

Abstract
HIF-1α plays an essential role in hemorrhagic shock-induced vasoconstriction. However, the underlying mechanisms remain poorly understood. Here, we studied both the role of HIF-1α in regulating vasodilatation, and the involvement of Cx40 in this process. We found that endothelium-dependent vasodilatation exhibited an overall decline after hemorrhagic shock: at the beginning of shock vasodilatation reactivity significantly decreased, followed by a slight increase from 0.5 h to 2 h after shock. After 2 h vasodilatation dropped again. Throughout this process, protein levels of HIF-1α gradually increased. In the late period of shock, vasodilatation reactivity was enhanced by oligomycin, an HIF-1α inhibitor, suggesting that HIF-1α may promote vasoconstriction. Moreover, in the late period of shock Cx40 levels gradually increased and exhibited a negative correlation with endothelium-dependent vasoconstriction reactivity. Furthermore, Cx40 AODN significantly improved vasoconstriction reactivity and could be regulated by either an HIF-1α inhibitor or an agonist. Together, these data suggest that HIF-1α may inhibit endothelium-dependent vasodilatation reactivity following hemorrhagic shock by up-regulating Cx40, especially in the late period of shock.
Keywords: HIF-1α, hemorrhagic shock, endothelium-dependent vasodilatation reactivity, Cx40
Introduction
Shock is a common and severe disease that accounts for about 50% of early death in trauma [1]. In the late period of shock, cardiovascular dysfunction may occur as a result of abnormal material and energy metabolism, and may be accompanied by systemic inflammatory response and multiple organ dysfunction. Additionally, hemorrhagic shock may result from shock-induced vascular hyporeactivity. Hypoxia-inducible factor 1α (HIF-1α) is the oxygen-regulated subunit of the transcriptional activator HIF-1, which accommodates cellular adaptation to low oxygen stress by regulating transcriptional processes in erythropoiesis, angiogenesis and metabolism [2]. HIF-1 is critical for the oxygen stress response and mediates changes in gene expression in response to cellular oxygen concentrations [3]. A study by Nagaraju GP et al. indicates that HIF-1α participates in tumor cell growth and metastasis by facilitating angiogenesis [4]. Likewise, Jiang H et al. show that HIF-1α is a critical regulator of gene expression under hypoxic and inflammatory conditions [5]. Our own previous study shows that HIF-1α plays an important regulatory role in hemorrhagic shock-induced vasoconstrictive hyporeactivity [6]. However, the role of HIF-1α in the regulation of vasodilative reactivity following hemorrhagic shock remains poorly understood.
Our previous study shows that connexin (Cx) 40, a myoendothelial gap junction (MEGJ) protein, may play an important role in the regulation of vascular reactivity after acute hemorrhagic shock [7]. Moreover, our recent study demonstrates a critical role for Cx40 in vascular constriction after hemorrhagic shock [8]. A study by Morton SK et al. investigated the role of Cx40 in the coordination of vasodilation in microcirculatory networks in hypertensive rats, and the results showed higher levels of Cx40 in all hypertensive animals [9]. Analogously, a study by Manuel HG investigated the role of Cx40 in the regulation of vascular reactivity in cirrhosis, and found similarly increased Cx40 levels during portal hypertension [10]. Both studies indicate that Cx40 may have an inhibitory effect on vascular relaxation. Nevertheless, it remains unclear whether HIF-1α regulates vasodilative reactivity following hemorrhagic shock.
In order to explore the regulatory effect of HIF-1α on vasodilative reactivity and the relationship to Cx40 expression following hemorrhagic shock, the current study utilized the hemorrhagic shock Sprague-Dawley rat model and hypoxia-treated blood vessels [superior mesenteric arteries (SMAs)]. The effects of oligomycin, an HIF-1α inhibitor, on vasodilative reactivity to Ach (acetylcholine; endothelium-dependent vasodilator) and SNP (sodium nitroprusside, endothelium-independent vasodilator) were observed, and the relationship between HIF-1α and Cx40 following hemorrhagic shock was investigated.
Materials and methods
Ethics approval
The present study was approved by the Research Council and Animal Care and Use Committee of the Research Institute of Surgery, Daping Hosptial, Third Military Medical University (Chongqing, China) and confirmed with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (8th edition, 2011). The protocol conformed with guidelines for the ethical use of animals. Efforts were made to minimize animal suffering and to reduce the number of animals used.
Reagents
Acetylcholine (Ach) and oligomycin (an HIF-1α inhibitor), were obtained from Sigma (St. Louis, MO). Sodium nitroprusside (SNP) was purchased from DCPC (Beijing, CHINA). EGb761 (EGb761: ginkgo biloba extract, a HIF-1α inducer) was purchased from Schwabe (Germany). Cx40 antibody was obtained from Alpha Diagnostics (San Antonio, USA). HIF-1α antibody was obtained from Novus (Carlsbad, USA). Cx40 antisense oligodeoxynucleotide (AODN) was purchased from Invitrogen (Carlsbad, CA). Horseradish peroxidase-conjugated secondary antibody, the enhanced chemiluminescence substrate kit, Western blot stripping buffer, and the bicinchoninic acid protein assay kit were obtained from Pierce (Appleton, WI).
Animals
Sprague-Dawley (SD) rats (200-250 g) were purchased from Animal Center of Research Institute of Surgery, the Third Military Medical University and fasted for 12 h, with the exception of water ad libitum before the experiment. On the day of the experiment, the rats were first anesthetized with sodium pentobarbital (30 mg/kg body weight, intraperitoneally) which was administered until the rats had no response to a needle stimulus, up to 50 mg/kg. The right femoral arteries of the rats were catheterized with polyethylene tubing (outer diameter, 0.965 mm; inner diameter, 0.58 mm) for monitoring the mean arterial pressure (MAP) and bleeding. To prevent clot formation, the tubing was filled with normal saline containing heparin (30 U/ml). After completion of catheterization, the rats were allowed to stabilize for 10 min and then hemorrhaged from the right femoral arterial catheter until the MAP dropped under 40 mmHg within 10 min. Rats were maintained at this MAP level for a certain time period predetermined in the experimental design.
Measurement of vasodilatation reactivity
SMAs were obtained from the rats. After removing the connective tissue, the SMAs were cut into 2-3 mm long arterial rings to measure the vasodilative response to serial concentrations of Ach and SNP (10-9, 10-8, 10-7, 10-6 and 10-5 mol/L). The reactivity was measured with an isolated vascular tension-measuring system (AD Instruments, Castle Hill, NSW, Australia) as described previously [11]. The Diastolic level (%)=Decreased tension value after stimulation with Ach or SNP/Increased tension value after NE pre-constriction (final concentration 10-6 mol/L) × 100%. The dose-response curves of SMA to Ach or SNP, and maximal contraction (Emax), were used to reflect the vascular vasodilative reactivity.
Anoxia treatment of arterial rings
The culture plates with SMA rings were placed in an anoxia incubator chamber (MIC-101; Billups-Rothenberg, Inc., Del Mar, Calif) under a humidified environment with 5% CO2 at 37°C. The chamber was bubbled with 95% N2/5% CO2 for 15 min, occluded for 10 min, and bubbled again. This procedure was repeated four times until the O2 concentration was <0.2%, a concentration which was actively maintained for a predetermined time according to experimental design [12].
mRNA expression of HIF-1α and Cx40
mRNA expression of HIF-1α and Cx40 was tested by reverse-transcription polymerase chain reaction (PCR). Each SMA sample was pooled in 1 ml tripure lysis solution (Roche, Shanghai, China). RNA extraction was conducted in accordance with the routine method. The extracted RNA was reverse transcribed as per the manufacturer’s specifications (Takara, Shiga, Japan). The primer pairs and PCR conditions of HIF-1α, Cx40 and β-actin are shown in Table 1. The PCR products were electrophoresed in 1.5% agarose gel and stained with ethidium bromide.
Table 1.
Primer pairs and PCR conditions of HIF-1α, Cx40 and β-actin
| Gene | Primer sequence | Degeneration | Amplification | Elongation | |
|---|---|---|---|---|---|
|
| |||||
| Amplification procedure | Cycle number | ||||
| HIF-1α | |||||
| Forward | 5’-GGTGCTAACAGATGATGGTGAC-3’ | 94°C, 2 min | 94°C, 30 s; | 30 | 72°C, 10 min |
| Reverse | 5’-GGCTCATAACCCATCAACTCAG-3’ | 58°C, 30 s | |||
| Cx40 | |||||
| Forward | 5’-GACAAGCACCAGCTTCTTGG-3’ | 94°C, 2 min | 94°C, 30 s; | 33 | 72°C, 10 min |
| Reverse | 5’-AGAGAAGGTGCTGAGGAAGG-3’ | 55°C, 30 s | |||
| β-actin | |||||
| Forward | 5’-GAGACCTTCAACACCCCAGCC-3’ | 94°C, 2 min | 94°C, 30 s; | 30 | 72°C, 10 min |
| Reverse | 5’-CCGTCAGGCAGCTCATAGCTG-3’ | 60°C, 30 s | |||
Oligomycin administration and Cx40 AODN treatment
For Oligomycin administration in vivo and vitro, rats in the oligomycin group were intraperitoneally injected with oligomvcin (9 μg/kg) 4 h before hemorrhaging, and SMA rings from the oligomycin group were cultured with oligomycin (10 ng/ml) for 12 h.
For Cx40 AODN treatment, Cx40 AODN (100 mM) was transfected into SMAs with transfection reagent (5:1 vol/vol; Qiagen, Montgomery County, Md) 12 h before treatment. Cx40 AODN was specifically complementary to the ribosome AUG translation start codon region of murine Cx40 mRNA. The sequence of Cx40 AODN was 5’-GTC ACCCATCTTGCCAAG-3’.
Statistical analysis
All data were expressed as mean ± SD and analyzed by SPSS 15.0 (Chicago, IL). The difference between multiple groups was analyzed by one-way ANOVA and post hoc test (Student-Newman-Keuls test). The difference between two groups was tested by independent t test. The Pearson correlation test was used to analyze dependability. P values <0.05 were considered significant, and P<0.01 was considered even more significant.
Results
Overall decline in vasodilatation reactivity during hemorrhagic shock
The vasodilative response of SMAs to Ach and SNP was tested in vivo and vitro in order to evaluate the changes in endothelium-dependent and -independent vasodilatation reactivity after shock. Forty-eight SD rats were randomly divided into six groups (n=8 rats/group): one control group and five hemorrhagic shock groups (MAP at 40 mmHg for 0.5 h, 1 h, 2 h, 3 h and 4 h). Two SMA rings were collected from each sample, one for Ach, one for SNP. Another forty-eight SD rats were also divided into similar groups but were instead treated with anoxia in vitro or hemorrhage in vivo.
As compared with the control group, the cumulative dose-response curves of SMA to Ach were significantly shifted to the right, and vasodilatation reactivity at 4 h was the lowest (Figure 1A). The Emax of vasodilatation reactivity to Ach was significantly decreased at the beginning of shock, slightly increased from 0.5 h to 2 h after shock, then continuously decreased to its lowest level (P<0.01) (Figure 1B, 1C). Endothelium-independent vasodilatation reactivity exhibited an obvious reduction at low concentrations (10-9, 10-8, 10-7 mol/L) of SNP stimulation (P<0.01), while there was no significant difference between groups under high concentration (10-6, 10-5 mol/L) SNP stimulation (P>0.05) (Figure 1B, 1C). The trend of vasodilatation reactivity to Ach in vitro after anoxia exhibited an overall decline, which meant the Emax of reactivity was significantly decreased at the beginning of shock, maintained steady levels between 0.5 h and 2 h after anoxia, and then continuously decreased to the end (Figure 1D-F). Not surprisingly, there was also no significant difference between groups under high concentrations of SNP (P>0.05) (Figure 1E, 1F).
Figure 1.
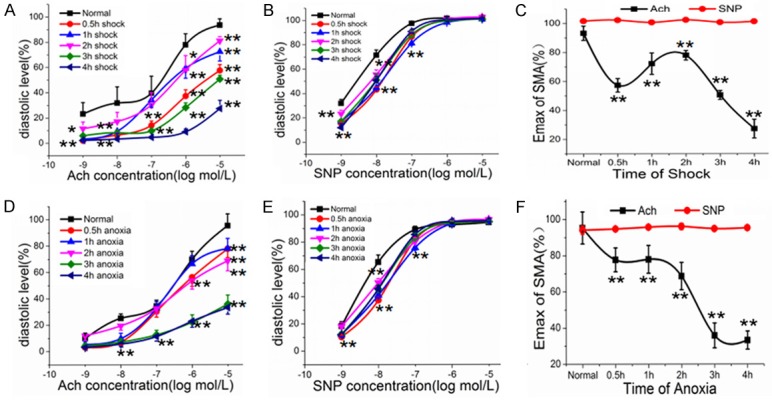
Changes in vasodilatation reactivity in hemorrhagic shock. A: The cumulative dose-response curves to Ach after shock. B: The cumulative dose-response curves to SNP after shock. C: The maximal vasodilatation reactivity (Emax) of SMA to Ach and SNP after shock. D: The cumulative dose-response curves to Ach after anoxia. E: The cumulative dose-response curves to SNP after anoxia. F: The maximal vasodilatation reactivity (Emax) of SMA after anoxia. *P<0.05, **P<0.01 as compared with control group.
Over-expression of HIF-1α after hemorrhagic shock
The rest of the samples of SMA in shock groups of Section 3.1 were tested for expression of HIF-1α mRNA. As compared with the sham-operated group, expression of HIF-1α mRNA was increased gradually after hemorrhagic shock and peaked at 4 h (Figure 2A, 2B), which was consistent with protein expression of HIF-1α after shock (Figure 2C, 2D). The trend was negatively correlated with endothelium-dependent vasodilatation reactivity after 2 h of hemorrhagic shock (Pearson=-0.999, P<0.05). However, there was no correlation between HIF-1α expression and SNP-induced endothelium-independent vasodilatation reactivity.
Figure 2.
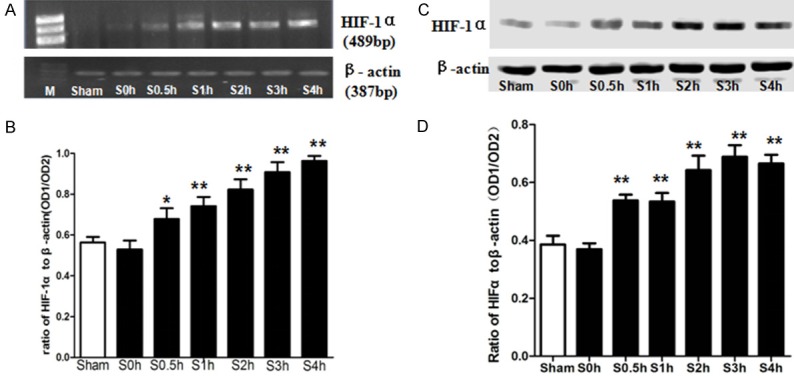
The expression of HIF-1α after hemorrhagic shock. A: Electrophoresis of HIF-1α mRNA and β-actin mRNA. B: Statistical graph of graph A. C: Western blot of HIF-1α after shock. D: Statistical graph of graph C. M, molecular marker; Sham, sham-operated group; S, shocked groups. *P<0.05, **P<0.01 as compared with the sham-operated group.
The inhibitory effect of HIF-1α on endothelium-dependent vasodilatation reactivity in the late period of shock
Ninety-six SD rats were randomly divided into twelve groups (n=8 rats/group) and the groups were similar to that in section 3.1 but were treated with oligomycin, an HIF-1α inhibitor. The vasodilatation reactivity to Ach significantly decreased in the beginning of shock but gradually increased in the late period (2 h-4 h after shock) (Figure 3A). Compared with the shock groups, oligomycin increased the diastolic Emax by 13.24% at 2 h after shock, 41.25% at 3 h after shock and 64.37% at 4 h after shock, which were significantly higher than those in the shock groups (P<0.01) (Figure 3B). The same trend was also found in vitro after treatment with anoxia (Figure 3C, 3D). However, after oligomycin treatment, the response of vasodilatation reactivity to SNP was not as obvious as that to Ach. Compared with shock groups, oligomycin decreased the reactivity to SNP before 2 h after shock (P<0.01) but had no effect on vasodilatation reactivity to SNP after 3 h (P>0.05) (Figure 3E, 3F).
Figure 3.
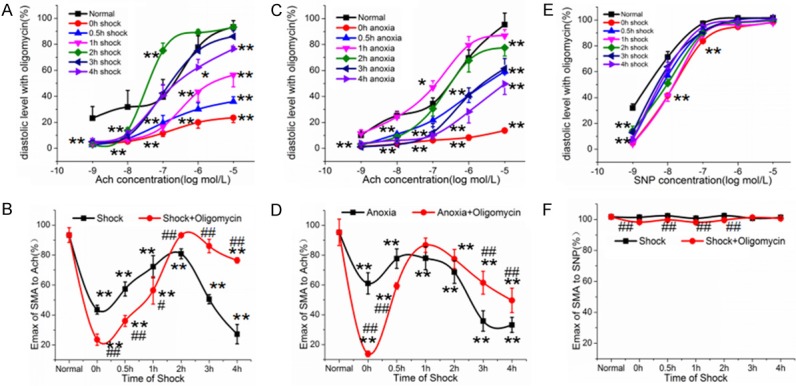
The effect of HIF-1α on vasodilatation reactivity in hemorrhagic shock. A: The cumulative dose-response curves to Ach after shock treated with oligomycin. B: The maximal vasodilatation reactivity (Emax) of SMA to Ach in shock group (data from section 3.1) and shock + oligomycin group. C: The cumulative dose-response curves to Ach after anoxia treated with oligomycin. D: The maximal vasodilatation reactivity (Emax) of SMA to Ach in anxoia group (data from section 3.1) and anoxia + oligomycin group. E: The cumulative dose-response curves to SNP after shock treated with oligomycin. F: The maximal vasodilatation reactivity (Emax) of SMA to SNP in shock group (data from section 3.1) and shock + oligomycin group. *P<0.05, **P<0.01 as compared with normal group. #P<0.05, ##P<0.01 as compared with the same shock time group.
Cx40 may inhibit endothelium-dependent vasodilatation reactivity in the late period of shock
Compared to the sham-operated group, Cx40 expression decreased gradually in the early period of shock (10 min-1 h after shock) and rose to a peak at 4 h after hemorrhagic shock (Figure 4A, 4B). This trend was negatively correlated with endothelium-dependent vasodilatation reactivity after hemorrhagic shock (Pearson=-0.816, P<0.05).
Figure 4.

The expression of Cx40 in hemorrhagic shock and the effect on vasodilatation reactivity after Cx40 AODN. A: Protein expression of Cx40 after shock. B: Statistical graph of graph A. C: The expression of Cx40 mRNA in control group and Cx40 AODN group. D: Statistical graph of graph C. E: The cumulative dose-response curves to Ach in normal conditions after Cx40 AODN. F: The cumulative dose-response curves to Ach in 1 h anoxia after Cx40 AODN. G: The cumulative dose-response curves to Ach in 4 h anoxia after Cx40 AODN. H: The maximal vasodilatation reactivity (Emax) of SMA to Ach after anoxia treated with Cx40 AODN. *P<0.05, **P<0.01 as compared with sham-operated group. #P<0.05, ##P<0.01 as compared with the same anoxia time group.
Forty-eight SD rats were randomly divided into three groups: non-anoxia (n=16), anoxia for 1 h (n=16) and anoxia for 4 h (n=16). Each group was also randomly divided into two subgroups: control group (n=8) and Cx40 AODN group (n=8). After treatment with Cx40 AODN, expression levels of Cx40 mRNA in SMAs were significantly decreased (P<0.01) (Figure 4C, 4D). Compared with the same anoxia-time groups, Cx40 AODN had no effect under normal conditions or 1 h after anoxia (P>0.05) (Figure 4E, 4F), but significantly improved vasodilatation reactivity at 4 h after anoxia (P<0.01) (Figure 4G, 4H). These results suggest that overexpression of Cx40 in the late period of shock may have an inhibitory effect on endothelium-dependent vasodilatation reactivity.
HIF-1α may up-regulate Cx40 expression after hemorrhagic shock
Thirty-six SD rats were randomly divided into three groups: a control group (n=12), EGb761 treatment (a HIF-1α agonist, n=12), and EGb761 + oligomycin treatment (n=12). Each group was divided into three subgroups: non-anoxia (n=4), anoxia for 1 h (n=4) and anoxia for 4 h (n=4). Compared with the control groups, EGb761 increased the expression of HIF-1α mRNA in the non-anoxia group (P<0.01) and the 4 h anoxia group (P<0.05) (Figure 5A, 5B). The expression of Cx40 mRNA significantly increased in the EGb761 group (P<0.01) compared to control groups, but decreased significantly after oligomycin treatment in the non-anoxia group and 4 h anoxia group (P<0.01) (Figure 5A, 5C). These results suggest that EGb761 may enhance the expression of Cx40 and oligomycin may inhibit Cx40 expression in the late period of shock, which indicates that HIF-1α inhibits vasodilatation response via up-regulating Cx40 expression, especially in the late period of shock.
Figure 5.
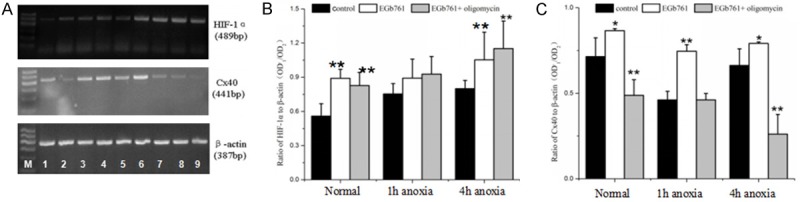
Relationship between HIF-1α and Cx40 in the regulation of vasodilatation reactivity after anoxia. A: RT-PCR electrophoregrams of HIF-1α, Cx40 and β-actin mRNA. M: DNA marker; 1: non-anoxia control; 2: 1 h anoxia control; 3: 4 h anoxia control; 4: non-anoxia + EGb761; 5: 1 h anoxia + EGb761; 6: 4 h anoxia + EGb761; 7: non-anoxia + EGb761 + oligomycin; 8: 1 h anoxia + EGb761 + oligomycin; 9: 4 h anoxia + EGb761 + oligomycin. B: Bar graph of HIF-1α RT-PCR. C: Bar graph of Cx40 RT-PCR. *P<0.05, **P<0.01 as compared with the same anoxia time control group.
Discussion
Vascular reactivity, including vasoconstriction and vasodilation, plays an essential role in the development of shock and may be regulated by many inducing factors. For example, high concentrations of catecholamine and cytokines, such as TNF-α and IL-1β, inhibit the transcription of adrenergic receptors and cause vascular hyporeactivity via receptor desensitization. After shock, large amounts of NO may induce the production of OONO-, and result in vascular hyporeactivity through increased opening of KATP channels and tyrosine phosphorylation of BKCa channels [13]. It has been demonstrated that PKC, RhoA, and Rac may regulate vascular reactivity through calcium desensitization mechanisms [14-16]. HIF-1α is a major regulator of cellular and systemic oxygen homeostasis, and mediates gene expression in response to hypoxia [17]. Our previous study shows that HIF-1α may regulate vasoconstrictive reactivity after hemorrhagic shock [18]. The results showed that HIF-1α, eNOS, iNOS and COX-2 mRNA expression increased after hemorrhagic shock, and oligomycin may down-regulate the expression of these genes and their products, NO, CO and PGI, to some extent. The mechanism by which HIF-1α regulates vasoconstrictive reactivity may be related to the regulatory effects of HIF-1α on the expression of eNOS, iNOS, HO-1, COX-2 as well as the production of NO, CO and PGI. This paper demonstrates that HIF-1α may also play an important role in the regulation of vasodilatation reactivity after hemorrhagic shock. Our current results showed that endothelium-dependent vasodilatation reactivity exhibited an overall decline after hemorrhagic shock, and oligomycin improved vasodilatation reactivity in the late period of shock. The mechanism by which HIF-1α regulates vasodilatation reactivity may be related to the activating effects of HIF-1α on Cx40, a MEGJ protein.
MEGJ is the gap junction between vascular endothelial cells (VECs) and vascular smooth muscle cells (VSMCs), which includes 21 connexin (Cx) members in the human gene profile and 20 Cx members in the mouse gene profile. Connexins allow the exchange of ions and small metabolites between neighboring cells as part of the regulation of cell functions. Data from Climent B et al. showed that VSMC may indirectly modulate VEC hyperpolarization and nitric oxide (NO) release via MEGJ [19]. It is reported that Cx37, Cx40, Cx43, Cx45 are mostly expressed in the cardiovascular system, and their expression levels vary with vascular territory and species [20]. Cx43 seems to be the predominant connexin isoform in aortic smooth muscle cells, while in endothelial cells, Cx40 expression is abundant [21].
Our previous study mainly focused on the relationship between vasoconstriction reactivity and MEGJ [12]. The results showed that mRNA and protein expression of Cx40 and Cx37 were negatively associated with vasoconstriction reactivity, while Cx43 played a positive role in vasoconstriction reactivity after shock. The mechanism may be related to their regulating effects on the calcium sensitivity of VSMCs. The present study clarifies the missing link between vasodilatation reactivity and MEGJ after hemorrhagic shock. Our results indicated that Cx40 gradually increased and correlated negatively with endothelium-dependent vasoconstriction reactivity in the late period of shock. The finding that Cx40 AODN significantly improved vasoconstriction reactivity and could be regulated by HIF-1α inhibitor/agonist also demonstrates the negative effect of Cx40 on HIF-1α-regulating vasoconstriction reactivity. A study by Manuel HG et al. investigated the relationship between Cx40 and vascular reactivity in cirrhosis, and the results showed that Cx40 was highly expressed in portal hypertension, which is consistent with our findings in hemorrhagic shock. However, Le Gal L et al. stressed the extrusive role of renin in mouse hypertension linked to loss of Cx40, which seems contradictory to our findings. However, further investigation showed that blood pressure was improved by restoration of Cx40 expression in renin-producing cells, but not in VECs [22].
However, it is worth noting that endothelium-dependent vasoconstriction reactivity is significantly decreased in the beginning of shock, especially in vivo, which seems to be inconsistent with the overall declining trend. We speculate that acute injuries of early shock may stimulate vascular tone and cause this discordant phenomenon. Subsequently, the organisms activate compensatory mechanisms and release protective factors, such as adenosine and HSP70, which may restore vasoconstriction reactivity from 0.5 h to 2 h after shock. The trends of vasoconstriction reactivity after 2 h of shock seem to truly reflect the effect of HIF-1α on endothelium-dependent vasodilatation reactivity in hemorrhagic shock.
The present study also had some limitations. It should be noted that the animal model we used, the rat, may turn out not to analogize well to humans in its response to shock. Second, only an essential MEGJ protein, Cx40, was investigated in the present study. It is not clear whether other Cx molecules participate in the regulation of HIF-1α-induced endothelium-dependent vasodilatation reactivity. Third, the mechanisms by which vasodilatation reactivity significant decreased in the beginning of shock and was restored again in early shock need further investigation. Last, it remains to be determined whether HIF-1α acts on Cx40 directly or indirectly, and whether the specific combination of HIF-1α and Cx40 is important.
In conclusion, this study suggests that HIF-1α plays an important role in the regulation of endothelium-dependent vasodilatation reactivity following hemorrhagic shock. The mechanism may be related to the up-regulation of Cx40 in direct or indirect ways. This finding suggests that the regulation of Cx40 may be a potential therapeutic target for HIF-1α-induced injuries following hemorrhagic shock.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (grant number 81270400, and the Program for Changjiang Scholars and Innovative Research Team in University (IRT1216).
Disclosure of conflict of interest
None.
References
- 1.Liu L, Tian K, Zhu Y, Ding X, Li T. Delta opioid receptor antagonist, ICI 174,864, is suitable for the early treatment of uncontrolled hemorrhagic shock in rats. Anesthesiology. 2013;119:379–388. doi: 10.1097/ALN.0b013e31829b3804. [DOI] [PubMed] [Google Scholar]
- 2.Wu D, Potluri N, Lu J, Kim Y, Rastinejad F. Structural integration in hypoxia-inducible factors. Nature. 2015;524:303–308. doi: 10.1038/nature14883. [DOI] [PubMed] [Google Scholar]
- 3.Semenza GL, Wang GL. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol. 1992;12:5447–5454. doi: 10.1128/mcb.12.12.5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nagaraju GP, Zhu S, Ko JE, Ashritha N, Kandimalla R, Snyder JP, Shoji M, El-Rayes BF. Antiangiogenic effects of a novel synthetic curcumin analogue in pancreatic cancer. Cancer Lett. 2015;357:557–565. doi: 10.1016/j.canlet.2014.12.007. [DOI] [PubMed] [Google Scholar]
- 5.Jiang H, Hu R, Sun L, Chai D, Cao Z, Li Q. Critical role of toll-like receptor 4 in hypoxia-inducible factor 1alpha activation during trauma/hemorrhagic shocky induced acute lung injury after lymph infusion in mice. Shock. 2014;42:271–278. doi: 10.1097/SHK.0000000000000212. [DOI] [PubMed] [Google Scholar]
- 6.Zhang Y, Ming J, Li T, Yang G, Xu J, Chen W, Liu L. regulatory effects of hypoxia-inducible factor 1alpha on vascular reactivity and its mechanisms following hemorrhagic shock in rats. Shock. 2008;30:557–562. doi: 10.1097/SHK.0b013e31816a2136. [DOI] [PubMed] [Google Scholar]
- 7.Liu L, Zhang J, Zhu Y, Xiao X, Peng X, Yang G, Zang J, Liu S, Li T. Beneficial effects of platelet-derived growth factor on hemorrhagic shock in rats and the underlying mechanisms. Am J Physiol Heart Circ Physiol. 2014;307:H1277–1287. doi: 10.1152/ajpheart.00006.2014. [DOI] [PubMed] [Google Scholar]
- 8.Zhang J, Yang GM, Zhu Y, Peng XY, Liu LM, Li T. Bradykinin induces vascular contraction after hemorrhagic shock in rats. J Surg Res. 2015;193:334–343. doi: 10.1016/j.jss.2014.06.033. [DOI] [PubMed] [Google Scholar]
- 9.Morton SK, Chaston DJ, Howitt L, Heisler J, Nicholson BJ, Fairweather S, Broer S, Ashton AW, Matthaei KI, Hill CE. Loss of functional endothelial connexin40 results in exercise-induced hypertension in mice. Hypertension. 2015;65:662–669. doi: 10.1161/HYPERTENSIONAHA.114.04578. [DOI] [PubMed] [Google Scholar]
- 10.Hernandez-Guerra M, Gonzalez-Mendez Y, de Ganzo ZA, Salido E, Garcia-Pagan JC, Abrante B, Malagon AM, Bosch J, Quintero E. Role of gap junctions modulating hepatic vascular tone in cirrhosis. Liver Int. 2014;34:859–868. doi: 10.1111/liv.12446. [DOI] [PubMed] [Google Scholar]
- 11.Li T, Liu L, Xu J, Yang G, Ming J. Changes of Rho kinase activity after hemorrhagic shock and its role in shock-induced biphasic response of vascular reactivity and calcium sensitivity. Shock. 2006;26:504–509. doi: 10.1097/01.shk.0000228796.41044.41. [DOI] [PubMed] [Google Scholar]
- 12.Ming J, Li T, Zhang Y, Xu J, Yang G, Liu L. Regulatory effects of myoendothelial gap junction on vascular reactivity after hemorrhagic shock in rats. Shock. 2009;31:80–86. doi: 10.1097/SHK.0b013e31817d3eF2-11. [DOI] [PubMed] [Google Scholar]
- 13.Duan C, Yang G, Li T, Liu L. Advances in Vascular Hyporeactivity After Shock: The Mechanisms and Managements. Shock. 2015;44:524–534. doi: 10.1097/SHK.0000000000000457. [DOI] [PubMed] [Google Scholar]
- 14.Hu Y, Li T, Tang XF, Chen K, Liu L. Effects of ischemic preconditioning on vascular reactivity and calcium sensitivity after hemorrhagic shock and their relationship to the RhoA-Rho-kinase pathway in rats. J Cardiovasc Pharmacol. 2011;57:231–239. doi: 10.1097/FJC.0b013e318204a910. [DOI] [PubMed] [Google Scholar]
- 15.Xu J, Li T, Yang GM, Liu LM. Protein kinase C isoforms responsible for the regulation of vascular calcium sensitivity and their relationship to integrin-linked kinase pathway after hemorrhagic shock. J Trauma. 2010;69:1274–1281. doi: 10.1097/TA.0b013e3181d74abe. [DOI] [PubMed] [Google Scholar]
- 16.Xu J, Yang G, Li T, Ming J, Liu L. Involvement of Cpi-17 and zipper-interacting protein kinase in the regulation of protein kinase C-alpha, protein kinase C-epsilon on vascular calcium sensitivity after hemorrhagic shock. Shock. 2010;33:49–55. doi: 10.1097/SHK.0b013e3181a76d77. [DOI] [PubMed] [Google Scholar]
- 17.Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 18.Gemel J, Levy AE, Simon AR, Bennett KB, Ai X, Akhter S, Beyer EC. Connexin40 abnormalities and atrial fibrillation in the human heart. J Mol Cell Cardiol. 2014;76:159–168. doi: 10.1016/j.yjmcc.2014.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Climent B, Schubert R, Stankevicius E, Garcia-Sacristan A, Simonsen U, Rivera L. Large conductance Ca2+-activated K+ channels modulate endothelial cell outward currents and nitric oxide release in the intact rat superior mesenteric artery. Biochem Biophys Res Commun. 2012;417:1007–1013. doi: 10.1016/j.bbrc.2011.12.076. [DOI] [PubMed] [Google Scholar]
- 20.Valiunas V, Doronin S, Valiuniene L, Potapova I, Zuckerman J, Walcott B, Robinson RB, Rosen MR, Brink PR, Cohen IS. Human mesenchymal stem cells make cardiac connexins and form functional gap junctions. J Physiol. 2004;555:617–626. doi: 10.1113/jphysiol.2003.058719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de Wit C, Roos F, Bolz SS, Kirchhoff S, Kruger O, Willecke K, Pohl U. Impaired conduction of vasodilation along arterioles in connexin40-deficient mice. Circ Res. 2000;86:649–655. doi: 10.1161/01.res.86.6.649. [DOI] [PubMed] [Google Scholar]
- 22.Le Gal L, Alonso F, Wagner C, Germain S, Nardelli Haefliger D, Meda P, Haefliger JA. Restoration of connexin 40 (Cx40) in Renin-producing cells reduces the hypertension of Cx40 null mice. Hypertension. 2014;63:1198–1204. doi: 10.1161/HYPERTENSIONAHA.113.02976. [DOI] [PubMed] [Google Scholar]