the injured segment and incubated for 15 min at room temperature. EBM-2 didn’t contain serum or any cytokines. After this 15-min incubation, the infusion cannula was removed. After the incubation, the blood flow to the common carotid was restored by releasing the ligatures, and the wound was then closed. No adverse neurological or vascular effects were observed in any animal that was subjected to this procedure. 3 days after the carotid artery injury, endothelial regeneration was evaluated by staining the denuded areas using 200 μL of a solution containing 5% Evans blue dye via tail vein injection. The re-endothelialized area was calculated as the difference between the blue-stained area and the injured area via computer-assisted morphometric analysis. Then, the mean values of the re-endothelialized area were used for analysis. Notably, this model has been demonstrated to facilitate accurate quantification of re-endothelialization.
Enzyme-linked immunosorbent assay (ELISA)
Cell culture supernatants were used for ELISA experiments. The supernatants of EPCs subjected to various treatments for 24 h were collected. ELISAs were prepared using VEGF and IL-8 Valukine ELISA Kits (R&D Systems, Minneapolis, MN, USA). The ELISA procedure was performed according to the recommended instructions. All analyses and calibrations were performed in duplicate.
Western blot analysis
Following treatment, the EPCs were washed with ice-cold PBS and lysed using lysis buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 1 mM EDTA, and 1 mM EGTA) supplemented with a protease inhibitor cocktail (Merck, Germany), 1 mM PMSF, 1 mM Na3VO4, and 10 mM NaF.
The protein concentration was determined using the Bradford method. After denaturation at 95°C for 5 min, 30 μg of protein was loaded in each lane, subjected to 10% sodium dodecylsulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and then transferred to a polyvinylidene difluoride (PVDF) membrane (Bio-Rad, Hercules, USA). The membranes were blocked using 5% nonfat milkand then incubated overnight in a primary antibody at the appropriate dilution, followed by incubation for 1 h in a secondary antibody conjugated to horseradish peroxidase (1:10000). After reaction with an enhanced chemiluminescence reagent (Amersham, Haemek, Israel), images of the membranes were captured using an LAS-4000 imaging system (Fujifilm, Tokyo, Japan).
RNA extraction and QRT-PCR analysis
RNA was extracted using Trizol isolation reagent (Invitrogen, USA). The RNA content of the samples was quantified by measuring their absorbance at 260 nm. Reverse-transcription (RT) was performed using a One Step RT-PCR Kit (Promega, USA) according to the manufacturer’s instructions. Primers were synthesized according to the specific motif: 5’-TCTTGGGTGCATTGGAGCCT-3’ (VEGF, sense), 5’-AGCTCATCTCTCCTATGTGC-3’ (VEGF, anti-sense); 5’-CCTGATTTCTGCA-GCTCTGT-3’ (IL-8, sense), 5’-AACTTCTCCACAACCCTCTG-3’ (IL-8, anti-sense); 5’- GGGTGTGAACCATGAGAAGT-3’ (GAPDH, sense), 5’-GACTGTGGTCATG-AGTCCT-3’ (GAPDH, anti-sense). Real-time PCR was performed using an ABI 7500 thermal cycler (Applied Biosystems, CA, USA) with SYBR green PCR mix (Takara) according to the manufacturer’s instructions. The fold-change in the relative expression level of VEGF and IL-8 was calculated using the 2-∆∆Ct method.
Intracellular fluorescence measurement of ROS
Intracellular ROS levels were measured via flow cytometry using the fluorescent probe H2DCF-DA, which, after crossing the plasma membrane due to its lipid permeability, is deacetylated to H2DCF and oxidized to the fluorescent compound DCF. EPCs were cultured for 7 days in EGM-2, treated with various drugs, and then loaded with 5 μM H2DCFH-DA in serum-free medium to exclude the interference of phenol red at 37°C for 30 min. After washing twice with PBS, the cells were immediately measured using a flow cytometer (FACSCalibur, Becton Dickinson, NJ, USA) at an excitation wavelength of 488 nm and an emission wavelength of 525 nm. The ROS levels were determined by quantifying the changes in fluorescence intensity compared to that of the control cells.
Determination of NADPH oxidase activity and anti-oxidase activity
The lucigenin-derived enhanced chemiluminescence assay was used to determine NADPH oxidase activity. Briefly, quiescent cells were treated as indicated and harvested. After low-speed centrifugation, the pellet was resuspended in ice-cold buffer, pH 7.0, containing 1 mmol/L ethylene glycol tetra-acetic acid (EGTA), 150 mmol/L sucrose, and protease inhibitor cocktail (Merck, Germany). Then, the cells were homogenized. The total protein concentration was determined via the Bradford assay and adjusted to 1 mg/mL. Each 200 μL protein sample treated with 500 μmol/L lucigenin, the electron acceptor, and 100 μmol/L NADPH, the substrate, was measured for 6 min in quadruplicate using a luminometer (Berthold Luminometer Centro LB 960, Germany). The data were collected at 2 min intervals to measure the relative changes in NADPH oxidase activity.
Small interference RNA (siRNA) transfection
siRNA duplexes were synthesized by GenePharma (Shanghai, China). The p67phox sense siRNA sequence is 5’-CAG GGA ACA UUG UCU UUG UdTT-3’; the p67phox anti-sense siRNA sequence is 5’-ACA AAG ACA AUG UUC CCU GdTT-3’. The Nox2 sense siRNA sequence is 5’-CUC UGC GAU UCA CAC CAU UdTT-3’; the Nox2 anti-sense siRNA sequence is 5’-AAU GGU GUG AAU CGC AGA GdTT-3’. The negative control (NC) sense siRNA sequence is 5’-UUC UCC GAA CGU GUC ACG UdTT-3’; the NC anti-sense siRNA sequence is 5’-ACG UGA CAC GUU CGG AGA AdTT-3’. Transfection of EPCs with siRNAs was performed using Hiperfect transfection reagent (QIAGEN) according to the manufacturer’s instructions. Briefly, 150 pmol siRNA specific to p67phox, Nox2 or NC was diluted in the appropriate volume of serum-free EBM-2 at a final volume of 500 ml. For complex formation, 15 mL of Hiperfect transfection reagent was added to the diluted siRNA, and the samples were incubated for 10 min at room temperature. After incubation of the cells with the transfection complexes for 5 h under normal growth conditions, 1 mL of fresh culture medium containing serum was added for further culture. The siRNA efficacy was verified via Western blot.
Statistical analysis
The obtained data are expressed as the means ± SEM from at least three independent experiments and were analyzed via unpaired Student’s t-test for comparisons between two groups or one-way ANOVA for multiple comparisons. P values <0.05 were considered to be statistically significant.
Results
In vivo re-endothelialization capacity of EPC-CM from Hcy-stimulated EPCs was severely reduced
EBM-2, which did not contain cytokines, had no effect on re-endothelialization after carotid injury. Transplantation of EPC-CM from untreated EPCs (n=6) markedly accelerated the re-endothelialization of denuded carotid arteries in SD rats. Notably, the in vivo re-endothelialization capacity of EPC-CM derived from HHcy-treated EPCs (n=5) was markedly reduced (Figure 1).
Figure 1.

Re-endothelialization capacity of EPC-CM was impaired by Hcy. A. Re-endothelialized area at day 3 after carotid injury in SD rats with EBM-2 (n=5), transplantation of EPC-CM from healthy EPCs (n=6), or EPC-CM from Hcy-pretreated EPCs (n=5; each CM were collected from 5×105 EPCs). B. Representative photographs. EPC-CM from healthy or Hcy-pretreated EPCs was infused into the injured segment. *P < 0.05 vs. EBM-2, **P < 0.05 vs. EPC-CM.
HHcy reduced VEGF and IL-8 secretion from EPCs
EPCs were isolated, cultured, and treated with Hcy to determine the effects of Hcy on VEGF and IL-8 production by cultured EPCs. The quantities of VEGF and IL-8 secreted from EPCs were determined via ELISA. High concentrations of Hcy (100-500 μmol/L) significantly reduced VEGF and IL-8 production. Treatment of the cells with 200 μmol/L Hcy induced a time-dependent decrease in VEGF and IL-8 secretion (Figure 2A, 2B).
Figure 2.

Effects of Hcy and PIO on the secretion and expression of VEGF and IL-8 in cultured human EPCs. A, B. Levels of VEGF and IL-8 in the supernatants of cultured EPCs were measured via ELISA after incubation in Hcy at the indicated concentrations and periods. C, D. EPCs were pre-incubated with PIO (10 μM) for 30 min prior to the addition of Hcy (200 μM). VEGF and IL-8 secretion and mRNA expression were assessed via ELISA and real-time RT-PCR, respectively. The data are represented as the means ± SEM from three independent experiments. *P<0.05 vs. untreated cells, **P<0.01 vs. untreated cells, †P<0.05 vs. untreated cells, ††P<0.05 vs. Hcy-treated cells, #P<0.01 vs. untreated cells, ##P<0.01 vs. Hcy-treated cells.
PIO attenuated the HHcy-induced reduction in VEGF and IL-8 secretion from EPCs
Addition of PIO at 10 μmol/L to high Hcy-treated EPCs attenuated the reduction in VEGF and IL-8 secretion in the supernatants of in vitro cultured cells. To determine whether PIO modulates the expression of VEGF and IL-8 mRNA, total RNA was isolated from EPCs co-treated with Hcy. After normalization against GAPDH mRNA, the real-time PCR assay revealed that pretreatment of EPCs with PIO significantly inhibited the Hcy-induced reduction of VEGF and IL-8 mRNA expression. Thus, this detrimental effect of HHcy was attenuated by PIO pretreatment (Figure 2C, 2D).
PIO inhibited the Hcy-mediated activation of the PKC pathway
PKC functions as a major signaling system in response to extracellular signals. We examined the phosphorylation level of PKC in EPCs treated with Hcy and PIO. The cells were treated with 200 μM Hcy for 7.5, 15, 30, 60 or 120 min. Western blot analysis revealed that the phosphorylation levels of PKC were significantly increased after Hcy treatment. Further investigation revealed that the isoform PKCβП was the primary target of Hcy. The phosphorylation of PKC was inhibited in EPCs pretreated with PIO (Figure 3).
Figure 3.
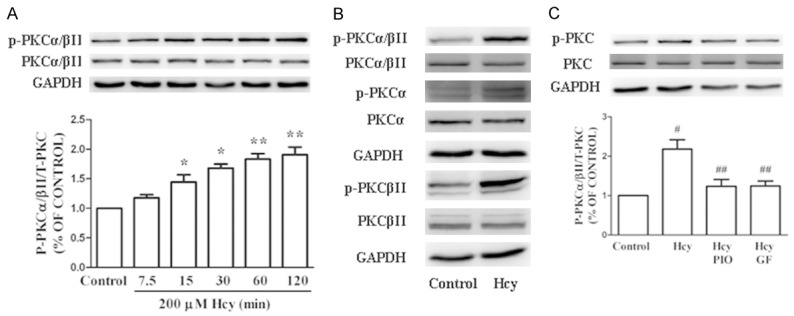
PIO inhibited the Hcy-induced activation of the PKC pathway. A. Hcy (200 μM) treatment time-dependently increased the phosphorylation of PKCα/βII. B. Treatment with Hcy for 1 h significantly enhanced the phosphorylation of PKCβII but not PKCα. C. EPCs were pre-incubated with PIO or GF for 1 h prior to the addition of Hcy. Hcy-stimulated PKC phosphorylation was inhibited by PIO and GF. Each bar represents the mean ± SEM of three independent experiments. *P<0.05 vs. untreated cells, **P<0.01 vs. untreated cells, #P<0.05 vs. untreated cells, ##P<0.05 vs. Hcy-treated cells. PIO: pioglitazone.
PIO inhibited Hcy-induced oxidative stress potentially via inhibition of PKC
We measured intracellular ROS levels using the ROS-sensitive fluorescent probe H2DCFH-DA via flow cytometry. We also measured NADPH oxidase activity via lucigenin-enhanced chemiluminescence in parallel. To determine whether the inhibitory effects of PIO in Hcy-induced oxidative stress involved PKC, we measured the effects of the PKC inhibitor GF109203X (GF, 5 μM) and a NADPH oxidase inhibitor (DPI, 5 μM). Both PIO and GF pretreatment significantly reduced Hcy-induced ROS production and NADPH oxidase activation, which was consistent with the results using DPI (Figure 4).
Figure 4.

PIO prevented Hcy-induced ROS production by NADPH oxidase. A. Hcy (200 μM) increased NADPH oxidase activity in a time-dependent manner. B. Pretreatment of EPCs with PIO (10 μM), DPI (5 μM), or GF (5 μM) for 30 min prior to the addition of Hcy (200 μM, 30 min) reduced NADPH oxidase activity. C, D. Representative experiment displayed ROS production after HHcy and PIO treatment. The relative fluorescence intensity of the cells was calculated relative to untreated control cells. The cells were pre-incubated with PIO (10 μM), DPI (5 μM), or GF (5 μM) for 30 min prior to the addition of 200 μM Hcy for 30 min and incubated in 10 μM H2DCF-DA for 30 min at 37°C. ROS production was inhibited by PIO, DPI, and GF. Each bar represents the mean ± SEM of five independent experiments. *P<0.05 vs. untreated cells, **P<0.05 vs. Hcy-treated cells. PIO: pioglitazone.
PIO may alleviate the Hcy-induced reduction in cytokine expression via inhibition of the PKC pathway and oxidative stress
EPCs were pre-incubated with PIO (10 μM), the PKC inhibitor GF (5 μM) and the NADPH oxidase inhibitor DPI (5 μM) for 30 min prior to the addition of Hcy (200 μM) for 24 h. The effects of the PKC inhibitor GF and the NADPH oxidase inhibitor DPI on both the protein and mRNA expression levels of VEGF and IL-8 in EPCs after co-treatment with Hcy were analyzed via ELISA and real-time RT-PCR, respectively. The results revealed that both DPI and GF prevented the Hcy-induced reduction in the expression of these two cytokines (Figure 5).
Figure 5.

PIO attenuated the Hcy-mediated impairments in EPC cytokine production via inhibition of NADPH oxidase and PKC. A, B. EPCs were pre-incubated with DPI (5 μM) or GF (5 μM) for 30 min prior to the addition of 200 μM Hcy. Both DPI and GF restored VEGF and IL-8 secretion and mRNA expression by EPCs based on ELISA and real-time RT-PCR, respectively. The data are presented as the means ± SEM from three independent experiments. *P<0.05 vs. untreated cells, **P<0.05 vs. Hcy-treated cells. PIO: pioglitazone.
Knockdown of Nox2 and p67phox expression inhibited the Hcy-induced dysfunction of EPCs
To further explore the underlying mechanism by which PIO restored Hcy-impaired EPC activity, transfection with Nox2 or p67phox siRNA were performed to downregulate the expression of these two NADPH subunits, which was confirmed via Western blot compared to negative control cells. Moreover, both Nox2 and p67phox siRNA transfection drastically abrogated the Hcy-mediated increase in ROS production and abolished the impairment of the paracrine function of EPCs. We also found that EPCs transfected with Nox2 and p67phox siRNA displayed apparently higher cell migration and adhesiveness than cells transfected with control siRNA when stimulated with Hcy (Figure 6).
Figure 6.
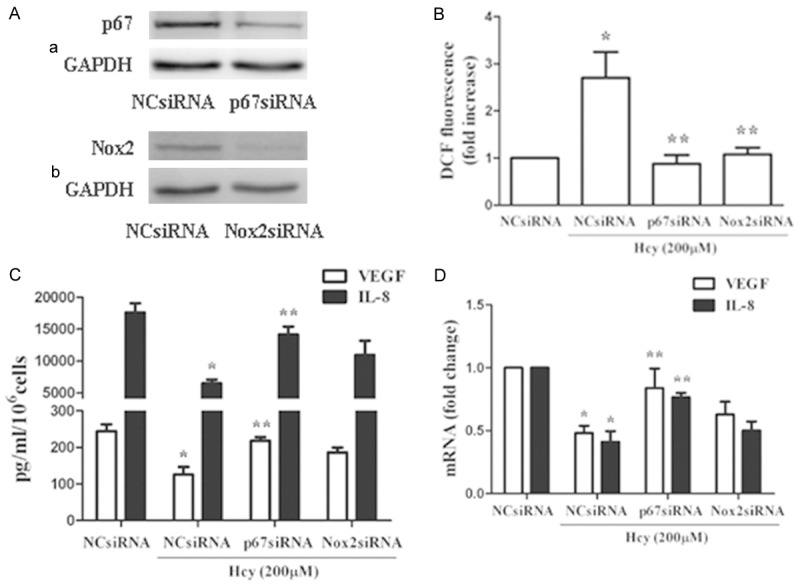
The NADPH oxidase subunits p67phox and Nox2 played an important role in the Hcy-induced EPC dysfunction of the paracrine. A. Western blot analysis revealed that transfection with p67 or Nox2 siRNA resulted in an approximately 75% decrease in p67 and Nox2 expression, respectively, compared to negative control cells. B. ROS production induced by Hcy treatment was abolished by p67 and Nox2 knockdown. C, D. The Hcy-induced decrease in VEGF and IL-8 production was abolished by treatment with p67phox siRNA. Nox2 siRNA ameliorated the decreases in cytokine secretion and expression. However, this protective effect of Nox2 siRNA was not statistically significant. *P<0.05 vs. NC siRNA transfection, **P<0.05 vs. NC siRNA transfection with Hcy (200 μM, 24 h) treatment. The VEGF- and IL-8-related comparisons were performed between each corresponding cytokine. PIO: pioglitazone.
PIO protected EPCs from Hcy in a PPAR-γ-independent manner
The effect of PIO is known to be mediated by two nuclear receptors, PPAR-γ and the retinoid X receptor. In the present study, we used GW9662 (5 μM) as a specific PPAR-γ blocker to determine whether PIO exerted its protective effect on EPCs against Hcy stimulation by activating PPAR-γ. The results revealed GW9662 did not block the PIO-mediated attenuation of the Hcy-induced reduction in VEGF and IL-8 production. This result suggested that the protective effect of PIO occurs via a PPAR-γ-independent pathway (Figure 7).
Figure 7.

PIO inhibited the decrease in VEGF and IL-8 secretion and expression via PPARγ-independent mechanisms. A, B. EPCs were pretreated with PIO and treated with Hcy in the presence or absence of the PPARγ antagonist GW9662. The addition of GW9662 did not abrogate the therapeutic effect of PIO on EPC cytokine secretion. *P<0.05 vs. untreated cells, **P<0.05 vs. Hcy treated cells. The VEGF- and IL-8-related comparisons were performed between each corresponding cytokine. PIO: pioglitazone.
Discussion
Mounting evidence has revealed that circulating EPCs do not contribute to regeneration of the endothelium. EPCs primarily act in a paracrine manner to promote re-endothelialization and angiogenesis by secreting angiogenic growth factors, such as VEGF and IL-8 [18]. The present study indicated for the first time that a high concentration of Hcy suppressed the secretion of VEGF and IL-8 in cultured human EPCs. Addition of PIO (10 μmol/L) to HHcy (200 μmol/L)-treated EPCs attenuated these reductions in VEGF and IL-8.
The plasma concentration of Hcy in patients suffering from homozygous homocystinuria is as high as 500 μmol/L. The results indicated that Hcy at 200 μmol/l (equivalent to severe elevation of plasma Hcy in humans) inhibited the cytokine production by EPCs for 24 h (approximately 50% reduction, P < 0.01). This Hcy concentration was in line with our previous studies [5,7]. Hyper-homocysteinemia has been linked clinically to coronary artery, cerebrovascular, and peripheral arterial occlusive diseases [19]. Previous studies demonstrated Hcy inhibited angiogenesis and re-endothelialization in vivo and in vitro. Hcy was proven to reduce the extent of IL-8 and IL-6 production by ECs and macrophages [20]. Moreover, the anti-angiogenic effects and mechanism of Hcy may be associated with VEGF/VEGFR inhibition in cultured human umbilical vein endothelial cells (HUVEC) [21]. In this study, we demonstrated that HHcy impaired the paracrine function of EPCs by inhibiting VEGF and IL-8 expression and secretion, which could be reversed by pre-treatment with PIO.
In a recent study, the combination of HHcy and hyperglycemia activated PKCβ2 in endothelial cells [22]. Hcy activated NADPH oxidase in monocytes via PKCβ [13]. Our results implied that activation of PKC and NADPH oxidase was involved in HHcy-induced EPC paracrine dysfunction. The phosphorylation levels of PKC, NADPH oxidase activity, and ROS level were significantly increased after HHcy treatment. NADPH oxidases are key generators of ROS in the blood vessel wall and other tissues during cardiovascular disease progression [23]. the NADPH complex consists of membrane-bound components, termed flavocytochrome b558 (p22phox and gp91phox subunits), and cytosolic components (p47phox, p67phox, p40phox, and Rac1) [24]. Our results further demonstrated that treatment with siRNA against the NADPH oxidase components p67phox and Nox2 suppressed Hcy-mediated oxidative stress. In addition, p67phox siRNA exerted a more significant reversal of Hcy-induced paracrine dysfunction than Nox2 siRNA. PKC inhibitor and NADPH-konckdown rescued reduction of VEGF and IL-8. We observed for the first time HHcy may impair EPC paracrine function via PKC/NADPH oxidase axis.
PIO is understood to function far beyond its original use as an insulin sensitizer and to display “pleiotropic effects”, including anti-apoptosis, anti-senescence [25], anti-oxidation and modulation of immune activation [26], stabilization of atherosclerotic plaques [27], and improvement of endothelial function. Furthermore, several studies have provided reports regarding the effect of PIO-restored cytokine production. PIO increased the serum levels of VEGF and IL-8 in patients suffering from type-2 diabetes [28]. PIO also restored the VEGF protein expression levels that were decreased in ischemic muscle and promoted angiogenesis in ischemic tissue of diabetic mice [29]. In the present study, we confirmed that PIO normalized the levels VEGF and IL-8 expression and secretion that were decreased by HHcy. We have also provided mechanistic insights regarding the PKC/NADPH oxidase signaling pathways responsible for the PIO-mediated therapeutic effect on the paracrine function of EPCs.
PPAR-γ may play an important role in regulating the induction and execution of immune responses [30]. PIO was assumed to restore the impaired angiogenesis in ischemic muscle of diabetic mice by stimulating the nuclear receptor PPARγ [29]. However, PIO can stimulate several PPARγ-independent pathways that are important for angiogenesis. For instance, PIO caused a relaxation of isolated blood vessels [31] and prevented apoptosis of EPCs [16] in a PPARγ-independent manner. In our study, combined treatment with PIO and the selective PPARγ inhibitor GW9662 did not abrogate the beneficial effect of PIO on the impaired paracrine function of EPCs. These results suggested that protective effect of PIO on cytokine secretion by EPCs could be unrelated to its activation of PPARγ.
There were some limitations in this study. The levels of VEGF and IL-8 in EPC-CM seemed to be elevated after stimulation with Hcy (50 μM). But there was no significant difference between 50 μM group and the control group. A previous study has reported that EPCs are more resistant to oxidative stress than differentiated ECs due to the elevated expression of intracellular antioxidant enzymes [32]. We speculated that higher ROS production mediated by HHcy could impair these anti-oxidative properties and thereby inhibit the paracrine activity of EPCs. Besides, focusing on the analysis of mechanism of HHcy and PIO action on PKC/NADPH oxidase pathway to explain their effects on IL-8 and VEGF gene expression may not be sufficiency. Other signalling pathways involved in the activation of IL-8 and VEGF genes were not investigated in this study and needed further research. Finally, we focused on mechanism of HHcy and PIO affecting EPC paracrine function, therefore the in vivo re-endothelialization data in Figure 1 was not followed up in other experiments. Not withstanding its limitation, this study did suggest HHcy impaired re-endothelialization capacity through inhibiting EPC paracrine function.
In conclusion, this study demonstrated that HHcy impaired the expression and secretion of VEGF and IL-8 by EPCs, which was evidently attenuated by PIO in a PPARγ-independent manner. The mechanism of PIO-mediated therapeutic effects on EPC paracrine action involved the inhibition of PKCβII and intracellular ROS production by NADPH oxidase. We further confirmed this mechanism via knockdown of p67phox and Nox2. Moreover, p67phox siRNA appeared to play a more important role than Nox2 siRNA in attenuating cytokine secretion and expression by HHcy-stimulated EPCs. These findings suggest a potential therapeutic role of PIO in EPC dysfunction under HHcy stimulation and warrant further investigation of EPC-based cytotherapy for ischemic vascular diseases.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grants 81500211), Natural Science Foundation of Zhejiang Province (Grants LQ14H020003), Projects of medicine and health technology plan in zhejiang province (Grants 2014KYB135).
Disclosure of conflict of interest
None.
Authors’ contribution
J.Z. designed and conducted experiments, and wrote the manuscript. S.X., Y.Z., L.Y., C.J., and F.D. performed the animal model and experiments. S.X. and G.F. analyzed data and interpreted the results.
References
- 1.Di Santo S, Yang Z, Wyler von Ballmoos M, Voelzmann J, Diehm N, Baumgartner I, Kalka C. Novel cell-free strategy for therapeutic angiogenesis: in vitro generated conditioned medium can replace progenitor cell transplantation. PLoS One. 2009;4:e5643. doi: 10.1371/journal.pone.0005643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Urbich C, Aicher A, Heeschen C, Dernbach E, Hofmann WK, Zeiher AM, Dimmeler S. Soluble factors released by endothelial progenitor cells promote migration of endothelial cells and cardiac resident progenitor cells. J Mol Cell Cardiol. 2005;39:733–742. doi: 10.1016/j.yjmcc.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 3.He T, Peterson TE, Katusic ZS. Paracrine mitogenic effect of human endothelial progenitor cells: role of interleukin-8. Am J Physiol Heart Circ Physiol. 2005;289:H968–972. doi: 10.1152/ajpheart.01166.2004. [DOI] [PubMed] [Google Scholar]
- 4.Abe Y, Ozaki Y, Kasuya J, Yamamoto K, Ando J, Sudo R, Ikeda M, Tanishita K. Endothelial progenitor cells promote directional three-dimensional endothelial network formation by secreting vascular endothelial growth factor. PLoS One. 2013;8:e82085. doi: 10.1371/journal.pone.0082085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen JZ, Zhu JH, Wang XX, Xie XD, Sun J, Shang YP, Guo XG, Dai HM, Hu SJ. Effects of homocysteine on number and activity of endothelial progenitor cells from peripheral blood. J Mol Cell Cardiol. 2004;36:233–239. doi: 10.1016/j.yjmcc.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 6.Zhu J, Wang X, Chen J, Sun J, Zhang F. Reduced number and activity of circulating endothelial progenitor cells from patients with hyperhomocysteinemia. Arch Med Res. 2006;37:484–489. doi: 10.1016/j.arcmed.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 7.Zhu JH, Chen JZ, Wang XX, Xie XD, Sun J, Zhang FR. Homocysteine accelerates senescence and reduces proliferation of endothelial progenitor cells. J Mol Cell Cardiol. 2006;40:648–652. doi: 10.1016/j.yjmcc.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 8.Bao XM, Wu CF, Lu GP. Atorvastatin inhibits homocysteine-induced oxidative stress and apoptosis in endothelial progenitor cells involving Nox4 and p38MAPK. Atherosclerosis. 2010;210:114–121. doi: 10.1016/j.atherosclerosis.2009.11.032. [DOI] [PubMed] [Google Scholar]
- 9.Latacha KS, Rosenquist TH. Homocysteine inhibits extra-embryonic vascular development in the avian embryo. Dev Dyn. 2005;234:323–331. doi: 10.1002/dvdy.20527. [DOI] [PubMed] [Google Scholar]
- 10.Zhang C, Hu JJ, Xia M, Boini KM, Brimson CA, Laperle LA, Li PL. Protection of podocytes from hyperhomocysteinemia-induced injury by deletion of the gp91phox gene. Free Radic Biol Med. 2010;48:1109–1117. doi: 10.1016/j.freeradbiomed.2010.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee I, Lee H, Kim JM, Chae EH, Kim SJ, Chang N. Short-term hyperhomocysteinemia-induced oxidative stress activates retinal glial cells and increases vascular endothelial growth factor expression in rat retina. Biosci Biotechnol Biochem. 2007;71:1203–1210. doi: 10.1271/bbb.60657. [DOI] [PubMed] [Google Scholar]
- 12.Jiang X, Yang F, Tan H, Liao D, Bryan RM Jr, Randhawa JK, Rumbaut RE, Durante W, Schafer AI, Yang X, Wang H. Hyperhomocystinemia impairs endothelial function and eNOS activity via PKC activation. Arterioscler Thromb Vasc Biol. 2005;25:2515–2521. doi: 10.1161/01.ATV.0000189559.87328.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Siow YL, Au-Yeung KK, Woo CW, O K. Homocysteine stimulates phosphorylation of NADPH oxidase p47phox and p67phox subunits in monocytes via protein kinase Cbeta activation. Biochem J. 2006;398:73–82. doi: 10.1042/BJ20051810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Werner C, Kamani CH, Gensch C, Bohm M, Laufs U. The peroxisome proliferator-activated receptor-gamma agonist pioglitazone increases number and function of endothelial progenitor cells in patients with coronary artery disease and normal glucose tolerance. Diabetes. 2007;56:2609–2615. doi: 10.2337/db07-0069. [DOI] [PubMed] [Google Scholar]
- 15.Spigoni V, Picconi A, Cito M, Ridolfi V, Bonomini S, Casali C, Zavaroni I, Gnudi L, Metra M, Dei Cas A. Pioglitazone improves in vitro viability and function of endothelial progenitor cells from individuals with impaired glucose tolerance. PLoS One. 2012;7:e48283. doi: 10.1371/journal.pone.0048283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gensch C, Clever YP, Werner C, Hanhoun M, Bohm M, Laufs U. The PPAR-gamma agonist pioglitazone increases neoangiogenesis and prevents apoptosis of endothelial progenitor cells. Atherosclerosis. 2007;192:67–74. doi: 10.1016/j.atherosclerosis.2006.06.026. [DOI] [PubMed] [Google Scholar]
- 17.Imanishi T, Kobayashi K, Kuroi A, Ikejima H, Akasaka T. Pioglitazone inhibits angiotensin II-induced senescence of endothelial progenitor cell. Hypertens Res. 2008;31:757–765. doi: 10.1291/hypres.31.757. [DOI] [PubMed] [Google Scholar]
- 18.Krenning G, van Luyn MJ, Harmsen MC. Endothelial progenitor cell-based neovascularization: implications for therapy. Trends Mol Med. 2009;15:180–189. doi: 10.1016/j.molmed.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 19.Cacciapuoti F. Hyper-homocysteinemia: a novel risk factor or a powerful marker for cardiovascular diseases? Pathogenetic and therapeutical uncertainties. J Thromb Thrombolysis. 2011;32:82–88. doi: 10.1007/s11239-011-0550-4. [DOI] [PubMed] [Google Scholar]
- 20.Dalal S, Parkin SM, Homer-Vanniasinkam S, Nicolaou A. Effect of homocysteine on cytokine production by human endothelial cells and monocytes. Ann Clin Biochem. 2003;40:534–541. doi: 10.1258/000456303322326452. [DOI] [PubMed] [Google Scholar]
- 21.Zhang Q, Li Q, Chen Y, Huang X, Yang IH, Cao L, Wu WK, Tan HM. Homocysteine-impaired angiogenesis is associated with VEGF/VEGFR inhibition. Front Biosci (Elite Ed) 2012;4:2525–2535. doi: 10.2741/e563. [DOI] [PubMed] [Google Scholar]
- 22.Cheng Z, Jiang X, Pansuria M, Fang P, Mai J, Mallilankaraman K, Gandhirajan RK, Eguchi S, Scalia R, Madesh M, Yang X, Wang H. Hyperhomocysteinemia and hyperglycemia induce and potentiate endothelial dysfunction via mu-calpain activation. Diabetes. 2015;64:947–959. doi: 10.2337/db14-0784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sirker A, Zhang M, Shah AM. NADPH oxidases in cardiovascular disease: insights from in vivo models and clinical studies. Basic Res Cardiol. 2011;106:735–747. doi: 10.1007/s00395-011-0190-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 25.Werner C, Gensch C, Poss J, Haendeler J, Bohm M, Laufs U. Pioglitazone activates aortic telomerase and prevents stress-induced endothelial apoptosis. Atherosclerosis. 2011;216:23–34. doi: 10.1016/j.atherosclerosis.2011.02.011. [DOI] [PubMed] [Google Scholar]
- 26.Yu HC, Feng SF, Chao PL, Lin AM. Anti-inflammatory effects of pioglitazone on iron-induced oxidative injury in the nigrostriatal dopaminergic system. Neuropathol Appl Neurobiol. 2010;36:612–622. doi: 10.1111/j.1365-2990.2010.01107.x. [DOI] [PubMed] [Google Scholar]
- 27.Yang HB, Zhao XY, Zhang JY, Du YY, Wang XF. Pioglitazone induces regression and stabilization of coronary atherosclerotic plaques in patients with impaired glucose tolerance. Diabet Med. 2012;29:359–365. doi: 10.1111/j.1464-5491.2011.03458.x. [DOI] [PubMed] [Google Scholar]
- 28.Vijay SK, Mishra M, Kumar H, Tripathi K. Effect of pioglitazone and rosiglitazone on mediators of endothelial dysfunction, markers of angiogenesis and inflammatory cytokines in type-2 diabetes. Acta Diabetol. 2009;46:27–33. doi: 10.1007/s00592-008-0054-7. [DOI] [PubMed] [Google Scholar]
- 29.Huang PH, Sata M, Nishimatsu H, Sumi M, Hirata Y, Nagai R. Pioglitazone ameliorates endothelial dysfunction and restores ischemia-induced angiogenesis in diabetic mice. Biomed Pharmacother. 2008;62:46–52. doi: 10.1016/j.biopha.2007.06.014. [DOI] [PubMed] [Google Scholar]
- 30.Hontecillas R, Bassaganya-Riera J. Peroxisome proliferator-activated receptor gamma is required for regulatory CD4+ T cell-mediated protection against colitis. J Immunol. 2007;178:2940–2949. doi: 10.4049/jimmunol.178.5.2940. [DOI] [PubMed] [Google Scholar]
- 31.Nomura H, Yamawaki H, Mukohda M, Okada M, Hara Y. Mechanisms underlying pioglitazone-mediated relaxation in isolated blood vessel. J Pharmacol Sci. 2008;108:258–265. doi: 10.1254/jphs.08117fp. [DOI] [PubMed] [Google Scholar]
- 32.Dernbach E, Urbich C, Brandes RP, Hofmann WK, Zeiher AM, Dimmeler S. Antioxidative stress-associated genes in circulating progenitor cells: evidence for enhanced resistance against oxidative stress. Blood. 2004;104:3591–3597. doi: 10.1182/blood-2003-12-4103. [DOI] [PubMed] [Google Scholar]