

Abstract
In this study, we aimed to identify potential microRNA (miRNA) regulators of angiotensin-converting enzyme 2 (ACE2) and to explore their roles in lipopolysaccharide (LPS)-induced acute lung injury (ALI). The expression of predicted miRNA regulators of ACE2 was examined in LPS-exposed pulmonary microvascular endothelial cells (PMVECs). Gain- and loss-of-function studies were performed to determine the functions of candidate miRNAs in LPS-induced PMVEC apoptosis and inflammatory response. The roles of the miRNAs in LPS-induced lung inflammation and permeability were investigated in a mouse model. Notably, LPS (1 μg/mL) significantly induced the expression of miR-1246 in PMVECs. ACE2 was validated as a target gene of miR-1246. Silencing of miR-1246 prevented LPS-induced inhibition of ACE2, which was accompanied by reduced apoptosis and production of IL-1β and TNF-α. In contrast, ectopic expression of miR-1246 triggered apoptosis in PMVECs and promoted IL-1β and TNF-α release. MiR-1246-mediated apoptosis of PMVECs was impaired by overexpression of ACE2. Depletion of miR-1246 attenuated lung inflammation, neutrophil infiltration, and vascular permeability and restored pulmonary expression of ACE2 in LPS-exposed mice. Taken together, miR-1246 meditates LPS-induced pulmonary endothelial cell apoptosis in vitro and ALI in mouse models, which are, at least partially, ascribed to repression of ACE2.
Keywords: ACE2, apoptosis, inflammation, microRNA, lung injury, target genes
Introduction
Acute lung injury (ALI) and its severe condition, acute respiratory distress syndrome (ARDS), has a high morbidity and mortality [1,2]. Pathogenic infections are a leading cause of ALI/ARDS [3]. The pulmonary endothelium plays a critical role in lung homeostasis [4]. In infection states such as sepsis and pneumonia, endothelial function and integrity are altered, consequently producing more inflammatory and chemotactic substances and leading to vascular leakage and lung edema [5]. Understanding of the mechanism for the regulation of endothelial responses is important to develop effective therapies for ALI/ARDS.
Angiotensin-converting enzyme (ACE) 2 is a membrane-associated aminopeptidase expressed in multiple lung cell types, such as alveolar epithelial cells and endothelial cells [6]. ACE2 is a homologue of ACE but shows distinct biological activities. While ACE is responsible for converting angiotensin (Ang) I (Ang I) to Ang II, ACE2 contributes to the conversion of Ang II to Ang-(1-7) [7]. Accumulating evidence indicates the implication of ACE2 in the pathogenesis of ALI/ARDS [8-10]. For example, ACE2 overexpression was found to prevent lipopolysaccharide (LPS)-induced ALI in rats [8]. ACE2 also counteracts LPS-induced apoptosis in pulmonary microvascular endothelial cells (PMVECs) [10].
microRNAs (miRNAs) are small, endogenous noncoding RNAs participating in various biological processes such as development, proliferation, survival, inflammation, tumorigenesis, and angiogenesis [11]. They show the ability to repress gene expression by incompletely binding to the 3’-untranslated region (UTR) of target mRNAs [12]. A few of miRNAs, e.g. miR-454 [13] and miR-125b [14] have been identified to reduce the severity of LPS-induced ALI. It has been reported that miR-421 is a negative regulator of ACE2 and overexpression of miR-421 reduced ACE2 expression in primary cardiac myofibroblasts [15]. However, few studies have investigated the miRNA regulators of ACE2 in ALI/ARDS.
Therefore, this study was undertaken to identify and functionally characterize potential miRNA regulators of ACE2, which would be involved in LPS-induced pulmonary endothelial cell death and ALI.
Materials and methods
Cell culture and LPS treatment
Human PMVECs were purchased from ScienCell Research Laboratories, Inc. (Carlsbad, CA, USA) and cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal calf serum (FCS; GE Healthcare HyClone, Logan, UT, USA). Cells were treated with LPS (1 μg/mL; Sigma-Aldrich, St. Louis, MO, USA) for 48 h [16] and examined for gene expression and apoptosis.
Quantitative real-time PCR analysis
Total RNA was isolated from cells using TRIzol Reagent according to the manufacturer’s instructions (Invitrogen, Carlsbad, CA, USA). Reverse transcription was performed using miRNA specific stem-loop primers (Applied Biosystems, Foster City, CA, USA). Mature miRNAs were detected by the TaqMan MicroRNA Assay, according to the manufacturer’s instructions (Applied Biosystems). U6 small nuclear RNA was used as an internal control.
Cell transfection
miR-1246 mimic, anti-miR-1246, and corresponding negative controls were obtained from Ambion (Austin, TX, USA). If not stated otherwise, these nucleotide molecules were used at a final concentration of 50 nM. PMVECs at ~80% confluence were tramsfected for 24 h using the RNAiMAX Transfection Reagent (Invitrogen) before LPS exposure. In rescue experiments, PMVECs were co-transfected with a miRNA-resistant variant of ACE2 (1 μg) and miR-1246 mimic (50 nM) prior to the treatment with LPS. The ACE2-expressing plasmid was generated by cloning human ACE2 cDNA lacking the 3’-UTR (Origene, Rockville, MD, USA) into pcDNA3.1(+) vector.
Luciferase reporter assay
Full-length ACE2 3’-UTR was amplified by PCR and cloned into the pMIR-REPORT Luciferase miRNA Expression Reporter Vector (ThermoFisher Scientific, Waltham, MA, USA). Site mutations were generated by PCR using the QuikChange site-directed mutagenesis kit (Stratagen, Santa Clara, CA, USA). For luciferase reporter assay, HEK-293T cells were seeded onto 24-well plates at a density of 3 × 104 cells per well and transfected with wild-type or mutated ACE2 3’-UTR reporter constructs (0.2 μg), miR-1246 mimic or control miRNA (50 nM), and the Renilla luciferase reporter plasmid pRL-TK (10 ng; Promega, Madison, WI, USA) using Lipofectamine 2000 reagent (Invitrogen). At 24 h after transfection, cells were lysed and luciferase activities were measured using the Dual Luciferase Reporter Assay System (Promega). The firefly luciferase activity was normalized to the Renilla luciferase activity.
Apoptosis analysis
Apoptosis was analyzed using a terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) assay kit following the manufacturer’s instructions (Roche Applied Science, Indianapolis, IN, USA). In brief, cells were fixed, permeabilized, and incubated with fluorescein isothiocyanate (FITC)-labeled dUTP and terminal deoxynucleotidyltransferase for 1 h at 37°C. Nuclei counterstaining was performed using DAPI (Molecular Probes, Eugene, OR, USA). Stained cells were examined under a fluorescence microscope and the percentage of TUNEL-positive cells was determined.
Animal experiments
A total of 32 C57BL/6 mice (5-6 week old; Shanghai Laboratory Animal Center, Shanghai, China) were used in this study and randomly assigned to the following 4 groups (n = 8 for each): control inhibitor + PBS, control inhibitor + LPS, anti-miR-1246 + PBS, and anti-miR-1246 + LPS. The mice were anesthetized and intratracheally administered with control inhibitor or anti-miR-1246 (2 mg/kg) [17]. Twenty-four hours later, LPS from Escherichia coli (Sigma-Aldrich; 1 mg/kg) or PBS was instilled intratracheally. After another 24 h, animals were sacrificed and bronchoalveolar lavage (BAL) fluid was collected. The trachea was cannulated with a 18-gauge polypropylene catheter and the lung was lavaged with 1 ml of PBS. BAL fluid was centrifuged at 1500 g for 10 min. The pellet was re-suspended in PBS and total cells were counted with a hemacytometer. For differential cell counting, cytospin slides were prepared and stained with DiffQuick (Baxter Scientific, Deerfield, IL, USA). The BALF supernatant was collected and examined for the levels of total protein and inflammatory cytokines. Protein quantification was achieved using the BCA Protein Assay Reagent kit (ThermoFisher Scientific). For assessment of lung injury, sections of lung tissues were stained with hematoxylin and eosin (H&E) and examined under a microscope. The degree of lung injury was semi-quantitatively scored in a blinded manner, as described previously [8]. All experiments involving animals were performed as per the approval by the Animal Care and Use Committee of Shanghai Jiaotong University (Shanghai, China).
Measurement of inflammatory cytokines by enzyme-linked immunosorbent assay (ELISA)
The levels of interleukin (IL)-1β and tumor necrosis factor alpha (TNF-α) in conditioned medium of PMVECs or BAL fluids were analyzed with corresponding ELISA kits (eBioscience, San Diego, CA, USA) according to the manufacturer’s instructions.
Evaluation of lung permeability
Lung permeability was assessed using the Evans blue dye extravasation method, as described previously [18]. In brief, Evans blue dye (Sigma-Aldrich; 30 mg/kg) was injected into the external jugular vein 1 h before euthanasia. The lung was perfused with PBS and Evans blue dye was extracted from the lung using formamide for 18 h at 60°C. Absorbance was measured at 620 nm. Results are normalized to the dry weight of the lung.
Myeloperoxidase activity assay
Lung tissues were homogenized and the supernatant was subjected to myeloperoxidase activity assay using a commercially available kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China). Absorbance was determined at 460 nm.
Western blot analysis
Protein samples from cells were prepared in radioimmune precipitation buffer containing 1 μg/L aprotinin, 10 μg/L leupeptin, and 1 mM phenylmethylsulfonyl fluoride (Sigma-Aldrich) and separated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis. The primary antibodies were as follows: rabbit anti-ACE2 polyclonal antibody (1:300 dilution; Abcam, London, UK), rabbit anti-Bcl-2 monoclonal antibody (1:200 dilution; Abcam), rabbit anti-Bcl-xL polyclonal antibody (1:500 dilution; Abcam), rabbit anti-Bax polyclonal antibody (1:300 dilution; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA), and rabbit anti-β-actin polyclonal antibody (1:1000 dilution; Santa Cruz Biotechnology, Inc.). After incubating with horseradish peroxidase-conjugated goat anti-rabbit IgG, protein bands were detected using an enhanced chemiluminescence detection kit (ThermoFisher Scientific). Quantification of the bands was performed using Quantity One software (Bio-Rad, Hercules, CA, USA).
Statistical analysis
Data are presented as the means ± standard deviation (SD). Statistical differences were analyzed using the Student’s t test or one-way analysis of variance (ANOVA) with the Tukey’s test. P values < 0.05 were considered significant.
Results
LPS induces miR-1246 to target ACE2 expression in PMVECs
To identify the miRNA mediator involved in LPS-induced suppression of ACE2 in PMVECs, we examined the effect of LPS treatment on the expression of a set of predicted miRNA regulators based on an online prediction software miRDB (http://mirdb.org/miRDB/). As shown in Figure 1A, there was a 3.6-fold increase in miR-1246 in PMVECs after treatment with LPS (1 μg/mL). However, the expression of miR-421, miR-574, or miR-936 was not altered upon LPS exposure (data not shown). To validate the downregulation of ACE2 by miR-1246, PMVECs were transfected with miR-1246 mimic and measured for expression of ACE2 protein. The results showed that delivery of miR-1246 mimic reduced the protein expression of ACE2 by about 70%, compared with control miRNA-transfected cells (Figure 1B). As expected, delivery of miR-1246 mimic had no significant impact on the expression of miR-574 and miR-936 in PMVECs. Luciferase reporter assays further demonstrated that miR-1246 repressed the expression of the luciferase reporter containing the wild-type but not mutated ACE2 3’-UTR (Figure 1C), indicating that miR-1246 directly targets the ACE2 3’-UTR. To confirm the hypothesis that LPS may suppress the expression of ACE2 via induction of miR-1246 expression, PMVECs were pretransfected with anti-miR-1246 inhibitors and then exposed to LPS. Western blot analysis revealed that LPS significantly inhibited ACE2 expression, which was almost completely blocked by delivery of anti-miR-1246 inhibitors (Figure 1D). Taken together, miR-1246 mediates the negative effect of LPS on ACE2 expression in PMVECs.
Figure 1.
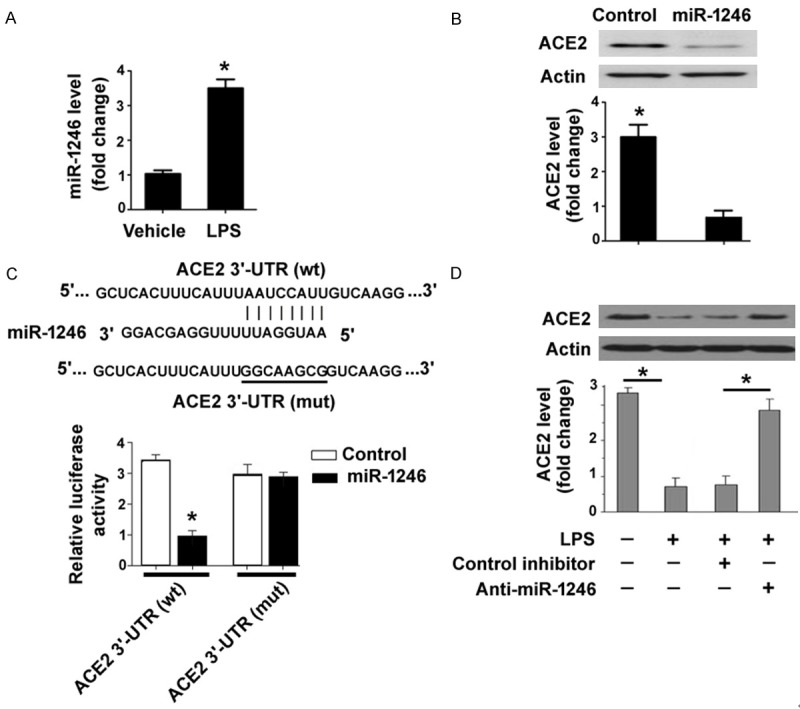
LPS induces miR-1246 to target ACE2 expression in PMVECs. A: qRT-PCR analysis of miR-1246 expression in PMVECs treated with LPS or vehicle control. *P < 0.05 vs. vehicle. B: Western blot analysis of ACE2 protein in PMVECs transfected with miR-1246 mimic or negative control miRNA. Bar graphs (bottom) show densitometric analysis of ACE2 protein. *P < 0.05 vs. vehicle. C: Luciferase reporter assay showed that miR-1246 repressed the expression of the luciferase reporter containing the wild-type (wt) but not mutated (mut) ACE2 3’-UTR. The predicted target site for miR-1246 in the ACE2 3’-UTR is shown in top panels. *P < 0.05 vs. vehicle. D: Western blot analysis of ACE2 protein in PMVECs pretransfected with anti-miR-1246 or control inhibitors before exposure to LPS. *P < 0.05.
miR-1246 is involved in LPS-triggered apoptosis and inflammatory response in PMVECs
We next explored whether miR-1246 is involved in the pro-apoptotic and pro-inflammatory activity of LPS. To this end, miR-1246 knockdown experiments were done before exposure to LPS. As shown in Figure 2A, LPS significantly caused apoptotic death in PMVECs, compared with the vehicle control (26.2 ± 2.5% vs. 4.8 ± 1.1%, P < 0.05). Pretransfection with anti-miR-1246 inhibitors led to ~45% inhibition of LPS-induced apoptosis. Moreover, delivery of anti-miR-1246 inhibitors reduced the production of IL-1β and TNF-α by about 42% and 60%, respectively, in LPS-treated PMVECs (Figure 2B). To further confirm the regulatory effects of miR-1246 on PMVECs, we performed gain-of-function studies. We found that transfection with miR-1246 mimic resulted in a 3-fold increase in the percentage of apoptosis in PMVECs, compared with control miRNA-transfected cells (Figure 2C). In addition, enforced expression of miR-1246 significantly promoted the secretion of IL-1β and TNF-α from PMVECs (Figure 2D). Overall, these data suggest that LPS-induced apoptotic and inflammatory responses in PMVECs involve elevation of miR-1246 expression.
Figure 2.
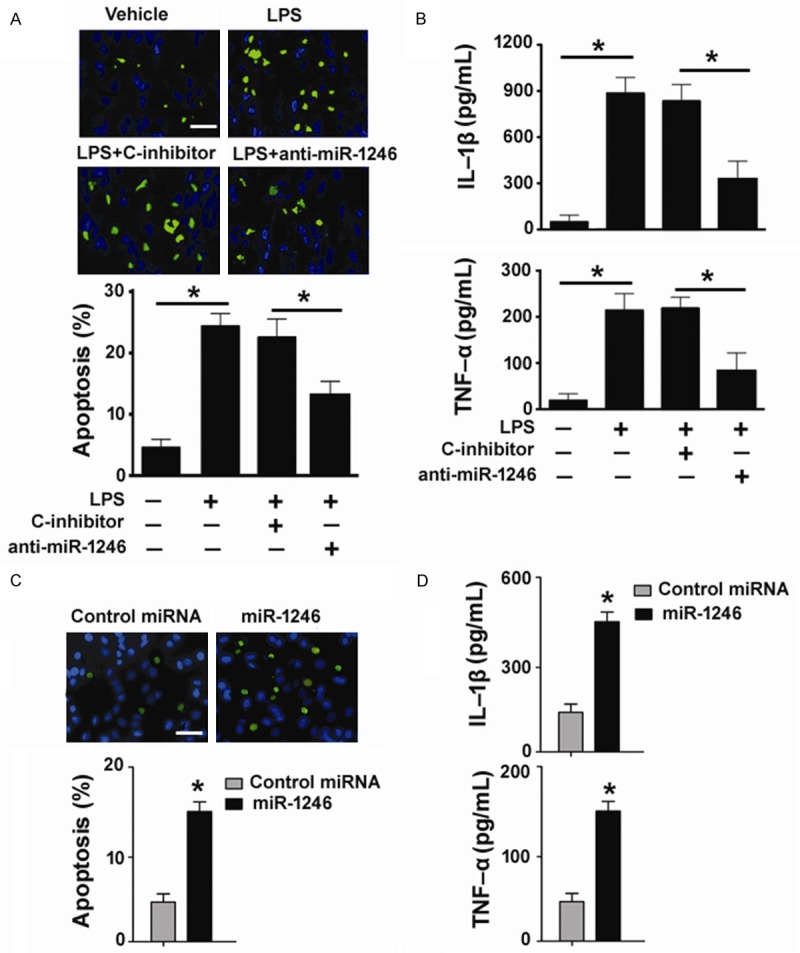
miR-1246 is involved in LPS-triggered apoptosis and inflammatory response in PMVECs. A: TUNEL staining of PMVECs pretransfected with anti-miR-1246 or control inhibitors (C-inhibitor) before exposure to LPS. Scale bar, 50 μm. *P < 0.05. B: ELISA analysis of IL-1β and TNF-α levels in conditioned medium from PMVECs with indicated treatments. *P < 0.05. C: TUNEL staining of PMVECs transfected with miR-1246 mimic or negative control miRNA. Scale bar, 50 μm. D: Measurement of IL-1β and TNF-α levels in conditioned medium from PMVECs transfected with miR-1246 mimic or negative control miRNA. *P < 0.05 vs. control.
miR-1246 promotes PMVEC apoptosis by repressing ACE2 expression
Next, we checked if the pro-apoptotic activity of miR-1246 in PMVECs is associated with targeting of ACE2. To address this issue, we performed rescue experiments with a miRNA-resistant variant of ACE2. Co-transfection with the ACE2-expressing plasmid restored the expression of ACE2 protein in miR-1246 mimic-transfected PMVECs (Figure 3A). Interestingly, enforced expression of ACE2 impaired apoptotic response induced by miR-1246 mimic in PMVECs (Figure 3B). Measurement of apoptosis-related proteins further demonstrated that miR-1246 mimic decreased the expression of anti-apoptotic proteins Bcl-2 and Bcl-xL and increased the expression of pro-apoptotic protein Bax (Figure 3C). However, such molecular changes were prevented by overexpression of ACE2.
Figure 3.
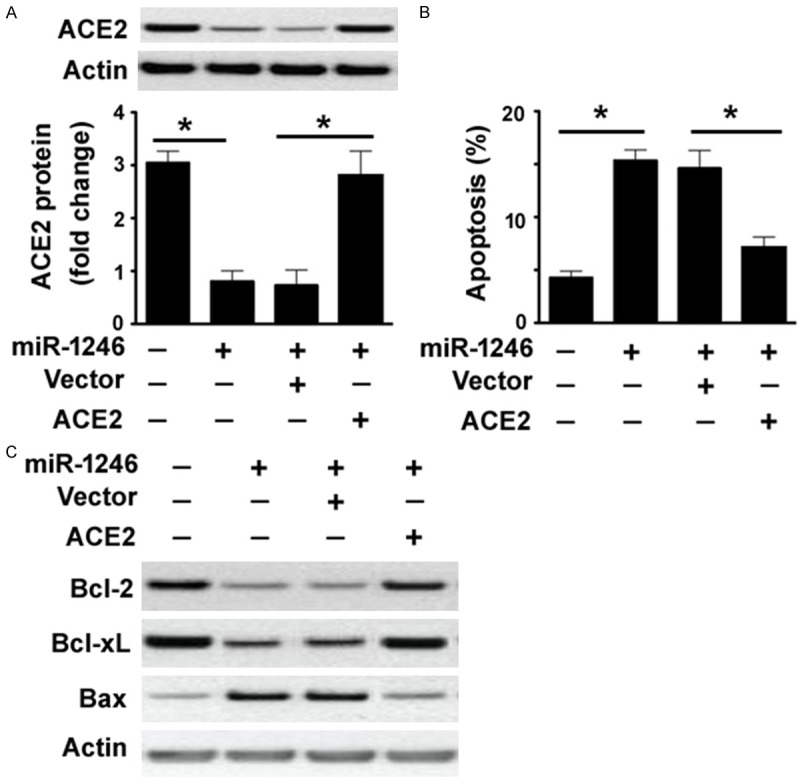
miR-1246 promotes PMVEC apoptosis by repressing ACE2 expression. A: Western blot analysis of ACE2 protein in PMVECs transfected with indicated constructs. B: Detection of apoptosis by TUNEL staining in PMVECs transfected with indicated constructs. *P < 0.05. C: Western blot analysis of Bcl-2, Bcl-xL, and Bax proteins in PMVECs transfected with indicated constructs.
Inhibition of miR-1246 attenuates LPS-induced lung inflammation
Having identified that miR-1246 is involved in LPS-induced endothelial cell apoptosis, we next sought to determine the effect of silencing of miR-1246 on LPS-induced lung injury. H&E staining of lung sections from mice injected with anti-miR-1246 inhibitors revealed a marked reduction of infiltrating inflammatory cells in response to LPS (Figure 4A). Lung injury scores were significantly lower in the LPS + anti-miR-1246 group than in the LPS + control inhibitor group (Figure 4B). However, administration of anti-miR-1246 inhibitors alone did not alter lung injury scores, compared with the control group. Myeloperoxidase activity is commonly used as a quantitative parameter for neutrophil infiltration into lung tissues (18). We found that the myeloperoxidase activity in the lung from LPS-exposed mice was significantly suppressed by anti-miR-1246 inhibitors (Figure 4C). To confirm that these lung pathological changes are due to inhibition of miR-1246, we examined the expression of miR-1246 in the lung. As expected, administration of anti-miR-1246 inhibitors restored the expression of ACE2 in the lung (Figure 4D). Taken together, these data suggest that miR-1246 is required for LPS-induced lung inflammation.
Figure 4.
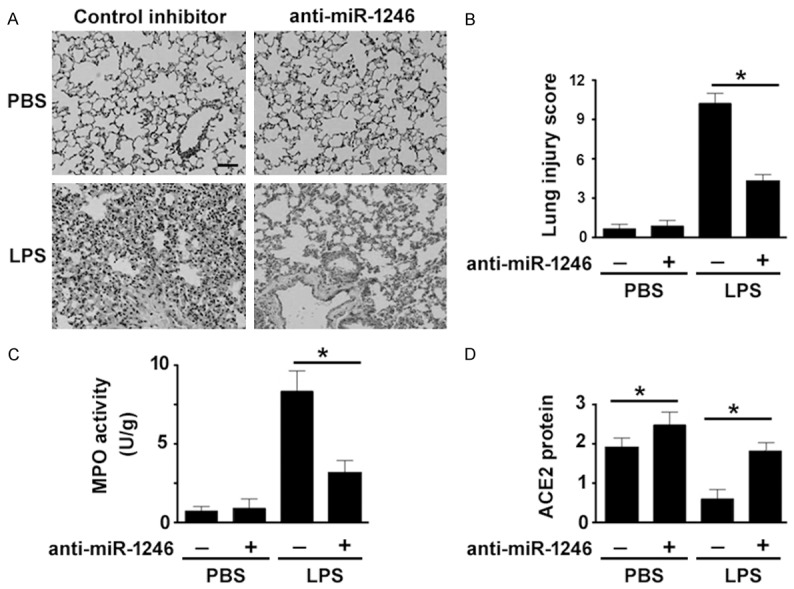
Inhibition of miR-1246 attenuates LPS-induced lung inflammation. Mice were intratracheally administered with control inhibitor or anti-miR-1246 (2 mg/kg) 24 h before exposure to LPS or PBS. A: Lung sections were stained with H&E. B: Lung injury scores were determined. C: Measurement of myeloperoxidase (MPO) activity in the lung. D: Western blot analysis of ACE2 protein in the lung. *P < 0.05.
Silencing of miR-1246 reduces LPS-induced lung permeability
We also investigated the effect of silencing of miR-1246 on lung vascular permeability. As determined by Evans blue dye extravasation (Figure 5A), LPS exposure resulted in a 3.2-fold increase in lung permeability, relative to the control group (P < 0.05). However, such increase was significantly blocked by depletion of miR-1246. Moreover, miR-1246 knockdown impaired the LPS-induced increase in total and neutrophil counts (Figure 5B), inflammatory cytokines (Figure 5C), and protein levels (Figure 5D) in the BAL fluid. Taken together, these observations underscore the involvement of miR-1246 in LPS-induced lung injury.
Figure 5.
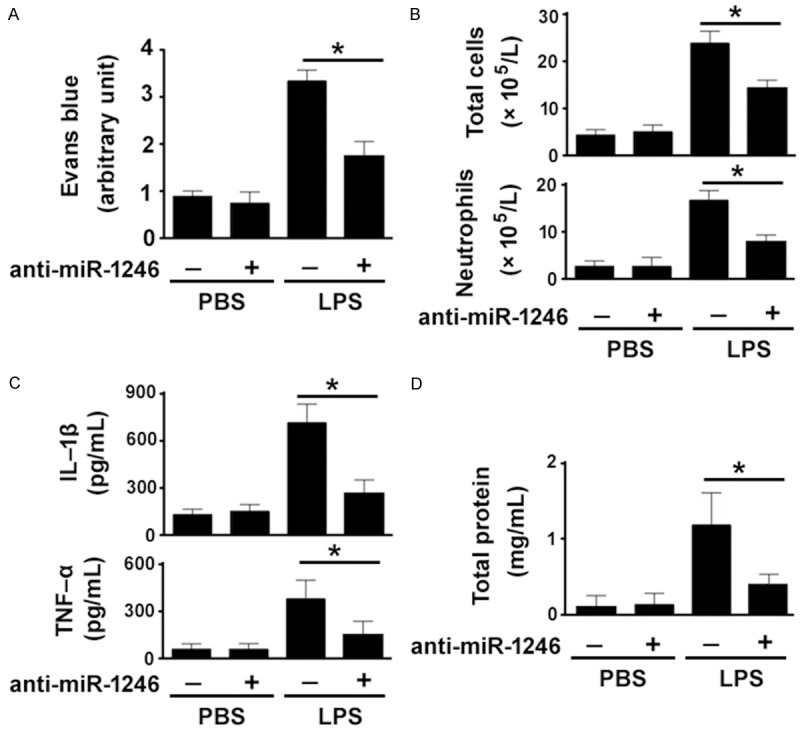
Silencing of miR-1246 reduces LPS-induced lung permeability. Mice were intratracheally administered with control inhibitor or anti-miR-1246 (2 mg/kg) 24 h before exposure to LPS or PBS. A: Evans blue dye extravasation assay revealed that LPS-induced lung permeability was significantly blocked by depletion of miR-1246. B: Total cells and neutrophils in BAL fluids were counted. C: TNF-α and IL-1β levels in BAL fluids were quantified by ELISA. D: Measurement of total protein levels in BAL fluids. *P < 0.05.
Discussion
ACE2 has been shown to affect multiple aspects of the pathogenesis of ALI/ARDS [8,10,19]. It has been documented that ACE2 can protect pulmonary endothelial cells from LPS- [10] or bleomycin-induced apoptosis [20]. Therefore, dysregulation of ACE2 may be an important mechanism contributing to ALI/ARDS. In this study, we focused on the miRNA regulators of ACE2. Our data showed that miR-1246 had the ability to repress ACE2 expression by binding to its 3’-UTR. Most interestingly, miR-1246 expression was elevated in PMVECs in response to LPS. A previous study has reported that LPS can suppress ACE2 expression in PMVECs [10]. Therefore, it is possible that LPS may inhibit ACE2 expression via induction of miR-1246 in PMVECs. This hypothesis was supported by the finding that delivery of anti-miR-1246 inhibitors almost completely reversed the suppression of ACE2 expression by LPS.
Although a few of miRNAs, e.g. miR-454 [13] and miR-125b [14] have been found to be implicated in ALI/ARDS, a large number of miRNAs are less characterized in this disease. In this study, we showed that silencing of miR-1246 significantly prevented LPS-induced apoptosis and production of inflammatory cytokines in PMVECs. Moreover, ectopic expression of miR-1246 phenocopied the effects of LPS treatment on PMVECs, leading to apoptotic death and increased secretion of inflammatory cytokines. These observations indicate that miR-1246 is responsible for LPS-induced endothelial cell inflammation and death, suggesting its potential implication in the pathogenesis of ALI/ARDS.
Similar to our findings in PMVECs, miR-1246 was reported to cause apoptotic death in HaCaT keratinocytes [21] and hepatocellular carcinoma cells [22]. Different target genes were suggested to mediate the pro-apoptotic activity of miR-1246. While inhibition of PTKN2 is responsible for miR-1246-mediated apoptosis in HaCaT keratinocytes [21], this miRNA triggers apoptotic response in hepatocellular carcinoma cells by targeting nuclear factor I/B [22]. In this study, we found that restoration of ACE2 attenuated apoptotic response in miR-1246-transfected PMVECs. Moreover, ACE2 overexpression reversed miR-1246-induced suppression of Bcl-2 and Bcl-xL expression and enhancement of Bax expression. These data provide evidence that targeting ACE2 is causally linked to miR-1246-induced apoptosis in PMVECs.
Functional alteration of pulmonary endothelium is critical role in the development of ALI/ARDS [4]. Given the findings in PMVECs, we hypothesized that miR-1246 might contribute to LPS-induced ALI. In line with this hypothesis, we found that delivery of anti-miR-1246 inhibitors hampered inflammatory cell infiltration to the lung and alleviated lung injury in LPS-exposed mice. Accumulation of activated neutrophils is a hallmark of ALI, which contributes to elevated production of inflammatory cytokines [23]. Recruitment of neutrophils in response to LPS has been reported to increase microvascular permeability during ALI [24]. Notably, we found that myeloperoxidase activity, an index of neutrophil activation was significantly reduced by administration of anti-miR-1246 inhibitors in LPS-exposed mice. Moreover, depletion of miR-1246 reduced LPS-induced lung permeability and production of inflammatory mediators in BAL fluids. At the molecular level, we confirmed that delivery of anti-miR-1246 inhibitors restored the expression of ACE2 in the lung from LPS-exposed mice. ACE2 has the ability to counteract pathological inflammatory responses [25,26]. ACE2 deficiency was found to increase neutrophil, macrophage, and T cell infiltration in the kidney and pro-inflammatory cytokine production in a mouse model of acute kidney injury [26]. Another study reported that ACE2 can attenuate vascular permeability during ALI [27]. These studies suggest that anti-miR-1246-mediated protection against LPS-induced inflammation and permeability is ascribed, at least partially, to induction of ACE2.
This work shows that miR-1246 mediates LPS-induced pulmonary endothelial dysfunction and apoptosis in vitro and lung inflammation and permeability in vivo by targeting ACE2. This miRNA may represent a potential therapeutic target for ALI/ARDS.
Disclosure of conflict of interest
None.
References
- 1.de la Vega MR, Dodson M, Gross C, Manzour H, Lantz RC, Chapman E, Wang T, Black SM, Garcia JG, Zhang DD. Role of Nrf2 and Autophagy in Acute Lung Injury. Curr Pharmacol Rep. 2016;2:91–101. doi: 10.1007/s40495-016-0053-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yadam S, Bihler E, Balaan M. Acute respiratory distress syndrome. Crit Care Nurs Q. 2016;39:190–195. doi: 10.1097/CNQ.0000000000000111. [DOI] [PubMed] [Google Scholar]
- 3.Fujishima S, Gando S, Daizoh S, Kushimoto S, Ogura H, Mayumi T, Takuma K, Kotani J, Yamashita N, Tsuruta R, Takeyama N, Shiraishi S, Araki T, Suzuki K, Ikeda H, Miki Y, Suzuki Y, Yamaguchi Y, Aikawa N Japanese Association for Acute Medicine Sepsis Registry (JAAM SR) Study Group. Infection site is predictive of outcome in acute lung injury associated with severe sepsis and septic shock. Respirology. 2016;21:898–904. doi: 10.1111/resp.12769. [DOI] [PubMed] [Google Scholar]
- 4.Millar FR, Summers C, Griffiths MJ, Toshner MR, Proudfoot AG. The pulmonary endothelium in acute respiratory distress syndrome: insights and therapeutic opportunities. Thorax. 2016;71:462–473. doi: 10.1136/thoraxjnl-2015-207461. [DOI] [PubMed] [Google Scholar]
- 5.Maniatis NA, Orfanos SE. The endothelium in acute lung injury/acute respiratory distress syndrome. Curr Opin Crit Care. 2008;14:22–30. doi: 10.1097/MCC.0b013e3282f269b9. [DOI] [PubMed] [Google Scholar]
- 6.Hamming I, Timens W, Bulthuis ML, Lely AT, Navis G, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. 2004;203:631–637. doi: 10.1002/path.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Santos RA, Ferreira AJ, Verano-Braga T, Bader M. Angiotensin-converting enzyme 2, angiotensin-(1-7) and Mas: new players of the renin-angiotensin system. J Endocrinol. 2013;216:R1–R17. doi: 10.1530/JOE-12-0341. [DOI] [PubMed] [Google Scholar]
- 8.Li Y, Zeng Z, Cao Y, Liu Y, Ping F, Liang M, Xue Y, Xi C, Zhou M, Jiang W. Angiotensin-converting enzyme 2 prevents lipopolysaccharide-induced rat acute lung injury via suppressing the ERK1/2 and NF-κB signaling pathways. Sci Rep. 2016;6:27911. doi: 10.1038/srep27911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang L, Li Y, Qin H, Xing D, Su J, Hu Z. Crosstalk between ACE2 and PLGF regulates vascular permeability during acute lung injury. Am J Transl Res. 2016;8:1246–1252. [PMC free article] [PubMed] [Google Scholar]
- 10.Li Y, Cao Y, Zeng Z, Liang M, Xue Y, Xi C, Zhou M, Jiang W. Angiotensin-converting enzyme 2/angiotensin-(1-7)/Mas axis prevents lipopolysaccharide-induced apoptosis of pulmonary microvascular endothelial cells by inhibiting JNK/NF-κB pathways. Sci Rep. 2015;5:8209. doi: 10.1038/srep08209. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 11.Paladini L, Fabris L, Bottai G, Raschioni C, Calin GA, Santarpia L. Targeting microRNAs as key modulators of tumor immune response. J Exp Clin Cancer Res. 2016;35:103. doi: 10.1186/s13046-016-0375-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rajasekaran S, Pattarayan D, Rajaguru P, Sudhakar Gandhi PS, Thimmulappa RK. MicroRNA regulation of acute lung injury and acute respiratory distress syndrome. J Cell Physiol. 2016;231:2097–2106. doi: 10.1002/jcp.25316. [DOI] [PubMed] [Google Scholar]
- 13.Tao Z, Yuan Y, Liao Q. Alleviation of Lipopolysaccharides-Induced Acute Lung Injury by MiR-454. Cell Physiol Biochem. 2016;38:65–74. doi: 10.1159/000438609. [DOI] [PubMed] [Google Scholar]
- 14.Guo Z, Gu Y, Wang C, Zhang J, Shan S, Gu X, Wang K, Han Y, Ren T. Enforced expression of miR-125b attenuates LPS-induced acute lung injury. Immunol Lett. 2014;162:18–26. doi: 10.1016/j.imlet.2014.06.008. [DOI] [PubMed] [Google Scholar]
- 15.Lambert DW, Lambert LA, Clarke NE, Hooper NM, Porter KE, Turner AJ. Angiotensin-converting enzyme 2 is subject to post-transcriptional regulation by miR-421. Clin Sci (Lond) 2014;127:243–249. doi: 10.1042/CS20130420. [DOI] [PubMed] [Google Scholar]
- 16.Sampath V, Radish AC, Eis AL, Broniowska K, Hogg N, Konduri GG. Attenuation of lipopolysaccharide-induced oxidative stress and apoptosis in fetal pulmonary artery endothelial cells by hypoxia. Free Radic Biol Med. 2009;46:663–671. doi: 10.1016/j.freeradbiomed.2008.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ying H, Kang Y, Zhang H, Zhao D, Xia J, Lu Z, Wang H, Xu F, Shi L. MiR-127 modulates macrophage polarization and promotes lung inflammation and injury by activating the JNK pathway. J Immunol. 2015;194:1239–1251. doi: 10.4049/jimmunol.1402088. [DOI] [PubMed] [Google Scholar]
- 18.Duan Y, Learoyd J, Meliton AY, Leff AR, Zhu X. Inhibition of Pyk2 blocks lung inflammation and injury in a mouse model of acute lung injury. Respir Res. 2012;13:4. doi: 10.1186/1465-9921-13-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Imai Y, Kuba K, Rao S, Huan Y, Guo F, Guan B, Yang P, Sarao R, Wada T, Leong-Poi H, Crackower MA, Fukamizu A, Hui CC, Hein L, Uhlig S, Slutsky AS, Jiang C, Penninger JM. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature. 2005;436:112–116. doi: 10.1038/nature03712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bao H, Gao F, Xie G, Liu Z. Angiotensin-converting enzyme 2 inhibits apoptosis of pulmonary endothelial cells during acute lung injury through suppressing MiR-4262. Cell Physiol Biochem. 2015;37:759–767. doi: 10.1159/000430393. [DOI] [PubMed] [Google Scholar]
- 21.Li W, Wu YF, Xu RH, Lu H, Hu C, Qian H. miR-1246 releases RTKN2-dependent resistance to UVB-induced apoptosis in HaCaT cells. Mol Cell Biochem. 2014;394:299–306. doi: 10.1007/s11010-014-2108-1. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Q, Cao LY, Cheng SJ, Zhang AM, Jin XS, Li Y. p53-induced microRNA-1246 inhibits the cell growth of human hepatocellular carcinoma cells by targeting NFIB. Oncol Rep. 2015;33:1335–1341. doi: 10.3892/or.2015.3715. [DOI] [PubMed] [Google Scholar]
- 23.Abraham E. Neutrophils and acute lung injury. Crit Care Med. 2003;31:S195–S199. doi: 10.1097/01.CCM.0000057843.47705.E8. [DOI] [PubMed] [Google Scholar]
- 24.Chignard M, Balloy V. Neutrophil recruitment and increased permeability during acute lung injury induced by lipopolysaccharide. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1083–L1090. doi: 10.1152/ajplung.2000.279.6.L1083. [DOI] [PubMed] [Google Scholar]
- 25.Xue T, Wei N, Xin Z, Qingyu X. Angiotensin-converting enzyme-2 overexpression attenuates inflammation in rat model of chronic obstructive pulmonary disease. Inhal Toxicol. 2014;26:14–22. doi: 10.3109/08958378.2013.850563. [DOI] [PubMed] [Google Scholar]
- 26.Fang F, Liu GC, Zhou X, Yang S, Reich HN, Williams V, Hu A, Pan J, Konvalinka A, Oudit GY, Scholey JW, John R. Loss of ACE2 exacerbates murine renal ischemia-reperfusion injury. PLoS One. 2013;8:e71433. doi: 10.1371/journal.pone.0071433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu X, Lin Q, Qin X, Ruan Z, Zhou J, Lin Z, Su Y, Zheng J, Liu Z. ACE2 antagonizes VEGFa to reduce vascular permeability during acute lung injury. Cell Physiol Biochem. 2016;38:1055–1062. doi: 10.1159/000443056. [DOI] [PubMed] [Google Scholar]