

Abstract
It has been reported that celecoxib, a cyclooxygenase-2 (COX-2)-selective nonsteroidal anti-inflammatory drug (NSAID), regulates the radiosensitivity of several cancer cells. BCCIP (BRCA2 and CDKN1A interacting protein) plays a critical role in maintaining the critical functions of p53 in tumor suppression and response to therapy. However, whether the effect of celecoxib on the radiosensitivity of colorectal cancer (CRC) cells is dependent on BCCIP is largely unclear. In this study, we found that celecoxib enhanced the radiosensitivity of HeLa (a human cervical carcinoma cell line), A549 (a human lung carcinoma cell line), and HCT116 cells (a human CRC cells line). Among these cells, COX-2 expression was undetected in HCT116 cells. Treatment with celecoxib significantly increased BCCIP expression in COX-2 negative HCT116 cells. Knockdown of BCCIP obviously abrogated the enhanced radiosensitivity of HCT116 cells induced by celecoxib. A combination of celecoxib and irradiation treatment induced much more γ-H2AX foci formation, higher levels of radiation injury-related proteins phosphorylation, G2/M arrest, apoptosis, and p53 and p21 expression, and lower levels of Cyclin B1 in HCT116 cells than those in cells treated with irradiation alone. However, these changes were undetected in BCCIP-silenced HCT116 cells. Therefore, these data suggest that BCCIP gene may be a radiosensitivity-related gene in CRC. Celecoxib affects the functions of p53 and inhibits the recovery from the irradiation-induced injury by up-regulating the expression of BCCIP, and subsequently regulates the expressions of genes such as p21 and Cyclin B1 to enhance the radiosensitivity of HCT116 cells in a COX-2 independent manner.
Keywords: Celecoxib, BCCIP, radiosensitivity, colorectal cancer cells, COX-2
Introduction
Colorectal cancer (CRC) is one of the most common malignant tumors and the third leading cause of cancer-related deaths worldwide [1,2]. The incidence of and mortality due to CRC are increasing every year by year in China, with the improvement in economy and the changes in eating habits. CRC is the third most common cancer and the fourth leading cause of mortality among all malignant tumors in China.
Locally advanced rectal cancer (LARC) often requires treatment using radiotherapy. Long course preoperative radiotherapy has been shown to be effective in downsizing LARC [3-5]. At present, the patients in T3 and T4 phases of cancer, as determined by imaging, are considered candidates for preoperative radiotherapy and chemotherapy. Previous studies showed considerable individual differences in the effects of treatment using preoperative radiotherapy and chemotherapy in patients with LARC, which varied from tumor complete disappearance of the tumor to no response. Moreover, the responses of rectal cancer to radiotherapy and chemotherapy, and the pathological remission rate were related to several factors, including tumor stage, operation time, drug factors, etc [6].
Celecoxib was the first COX-2-selective non-steroidal anti-inflammatroy drug (NSAID). This drug was used clinically both for patients with osteoarthritis and for those with articular rheumatism for pain relief, and for patients with familial adenomatous polyposis, as an adjuvant to chemotherapy [7]. It was demonstrated to possess potent anti-cancer activities against various human cancers [8-10]. Recently, increasing evidence has shown that celecoxib enhances the radiosensitivity of several cancer cell lines [11,12]. Compared to HT-29 cells (with wild-type COX-2), HCT116 cells are COX-2-deficient human CRC cells [13]. Therefore, whether and how celecoxib regulates the radiosensitivity of HCT116 cells are unclear.
BCCIP is a p21- and BRCA2-interacting protein that has been shown to play roles in both cell cycle arrest and DNA repair [14-17]. Owing to alternative pre-mRNA splicing, human tissues have two major isoforms, BCCIPα and BCCIPβ [18]. In most human tissues and cell lines, BCCIPβ is the major expressed isoform. However, it appears that mice have only the BCCIPβ isoform. A recent study showed that BCCIP was positively expressed in CRC tissues [19]. However, the expression and role of BCCIP in CRC cells are largely unclear.
Therefore, the present study aimed to investigate whether celecoxib regulates the radiosensitivity of HCT116 cells by modulating the expression of BCCIP, and to further analyze the underlying regulation mechanisms in vitro.
Materials and methods
Reagents
Celecoxib, 4-[5-(4-methylphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]benzenesulfonamide, was purchased from Pfizer Co., Ltd., (Groton, CT, USA), dissolved in dimethyl sulfoxide (DMSO; Sigma-Aldrich, Boston, MA, USA) at 10 mM and diluted immediately before each experiment.
Cell lines
Human CRC cell line (HCT116), human cervical carcinoma cell line (HeLa), and human lung carcinoma cell line (A549) were purchased from the American Type Culture Collection (ATCC, USA). All of the culture media (Gibco, USA) were supplemented with 10% fetal bovine serum (FBS; Hyclone, Logan, UT, USA). These cells were incubated at 37°C in a humidified atmosphere containing 5% CO2.
Irradiation
Cells were vertically irradiated with 6MV X-rays (P-M type Medical-Linear Accelerator; SIEMENS, AG, Erlangen, Germany). The irradiation method was as follows: the dose rate was 200 cGy/min, the source skin distance (SSD) was 100 cm, and the field was 10 cm * 10 cm. The doses used were as follows: 2, 4, 6, 8, or 10 Gy for clonogenic survival assays and cell cycle analysis, and 6 Gy for the γ-H2AX foci formation, detecting apoptosis, and immunoblotting analysis. The dose of 6 Gy was selected on the basis of the results of a clonogenic survival assay, as noted in the Results.
Cell-counting kit-8 (CCK-8) assay
HeLa, A549, and HCT116 cells were seeded in 96-well plates (5 × 103 cells/well), and incubated with different concentrations of celecoxib (2.5, 5, 10, 20, 40, 50, or 100 μM) for 3, 6, 12, 24, 48 or 72 h, respectively. Subsequently, the viability of these cells was measured and analyzed using the CCK-8 assay (Dojindo, Japan). The CCK-8 reagent was added to each well and the cells were incubated at 37°C for 2 h, according to the manufacturer’s protocol. The absorbance at 450 nm was measured and used to represent the viability of cells. Each experiment was performed in six parallel wells, and repeated three times. The inhibition ratio of cell proliferation (%) = (Ac-At)/(Ac-Ab) * 100%, where Ac is the absorbance of control groups (treatment with 1‰ DMSO), At is the absorbance of celecoxib treatment groups, and Ab is the absorbance of blank holes. In subsequent trials, the concentration of celecoxib was 30 μM.
Knockdown of BCCIP in HCT116 cells
The shRNAs for targeting BCCIP were designed and synthesized by Invitrogen (Shanghai, China). The sequences were: 1) For Sh1: forward, 5’-CCG GGT GTG ATT AAG CAA ACG GAT GTC AAG AGC ATC CGT TTG CTT AAT CAC AC-3’, and reverse, 5’-GTG TGA TTA AGC AAA CGG ATG CTC TTG ACA TCC GTT TGC TTA ATC ACA CTT AA-3’; 2) For Sh2: forward, 5’-GAT CGC CAT GTG GGA AGT GCT ACT CAA GAG GTA GCA CTT CCC ACA TGG C-3’, and reverse, 5’-GCC ATG TGG GAA GTG CTA CCT CTT GAG TAG CAC TTC CCA CAT GGC TCG A-3’; 3) For Sh3: forward, 5’-GAT CGC TGC GTT AAT GTT TGC AAA TTC AAG AGA TTT GCA AAC ATT AAC GCA GC-3’, and reverse, 5’-GCT GCG TTA ATG TTT GCA AAT CTC TTG AAT TTG CAA ACA TTA ACG CAG CTC GA-3’; 4) For ShNC, forward, 5’-ACT ACC GTT GTT ATA GGT GTC AAG ACA CCT ATA ACA ACG GTA GA-3’, and reverse, 5’-ACT ACC GTT GTT ATA GGT GCT CTT GAC ACC TAT AAC AAC GGT AGA-3’. Subsequently, we constructed the plasmids expressing these shRNAs were constructed (pPUR/U6-NC, pSilencer2.1Hyg-NC, pPUR/U6-sh1, pSilencer2.1Hyg-sh2, pSilencer2.1Hyg-sh3, pPUR/U6-sh1+pSilencer2.1Hyg-sh2, pPUR/U6-sh1+pSilencer2.1Hyg-sh3), and verified the BCCIP knockdown was verified by qRT-PCR. The BCCIP stably silenced HCT116 cell line (S-BCCIP) and the corresponding negative control cell line (NC) were established by transfection with the plasmid combinations (pPUR/U6-sh1+pSilencer2.1Hyg-sh3, pPUR/U6-shNC+pSilencer2.1Hyg-shNC) and antibiotic screening, respectively. The efficiency of BCCIP silencing in S-BCCIP was verified by western blotting.
Clonogenic survival assay
A standard colony formation assay was performed immediately after exposing the HCT116 cells to X-rays. HCT116 cells were treated with or without 30 μM celecoxib for 24 h and further exposed to irradiation (cells were coated with 1.5 cm of tissue equivalent filler). After irradiation, the cells were cultured at 37°C in a humidified at mosphere containing 5% CO2. After 10-12 days, the colonies were fixed and stained. Three replicate dishes were used for each dose, and colonies (>50 cells) were scored as survivors [20]. The plating efficiency (PE %) was determined as the number of colonies formed/the number of seeded cells * 100%. The survival fraction = PE of trial groups/PE of the control group. The doses used were 0, 1, 2, 4, 6, and 8 Gy. Three independent experiments were performed for each dose.
Western blotting
Cells were harvested and lysed at room temperature in RIPA lysis and extraction buffer (Thermo Fisher Scientific, Waltham, MA, USA) with a protease inhibitor cocktail (Halt Protease Inhibitor Cocktail; Thermo Fisher Scientific). Protein concentrations were determined using a 660-nm protein assay kit (Thermo Fisher Scientific) containing Ionic Detergent Compatibility Reagent (Thermo Fisher Scientific). Samples that contained the same amounts of protein were separated using 12.5%-15% SDS-PAGE electrophoresis, followed by electrotransferred onto polyvinylidene fluoride (PVDF) membranes (Bio-Rad Laboratories, USA). These blots were incubated with the following antibodies under conditions recommended by the manufacturers. The primary antibodies used were anti-COX-2 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-BCCIP (Abcam, USA), anti-γ-H2AX (Cell Signaling Technology, USA), anti-p-Chk2 (Cell Signaling Technology), antip-ATM (Cell Signaling Technology), anti-p53 (Santa Cruz Biotechnology), anti-p21 (Santa Cruz Biotechnology), anti-Cyclin B1 (Santa Cruz Biotechnology), anti-Cdc2 (Santa Cruz Biotechnology), anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Cell Signaling Technology), anti-tubulin (Santa Cruz Biotechnology), and anti-β-actin (Sigma). Horseradish peroxidase-conjugated anti-mouse (Santa Cruz Biotechnology) or anti-rabbit antibody (Santa Cruz Biotechnology) was used as the secondary antibody. Signals were detected using an enhanced chemiluminescence system (Millipore, Billerica, MA, USA).
RNA extraction, RT, and PCR
After culture and experimental treatment, the cell culture medium was removed, cells were washed with PBS, and RNA was extracted using TRizol (Invitrogen). RNA concentration was measured based on the absorbance at 260 nm, and RNA was reverse transcribed onto cDNA using a RT Kit (Fermentas, Pittsburgh, PA, USA). The primers used for qualitative PCR were designed by Sangon Biotech Company (Shanghai, China). Qualitative PCR was performed according to the manufacturer’s instructions and DNA products were detected by 1% agarose electrophoresis. The values of the band intensities were determined using the Image J Software (National Institute of Mental Health, Bethesda, MD, USA). The sequences of the primers are described in Table 1. All assays were performed in triplicate.
Table 1.
Sequences of the primers of BCCIP and GAPDH
| Gene | Primer sequence |
|---|---|
| BCCIP | Forward, 5’-AGA ACC ATA TTG GGA GTG-3’; |
| Reverse, 5’-TCT CAC AGA AGC GTA GAA C-3’ | |
| GAPDH | Forward, 5’-GTC AAC GGA TTT GGT CTG TAT T-3’; |
| Reverse, 5’-AGT CTT CTG GGT GGC AGT GAT-3’ |
Immunofluorescence (IF)
After celecoxib treatment and irradiation, monolayers of HCT116 cells were fixed with 4% (vol/vol) paraformaldehyde for 10 min at room temperature, followed by 10-min incubation with 0.2% (vol/vol) Triton X-100. After washing with PBS, nonspecific binding was blocked with 10% FBS in PBS. Anti-γ-H2AX rabbit polyclonal antibodies (1:50; Cell Signal Technology) diluted in PBS were added for 1 h, avoiding light; and then the slides were then mounted in 0.1% (wt/vol) 4,6-diamidino-2-phenylindole (DAPI; 1:50; Invitrogen) for nuclear counterstaining for 5 min at room temperature and observed under an Olympus BX51 fluorescence microscope (Olympus). The secondary antibodies were Texas Red-conjugated anti-rabbit (1:250; Rockland) antibodies were used as secondary antibodies. The experiments were carried out in triplicate and repeated three times.
Cell cycle analysis
The HCT116 cells were plated in 6-well flatbottom microplates, treated with celecoxib (30 μM) and irradiation, and then harvested, fixed in 70% v/v ethanol and stored at -20°C. Cells were then washed twice with ice-cold PBS, and incubated with RNase and the DNA-intercalating dye propidium iodide (PI) (Beyotime). Cell-cycle phase analysis was performed by using a Becton Dickinson Facstar flow cytometer (Becton Dickson, Waltham, MA, USA) equipped with a Becton DickinsonCellFitTM software (Becton Dickson).
Apoptosis assay
The apoptosis level of HCT116 cells was analyzed using Annexin V-FITC apoptosis assay (Invitrogen), according to the manufacturer’s protocol.
Statistics
All values are shown as the mean ± SD. Data were analyzed using GraphPad Prism version 5.0 (GraphPad software, San Diego, CA, USA) by t-test or analysis of variance (ANOVA). Differences were considered statistically significant at P<0.05.
Results
Celecoxib enhances the radiosensitivity of HCT116 cells in a COX-2 independent manner
In order to evaluate the expression of COX-2 in different cancer cells, we measured the expression of COX-2 by western blotting in HeLa, A549, and HCT116 cells. As shown in Figure 1, both HeLa and A549 cells expressed high levels of COX-2 (Figure 1A). However, HCT116 cells showed negative expression of COX-2 (Figure 1A); these results were consistent with a previous report [13]. Further analysis showed that celecoxib significantly inhibited the proliferation of these three cell lines in both dosage and time-dependent manners (P<0.05, P<0.01 or P<0.001) (Figure 1B). The optimal dose of celecoxib for cell inhibition was about 30 μM (P<0.001) (Figure 1B). Subsequently, the effect of celecoxib on the radiosensitivity of these three cells lines was detected by colony formation assay. The results in Figure 1C showed that 30 μM celecoxib could obviously decreased the survival fraction of these three cell lines. These data suggest that celecoxib enhance the radiosensitivity of these three cell lines, and that these effects are independent of COX-2.
Figure 1.
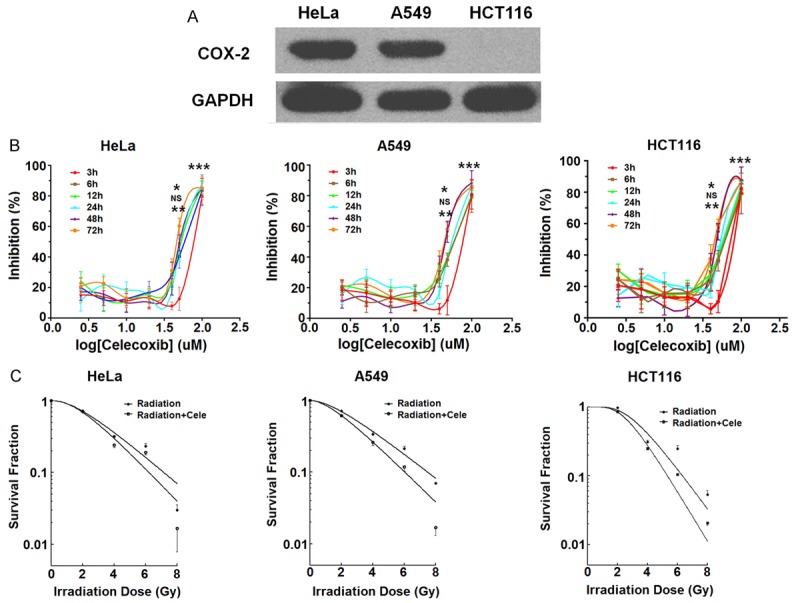
Celecoxib enhances the radiosensitivity of HCT116 cells in a COX-2 independent manner. A. Western blotting analysis for COX-2 expression in HeLa, A549, and HCT116 cells. B. After stimulation with different concentrations of celecoxib (2.5, 5, 10, 20, 40, 50 or 100 μM) for 3, 6, 12, 24, 48, or 72 h, respectively, the viability of HeLa, A549 and HCT116 cells was analyzed by the CCK-8 assay. C. Clonogenic survival assay was performed to evaluate the radiosensitivity of HeLa, A549 and HCT116 cells after pre-treatment with celecoxib (30 μM) and/or irradiation (0, 1, 2, 4, 6, or 8 Gy). Irradiation+cele: combination of irradiation and celecoxib treatment. The data are presented as mean ± SD. *P<0.05, **P<0.01 and ***P<0.001 (one-way ANOVA). NS: no statistically significant difference.
Celecoxib up-regulated the expression of BCCIP in HCT116 cells
BCCIP has been considered as a prognostic marker for radiotherapy for laryngeal cancer [21]. To further explore the potential mechanism of celecoxib on radiosensitivity regulation in HCT116 cells, we analyzed the expression of BCCIP in these three cell lines. As shown, all these cells expressed BCCIP (Figure 2A). Moreover, treatment with 30 μM celecoxib led to varying degress of increased expressions of BCCIP in these cells (Figure 2B). Among these, the up-regulation of BCCIP protein was observed in HCT116 cells with negative COX-2 expression after incubating with 30 μM celecoxib for 6 h.
Figure 2.
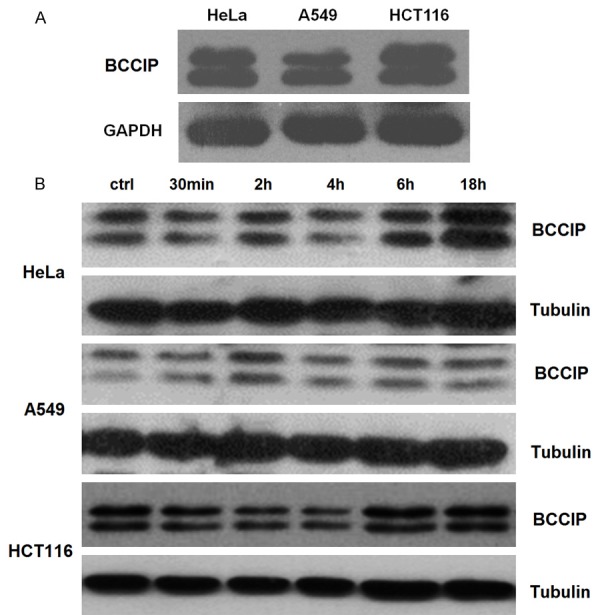
Celecoxib up-regulated the expression of BCCIP in HCT116 cells. A. Western blotting analysis for BCCIP expression in HeLa, A549, and HCT116 cells. B. After stimulation with celecoxib (30 μM) for 30 min, 2, 4, 6, or 18 h, respectively. Then, the expression of BCCIP in HeLa, A549, and HCT116 cells was analyzed by western blotting.
Enhanced radiosensitivity of HCT116 cells induced by celecoxib is dependent on BCCIP
To investigate whether the effect of celecoxib on the radiosensitivity of HCT116 cells is dependent on BCCIP, we constructed the plasmids expressing shRNAs to silence the expression of BCCIP (pPUR/U6-NC, pSilencer2.1Hyg-NC, pPUR/U6-sh1, pSilencer2.1Hyg-sh2, pSilencer2.1Hyg-sh3, pPUR/U6-sh1+pSilencer2.1Hyg-sh2, pPUR/U6-sh1+pSilencer2.1Hyg-sh3) (Figure 3A). After transfection, all silenced plasmids could significantly knock down the expression of BCCIP in HCT116 cells (P<0.01 or P<0.001) (Figure 3B), especially pPUR/U6-sh1+pSilencer2.1Hyg-sh3 (RNAi1+3) (P<0.001) (Figure 3B). Next, we established BCCIP stably silenced HCT116 cell line (S-BCCIP) by transfection with the plasmid combination RNAi1+3, and the corresponding control stable cell line (NC). The efficiency of silencing was verified by western blotting (Figure 3C). Unlike NC cells, further study showed that there was no obvious difference in the survival fraction between irradiation group and the combination of irradiation and celecoxib group (irradiation + celecoxib) (Figure 3D), suggesting that the absence of BCCIP abrogate the enhanced radiosensitivity of HCT116 cells induced by celecoxib.
Figure 3.

The enhanced radiosensitivity of HCT116 cells induced by celecoxib is dependent on BCCIP. A, B. After transfection with the plasmids (pPUR/U6-NC, pSilencer2.1Hyg-NC, pPUR/U6-sh1, pSilencer2.1Hyg-sh2, pSilencer2.1Hyg-sh3, pPUR/U6-sh1+pSilencer2.1Hyg-sh2 or pPUR/U6-sh1+pSilencer2.1Hyg-sh3), qRT-PCR was used to evaluate the transcriptional level of BCCIP in HCT116 cells. Blank: no plasmid transfection; NC: transfection with pPUR/U6-NC; RNAi1: transfection with pPUR/U6-sh1; RNAi2: transfection with pSilencer2.1Hyg-sh2; RNAi3: transfection with pSilencer2.1Hyg-sh3; RNAi1+2: transfection with pPUR/U6-sh1 and pSilencer2.1Hyg-sh2; RNAi1+3: transfection with pPUR/U6-sh1 and pSilencer2.1Hyg-sh3. C. BCCIP stably silenced HCT116 cell line (S-BCCIP) was established by transfection with the plasmid combination (pPUR/U6-sh1+pSilencer2.1Hyg-sh3) and the subsequent antibiotic screening. Meanwhile the corresponding NC cell line was established. The efficiency of BCCIP silencing was verified by western blotting. D. Clonogenic survival assay was performed to evaluate the radiosensitivity of NC and S-BCCIP cells after pre-treatment with celecoxib (30 μM) and/or irradiation (0, 1, 2, 4, 6, or 8 Gy). The data are presented as mean ± SD. **P<0.01 and ***P<0.001 (one-way ANOVA).
Celecoxib/BCCIP signaling aggravates irradiation-induced injury
γ-H2AX is currently recognized as a molecular marker of DNA double strand breaks (DSBs). It has been confirmed that the analysis of γ-H2AX level in human peripheral blood lymphocytes is feasible for directing the selection of radiation biodosimeter [22-25]. Therefore, we analyzed the level of DNA repair-related foci of γ-H2AX, p-ATM, and p-Chk2 were detected with immunofluorescence staining and/or western blotting at different time points after irradiation, respectively. As shown, the γ-H2AX foci formation in the group of irradiation + celecoxib was higher than that of irradiation group in NC cells (P<0.05) (Figure 4A and 4B). However, a simiar results was not observed in S-BCCIP cells (P>0.05) (Figure 4A and 4B). Subsequently, the results of western blotting showed that celecoxib could further up-regulate the stimulatory effect of irradiation on the expression of γ-H2AX, p-ATM and p-Chk2 in NC rather than S-BCCIP cells (Figure 4C). These data indicate that celecoxib aggravates the irradiation-induced injury in HCT116 cells through up-regulating BCCIP expression.
Figure 4.
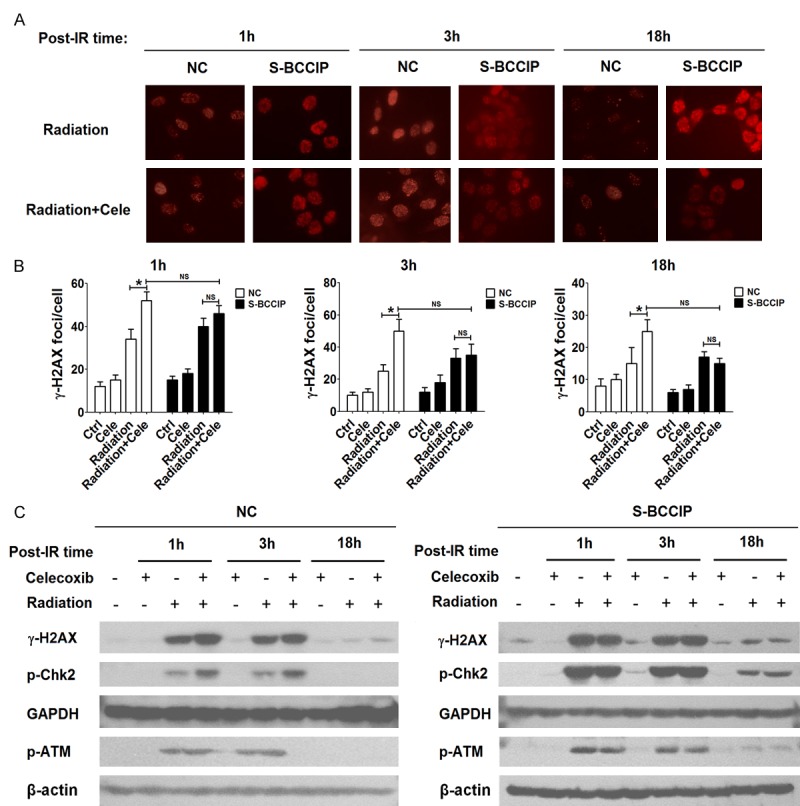
Celecoxib/BCCIP signaling amplifies irradiated-induced injury. A, B. Immunofluorescence staining was performed to analyze the γ-H2AX foci formation in NC and S-BCCIP cells at different time points of after irradiation (6 Gy) and/or pre-treatment with celecoxib (30 μM). C. NC and S-BCCIP cells were pre-treated with celecoxib (30 μM) and/or irradiation (6 Gy) for 1, 3, or 18 h, and then the protein levels of γ-H2AX, p-ATM and p-Chk2 in NC and S-BCCIP cells were detected by western blotting.
Celecoxib promotes irradiation-induced G2/M arrest and apoptosis in HCT116 cells by BCCIP
To analyze the effect of celecoxib and irradiation on G2/M arrest and apoptosis in HCT116 cells, the cell cycle distributions and apoptosis levels of NC and S-BCCIP cells after celecoxib and/or irradiation were evaluated by flow cytometry analysis and apoptosis assay. As shown, pre-treatment with celecoxib markedly increased G2/M arrest in NC cells induced by irradiation (P<0.01) (Figure 5A-C). However, this difference was not observed in S-BCCIP cells (P>0.05) (Figure 5A-C).
Figure 5.

Celecoxib promotes irradiation-induced G2/M arrest in HCT116 cells by BCCIP. (A-C) After pre-treatment with celecoxib (30 μM) and/or irradiation (0, 1, 2, 4, 6, 8 or 10 Gy), the cell cycle distributions (A, B) and percentage of cells arrested in the G2/M phase (C) of both NC and S-BCCIP cells were analyzed by flow cytometry analysis. The data are presented as the mean ± SD. **P<0.01 (t-test).
Cell apoptosis could be induced by irradiation in both NC and S-BCCIP cells (P<0.01) (Figure 6A), and it further increased in NC cells when cells were pre-treated with celecoxib (P<0.05) (Figure 6A), while the increase of apoptosis was limited in S-BCCIP cells subjected to the same treatment (P>0.05) (Figure 6A). In addition, treatment with celecoxib further up-regulated the expression of BCCIP, p53 and p21, while it down-regulated the expression of Cyclin B1 in NC but not S-BCCIP cells induced by irradiation (Figure 6B); however, the level of Cdc2 was not changed. Together, these results suggest that the celecoxib/BCCIP signaling further promote G2/M arrest and apoptosis in HCT116 cells induced by irradiation.
Figure 6.
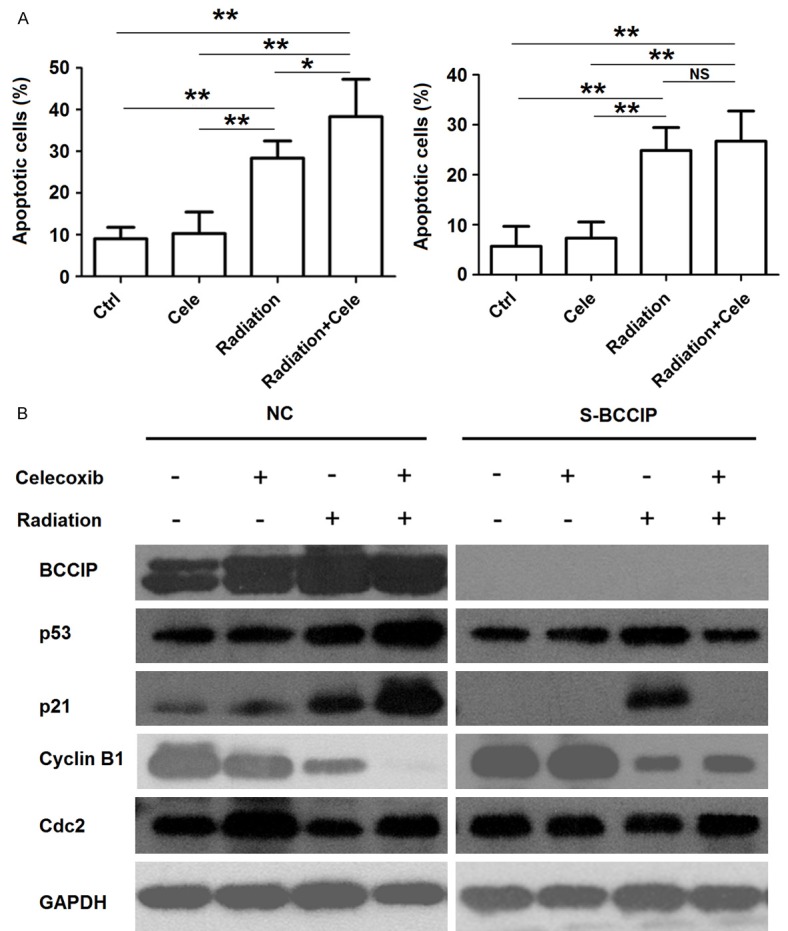
Celecoxib promotes irradiation-induced apoptosis in HCT116 cells by BCCIP. (A, B) After pre-treatment with celecoxib (30 μM) and/or irradiation (6 Gy), the apoptosis (A) and the protein levels of BCCIP, p53, p21, and Cyclin B1 (B) in both NC and S-BCCIP cells were analyzed by apoptosis assay and western blotting, respectively. The data are presented as mean ± SD. *P<0.05 and **P<0.01 (one-way ANOVA).
Discussion
Radiotherapy has been an accepted treatment for LARC (T3/4N1/2) since the 1990s, and preoperative chemoradiotherapy (CRT) is recognized as the standard therapy today [26]. At present, more than 70% of cancer patients with cancer require radiotherapy during the course of their entire treatment. With the development of precise radiotherapy, the normal tissues around the tumor were protected well, and the radiation dose delivered to the tumor increased. However, since most tumors show radiation resistance to some extent, the control rate of the tumor is limited, and a risk of recurrence and metastasis still exist. Therefore, improvement of the radiosensitivity of tumor cells is directly related to the effectiveness of radiation therapy. As an important means to increase the sensitivity of tumors, reverse the radiation resistance and improve the curative effect of radiotherapy, the radio-sensitizer has been a research focus in the field of radiation biology.
Several studies have suggested that resistance to radiation could be attributed to elevated COX-2 expression in cancer cells is the cause of radiation resistance [27], and the application of COX-2 inhibitors can improve the sensitivity of tumors to irradiation by down-regulating the expression of COX-2 [28-30]. Celecoxib, a selective inhibitor of COX-2 with high cardiovascular safety, is now widely used in the clinical setting. Studies have shown that celecoxib can play inhibitory effects on cell proliferation and stimulation of apoptosis at lower concentrations than those required for COX-2 inhibition [31]. Therefore, serveral studies showed the anti-tumor effects of celecoxib functions in both COX-2 dependent and independent manners. In the present study, we also found that celecoxib inhibited the proliferation of HeLa, A549 and HCT116 cells although HCT116 were negative for COX-2 expression. In addition, celecoxib could enhance the radiosensitivity of these three kinds of cells in vitro. These data suggest that the enhancement of radiosensitivity of HCT116 cells triggered by celecoxib is likely mediated by other molecules rather than COX-2.
Originally, BCCIP was identified as a BRCA2 and CDKN1A (Cip1/waf1/p21)-interacting protein [15]. Silencing of BCCIP leads to significantly reduced of homologous recombination [32], spontaneous chromatid aberrations including single chromatid breaks and sister chromatid unions [33], cell cycle dysregulation [18,34], and cytokinesis failure [35]. Moreover, BCCIP is an essential gene. Persistent absence of BCCIP leads to the proliferation defects and mitosis failure [33,35]. Down-regulation of BCCIP expression has been observed in some human cancers [21,36,37], but the significant mutations of BCCIP have not been reported in cancer tissues. In this study, we observed that all these three cell lines expressed BCCIP, and celecoxib obviously up-regulated the expression of BCCIP in all these cells. Recently, BCCIP has been considered as a prognostic marker for radiotherapy for laryngeal cancer, and the levels of functional BCCIP and p53 have been shown to be positively correlated with the radiosensitivity of cancer tissues [21]. These reports inspired us to further study the relationship between celecoxib and BCCIP in the regulation of radiosensitivity of HCT116 cells.
Subsequently, we constructed the stable BCCIP-silenced HCT116 cells (S-BCCIP), and found that the radiosensitizing effect induced by celecoxib in S-BCCIP cells in vitro disappeared. These results confirm the key role of BCCIP in the effect of celecoxib on the radiosensitivity of HCT116 cells. Histone H2AX is a minor component of nuclear histone H2A. The phosphorylation of histone H2AX at Ser 139, named γ-H2AX, was originally identified as an early event after the formation of DSBs induced by ionizing radiation [38]. As the number of foci is reported to be proportional to the number of DSBs [39], many investigators performed immunofluorescence staining for γ-H2AX, and counted the number of foci and percentages of γ-H2AX-positive cells as a marker for DSBs. In the present study, knock-down of BCCIP in irradiated HCT116 cells also obliterated the increase of celecoxib-induced γ-H2AX foci formation. Considering the importance of γ-H2AX-ATM-Chk2 checkpoint activation in DNA replication arrested fork [40], we further analyzed the levels of γ-H2AX, p-ATM, and p-Chk2 in NC and S-BCCIP cells after celecoxib and/or irradiation, and observed no significant increases of γ-H2AX, p-ATM, and p-Chk2 levels in S-BCCIP cells compared with NC cells. These data above indicate that the celecoxib/BCCIP signaling can promote the DNA repair induced by irradiation, possibly through activation of the γ-H2AX-ATM-Chk2 checkpoint.
It is well known that cells in the G2-M phase are the most sensitive to radiation, while those in the G1 and S phase are the most insensitive [41,42]. Results from a study by Shin et al., [44] suggest that DNA damage induced by radiation may result in changes in the G2-M regulatory site. Celecoxib may exert an inhibitory effect on enhanced radiation-induced G2-M arrest in the COX-2-overexpressing cells, which may allow the arrested cells to enter mitosis and die after radiation, but may also further enhance radiation-induced G2-M arrest in the COX-2 low-expressing cells in an unknown manner [43]. Here, celecoxib was shown to promote the G2-M phase arrest induced by irradiation by up-regulation of BCCIP in HCT116 cells.
Further analysis showed that celecoxib led to increased expression of BCCIP, p53 and p21, and decreased expression of Cyclin B1. Besides, the apoptosis rate of HCT116 cells after irradiation was observed upon celecoxib treatment. p53 inactivation has been shown to be associated with decreased radiation sensitivity and apoptotic cell death [18]. It has recently been shown that BCCIP is required for the transactivation activity of wild-type p53 [14]. In p53 wild-type cells, knock-down of BCCIP diminished the transactivation activity of p53, inhibited the binding of p53 to promoters of p53 target genes p21 and HDM2, and reduced the tetrameric formation of p53 [44]. Therefore, the defects of BCCIP override the transactivation function of wild-type p53, suggesting a critical role of BCCIP in maintaining the functions of p53 in tumor suppression and response to therapy. From our results, it can be concluded that the enhanced effect of celecoxib on irradiation-induced apoptosis is dependent on the BCCIP/p53 signaling pathway.
Collectively, based on our results and those of other reports, celecoxib enhances the radiosensitivity of HCT116 cells by up-regulating BCCIP expression. The possible mechanisms underlying celecoxib/BCCIP signaling in the regulation of radiosensitivity are as follows: 1) promotion of DNA repair induced by irradiation by the γ-H2AX-ATM-Chk2 checkpoint activation; 2) enhancement of radiation-induced G2-M arrest; and 3) aggravation of irradiation-induced apoptosis by the BCCIP/p53 signaling. These data suggest that BCCIP should be a target gene related to the radiation sensitivity of CRC cells, and that targeting BCCIP could be a potential method for radiosensitization in patients with CRC.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (NSFC) (No.81302384) and the Science Foundation of Jiangsu Province (BK20140007), a Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD) and Jiangsu Provincial Key Laboratory of Radiation Medicine and Protection.
Disclosure of conflict of interest
None.
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 3.Improved survival with preoperative radiotherapy in respectable rectal cancer. Swedish rectal cancer trial. N Engl J Med. 1997;336:980–987. doi: 10.1056/NEJM199704033361402. [DOI] [PubMed] [Google Scholar]
- 4.Bosset JF, Collette L, Calais G, Mineur L, Maingon P, Radosevic-Jelic L, Daban A, Bardet E, Beny A, Ollier JC EORTC Radiotherapy Group Trial 22921. Chemotherapy with preoperative radiotherapy in rectal cancer. N Engl J Med. 2006;355:1114–1123. doi: 10.1056/NEJMoa060829. [DOI] [PubMed] [Google Scholar]
- 5.Gerard JP, Conroy T, Bonnetain F, Bouché O, Chapet O, Closon-Dejardin MT, Untereiner M, Leduc B, Francois E, Maurel J, Seitz JF, Buecher B, Mackiewicz R, Ducreux M, Bedenne L. Preoperativeradiotherapy with or without concurrent fluorouracil and leucovorin in T3-4 rectal cancers: results of FFCD 9203. J. Clin. Oncol. 2006;24:4620–4625. doi: 10.1200/JCO.2006.06.7629. [DOI] [PubMed] [Google Scholar]
- 6.Rega D, Pecori B, Scala D, Avallone A, Pace U, Petrillo A, Aloj L, Tatangelo F, Delrio P. Evaluation of tumor response after short-course radiotherapy and delayed surgery for rectal cancer. PLoS One. 2016;11:e0160732. doi: 10.1371/journal.pone.0160732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Steinbach G, Lynch PM, Phillips RK, Wallace MH, Hawk E, Gordon GB, Wakabayashi N, Saunders B, Shen Y, Fujimura T, Su LK, Levin B, Godio L, Patterson S, Rodriguez-Bigas MA, Jester SL, King KL, Schumacher M, Abbruzzese J, DuBois RN, Hittelman WN, Zimmerman S, Sherman JW, Kelloff G. The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. New Engl J Med. 2000;342:1946–1952. doi: 10.1056/NEJM200006293422603. [DOI] [PubMed] [Google Scholar]
- 8.Kawamori T, Rao CV, Seibert K, Reddy BS. Chemopreventive activity of celecoxib, a specific cyclooxygenase-2 inhibitor, against colon carcinogenesis. Cancer Res. 1998;58:409–412. [PubMed] [Google Scholar]
- 9.Pakos EE, Ioannidis JP. Radiotherapy vs. nonsteroidal anti-inflammatory drugs for the prevention of heterotopic ossification after major hip procedures: a meta-analysis of randomized trials. Int J Radiat Oncol Biol Phys. 2004;60:888–895. doi: 10.1016/j.ijrobp.2003.11.015. [DOI] [PubMed] [Google Scholar]
- 10.Shi S, Klotz U. Clinical use and pharmacological properties of selective COX-2 inhibitors. Eur J Clin Pharmacol. 2008;64:233–252. doi: 10.1007/s00228-007-0400-7. [DOI] [PubMed] [Google Scholar]
- 11.Suzuki K, Gerelchuluun A, Hong Z, Sun L, Zenkoh J, Moritake T, Tsuboi K. Celecoxib enhances radiosensitivity of hypoxic glioblastoma cells through endoplasmic reticulum stress. Neuro Oncol. 2013;15:1186–1199. doi: 10.1093/neuonc/not062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang MY, Lee HT, Chen CM, Shen CC, Ma HI. Celecoxib suppresses the phosphorylation of STAT3 protein and can enhance the radiosensitivity of medulloblastoma-derived cancer stem-like cells. Int J Mol Sci. 2014;15:11013–11029. doi: 10.3390/ijms150611013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Semaan J, Pinon A, Rioux B, Hassan L, Limami Y, Pouget C, Fagnère C, Sol V, Diab-Assaf M, Simon A, Liagre B. Resistance to 3-HTMC-induced apoptosis through activation of PI3K/Akt, MEK/ERK and p38/COX-2/PGE2 pathways in human HT-29 and HCT116 colorectal cancer cells. J Cell Biochem. 2016;117:2875–2885. doi: 10.1002/jcb.25600. [DOI] [PubMed] [Google Scholar]
- 14.Ono T, Kitaura H, Ugai H, Murata T, Yokoyama KK, Iguchi-Ariga SM, Ariga H. TOK-1, a novel p21Cip1-binding protein that cooperatively enhances p21-dependent inhibitory activity toward CDK2 kinase. J Biol Chem. 2000;275:31145–31154. doi: 10.1074/jbc.M003031200. [DOI] [PubMed] [Google Scholar]
- 15.Liu J, Yuan Y, Huan J, Shen Z. Inhibition of breast and brain cancer cell growth by BCCIPalpha, an evolutionarily conserved nuclear protein that interacts with BRCA2. Oncogene. 2001;20:336–345. doi: 10.1038/sj.onc.1204098. [DOI] [PubMed] [Google Scholar]
- 16.Meng X, Liu J, Shen Z. Inhibition of G1 to S cell cycle progression by BCCIP beta. Cell Cycle. 2004;3:343–348. [PubMed] [Google Scholar]
- 17.Fan J, Wray J, Meng X, Shen Z. BCCIP is required for the nuclear localization of the p21 protein. Cell Cycle. 2009;8:3019–3024. [PMC free article] [PubMed] [Google Scholar]
- 18.Meng X, Liu J, Shen Z. Genomic structure of the human BCCIP gene and its expression in cancer. Gene. 2003;302:139–146. doi: 10.1016/s0378-1119(02)01098-3. [DOI] [PubMed] [Google Scholar]
- 19.Liu X, Cao L, Ni J, Liu N, Zhao X, Wang Y, Zhu L, Wang L, Wang J, Yue Y, Cai Y, Jin J. Differential BCCIP gene expression in primary human ovarian cancer, renal cell carcinoma and colorectal cancer tissues. Int J Oncol. 2013;43:1925–1934. doi: 10.3892/ijo.2013.2124. [DOI] [PubMed] [Google Scholar]
- 20.Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro. Nat Protoc. 2006;1:2315–2319. doi: 10.1038/nprot.2006.339. [DOI] [PubMed] [Google Scholar]
- 21.Rewari A, Lu H, Parikh R, Yang Q, Shen Z, Haffty BG. BCCIP as a prognostic marker for radiotherapy of laryngeal cancer. Radiother Oncol. 2009;90:183–188. doi: 10.1016/j.radonc.2008.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Redon CE, Dickey JS, Bonner WM, Sedelnikova OA. γ-H2AX as a biomarker of DNA damage induced by ionizing radiation in human peripheral blood lymphocytes and artificial skin. Adv Space Res. 2009;43:1171–1178. doi: 10.1016/j.asr.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roch-Lefèvre S, Mandina T, Voisin P, Gaëtan G, Mesa JE, Valente M, Bonnesoeur P, García O, Voisin P, Roy L. Quantification of γ-H2AX foci in human lymphocytes: a method for biological dosimetry after ionizing radiation exposure. Radiat Res. 2010;174:185–194. doi: 10.1667/RR1775.1. [DOI] [PubMed] [Google Scholar]
- 24.Rothkamm K, Horn S. γ-H2AX as protein biomarker for radiation exposure. Ann Ist Super Sanita. 2009;45:265–271. [PubMed] [Google Scholar]
- 25.Goodarzi AA, Jeggo PA. Irradiation induced foci (IRIF) as a biomarker for radiosensitivity. Mutat Res. 2012;736:39–47. doi: 10.1016/j.mrfmmm.2011.05.017. [DOI] [PubMed] [Google Scholar]
- 26.Glynne-Jones R, Hall M. Radiotherapy and locally advanced rectal cancer. Br J Surg. 2015;102:1443–1445. doi: 10.1002/bjs.9930. [DOI] [PubMed] [Google Scholar]
- 27.Terakado N, Shintani S, Yano J, Chunnan L, Mihara M, Nakashiro K, Hamakawa H. Overexpression of cyclooxygenase-2 is associated with radioresistance in oral squamous cell carcinoma. Oral Oncology. 2004;40:383–389. doi: 10.1016/j.oraloncology.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 28.Raju U, Nakata E, Yang P, Newman RA, Ang KK, Milas L. In vitro enhancement of tumor cell radiosensitivity by a selective inhibitor of cyclooxygenase-2 enzyme: mechanistic considerations. Int J Radiation Oncology Biol Phys. 2002;54:886–894. doi: 10.1016/s0360-3016(02)03023-7. [DOI] [PubMed] [Google Scholar]
- 29.Pyo H, Choy H, Amorino GP, Kim JS, Cao Q, Hercules SK, DuBois RN. A selective cyclooxygenase-2 inhibitor, NS-398, enhances the effect of radiation in vitro and in vivo preferentially on the cells that express cyclooxygenase-2. Clin Cancer Res. 2001;7:2998–3005. [PubMed] [Google Scholar]
- 30.Petersen C, Petersen S, Milas L, Lang FF, Tofilon PJ. Enhancement of intrinsic tumor cell radiosensitivity induced by a selective cyclooxygenase-2 inhibitor. Clin Cancer Res. 2000;6:2513–2520. [PubMed] [Google Scholar]
- 31.Schönthal AH. Antitumor properties of dimethyl-celecoxib, a derivative of celecoxib that does not inhibit cyclooxygenase-2: implications for glioma therapy. Neurosurg Focus. 2006;20:E21. doi: 10.3171/foc.2006.20.4.14. [DOI] [PubMed] [Google Scholar]
- 32.Lu H, Yue J, Meng X, Nickoloff JA, Shen Z. BCCIP regulates homologous recombination by distinct domains and suppresses spontaneous DNA damage. Nucleic Acids Res. 2007;35:7160–7170. doi: 10.1093/nar/gkm732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lu H, Huang YY, Mehrotra S, Droz-Rosario R, Liu J, Bhaumik M, White E, Shen Z. Essential roles of BCCIP in mouse embryonic development and structural stability of chromosomes. PLoS Genet. 2011;7:e1002291. doi: 10.1371/journal.pgen.1002291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meng X, Yue J, Liu Z, Shen Z. Abrogation of the transactivation activity of p53 by BCCIP down-regulation. J Biol Chem. 2007;282:1570–1576. doi: 10.1074/jbc.M607520200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meng X, Fan J, Shen Z. Roles of BCCIP in chromosome stability and cytokinesis. Oncogene. 2007;26:6253–6260. doi: 10.1038/sj.onc.1210460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu J, Lu H, Ohgaki H, Merlo A, Shen Z. Alterations of BCCIP, a BRCA2 interacting protein, in astrocytomas. BMC Cancer. 2009;9:268. doi: 10.1186/1471-2407-9-268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roversi G, Pfundt R, Moroni RF, Magnani I, van Reijmersdal S, Pollo B, Straatman H, Larizza L, Schoenmakers EF. Identification of novel genomic markers related to progression to glioblastoma through genomic profiling of 25 primary glioma cell lines. Oncogene. 2006;25:1571–1583. doi: 10.1038/sj.onc.1209177. [DOI] [PubMed] [Google Scholar]
- 38.Ibuki Y, Toyooka T. Evaluation of chemical phototoxicity, focusing on phosphorylated histone H2AX. J Radiat Res. 2015;56:220–228. doi: 10.1093/jrr/rru105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sedelnikova OA, Rogakou EP, Panyutin IG, Onner WM. Quantitative detection of (125)IdU-induced DNA double-strand breaks with γ-H2AX antibody. Radiat Res. 2002;158:486–492. doi: 10.1667/0033-7587(2002)158[0486:qdoiid]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 40.Rao VA, Conti C, Guirouilh-Barbat J, Nakamura A, Miao ZH, Davies SL, Saccá B, Hickson ID, Bensimon A, Pommier Y. Endogenous gamma-H2AX-ATM-Chk2 checkpoint activation in Bloom’s syndrome helicase deficient cells is related to DNA replication arrested forks. Mol Cancer Res. 2007;5:713–724. doi: 10.1158/1541-7786.MCR-07-0028. [DOI] [PubMed] [Google Scholar]
- 41.Pawlik TM, Keyomarsi K. Role of cell cycle in mediating sensitivity to radiotherapy. Int J Radiat Oncol Biol Phys. 2004;59:928–942. doi: 10.1016/j.ijrobp.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 42.Geldof AA, Plaizier MA, Duivenvoorden I, Ringelberg M, Versteegh RT, Newling DW, Teule GJ. Cell cycle perturbations and radiosensitization effects in a human prostate cancer cell line. J Cancer Res Clin Oncol. 2003;129:175–182. doi: 10.1007/s00432-002-0412-8. [DOI] [PubMed] [Google Scholar]
- 43.Shin YK, Park JS, Kim HS, Jun HJ, Kim GE, Suh CO, Yun YS, Pyo H. Radiosensitivity enhancement by celecoxib, a cyclooxygenase (COX)-2 selective inhibitor, via COX-2-dependent cell cycle regulation on human cancer cells expressing differential COX-2 levels. Cancer Res. 2005;65:9501–9509. doi: 10.1158/0008-5472.CAN-05-0220. [DOI] [PubMed] [Google Scholar]
- 44.Huang YY, Lu H, Liu S, Droz-Rosario R, Shen Z. Requirement of mouse BCCIP for neural development and progenitor proliferation. PLoS One. 2012;7:e30638. doi: 10.1371/journal.pone.0030638. [DOI] [PMC free article] [PubMed] [Google Scholar]