

Abstract
The aim of this study is to examine the role of thioredoxin-2 (Trx2) in autophagy and apoptosis during myocardial ischemia-reperfusion (I/R) injury in vitro. We employed the oxygen-glucose deprivation and reperfusion (OGD/R) model of H9c2 cells and used lentiviral infection to overexpress Trx2. H9c2 cell viability and injury assays were conducted using a Cell Counting Kit-8 (CCK-8) and alactate dehydrogenase (LDH) kit. The effects of Trx2 on autophagy and apoptosis were measured by transmission electron microscopy (TEM), western blot, and flow cytometry. Our results showed that the expression of Trx2 was significantly decreased at reperfusion 6 h after OGD 12 h treatment. Trx2 overexpression inhibited autophagy in H9c2 cells subjected to OGD/R. As the underlying mechanisms, both Akt kinase/the mammalian target of rapamycin (Akt/mTOR) and AMP-activated protein kinase (AMPK)/mTOR signaling pathways were involved in the regulation of Trx2 during autophagy, which was also mediated by reactive oxygen species (ROS). 3-methyladenine (3-MA), an inhibitor of autophagy, not only suppressed OGD/R-induced autophagy but also decreased apoptosis. As a classical autophagy sensitizer, rapamycin (Rapa) augmented autophagy as well as apoptosis. Additionally, we further demonstrated that Trx2 could alleviate OGD/R-induced apoptosis via mitochondrion-mediated intrinsic apoptotic pathway. In summary, our data indicated that Trx2 protects cardiomyocytes under OGD/R by inhibiting autophagy and apoptosis. Trx2 may be a crucial regulatory protein during I/R-induced cardiomyocyte injury and death.
Keywords: Trx2, autophagy, apoptosis, oxygen-glucose deprivation and reperfusion
Introduction
Ischemic heart disease is a major cause of death worldwide [1]. Medical and surgical reperfusion are the main therapeutic strategies used to treat ischemic heart disease [2]. However, accumulating evidence demonstrates that myocardial ischemia-reperfusion (I/R) injury can be induced during the recovery of blood flow to the ischemic myocardium [3,4]. Therefore, pivotal strategies should be considered to prevent and reduce myocardial I/R injury.
There are multiple biological processes and cell signaling pathways involved in myocardial I/R injury. Emerging studies suggest that autophagy is implicated in myocardial I/R and that the results of myocardial I/R injury are affected by regulating autophagy [5-7]. As a conserved and catabolic lysosomal degradation pathway, autophagy is a dynamic process that degrades and recycles old or damaged proteins and organelles and serves a crucial function under both physiological and pathological circumstances by maintaining cellular homoeostasis [7,8]. Moderate autophagy may be protective for cell survival as an adaptive response of myocardial cells, while excessive autophagy may result in the acceleration of cell death. Some investigators have shown that up-regulation of autophagy helps protect myocardial cells by advancing protein decomposition and organelle turnover during nutrient deficiencies, such as hypoxia or ischemia [9,10]. However, most lines of evidence illustrate that autophagy is further induced and detrimental during myocardial reperfusion injury [5,6,11].
Thioredoxins are small proteins that are crucial for maintaining the balance of cellular redox and are involved in the control of oxidative stress and cell death [12]. Thioredoxin-2 (Trx2) is a mitochondrial form of the thioredoxin superfamily and is highly expressed in heart and other tissues of high metabolic demand, such as the brain and liver [13]. Myocardial mitochondria are considered a major site for reactive oxygen species (ROS) production [14]. Trx2 can directly catalyze mitochondrial thiol-disulfide exchanges and scavenge cellular ROS [15,16]. Previous studies have demonstrated that Trx2 is critical for maintaining cardiac function by suppressing ROS production, preserving mitochondrial integrity, and preventing apoptosis [13,16]. However, the relationship between Trx2 and autophagy remains uncertain.
In this study, we used the oxygen-glucose deprivation and reperfusion (OGD/R) model of the H9c2 culture line to imitate myocardial I/R conditions and investigated whether the protective effects of Trx2 are involved in inhibiting autophagy and apoptosis to alleviate myocardial I/R injury in vitro.
Materials and methods
Cell culture and the induction of OGD/R
The H9c2 cardiomyocyte cell line (rat embryonic ventricular myocytes) was purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). The H9c2 cells were maintained in DMEM high-glucose medium (HyClone, Beijing, China, Cat. No. SH30243.01FS) and supplemented with 10% fetal bovineserum (FBS, Gibco, Carlsbad, CA, USA, Cat. No. 10099141) and 1% penicillin-streptomycin (Gibco, Cat. No. C0222) in a humidified atmosphere containing 5% CO2 at 37°C. For the OGD/R experiments, OGD was conducted by incubating the cells in glucose-free DMEM medium (Gibco, Cat. No. 11966025) within an anaerobic chamber (1% O2, 5% CO2) at 37°C for 12 h to imitate ischemic injury. Then the cells were incubated with normal culture medium under normoxic conditions (95% air, 5% CO2) for 3, 6, 12, and 24 h at 37°C (reperfusion, R). The glucose and oxygen conditions described approximates the reperfusion process in vivo. Normoxic control cells were treated identically without OGD/R.
H9c2 viability assay and lactate dehydrogenase (LDH) secretion
To evaluate the OGD/R model, the H9c2 cells were seeded in 96-well plates, and cell viability was assayed by Cell Counting Kit-8 (CCK-8) (DOJINDO, Kumamoto, Japan, Cat. No. AY-4710P) after OGD/R treatments. To assay the degree of cell injury induced by OGD/R protocol, LDH released into culture media was measured using a LDH Activity Assay kit (Beyotime Institute of Biotechnology, Shanghai, China, Cat. No. C0016). At the reperfusion time points indicated previously, cell viability and injury were assayed according to the manufacturer’s instructions.
Lentiviral overexpression of Trx2 (Lv-Trx2) and Lv-Trx2 H9c2 cell line construction
The virus packaging system contains three plasmid vectors. EcoRI and BamHI are insertion sites. The pLenO-CMV-MCS-EF1α-Puro vector contains a CMV promoter to initiate the expression of target genes and a puromycin resistance gene to screen the cell clones expressing the target gene. Briefly, Lv-Trx2 was produced by three-plasmid co-transfection of 293T cells. The viruses were concentrated by ultracentrifugation and titered by transduction of confluent 293T cells. The Lv-Trx2 H9c2 cell line was constructed by Lv-Trx2 infection and puromycin screening. Lv-vector H9c2 cells were treated as control cells.
Quantitative real-time PCR (qRT-PCR)
Total RNA was extracted from normal and Lv-Trx2 H9c2 cells using TRIzol Reagent (Takara, Dalian, China, Cat. No. Cat. No.RR036A). cDNA was synthesized via reverse transcription reactions with PrimeScript RT Master Mix (Takara). qRT-PCR analysis was performed using SYBR Premix Ex Taq (TaKaRa, Cat. No. Cat. No. RR420A) according to the manufacturer’s instructions. β-actin was used for normalization. Reactions were conducted using the ABI Prism 7500 Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). The primers used in this study for qRT-PCR were as follows:
Forward 5’-TGCTGGTGGTCTAACTGGAAC-3’ and Reverse 5’-TCAATGGCAAGGTCTGTGTG-3’ for Trx2; Forward 5’-AGAGCCTCGCCTTTGCCGAT-3’ and Reverse 5’-TGCCAGATTTTCTCCATGTCGT-3’ for β-actin.
Drug treatment
3-methyladenine(3-MA, Cat. No. M9281), rapamycin (Rapa, Cat. No. V900930) and N-acetyl-L-cysteine (NAC, Cat. No. A7250) were purchased from Sigma-Aldrich (St. Louis, MO, USA). 3-MA (final concentration of 10 mM) and Rapa (final concentration of 100 nM) were dissolved in DMSO and administered to the medium at the onset of reperfusion. All cells were compared with a control group that received the same amount of DMSO. NAC (final concentration of 500 μM) was dissolved in sterile double-distilled water and administered to the medium for 60 min of incubation before and during OGD/R. The cells were harvested for subsequent experiments.
Western blot
Proteins were extracted from H9c2 cells that had been treated with the conditions indicated. The measurement of protein concentrations in the lysates was performed with a BCA assay kit (Beyotime Institute of Biotechnology, Shanghai, China, Cat. No. P0010). For western blot analysis, equal amounts of protein (30 μg/lane) were subjected to a 6% or 12% SDS-PAGE. The proteins were then blotted onto polyvinylidenedifluoride (PVDF) membranes (Millipore), which were then blocked with 5% nonfat milk for 2 h at room temperature. The membranes were incubated with the corresponding primary antibodies at 4°C overnight. Antibodies included: rabbit anti-Trx2 (1:10000, Abcam, Cambridge, UK, Cat. No. ab185544), rabbit anti-LC3A/B (1:1000, Abcam, Cat. No. ab48394), rabbit anti-APG5L/ATG5 (1:1000, Abcam, Cat. No. ab108327), rabbit anti-Beclin-1 (1:1000, Cell Signaling Technology, Danvers, MA, USA, Cat. No. 3495), rabbit anti-phospho-mTOR (1:1000, Cell Signaling Technology, Cat. No. 2971), rabbit anti-mTOR (1:1000, Cell Signaling Technology, Cat. No. 2983), rabbit anti-phospho-ERK1/2 (Thr202/Tyr204) (1:1000, Cell Signaling Technology, Cat. No. 4094), rabbit anti-phospho-Akt (1:1000, Cell Signaling Technology, Cat. No. 4060), rabbit anti-Akt (1:1000, Cell Signaling Technology, Cat. No. 4691), rabbit anti-phospho-AMPK (1:1000, Cell Signaling Technology, Cat. No. 2535), rabbit anti-AMPK (1:1000, Cell Signaling Technology, Cat. No. 5832), rabbit anti-cleaved caspase3 (1:1000, Cell Signaling Technology, Cat. No. 9664), rabbit anti-Bcl-2 (1:1000, Abcam, Cat. No. ab59348), rabbit anti-Bax (1:5000, Abcam, Cat. No. ab32503), and mouse anti-β-actin (1:5000, Abcam, Cat. No. ab8226). The PVDF membranes were washed and then incubated with appropriate secondary antibodies for 1 h at room temperature. Protein bands were visualized using a chemiluminescence imaging analysis system, and quantitation was analyzed using ChemiDoc XRS+ software (Bio-Rad Laboratories, Hercules, CA, USA).
Transmission electron microscopy (TEM) assay
H9c2 cell samples were treated for TEM using routine procedures [17]. Briefly, H9c2 cells were washed with phosphate buffer saline (PBS, Gibco, Cat. No. 8115178) and then fixed in glutaraldehyde (2%) for 2 h at 4°C. The cells were collected and the process of treatment passed through post fixation, dehydration, embedding, and solidification in turn. Thin sections were obtained and examined with a Morgagni 286 transmission electron microscope (FEI Company, Eindhoven, Netherlands).
ROS production assay
The production of cellular ROS was assayed with a ROS assay kit (Beyotime Institute of Biotechnology, China, Cat. No. S0033). H9c2 cells had been treated with the conditions indicated and then incubated with 10 μM 2’,7’-dichlorodihydrofluoresceindiacetate (DCFH-DA) for 20 min in the dark at 37°C and washed three times with serum-free medium. The cells were then collected into the flow tubes, and DCFH-DA fluorescence was detected at 525 nm by flow cytometry.
Flow cytometry analysis of apoptosis
For apoptosis analysis, an Annexin V-PE/7-AAD apoptosis assay kit (eBioscience, California, America, Cat. No. 559763) was used following the manufacturer’s instructions. In brief, H9c2 cells were harvested after treatment, washed twice with PBS, and then resuspended in 100 μL of binding buffer after staining with Annexin V-PE/7-AAD in the dark for 15 min. The samples were immediately mixed with 400 μL of binding buffer and then analyzed by flow cytometer (Becton-Dickinson, Franklin Lakes, NJ, USA).
Statistical analysis
All experiments were repeated at least three times independently, and the data were presented as the means ± SD. All statistical tests were analyzed with GraphPad Prism 6. Statistical comparisons were conducted using Student’s t test or a one-way analysis of variance (ANOVA). P < 0.05 was considered statistically significant.
Results
Trx2 expression was reduced during OGD/R injury
OGD/R-induced myocardial cell injury acts as an in vitro model of myocardial I/R injury. In our study, H9c2 cells were subjected to 12 h of OGD followed by reperfusion for 0, 3, 6, 12, 24 h, at which times cell viability and LDH leakage were detected. We found that the viability of H9c2 cells decreased markedly with prolonged reperfusion time, with a little increase at reperfusion 24 h (Figure 1A). Only 51.6% of H9c2 cells remained viable at 6 h. Meanwhile, LDH release increased significantly from OGD 12 h to 24 h after reperfusion (Figure 1B). Moreover, western blot analysis showed that the expression of Trx2 declined after OGD/R and reached a nadir at reperfusion 6 h, as shown in Figure 1C.
Figure 1.
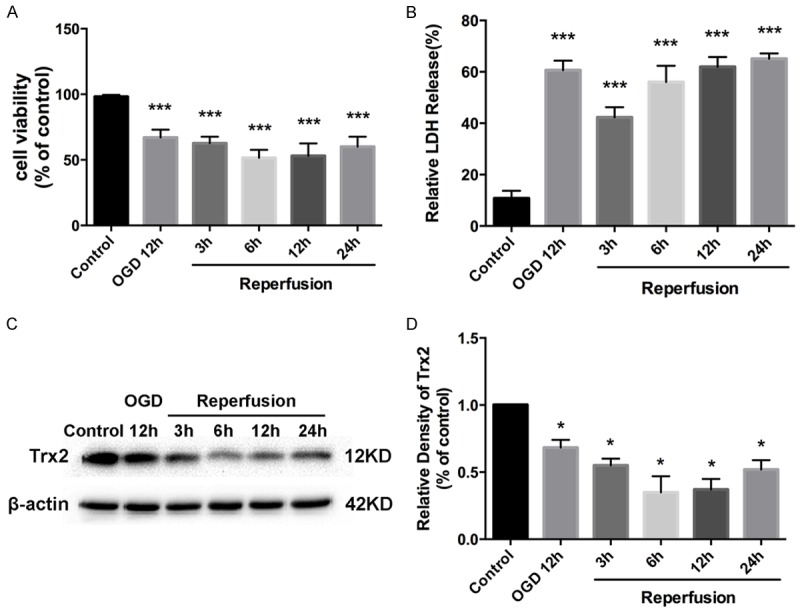
H9c2 cells were subjected to 12 h oxygen-glucose deprivation (OGD) followed by reperfusion for 3 h, 6 h, 12 h, 24 h. A. The cell viability of H9c2 cells after oxygen-glucose deprivation and reperfusion (OGD/R) assayed by Cell Counting Kit-8 (CCK-8). B. OGD/R induced H9c2 cell injury evaluated by lactate dehydrogenase (LDH) assay. C, D. Western blot assay showed the expression of thioredoxin-2 (Trx2) at the indicated time points during OGD/R. *P < 0.05 vs control, ***P < 0.001 vs control.
Trx2 overexpression alleviates OGD/R injury
Because the expression of Trx2 declined to a nadir at reperfusion 6 h, we chose this time point for the following study. To clarify whether Trx2 plays an important role in protecting myocardial OGD/R injury, we constructed an H9c2 cell line encoding lentivirus-mediated Trx2 overexpression (Lv-Trx2 H9c2). Lv-vector H9c2 cells were treated as control cells. qRT-PCR and western blot analysis showed that significant Trx2 overexpression was observed in the Lv-Trx2 H9c2 cell line compared to the Lv-vector group under normoxic conditions (Figure 2A, 2B). After OGD/R injury, the expression level of Trx2 protein was reduced but remained markedly higher than in the Lv-vector group (Figure 2B). The effect of Trx2 on OGD/R injury in H9c2 cells was examined by CCK-8 and LDH assay. As shown in Figure 2C, 2D, Trx2 overexpression decreased the susceptibility of H9c2 cells to OGD/R injury, resulting in increased cell viability and decreased LDH leakage compared with Lv-vector group.
Figure 2.

Effects of Trx2 overexpression in Lv-Trx2 H9c2 cells with OGD/R model. A. Quantitative real-time PCR (qRT-PCR) analysis of Trx2 mRNA expression. B. Western blot result showing Trx2 expression. A, B. *P < 0.05 vs corresponding normoxia group, #P < 0.05 vs corresponding Lv-vector group. C. The cell viability measured by CCK-8 assay. D. Cell injury evaluated by LDH kit. C, D. **P < 0.01 vs control group, @P > 0.05 vs OGD/R group, #P < 0.05 vs Lv-vector group.
Trx2-mediated protection involved the inhibition of autophagy in H9c2 cells during OGD/R injury
Autophagy activation was confirmed by TEM. The presence of typical autophagic vacuoles were observed in H9c2 cells after OGD/R, compared with few vacuoles in control cells (Figure 3A, 3B). These autophagic vacuoles included numerous double membrane-bound bodies and contained degenerated organelles. Less accumulation of autophagic vacuoles was observed in Lv-Trx2 H9c2 cell line (Figure 3A, 3B). Moreover, we further determined the expression of autophagy-related proteins during OGD/R. Microtubule-associated protein light chain 3 II (LC3 II) is the lipid form of LC3 and an acceptable marker for autophagy initiation. Western blot analysis showed that the expression of LC3 II was increased after OGD/R and peaked at reperfusion 6 h (Figure 3C, 3D). Another two autophagy-related markers, ATG5 and beclin1, increased from 12 h OGD to 24 h after reperfusion compared with the control cells (Figure 3C, 3D). To better explore the effect of Trx2 on autophagy, we choose two time points (at reperfusion 6 h and 12 h) to confirm the effect of Trx2 on the expression of autophagy-related proteins after OGD/R injury. Compared with the Lv-vector group, Lv-Trx2 H9c2 cells decreased the accumulation of LC3 II (Figure 4A, 4B). Also, a significant reduction in the expression of Beclin-1 and ATG5 was observed at 6 h and 12 h after reperfusion in Lv-Trx2 H9c2 cells (Figure 4A, 4B).
Figure 3.

Effects of Trx2 on OGD/R-induced autophagy in H9c2 cells. (A) Representative autophagosome images of the electron microscopic analysis in H9c2 cells (scale bar, 1 μm). (B) Quantification of the number of autophagosome from (A). (C, D) Western blot showed that OGD/R induced the expression of autophagy-related proteins microtubule-associated protein light chain 3 (LC3), ATG5, and Beclin-1. *P < 0.05 vs control, @P > 0.05 vs OGD/R group, #P < 0.05 vs LV-vector group.
Figure 4.

Trx2 inhibited autophagy through the mammalian target of rapamycin (mTOR)-dependent pathway in H9c2 cells subjected to OGD/R. A, B. Western blot results of autophagy-related proteins at reperfusion 6 h and 12 h. C, D. Western blot showed the effects of Trx2 on the expression of mTOR, extracellular signal-regulated kinase 1/2 (ERK1/2), the serine/threonine kinase Akt (Akt), and AMP-activated protein kinase (AMPK) at 6 h and 12 h after reperfusion. *P < 0.05 vs corresponding Lv-vector group, @P > 0.05 vs corresponding Lv-vector group.
Potential molecular mechanisms of Trx2 regulation during autophagy
We explored the potential molecular mechanisms underlying the regulation of Trx2 on autophagy. Figure 4C, 4D showed that the level of phospho-mTOR (p-mTOR) increased significantly in the Lv-Trx2 H9c2 cell line compared to the Lv-vector group under the context of OGD/R, while the total mTOR level was nearly unchanged. To further explore the upstream signal targeting mTOR, we examined whether three key mediators of mTOR-AMP-activated protein kinase (AMPK), serine/threonine kinase Akt, and extracellular signal-regulated kinase 1/2 (ERK1/2)- were involved in the mTOR-dependent pathway. We found that Trx2 overexpression induced heightened expression of p-Akt and decreased the activation of p-AMPK. There was no obvious change trend for p-ERK1/2 between the two groups (Figure 4C, 4D).
Trx2 regulated OGD/R-induced autophagy by reducing ROS accumulation
As an important mediator of autophagy, cellular ROS was detected by flow cytometry using DCFH-DA. H9c2 cells were subjected to NAC prior to OGD/R treatment, and the level of LC3 II was determined by western blot analysis to explore the role of ROS during Trx2-regulated autophagy. As shown in Figure 5A, 5B, OGD/R injury caused ROS accumulation in H9c2 cells while the pre-treatment of NAC and Trx2 overexpression both reduced ROS accumulation. In particular, we also found that the pre-treatment of NAC and the overexpression of Trx2 both rescued LC3-II accumulation induced by OGD/R injury (Figure 5C, 5D).
Figure 5.
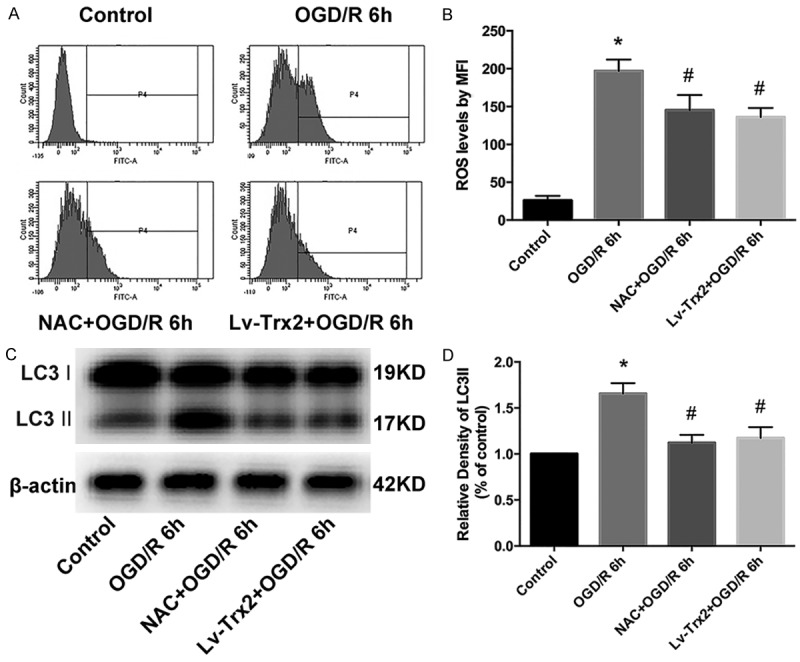
Trx2-regulated autophagy was mediated by reactive oxygen species (ROS). A. The level of ROS was determined by flow cytometry assay. B. The mean fluorescence intensity (MFI) of 2’,7’-dichlorodihydroflurescein (DCF) was used to reflect cellular ROS levels. C, D. LC3-II accumulation was analyzed by western blotting analysis. *P < 0.05 vs control, #P < 0.05 vs OGD/R group.
Apoptosis induced by OGD/R is related to autophagy
As indicated, we hypothesized that Trx2 inhibits autophagy, providing a protective effect during OGD/R injury. 3-MA, a classical autophagy inhibitor, and Rapa, a classical autophagy sensitizer, were employed to further verify our hypothesis. As shown in Figure 6, the expression levels of LC3 II and cleaved caspase-3 (c-cas3) decreased when administrated with 3-MA, while the application of Rapa sharply enhanced LC3-II as well as c-cas3 protein expression.
Figure 6.
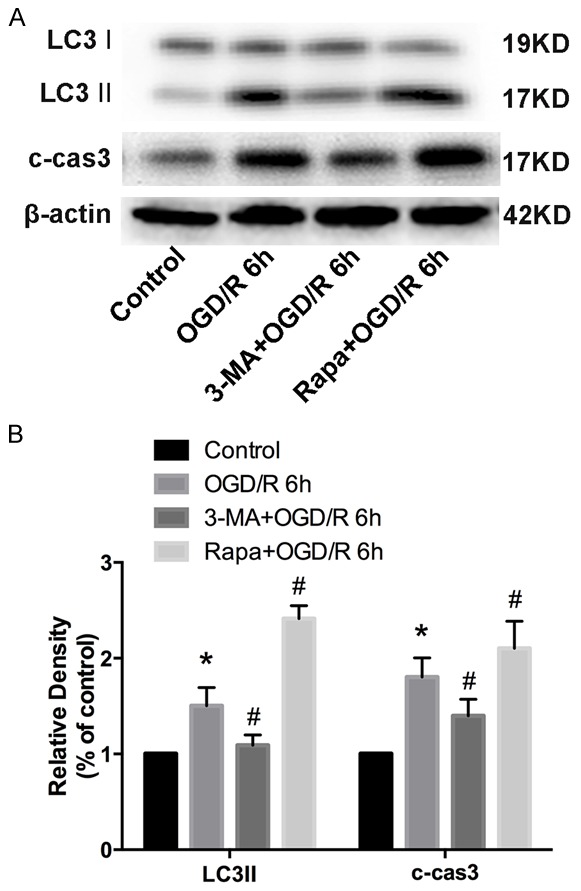
The apoptosis induced by OGD/R was related to autophagy. A, B. Western blot analysis showed the expression level of LC3 and cleaved caspase3 (c-cas3) treated with3-methyladenine (3-MA) and rapamycin (Rapa). *P < 0.05 vs control, #P < 0.05 vs OGD/R group.
Trx2 decreased apoptosis induced by OGD/R in H9c2 cells
Flow cytometry analysis indicated that Trx2 attenuated OGD/R-induced apoptosis in H9c2 cells (Figure 7A, 7B). In addition, we examined the expression levels of apoptosis-related proteins using western blot analysis. The results indicated that the activation of c-cas3 was induced significantly during OGD/R injury, while Trx2 overexpression could reduce this tendency (Figure 7C, 7D). Additionally, we found that Trx2 increased the downregulation tendency of Bcl-2 and alleviated the upregulation tendency of Bax in the H9c2 cells subjected to OGD/R (Figure 7C, 7D).
Figure 7.

Trx2 decreased OGD/R-induced apoptosis in H9c2 cells. A, B. Flow cytometry analysis showed apoptosis induced by OGD/R was inhibited by Trx2 overexpression. C, D. Western blot analysis showed the expression levels of cleaved caspase-3 (c-cas3), Bcl-2, and Bax. *P < 0.05 vs control, @P > 0.05 vs OGD/R group, #P < 0.05 vs Lv-vector group.
Discussion
Cell death plays an essential role in ischemic reperfusion pathogenesis. For several decades, apoptosis, known as Type I cell death, has been identified as the principal mechanism for programmed cell death and extensively studied in mammalian tissues [18]. However, the process of cell death is complex and well controlled. Recently, mounting studies have reported autophagy activation in myocardial ischemia and reperfusion in vitro and in vivo [5,9,19]. Excessive or dysregulated autophagy leads to cardiomyocyte damage or even death, also known as Type II programmed cell death [5,20]. In our study, we used a cardiomyocyte OGD/R model to demonstrate that the overexpression of Trx2 via a recombinant lentiviral vector is a promising cardioprotective strategy to reduce autophagic and apoptotic effects in vitro. Firstly, we showed that lentiviral infection effectively led to the overexpression of Trx2 in H9c2 cells, which significantly alleviated OGD/R injury. Secondly, we proved that Trx2-mediated protection involved the inhibition of autophagy through the mTOR pathway. Finally, we further demonstrated that Trx2 could alleviate apoptosis induced by OGD/R via the mitochondrion-mediated intrinsic apoptotic pathway. Taken together, these data strongly confirm the cardioprotective role of Trx2 in vitro.
There are many signaling cascades involved in autophagy regulation in response to different stimuli in cells’ internal and external environments [21]. However, a master regulator, serine/threonine kinase mTOR, is known as the classical pathway [22]. The mTOR pathway includes two functional complexes: one is mTORC1 (the mTOR complex 1), which is rapamycin-sensitive and regulates autophagy negatively, and the other is mTORC2 (the mTOR complex 2), which cannot regulate autophagy directly [20]. The activity of mTOR is regulated by ERK1/2, Akt, and AMPK, key upstream signaling molecules of mTOR [21]. They converge on mTOR to regulate downstream molecules and then regulate the autophagy process. The Ras/Raf1/ERK1/2 pathway is reported to positively regulate autophagy in response to nutrient starvation via inhibition of mTOR in the cell [18,23]. As a key mediator of mTOR, AMPK has demonstrated positive regulation of autophagy by negatively regulating mTOR activity [24-27]. Akt is a downstream regulator of PI3K, and the PI3K/Akt/mTOR pathway plays an important part in cell autophagy [28]. The activation of Akt could decrease autophagy by activating mTOR [29]. We found that Trx2 enhanced the activation of mTORC1, which resulted in decreased autophagy. Our results indicate that Trx2 decreased the activation of AMPK and heightened the expression of p-Akt. However, there was no obvious change trend for the ERK1/2 pathway. These results indicate that Akt/mTOR and AMPK/mTOR signaling pathways were involved in Trx2-regulated autophagy. In addition, a growing number of studies in recent years have demonstrated that ROS are important mediators of autophagy [30]. Trx2, as a mitochondrial form of the Trx system, is critical for maintaining cellular redox balance, thus controlling cellular oxidative stress [12]. NAC, a ROS scavenger, can markedly block intracellular ROS, which plays a crucial role in the activation of autophagy. Our data showed that inhibition of ROS by NAC and Trx2 overexpression can decrease not only ROS levels but also the level of LC3 II, which suggests that ROS could mediate the induction of autophagy. In addition, it is possible that a more complex regulation network is involved in Trx2-regulated autophagy in the content of OGD/R of H9c2 cells. The detailed roles of the network of regulation in Trx2-regulated autophagy remain to be further explored.
Previous studies reported that Trx2 is involved in the mitochondrial apoptosis signaling pathway, which is induced in response to various stimuli, including oxidative stress, and is regulated by the Bcl-2 family [16,31]. The main pathway of cell apoptosis is dependent on the caspase pathway, and caspase3 is the final executor of cell apoptosis [32]. In this study, flow cytometry assay showed that Trx2 attenuates apoptosis. Western blot analysis demonstrated that Trx2 could inhibit c-cas3 protein expression. Moreover, Trx2 could decrease the expression of Bax and enhance Bcl-2 protein level, which showed that the regulation of Trx2 on OGD/R-induced apoptosis is via mitochondrion-mediated intrinsic apoptotic pathway. Booth and co-workers reported that the boundary is not completely clear between Type I cell death and Type II cell death because apoptosis possibly begins with autophagy, and autophagy usually ends with apoptosis [18]. Moreover, several studies suggest that autophagy may enhance cell apoptosis, and inhibition of autophagy could reduce apoptosis [33,34]. Our data demonstrated that the autophagy inducer Rapa increased apoptosis, and the autophagy inhibitor 3-MA promoted cell survival against OGD/R in the H9c2 cells. As distinct cellular processes, autophagy and apoptosis have different morphological and biochemical features; however, the protein networks between them can be highly interconnected. In this respect, more research is needed to determine the complex interplay between autophagic and apoptotic proteins.
In summary, data from our study revealed that Trx2 provided cardio protection against OGD/R injury by inhibiting autophagy and apoptosis in vitro. Our current knowledge will provide a theoretical foundation for further studying the effect of Trx2 in vivo and a new insight into the pathogenesis of myocardial I/R.
Acknowledgements
This work was supported by Nature Science Foundation of China Grant (NO. 81170124).
Disclosure of conflict of interest
None.
References
- 1.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Magid D, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER, Moy CS, Mussolino ME, Nichol G, Paynter NP, Schreiner PJ, Sorlie PD, Stein J, Turan TN, Virani SS, Wong ND, Woo D, Turner MB. Heart disease and stroke statistics--2013 update: a report from the American Heart Association. Circulation. 2013;127:e6–e245. doi: 10.1161/CIR.0b013e31828124ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nielsen PH, Maeng M, Busk M, Mortensen LS, Kristensen SD, Nielsen TT, Andersen HR. Primary angioplasty versus fibrinolysis in acute myocardial infarction: long-term follow-up in the Danish acute myocardial infarction 2 trial. Circulation. 2010;121:1484–1491. doi: 10.1161/CIRCULATIONAHA.109.873224. [DOI] [PubMed] [Google Scholar]
- 3.Hausenloy DJ, Yellon DM. Targeting myocardial reperfusion injury--the search continues. N Engl J Med. 2015;373:1073–1075. doi: 10.1056/NEJMe1509718. [DOI] [PubMed] [Google Scholar]
- 4.Ovize M, Baxter GF, Di Lisa F, Ferdinandy P, Garcia-Dorado D, Hausenloy DJ, Heusch G, Vinten-Johansen J, Yellon DM, Schulz R. Postconditioning and protection from reperfusion injury: where do we stand? Position paper from the Working Group of Cellular Biology of the Heart of the European Society of Cardiology. Cardiovasc Res. 2010;87:406–423. doi: 10.1093/cvr/cvq129. [DOI] [PubMed] [Google Scholar]
- 5.Wang B, Zhong S, Zheng F, Zhang Y, Gao F, Chen Y, Lu B, Xu H, Shi G. N-n-butyl haloperidol iodide protects cardiomyocytes against hypoxia/reoxygenation injury by inhibiting autophagy. Oncotarget. 2015;6:24709–24721. doi: 10.18632/oncotarget.5077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zeng C, Li H, Fan Z, Zhong L, Guo Z, Guo Y, Xi Y. Crocin-elicited autophagy rescues myocardial ischemia/reperfusion injury via paradoxical mechanisms. Am J Chin Med. 2016;44:515–530. doi: 10.1142/S0192415X16500282. [DOI] [PubMed] [Google Scholar]
- 7.Sciarretta S, Hariharan N, Monden Y, Zablocki D, Sadoshima J. Is autophagy in response to ischemia and reperfusion protective or detrimental for the heart? Pediatr Cardiol. 2011;32:275–281. doi: 10.1007/s00246-010-9855-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Galluzzi L, Pietrocola F, Levine B, Kroemer G. Metabolic control of autophagy. Cell. 2014;159:1263–1276. doi: 10.1016/j.cell.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu L, Wu Y, Huang X. Orientin protects myocardial cells against hypoxia-reoxygenation injury through induction of autophagy. Eur J Pharmacol. 2016;776:90–98. doi: 10.1016/j.ejphar.2016.02.037. [DOI] [PubMed] [Google Scholar]
- 10.Dosenko VE, Nagibin VS, Tumanovska LV, Moibenko AA. Protective effect of autophagy in anoxia-reoxygenation of isolated cardiomyocyte? Autophagy. 2006;2:305–306. doi: 10.4161/auto.2946. [DOI] [PubMed] [Google Scholar]
- 11.Sadoshima J. The role of autophagy during ischemia/reperfusion. Autophagy. 2008;4:402–403. doi: 10.4161/auto.5924. [DOI] [PubMed] [Google Scholar]
- 12.Lu J, Holmgren A. Thioredoxin system in cell death progression. Antioxid Redox Signal. 2012;17:1738–1747. doi: 10.1089/ars.2012.4650. [DOI] [PubMed] [Google Scholar]
- 13.Huang Q, Zhou HJ, Zhang H, Huang Y, Hinojosa-Kirschenbaum F, Fan P, Yao L, Belardinelli L, Tellides G, Giordano FJ, Budas GR, Min W. Thioredoxin-2 inhibits mitochondrial reactive oxygen species generation and apoptosis stress kinase-1 activity to maintain cardiac function. Circulation. 2015;131:1082–1097. doi: 10.1161/CIRCULATIONAHA.114.012725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lesnefsky EJ, Chen Q, Hoppel CL. Mitochondrial metabolism in aging heart. Circ Res. 2016;118:1593–1611. doi: 10.1161/CIRCRESAHA.116.307505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.He M, Cai J, Go YM, Johnson JM, Martin WD, Hansen JM, Jones DP. Identification of thioredoxin-2 as a regulator of the mitochondrial permeability transition. Toxicol Sci. 2008;105:44–50. doi: 10.1093/toxsci/kfn116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tanaka T, Hosoi F, Yamaguchi-Iwai Y, Nakamura H, Masutani H, Ueda S, Nishiyama A, Takeda S, Wada H, Spyrou G, Yodoi J. Thioredoxin-2 (TRX-2) is an essential gene regulating mitochondria-dependent apoptosis. EMBO J. 2002;21:1695–1703. doi: 10.1093/emboj/21.7.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saito T, Asai K, Sato S, Hayashi M, Adachi A, Sasaki Y, Takano H, Mizuno K, Shimizu W. Autophagic vacuoles in cardiomyocytes of dilated cardiomyopathy with initially decompensated heart failure predict improved prognosis. Autophagy. 2016;12:579–587. doi: 10.1080/15548627.2016.1145326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Booth LA, Tavallai S, Hamed HA, Cruickshanks N, Dent P. The role of cell signalling in the crosstalk between autophagy and apoptosis. Cell Signal. 2014;26:549–555. doi: 10.1016/j.cellsig.2013.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu X, Zhang C, Zhang C, Li J, Guo W, Yan D, Yang C, Zhao J, Xia T, Wang Y, Xu R, Wu X, Shi J. Heat shock protein 70 inhibits cardiomyocyte necroptosis through repressing autophagy in myocardial ischemia/reperfusion injury. In Vitro Cell Dev Biol Anim. 2016;52:690–698. doi: 10.1007/s11626-016-0039-8. [DOI] [PubMed] [Google Scholar]
- 20.Lv S, Xu QY, Sun EC, Zhang JK, Wu DL. Dissection and integration of the autophagy signaling network initiated by bluetongue virus infection: crucial candidates ERK1/2, Akt and AMPK. Sci Rep. 2016;6:23130. doi: 10.1038/srep23130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jung CH, Ro SH, Cao J, Otto NM, Kim DH. mTOR regulation of autophagy. FEBS Lett. 2010;584:1287–1295. doi: 10.1016/j.febslet.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cagnol S, Chambard JC. ERK and cell death: mechanisms of ERK-induced cell death--apoptosis, autophagy and senescence. FEBS J. 2010;277:2–21. doi: 10.1111/j.1742-4658.2009.07366.x. [DOI] [PubMed] [Google Scholar]
- 24.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 25.Shang L, Wang X. AMPK and mTOR coordinate the regulation of Ulk1 and mammalian autophagy initiation. Autophagy. 2011;7:924–926. doi: 10.4161/auto.7.8.15860. [DOI] [PubMed] [Google Scholar]
- 26.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–590. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 28.Martelli AM, Chiarini F, Evangelisti C, Grimaldi C, Ognibene A, Manzoli L, Billi AM, McCubrey JA. The phosphatidylinositol 3-kinase/AKT/mammalian target of rapamycin signaling network and the control of normal myelopoiesis. Histol Histopathol. 2010;25:669–680. doi: 10.14670/HH-25.669. [DOI] [PubMed] [Google Scholar]
- 29.Dibble CC, Cantley LC. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 2015;25:545–555. doi: 10.1016/j.tcb.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Navarro-Yepes J, Burns M, Anandhan A, Khalimonchuk O, del Razo LM, Quintanilla-Vega B, Pappa A, Panayiotidis MI, Franco R. Oxidative stress, redox signaling, and autophagy: cell death versus survival. Antioxid Redox Signal. 2014;21:66–85. doi: 10.1089/ars.2014.5837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schwarz M, Andrade-Navarro MA, Gross A. Mitochondrial carriers and pores: key regulators of the mitochondrial apoptotic program? Apoptosis. 2007;12:869–876. doi: 10.1007/s10495-007-0748-2. [DOI] [PubMed] [Google Scholar]
- 32.Tait SW, Green DR. Caspase-independent cell death: leaving the set without the final cut. Oncogene. 2008;27:6452–6461. doi: 10.1038/onc.2008.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, Levine B, Sadoshima J. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res. 2007;100:914–922. doi: 10.1161/01.RES.0000261924.76669.36. [DOI] [PubMed] [Google Scholar]
- 34.Wang ZG, Wang Y, Huang Y, Lu Q, Zheng L, Hu D, Feng WK, Liu YL, Ji KT, Zhang HY, Fu XB, Li XK, Chu MP, Xiao J. bFGF regulates autophagy and ubiquitinated protein accumulation induced by myocardial ischemia/reperfusion via the activation of the PI3K/Akt/mTOR pathway. Sci Rep. 2015;5:9287. doi: 10.1038/srep09287. [DOI] [PMC free article] [PubMed] [Google Scholar]