

Abstract
The design of enzyme-like complexity within metal–organic frameworks (MOFs) requires multiple reactions to be performed on a MOF crystal without losing access to its interior. Here, we show that seven post-synthetic reactions can be successfully achieved within the pores of a multivariate MOF, MTV-IRMOF-74-III, to covalently incorporate tripeptides that resemble the active sites of enzymes in their spatial arrangement and compositional heterogeneity. These reactions build up H2N-Pro-Gly-Ala-CONHL and H2N-Cys-His-Asp-CONHL (where L = organic struts) amino acid sequences by covalently attaching them to the organic struts in the MOFs, without losing porosity or crystallinity. An enabling feature of this chemistry is that the primary amine functionality (−CH2NHBoc) of the original MOF is more reactive than the commonly examined aromatic amines (−NH2), and this allowed for the multi-step reactions to be carried out in tandem within the MOF. Preliminary findings indicate that the complexity thus achieved can affect reactions that were previously accomplished only in the presence of enzymes.
The ability to perform post-synthetic covalent reactions on metal–organic frameworks (MOFs) with the same precision being practiced in molecular chemistry has led to MOFs capable of highly selective gas adsorption.1 A longstanding objective has been to design molecularly defined systems in which functional groups are covalently incorporated in a manner akin to the active sites of enzymes. This task inevitably requires multiple reactions to be carried out on a specific functionality covalently attached within the pores of a MOF to build up units of the desired size, spatial orientation, complexity, and compositional heterogeneity. In this Communication, we present the results of seven post-synthetic covalent reactions in tandem within the pore of a MOF without loss of structural integrity, order, or porosity of the extended structure.2 These reactions also give the pores multivariate (MTV) functionality, leading to our preliminary findings that they can affect reactions only known for enzymes.
Specifically, we chose a functionalized IRMOF-74-III (Scheme 1), constructed from magnesium oxide rods joined by terphenylene organic struts [Mg2(L), where H4L = 3,3″-dihydroxy-(1,1′:4′,1″-terphenyl)-4,4″-dicarboxylic acid], to make an extended structure based on the parent structure of MOF-74, but having 25 Å one-dimensional channels.3 The highly porous IRMOF-74-III allows for facile diffusion of reactants into the pore interiors and the targeting of specific sites within the MOF.
Scheme 1. Seven Post-synthetic Reactions to Achieve Enzyme-like Complexity in the Pores of MTV-IRMOF-74-III-(CH3)0.6(CH2NHBoc)0.4a.
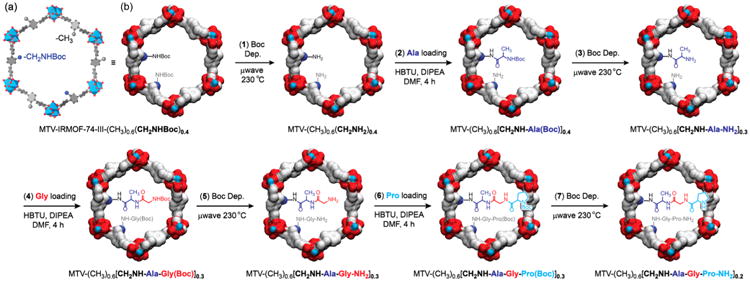
a(a) Polyhedral drawing of −CH2NHBoc functionalized MOF pore. (b) Post-synthetic reactions (1)–(7) are illustrated using a van der Waals surface: thermal Boc deprotections for (1), (3), (5), and (7); amino acid loading steps for (2), (4), and (6) (Ala, Gly, and Pro, respectively). The evolution of one potential reaction byproduct (i.e., dipeptide H2N-Pro-Gly-CONHL) is represented in gray. These byproducts, due to incomplete post-synthetic transformations, also affect the molecular formula of the compounds, as can be noted in the scheme.
We prepared a series of MTV-IRMOF-74-III materials using two types of organic struts, H4L-CH3 and H4L-CH2NHBoc (hereafter, MTV-(CH3)(1-x)(CH2NHBoc)x, x = 0.2–0.8; Boc = tert-butyloxycarbonyl), to study the optimal concentration of reactive sites for carrying out tandem post-synthetic reactions. We found that among these, MTV-(CH3)0.6(CH2NHBoc)0.4 led to efficient reactant diffusion into the MOF pores and, concomitantly, better overall yield. Additionally, we compared the reactivity of MTV-(CH3)(1-x)(CH2NHBoc)x, functionalized with primary amines [MTV-(CH3)0.6(CH2NH2)0.4], to that of the analogous material functionalized with aromatic amines [MTV-(CH3)0.6(NH2)0.4], toward peptide bond formation starting with alanine (Ala). We show that, under the same reaction conditions, primary amine nucleophiles lead to 97% greater reaction yield compared to their aromatic amine counterparts. We further demonstrate that MTV-IRMOF-74-III-(CH3)0.6(CH2NHBoc)0.4 can be used as a reactant for the sequential loading of tripeptides (H2N-Pro-Gly-Ala-CONHL and H2N-Cys-His-Asp-CONHL, with Gly = glycine, Pro = proline, Asp = aspartic acid, His = histidine, Cys = cysteine, and L = organic struts) through seven tandem reactions. Preliminary results showed that the MTV functionality in the pores of MTV-IRMOF-74-III-tripeptides can catalyze stereoselective chlorinations (H2N-Pro-Gly-Ala-CONHL), and even sequence-specific peptide bond cleavage (H2N-Cys-His-Asp-CONHL), as the enzyme tobacco etch virus (TEV) endopeptidase.4
For clarity, the post-synthetic strategy and its implementation, results, and catalytic activity are covered in several sections below to show that (i) a linear relationship exists between input and output of the variously functionalized organic struts in the synthesis of MTV-IRMOF-74-III series, (ii) precise control is exercised over the type of functionalities and their ratio being incorporated within the MOF pores, thus allowing for desirable yields of post-synthetic reactions, (iii) achieving high-yielding reactions, especially in the initial post-synthetic reactions, coupled with the preservation of high porosity led to the realization of multiple reactions to be carried out in tandem without the challenge of low yields or vanishing porosity as further reactions are carried out, and (iv) the complexity built into the pores leads to evidence of highly selective catalytic activity.
Synthesis of MTV-IRMOF-74-III Series and Control of Functionality Ratio
The organic struts of MTV-IRMOF-74-III were functionalized with Boc-protected primary amines and methyl groups, according to reported methodology.1 It is known that reactions performed in fully functionalized MOFs lead to drastic loss of their accessible pore volume, making it impossible to perform further tandem transformations.5 In order to overcome this challenge, we utilized the MTV approach, whereby the unfunctionalized strut is used to dilute the struts having the reactive groups in the MOF, and investigate its effect on the reaction yield.6 Hence, we prepared a series of MTV-(CH3)(1-x)(CH2NHBoc)x by a procedure analogous to that reported in literature.3 The solvothermal reaction of Mg(NO3)2·6H2O with different ratios of H4L-CH3 and H4L-CH2NHBoc allowed us to prepare MTV-IRMOF-74-III (x = 0.2, 0.4, 0.6, and 0.8) [for details see Supporting Information (SI), Section S1]. Based on the acid digestion 1H NMR spectra of guest-free MOF sample, the ratio of the struts present in the resulting structure (output ratio) was estimated. Unlike MTV-MOF-5 and MTV-MOF-177,6,7 the output ratio is nearly identical to the input ratio (the quantity of struts used in the synthesis; see Figure S1, SI).
Reactivity and Control of Specific Functionality in MTV-IRMOF-74-III
The MTV-(CH3)(1-x)(CH2NHBoc)x series was employed toward post-synthetic peptide bond formation using protected Ala (Scheme 1).1 In a typical example, microwave-treated MTV-(CH3)0.6(CH2NHBoc)0.4 sample showed quantitative cleavage of the Boc group, which was confirmed by the disappearance of its representative singlet (δ = 1.31 for CH3 group) in the 1H NMR spectrum of the digested sample (Section S4, SI). Deprotected MTV-(CH3)0.6-(CH2NH2)0.4 was then used to covalently bind Ala [step (2), Scheme 1; Section S2.9, SI]. The obtained crystalline powder sample was analyzed by powder X-ray diffraction (PXRD), 1H NMR spectroscopy, FT-IR, and HPLC (Sections S3, S4, S5, and S8, respectively, SI). We found that the yield of peptide bond formation with Ala was nearly quantitative (97%) when the ratio of CH2NHBoc was below 0.6 (Figure S2, SI). It is presumed that higher concentration of primary amines in IRMOF-74-III might hamper the fast diffusion of reagents to the pore interiors. Thus, we chose MTV-(CH3)0.6-(CH2NHBoc)0.4 as starting material to carry out consecutive post-synthetic reactions, in order to mitigate potential guest diffusion problems and steric repulsion between functionalities in the pores.
Aromatic amines are by far the most studied nucleophiles in post-synthetic functionalization of MOFs.8 Therefore, the reactivity of aromatic amines was compared with that of the primary amines. We prepared the analogous compound MTV-(CH3)0.6(NH2)0.4 and performed the same post-synthetic peptide bond formation reaction with Boc-protected Ala. 1H NMR analysis after the reaction indicates no Ala loading, as evidenced by the absence of the signal corresponding to the singlet of the Boc group (δ = 1.35, Figure S32, SI). This observation highlights the importance of having highly reactive functionalities (i.e., primary amines) in the pores of MOFs to perform efficient post-synthetic reactions.
Post-synthetic Reactions Steps (3)–(7)
Quantitative loading of Ala in MTV-(CH3)0.6(CH2NH2)0.4 prompted us to proceed to the third reaction [(3), Scheme 1] by cleaving the Boc-protecting groups at the N-terminal of Ala by microwave heating. From 1H NMR analysis of the microwave-treated material, we confirmed a 76% yield in the deprotection reaction, as indicated by the downfield shift and integral of the Ala methyl group doublet (from δ = 1.19 to 1.33; Figure S34, SI). Similar to the reactions shown in steps (1) and (2), Boc-deprotections [(3), (5), and (7)] and amino acid loadings [(4) and (6)] were performed to incorporate H2N-Pro-Gly-Ala-CONHL peptide into the pores of MTV-IRMOF-74-III (Scheme 1). It is worth noting that the maximum number of post-synthetic modifications in tandem previously reported in MOFs is four.2
PXRD analyses confirmed that the crystallinity and structure remain intact throughout the seven reactions by the coincidence of their profile with the pattern calculated for unfunctionalized IRMOF-74-III (Figure 1a). The ratio of integrals for the methyl group in L-CH3, and for each of the loaded amino acids in the acid-digested 1H NMR spectra, was used to quantify the yield of each performed reaction. The tripeptide H2N-Pro-Gly-Ala-CONHL was covalently bound to the MOF with an overall yield of 57% (average of 93% yield per step, Section 2, SI).
Figure 1.
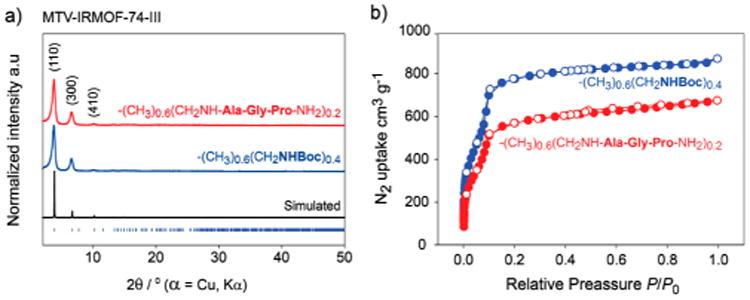
(a) PXRD patterns for simulated IRMOF-74-III (black), starting material MTV-(CH3)0.6(CH2NHBoc)0.4 (blue), and product after seven post-synthetic reactions, [MTV-(CH3)0.6(CH2NH-Ala-Gly-Pro-NH2)0.2 (red). (b) N2 isotherms at 77 K for MTV-(CH3)0.6-(CH2NHBoc)0.4 (blue) and MTV-(CH3)0.6(CH2NH-Ala-Gly-Pro-NH2)0.2 (red).
After the removal of guest molecules from the pores (Section S1.4, SI), the porosity of the product of the seven post-synthetic reactions was evaluated by measurement of the N2 adsorption isotherm at 77 K. A steep N2 uptake was observed below P/P0 = 0.1, which is indicative of the presence of permanent porosity even after the seven reactions (Figure 1b). The observed Brunauer–Emmett–Teller (BET) surface area of the tripeptide-loaded sample (1760 m2 g−1) is lower than that of the starting material MTV-(CH3)0.6(CH2NHBoc)0.4 (2330 m2 g−1). This is because of the covalently attached tripeptide, which occupies a significant amount of the pore space.
Building Complexity in the Pores
Encouraged by the successful sequential loading of tripeptide in the MOF pores, we proceeded to covalently bind another peptide sequence of acidic and basic amino acids. To achieve this goal, orthogonal protection of these functionalities is necessary to avoid side reactions. Accordingly, fluorenylmethyloxycarbonyl (Fmoc) protection and deprotection chemistry9 was used to incorporate the tripeptide H2N-Cys-His-Asp-CONHL into MTV-(CH3)0.6(CH2NH2)0.4. This specific sequence was selected since it has been shown that the amino acids Asp, His, and Cys (and their proximity in the active site) are responsible for the unique catalytic activity of the enzyme TEV.10 In seven post-synthetic reactions, we prepared MTV-(CH3)0.6(CH2NH-Asp-His-Cys-NH2)0.1, by loading Fmoc-Asp(tBu)-OH, Fmoc-His-(Boc)-OH, and Boc-Cys(fm)-OH (fm = fluorenylmethyl), where Fmoc or Boc groups were used to protect N-terminal functionalities, and tert-butyl ester (tBu), Boc, or fm groups were used to protect the amino acid side chains (in Asp, His, and Cys, respectively). After the Asp and His loadings [steps (2) and (4), Scheme S2, SI], the MOFs were washed with DMF, and the Fmoc groups were removed under basic conditions (Section S2.2, SI). MTV-(CH3)0.6(CH2NH-Asp-His-Cys-NH2)0.1 was obtained with an overall yield of 20% (Section S2, SI). 1H NMR and HPLC of the digested samples were used to follow the reactions (Figures S35 and S44, SI). The presence of H2N-Cys-His-Asp-CONHL tripeptide in the pores of IRMOF-74-III was confirmed by the mass of the parent fragment found by ESI-MS analysis of the digested sample (calcd m/z 734.2 Da, found 734.4 Da). PXRD analysis and N2 isotherms at 77 K showed that MTV-(CH3)0.6-(CH2NH-Asp-His-Cys-NH2)0.1 retained crystallinity and porosity (BET = 1920 m2 g−1) after seven post-synthetic covalent reactions (Figures S17 and S41, SI).
Selective Catalytic Activity of the MTV-MOF
Amino acids and short peptides are known to be active organo-catalysts.11 We chose to evaluate our MTV-IRMOF-III tripeptides in two different catalytic reactions that strongly depend on the complexity of the pores. MTV-(CH3)0.6-(CH2NH-Ala-Gly-Pro-NH2)0.2 was evaluated as catalyst in the α-chlorination of butyraldehyde, which is known to be catalyzed by proline derivatives [lit. 99% conv, 2% enantio-selectivity (ee)].12 We sought to take advantage of the molecular control in the pores of IRMOF-74-III, to study the effect of pore constraints on this reaction. Hence, we prepared MTV-(CH3)0.8(CH2NH-Pro-NH2)0.2 (Cat. A, Scheme S4, SI) as a control catalyst and compared its activity with that of MTV-(CH3)0.6(CH2NH-Ala-Gly-Pro-NH2)0.2 (Cat. B). N2 isotherms for both materials showed a 17% decrease in the BET surface area of Cat. B compared to Cat. A (1760 m2 g−1 to 2120 m2 g−1, respectively; Section S6, SI). This decrease is related to the bulkiness of the tripeptide, H2N-Pro-Gly-Ala-CONHL, compared to a simple amino acid, Pro, in the pores. Under the same conditions, Cat. A was found to provide 69% more of the desired product compared to Cat. B (94% vs 25%, Figure S48, SI). This is expected since there is less available pore volume in Cat. B.
Notably, a significant increase in the ee of this α-chlorination reaction was observed when functionalized MOFs were used as catalysts compared to the reported homogeneous-phase proline catalysis (20% ee vs 2% ee). This increased asymmetric induction, which is likely due to the increased stereochemical constraints within the congested MOF framework,13 is especially striking, given that the catalytic sites are not entirely uniform (Scheme 1). Ultimately, these results demonstrate the exciting potential of imparting control of catalyst reactivity and selectivity by the molecular tuning of the MOF architecture and the active site.
Next, we turned our attention to recreating the high selectivity exhibited by the enzyme TEV endopeptidase within a synthetically modified MOF framework. Specifically, MTV-(CH3)0.6(CH2NH-Asp-His-Cys-NH2)0.1 was studied as a heterogeneous catalyst in sequence-specific peptide cleavage due to its TEV-like peptide sequence (Figure 2a).
Figure 2.
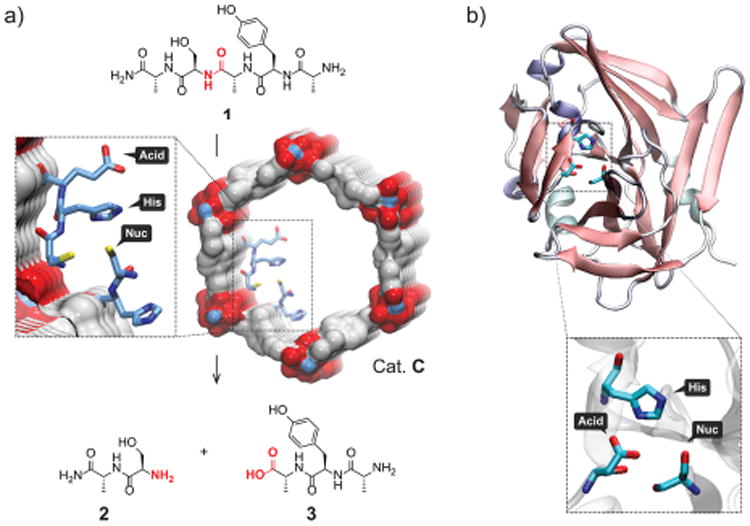
(a) Catalytic cleavage of pentapeptide 1 by Cat. C [MTV-IRMOF-74-III-(CH3)0.6(CH2NH-Asp-His-Cys-NH2)0.1] in the specific sequence containing serine. (b) Cartoon representation of the enzyme TEV endoprotease, highlighting the three amino acids that participate in the catalysis.
TEV protease is known to recognize selectively the amino acid serine (Ser) in a peptide substrate and cleave the amide bond in which Ser is involved. In order to achieve such precision, three amino acids in its active site, Asp, His, and Cys/Ser, have been proposed to participate cooperatively (Figure 2b). It is believed that the acid polarizes His, which simultaneously activates the nucleophile, Cys, for attack on the carbonyl carbon of the peptide bond. Thus, the proximity of all three amino acids is of paramount importance for successful catalysis.
Similarly, the pores of MTV-(CH3)0.6(CH2NH-Asp-His-Cys-NH2)0.1 contain not only the target tripeptide but also a complex MTV mixture of “byproducts” remaining from the sequential loading of amino acids such as H2N-Cys-His-CONHL (Figure 2a). We anticipated that this complexity, and the steric constraints it provides within the pores of MTV-IRMOF-74-III, would help to induce the substrate conformation necessary to recreate the active site of the TEV endopeptidase.
The catalytic activity of MTV-(CH3)0.6(CH2NH-Asp-His-Cys-NH2)0.1 (Cat. C) was investigated for selective cleavage of the peptide bond of serine in the pentapeptide H2N-Ala-Tyr-Ala-Ser-Ala-CONH2 (1, Figure 2a; Tyr = tyrosine). After 24 h at room temperature, we observed the presence of the expected cleavage product H2N-Ala-Tyr-Ala-CO2H (3) (Figure 2a; calcd m/z 323.2 Da, found 323.9 Da) by ESI-MS analysis of the reaction supernatant. The conversion was found to be approximately 5% by HPLC analysis (Figures S47 and S51, SI). Under the same conditions, neither of the performed control reactions using the molecular analogue of the tripeptide, H2N-Cys-His-Asp-CONH2, nor the unfunctionalized MOF showed the formation of the cleavage product. Work is ongoing to improve conversion; however, this preliminary result shows that molecular control of the sequence of peptides in the MTV functionalized MOF allows for the recreation of complex, spatially induced catalytic transformations. We further observed that the PXRD pattern of the sample after these catalytic reactions (chlorinations or peptide cleavage) was identical to that of the activated sample, thus indicating full preservation of MTV-(CH3)0.6(CH2NH-Ala-Gly-Pro-NH2)0.2 and MTV-(CH3)0.6(CH2NH-Asp-His-Cys-NH2)0.1 structures (Figures S18 and S19, SI).
Supplementary Material
Acknowledgments
This work was partially supported for synthesis by BASF SE (Ludwigshafen, Germany), catalytic reactions by U.S. Department of Defense, Defense Threat Reduction Agency (HDTRA 1-12-1-0053), and gas adsorption studies by the Center for Gas Separations Relevant to Clean Energy Technologies, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Basic Energy Sciences (DE-SC0001015). D.A.N. was supported by the National Institutes of Health under a Kirschtein National Service Award (F32GM097956).
Footnotes
Supporting Information: The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b04204.
Notes: The authors declare no competing financial interest.
References
- 1.Fracaroli AM, Furukawa H, Suzuki M, Dodd M, Okajima S, Gándara F, Reimer JA, Yaghi OM. J Am Chem Soc. 2014;136:8863. doi: 10.1021/ja503296c. [DOI] [PubMed] [Google Scholar]
- 2.Up to four post-synthetic reactions have been carried out on a MOF. Garibay SJ, Wang Z, Tanabe KK, Cohen SM. Inorg Chem. 2009;48:7341. doi: 10.1021/ic900796n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deng H, Grunder S, Cordova KE, Valente C, Furukawa H, Hmadeh M, Gándara F, Whalley AC, Liu Z, Asahina S, Kazumori H, O'Keeffe M, Terasaki O, Stoddart J, Yaghi OM. Science. 2012;336:1018. doi: 10.1126/science.1220131. [DOI] [PubMed] [Google Scholar]
- 4.Carrington JC, Dougherty WG. Proc Natl Acad Sci U S A. 1988;85:3391. doi: 10.1073/pnas.85.10.3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Canivet J, Aguado S, Schuurman Y, Farrusseng D. J Am Chem Soc. 2013;135:4195. doi: 10.1021/ja312120x. [DOI] [PubMed] [Google Scholar]
- 6.Deng H, Doonan CJ, Furukawa H, Ferreira RB, Towne J, Knobler CB, Wang B, Yaghi OM. Science. 2010;327:846. doi: 10.1126/science.1181761. [DOI] [PubMed] [Google Scholar]
- 7.Zhang YB, Furukawa H, Ko N, Nie W, Park HJ, Okajima S, Cordova KE, Deng H, Kim J, Yaghi OMJ. Am Chem Soc. 2015;137:2641. doi: 10.1021/ja512311a. [DOI] [PubMed] [Google Scholar]
- 8.(a) Cohen SM. Chem Rev. 2012;112:970. doi: 10.1021/cr200179u. [DOI] [PubMed] [Google Scholar]; (b) Tanabe KK, Cohen SM. Chem Soc Rev. 2011;40:498. doi: 10.1039/c0cs00031k. [DOI] [PubMed] [Google Scholar]; (c) Garibay SJ, Cohen SM. Chem Commun. 2010;46:7700. doi: 10.1039/c0cc02990d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Lun DJ, Waterhouse GIN, Telfer SG. J Am Chem Soc. 2011;133:5806. doi: 10.1021/ja202223d. [DOI] [PubMed] [Google Scholar]; (e) Tanabe KK, Cohen SM. Peptides were introduced into MOFs by one post-synthetic modification step Angew Chem, Int Ed. 2009;48:7424. [Google Scholar]; (f) Bonnefoy J, Legrand A, Quadrelli EA, Canivet J, Farrusseng D. J Am Chem Soc. 2015;137:9409. doi: 10.1021/jacs.5b05327. [DOI] [PubMed] [Google Scholar]
- 9.(a) Sheppard R. J Pept Sci. 2003;9:545. doi: 10.1002/psc.479. [DOI] [PubMed] [Google Scholar]; (b) Behrendt R, White P, Offer J. J Pept Sci. 2016;22:4. doi: 10.1002/psc.2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Phan J, Zdanov A, Evdokimov AG, Tropea JE, Peters HK, Kapust RB, Li M, Wlodawer A, Waugh DS. J Biol Chem. 2002;277:50564. doi: 10.1074/jbc.M207224200. [DOI] [PubMed] [Google Scholar]
- 11.(a) Gustafson JL, Lim D, Miller SJ. Science. 2010;328:1251. doi: 10.1126/science.1188403. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) MacMillan DWC. Nature. 2008;455:304. doi: 10.1038/nature07367. [DOI] [PubMed] [Google Scholar]; (c) Mukherjee S, Yang JW, Hoffmann S, List B. Chem Rev. 2007;107:5471. doi: 10.1021/cr0684016. [DOI] [PubMed] [Google Scholar]; (d) Bertelsen S, Jørgensen KA. Chem Soc Rev. 2009;38:2178. doi: 10.1039/b903816g. [DOI] [PubMed] [Google Scholar]
- 12.Brochu MP, Brown SP, MacMillan DWC. J Am Chem Soc. 2004;126:4108. doi: 10.1021/ja049562z. [DOI] [PubMed] [Google Scholar]
- 13.Zhao C, Toste FD, Raymond KN, Bergman RG. J Am Chem Soc. 2014;136:14409. doi: 10.1021/ja508799p. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.