

Abstract
Polypharmacology is beginning to emerge as an important concept in the field of drug discovery. However, there are no established approaches to either select appropriate target sets or design polypharmacological drugs. Here, we propose a structural-proteomics approach that utilizes the structural information of the binding sites at a genome-scale obtained through in-house algorithms to characterize the pocketome, yielding a list of ligands that can participate in various biochemical events in the mycobacterial cell. The pocket-type space is seen to be much larger than the sequence or fold-space, suggesting that variations at the site-level contribute significantly to functional repertoire of the organism. All-pair comparisons of binding sites within Mycobacterium tuberculosis (Mtb), pocket-similarity network construction and clustering result in identification of binding-site sets, each containing a group of similar binding sites, theoretically having a potential to interact with a common set of compounds. A polypharmacology index is formulated to rank targets by incorporating a measure of druggability and similarity to other pockets within the proteome. This study presents a rational approach to identify targets with polypharmacological potential along with possible drugs for repurposing, while simultaneously, obtaining clues on lead compounds for use in new drug-discovery pipelines.
Terms such as ‘potentiation’, ‘synergistic action’, ‘adverse effects or side effects’ or ‘idiopathic and idiosyncratic effects’, are encountered very frequently in the field of pharmacology. It has widely been recognized that many drugs exhibit complex pharmacologies1,2. However complete molecular bases for many of these are not as yet delineated. Complexity in drug action is best explained by appreciating that drug molecules often interact with multiple proteins3,4. While many unintended interactions lead to adverse pharmacological effects and are hence undesirable5, some others exhibit beneficial and synergistic effects and are therefore highly desirable6. While it is common knowledge that drugs cause adverse effects or side effects, beneficial effects due to plurality of interactions by drug molecules with selected targets are only now beginning to be appreciated. As a result, the term ‘polypharmacology’ has been coined3 which aims to achieve the desired therapeutic effect through modulation of more than one target typically by a single drug, in contrast to the term ‘polypharmacy’ which refers to achieving the therapeutic effect through a combination of drugs acting on different targets. A systems perspective is essential to understand polypharmacology, since it is essentially an emergent property of the system as a whole7. Examples of drugs affecting multiple targets include clozapine; a drug used to treat schizophrenia, also known to interact with both dopaminergic and serotonergic receptors8; methadone, a known μ-opioid receptor agonist that also inhibits NMDA leading to more effective action against neuropathic pain9. Similarly, imatinib (Gleevec), a widely used anticancer drug designed to inhibit BCR-Abl1, a defective tyrosine-kinase protein expressed in chronic myelogenous leukemia (CML) condition due to abnormal chromosomal rearrangement, is also known to affect receptor tyrosine kinases (RTK), that include platelet derived growth factor receptor (PDGFR) and c-kit transmembrane kinase, perhaps contributing to its efficacy10. A similar effect is observed in case of valproic acid, an approved drug to treat bipolar disorders by possibly acting on voltage-dependent sodium channels. It is additionally known to inhibit histone deacetylases, gamma-amino butyric acid receptors, possibly also cyclooxygenase and effective for treatment of tumors and Alzheimer's disease as well11,12.
It is clear from these examples that targeting multiple targets simultaneously holds promise for achieving higher therapeutic efficacy than with one best target at a time for treatment of multi- factorial diseases. The problem then can be translated to selecting multiple targets that are amenable for manipulation with a single drug and then rationally design polypharmacological drugs. Identification of such target sets poses several challenges. An approach capturing both global perspectives using systems biology methods and simultaneously atomic level detail of individual molecules in the proteome using structural analyses is necessary to first understand the basis for polypharmacology and then to predict or even design such behavior in new drugs. As a case study, for M. tuberculosis, the causative organism of tuberculosis, we illustrate how concepts of polypharmacology can be understood and applied in a systematic manner by adopting a structural proteomics approach that analyses binding sites at a genome scale and identifies promising drug candidates from approved and potential drug databases.
Tuberculosis (TB) causes around 8.6 million new infections and 1.3 million deaths every year and has been one of the largest killers among infectious diseases for several decades now, despite the availability of a handful of chemotherapeutic agents, the BCG vaccine and an extensive effort by the medical community to tackle the disease13. The situation warrants discovery of newer drugs to combat the causative pathogen Mycobacterium tuberculosis (Mtb). Important problems confronting treatment of tuberculosis are prolonged therapy, emergence of drug resistance, and co-morbidity with immunosuppressive diseases, such as HIV. This is in addition to the problem of the latency, which refers to the ability of the pathogen to enter and reside in a dormant state, inaccessible to conventional therapy that can reactivate to an infectious state, even after several decades. A survey of the mechanism of action of clinically used drugs currently indicates that these drugs act through only a handful of target proteins, covering only a small percentage of biological processes in the microbe. Several studies have indicated that there are many more proteins and processes that could be potentially targeted, but are yet to be exploited systematically14,15,16,17,18,19,20.
The objective of this study is to detect and characterize the pocketome21 of Mtb with a structural perspective so as to identify strategic target sets for polypharmacology. The pocketome here refers to the sum total of all the putative binding sites within the proteome and a ‘target set’ refers to the groups of targets sharing similarity in their ligand binding pockets. To address these aspects, two critical inputs are required. First, structural models at a proteome scale and second, powerful and efficient computational tools to mine such proteome scale structural data are both required. Recently, we have built structural models covering 70% of the Mtb proteome, which we utilize for this study. We also take advantage of the suite of computational algorithms that have been recently developed in our laboratory for binding site detection22, comparison23,24 and functional characterization25. Using automated methods for comparing binding pockets23,24, a first level function annotation was obtained in that study26. Here, we report (a) characterizing the pocketome to obtain proteome-wide ligand associations, (b) identifying number of pocket types present in Mtb proteome, (c) identifying clusters of similar pockets in the proteome, (d) estimating druggability among such pocket clusters and (e) identifying target sets with a polypharmacological potential. We also take advantage of an earlier study in the laboratory that identified potential drug targets using a multilevel pipeline19 integrating systems level interactome analysis, sequence and structural uniqueness as compared to the host genome along with experimentally derived gene essentiality data27,28. The 451 targets identified through these multiple filters are now assessed for their polypharmacological potential and for estimating druggability with a new perspective. We thus, develop a novel approach to identify target sets suitable for polypharmacological intervention and demonstrate that rational selection of polypharmacological targets is theoretically possible, which holds promise for rational design of polypharmacological drugs. The approach is generic and has the potential to be applied widely in drug discovery.
Methods
Proteome-scale structural models and pocketome detection
Structural models of the Mtb proteome were obtained from a recent study by our group26. Crystal structures of 324 Mtb proteins available in PDB and 2737 comparative models that we generated in that study together account for about 70% of the proteome. Since the reliability of the protein structural models is central to all the analysis being performed in this study, utmost care was taken to choose only reliable protein structure models. Methods for structure verification included statistical scoring potential29,30, secondary structure compatibility31 and stereochemical quality check32. Multi-domain protein structures were also included wherein the models of various regions of proteins are present. However only those binding sites that were largely contained within the domains are analyzed here, which leaves out those sites that may be present at the interfaces. This holds for oligomeric proteins as well where each subunit is modeled separately and a template for the whole complex is unavailable. More information on these structures can be found at http://proline.biochem.iisc.ernet.in/mtbpocketome/materials.php.
Identification of binding sites
Different algorithms are available for detection of binding sites in protein structures. Consensus identification from different methods was used to detect the high-confidence pockets from the proteome. The individual methods used for this are PocketDepth (PD)22, a grid-based geometric method, Ligsite33, that captures evolutionary information and SiteHound34, an energy based method. PD, an in-house method that uses a depth-based clustering algorithm for detecting putative binding sites in the given protein structures, where a notion of centrality of empty subspaces in the protein defines depth, was initially used to obtain pockets. This algorithm was combined with LIGSITEcsc33, which captures surface-solvent-surface events involving grooves using Connolly's surface35 and maps the degree of conservation of the residues in the selected surface to detect binding sites in a given protein. In addition to the pockets identified by these methods, binding sites were also selected based on the experimental information available directly for that protein or inferred from its homologues. For this, database entries were mined using the respective general feature format files (GFF) obtained from Uniprot database36 (workflow in Figure S1). Finally, known binding motifs documented in Prosite37 were scanned against each protein sequence in the proteome to identify possible binding sites.
Genome-wide binding site comparisons
The binding sites obtained were compared using an in-house algorithm – PocketMatch (PM)23. PM computes shape descriptors of the pockets and compares sorted arrays of all-pair distance elements grouped into 90 combinations of chemical type pairs to calculate a combined similarity score between pairs of binding sites. All–pair combinations of 13858 binding sites that involved over 192 million comparisons could be accomplished using PM on a Intel(R) Core(TM) x86_64 i7-2600 CPU @ 3.40 GHz with Linux Mint 14 platform. Two types of scores are reported from pair-wise comparison of binding sites – PMIN, that captures local similarities and the PMAX score that captures global similarities of the pocket as a whole, along with a measure of statistical significance for each score. From our previous studies we know that PMAX score of ≥0.4 reflects meaningful similarities in binding sites, while PMAX ≥ 0.6 denotes meaningful and significant levels of similarities25. A default cut-off of PMAX ≥ 0.6 is used in this study. However, depending upon the question addressed, and the level of stringency required at a particular step the threshold has been varied for specific analyses, and explicitly stated in the relevant sections (Table S1). A statistical significance is also computed for each comparison as described by us previously25. A p-value threshold of 1E-04 has been adopted to identify statistically significant similarities.
Binding site similarity network construction and clustering
To represent similarities in the pocketome, a network formulation was used. Each binding site in the pocketome is represented as a node whereas similarities between pairs of sites are represented as edges (Network-type 1, Table S2). Clustering is performed on this network to group similar binding sites. MCODE algorithm38, is a well-known automated method to detect highly interconnected subgraphs/clusters within a given network (node score cutoff = 0.2, K-core value = 2 and max depth = 100) through a Cytoscape39 plugin. Each cluster obtained from this analysis is referred to as sets. Although there exist many tools for obtaining the highly connected subcomponents from the network40,41, many of these including MCODE face the problem of resolution level of clusters42. This problem can be alleviated to some extent in this case of binding site similarity network by increasing the cut-off, which has been set to PMAX ≥ 0.7. Invariably exact number of clusters obtained is dependent on the thresholds used irrespective of the clustering method. With a threshold set at such high level, the clusters identified are of high confidence although it comes at the cost of losing some information on site similarity below the threshold. In addition we establish the biological significance of the threshold used by carrying out the same analysis on the PDB pockets derived from MOAD database which resulted in obtaining meaningful clusters with highly similar ligands as judged by average Tanimoto chemical similarity ~ 0.8, and hence we proceeded with the workflow. Other network properties such as disconnected components, degree distribution, clustering coefficient, betweenness and Eigen centrality are calculated using the igraph package43. To answer the precise question being addressed, a suitable network formulation is used. Description of the network variants constructed in this study along with the specific purpose is given in Table S2.
Sequence-structure-pocket comparisons
Pairwise structure comparison was carried out using TM-Align software44. TM-Align compares a given pair of folds and reports an optimal alignment. Sequence similarity for each pair was then computed from the corresponding sequences using BLAST245, a widely used tool for alignment of sequences. Sequence and structure similarity scores were calculated only for pairs of proteins with significant pocket similarities (PMAX of ≥0.60). The pocket-similarity score; sequence-similarity score and structure-similarity score (TM Score) were then used as axes to plot a 3D scatterplot. A TM-score of ≥0.4 is known to indicate significant simlarity44 and is the suggested cut-off for this algorithm. The data points were manually binned into three-different categories: (a) low structural and low sequence similarity (TMScore < 0.4 and sequence identity < 30%), (b) high structural but low sequence similarity (TMScore > 0.4 and sequence identity < 30%) and (c) high structural and high sequence similarity (TMScore > 0.4 and sequence identity > 30%).
Drug binding sites
A combined list of drugs or drug-like compounds was prepared from DrugBank46 and DrugPort (http://www.ebi.ac.uk/thornton-srv/databases/drugport/). These included approved drugs, experimental drugs and nutraceuticals. The binding sites were then extracted from these complexes by considering complete residues of all atoms that lie within 4.5Å of any atom from the drug molecule. 10658 drug-binding sites reported in Drugbank and 2516 reported in Drugport were obtained from PDB through this process (full list is provided at http://proline.biochem.iisc.ernet.in/mtbpocketome/methods.php) and is referred to as ‘knowndrug-sites_DB’ here after. These known drug-binding sites were then scanned for similarities against different binding site clusters and also against high-confidence targets from Mtb. A subset of ‘approved drug-sites’ containing 399 compounds and 3112 binding sites is also derived from ‘knowndrug-sites_DB’.
Polypharmacological index (PPI)
A polypharmacology druggability score referred to as PPI was computed for each binding site by considering three aspects: (a) to score positively the similarity of the sites to other sites in the pocketome and thus contribute to polypharmacological profile of the target, (b) to score positively for those sets of sites that resemble any ‘approved drug-sites’ and thus captures druggability and (c) to penalize those sites that exhibit similarity to cofactor binding sites since that would increase chances of adverse polypharmacology7,47, thus capturing specificity. The first and second aspects refer to desired attributes and hence get a positive score, while the third aspect is penalized so as to improve specificity. For this, a separate dataset of 29215 cofactor-binding sites was created from PDB (http://proline.biochem.iisc.ernet.in/mtbpocketome/methods.php). The list of cofactors were manually extracted from PDB and mined from the Cofactor Database48. Each pocket was compared to these cofactor-binding sites using PM. Each pocket was then scanned against ‘approved-drug binding sites’ described earlier. A scoring scheme was generated to rank the pockets for their druggability such that the score:
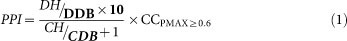 |
In equation (1), DH = No. of drug binding site hits ≥ PMAX 0.5, DDB = Size of drug binding site database (~3112), CH = No. of cofactor binding sites hits ≥ PMAX 0.6 and P-value ~ 1E-04, CDB = Size of cofactor binding sites database and CCPMAX≥0.6 is clustering coefficient of binding site derived from binding site similarity network at PMAX ≥ 0.60.
Validation
A validation component was included for each of the aspects involved in this study. Different prediction steps that have been validated are as follows: (i) pocket detection using the consensus approach, (ii) ligand associations through PM scores, (iii) clustering of similar sites from networks, and (iv) inferring drug binding from pocket level similarities. Both PD and PM algorithm have already been extensively validated through use of appropriate datasets (PD22 and PM23). Further, in this study a large-scale comparison with the crystallographically derived sites from the Procognate database49 has been carried out. In 2442 of 3209 complexes, a pocket at a similar location as well as the same ligand association is predicted. The entire site-based function annotation pipeline has also been validated in PocketAnnotate25 using apo-holo protein datasets. Put together, it is clear that methods used for binding site identification, measuring similarities between binding sites and obtaining ligand associations based on binding site similarities are sufficiently reliable.
To validate the method of clustering, a binding-site similarity network of protein-ligand complexes from PDB was constructed (Network-type 2, Table S2). The protein-ligand complexes were obtained from BindingMOAD database50 that stores the information of binding sites in the PDB. Around 16275 binding sites were derived from the database and all-versus-all (that amounts to ~132 million) comparisons were carried out using PM. A binding-site similarity network was constructed with similar cut-offs as used for Mtb pocketome (Network-type 1, Table S2) and identical protocol was followed for clustering. Around 1777 clusters were found and majority of the clusters contained binding sites specific for similar ligands as judged from their Tanimoto scores calculated from open Babel toolbox51. As many as 1410 of these clusters show an average Tanimoto score of more >0.8 for the chemical fingerprints of the ligands associated with them, reflecting that not only are the sites similar to each other within each cluster, but the ligands they bind to are also very similar. This validates the clustering algorithm as the binding sites that interact with chemically similar ligands are grouped into the same cluster.
Finally, for the predicted associations, a validation exercise was carried out to test the geometric compatibility and energetic feasibility of binding of that drug to the corresponding pocket. To do this, the predicted drug was docked onto its corresponding target using Autodock Vina52 and the intermolecular binding energy computed. Since drug associations are predicted from binding sites derived from experimentally resolved protein-drug complexes, intermolecular energies between the drug and the corresponding protein in PDB from which the binding site was derived could also be easily calculated. The intermolecular energies from the docked complexes were then systematically compared with the corresponding experimental complexes and a ratio of the two was computed in each case. 1337 docking exercises were performed and of these, in around 87% of the cases, the interaction scores obtained were similar (ratio of scores > 0.7) to that of native drug complexes. This serves to independently verify our drug association method based on binding site similarities, as it estimates the feasibility of predicted interactions in the Mtb pockets.
Complete datasets along with supporting files for this section is made available at http://proline.biochem.iisc.ernet.in/mtbpocketome/methods.php. A detailed list of all the tools and the databases used in the workflow is also listed in supplementary (Table S3).
Results
This study has resulted in obtaining a global perspective of small molecule binding sites in the proteome of Mtb. Most notably, through a single study of the pocketome, hundreds of binding sites are analyzed in detail, obtaining possible drug associations for the entire set of promising targets in Mtb, as described in the following sections. To the best of our knowledge, this is the first study that comprehensively characterizes the pocketome and the binding site similarities within it, at a genome scale for any organism.
Mapping the small molecule binding pocket space in Mtb: characterization of the Mtb pocketome
Availability of protein structures at a proteome scale and well-validated methods to identify small molecule ligand binding pockets renders it feasible to map the binding pocket space in the organism. Understanding the pocketome of Mtb provides ready answers to several questions such as (a) how many pocket or site-types are present in Mtb, (b) what are the small molecule ligands recognized by the proteome, (c) what are the relative frequencies of occurrence of sites for different small molecule ligands; (d) for how many known ligands, can sites be recognized in Mtb, (e) how many binding sites in Mtb are unique as compared to known binding pockets in PDB and (f) how does site-typing relate to sequence or structural fold based classification.
From the three site detection algorithms, 9029 pockets from 2809 proteins were chosen as consensus pockets. To this, 801 new binding pockets were added based on prior annotation in sequence databases. In addition, 4240 new sites from sequence motif searches in Prosite were also added. It must be noted that most sites added on from sequence based searches were also identified from structure based approaches but were not selected at that stage itself since they were not consensus predictions, meaning that at least one computational method failed to identify them. Full lists of Mtb protein structures and sites identified through different methods and information on other resources used are made available through a website - http://proline.biochem.iisc.ernet.in/mtbpocketome/materials.php. Overall, 13858 high confidence pockets were derived from the structural information currently available on all the proteins in Mtb. This includes 2877 sites, one each from the protein structural models of Mtb, that was recently studied in our laboratory, with a goal of obtaining structural annotation of the proteome26. Since the objective here is to define and characterize the pocketome, all consensus pocket predictions as well as all those with different experimental direct and indirect clues are included, which average to about 4 pockets per protein.
The pockets thus obtained are then analyzed for their ligand recognition properties by comparing them to known binding sites derived from PDB. Around 6906 pockets exhibited significant similarity in the entire pockets to some or the other known binding site in PDB (Table S4), leading to deriving ligand associations for about 50% of the pocketome. In addition partial similarity is observed for 4695 more pockets as judged by PMIN score > 0.5, together covering about 84% of the pocketome. Not surprisingly, these ligand associations capture most of the reported biochemical reactions in Mtb. Figure 1A describes the coverage of the structural information available for proteins and the ligand annotations obtained for them in terms of KEGG pathways. Figure 1B depicts the complete metabolic map currently known for Mtb from KEGG53,54,55. Highlighted in this map are proteins for which (a) structural models are available (edges colored in black) and (b) ligands whose associations with the proteins are characterized (red). The coverage of the reactome from this approach is seen to be high, indicating that most of the enzymes participating in cellular metabolism has been sufficiently captured in terms of enzyme structures that catalyze different reactions along with the information of binding site residues that could be involved in the molecular recognition of corresponding ligands. These can be interactively explored at http://proline.biochem.iisc.ernet.in/mtbpocketome/pathways.php.
Figure 1. An overview of the characterization of the enzymes in the Mtb pocketome, in terms of binding site analysis.

(A) A stacked bar plot showing the coverage of protein structures and the confident ligand associations available with respect to the KEGG pathways. For each pathway the lower most bar in the stack corresponds to the number of genes or proteins in the pathway, the middle bar indicates the number of structural models available for the pathway and the top most stack indicates the number of proteins for which ligand annotations are made based on binding site structures. Each stack corresponds to one KEGG pathway in Mtb. (B) Metabolic map of central metabolism in Mtb, indicating extensive coverage of ligand annotation in the Mtb reactome from this study. The edges colored in black indicates the availability of protein structure catalyzing the reaction and the nodes colored in red represent the small ligand molecules taking part in the reaction for which the binding site has been mapped onto the respective protein structure.
Given that this analysis is carried out at a genome scale, it is possible to analyze the frequency of occurrence of different ligand binding sites. Fig. 2, which illustrates this, serves qualitatively as a computational equivalent of a metabolome spectrum that can be obtained from a mass spectrometer for unit abundances of each binding site. The ligands are arranged according to their molecular weight on the x-axis. However, it must be noted that Figure 2 is derived using a novel methodology using structure-based function annotation concepts. The most frequently observed ligands through this approach turn out to be NAD followed by ADP, FAD and ATP. Ligands that can bind to the pocketome span a wide range of sizes, the smallest detected being 74 Da (tertiary-butyl alcohol) to about 1416 Da (bleomycin).
Figure 2. An illustration of ligand associations for Mtb pocketome.

Distribution of different ligand hits obtained for the predicted pockets in the proteome. The ligands are ordered by their molecular weights. The frequency on the Y-axis indicates the number of occurrences of the binding site of that ligand in the Mtb pocketome. This spectrum is qualitatively equivalent to the mass spectrum of the Mtb metabolome for unit protein abundances.
Binding site space of Mtb proteome is much higher than the sequence or the fold space
Next, to determine how many unique pocket types are present in Mtb proteome, we compared all detected binding pockets of Mtb to each other. We construct a binding site similarity network56,57 with binding sites as nodes that are connected by edges only if the corresponding pair shared a similarity (Network-type 1, Table S2). The sets of highly connected components were then identified from the network, through MCODE algorithm38. This exercise yielded 29 clusters from the connected components in the network (Figure 3A), while the remaining proteins that shared no similarity with any other in the proteome were all singletons. Validation of our approach involving measure of binding site similarities, network construction and clustering, was carried out by applying an identical protocol to the MOAD dataset, a subset of protein-ligand complexes in PDB, from which expected clustering pattern was obtained as shown in Figure 3B (see methodology section on validation). We refer to the 29 clusters obtained from the Mtb pocketome as ‘sets’ of proteins containing similar binding sites within each. By considering one representative of each set and adding onto the singletons, we find that the pocketome contains 6584 different types of pockets. We note that the exact number of unique site types is critically dependent on the site-similarity cut-off that is used. If the PMAX cut-off is lowered, the number of site-types become fewer due to partial similarities, but reduces sensitivity of the typing. If the PMAX cut-off is increased, the number of unique types increases significantly, leaning towards placing individual sites as singletons and thus of little use for understanding similarities. Since the purpose of site typing in this study is to identify group of sites that can bind to similar type of ligands, we use a cut-off of 0.6 PMAX, which we know from our earlier benchmarking analysis (PM), to be a cut-off that implies a high possibility of two sites recognizing a same ligand. In any case, all-pair similarities at different cut-offs are captured in Figure 4. Since an all vs all comparison of binding sites resulted in about 96 million comparisons, its visualization and interpretation became challenging. To capture the essence of pocketome-wide comparisons, we have utilized the hexbin density plot (Figure 4A) for visualization that illustrates the density distribution of PM global (PMAX) versus local similarity scores (PMIN) of all comparisons (Figure 4A and 4B). We observe that of the 96 million unique pairs (of 192 million pair combinations), only a tiny fraction (Figure 4D) - 0.4%, resemble each other closely in their entire sites and about 60% more exhibit part-similarity (PMIN > 0.5) to each other. The fact that these pockets group into 6584 unique site-types indicate that the proteome is capable of atleast 6584 binding modes of small molecule recognition (Supplementary Text 1).
Figure 3. Binding Site Similarity networks.

(A) The binding site similarity network obtained for Mtb Pocketome. Each node represents the predicted binding site and an edge between two nodes represents high similarity shared (PMAX ≥ 0.7) between them. The colors represent different clusters or sets of binding sites predicted by MCODE algorithm. (B) Binding site similarity network of pockets obtained from MOAD dataset, carried out as a validation exercise. The color of the nodes again depicts set of similar binding sites obtained from MCODE algorithm. Three such example clusters binding to ATP, heme and phosphoglycerate respectively are shown in enlarged version.
Figure 4.

An overview of all-pair binding site similarities in Mtb Pocketome representing the results of 96 million comparisons (A) Hexbin plot depicts the distribution of all-pair similarity scores obtained using PocketMatch. The y-axis depicts the local or partial binding site similarity scores (PMIN) and the x-axis depicts the global-similarity scores (PMAX). The color of the hexbin represents the density of the scores obtained and is shown in the legend next to the plot. (B) Distribution of all-pair PMIN scores. (C) Distribution of all-pair PMAX scores. (D) Degree distribution of the sites in Mtb binding site similarity network, indirectly capturing number of similar sites.
Typing of binding sites immediately begs a question as to whether these could be detected by sequence and fold analyses alone. In order to see how many sequence types and similarly how many fold types constitute the Mtb proteome, all-pair sequence and fold comparisons were carried out. For each of the pocket pairs with significant similarity, values obtained from their corresponding sequence and fold level comparisons are plotted in Figure 5. It can be seen from the figure that protein pairs exhibiting similar sites do not in many cases share either sequence or fold level similarities. Hence identifying similar ligand binding properties in pairs of proteins are not obvious from sequence and fold comparisons in many cases. The fact that Mtb proteome consists of around 1831 unique sequences in terms of Pfam domains58, ~400 unique structural folds and about 1213 ligands, but 6584 binding site types, clearly indicate that the binding site space is much larger than the sequence or the fold space. The 6584 site types bind to the 1213 ligands and probably more yet to be characterized. Observation of these many different pocket types is suggestive of different modes of ligand recognition evolved to cater to specific functional requirements. Such fine-grained typing helps to understand specific ligands of the same class that the proteins can discriminate against. Mtb is known to be a highly redundant genome, with several paralogues for many proteins. Observations of subtle differences in binding sites are clearly indicative of the fine modulation of the ligand varieties required for specific molecular recognition.
Figure 5. An illustration of the structure-sequence-pocket space relationships in Mtb proteome.

The 3D scatterplot depicts the distribution of high similarity pockets with respect to sequence and structural similarity scores obtained for the corresponding proteins. The color represents different categories of sequence-structure relationship and an example is highlighted from each of these categories with the depiction of proteins and pockets similarity.
Figure 5 also shows illustrative examples for binding site similarities observed at three different cases:(a) high sequence similarity and same-fold pairs representing the paralogue pairs in Mtb (b) low sequence similarity and same-fold pairs, and (c) low sequence similarity and different-fold pairs. The first case has been illustrated with an example of fibronectin proteins. There are three fibronectin binding proteins within Mtb (FbpA, FbpB and FbpC), all known to have mycolyl-transferase activity involving transfer of long-chain fatty acids to trehalose derivatives, resulting in high affinity of mycobacteria towards fibronectin. The structural superposition of two such proteins – FbpA(Rv1886c) and FbpB(Rv3804c), along with their pocket alignment is illustrated in Figure 5, showing high similarity in their pockets as might be expected and in a way serves as a positive control for the analysis. The second case involving pair of proteins sharing high structural and pocket similarity despite low sequence identity has been illustrated by an example of MbtB, a phenyloxazoline synthetase and Rv3087, a possible triacylglycerol synthase. Both these proteins are predicted to adopt CoA-dependent acyl transferase fold and further share similarity between correspondingly predicted pockets as depicted in Figure 5. In the third case, high pocket similarity scores were observed for protein-pairs with no sequence or structural similarity. As an example illustrated here, a pocket in farnesyl pyrophosphate synthase (Rv1086) was found to share a significant similarity with another pocket in glycerol-3-phosphate dehydrogenase (Figure 5). Both these genes are indirectly involved in lipid metabolism, and this similarity can possibly be exploited in structure-based drug discovery, as lipid metabolism is crucial for survival of Mtb.
Identifying polypharmacological target sets from Mtb binding site similarity network
Analysis so far has identified binding pockets in Mtb proteome, estimated all-pair similarities among then and clustered sets of sites with significant similarities. The 29 binding site sets, thus identified, presents an opportunity to rationally select polypharmacological targets among them. It must be noted that the number of sets obtained can vary with the clustering algorithm used and PMAX cut-off defined to draw the edge due to inherent property of similarity network and the cluster resolution. Higher confidence is more important than the precise number of clusters. Hence we err on the side of caution and use a stringent threshold for deriving clusters. The proposed threshold was validated by carrying out similar analysis on pockets derived from MOAD dataset that resulted in obtaining sets containing highly similar chemical entities (average Tanimoto chemical similarity of ~0.8). Hence, we are confident about the similarity relationship that exists within the derived 29 sets through this workflow. Figure 3A illustrates binding site similarity network and 29 distinct sets highlighted in different colors containing at least three sites in each set (superposition of binding sites within sets – Figure S2). Functional enrichment analysis carried out for each of these sets, indicate that proteins in these sets are well distributed across eight Tuberculist59 functional classes and across 80 functional ontological terms, implying that these sites mediate a variety of functions (Table 1). The most abundant tuberculist category in the list is of intermediary metabolism and respiration, cell wall and cell processes followed by lipid metabolism.
Table 1. Binding Site Sets: A list of the proteins in the 29 binding sites sets, along with ontological terms associated with them. For each set, high scoring Drugbank hits are also listed. The proteins recognized as targets from targetTB study are highlighted in blue.
| Binding Site Set | Accession Numbers | Ontology Terms | DrugBank Hit |
|---|---|---|---|
| Set 1 | Rv0015c;Rv0480cRv0973c;Rv0980cRv1004c;Rv1158cRv1230c;Rv1747Rv2407;Rv2589Rv2908c;Rv3629c | negative regulation of lipid biosynthetic process;protein phosphorylation;arylsulfatase activity;positive regulation of DNA binding;regulation of cell shape;plasma membrane;methylcrotonoyl-CoA carboxylase activity;negative regulation of catalytic activity;positive regulation of catalytic activity;protein serine/threonine kinase activity;protein kinase activity;protein autophosphorylation;protein binding;growth;cytosol;cell wall;extracellular region; | Indolacetamide derivative (IOK); CO-5-Methoxybenzimidazolylcobamide (B1M); |
| Set 2 | Rv0620;Rv1276cRv1317c;Rv2524c | galactokinase activity;cell wall;cytosol;plasma membrane;growth; | Progesterone (1CA);Benzenoid derivative (FHM);Colchicine (LOC);Quinolinone derivate (LGD);Benzonitrile (LGB); |
| Set 3 | Rv1272c;Rv2192cRv2276;Rv2690c | anthranilate phosphoribosyltransferase activity;tryptophan biosynthetic process;carbon monoxide binding;heme binding;oxidoreductase activity;oxidation-reduction process;plasma membrane;growth;extracellular region; | Bisadenosine-pentaphosphate (AP5);Biliverdine (BLA);Molybdenum (PCD);Pterine cytosine nucleotide (MCN);Succinyl coA (SCA); |
| Set 4 | Rv0089;Rv0761cRv0849;Rv1761cRv1863c;Rv2194Rv2679;Rv2728c | heme binding;electron carrier activity;enoyl-CoA hydratase activity;electron transport;iron ion binding;plasma membrane;integral to plasma membrane;growth;cell wall; | Cyanocobalamin (CNC);FAD-isobutylketone (FAB);Pyrimidine ethanone adduct (TDT);HydroxyFAD (6FA); |
| Set 5 | Rv0527;Rv3319Rv3541c | cytochrome complex assembly;succinate dehydrogenase activity;membrane;plasma membrane;growth; | Phosphatidylethanolamine (PEE);Progesterone (1CA); Stearic acid (OLA);Colchicinoid derivative (CN2);Hexadecanal (PLM); |
| Set 6 | Rv2897c;Rv3521Rv3825c;Rv3871 | fatty acid biosynthetic process;cellular response to iron ion starvation;response to host immune response;plasma membrane;cell wall;cytosol; | FAD-isobutylketone (FAB);Adenosine-pentaphosphate (UP5);Uridine-diPO4-acetylgulcosamine(UD1);Demethyl-dimethylamino FAD(RFL);4-oxo NAD-phosphate(ODP); |
| Set 7 | Rv0435c;Rv0941cRv1601;Rv3206cRv3791 | cysteine biosynthetic process;sulfotransferase activity;catalytic activity;active evasion of host immune response;plasma membrane;growth; | Cyclohexyl-beta-d-maltoside (MA4);Metylisoxazole (W35);Stearic acid (OLA); myristic acid (MYR); Phosphatidyl ethalnolamine (EPH); |
| Set 8 | Rv0538;Rv2393Rv2813;Rv3242cRv3923c | ribonuclease P activity;sirohydrochlorin ferrochelatase activity;growth; | 3-Acetylpyridine adenine dinucleotide (A3D);3-fluoro-5-morpholin-4-yl-N-benzamide (WBT);Nicotinamide Adenine Dinucleotide cyclohexanone (NDC);Lapatinib (FMM);Eicosapentanoic acid (EPA); |
| Set 9 | Rv0166;Rv0732Rv0978c;Rv1457cRv1795;Rv1941Rv2209 | NA | 3-Acetylpyridine adenine dinucleotide (A3D);Spironolactone (SNL);Aleglitazar (RO7); Nicotinamide Dinucleotide cyclohexanone (NDC);Quinolinone derivative (LGD); |
| Set 10 | Rv0274;Rv1141cRv2842c;Rv3011c | enoyl-CoA hydratase activity;plasma membrane; | 3-Acetylpridine adenine dinculeotide (A3D);Analogue of Indinavir (XN2);Pyrimidine ethanone adduct (TDT);Demethyl-dimethylamino FAD (RFL);5,6-dihydroxy-NADP (NZQ); |
| Set 11 | Rv0194;Rv0343Rv1675c;Rv1838c | efflux transmembrane transporter activity;regulation of transcriptionDNA-dependent;regulation of growth;DNA binding;plasma membrane;cell wall;cytosol; | Cyclohexyl-beta-d-maltoside (MA4);Oleic acid (OLA);Isoquinolinone derivative (IZA);Isoindoloindolone derivative (CZA); Ethoxypyridine acetamide derivative (877); |
| Set 12 | Rv0687;Rv2158cRv2629;Rv3072c | response to hypoxia;cytosol;plasma membrane; | FAD-isobutylketone (FAB); Analogue of Indinavir (XN2);Tipranavir (TPV);Pyrimidine ethanone adduct (TDT); Carmbamic acid tert-butyl ester (Q50); |
| Set 13 | Rv0904c;Rv1358Rv2988c;Rv3700c | mycolic acid biosynthetic process;protein binding;plasma membrane; | Retronavir (RIT);Lopinavir (AB1); Analogue of Indinavir (XN2);3-fluoro-5-morpholin-4-yl-N-benzamide (WBT); Benzamide derivative (P40); |
| Set 14 | Rv0148;Rv0291Rv0557;Rv1863cRv1918c;Rv3910 | integral to plasma membrane;growth;plasma membrane;extracellular region; | Oleic acid (OLA); Adenosine-5-Diphosphoribose (APR); Colchicinoid derivative (CN2);Dimethyl phenyln propanamide derivative (G1L); Pyridine acetamide derivative (877); |
| Set 15 | Rv1086;Rv2383cRv2982c | Z-farnesyl diphosphate synthase activity;di-transpoly-cis-decaprenylcistransferase activity;glycerol-3-phosphate dehydrogenase [NAD(P)+] activity;phosphopantetheine binding;polyprenol biosynthetic process;acyl carrier activity;siderophore biosynthetic process;cellular response to iron ion starvation;manganese ion binding;plasma membrane;magnesium ion binding;response to host immune response;pathogenesis;cytosol; | Phosphatidyl ethanolamine (PEE); Colchicinoid derivative (CN2);Oleic acid (OLA); Progesterone (1CA); Propanamide derivative (FHM); |
| Set 16 | Rv1125;Rv1248cRv3729 | 2-oxoglutarate decarboxylase activity;oxoglutarate dehydrogenase (succinyl-transferring) activity;pyruvate dehydrogenase complex;tricarboxylic acid cycle;electron carrier activity;oxidation-reduction process;plasma membrane;magnesium ion binding;protein binding;growth; | Petrin cytosine dinucleotide (MCN);Molybdenum cofactor (PCD); FAD-isobutylketone (FAB); Co-cyanocobalamin (CNC); Bis-adenosine 5-pentaphosphate (AP5); |
| Set 17 | Rv0070c;Rv2381cRv3029c | glycine hydroxymethyltransferase activity;electron carrier activity;electron transport;plasma membrane;cytosol;growth; | Cyclohexyl-beta-d-maltoside (MA4); Isoindoloindolone derivative (CZA); Isoquinolinone derivative (IZA); Carbothiomide derivative (DKI); Pyridine acetamide derivative (877); |
| Set 18 | Rv1400c;Rv1683Rv2029c | poly-hydroxybutyrate biosynthetic process;acyltransferase activity;growth;plasma membrane; | Cyclohexyl-beta-d-maltoside (MA4); Pyridine carboxamide derivative (ZZY); Thiazolone derivative (ZMG); Purinamine derivative (ZIP); Stearic acid (STE); |
| Set 19 | Rv0125;Rv0304cRv2683 | extracellular region; | Cyclohexyl-beta-d-maltoside (MA4); Isoindoloindolone derivative (CZA); Hydroxy ocatadeca-didenoic acid (243); Dihydropyrimidine amine derivative (YF4); Hydroxy-3-methoxybenzoate (VNL); |
| Set 20 | Rv0651;Rv3448Rv3573c | ribosome biogenesis;intracellular;plasma membrane;growth; | Oleci acid (OLA); Ethylpyrimidine diamine derivative (N22); Cyclohexyl-beta-d-maltoside (MA4); Methylpyridazine piperidine propyloxyphenylacetate (J78); Phosphotidyl ethanolamine derivative (EPH); |
| Set 21 | Rv0512;Rv1066Rv2075c | NA | Imatinib (STI); Lopinavir (AB1); Tipranavir (TPV); Ritonavir (RIT); Raloxifine (RAL); |
| Set 22 | Rv1666c;Rv2383cRv3087 | phosphopantetheine binding;acyl carrier activity;siderophore biosynthetic process;cellular response to iron ion starvation;response to host immune response;pathogenesis;plasma membrane; | Co-cyanocobalamin (CNC); Pyrimidine ethanone adduct (TDT); Phosphatidylethanolamine (PEE); PentadecylcoA (NHM); TetradecanoylcoA (MYA); |
| Set 23 | Rv0702;Rv3224Rv3702c | response to iron ion;cytosol;plasma membrane; | Biliveridine (BLA); Phenyl acetic acid derivative (965); FAD-isobutylketone (FAB); Analogue of Indinavir (XN2);3-fluoro-5-morpholin-4-yl-N-benzamide (WBT); |
| Set 24 | Rv0767c;Rv1181Rv3158 | NADH dehydrogenase (ubiquinone) activity;integral to plasma membrane;extracellular region;cell wall;plasma membrane; | Biliverdine (BLA); Succinyl CoA (SCA); 3-Acetylpyridine adenine dinucleotide (A3D);4-oxo NADP (ODP); 5,6-dihydroxy NADP (NZQ); |
| Set 25 | Rv0843;Rv1877Rv3757c | binding;glycine betaine transport;response to host;transporter activity;transport;membrane;extracellular region; | Cyclohexyl-beta-d-maltoside (MA4); CZA (Isoindoloindolone derivative); Isoindoloindolone derivative (IZA); Ethoxypyridine acetamide derivative (877); Crizotinib (VGH); |
| Set 26 | Rv1181;Rv1979cRv3825c | fatty acid biosynthetic process;cellular response to iron ion starvation;cell wall;response to host immune response;plasma membrane;cytosol; | Molybdenum (PCD); Bilivridine (BLA); Pterin cytosine dinucleotide (MCN); Co-cyanocobalamin (CNC); FAD-isobutylketone (FAB); |
| Set 27 | Rv1361c;Rv1800Rv3144c | response to host immune response; | Imatinib (STI); Biliverdine (BLA); Digalactosyl diacy glycerol (DGD); Eicosapentaenoic acid (EPA); Arachidonic acid (ACD); |
| Set 28 | Rv1438;Rv3021cRv3585 | DNA helicase activity;DNA replication;triose-phosphate isomerase activity;plasma membrane;ATP binding;protein homodimerization activity;cytosol;growth;extracellular region; | Cyclohexyl-beta-d-maltoside (MA4);Crizotinib (VGH); Pyrazoloquinolinone (73Q); Pyridine carboxamide derivative (ZZY); Diphenyl indole carboxylic acid (VX3); |
| Set 29 | Rv1832;Rv2677cRv3578 | glycine dehydrogenase (decarboxylating) activity;oxygen-dependent protoporphyrinogen oxidase activity;cell wall;plasma membrane;growth; | Biliverdine (BLA); Molybdenum (PCD); Pterine cytosine nucleotide (MCN); Succinyl CoA (SCA); Phosphatidylethanolamine (PEE); |
A set of proteins that can bind to the same drug and have the properties desired in drug targets60,61, would constitute first lists of polypharmacological targets. An ideal drug target needs to satisfy many criteria60, many of which have already been studied previously in our laboratory. We therefore use the list of 451 drug targets identified as a high-confidence list derived from our previous study- targetTB19. This study incorporated a multi-level pipeline to identify proteins that have several qualities desired in ideal drug targets. The pipeline has several steps of filtering using systems level reactome and interactome analysis, sequence level comparative genomics with the host and a structure level assessment of druggability. The reactome and interactome analyses capture essentiality, while sequence and structural analyses capture specificity. The targetTB pipeline yielded prediction of 451 proteins as high confidence drug targets, some of which were already known in literature and many were new identifications. 20 of these targets in fact appear in 18 sets identified here. Table 1 lists the sets and highlights those that are identified as promising drug targets in targetTB.
Identifying similarities to known drug binding sites
Our next goal was to screen the known drug binding sites (knowndrug-sites_DB) to identify site similarities with any of the shortlisted Mtb pockets. Any significant hit against the binding site sets would also be a good clue for being a possible lead with a polypharmacological profile. The database consisted of about 10658 binding sites for 1541 FDA-approved small molecule drugs, 150 FDA-approved biotech (protein/peptide) drugs, 86 nutraceuticals and 5082 experimental drugs. Interestingly, we observe at least one hit for most of the 29 sets. In all, 189 hits were obtained against ‘known drug-site_DB’. Figure 6 illustrates the set of top ranked drug hits that can be associated to each set (Network-type 3, Table S2). Some of the associated molecules from the Drugbank indeed correspond to the approved drugs subset, which are highlighted in Figure 6.
Figure 6. Drug-hits for Polypharmacological targets.

Each disconnected component represents a set of polypharmacological targets obtained from Mtb binding site similarity network. Two type of nodes are present in the network, the predicted binding sites are shown as spheres and the drugs sharing a binding site similarity are shown as triangles. The red colored circular nodes represent binding sites of high-confidence targets. Approved drugs are also highlighted in red.
Ranking the sites in the pocketome through polypharmacological potential index (PPI)
In order to pick only those sites that are specifically druggable among the shortlisted proteins, we compute a polypharmacological index for each predicted binding site. Three aspects are considered in computing this index, which are, number of similar sites in the Mtb pocketome, implying polypharmacological possibility, number of drug clues obtained implying druggability and extent of specificity through number of druggable binding sites as compared to cofactor binding sites. The index thus (a) scores positively for the similarity of the sites to other sites in the pocketome and thus contribute to polypharmacological profile of the target (b) scores positively for those sets of sites that resemble any approved drug's known binding site and thus, indirectly implies druggability and (c) penalizes those sites that exhibit similarity to cofactor binding sites since that would increase chances of adverse polypharmacology7,47. Using this index, as described in equation (1), we rank list the sets based on the polypharmacological indices of the individual sites contained in each set and observe that set12 and set13 are the top ranking sets. Set12 contains a binding site from a protein - Rv0687, a probable short-chain dehydrogenase/reductase, possibly involved in cellular metabolism and is found to be an essential gene through transposon site hybridization experiments28. Similarly, set13 also contains binding site of AccD3 (Rv0904c), a putative acetyl coenzyme A carboxylase that has been listed as essential for Mtb through various analyses19. Proteins containing sites with the highest PPI can be regarded to be most promising candidate sites for design of specific inhibitors, thus providing a list of possible polypharmacological drug targets. The top 20 high-scoring sites are derived from proteins that include - Pks2(Rv3825c), a polyketide synthase, PknD (Rv0931c), a transmembrane serine/threonine protein-kinase, which are already good targets as listed in targetTB 13 of these are also included in the TDR database14 with a druggability score. Full list of targets containing the information on cofactor hits, drug hits, clustering-coefficient, PPI Score and normalized degree have been enlisted in supplementary table (Table S5). Targeting each set with a single drug can theoretically be envisaged to result in binding to and possibly modulating the function of all the members in that set simultaneously. Those that exhibited low PPI score are not considered as good polypharmacological targets by default. One reason for this could be that the binding sites in these resemble cofactor-binding sites and hence have a high frequency of occurrence. However, it must be noted that there are reports in literature which indicate successes for targeting cofactor-binding sites62,63. Careful design could achieve specific binding to the required sites.
Leads for polypharmacology of high confidence targets and clues for drug repurposing
We systematically analyzed the subset of approved drug-sites from ‘knowndrug-site_DB’ that could serve as clues for lead design or drug repurposing, through construction of a bipartite network (Network-type 4, Table S2) consisting of binding sites from 451 targetTB19 drug targets and their similarities with binding sites of approved drugs. A bipartite network provides ready insights on two fronts, (a) rank list of drugs based on their clustering coefficient depicting the number of associations to different putative targets in Mtb, (b) rank list of proteins based on their clustering coefficient depicting the number of associations to approved drugs. While the first results in identification of polypharmacological sets, the second is useful for short-listing candidates for drug repurposing involving any drug target in Mtb. Since the same analysis can provide useful information to infer drug associations for all promising targets, in this exercise we do not restrict the analysis only to polypharmacological targets, instead we include all the targets identified from targetTB. Supplementary Figure (Figure S4) illustrates the network, provides information about the list of targets for which a significant drug association is made and conversely a list of drugs for which a putative target in Mtb is identified. The clustering coefficient (CCbp) derived here for both protein and the drug is through projection of bipartite network onto corresponding single mode networks using tnet algorithm64. These promising drug associations are further verified by estimating the energetic feasibility of their binding at the given site through molecular docking (see Methodology section). Among the highly connected drugs are atazanavir, indinavir, lopinavir - antiretroviral drugs whose binding site in HIV virus bears similarity to proteins - PpsE(Rv2935), Rv2842c(conserved proteins), Rv2689c(conserved alanine and glycine rich protein), MurE(Rv2158c). These antiretroviral drugs were reported by Kinnings et.al in their TBDrugome65 study as well. Further, there is indirect support from literature for the identification of ivermectin, another highly connected drug. Lim et.al have reported that it has antimycobacterial properties through the study of its effect on Mtb cultures of clinical strains and multidrug resistant strains66. Ivermectin is observed to have a high clustering coefficient in the network showing associations with SecY(Rv0732), MycP(Rv0291), DapD(Rv1201c) all identfiied as essential genes in Mtb. The whole set of associations obtained here can be regarded as ready shortlists for experimental testing. Targets with highest clustering coefficient in this network correspond to the most druggable targets. These include MurE protein, followed by LpdA protein. MurE is involved in cell wall formation and peptidoglycan biosynthesis, which is essential for mycobacterial survival while LpdA is a probable quinonereductase, already known to contribute towards virulence of Mtb. A full list of possible repurposable drugs and targets are listed in Table 2 and Table 3. Table 2 also lists down the essentiality criteria determined for each target obtained from our previous study20 that incorporated analysis of microarray expression profiles, flux-balance analysis, protein-protein interaction network, phyletic retention and available literature on transposon site hybridization (TraSH) experiments.
Table 2. Prioritized Drug Targets: Ranking putative drug targets from targetTB H-list, based on the number of connections to approved drugs from the databases used in this study. The description of each protein along with its clustering coefficient (CC) value in the bipartite network has been listed. The essentiality inference of the targets obtained from Ghosh et.al 2013, has also been indicated, these include (A) Microarray analysis, (B) Flux balance analysis, (C) Protein-protein interaction analysis, (D) Phyletic retention analysis and (E) Transposon hybridization experiments.
| Protein | Gene Name | CCbp | Essentiality Inference | Protein Description |
|---|---|---|---|---|
| Rv2158c | murE | 0.77 | A; B; C; D; E | ProbableUDP-N-acetylmuramoylalanyl-D-glutamate-2,6-diaminopimelate ligase MurE |
| Rv2842c | Rv2842c | 0.72 | A | Conserved hypothetical protein |
| Rv3303c | lpdA | 0.71 | A; D; E | NAD(P)H quinone reductase LpdA |
| Rv2335 | cysE | 0.66 | A; B; C; E | Probable serine acetyltransferase CysE (sat) |
| Rv2398c | cysW | 0.63 | A; D; E | Probable sulfate-transport integral membrane protein ABC transporter CysW |
| Rv2689c | Rv2689c | 0.61 | A; D | Conserved alanine and valine and glycine rich protein |
| Rv3139 | fadE24 | 0.59 | A; D; E | Probable acyl-CoA dehydrogenase FadE24 |
| Rv2935 | ppsE | 0.58 | A; B | Phenolpthiocerol synthesis type-I polyketide synthase PpsE |
| Rv2026c | Rv2026c | 0.58 | A; D; E | Conserved hypothetical protein |
| Rv0914c | Rv0914c | 0.55 | A; D | Possible lipid carrier protein or keto acyl-CoA thiolase |
| Rv3343c | ppe54 | 0.52 | A; E | PPE family protein PPE54 |
| Rv2388c | hemN | 0.52 | A; B; C; D | Probable oxygen-independent coproporphyrinogen III oxidase HemN (coproporphyrinogenase) (coprogen oxidase) |
| Rv0542c | menE | 0.48 | A; C; D; E | Possible O-succinylbenzoic acid--CoA ligase MenE (OSB-CoA synthetase) (O-succinylbenzoate-CoA synthase) |
| Rv3245c | mtrB | 0.48 | A; D; E | Two component sensory transduction histidine kinase MtrB |
| Rv2900c | fdhF | 0.46 | A | Possible formate dehydrogenase H FdhF (formate-hydrogen-lyase-linked, selenocysteine-containing polypeptide) (formate dehydrogenase-H alpha subunit) (FDH-H) |
| Rv0220 | lipC | 0.45 | A; D | Probable esterase LipC |
| Rv1220c | Rv1220c | 0.45 | A | Probable methyltransferase |
| Rv3801c | fadD32 | 0.43 | A; B; D; E | Fatty-acid-AMP ligase FadD32 (fatty-acid-AMP synthetase) (fatty-acid-AMP synthase). Also shown to have acyl-ACP ligase activity. |
| Rv1613 | trpA | 0.43 | A; B; E | Probable tryptophan synthase, alpha subunit TrpA |
| Rv2573 | Rv2573 | 0.42 | A; B; C; E | Conserved hypothetical protein |
| Rv2678c | hemE | 0.42 | A; B; C; E | Probable uroporphyrinogen decarboxylase HemE (uroporphyrinogen III decarboxylase) (URO-D) (UPD) |
Table 3. Approved Drugs with potential for repurposing in tuberculosis. Identified hits from the list of Approved drugs, listed in the order of clustering coefficient (CC) in the bipartite network. Inferred Mtb targets for the corresponding drug based upon the associations in the bipartite network have also been listed.
| Drug Name | CCbp | Inferred targets in Mtb | Known pharmacological action |
|---|---|---|---|
| Atazanavir (DB01072;DR7) | 1 | Rv2026c; Rv2335; Rv1220; Rv2935 | Used in combination with other antiretroviral agents for the treatment of HIV-1 infection, as well as postexposure prophylaxis of HIV infection in individuals who have had occupational or nonoccupational exposure to potentially infectious body fluids of a person known to be infected with HIV when that exposure represents a substantial risk for HIV transmission. |
| Indinavir (DB00224;MK1) | 0.95 | Rv2158c; Rv2026; Rv2335; Rv2842c; Rv1220c; Rv2689c; Rv2935 | Indinavir is an antiretroviral drug for the treatment of HIV infection. |
| Lopinavir (DB01601;AB1) | 0.86 | Rv0542c; Rv1663; Rv2678c; Rv2842c; Rv2689c; Rv2935 | Indicated in combination with other antiretroviral agents for the treatment of HIV-infection |
| Saquinavir (DB01232;ROC) | 0.81 | Rv2158c; Rv0904c; Rv2026c; Rv3303c; Rv2510c; Rv0627; Rv1663; Rv2335; Rv2946c; Rv2996c; Rv2678c; Rv2842c; Rv0237c; Rv1220c′ Rv2689c; Rv1505c; Rv2935; Rv2931 | For the treatment of HIV-1 with advanced immunodeficiency together with antiretroviral nucleoside analogues. |
| Nelfinavir (DB00220;1UN) | 0.77 | Rv0237; Rv1663; Rv1843c; Rv2026c; Rv2678c; Rv2689c; Rv2842c; Rv2935; Rv3303c; Rv3882c | Used in combination with other antiviral drugs in the treatment of HIV in both adults and children |
| Nilotinib (DB04868;NIL) | 0.7 | Rv0237; Rv1663; Rv1843c; Rv2026c; Rv2678c; Rv2689c; Rv2842c; Rv2935; Rv3303c; Rv3882c; | For the potential treatment of various leukemias, including chronic myeloid leukemia (CML) |
| Tipranavir (DB00932;TPV) | 0.69 | Rv0555; Rv0904c; Rv1127c; Rv1550; Rv1663; Rv2026c; Rv2153c; Rv2388c; Rv2678c; Rv2689c; Rv2726c; Rv3825c; Rv3883c; Rv3886c; | For combination antiretroviral treatment of HIV-1 infected adult patients with evidence of viral replication, who are highly treatment-experienced or have HIV-1 strains resistant to multiple protease inhibitors |
| Ritonavir (DB00503;RIT) | 0.68 | Rv0511; Rv0627; Rv0904c; Rv1092c; Rv1220c; Rv1237; Rv1663; Rv1843c; Rv2026c.; Rv2335; Rv2388c; Rv2678c; Rv2689c; Rv2842c; Rv2935; Rv3303c; Rv3509c; Rv3582c; Rv3825c; Rv3886c; | Indicated in combination with other antiretroviral agents for the treatment of HIV-infection. |
| Rosiglitazone (DB00412;BRL) | 0.66 | Rv0755c; Rv1753c; Rv1886c; Rv3804c; | For the treatment of Type II diabetes mellitus |
| Amprenavir (DB00701;478) | 0.64 | Rv0085; Rv0237; Rv0478; Rv0635; Rv1127c; Rv1237; Rv1505c; Rv1542c; Rv1655; Rv1663.2; Rv1843c; Rv1850; Rv2153c; Rv2524c; Rv2524c; Rv2613c; Rv2689c; Rv2931; Rv2938; Rv2939; Rv3135; Rv3245c; Rv3804c; Rv3882c; Rv3883c; Rv3886c; Rv3887c; | For the treatment of HIV-1 infection in combination with other antiretroviral agents. |
| Gefitinib (DB00317;IRE) | 0.61 | Rv0085; Rv0291; Rv1287; Rv1836c; Rv2156c; Rv2296; Rv2869c; Rv2889c; Rv3737; | For the continued treatment of patients with locally advanced or metastatic non-small cell lung cancer after failure of either platinum-based or docetaxel chemotherapies. |
| Tamoxifen (DB00675;CTX) | 0.61 | Rv0228; Rv0237; Rv0290; Rv0527; Rv0545c; Rv1650; Rv1836c; Rv2281; Rv2439c; Rv2678c; Rv2986c; Rv3722c; Rv3737; | For the treatment of breast cancer. |
| Dasatinib (DB01254;1N1) | 0.61 | Rv2069; Rv2202c; Rv2231c; Rv2727c; Rv3372; Rv3886c; | For the treatment of adults with chronic, accelerated, or myeloid or lymphoid blast phase chronic myeloid leukemia with resistance or intolerance to prior therapy. |
| Darunavir (DB01264;017) | 0.6 | Rv0542c; Rv1237; Rv1409; Rv1663; Rv2026c; Rv2153c; Rv2158c; Rv2613c; Rv2678c; Rv2689c; Rv2842c; Rv2897c; Rv2935; Rv3255c; Rv3825c; Rv3886c; | Darunavir, co-administered with ritonavir, and with other antiretroviral agents, is indicated for the treatment of human immunodeficiency virus (HIV) infection in antiretroviral treatment-experienced adult patients, such as those with HIV-1 strains resistant to more than one protease inhibitor. |
| Adenosine (DB00640;ADN) | 0.59 | Rv0032; Rv0291; Rv0479c; Rv0509; Rv0545c; Rv1201c; Rv1650; Rv1836c; Rv1843c; Rv2296; Rv2482c; Rv2869c; Rv3037c; Rv3255c; Rv3579c; Rv3737; Rv3740c; Rv3825c; Rv3909; | Used as an initial treatment for the termination of paroxysmal supraventricular tachycardia (PVST), including that associated with accessory bypass tracts, and is a drug of choice for terminating stable, narrow-complex supraventricular tachycardias (SVT). |
| Vinblastine (DB00570;VLB) | 0.59 | Rv0914c; Rv2196; Rv2398c; Rv3882c; | For treatment of breast cancer, testicular cancer, lymphomas, neuroblastoma, Hodgkin's and non-Hodgkin's lymphomas, mycosis fungoides, histiocytosis, and Kaposi's sarcoma. |
| Spironolactone (DB00421;SNL) | 0.59 | Rv0732; Rv0914c; Rv1181.4; Rv1795; Rv2113; Rv2196; Rv2398c; Rv3801c; Rv3825c; Rv3882c; | Used primarily to treat low-renin hypertension, hypokalemia, and Conn's syndrome. |
| Colchicin (DB01394;LOC) | 0.57 | Rv0032; Rv0474; Rv0570; Rv0732; Rv1181; Rv1296; Rv1300; Rv1380; Rv1655; Rv2113; Rv2156c; Rv2178c; Rv2281; Rv2524c; Rv3037c; Rv3712; Rv3882c; Rv3886c; | For treatment and relief of pain in attacks of acute gouty arthritis. |
| Podofilox (DB01179;POD) | 0.56 | Rv0474; Rv0570; Rv0732; Rv0904c; Rv1181; Rv1296; Rv1300; Rv1550; Rv1650; Rv1795; Rv2113; Rv2156c; Rv2178c; Rv2259; Rv2281; Rv3015c; Rv3037c; Rv3712; Rv3790; Rv3882c; Rv3886c; | For treatment of external genital warts (Condyloma acuminatum). |
| Lapatinib (DB01259;FMM) | 0.56 | Rv0237; Rv0511; Rv0527; Rv0545c; Rv0554; Rv0555; Rv0570; Rv0724; Rv0732; Rv0904c; Rv1127c; Rv1181; Rv1220c; Rv1485; Rv1505c; Rv1530; Rv1650; Rv1663; Rv1843c; Rv1872c; Rv1905c; Rv2165c; Rv2231c; Rv2439c; Rv2524c; Rv2610c; Rv2613c; Rv2623; Rv2678c; Rv2897c; Rv2902c; Rv2941; Rv3255c; Rv3405c; Rv3582c; Rv3585; Rv3635; Rv3737; Rv3825c; Rv3871; Rv3882c; Rv3886c; Rv3887c; | Indicated in combination with capecitabine for the treatment of patients with advanced or metastatic breast cancer whose tumors overexpress the human epidermal receptor type 2 (HER2) protein and who have received prior therapy including an anthracycline, a taxane, and trastuzuma. |
| Imatinib (DB00619;STI) | 0.55 | Rv0120c; Rv0627; Rv0843; Rv0904c; Rv1078; Rv1127c; Rv1237; Rv1287; Rv1293; Rv1409; Rv1459c; Rv1492; Rv1542c; Rv1843c; Rv2153c; Rv2170; Rv2231c; Rv2610c; Rv2613c; Rv2623; Rv2869c; Rv2996c; Rv3132c; | For the treatment Philadelphia chromosome positive chronic myeloid leukemia (CML) and malignant gastrointestinal stromal tumors (GIST) |
| Bexarotene (DB00307;9RA) | 0.55 | Rv0755c; Rv1237; Rv1753c; Rv1886c; Rv3804c; | Used orally for the treatment of skin manifestations of cutaneous T-cell lymphoma (CTCL) in patients who are refractory to at least one prior systemic therapy. |
| Raloxifene (DB00481;RAL) | 0.54 | Rv0237; Rv0627; Rv0904c; Rv1204c; Rv1237; Rv1277; Rv1459c; Rv1505c; Rv1650; Rv1653; Rv1663; Rv2026c; Rv2124c; Rv2388c; Rv2439c; Rv2524c; Rv2573; Rv2613c; Rv2678c; Rv2935; Rv2938; Rv2946c; Rv3097c; Rv3132c; Rv3405c; Rv3585; Rv3800c; Rv3804c;Rv3825c | For the prevention and treatment of osteoporosis in post-menopausal women, as well as prevention and treatment of corticosteroid-induced bone loss. |
| Prednisone (DB00635;PDN) | 0.53 | Rv0256c; Rv1237; Rv1613; Rv1886c; Rv2900c; Rv3245c; Rv3343c; | For the treatment of drug-induced allergic reactions, perennial or seasonal allergic rhinitis, serum sickness, giant cell arteritis acute rheumatic or nonrheumatic carditis, systemic dermatomyositis, systemic lupus erythematosus, atopic dermatitis, contact dermatitis, exfoliative dermatitis. |
| Erythromycin (DB00199;ERY) | 0.53 | Rv1181; Rv1257c; Rv1589; Rv2243; Rv3712; Rv3793; Rv3801c; Rv3886c; | For use in the treatment of infections caused by susceptible strains of microorganisms in the following diseases: respiratory tract infections (upper and lower) of mild to moderate degree, pertussis (whooping cough), as adjunct to antitoxin in infections due to Corynebacterium diphtheriae. |
| Progesterone (DB00396;STR) | 0.53 | Rv0220; Rv0724; Rv1237; Rv1613; Rv1753c; Rv1886c; Rv2900c; Rv2938; Rv3245c; Rv3343c; Rv3801c; Rv3804c; Rv3825c; | For progesterone supplementation or replacement as part of an Assisted Reproductive Technology (ART) treatment for infertile women with progesterone deficiency |
| Ivermectin (DB00602;IVM) | 0.53 | Rv0032; Rv0256c; Rv0291; Rv0527; Rv0545c; Rv0585c; Rv0732; Rv1201c; Rv1385; Rv1613; Rv1714; Rv2113; Rv2938; Rv3255c; Rv3825c; | For the treatment of intestinal (i.e., nondisseminated) strongyloidiasis due to the nematode parasite Strongyloides stercoralis. Also for the treatment of onchocerciasis (river blindness) due to the nematode parasite Onchocerca volvulus. Can be used to treat scabies caused by Sarcoptes scabiei. |
Agreement with previously reported drug associations
One way of validating our approach is to analyze whether previously characterized associations from literature are identified in this approach. Isoniazid adduct, a front-line clinical drug for TB is well known to bind to its target InhA, an enoyl reductase. In addition, a crystallographic study also identifies its binding with DHFR67. It was indeed gratifying to observe that the binding sites of both InhA and DHFR were found to be similar with a PMAX of 0.52 (P-value = 8.4e-03) and PMIN score of 0.73 (Figure 7).
Figure 7.

A selected example of similar binding sites in different proteins predicted in this study matching with crystal structures available in literature (A) Structural superposition of dihydrofolate reductase (red cartoon) and InhA (blue cartoon, PDBID: 1ZID) protein based on the similarity of the binding sites (shown as sticks). The inset shows the similarity of the binding sites with the isoniazid adduct shown in ball and stick representation. (B) Crystal structure of dihydrofolate reductase with characterization of binding site for isoniazid adduct (PDBID: 2CIG).
A pull-down assay reported in literature by Argyrou et.al68 independently identified 18 other proteins that bind to isoniazid adduct, which are perhaps secondary targets of this drug. We observe that 10 of the 18 proteins identified through this study (listed in Table S6), including dihydrofolate reducatse, have binding sites similar to that in InhA, explaining the basis of such cross-reactivity.
A similar exercise was carried out for all the clinically used anti-tubercular drugs whose targets are well defined and where structural models are available of the complexes. The availability of these complexes enables us to extract out the binding site and compare against the pocketome. These drugs include cycloserine (DCS), para-amino salicylic acid (BHA), kanamycin (KAN), isoniazid (ISZ), rifampicin (RFP), rifabutin (RBT) and streptomycin (SRY). Figure S3 summarizes the results obtained for this analysis. Our analysis supports recently obtained experimental evidence on para-amino salicylic acid influencing the enzymes of the folate pathway69,70,71 as many pockets belonging to the proteins in this pathway seem to have significant similarity to known binding site of PAS from p-hydroxybenzoate hydrolase (Table S7).
An additional type of validation is to identify similarities in pairs of proteins previously reported in literature. A study by Kinnings et al., called TBDrugome65, using structural models of about 1097 proteins, corresponding to about a third of our data, predicted pockets using a different algorithm and subsequent binding site comparisons also by a different method65, has reported some drug associations with Mtb proteins. We have performed a systematic comparison of the drug associations obtained in this study with that reported in the study. Out of 1097 cases. 662 pockets were detected with the same ligand association (PMAX ≥ 0.4). It must be noted that in present work, the coverage of the proteome is much larger; binding pocket identification is much more rigorous involving multiple approaches and pocket comparison is carried out using a different algorithm that has been extensively validated against PDB. Given that detecting and comparing binding sites is a far from trivial exercise and is sensitive to the algorithm used, it is useful to have a comparison using two different approaches (Supplementary Text 2). Our observation that many of the associations reported by TBDrugome65 study forms a subset of our results serve to validate each other enhancing confidence for the whole set of drug associations.
Discussion
Advances in genomics and related technologies are pushing the boundaries of the scale and resolution at which any organism or a given biological process is understood. This study, comprehensively studies a pocketome at the structural level, illustrating that the ligand binding space of the proteome can be probed algorithmically and utilized to obtain high resolution insights into several newly pursued aspects of drug discovery including polypharmacological target selection, combination targets and possible drug repurposing. Characterizing the pocketome at the structural level in M.tuberculosis appears to be among the first to study ligand-binding space comprehensively at the structural level in any organism. The novelty of the approach used in this study is to probe the entire set of small molecule binding sites in the organism through protein structures and the sub-structure comparisons in them. The workflow incorporates stringent steps of filtering and validation at each step. First, the structural models of the proteome used are already validated in our previous study through various stereochemical parameters and energetic considerations including secondary structure compatibility, informatics based statistical scoring of neighborhoods of each amino acid in each protein. Binding sites are picked based on a consensus prediction by three orthogonal binding site detection algorithms that capture residue conservation or evolutionary information, geometric parameters consistent with known binding sites and energetically favorable locations in the protein for ligand binding. Since individual methods have their own advantages as well as limitations, deploying a consensus approach is useful for overcoming individual limitations and hence enhances confidence. A large scale comparison with binding site residues known from sequence motifs or individual molecular biology experiments available in literature documented in databases, places the binding site predictions from this study in context of all available data in literature, making it possible to comprehend different evidences for ligand binding sites and hence ligand associations in a unified manner. Systematic comparisons to KEGG, PDB and Procognate ligand associations, are provided as a comprehensive resource through a web-accessible database. Such large-scale comparisons showing good agreement with different approaches automatically serves to validate currently used methodology themselves.
Binding site comparisons, which form the next step in the workflow, are carried out using home-grown algorithms previously reported and made available in literature. Tuning the algorithms for high-performance has rendered it feasible to carry out much of the analyses reported here, amounting to about 192 million comparisons, all at the structural level. Representing, analyzing and interpreting data from such large-scale comparisons presents the next challenge in the workflow, which has been addressed using network approaches. Network abstractions make a large amount of data computationally tractable using graph theoretical methods, an approach increasingly being used to analyze biological data3,56,57,60. Binding site networks constructed from all-pair comparisons include only those pairs that are sufficiently similar in their binding sites in the network. An algorithmic approach such as this allows for construction and probing of the network with different thresholds reflecting different stringencies, to cater to the specific question being asked of the network. Where drug associations are made for possible repurposing, a higher threshold is more meaningful so that associations made are of high confidence. This would mean that some associations that are still significant but below the threshold are missed out. Similarities at a lower threshold can still provide important clues for possible lead compounds, which can be obtained fairly easily with the network data obtained from this study. All data is therefore made available as a web accessible resource that is expected to be useful to the drug discovery community.
Network abstractions enable delineation of closely related communities or clusters reflecting sets of highly similar binding sites. Clustering methodology on a network such as this has been validated by performing a similar exercise on a dataset termed as MOAD, which contains well-curated high-resolution protein-ligand complexes from PDB. Obtaining separate clusters, each cluster containing chemically similar ligands indeed illustrates the capability of the approach to identify these clusters from a large data set.
Obtaining a definition of the pocketome in Mtb provides an unique opportunity to understand the range of binding sites present in the cell, set of possible ligands recognized by the cell, structural profile of the sites and the list of unique sites, leading to an understanding of the cellular functioning in terms of structural scaffolds that facilitate the underlying molecular recognition events. Knowledge of the binding sites at the structural level in each protein in the proteome provides a novel high-resolution approach for obtaining the predicted set of small molecules that participate in biochemical events in the cell or in other words a computational equivalent of a metabolome. Observation of recognizable binding sites in a number of conserved hypotheticals also provides significant clues to their possible functional roles, leading to new annotations. Ability to compare all-pair pockets at the structural level provides another new opportunity to identify number of unique binding site types represented by the genome. Observation that proteins similar even in sequence space and fold space exhibit significant differences in their site types point to the fact that the pocketome space is much larger than the sequence and fold space of the genome, suggesting that evolution of finer features of function, generation of ligand specificities and affinities has emerged through site variation alone.
Knowledge of the pocketome, similarities and differences among the individual sites in them has large implications for drug discovery. The importance of the right choice of the target protein, right at the start of the discovery pipeline, has been well recognized. Choice of target proteins have typically been largely guided by some prior knowledge of the protein or prior success with a related protein in a different condition and has not in many cases have had the advantage of a systematic exploration of the target space available for that condition. Selecting sets of proteins that share high similarity in their binding sites paves a well-lit path to identify polypharmacological targets. Abstraction of all-pair comparisons as networks facilitates identification of highly connected components or clusters in the networks, each cluster capturing one set of possible polypharmacological proteins. When this is integrated with knowledge from previous studies of drug target identification, it results in picking high-confidence targets that have additional criteria of being possible polypharmacological targets. Since databases such as Drugbank contain information about approved drugs and binding sites in their corresponding targets, it has become feasible to compare them with the pocketome of Mtb using the high-performance algorithms. The workflow in this study has yielded a ready shortlist of sets of promising drug targets with polypharmacological possibilities and at the same time has identified possible drug candidates either directly for repurposing or at the least as significant lead clues that can be used to design new drug molecules against the entire group of proteins in each set. In other words, it also identified compounds that have the potential to act as polypharmacological drugs. The PPI computed here captures this in a systematic manner at the same time ensuring that those sites such as cofactor binding sites seen often in the pocketome are filtered out.
In summary, this work defines the pocketome of Mtb by structural level characterization of the binding sites at a genome-scale, mapping ligands onto individual sites, which has lead to an understanding of the available pocketome space. The pocketome space is seen to be much larger then the sequence or the fold space, suggestive of the wide repertoire of specific functional roles achieved by the cell. On the other hand, the binding-site similarity network constructed has indicated the presence of about 29 sets together comprising about 121 proteins that share significant similarities within each set. These sets can now be exploited as possible polypharmacological target sets. A bipartite network derived by comparing known and approved drug binding sites to the pocketome has provided several significant drug associations for potential drug targets and thus important clues for possible drug repurposing. A list of approved drugs that could have new targets in Mtb is also obtained from the study. The approach used here is fairly generic and can be applied to other organisms as well, and can be incorporated in many drug discovery programmes.
Supplementary Material
Supplementary Information
Acknowledgments
We thank the Department of Biotechnology (DBT), Government of India and Open Source Drug Discovery Consortium for financial support. We would like to thank Prof. Sir Tom Blundell, University of Cambridge, Prof. Srinivasan, Indian Institute of Science, Prof. Sowdhamini, National Center of Biological Sciences and Prof. Samir Brahmachari from Institute of Genomics and Integrative Biology, for helpful discussions.
Footnotes
The authors declare no competing financial interests.
Author Contributions Conceived and designed the experiments: N.S.C. Performed the experiments: P.A. Analyzed the data: N.S.C. and P.A. Wrote the paper: N.S.C. and P.A. Website design and implementation: P.A.
References
- Konopa K. & Jassem J. The role of pemetrexed combined with targeted agents for non-small cell lung cancer. Curr Drug Targets 11, 2–11 (2010). [DOI] [PubMed] [Google Scholar]
- Winum J. Y., Maresca A., Carta F., Scozzafava A. & Supuran C. T. Polypharmacology of sulfonamides: pazopanib, a multitargeted receptor tyrosine kinase inhibitor in clinical use, potently inhibits several mammalian carbonic anhydrases. Chem Commun (Camb) 48, 8177–9 (2012). [DOI] [PubMed] [Google Scholar]
- Hopkins A. L. Network pharmacology: the next paradigm in drug discovery. Nat Chem Biol 4, 682–90 (2008). [DOI] [PubMed] [Google Scholar]
- Csermely P., Korcsmaros T., Kiss H. J., London G. & Nussinov R. Structure and dynamics of molecular networks: a novel paradigm of drug discovery: a comprehensive review. Pharmacol Ther 138, 333–408 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S. et al. Systems pharmacology of adverse event mitigation by drug combinations. Sci Transl Med 5, 206ra140 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besnard J. et al. Automated design of ligands to polypharmacological profiles. Nature 492, 215–20 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boran A. D. & Iyengar R. Systems approaches to polypharmacology and drug discovery. Curr Opin Drug Discov Devel 13, 297–309 (2010). [PMC free article] [PubMed] [Google Scholar]
- Gobbi G. & Janiri L. Clozapine blocks dopamine, 5-HT2 and 5-HT3 responses in the medial prefrontal cortex: an in vivo microiontophoretic study. Eur Neuropsychopharmacol 10, 43–9 (1999). [DOI] [PubMed] [Google Scholar]
- Sotgiu M. L., Valente M., Storchi R., Caramenti G. & Biella G. E. Cooperative N-methyl-D-aspartate (NMDA) receptor antagonism and mu-opioid receptor agonism mediate the methadone inhibition of the spinal neuron pain-related hyperactivity in a rat model of neuropathic pain. Pharmacol Res 60, 284–90 (2009). [DOI] [PubMed] [Google Scholar]
- Nagar B. c-Abl tyrosine kinase and inhibition by the cancer drug imatinib (Gleevec/STI-571). J Nutr 137, 1518S–1523S; discussion 1548S (2007). [DOI] [PubMed] [Google Scholar]
- Venkataramani V. et al. Histone deacetylase inhibitor valproic acid inhibits cancer cell proliferation via down-regulation of the alzheimer amyloid precursor protein. J Biol Chem 285, 10678–89 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X. Z., Li X. J. & Zhang H. Y. Valproic acid as a promising agent to combat Alzheimer's disease. Brain Res Bull 81, 3–6 (2010). [DOI] [PubMed] [Google Scholar]
- Zumla A., Nahid P. & Cole S. T. Advances in the development of new tuberculosis drugs and treatment regimens. Nat Rev Drug Discov 12, 388–404 (2013). [DOI] [PubMed] [Google Scholar]
- Aguero F. et al. Genomic-scale prioritization of drug targets: the TDR Targets database. Nat Rev Drug Discov 7, 900–7 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowther G. J. et al. Identification of attractive drug targets in neglected-disease pathogens using an in silico approach. PLoS Negl Trop Dis 4, e804 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farhat M. R. et al. Genomic analysis identifies targets of convergent positive selection in drug-resistant Mycobacterium tuberculosis. Nat Genet 45, 1183–9 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan S., Daugelat S., Rao P. S. & Schreiber M. Prioritizing genomic drug targets in pathogens: application to Mycobacterium tuberculosis. PLoS Comput Biol 2, e61 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Jimenez F. et al. Target Prediction for an Open Access Set of Compounds Active against Mycobacterium tuberculosis. PLoS Comput Biol 9, e1003253 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman K., Yeturu K. & Chandra N. targetTB: a target identification pipeline for Mycobacterium tuberculosis through an interactome, reactome and genome-scale structural analysis. BMC Syst Biol 2, 109 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S., Baloni P., Mukherjee S., Anand P. & Chandra N. A multi-level multi-scale approach to study essential genes in Mycobacterium tuberculosis. BMC Syst Biol 7, 132 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- An J., Totrov M. & Abagyan R. Pocketome via comprehensive identification and classification of ligand binding envelopes. Mol Cell Proteomics 4, 752–61 (2005). [DOI] [PubMed] [Google Scholar]
- Kalidas Y. & Chandra N. PocketDepth: a new depth based algorithm for identification of ligand binding sites in proteins. J Struct Biol 161, 31–42 (2008). [DOI] [PubMed] [Google Scholar]
- Yeturu K. & Chandra N. PocketMatch: a new algorithm to compare binding sites in protein structures. BMC Bioinformatics 9, 543 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeturu K. & Chandra N. PocketAlign a novel algorithm for aligning binding sites in protein structures. J Chem Inf Model 51, 1725–36 (2011). [DOI] [PubMed] [Google Scholar]
- Anand P., Yeturu K. & Chandra N. PocketAnnotate: towards site-based function annotation. Nucleic Acids Res 40, W400–8 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand P. et al. Structural annotation of Mycobacterium tuberculosis proteome. PLoS One 6, e27044 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin J. E. et al. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog 7, e1002251 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassetti C. M., Boyd D. H. & Rubin E. J. Comprehensive identification of conditionally essential genes in mycobacteria. Proc Natl Acad Sci U S A 98, 12712–7 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen M. Y. & Sali A. Statistical potential for assessment and prediction of protein structures. Protein Sci 15, 2507–24 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colovos C. & Yeates T. O. Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci 2, 1511–9 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mereghetti P., Ganadu M. L., Papaleo E., Fantucci P. & De Gioia L. Validation of protein models by a neural network approach. BMC Bioinformatics 9, 66 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski R. A., Rullmannn J. A., MacArthur M. W., Kaptein R. & Thornton J. M. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR 8, 477–86 (1996). [DOI] [PubMed] [Google Scholar]
- Huang B. & Schroeder M. LIGSITEcsc: predicting ligand binding sites using the Connolly surface and degree of conservation. BMC Struct Biol 6, 19 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghersi D. & Sanchez R. EasyMIFS and SiteHound: a toolkit for the identification of ligand-binding sites in protein structures. Bioinformatics 25, 3185–6 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly M. L. The molecular surface package. J Mol Graph 11, 139–41 (1993). [DOI] [PubMed] [Google Scholar]
- Bairoch A. et al. The Universal Protein Resource (UniProt). Nucleic Acids Res 33, D154–9 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigrist C. J. et al. New and continuing developments at PROSITE. Nucleic Acids Res 41, D344–7 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader G. D. & Hogue C. W. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics 4, 2 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13, 2498–504 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nepusz T., Yu H. & Paccanaro A. Detecting overlapping protein complexes in protein-protein interaction networks. Nat Methods 9, 471–2 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris J. H. et al. clusterMaker: a multi-algorithm clustering plugin for Cytoscape. BMC Bioinformatics 12, 436 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arenas A., Fernández A. & Gómez S. Analysis of the structure of complex networks at different resolution levels. New Journal of Physics 10, 053039 (2008). [Google Scholar]
- Csardi G. & Nepusz T. The igraph software package for complex network research. InterJournal Complex Systems 1695, 1695 (2006). [Google Scholar]
- Zhang Y. & Skolnick J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res 33, 2302–9 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusova T. A. & Madden T. L. BLAST 2 Sequences, a new tool for comparing protein and nucleotide sequences. FEMS Microbiol Lett 174, 247–50 (1999). [DOI] [PubMed] [Google Scholar]
- Knox C. et al. DrugBank 3.0: a comprehensive resource for ‘omics’ research on drugs. Nucleic Acids Res 39, D1035–41 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S. & Iyengar R. Systems pharmacology: network analysis to identify multiscale mechanisms of drug action. Annu Rev Pharmacol Toxicol 52, 505–21 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer J. D., Holliday G. L. & Thornton J. M. The CoFactor database: organic cofactors in enzyme catalysis. Bioinformatics 26, 2496–7 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashton M., Nobeli I. & Thornton J. M. PROCOGNATE: a cognate ligand domain mapping for enzymes. Nucleic Acids Res 36, D618–22 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson M. L. et al. Binding MOAD, a high-quality protein-ligand database. Nucleic Acids Res 36, D674–8 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Boyle N. M. et al. Open Babel: An open chemical toolbox. J Cheminform 3, 33 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O. & Olson A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31, 455–61 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M., Goto S., Kawashima S., Okuno Y. & Hattori M. The KEGG resource for deciphering the genome. Nucleic Acids Res 32, D277–80 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M. et al. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res 42, D199–205 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M. & Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28, 27–30 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park K. & Kim D. Binding similarity network of ligand. Proteins 71, 960–71 (2008). [DOI] [PubMed] [Google Scholar]
- Zhang Z. & Grigorov M. G. Similarity networks of protein binding sites. Proteins 62, 470–8 (2006). [DOI] [PubMed] [Google Scholar]
- Finn R. D. et al. Pfam: the protein families database. Nucleic Acids Res 42, D222–30 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lew J. M., Kapopoulou A., Jones L. M. & Cole S. T. TubercuList--10 years after. Tuberculosis (Edinb) 91, 1–7 (2011). [DOI] [PubMed] [Google Scholar]
- Chandra N. & Padiadpu J. Network approaches to drug discovery. Expert Opin Drug Discov 8, 7–20 (2013). [DOI] [PubMed] [Google Scholar]
- Farkas I. J. et al. Network-based tools for the identification of novel drug targets. Sci Signal 4, pt3 (2011). [DOI] [PubMed] [Google Scholar]
- Cai S. et al. The rationale for targeting the NAD/NADH cofactor binding site of parasitic S-adenosyl-L-homocysteine hydrolase for the design of anti-parasitic drugs. Nucleosides Nucleotides Nucleic Acids 28, 485–503 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright H. T. Cofactors in fatty acid biosynthesis-active site organizers and drug targets. Structure 12, 358–9 (2004). [DOI] [PubMed] [Google Scholar]
- Opsahl T. Triadic closure in two-mode networks: Redefining the global and local clustering coefficients. Social Networks 35, 159–167 (2013). [Google Scholar]
- Kinnings S. L., Xie L., Fung K. H., Jackson R. M. & Bourne P. E. The Mycobacterium tuberculosis drugome and its polypharmacological implications. PLoS Comput Biol 6, e1000976 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim L. E. et al. Anthelmintic avermectins kill Mycobacterium tuberculosis, including multidrug-resistant clinical strains. Antimicrob Agents Chemother 57, 1040–6 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argyrou A., Vetting M. W., Aladegbami B. & Blanchard J. S. Mycobacterium tuberculosis dihydrofolate reductase is a target for isoniazid. Nat Struct Mol Biol 13, 408–13 (2006). [DOI] [PubMed] [Google Scholar]
- Argyrou A., Jin L., Siconilfi-Baez L., Angeletti R. H. & Blanchard J. S. Proteome-wide profiling of isoniazid targets in Mycobacterium tuberculosis. Biochemistry 45, 13947–53 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty S., Gruber T., Barry C. E. 3rd, Boshoff H. I. & Rhee K. Y. Para-aminosalicylic acid acts as an alternative substrate of folate metabolism in Mycobacterium tuberculosis. Science 339, 88–91 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao F. et al. Binding pocket alterations in dihydrofolate synthase confer resistance to para-aminosalicylic acid in clinical isolates of Mycobacterium tuberculosis. Antimicrob Agents Chemother 58, 1479–87 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J. et al. para-Aminosalicylic acid is a prodrug targeting dihydrofolate reductase in Mycobacterium tuberculosis. J Biol Chem 288, 23447–56 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information