

Abstract
Phosphorylation of the C-terminal domain of RNA polymerase II (CTD) plays an essential role in eukaryotic transcription by recruiting transcriptional regulatory factors to the active polymerase. However, the scarcity of basic residues and repetitive nature of the CTD sequence impose a huge challenge for site-specific characterization of phosphorylation, hindering our understanding of this crucial biological process. Herein, we apply LC-UVPD-MS methods to analyze post-translational modification along native sequence CTDs. Application of our method to the Drosophila melanogaster CTD reveals the phosphorylation pattern of this model organism for the first time. The divergent nature of fly CTD allows us to derive rules defining how flanking residues affect phosphorylation choice by CTD kinases. Our data support the use of LC-UVPD-MS to decipher the CTD code and determine rules that program its function.
Graphical abstract

RNA polymerase II (RNAP II) is the eukaryotic multiprotein complex responsible for the generation of messenger and many noncoding RNAs.1–4 In vivo these processes require the coordinated phosphorylation and dephosphorylation of the carboxyl-terminal domain (CTD) of RNAP II’s largest subunit, Rpb1.5 The CTD is an evolutionarily conserved domain composed of a species-specific number of repeats of the consensus amino acid heptad, Tyr1-Ser2-Pro3-Thr4-Ser5-Pro6-Ser7.6 Site-specific phosphorylation of the CTD consensus heptad (i.e., at Tyr1, Ser2, Thr4, Ser5, or Ser7) results in the orchestrated recruitment of protein factors that influence transcription at different stages and promote RNA processing, gene regulation, and accurate initiation and termination of transcription.5 This series of post-translational modifications and the protein factors they recruit constitute the “CTD Code” for eukaryotic transcription.7
Specific monoclonal antibodies against modifications of the consensus heptad have broadened our understanding of CTD phosphorylation and helped characterize the temporal association of CTD modifying proteins by different modifications during the transcription cycle.5 However, antibodies suffer from several inherent limitations. First, the available antibodies have been raised against consensus CTD sequences that are not present in all species. Important model systems like Drosophila melanogaster and Homo sapiens contain a number of heptads that diverge from the consensus sequence and cannot be confidently characterized by these tools. Second, the ability of other marks within CTD heptads to interfere with antibody/epitope recognition is not completely known.8 This results in an inability to reliably quantify the total phosphorylation of the CTD. Third, antibodies cannot localize phosphorylation marks along the sprawling CTD. Finally, due to the similarity of phosphate-accepting motifs (i.e., YS2P vs TS5P vs PS7Y) the potential for cross-reactivity of these antibodies cannot be ignored.
Tandem mass spectrometry methods can map the location of post-translational modifications in high resolution and, therefore, are considered the gold standard for such analysis.9 However, the highly repetitive heptapeptide sequence of RNAP II CTD is especially challenging for LC-MS/MS analysis. Recently, important strides have been made in the study of the CTD with the development of mass spectrometry approaches to analyze the CTD in yeast10 and human cells11 by introducing mutations to the CTD sequence to facilitate proteolytic digestion and modification site localization. By expressing these mutant constructs in cells and analyzing resultant modifications with collision-induced dissociation (CID), several conclusions were made. First, CTD phosphorylation is evenly dispersed throughout the length of the CTD and is not localized proximal or distal to the catalytic core of RNAP II. Second, within individual heptad repeats, Ser2 and Ser5 phosphorylations are significantly more abundant compared to other modification sites (i.e., phosphorylation of Tyr1, Thr4, and Ser7). Finally, the phosphorylation along the CTD is significantly less dense than once thought, with the majority of heptads only accepting one phosphate. These findings establish mass spectrometry as an indispensable tool for studying CTD biology and lay the groundwork for further technological advancement.
Developing methodologies to analyze the CTD without the necessity to change the original sequence would be an important step toward understanding the function of the CTD. However, the scarcity of basic sites prevents tryptic digestion and also suppresses effective protonation, which is critical for analysis using conventional positive mode MS/MS techniques including CID and electron activation methods. Phosphorylation increases the acidity of the consensus sequence and further decreases the efficiency of protonation and the success of subsequent positive mode analysis. Negative polarity peptide analysis generally results in uninformative CID mass spectra comprised mostly of neutral phosphate losses, precluding both identification and characterization of peptides.12,13 Ultraviolet photodissociation (UVPD) using 193 nm photons is an alternative to existing collision- and electron-based activation methods that offers several advantages for phosphorylation site mapping in the CTD. Charge state bias, which is a limitation for traditional methods, is largely overcome using UVPD, and high sequence coverage has been demonstrated even for singly charged precursor ions.14–17 The high energy deposition (6.4 eV) that is achieved upon absorption of a 193 nm photon permits access to fragmentation pathways that are not available using traditional methods, ultimately leading to the formation of a, b, c, x, y, and z ions which account for cleavage of each bond in the peptide backbone.18 The greater number of product ions obtained using UVPD increases the confidence of peptide sequencing results while also improving the ability to pinpoint sites of modification. Another merit of UVPD is the ability to generate diagnostic product ions from peptide anions.19–21 Although peptide analysis is generally undertaken in the positive ion mode based on greater sensitivity and number of applicable MS/MS techniques, the negative mode offers unique benefits for certain types of peptides. This is especially true for characterization of labile PTMs including phosphorylation, sulfation, and O-glycosylation, all of which exhibit superior retention using negative mode UVPD.20,22–25 Furthermore, alternating between positive and negative electrospray ionization modes is easily done, even within a single LC-MS run, further increasing the versatility of UVPD-MS.
In the present study, UVPD-MS is used to its full advantage in both positive and negative modes to analyze CTD modifications in unprecedented detail. We investigate two highly dissimilar eukaryotic CTDs with no introduced mutations: yeast (Saccharomyces cerevisiae) that is composed almost entirely of consensus heptads and fruit fly (Drosophila melanogaster) that diverges greatly from consensus sequence. In the yeast CTD (yCTD), we phosphorylate GST-yCTD constructs with two physiologically relevant CTD kinases, TFIIH and Erk2, and pinpoint the resultant phosphorylation sites. To further test the power of UVPD in detecting CTD modification, we use the Drosophila melanogaster CTD (DmCTD), which has a highly divergent sequence with only 2 of its approximately 45 heptad repeats matching the consensus sequence. By localizing the phosphorylation sites introduced by Erk2, our results reveal how the flanking residues affect the phosphorylation choice by this kinase. The novel strategy establishes UVPD-MS in combination with alternative proteases as a powerful way to investigate native CTD modifications across species.
RESULTS AND DISCUSSION
Analysis of Saccharomyces cerevisiae CTD
The identification of CTD modifications in the context of the wild-type amino acid sequence is obviously ideal for drawing physiologically relevant conclusions. However, the lack of basic residues in the consensus CTD sequence makes it inapplicable to established trypsin digestion methods for mass spectrometric analysis which previously necessitated the introduction of lysine or arginine residues at the Ser7 position for yeast CTD analysis.10,11 To investigate native yeast CTD modification we turned to alternative protease digestion strategies. Although yCTD lacks basic residues, it is highly enriched in the residue tyrosine that is found in the consensus sequence. The proteases chymotrypsin and proteinase K typically cleave peptide bonds flanked by aromatic and, to a lesser extent, aliphatic residues making the CTD an ideal substrate for these enzymes.
To investigate the efficacy of these proteases in LC-UVPD-MS analysis of yCTD we generated a GST-yCTD construct and purified recombinant protein to homogeneity. The recombinant protein was initially treated with trypsin overnight to digest and remove the GST tag. The intact yCTD portion was purified from GST peptides by passing the digest through a 10 kDa molecular weight cut off (MWCO) filter, which retained yCTD. The yCTD sample was then treated with either chymotrypsin or proteinase K. The final digests were then analyzed using LC-UVPD-MS. Varying numbers of missed cleavages by chymotrypsin resulted in a more complex peptide mixture compared to using proteinase K which provided more efficient digestion and generated just two dominant heptad peptides with sequences YSPTSPS and SPSYSPT (Figure 1A). While the observed proteinase K cleavages that occurred C-terminal to Thr4 and Ser7 deviated from the expected specificity C-terminal to tyrosine, the cleavage pattern was reproducible and thus proteinase K was considered the more optimal protease for yCTD digestion (for a schematic of the finalized work-flow, see Supplemental Figure 1). Ultraviolet photodissociation at 193 nm and CID were evaluated for sequencing the heptads generated by proteinase K digestion, and both identified two peptides with sequences YSPTSPS and SPSYSPT (Figure 1A). Using UVPD more extensive fragmentation including the production of a- and x-type sequence ions was achieved (Figure 1C and 1E), while the abundance of less informative water loss ions was decreased relative to the analogous CID spectra (Figure 1B and 1D). Additionally, improved phosphate retention on product ions has been demonstrated for UVPD,26 making it ultimately better suited for CTD phosphorylation analysis following kinase treatment. Both peptides contain a heptad repeat but the location of bond cleavage varies. For clarity, we will analyze and discuss the residues from this and subsequent analyses according to their position in the established CTD naming and numbering convention (i.e., Tyr1, Ser2, Pro3, Thr4, Ser5, Pro6, Ser7) though their positions in generated peptides may vary.
Figure 1.
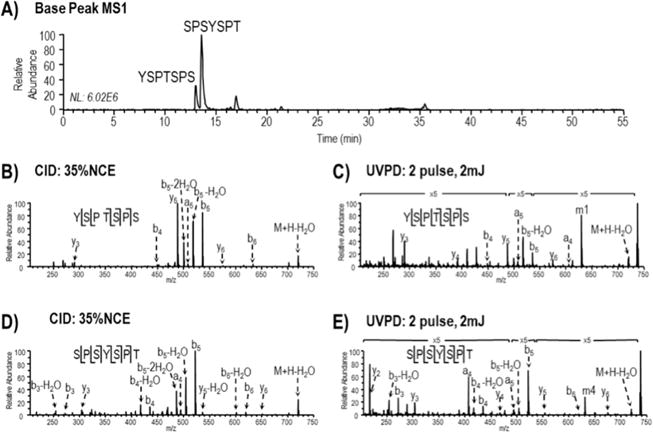
LC-MS base peak MS1 chromatogram and MS/MS spectra of unmodified yeast CTD heptads following digestion with trypsin and proteinase K. (A) LC-MS base peak MS1 chromatogram with peaks corresponding to the elution of unmodified heptad peptides (m/z 738) detected at time 13–14 min. (B and C) MS/MS (CID and UVPD) mass spectra acquired for protonated YSPTSPS. (D and E) MS/MS (CID and UVPD) mass spectra acquired for protonated SPSYSPT. Tyr side chain losses generated by UVPD are denoted m1 and m4 for YSPTSPS and SPSYSPT, respectively.
We next sought to apply our LC-UVPD-MS workflow to phosphorylated yCTD. To generate phosphorylated yCTD we treated GST-yCTD with TFIIH, a multiprotein complex that is required for the phosphorylation of the CTD at Ser5 during the initiation of RNAP II transcription.27 This reaction is catalyzed by the kinase subunit Kin28, in yeast, or Cdk7, in multicellular organisms.28,29 Given that nearly half of the heptads in the mammalian CTD match the consensus found in the yeast CTD,5 we utilized commercially available human TFIIH complex to investigate its patterning along the yCTD. Upon UVPD-MS analysis of TFIIH treated GST-yCTD, a peptide (ion of m/z 818, 1+) corresponding to the mass of the consensus heptad plus one phosphorylation was observed in the LC-MS chromatogram (Figure 2A), in addition to the two previously detected unmodified heptad peptides of m/z 738 corresponding to YSPTSPS and SPSYSPT. The MS/MS spectra acquired during the elution of the phospho-heptad showed distinctive variations at different elution time-points, thus revealing the presence of two isomers (Figure 2B). Targeted LC-UVPD-MS runs allowed better characterization of the two isomers. Two abundant UVPD product ions which were unique to the early (m/z 407, a4 from SPSYpSPT) and late (m/z 594, x5 from YSPTpSPS) portions of the elution profile were identified. Extracted ion chromatograms (Figure 2B) revealed that the two ions matched to different heptad peptides with phosphorylation at different positions as defined by the consensus sequence (YSPTSPS). The UVPD mass spectra confirmed the sequences as SPSYpSPT with phosphorylation on Ser2 (Figure 2C) and YSPTpSPS with phosphorylation on Ser5 (Figure 2D). In addition to the unique a4 (SPSYpSPT) and x5 (YSPTpSPS) ions, other diagnostic ions that confidently differentiated each of the two phosphopeptides were y2 and a2 and the presence or absence of y6. These findings demonstrate the ability of TFIIH to phosphorylate both Ser2 and Ser5 of the yCTD. Based on the integrated chromatographic peak areas attributed to the SPSYpSPT peptide versus the YSPTpSPS peptide in Figure 2B (and considering the similar ionization efficiencies and fragmentation efficiencies of these two heptads), it appears that phosphorylation of Ser5 may be more prominent than phosphorylation of Ser2. More accurate quantitation is difficult given that the extracted LC traces are based on single unique fragment ions for each peptide, but it is reasonable to estimate that phosphorylation of the Ser5 is favored over phosphorylation of the Ser2. No phosphorylations of Ser7, Thr4, or Tyr1 were detected.
Figure 2.
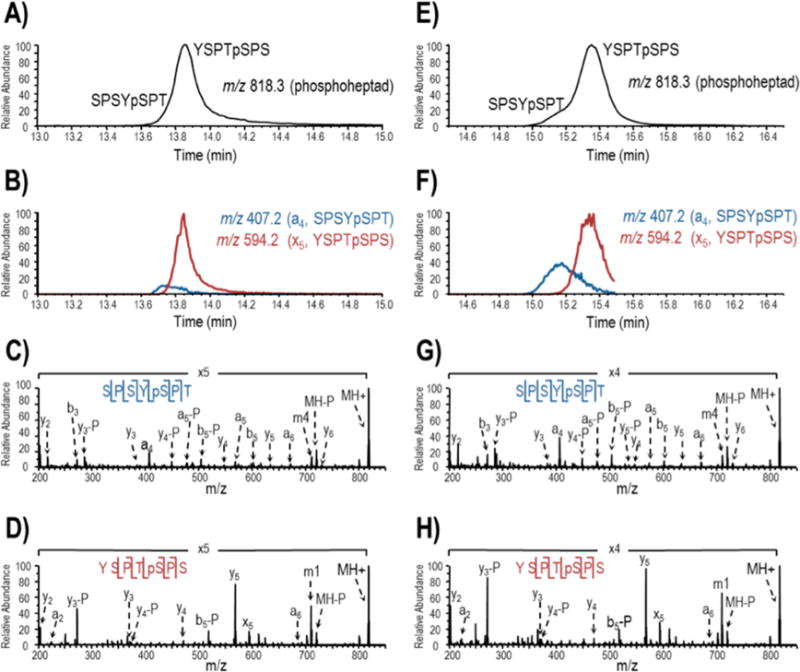
LC-UVPD-MS analysis of TFIIH and Erk2 treated yeast GST-CTD digested with trypsin and proteinase K. The base peak MS1 chromatograms (0–45 min) are provide in Supplemental Figure 2. Two singly phosphorylated heptads, m/z 818.3, are partially resolved in the base peak MS1 chromatogram (A for TFIIH and E for Erk2). In subsequent LC-MS analysis, m/z 818.3 was targeted for activation during the course of elution and extracted ion chromatograms (XICs) for distinguishing product ions, a4 from protonated SPSYpSPT and x5 from protonated YSPTpSPS, were generated to track the isomeric peptides (B for TFIIH and F for Erk2). UVPD using two 2 mJ pulses was used to sequence the heptad peptides and localize the sites of phosphorylation (C and D for TFIIH; G and H for Erk2). Ions that have undergone phosphate neutral loss are denoted with “-P”. Tyr side chain losses generated by UVPD are denoted m1 and m4 for YSPTSPS and SPSYSPT, respectively.
Recently, the physiological role of mitogen-activate protein kinase 1 (MAPK1) or Erk2 in phosphorylating the CTD of poised RNA polymerase II at developmentally important genes has been reported.30 In mouse embryonic stem cells, Erk2 associates with a transcriptionally poised RNAP II and phosphorylates it at Ser5. Upon specific developmental signals, occupancy by Erk2 and associated proteins gradually diminishes and these regions of the chromatin become transcriptionally active to direct cells down specific developmental paths. To identify phosphorylation targets of Erk2 within the consensus CTD, we took advantage of our LC-UVPD-MS approach to obtain high-resolution information about Erk2 patterning along the yCTD.
GST-yCTD was phosphorylated by Erk2 kinase and analyzed by the protocol used for TFIIH analysis (Supplemental Figure 1). Intriguingly, the LC-UVPD-MS results obtained for GST-yCTD treated with Erk2 kinase mirrored those observed with TFIIH in terms of the detection and differentiation of the same two phosphorylated heptads (Figure 2E and 2F). Again, two monophosphorylated species containing the consensus sequence were observed with phosphorylation confirmed at Ser2 or Ser5 upon Erk2 treatment of the CTD based on the UVPD mass spectra (Figure 2G and 2H). The YSPTpSPS phosphopeptide appears to be more prominent compared to the SPSYpSPT for the Erk2 treatment based on comparison of the abundance of the x5 and a4 ions for each peptide, respectively. Thus, preferential phosphorylation of Ser5 over Ser2 mirrors the trend observed for the TFIIH-treated yCTD. Phosphorylation was not detected at Ser7, Thr4 or Tyr1 in our spectra.
Despite the reactions occurring under conditions of excess ATP to GST-yCTD substrate, in the presence of large amounts of kinase, and during long incubation times, we did not observe simultaneous phosphorylation of Ser2 and Ser5 within a single heptad span. This is consistent with recent analysis of yCTD10 in that phosphorylation marks are not placed as densely as previously thought.
Our analysis provides the first evidence to support the similarity in CTD patterning between two kinases TFIIH and Erk2. Both kinases can phosphorylate Ser2 and Ser5, with a clear preference for Ser5. The preferential phosphorylation of Ser5 over Ser2 is more pronounced for the TFIIH than for Erk2. Furthermore, these findings fall in line with existing CTD MS data that suggests the CTD code is simplistic and trends toward single phosphorylation of a given heptad span.10,11
Analysis of Drosophila melanogaster CTD
As described above, our innovative LC-UVPD-MS approach proved applicable to the study of modifications within the heptads of native yCTD. An exciting benefit of this technology is it should be widely applicable to other organisms regardless of divergence from consensus sequence. A prime example for such a CTD is that of Drosophila melanogaster (fruit fly) in which only two of its forty-five heptad repeats faithfully recreate the consensus heptad, thus precluding analysis by antibodies generated against consensus heptads. Due to the heterogeneity of the DmCTD sequence, these analyses have the additional benefit of permitting us to map phosphorylations along the full-length DmCTD. This inherent variation from consensus sequence allows us to interrogate the role of flanking residues within the same heptad on the preferential site of phosphorylation.
Drosophila melanogaster is an important model system in the study of transcription, with many of the conclusions being drawn from this model organism extending across eukaryotes.31–35 Most core developmental mechanisms are highly conserved33 including JAK-STAT,31 Wnt signaling,32 Hedgehog signaling pathways36 and recently characterized Jarid2-Polycomb Repressive Complex 2 (PRC2) pathway.37 Jarid2 is the founding member of the JmjC domain family of proteins and has been characterized as a transcriptional corepressor.38,39 PRC2 is a multiprotein complex that catalyzes H3K27me3 methylation, a histone mark associated with gene repression.39 In flies and mouse embryonic stem cells (ESC), Jarid2 and PRC2 associate in vivo along chromatin at a subset of developmentally important genes37,39 and contribute to gene silencing by propagating H3K27me3 marks and maintaining RNAPII in the transcriptionally poised state.39 These poised RNAP II complexes are activated upon specific signals to induce rapid transcription and solidify cells down specific developmental paths. Poised RNAP II stands as a conserved developmental mechanism across metazoans.40
Recently, work in mouse ESC has demonstrated a physiological role for Erk2 as a bona fide CTD kinase for transcriptionally poised RNAPII at Jarid2-PRC2 targeted genes.30 This phosphorylation contributes to the poised RNAP II state and informs the subsequent transition to an active gene state characterized by replacement of poised RNAP II factors with canonical transcription machineries. The developmental role and mechanism of Jarid2-PRC2 is conserved between flies and mammals,37,39 and supports a yet to be explored role of Erk2 in fly development. To better understand the phosphorylation of Drosophila melanogaster CTD by Erk2, we endeavored to analyze the Erk2 phosphorylation pattern using our novel LC-UVPD-MS approach.
We generated GST fusion constructs of full-length Drosophila melanogaster CTD (GST-DmCTD). Multiple GST-DmCTD truncation constructs were also produced to reduce the overall complexity in each LC-MS run and facilitate complete sequence coverage. The sequences of the various DmCTD constructs are summarized in Supplemental Table 1, with heptads numbered as describe in Figure 3A. The CTD1 construct contains full-length DmCTD. CTD2 and CTD3 included the N-terminal region of the protein from heptad 1–16 and 1–25, respectively, whereas CTD4 included the C-terminal region from heptad 26–45.41 CTD5 covered an interior region of the protein spanning heptads 16–24. To digest GST-CTD1, trypsin, proteinase K, and chymotrypsin were considered. The low frequency of trypsin cleavage sites throughout the DmCTD prevents tryptic digestion into appropriately sized peptides for bottom-up LC-MS analysis. Proteinase K was rejected for its poorly defined cleavage specificity,42 a factor that would confound prediction of products within the more variable DmCTD sequence and increase the complexity of an already complex digest. Therefore, chymotrypsin was chosen as it more consistently cleaves C-terminal to aromatic residues with additional lower activity cleavage C-terminal to methionine, leucine, and histidine residues. LC-UVPD-MS analysis of unmodified GST-DmCTD chymotrypsin digests revealed peptides applicable to LC-MS and high coverage of the full-length as well as truncated DmCTD constructs.
Figure 3.
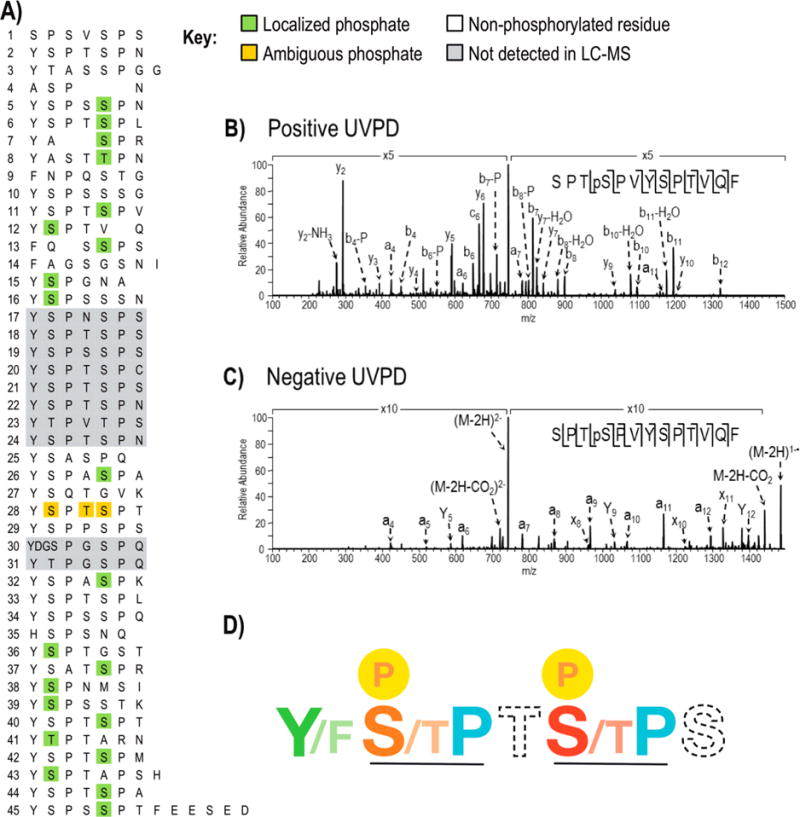
Phosphorylations identified in Drosophila melanogaster CTD following treatment with Erk2 using LC-UVPD-MS. A) The site of phosphorylation in Drosophila melanogaster CTD where confirmed sites are highlighted in green. One peptide (repeat 28) shows a single phosphorylation but the position of the phosphate could not be distinguished among three sites (shown in gold). Regions of the protein in gray were not detected in the Erk2 treated or control CTD samples. The phosphorylation map is the composite of sites identified using positive mode and negative mode LC-UVPD-MS. Representative UVPD mass spectra from positive mode (B) and negative mode (C) analysis are shown for the chymotryptic peptide SPTpSPVYSPTVQF which covers heptads 11 and 12. In both polarities, the doubly charged ions of m/z 745.3 (for positive mode) and m/z 743.3 (for negative mode) were activated using 2 pulses at 2 mJ. Ions that are detected following phosphate neutral loss are denoted by “-P”. D) The “rule book” for CTD phosphorylation by Erk2. SP motifs are recognized with the strict requirement for proline (blue) following serine/threonine (orange and red). Ser5 (S, red) is favored over Ser2 (S, orange) during phosphorylation by Erk2. Thr4 (T) and Ser7 (S) shown in dashed font had little impact on the phosphorylation outcome. An aromatic residue such as tyrosine (Y) or phenylalanine (F) is required (colored green) for phosphorylation.
To analyze phosphorylation patterning along GST-DmCTD, constructs were treated with Erk2 kinase and subjected to LC-UVPD-MS analysis (Figure 3A). Both positive and negative ionization modes were used to complement one another. Usually positive mode ionization affords high coverage with ample signal/noise ratio for standard peptides comprised of amino acids with basic side-chains (i.e., Lys, Arg). Analyzing peptides in negative mode using UVPD helps to ensure the detection of multiply phosphorylated peptides, which typically ionize more readily as anions than as protonated cations. In this way, negative mode UVPD analysis accounts for peptides in higher phosphorylation states which may arise from phosphorylation of neighboring heptads in multiheptad long peptides formed by chymotrypsin missed cleavages. Representative spectra for UVPD analysis are shown in Figure 3B (positive mode) and 3C (negative mode), each displaying extensive sequence coverage and confident phosphate localization. By analyzing peptides in both positive and negative ion modes, we ensured identification of the maximum number of phosphorylation sites for DmCTD. Indeed, positive mode UVPD analysis provided the best overall sequence coverage of full-length DmCTD. Following treatment with Erk2, 22 phosphopeptides were identified accounting for 20 unique phosphosites from 20 individual heptads (Table 1A). Phosphorylation of two additional heptads was also detected, but could not be confidently localized to a specific serine, threonine, or tyrosine (Table 1B). Fewer overall phosphopeptides were identified using negative mode UVPD (Table 2A and B); however, one phosphosite from heptad 5 that could not be pinpointed by positive mode UVPD was confidently localized upon negative mode UVPD analysis. All other site assignments agreed based on the UVPD spectra acquired for the protonated and deprotonated phosphopeptides.
Table 1.
(A) CTD Peptides with Localized Phosphosites from Positive Mode UVPD Analysisa and (B) CTD Peptides with Ambiguous Phosphosites from Positive Mode UVPD Analysis
| A | ||
|---|---|---|
| sequence | heptad # (phosphosite in heptad) | construct |
| TASSPGGASPNYSPSSPNYSPTpSPLY | 6 (S5) | full |
| YApSPRYASTTPNFNPQSTGY | 7 (S5) | full |
| ASPRYASTpTPNFNPQSTGY | 8 (T5) | full |
| SPTpSPVYpSPTVQF | 11 (S5), 12 (S2) | full |
| QSpSPSFAGSGSNIY | 13 (S5) | full |
| QSSPSFAGSGSNIYpSPGNAY | 15 (S2) | full |
| SPGNAYpSPSSSNY | 16 (S2) | full |
| SASPQYSPApSPAYSQTGVKY | 26 (S5) | full |
| SPApSPAYSQTGVKY | 26 (S5) | full |
| SPApSPKYSPTSPL | 32 (S5) | CTD4 |
| YSPSSPQHSPSNQYpSPTGSTY | 36 (S2) | full |
| SATpSPRYSPNMSIYSPSSTKY | 37 (S5) | full |
| SATSPRYpSPNMSIY | 38 (S2) | full, CTD4 |
| pSPSSTKY | 39 (S2) | full |
| SPTpSPTYpTPTARNY | 40 (S5), 41 (T2) | full |
| SPTSPTYpTPTARNY | 41 (T2) | full, CTD4 |
| SPTpSPMYSPTAPSHY | 42 (S5) | full, CTD4 |
| SPTpSPMYpSPTAPSHY | 42 (S5), 43 (S2) | full |
| SPTpSPAYSPSpSPTFEESED | 44 (S5), 45 (S5) | full |
| B | ||
|---|---|---|
| sequence | heptad # (phosphosite in heptad) | construct |
| TASSPGGASPNYp(SPSS) PNYSPTpSPLY | 5 (S2/S4/S5), 6 (S5) | Full |
| p(SPTS)PTYSPPSPSY | 28 (S2/T4/S5) | CTD4 |
| p(SPTS)PAYSPSSPTFEESED | 44 (S2/T4/S5) | Full, CTD4 |
A sequence coverage map is provided in Supporting Information Table 2.
Table 2.
(A) CTD Peptides with Localized Phosphosites from Negative Mode UVPD Analysisa and (B) CTD Peptides with Ambiguous Phosphosites from Negative Mode UVPD Analysis
| A | ||
|---|---|---|
| sequence | heptad # (phosphosite in heptad) | construct |
| SPSpSPNYSPTpSPLY | 5 (S5), 6 (S5) | Full, CTD2, CTD3 |
| SPSSPNYSPTpSPLY | 6 (S5) | Full, CTD2, CTD3 |
| SPTpSPVYpSPTVQF | 11 (S5), 12 (S2) | Full, CTD2, CTD3 |
| SPTSPVYpSPTVQF | 12(S2) | Full, CTD2, CTD3 |
| QSpSPSFAGSGSNIY | 13 (S5) | Full, CTD2, CTD3 |
| QSSPSFAGSGSNIYpSPGNAY | 15 (S2) | Full |
| SPApSPAYSQTGVKY | 26 (S5) | Full, CTD4 |
| SPApSPKYSPTSPL | 32 (S5) | CTD4 |
| SIYpSPSSTKY | 39 (S2) | CTD4 |
| SPTSPMYpSPTAPSHY | 43 (S2) | Full, CTD4 |
| SPTpSPAYSPSpSPTFEESED | 44 (S5), 45 (S5) | CTD4 |
| SPTSPAYSPSpSPTFEESED | 45 (S5) | Full, CTD4 |
| B | ||
|---|---|---|
| sequence | heptad # (phosphosite in heptad) | construct |
| ASp(TT)PNFNPQSTGY | 8 (T4/T5) | Full, CTD2, CTD3 |
| SPp(TS)PVYSPTVQF | 11 (T4/S5) | Full, CTD2, CTD3 |
| SAp(TS)PRYpSPNMSIY | 37 (T4/S5), 38 (S2) | CTD4 |
| SPp(TS) PAYSPSSPTFEESED | 44 (T4/S5) | Full, CTD4 |
A sequence coverage map provided in Supporting Information Table 2.
Our use of truncated DmCTD constructs (CTD2–4) proved beneficial in both confirming phosphosites identified and revealing phosphosites not observed in full-length DmCTD1. Identical phosphorylation sites were detected in CTD2 and CTD3 versus full length CTD1 (Table 2). CTD4, which covers the distal region of the DmCTD, shows several additional sites in negative mode analysis (Table 2A) that were then confirmed in positive mode analysis (Table 1A). A single unique phosphopeptide, SPApSPKYSPTSPL, was identified in both positive and negative modes only in CTD4 constructs, supporting the benefit of decreasing LC-MS complexity to identify all possible phosphorylation sites.
Despite the additional measures taken to facilitate full characterization of DmCTD, two regions lacked sequence coverage. These two regions, heptads 17–24 and heptads 30–31 (Figure 3A), were not detected in positive or negative mode or in any of the truncation constructs that contained them. These regions were also not detected in untreated GST-DmCTD constructs. These findings suggest the lack of coverage is not a result of phosphorylation of these heptads but rather is a characteristic inherent to this region. A smaller construct CTD5, which includes repeats 16–24, was made to allow focused study in this region, but it proved to be consistently resistant to digestion using chymotrypsin and resulted in subpar MS/MS analysis. A possible culprit is the cysteine located in the Ser7 position of heptad 20 which may impede ionization based on interprotein disulfide linkages, although reduction and alkylation of the CTD5 construct did not appear to improve the sequence coverage. Furthermore, the solubility of CTD5, once cleaved from the GST-tag, appears to be low and results in a low retention in solution upon GST tag cleavage. Instead of employing the bottom-up approach, an alternative intact protein strategy was pursued to characterize this region, which is described in the next section. Despite these uncovered regions of the DmCTD sequence, the majority of DmCTD exhibits high coverage upon LC-UVPD-MS analysis, and our findings can be summarized to derive conclusions about the distribution of phosphorylation along DmCTD and the influence of neighboring residues within heptads on Erk2 phosphorylation preference (Figure 3A).
Upon examination of DmCTD modifications several characteristics of Erk2 phosphorylation become apparent (Figure 3D). As expected for serine/threonine kinases such as Erk2, phosphorylation occurs exclusively on serine and threonine residues. Interestingly, other residues within the heptad seem to govern phosphorylation choice. First, all observed phosphorylations occur on serine/threonine residues flanked in the +1 (succeeding) position by proline. In nonconsensus heptads when proline is not present next to serine, the serine is not subject to phosphorylation as observed for the Ser5 position of repeats 9 and 10. When there is no serine/threonine at the fifth position of the consensus sequence or if the fifth residue serine/threonine is not flanked by proline at +1 position as found in heptads 16, 36, 38, 39, 41, and 43, phosphorylation at the Ser2 position can occur, given a suitable S/T-P motif is present (Figure 3A). This is in line with the role of Erk2 as a mitogen-activated protein kinase that phosphorylates serine/threonine residues flanked on their C-terminus by proline. We observe phosphorylation only at the Ser2 and Ser5 positions with a preference for Ser5 and no phosphorylation observed at the Thr4 or Ser7 position. Second, in contrast to the strict requirement of C-terminal proline after serine/threonine, residues at the 4 and 7 position of the heptad appear inconsequential for phosphorylation choice. The fourth position in the phosphorylated heptads can be occupied by serine (S), threonine (T), glycine (G), alanine (A), or asparagine (N). No correlation between the identity of the fourth position residue and the site of phosphorylation by Erk2 was identified. The seventh position in the heptad is even more divergent with residues asparagine (N), leucine (L), arginine (R) valine (V), glutamine (Q), serine (S), threonine (T), lysine (K), isoleucine (I), and methionine (M) all occupying phosphorylated heptads. Furthermore, the absence of these amino acids at the 4 and 7 position does not abolish phosphorylations within heptads (i.e., heptads 7 and 15). Taken together, these results suggest the requirements for residues at positions 4 and 7 are not stringent and have little impact on phosphorylation choice. Third, an aromatic residue like tyrosine (Y) or phenylalanine (F) is always present in the first position in phosphorylated heptads. This position is the best conserved within the consensus sequence. Substitution by alanine (A) (heptad 4) or histidine (H) (heptad 35) does not result in phosphorylation despite the presence of suitable SP motifs.
As observed in yCTD, phosphorylation of DmCTD by a single kinase results in a single phosphorylation within a heptad of conventional numbering with tyrosine (Y) as the first residue. Double phosphorylation (e.g., both Ser2/Ser5) within the same consensus heptad with Ser5 phosphorylation immediately C-terminal to Ser2 phosphorylation (i.e., YpSPTpSPS) was not observed, despite the clear ability of Erk2 to phosphorylate both sites in yCTD. Interestingly, we do observe Ser2 phosphorylation immediately following Ser5 phosphorylation (i.e., TpSPSYpSP) resulting in double phosphorylation within a seven-residue frame. In all these scenarios (repeat 11/12, 40/41, 42/43) (Table 1), the second repeat does not exhibit phosphorylation at position 5, either due to the absence of serine or threonine at the fifth position or the absence of proline at the sixth position, resulting in the phosphorylation Ser2 instead. Therefore, in the phosphorylation patterns of DmCTD, doubly phosphorylated Ser5/Ser2 is possible in addition to the singly phosphorylated Ser2 or Ser5 within a seven amino acid span.
One region of DmCTD (repeat 17–24) was consistently not covered in our bottom-up analysis of full-length (CTD1) and minimal-length (CTD5 including repeat 16–24) DmCTD recombinant protein (Figure 3A). However, its sequence does not deviate greatly from consensus sequence, making it a potentially good substrate for Erk2 based on our observed rulebook. Therefore, we used alternative MS approaches to detect phosphorylation. To overcome the suspected low solubility of CTD5 in the absence of the recombinant GST-tag, we used an intact protein MS strategy and measured the overall mass of the GST-CTD5 construct before and after kinase treatment. The mass spectrum of the intact wild-type GST-CTD5 exhibits two additional products after 1 h of kinase treatment, each consistent with addition of one or two phosphates (Figure 4A). Therefore, this region of DmCTD is subject to phosphorylation.
Figure 4.
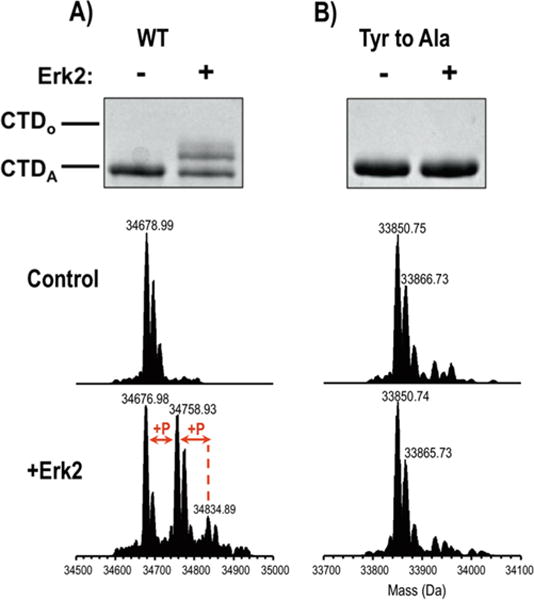
Gel shift assay and intact mass analysis results for CTD5 before and after treatment with Erk2. A) wild-type or native sequence and B) all tyrosine residues (Tyr) were mutated to alanine (Ala). A portion of the mass spectra of the intact proteins is shown to illustrate the addition of phosphate groups upon treatment with Erk2.
Tyrosine 1 is required for CTD phosphorylation by Erk2
Our observed rulebook suggests a previously unexplored role of tyrosine 1 in directing CTD phosphorylation. Erk2 phosphorylation at Ser5 and Ser2 appears to require a tyrosine or phenylalanine in the first position of the heptad. To test this observation and demonstrate our UVPD-MS approaches’ ability to generate hypotheses we replaced tyrosine in each repeat of the CTD5 construct with alanine (YtoA). This and the wild-type construct were phosphorylated with Erk2 and analyzed by SDS-PAGE gel shift and intact mass analysis (Figure 4). Consistent with our analysis for full-length DmCTD, the YtoA mutation abolishes phosphorylation in both gel shift and MS assays (Figure 4B). In fact, the untreated and Erk2-treated YtoA construct gives nearly identical mass spectra. This observation further supports the requirement for tyrosine or similarly bulky hydrophobic residue at the first position of the heptad. This requirement may contribute to the high conservation of tyrosine at this position in even the most divergent CTD sequences.5
Discussion
Our novel LC-UVPD-MS strategy provides high-resolution PTM identification and localization along native CTD sequences and across species. In addition to attaining residue level modification information, we have overcome previous limitations to MS/MS analysis of the CTD by utilizing alternative proteases and peptide fragmentation via UVPD. These innovations allow us to analyze native CTD sequence without the need to introduce protease sites. Additionally, UVPD is applicable to both positive and negative mode analysis, facilitates the detection of PTMs easily lost in other fragmentation methods, and generates a greater number of diagnostic fragment ions. Because of these factors, LC-UVPD-MS localizes phosphorylation marks with greater sensitivity and confidence than other fragmentation methods currently reported for CTD MS. Impressively, we were able to identify 22 phosphorylation sites in the DmCTD.
The major determinant for the phosphorylation state of the CTD appears to be the identity and positioning of flanking residues. Using the highly divergent DmCTD, we were able to interrogate how flanking residues affect Ser2 and Ser5 phosphorylation by Erk2 (Figure 3D). The strictest rule demands that a proline residue follow the serine/threonine subject to phosphorylation. Interestingly previous work from us and other laboratories has shown that this proline is also essential for dephosphorylation, through both its identity and isomerization state.43–45 Proline is unique among the natural amino acids because it can stably assume the cis-isomer conformation about its peptide bond, which is required for recognition by the essential CTD phosphatase Ssu72.43 Therefore, the presence of proline directs both the phosphorylation and dephosphorylation of CTD and the isomerization state informs the half-life of such marks in the context of eukaryotic transcription. In contrast, residues at the 4 and 7 position of the consensus heptad have little bearing on phosphorylation choice. Positions 4 and 7 can be occupied by a variety of amino acids or completely absent with no obvious impact on phosphorylation. In light of this, the recent MS analysis of yeast10 and human11 CTD, which incorporate Ser7 position mutations, is likely physiologically relevant for at least some CTD kinase phosphorylation patterns. For potential phosphorylation sites, phosphorylation at the 5 position is favored by Erk2 unless a suitable S/T-P motif is absent. In such situations, phosphorylation in the 2 position can occur when a suitable S/T-P motif is present. In addition to the strict requirement of S/T-P motif, our results highlight the requirement of an aromatic residue at the first position of the heptad repeat for the phosphorylation of residues at the Ser2 or Ser5 position. This might explain the observation that of the seven residues of the consensus heptad, tyrosine at the 1 position appears to be the best conserved even in highly divergent sequences like DmCTD.5
The rules established for CTD kinase patterning in our analysis open interesting avenues for future research. The importance of some of the residues within the consensus CTD sequence has been known for some time.46,47 However, the influence that alternative flanking residues can have on CTD phosphorylation choice has not been previously elucidated in such high resolution or in the context of the full-length native sequence CTD. Importantly, our approach requires no mutation of the native CTD sequence for analysis. Because of this, RNAP II from cell lines exhibiting transcription defects, for instance due to the knock-down of certain transcription factors, can be purified directly and subject to MS analysis to determine CTD phosphorylation pattern. Implementation of such methods allows for the direct correlation of the PTM state of the CTD to transcriptional status and represents a critical step toward a full understanding of the CTD code.
Supplementary Material
Acknowledgments
M. Rachel Mehaffey is acknowledged for her assistance with intact protein MS analysis and Bede Portz for his helpful scientific discussion. This work is supported by grants from the National Institutes of Health (R01 GM104896 to Y.J.Z., R21EB018391 to J.S.B., and R01 GM47477 to D.S.G.) and Welch Foundation (F-1778 to Y.J.Z. and F-1155 to J.S.B.). Funding from the UT System for support of the UT System Proteomics Core Facility Network is gratefully acknowledged.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.6b00729.
Experimental methods and materials, workflow figures, protein construct sequences, and Supporting Information references (PDF)
Notes
The authors declare no competing financial interest.
References
- 1.Steinmetz EJ, Warren CL, Kuehner JN, Panbehi B, Ansari AZ, Brow DA. Genome-wide distribution of yeast RNA polymerase II and its control by Sen1 helicase. Mol Cell. 2006;24:735–746. doi: 10.1016/j.molcel.2006.10.023. [DOI] [PubMed] [Google Scholar]
- 2.Koch F, Jourquin F, Ferrier P, Andrau JC. Genome-wide RNA polymerase II: not genes only! Trends Biochem Sci. 2008;33:265–273. doi: 10.1016/j.tibs.2008.04.006. [DOI] [PubMed] [Google Scholar]
- 3.Koch F, Fenouil R, Gut M, Cauchy P, Albert TK, Zacarias-Cabeza J, Spicuglia S, de la Chapelle AL, Heidemann M, Hintermair C, Eick D, Gut I, Ferrier P, Andrau JC. Transcription initiation platforms and GTF recruitment at tissue-specific enhancers and promoters. Nat Struct Mol Biol. 2011;18:956–963. doi: 10.1038/nsmb.2085. [DOI] [PubMed] [Google Scholar]
- 4.Jeronimo C, Bataille AR, Robert F. The writers, readers, and functions of the RNA polymerase II C-terminal domain code. Chem Rev. 2013;113:8491–8522. doi: 10.1021/cr4001397. [DOI] [PubMed] [Google Scholar]
- 5.Eick D, Geyer M. The RNA polymerase II carboxy-terminal domain (CTD) code. Chem Rev. 2013;113:8456–8490. doi: 10.1021/cr400071f. [DOI] [PubMed] [Google Scholar]
- 6.Chapman RD, Heidemann M, Hintermair C, Eick D. Molecular evolution of the RNA polymerase II CTD. Trends Genet. 2008;24:289–296. doi: 10.1016/j.tig.2008.03.010. [DOI] [PubMed] [Google Scholar]
- 7.Buratowski S. The CTD code. Nat Struct Biol. 2003;10:679–680. doi: 10.1038/nsb0903-679. [DOI] [PubMed] [Google Scholar]
- 8.Heidemann M, Hintermair C, Voss K, Eick D. Dynamic phosphorylation patterns of RNA polymerase II CTD during transcription. Biochim Biophys Acta, Gene Regul Mech. 2013;1829:55–62. doi: 10.1016/j.bbagrm.2012.08.013. [DOI] [PubMed] [Google Scholar]
- 9.Riley NM, Coon JJ. Phosphoproteomics in the Age of Rapid and Deep Proteome Profiling. Anal Chem. 2016;88:74–94. doi: 10.1021/acs.analchem.5b04123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Suh H, Ficarro SB, Kang UB, Chun Y, Marto JA, Buratowski S. Direct Analysis of Phosphorylation Sites on the Rpb1 C-Terminal Domain of RNA Polymerase II. Mol Cell. 2016;61:297–304. doi: 10.1016/j.molcel.2015.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schuller R, Forne I, Straub T, Schreieck A, Texier Y, Shah N, Decker TM, Cramer P, Imhof A, Eick D. Heptad-Specific Phosphorylation of RNA Polymerase II CTD. Mol Cell. 2016;61:305–314. doi: 10.1016/j.molcel.2015.12.003. [DOI] [PubMed] [Google Scholar]
- 12.Palumbo AM, Smith SA, Kalcic CL, Dantus M, Stemmer PM, Reid GE. Tandem mass spectrometry strategies for phosphoproteome analysis. Mass Spectrom Rev. 2011;30:600–625. doi: 10.1002/mas.20310. [DOI] [PubMed] [Google Scholar]
- 13.Brown R, Stuart SS, Houel S, Ahn NG, Old WM. Large-Scale Examination of Factors Influencing Phosphopeptide Neutral Loss during Collision Induced Dissociation. J Am Soc Mass Spectrom. 2015;26:1128–1142. doi: 10.1007/s13361-015-1109-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thompson MS, Cui W, Reilly JP. Factors that impact the vacuum ultraviolet photofragmentation of peptide ions. J Am Soc Mass Spectrom. 2007;18:1439–1452. doi: 10.1016/j.jasms.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 15.Madsen JA, Boutz DR, Brodbelt JS. Ultrafast ultraviolet photodissociation at 193 nm and its applicability to proteomic workflows. J Proteome Res. 2010;9:4205–4214. doi: 10.1021/pr100515x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robinson MR, Madsen JA, Brodbelt JS. 193 nm ultraviolet photodissociation of imidazolinylated Lys-N peptides for de novo sequencing. Anal Chem. 2012;84:2433–2439. doi: 10.1021/ac203227y. [DOI] [PubMed] [Google Scholar]
- 17.Greer SM, Parker WR, Brodbelt JS. Impact of Protease on Ultraviolet Photodissociation Mass Spectrometry for Bottom-up Proteomics. J Proteome Res. 2015;14:2626–2632. doi: 10.1021/acs.jproteome.5b00165. [DOI] [PubMed] [Google Scholar]
- 18.Brodbelt JS. Photodissociation mass spectrometry: new tools for characterization of biological molecules. Chem Soc Rev. 2014;43:2757–2783. doi: 10.1039/c3cs60444f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Greer SM, Cannon JR, Brodbelt JS. Improvement of shotgun proteomics in the negative mode by carbamylation of peptides and ultraviolet photodissociation mass spectrometry. Anal Chem. 2014;86:12285–12290. doi: 10.1021/ac5035314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Madsen JA, Kaoud TS, Dalby KN, Brodbelt JS. 193-nm photodissociation of singly and multiply charged peptide anions for acidic proteome characterization. Proteomics. 2011;11:1329–1334. doi: 10.1002/pmic.201000565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shaw JB, Madsen JA, Xu H, Brodbelt JS. Systematic comparison of ultraviolet photodissociation and electron transfer dissociation for peptide anion characterization. J Am Soc Mass Spectrom. 2012;23:1707–1715. doi: 10.1007/s13361-012-0424-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Han SW, Lee SW, Bahar O, Schwessinger B, Robinson MR, Shaw JB, Madsen JA, Brodbelt JS, Ronald PC. Tyrosine sulfation in a Gram-negative bacterium. Nat Commun. 2012;3:1153. doi: 10.1038/ncomms2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luo Y, Yogesha SD, Cannon JR, Yan W, Ellington AD, Brodbelt JS, Zhang Y. novel modifications on C-terminal domain of RNA polymerase II can fine-tune the phosphatase activity of Ssu72. ACS Chem Biol. 2013;8:2042–2052. doi: 10.1021/cb400229c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Madsen JA, Ko BJ, Xu H, Iwashkiw JA, Robotham SA, Shaw JB, Feldman MF, Brodbelt JS. Concurrent automated sequencing of the glycan and peptide portions of O-linked glycopeptide anions by ultraviolet photodissociation mass spectrometry. Anal Chem. 2013;85:9253–9261. doi: 10.1021/ac4021177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Robinson MR, Moore KL, Brodbelt JS. Direct identification of tyrosine sulfation by using ultraviolet photodissociation mass spectrometry. J Am Soc Mass Spectrom. 2014;25:1461–1471. doi: 10.1007/s13361-014-0910-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fort KL, Dyachenko A, Potel CM, Corradini E, Marino F, Barendregt A, Makarov AA, Scheltema RA, Heck AJ. Implementation of Ultraviolet Photodissociation on a Benchtop Q Exactive Mass Spectrometer and Its Application to Phosphoproteomics. Anal Chem. 2016;88:2303–2310. doi: 10.1021/acs.analchem.5b04162. [DOI] [PubMed] [Google Scholar]
- 27.Thomas MC, Chiang CM. The general transcription machinery and general cofactors. Crit Rev Biochem Mol Biol. 2006;41:105–178. doi: 10.1080/10409230600648736. [DOI] [PubMed] [Google Scholar]
- 28.Jeronimo C, Robert F. Kin28 regulates the transient association of Mediator with core promoters. Nat Struct Mol Biol. 2014;21:449–455. doi: 10.1038/nsmb.2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mayfield JE, Burkholder NT, Zhang YJ. Dephosphorylating eukaryotic RNA polymerase II. Biochim Biophys Acta, Proteins Proteomics. 2016;1864:372–387. doi: 10.1016/j.bbapap.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tee WW, Shen SS, Oksuz O, Narendra V, Reinberg D. Erk1/2 activity promotes chromatin features and RNAPII phosphorylation at developmental promoters in mouse ESCs. Cell. 2014;156:678–690. doi: 10.1016/j.cell.2014.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arbouzova NI, Zeidler MP. JAK/STAT signalling in Drosophila: insights into conserved regulatory and cellular functions. Development. 2006;133:2605–2616. doi: 10.1242/dev.02411. [DOI] [PubMed] [Google Scholar]
- 32.Nusse R. Wnt signaling in disease and in development. Cell Res. 2005;15:28–32. doi: 10.1038/sj.cr.7290260. [DOI] [PubMed] [Google Scholar]
- 33.Pires-daSilva A, Sommer RJ. The evolution of signalling pathways in animal development. Nat Rev Genet. 2003;4:39–49. doi: 10.1038/nrg977. [DOI] [PubMed] [Google Scholar]
- 34.Reiter LT, Potocki L, Chien S, Gribskov M, Bier E. A systematic analysis of human disease-associated gene sequences in Drosophila melanogaster. Genome Res. 2001;11:1114–1125. doi: 10.1101/gr.169101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rubin GM, Yandell MD, Wortman JR, Gabor Miklos GL, Nelson CR, Hariharan IK, Fortini ME, Li PW, Apweiler R, Fleischmann W, Cherry JM, Henikoff S, Skupski MP, Misra S, Ashburner M, Birney E, Boguski MS, Brody T, Brokstein P, Celniker SE, Chervitz SA, Coates D, Cravchik A, Gabrielian A, Galle RF, Gelbart WM, George RA, Goldstein LS, Gong F, Guan P, Harris NL, Hay BA, Hoskins RA, Li J, Li Z, Hynes RO, Jones SJ, Kuehl PM, Lemaitre B, Littleton JT, Morrison DK, Mungall C, O’Farrell PH, Pickeral OK, Shue C, Vosshall LB, Zhang J, Zhao Q, Zheng XH, Lewis S. Comparative genomics of the eukaryotes. Science. 2000;287:2204–2215. doi: 10.1126/science.287.5461.2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ingham PW, Nakano Y, Seger C. Mechanisms and functions of Hedgehog signalling across the metazoa. Nat Rev Genet. 2011;12:393–406. doi: 10.1038/nrg2984. [DOI] [PubMed] [Google Scholar]
- 37.Herz HM, Mohan M, Garrett AS, Miller C, Casto D, Zhang Y, Seidel C, Haug JS, Florens L, Washburn MP, Yamaguchi M, Shiekhattar R, Shilatifard A. Polycomb repressive complex 2-dependent and -independent functions of Jarid2 in transcriptional regulation in Drosophila. Mol Cell Biol. 2012;32:1683–1693. doi: 10.1128/MCB.06503-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klose RJ, Kallin EM, Zhang Y. JmjC-domain-containing proteins and histone demethylation. Nat Rev Genet. 2006;7:715–727. doi: 10.1038/nrg1945. [DOI] [PubMed] [Google Scholar]
- 39.Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–349. doi: 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Price DH. Poised polymerases: on your mark⋯get set⋯go! Mol Cell. 2008;30:7–10. doi: 10.1016/j.molcel.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 41.Zhang Z, Gilmour DS. Pcf11 is a termination factor in Drosophila that dismantles the elongation complex by bridging the CTD of RNA polymerase II to the nascent transcript. Mol Cell. 2006;21:65–74. doi: 10.1016/j.molcel.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 42.Betzel C, Bellemann M, Pal GP, Bajorath J, Saenger W, Wilson KS. X-ray and model-building studies on the specificity of the active site of proteinase K. Proteins: Struct Funct, Genet. 1988;4:157–164. doi: 10.1002/prot.340040302. [DOI] [PubMed] [Google Scholar]
- 43.Mayfield JE, Fan S, Wei S, Zhang M, Li B, Ellington AD, Etzkorn FA, Zhang YJ. Chemical tools to decipher regulation of phosphatases by proline isomerization on eukaryotic RNA polymerase II. ACS Chem Biol. 2015;10:2405–2414. doi: 10.1021/acschembio.5b00296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Werner-Allen JW, Lee CJ, Liu P, Nicely NI, Wang S, Greenleaf AL, Zhou P. cis-Proline-mediated Ser(P)5 dephosphorylation by the RNA polymerase II C-terminal domain phosphatase Ssu72. J Biol Chem. 2011;286:5717–5726. doi: 10.1074/jbc.M110.197129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu YX, Hirose Y, Zhou XZ, Lu KP, Manley JL. Pin1 modulates the structure and function of human RNA polymerase II. Genes Dev. 2003;17:2765–2776. doi: 10.1101/gad.1135503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.West ML, Corden JL. Construction and analysis of yeast RNA polymerase II CTD deletion and substitution mutations. Genetics. 1995;140:1223–1233. doi: 10.1093/genetics/140.4.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schwer B, Shuman S. Deciphering the RNA polymerase II CTD code in fission yeast. Mol Cell. 2011;43:311–318. doi: 10.1016/j.molcel.2011.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.