

Abstract
Zoonotic cutaneous Leishmaniasis caused by Leishmania (L.) major parasites affects urban and suburban areas in the center and south of Tunisia where the disease is endemo-epidemic. Several cases were reported in human patients for which infection due to L. major induced lesions with a broad range of severity. However, very little is known about the mechanisms underlying this diversity. Our hypothesis is that parasite genomic variability could, in addition to the host immunological background, contribute to the intra-species clinical variability observed in patients and explain the lesion size differences observed in the experimental model. Based on several epidemiological, in vivo and in vitro experiments, we focused on two clinical isolates showing contrasted severity in patients and BALB/c experimental mice model. We used DNA-seq as a high-throughput technology to facilitate the identification of genetic variants with discriminating potential between both isolates. Our results demonstrate that various levels of heterogeneity could be found between both L. major isolates in terms of chromosome or gene copy number variation (CNV), and that the intra-species divergence could surprisingly be related to Single Nucleotide Polymorphisms (SNPs) and Insertion/Deletion (InDels) events. Interestingly, we particularly focused here on genes affected by both types of variants and correlated them with the observed gene CNV. Whether these differences are sufficient to explain the severity in patients is obviously still open to debate, but we do believe that additional layers of -omic information is needed to complement the genomic screen in order to draw a more complete map of severity determinants.
Keywords: Leishmania major, zoonotic cutaneous leishmaniasis, severity, clinical isolates, high-throughput genomic screening, genomic variability
1. Introduction
Leishmania (L.) is a vector-borne protozoan parasite responsible for severe human diseases collectively termed leishmaniasis, ranging from auto-resolving cutaneous (CL) and muco-cutaneous forms to possibly lethal visceral forms (McGwire and Satoskar, 2014). Despite huge community efforts to minimize their invasiveness and control their spread, these parasites still circulate in 98 countries where they affected 12 million people and maintain 397 million at risk (Alvar et al., 2012). Ecological and environmental changes, population displacements, mobilization of water resources, and establishment of new irrigated zones through newly built dams, in addition to the absence of effective vaccine, and drug-resistant populations mainly explain the persistence of this disease as one of the 17 neglected tropical diseases (NTDs) prioritized by the WHO for intensified measures, investments, and investigation (WHO, 2010).
L. major parasites are responsible for the so-called human “zoonotic” cutaneous leishmaniasis (ZCL), a form of the disease that usually results in benign auto-resolutive cutaneous sore(s) or to more extensive lesion(s) causing severe disfiguring skin scars (Olliaro et al., 2013). In Tunisia, L. major is the most prevalent dermotropic species causing the heaviest burden with an incidence of few thousands of cases each year (Aoun and Bouratbine, 2014). Severe forms constitute the major public health problem in terms of case management and treatment, disability-adjusted life years (DALYs) lost and economic productivity reduction, in addition to the social stigma associated with deformities and disfiguring scars (Alvar et al., 2012).
Mechanisms and determinants of clinical severity and pathogenicity are not fully understood and might be determined by factors related to the parasite (Banuls et al., 2011; Stuart et al., 2008), the vector (Murray et al., 2005; Rohousova and Volf, 2006) and the host genetic background and immune responses (Kane and Mosser, 2000; Sakthianandeswaren et al., 2009; Schriefer et al., 2008). In Tunisia, a couple of studies showed that various L. major isolates differ in their experimental pathogenicity when injected into the footpad of the genetically susceptible BALB/c mice (Kebaier et al., 2001 and Attia, unpublished data). For human patients, understanding how intra-species parasite factors could influence the expression of disease is particularly important when a highly differential clinical severity of the lesions is observed between patients from closely related geographical areas. Disease polymorphism could be defined according to lesion size, clinical presentation and time to heal (Olliaro et al., 2013). These criteria might serve to classify L. major isolated from active lesions expressing functional phenotypic heterogeneity according to their induced severity in humans.
Although genomic differences could be readily detected between parasites belonging to different species (Rogers et al., 2011) that induce contrasted clinical pictures (inter-species diversity), these differences were less obvious when dealing with isolates belonging to the same species. Genomic variability encompasses not only variable number of repeats (e.g., microsatellites), but also a wide range of alterations such as single nucleotide polymorphisms (SNPs), transposable elements (e.g., Alu elements), single or multiple base mutations (e.g. insertions, deletions or their combination in the form of InDels), as well as larger structural variations (e.g., gene or chromosome copy number variations). The recent accumulation of information, due to the availability of high-throughput genomic sequencing, has eased the identification of such genetic variants at the whole genome level (Pavlopoulos et al., 2013), and even opened up new powerful clinical opportunities via the integration of diagnostic genome care (Vrijenhoek et al., 2015).
Since the completion of the first Leishmania genome (Ivens et al., 2005), and the subsequent availability of many other annotated or draft Leishmania genomes, omics technologies have participated to the understanding of Leishmania biology (Cantacessi et al., 2015), easing the identification of variants not previously detected by multilocus typing such as chromosome somy, gene dosage and SNPs within i) clinical isolates of the same species e.g., L. donovani (Downing et al., 2011), ii) closely related species of the same complex (Valdivia et al., 2015) or iii) between different species and strains (Rogers et al., 2011). It seems that these parasites have a remarkable ability to adapt to environmental changes through their genome plasticity, highlighting for instance a capacity to test for the essentiality of genes (Cruz et al., 1993), and giving novel routes to the molecular understanding of resistance to drugs (Imamura et al., 2016; Leprohon et al., 2009).
Genomic deep-sequencing approach hence successfully unraveled the genomic basis for diversity of clinical Leishmania populations and their evolvability during treatment. Indeed, the analysis of single-nucleotide diversity showed evidence of selection acting in a range of surface- and transport-related genes, including genes associated with drug resistance (Downing et al., 2011) and a two base-pair insertion in the aqua-glyceroporin gene responsible for antimonial resistance in L. donovani isolates (Imamura et al., 2016), whereas whole genome sequencing identified genomic markers associated with miltefosine resistance in L. major strains (Coelho et al., 2012). In L. infantum species, antimony resistant mutants showed specific amplified regions derived from two different chromosomes, SNPs, telomeric gene deletion (Brotherton et al., 2013; Mukherjee et al., 2013) and gene amplification and point mutations detected in a 5-Fluorouracil-resistant isolate (Ritt et al., 2013). Except for drug resistance, very few studies addressed the relationship between genomic variability, parasite virulence and clinical outcome diversity. Differences in gene and chromosome copy numbers and extensive nucleotide polymorphisms detected in L. (Viannia) peruviana, compared to L. (Viannia) braziliensis, have been described as contributing to some biological features of the former species, including a lower pathology and lack of mucosal development (Valdivia et al., 2015). Another study elegantly showed, using genome sequencing and experimental models, that L. donovani tropism might be due to mutations and/or amplification of a few genes (Zhang et al., 2014), although genomic events such as gene amplification have been described previously (Coderre et al., 1983).
We believe that despite sharing high genomic similarities (Attia et al., 2016), L. major parasites could exhibit subtle divergent genomic variations. Our hypothesis is that this intra-species variation among L. major isolates may, to some extent, contribute in an endemic area for ZCL to the clinical polymorphism of disease and to the outcome of infection. In an attempt to correlate parasite genomic diversity with disease outcome, we used DNA high-throughput sequencing approach to compare the genome of two L. major field isolates selected on the basis of their contrasted virulence and experimental pathogenicity. We focused specifically on the large-scale screening of genetic factors at the level of structural variations and single point mutations.
2. Material and Methods
2.1. Ethics statement
The study protocol, consent and ascent forms and procedures were reviewed and approved by the Institut Pasteur de Tunis Ethical Review Board. Patients, from whom parasites where obtained (or their parents or legal guardians in case of minors), have provided written informed consent for the collection of isolates and their subsequent analysis.
2.2. Sample collection from human patients and follow-up
A set of 35 L. major isolates were grown from ZCL lesions of patients living in five villages (i.e., Mnara, Mbarkia, Dhouibet, Ksour, Msadia) in the governorates of Sidi Bouzid and Kairouan, central Tunisia, an endemic area of cutaneous leishmaniaisis caused by L. major (Bettaieb et al., 2014). Tissue fragments from active lesion sites were inoculated on bi-phasic medium for parasite growth. Grown promastigotes were then maintained in RPMI 1640 (Sigma) medium supplemented with 10% of heat-inactivated fetal calf serum (FCS), 2 mM of L-glutamine, 100 U/ml of penicillin and 100 U/ml of streptomycin. Parasites were harvested within the shortest time after successful growth (not exceeding two in vitro passages); in order to avoid changes induced by prolonged in vitro cultures, deep frozen or pelleted and conserved at −20°C for subsequent DNA extraction. Patients were followed up during 180 days on a weekly basis to monitor the evolution of lesions, through time, until cure. Lesion severity was then calculated and corresponds to the integral of the approximation of the evolution of the lesion’s area over time, reported from onset to cure dates.
2.3. Molecular typing
DNA extraction was performed for all promastigotes isolates using the QIAamp DNA Mini Kit (Qiagen) following the manufacturer’s protocol. Leishmania parasites were subjected to molecular typing by targeting amplification of Internal Transcribed Spacer 1 (ITS1) region as previously described (Schonian et al., 2003). DNA from well established Tunisian Leishmania species (L. major, L. tropica, and L. infantum) and double-distilled water (DdH2O) were used as controls. DNA amplicons were visualized on a 2% agarose gel. For samples showing amplified DNA, 17 μl of the PCR product were further digested in the presence of 5 μl reaction volume consisting of 2.5 μl of ddH2O, 1.5 μl of 10X NE Buffer 2, and 10 U of endonuclease Hae III. Sizes of restriction fragments were controlled on a 2% Metaphor Agarose gel.
2.4. Experimental pathogenicity
Six- to eight-week old female BALB/c mice were infected with 2×106 metacyclic Peanut agglutinin (PNA)- L. major parasites by subcutaneous (s.c.) route in the left footpad in 50μl of saline solution. Infection was assessed once a week, for eight weeks, by measuring the inflammation volume of foot lesions with a plethysmometer. The lesion size was defined as the increase in the footpad volume after subtracting the size of the contra-lateral uninfected footpad.
2.5. DNA-seq library preparation
DNA-seq library preparation were performed on promastigotes of two selected isolates 1948 and 2938: one hypo-virulent (noted 1948_hV) and one hyper-virulent (noted 2938_HV) according to human lesion size from whom they were obtained and to their pathogenicity in infected BALB/c mice. DNA was extracted as described above. The genomic DNA (gDNA) was cleaned from any trace of contaminants using size selection by Agencourt AMPure beads XP (Beckman Coulter) with a bead:DNA ratio of 0.5X. Libraries were prepared using the KAPA Hyper Prep Kit used for Illumina platforms (KAPA Biosystems) following the manufacturer’s protocol. Physical fragmentation of DNA used the Covaris® instrument (Woburn, MA) as a sonication device using the Adaptive Focused Acoustics™ (AFA) technology. Briefly, a starting amount of 0.3–0.6 μg of sheared gDNA was end-repaired and A-tailed in a one step reaction. Ligation to specific Illumina indexed paired-end adapters was performed following an adapter:insert molar ratio of 40:1, followed by a post-ligation cleanup step with 0.8X SPRI cleanup combining the previous reaction to Agencourt AMPure XP reagent (Beckman Coulter) to remove unligated adapters. The AMPure XP beads were also used for gDNA library size selection of 220–550 bp inserts. Library quantification was performed using the qPCR-based Library Quantification Kit (Kapa Biosystems) to evaluate the molar concentration to be used in the sequencing runs. Libraries were sequenced using the TruSeq SBS Kit v3-HS (Illumina Inc.), compatible with both PCR-free and PCR-amplified libraries, with a standard protocol ensuring a paired-end run of 2 ×101-bp long reads on a HiSeq 2000 flow cell v3 (Illumina Inc.) based on sample multiplexing. The recorded output reached a maximum of 6,326 Gb for the highest yield. The primary analysis software Real Time Analysis (RTA 1.13.48) was used according to manufacturer’s protocol to process image analysis, base calling and base quality scoring of the run and followed by generation of fastq sequence files by CASAVA (v1.8).
2.6. Reference Genome and reference files
Illumina reads alignment was performed using the L. major Friedlin reference Genome (version 8.1). Both fasta (TriTrypDB-8.1_LmajorFriedlin.fa) and gff (TriTrypDB-8.1_LmajorFriedlin.gff) files from TritrypDB (http://tritrypdb.org/common/downloads/release-8.1/LmajorFriedlin/) were used respectively for further genome indexing and gene copy number variations (CNVs), single nucleotide polymorphisms (SNPs) and insertions/deletions (InDels) callings.
2.7. DNA-seq Illumina reads alignment
Taking into account the paired-end sequencing for each isolate, we obtained four DNA-seq paired-end libraries. We obtained 62,624,888 and 42,245,988 reads for 2938_HV and 1948_hV isolates respectively. The coverage was estimated by calculating the (read count/read length) divided by the total genome size. Raw fastq files were quality-checked using FastQC v0.11.4 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Reads were aligned to the reference genome using bowtie2 v2.2.5 in very sensitive local alignment mode (Langmead and Salzberg, 2012). Reads that mapped to multiple loci were kept but only a single alignment was reported for each read (default, -k 1) and showed a high percentage of uniquely mapped reads. File conversions were performed using samtools v1.2 (http://samtools.sourceforge.net) (Li et al., 2009). Coverage calculations were performed using bedtools v2.21.0 (Quinlan, 2014). Potential PCR duplicates were marked and removed thanks to Picard v1.135. Bam files local realignment was performed using GATK v3.4 (https://www.broadinstitute.org/gatk/).
2.8. Detection of structural variations, variant calling, and variant filtering
Somy corresponds to the median coverage per chromosome divided by the median coverage for the genome and was calculated based on the number of reads mapping to each base position from which was extrapolated a median read depth for each chromosome. The later was normalized to 2 to represent a base state of diploidy for the L. major genome. CNV of each gene was calculated considering only reads mapping to coding sequences (CDS). Fragments per kilobase of exon per million fragments mapped (FPKM) values were generated for each gene using Cufflinks v2.2.1 (Trapnell et al., 2010) and a median FPKM was evaluated for each chromosome. Gene CNVs were calculated using the method described by Rogers, Hiley and Dickens (Rogers et al, 2011). SNPs and InDels events were evaluated using GATK v3.4 with default options. In the analysis we used a stringent process of quality trimming of input data (fastq files) before the alignment, as the probability of an incorrect call was correlated with a drop in phred score. We also employed a stringent post-filtering of variant calls as described in GATK best practice documents. Using default parameters, SNPs were filtered considering the strand bias, the position of the SNP in the read, and the haplotype score, and InDels were filtered by read position bias and strand bias. No further filtering was done, as quality-based metrics are not useful with bowtie2 and local alignments, and also to maintain maximum sensitivity to detect variants for comparison. SnpEff v2.0.5 (http://snpeff.sourceforge.net/) (Cingolani et al., 2012) was used for genetic variants annotation and effect prediction. The variants in the resulting variant calling format (vcf) files were filtered using SnpSift v4.1 (http://snpeff.sourceforge.net/SnpSift.html).
2.9. Gene Ontology enrichment
In order to help interpretation, Gene Ontology (GO) annotations of the sets of identified genes were extracted via the TriTrypDB release 28, using the Benjamini-Hochberg false discovery rate (FDR).
3. Results and Discussion
3.1. The contrasted virulence associated to isolates has been characterized by in vivo monitoring and in vitro experiments
Thirty-five clinical L. major isolates were collected from ZCL patients during a two-years epidemiological follow-up study conducted in Tunisia. Parasites were obtained during the transmission season of L. major parasites in well-characterized endemic zones ((Bettaieb et al., 2014), Figure 1,A). A molecular typing based on ITS1 region amplification characterized isolates as belonging to the L. major species (data not shown). Two isolates showing the most contrasted virulence profiles were selected based on two criteria: i) the clinical severity of skin lesion as assessed by induration and ulceration sizes of the ZCL lesion, as well as its time to heal over a follow-up period of 180 days, and ii) the in vivo differential pathogenicity in susceptible BALB/c mice (n=6 per group), during eight-week follow-up after infection. For the two selected isolates (2938 and 1948), our results clearly define phenotypically distinct intra-species characteristics i) of severity in human ZCL patients (Figure 1,B), and ii) of lesion development in infected BALB/c mice (Figure 1,C). Patients’ characteristics are described in Supplementary Table 1.
Figure 1. Geographical, genetic and phenotypic characteristics of 2938_HV and 1948_hV samples.
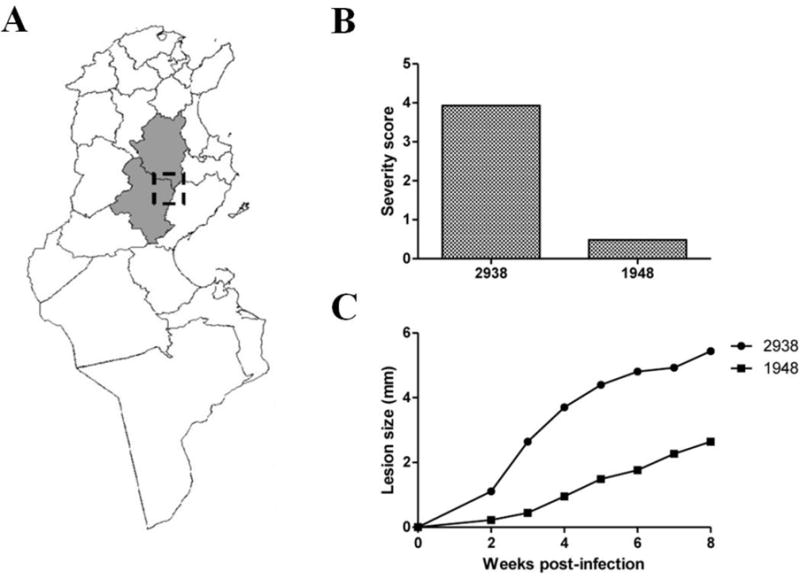
(A) Endemic foci in Tunisia from which the clinical isolates used in this study are originating. (B) Differential severity in Human patients is expressed as a score taking into account the lesion size (induration and ulceration) and the time to heal. (C) Differential pathogenicity in mice. The size and thickness at the level of the footpad lesion is measured in BALB/c mice during an 8-weeks follow-up after infection with peanut agglutinin negatively selected (PNA-) 2938_HV or 1948_hV parasites.
Indeed, and according to the follow-up of patients for 180 days, the isolate 2938 shows a severity score of four arbitrary units whereas the severity score of the isolate 1948 is less than one arbitrary unit (Figure 1,B). The experimental model of BALB/c mice shows that isolate 2938 induces a footpad lesion increase starting for week-2 upon infection whereas the isolate 1948 induces the same size lesion five weeks after infection. At the end of the experimental protocol (eight weeks upon infection), lesions induced by isolate 2938 at the inoculation site are almost three times bigger than those induced by isolate 1948 (Figure 1,C).
It is unlikely that any potential genetic difference between the two selected strains can be related to their geographical origin or the demographic and socioeconomic characteristics of patients from whom they were isolated, but it is likely related to their differential pathogenicity, among other factors (e.g., vector pressure). Indeed, both isolates were obtained during the same season from the same endemic area in two very close villages (Ksour and Msadia) both considered as “new and emerging” foci on the basis of case notification data in the district epidemiological surveillance system. The two villages share the same topography and climate, and there are no significant differences between the study villages regarding demographic and socioeconomic characteristics of households (Bettaieb et al, 2014). Contrasted clinical outcomes might also be related to qualitative and quantitative differences of host immune responses towards Leishmania infection induced by potentially the same parasite. However, retrieving the exact same contrasted trends in inbred experimental mice model shows this characteristic is likely related to intrinsic parasite determinants.
Although limited to only two representative strains, these features both in humans and murine model of the two selected isolates allow us to hypothesize that their variable severity is a product of parasite genomic variation and is independent from the host immunological background or environmental characteristics. For the sake of clarity, the strains discussed will be noted from here on as 2938_HV for the hyper-virulent isolate and 1948_hV for the hypo-virulent isolate.
3.2. Genome sequencing/coverage
DNA libraries generation and paired-end sequencing on an Illumina platform (HiSeq2000 device) was performed for both 2938_HV and 1948_hV. Samples sequencing yielded a total of about 62,6 million and 42,2 million reads respectively (Table 1).
Table 1.
Summary of DNA-seq reads quality control and mapping.
| 2938_HV | 1948_hV | |
|---|---|---|
| Total number of reads | 62,624,888 | 42,245,988 |
| Total number of mapped reads | 57,752,407 (92.22%) |
35,437,163 (83.88%) |
| Total number of properly paired reads | 57,426,006 (91.70%) |
35,093,060 (83.07%) |
| Total number of CDS in reference | 8,850 | 8,850 |
| Total number of CDS covered in samples | 8,850 | 8,850 |
Assessment of reads quality and their alignment to the Friedlin reference genome (TriTrypDB version 8.1) showed an overall proportion of properly mapped reads of about 92 and 83% respectively. The majority of reads are aligned on their entire length with a median mapping quality >28, implying that the subsequent trimming did not affect the results.
In order to check the range and uniformity of sequencing coverage, we estimated the overall coverage distribution for 2938_HV and 1948_hV by displaying the mapped reads depth at all base positions and the fraction of reads associated to each position covered (Supplementary Figure 1). The fraction at coverage clusters around a dominant unimodal distribution, associated to a small standard deviation, indicating that the whole genome is homogeneously covered. Moreover, the plot displays as a Gaussian distribution with a dominant frequency at around 100X as established by the calculated coverage formula, indicating a depth of coverage allowing to reliably detect heterozygous variants. All conclusions drawn from this study hence assume that reads are randomly distributed across the Leihsmania genome and that the ability to detect them is constant within the sequencing runs and throughout the whole genome. FPKM normalized coverage across each chromosome showed a complete genomic coverage of the annotated CDS reported in the reference genome for both samples (data not shown). All these results show no biased coverage towards any chromosomal, sub-chromosomal, or genic region, allowing the comparability of datasets from both samples. Only protein-coding CDS will be considered for the ensuing analyses.
3.3. Chromosome and gene CNVs
Leishmania is a versatile parasite with a fascinating ability to swiftly adapt to changes during its complex life cycle and survival under external sources of pressure. Its genome plasticity reflects an ability to adapt to environmental changes, and turns up to be a dynamic characteristic under various conditions. In order to test for the differential genome plasticity of the in vitro short-term cultured 2938_HV and 1948_hV promastigotes, we estimated chromosome and gene CNVs by averaging the reads density and calculating the median read depth across the chromosomes and the CDS respectively.
3.3.1. Chromosome CNVs
For all chromosomes, read depth showed disomy, trisomy or tetrasomy of chromosomes, as confirmed by allele frequency counts (Table 2). The absence of discordance between coverage and allele frequency is indicative of chromosomes that are mainly fully multisomic, and argues in favor of a small contribution of chromosomal mosaicism in the population, a common phenomenon in Leishmania generally characterized by mixed chromosomal profiles (Sterkers et al., 2012). A common observation on studied samples is that they displayed an overall diploid status, except for their chromosome 31 that was tetrasomic for both (Figure 2). This is not due to partial somy but rather to a whole chromosomal event, as no biased depth of coverage could be seen along the chromosomes. Chromosome 31 is indeed the only chromosome shown to be supernumerary in several Leishmania species (Downing et al., 2011; Rogers et al., 2011) including L. major (Akopyants et al., 2009; Ravel et al., 1998). Interestingly, an ontology analysis of chromosome 31 revealed its enrichment in genes related to iron metabolism, pointing out to the significance of its supernumerary maintenance to facilitate iron uptake under oxidative stressed environment (Valdivia et al., 2015).
Table 2.
Allele frequency counts for heterozygous and homozygous SNPs.
| 2938_HV | 1948_hV | |
|---|---|---|
| Homozygous SNPs (1.00) | 16,852 | 17,639 |
| Heterozygous SNPs (0.5) | 2,092 | 3,082 |
| Heterozygous SNPs (0.750/0.250) | 21/59 (LmjF.31) |
0/19 (LmjF.31) |
| Heterozygous SNPs (0.333/0.666) | 0/0 | 88/0 (LmjF.05, LmjF.07, LmjF.12, LmjF.17, LmjF.20, LmjF.23) |
Figure 2. Chromosome copy number variation (CNV) between 2938_HV and 1948_hV L. major isolates.
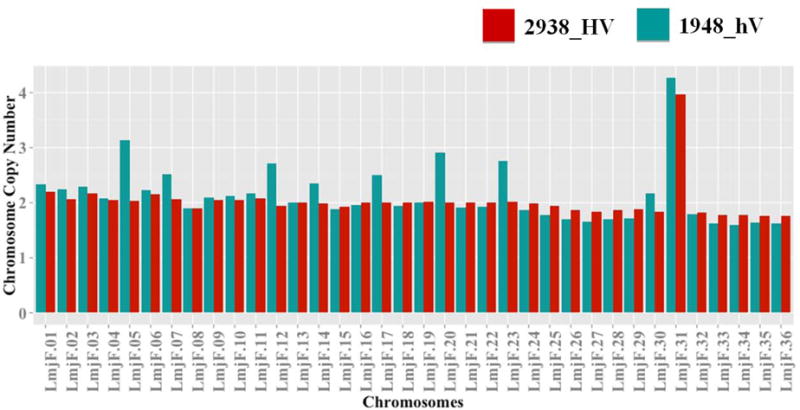
DNA libraries from both isolates were subjected to Illumina high-throughput sequencing. Reads were aligned on the reference L. major Friedlin genome v8.1. (A) Comparison of L. major chromosome CNV between isolates. The read depth was normalized across the genome. 2938_HV isolate is indicated in red and 1948_hV isolate in green.
Interestingly, whereas 2938_HV constantly maintains overall its diploid status, we observed a trisomic status for chromosomes 5, 7, 12, 17, 20, 23 in the 1948_hV. The ontology related to the supernumerary chromosomes in the hypo-virulent isolate showed enrichment of chromosome 5 in cation and proton transport, as well as energy and ATP synthesis coupled proton transport; for chromosome 7 in cation transport; for chromosome 12 in catabolic processes; for chromosome 17 in cyclic nucleotide metabolic process and translational elongation, cellular nitrogen compound biosynthetic process, cellular response to stimulus, cell communication or intracellular signal transduction; for chromosome 20 in cellular component biogenesis, protein complex biogenesis, or cell motility ; and finally for chromosome 23 mainly in acetyl-CoA metabolic process. Many of these pathways have already been described as accompanying the infectious process and parasite intracellular survival (Naderer and McConville, 2011). However, the significance of having extra-copies of these particular chromosomes in the hypo-virulent isolate needs to be confirmed by controlling the maintenance of somy after serial passages during in vitro growth in order to establish a strong correlation with the hypo-virulent phenotype of this 1948_hV isolate. This remarkable tolerance of Leishmania parasites for somy is not maintained for chromosomes across the literature (Sterkers et al., 2012) except for chromosome 31. It is indeed extensively described that almost all chromosomes could show various levels of somy in different Leishmania species (Sterkers et al., 2012; Valdivia et al., 2015). Chromosome somy have been shown to occur after experimental genetic manipulation such as gene knockouts (Cruz et al., 1993; Martinez-Calvillo et al., 2005), in drug resistant strains (Imamura et al., 2016; Leprohon et al., 2009; Mukherjee et al., 2013), or as a consequence of long-term in vitro culture (Martinez-Calvillo et al., 2005). Partial or full somy variation could lead to variation in heterozygosity and gene dose, a mechanism that might be related to prompt parasite adaptation (Imamura et al., 2016; Sterkers et al., 2012). These changes have been suggested not only as a way to test for gene essentiality during in vitro growth (Martinez-Calvillo et al., 2005), but also to modulate gene expression in response to environmental stress, suggesting a genetic contribution to disease tropism (Rogers et al., 2011).
3.3.2. Gene CNVs
Parasite ability to rapidly adapt to a changing environment relies also on the modulation of gene expression, which depends not only on aneuploidy but also on gene CNVs (Leprohon et al., 2009). Although not yet described in parasites, several studies support the concept of CNVs regulating RNA levels in humans (Haraksingh and Snyder, 2013; Mehta et al., 2014) and cattle (Zhou et al., 2016). However, this phenomenon has to be taken with caution in Leishmania parasites, where genes are not transcriptionally regulated by individual promoters. There are clear cases in Leishmania sp. where gene CNV has been linked to changes in gene expression, for example a study showing the presence of more copies of the A2 genes in the genome of Sri Lankan strain inducing visceral leishmaniasis than in the genome of a strain causing cutaneous leishmaniasis (Zhang et al., 2014). The A2 gene family, known to constitute stress-inducible factors necessary for visceral infection, shows higher transcript and protein levels in visceral strain. The same study described on the other hand higher copy numbers within the genome of cutaneous strain comparatively to the visceral strain, in regions of chromosome 1, 19, 23 and 29 that contain several genes e.g., ABC thiol transporters, eukaryotic initiation factor 4a, glycerol uptake proteins (Zhang et al., 2014). We compared protein-coding gene CNVs between both Tunisian isolates considering only the haploid gene counts and subtracting the reference haploid copy numbers. A total of 226 gene CNVs were detected, among which 84 were different between the two isolates. This set contains 61 non-hypothetical (Supplementary Table 2) and 23 hypothetical proteins.
Our analysis testing for differential non hypothetical gene CNVs between 2938_HV and 1948_hV revealed 16 orthologous groups of genes to be unique to 2938_HV, and 25 unique to 1948_hV. Gene CNVs were considered unique for one isolate when the second isolate has the same number of CNVs as the reference.
Gene tandem arrays that are over-represented at different levels of variation in both isolates comparatively to Friedlin reference strain include the elongation factor 1-alpha (EF1a), the promastigote surface antigen protein (PSA), and the class-I nuclease protein. EF1a is involved in many cellular functions related to translation, degradation or cytoskeletal movements, is known to be essential for parasite survival by the induction of macrophage deactivation (Nandan et al., 2002) and has been proposed as a drug target candidate (Nandan et al., 2003). Although this gene was highly overexpressed in both isolates compared to the reference, it was noticeable to find a higher gene copy number in the hypo-virulent isolate. This overexpression has also been described in other eukaryotes where extra-copies of EF1a were associated to a gene dosage effect in Plasmodium parasites (Vinkenoog et al., 1998). PSA is described as overexpressed in metacyclic promastigotes (Devault and Banuls, 2008). Interestingly, the progressive reduction of its mRNA and proteins levels during serial in vitro culture passages is associated to loss of virulence (Beetham et al., 2003). This phenomenon is concordant with our analysis showing that PSA possesses a less gene copy number in the hypo-virulent strain. The class-I nuclease protein was previously described as the tandem array containing the largest number of gene copies in L. major Friedlin as a peculiarity of this species (Rogers et al., 2011), and characterized as a hyper-inducible surface protein in trypanosomatids (Yamage et al., 2000).
Markedly, Hsp70/83 families showed a lower copy number in 1948_hV hypo-virulent isolate, compared to the hyper-virulent one. These proteins are known to have chaperone function upon stress conditions in amastigotes (Morales et al., 2010). Strikingly, genes such as heat shock proteins, tryparedoxin, or the tryparedoxin peroxidase related to oxidative/nitrosative stress (Teixeira et al., 2015) and known as virulence factors (Iyer et al., 2008), showed missing copies compared to the reference genome, although differentially present between our two isolates. Interestingly, the ATP-binding cassette protein subfamily A (ABCA) showed an expansion of gene copies in the 2938_HV genome. This gene family was previously described as overexpressed in antimony-resistant strains (Leprohon et al., 2006). These results strongly suggest that various levels of gene copy numbers and duplicated regions could hence be potentially related to the differential parasite virulence feature. This also highlights the importance of controlling chromosome and gene CNVs in Leishmania parasites before any gene family studies or genetic manipulation such as gene knockout (Rogers et al., 2011).
3.4. SNPs and InDels
Since variation in somy, affecting all chromosomes in L. donovani clinical isolates, has been described as leading to heterozygosity (Imamura et al., 2016), we aimed to analyze SNPs and InDels in our isolates. Our results showed that allele frequency counts corroborate the overall disomic status with a mixed allelic profile in chromosomes (Table 2). The observed high proportion of homozygous SNPs was expected as L. major genomic stability is relatively high compared to other Leishmania species.
Deleterious mutations (e.g., non-synonymous SNPs) are believed to make a significant contribution to phenotypic variation within populations (Chun and Fay, 2009). We thus considered here genes having both SNPs and InDels, two types of nucleotide-level variants that could drastically affect transcripts and proteins. Both types of variants in 2938_HV and 1948_hV isolates commonly affect about 91% of genes. For both isolates, SNPs and InDels appeared to be uniformly distributed across the genome. In addition, considering the total number of variants, our results showed no bias in i) over-representation related to the chromosome somy, and ii) distribution of affected genes related to chromosome size (Figure 3,A).
Figure 3. Number and chromosomal distribution of common or unique genes having both SNPs and InDels in 2938_HV and 1948_hV L. major isolates.
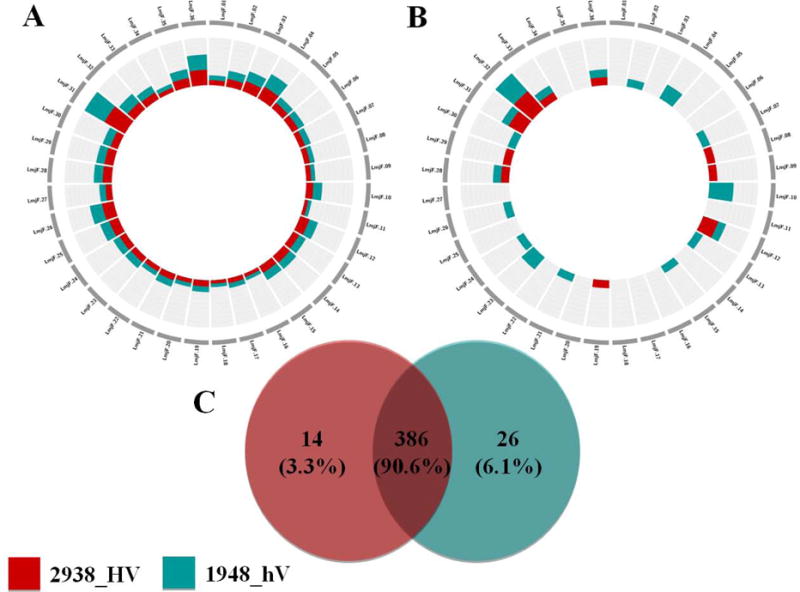
The plots show the number of variants associated to each chromosome. The chromosomes are not drawn to scale. The inner scale is adjusted to optimize the number of genes for each plot independently. (A) Chromosomal distribution of the total number of genes having both SNPs and Indels. (B) Chromosomal distribution of genes having both SNPs and Indels and that are unique to each isolate. (C) Venn diagram showing common and unique features after comparing genes having both SNPs and InDels in 2938_HV to genes having both SNPs and InDels in 1948_hV. 2938_HV isolate is indicated in red and 1948_hV isolate in green.
Adversely, when considering SNPs and InDels variants unique to each isolate, results highlight generally different chromosomes to be targeted by variation (Figure 3,B). Interestingly, about half the chromosomes did not show any SNPs or InDels pointing out to potential cold spots of mutation, or regions of high conservation that would require further functional analysis.
Figure 3,C shows a total of 14 and 26 genes that are uniquely affected in 2938_HV and 1948_hV respectively. The majority of these genes were associated to hypothetical functions. Among genes containing SNPs and InDels in the 2938_HV isolate, we report two surface antigen protein 2, a glycerol uptake protein, and two heat-shock proteins (Hsp70 and Hsp83) genes related to response to stress and protein folding biological processes. It is noteworthy that the copy numbers of both Hsp genes were also higher in this hyper-virulent isolate compared to the 1948_hV one. Hsp gene sequences have such a high conservation throughout evolution (Folgueira and Requena, 2007), that our finding raises the question of the impact of such variants on disease severity.
Two Hsp83 genes, among the 17 annotated L. major ones, have been described as having a single codon less than the remaining 15 (Folgueira and Requena, 2007). The fact that one (LmjF.33.0318) of these two shorter genes contains SNPs and InDels in our 2938_HV isolate might indicate that it is more prone to mutations.
Genes showing SNPs and InDels variation in hypo-virulent isolate have functions of glycerate kinase, nucleolar RNA-binding protein, leishmanolysin GP63, tryptophanyl-tRNA synthetase, microtubule-associated protein, and histidine secretory acid phosphatase; functions that are partly associated to the modulation of host protein kinase-mediated signal transduction.
The role of GP63, for example, as a virulence factor that affects host cell signaling mechanisms is now well established (Isnard et al., 2012). The lower number of mutations in the 2938_HV isolate indicates that GP63 gene might be under relaxed selective pressure. Indeed, GP63 protein is essential for C3bi conversion through its binding to the complement receptor 3 (CR3), a process known to circumvent the host intracellular oxidative burst and enhance parasite infectivity (Olivier et al., 2005). It is worthy to note that while genome plasticity is known to help pathogen adaptation, the sequence conservation of genes with pivotal role during the infectious process might be necessary to maintain, as described for the pe_pgrs genes of Mycobacterium tuberculosis (Copin et al., 2014).
Considering the potential effect of SNPs and InDels on gene function, we observed that their vast majority carries synonymous variants (Table 3). These variants are generally expected to have insignificant effects on gene function since the primary sequences of the genes are conserved. Notwithstanding, synonymous mutations have unexpectedly been shown to reshape the structure of proteins and hence their function (Hunt et al., 2009). Their significant contribution to several diseases (Sauna and Kimchi-Sarfaty, 2011) renders appealing to consider also their role in parasitic diseases.
Table 3.
Selected set of SNPs and InDels with synonymous variant effect or relevant impact on gene function.
| 2938_HV | 1948_hV | |||
|---|---|---|---|---|
|
| ||||
| SNPs | Indels | SNPs | Indels | |
| Synonymous variant | 9,144 | 0 | 10,054 | 0 |
| Splice region | 19 | 11 | 25 | 5 |
| Stop lost | 22 | 21 | 26 | 20 |
| Frameshift variant | 0 | 375 | 0 | 371 |
| start_ lost | 13 | 4 | 16 | 4 |
| Inframe insertion | 0 | 174 | 0 | 173 |
|
| ||||
| Total | 9,198 | 585 | 10,121 | 573 |
Interestingly, we also identified SNPs and InDels that can affect start and stop positions. The chromosomal distribution of high impact SNPs (Figure 4,A) does not necessarily correspond to that observed for high impact InDels (Figure 4,B). The circos plots show that these high impact variants did not exclusively affect the same chromosomes. Except for rare cases where chromosomes are more or exclusively targeted for variation in 1948_hV (chromosomes 6, 12, 13, 19) or in 2938_HV (chromosome 10, 34) isolates, selective SNPs pressure seems to be exerted on the same chromosomes (Figure 4,A). InDels are more scattered than SNPs all over the genome, except for chromosome 9. Number (Figure 4,C), chromosomal distribution (Figure 4,D), and function of genes bearing high impact SNPs and InDels variants highlight the common alteration of i) seven genes in both isolates, ii) one gene in 2938_HV, and iii) four genes in 1948_hV. Grippingly, the unique gene specifically altered in 2938_HV isolate corresponds to a tuzin pseudogene, whereas all four genes retailored in 1948_hV isolate correspond to hypothetical genes, with a significant frameshift variation inducing either a stop gain or a start loss. These genes deserve future investigation to unveil their potential involvement in disease severity.
Figure 4. Chromosomal distribution and number of genes having high impact SNPs or high impact InDels or both variants in 2938_HV and 1948_hV L. major isolates.
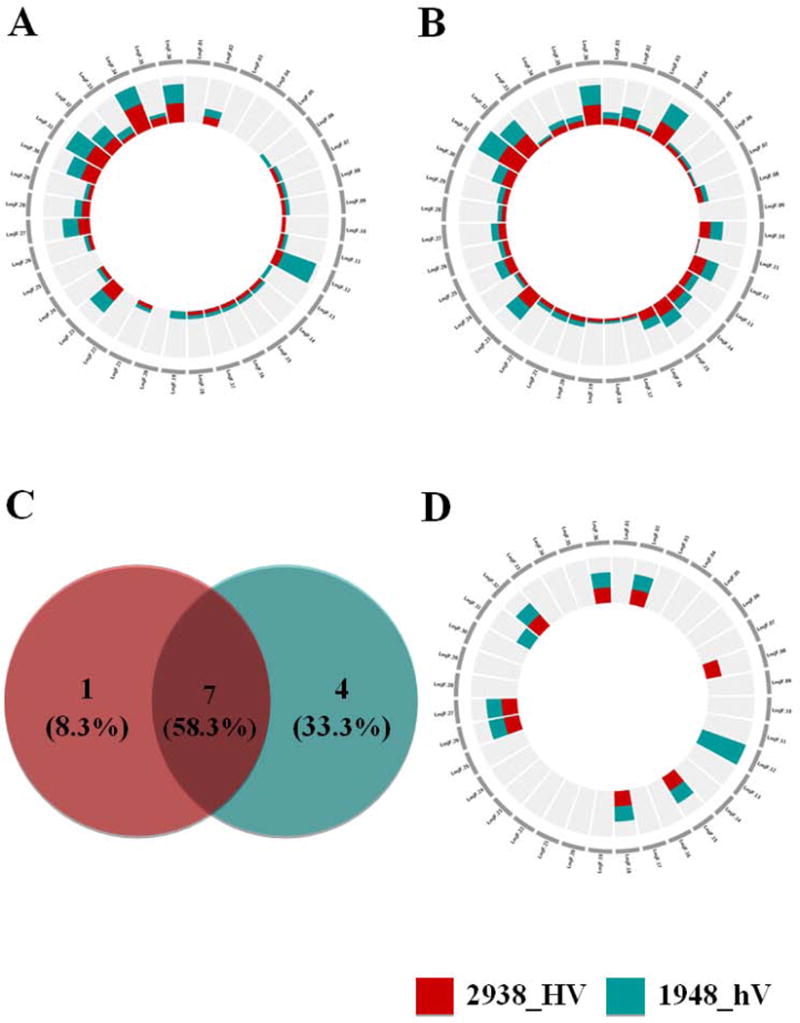
The plots show the number of variants associated to each chromosome. The chromosomes are not drawn to scale. The inner scale is adjusted to optimize the number of genes for each plot independently. (A) Chromosomal distribution of genes affected by high impact SNPs (B) Chromosomal distribution of genes affected by high impact Indels. (C) Venn diagram showing common and unique features after comparing genes having both high impact SNPs and high impact InDels in 2938_HV to genes having both high impact SNPs and high impact InDels in 1948_hV. (D) Chromosomal distribution of genes having both high impact SNPs and high impact Indels for genes that are unique to each isolate. 2938_HV isolate is indicated in red and 1948_hV isolate in green.
3.5. Genes affected by CNVs, SNPs and InDels
Correlating SNPs and InDels variants to gene CNVs could point out to genes under combined layers of selective pressure. Results showed in figure 5 highlight the total numbers of genes affected by CNVs, SNPs and InDels for 1948_hV (Figure 5,A) and 2938_HV (Figure 5,B) isolates. Comparisons indicate that only 14 genes are prone to this extensive variation, among which 11 are common to both isolates, while three are unique to each isolate (Figure 5,C).
Figure 5. Venn diagrams showing the number of genes affected by CNVs, SNPs and InDels in 2938_HV and 1948_hV L. major isolates.
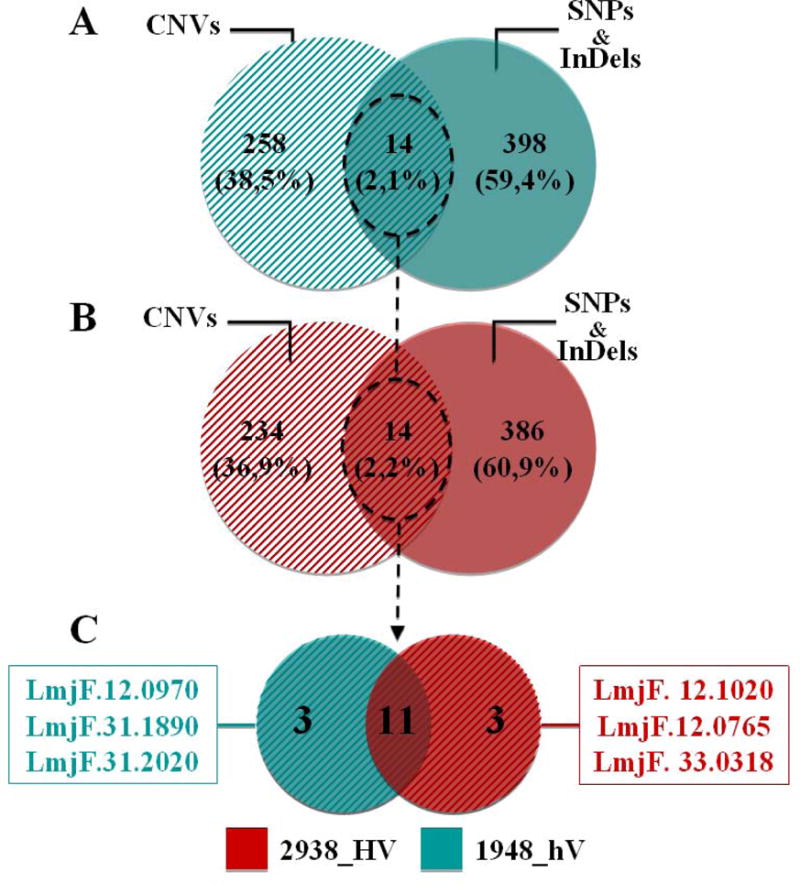
Venn diagrams show the number of genes affected by CNVs, the number of genes affected by both SNPs and InDels, and their comparisons for 2938_HV (A) and 1948_hV (B), and the comparison of common variants between 2938_HV and 1948_hV isolates (C). These comparisons take into account the total number of protein-coding genes. 2938_HV isolate is indicated in red and 1948_hV isolate in green.
Interestingly, among the 11 common genes, one is coding for a receptor-type adenylate cyclase b, four for members of the class-I nuclease-like protein, two for hsp70-related proteins-1, and four for hypothetical proteins. The fact that same genes are targeted by different variants in both isolates may indicate that the same genes could be under different regulatory mechanisms. Indeed, such phenomenon has been elegantly described in a study aiming to identify genes involved in Leishmania tropism (Zhang et al., 2008). Authors have identified a L. infantum and L. donovani gene that was thought to be only present in visceralizing Leishmania species, but ended up with the conclusion that levels of expression rather than sequence variation alone in genes ubiquitously shared between viscerotropic and dermotropic Leishmania species have the potential to significantly influence tissue tropism and clinical severity.
The three genes that are unique to 1948_hV code for a hypothetical protein (LmjF.12.0970), a peptidase m20/m25/m40 family-like protein (LmjF.31.1890), and a succinyl-diaminopimelate desuccinylase-like protein (LmjF.31.1890), whereas the three unique to 2938_HV are coding for Hsp83 (LmjF.33.0318) and two surface antigen protein 2 (LmjF.12.0765, LmjF.12.1020), genes that we previously described. The exact role of all these genes in disease severity deserves extensive functional analysis. Due to their involvement in many cellular processes (Nollen and Morimoto, 2002), genes such as the heat-shock family should be cautiously related to their regulation through various described mechanisms such as phosphorylation at the pathogenic stage (Morales et al., 2010), changes in their secondary RNA structure (David et al., 2010), increase of their mRNA levels (Shapira et al., 1988), and post-transcriptional mechanisms (Shapira et al., 2001). Other DNA-based methods have proven their efficiency in the intra-species screen for diversity. Our group recently reported that bearing a 58-bp microsatellite allele at the 71AT locus tends to be correlated with resistant feature of promastigotes to in vitro complement lysis, whereas parasites bearing the 64-bp allele at the same locus are less resistant (Attia et al., 2016). Interestingly, the same study showed that 1948_hV isolate bears the 64-bp microsatellite allele and is less resistant than the 2938_HV isolate, which bears the 58-bp microsatellite allele at the 71AT locus (Attia et al., 2016).
Taken altogether, our results (this present study and (Attia et al., 2016)) clearly indicate that differences at genomic level between differentially virulent L. major isolates may contribute to the contrasted lesion severity observed in ZCL patients and BALB/c animal model.
4. Conclusion
With its extraordinary genome plasticity, Leishmania is able to face and adapt to drastic environmental changes, and this behavior is species-, isolate-, or population-specific. This study relates a sum of genetic differences between parasites isolated from patients with ZCL living in an endemic country and causing differential and contrasted clinical outcomes, but also contrasted pathogenicity in BALB/c experimental model. The reported differences point out to their possible relationship to parasite pathogenicity. However, applying additional high-throughput approaches would help draw a systems-level view complementing the present whole-genome sequencing results. Because many confounding factors exist for disease severity, exploring these additional layers of regulation as well as identifying functional parasite biomarkers are required for a stronger proof for the direct association between genetic background and clinical severity. Future work should focus on integrating more studies on field isolates in order to enrich genomic variants analysis with population frequencies.
Supplementary Material
Highlights.
Genetic variants show discriminative potential between Leishmania major clinical isolates
Chromosomes CNVs, genes CNVs SNPs and InDels could be determinants of disease severity
Genes affected by SNPs, InDels and CNV with potential interests are identified
Acknowledgments
We are grateful to Prof. Afif Ben Salah, Mr Adel Gharbi and Mr Amor Zaatour for kindly providing us with L. major isolates from the field, all members of the Medical Epidemiology team in Institut Pasteur de Tunis for collecting these isolates and for the follow-up of lesions sizes and data collection, and the Tunisian patients for their kind help and consent. This work was funded as part of LeiSHield consortium by contribution of the Institut Pasteur International Direction and was partly supported by a NIAID/NIH Grant Number 5P50AI074178 and by Wellcome Trust (104111) Core Funding for the WTCMP. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests
The authors declare that they have no competing interests.
References
- Akopyants NS, Kimblin N, Secundino N, Patrick R, Peters N, Lawyer P, Dobson DE, Beverley SM, Sacks DL. Demonstration of genetic exchange during cyclical development of Leishmania in the sand fly vector. Science. 2009;324:265–268. doi: 10.1126/science.1169464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvar J, Velez ID, Bern C, Herrero M, Desjeux P, Cano J, Jannin J, den Boer M. Leishmaniasis worldwide and global estimates of its incidence. PLoS One. 2012;7:e35671. doi: 10.1371/journal.pone.0035671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoun K, Bouratbine A. Cutaneous leishmaniasis in North Africa: a review. Parasite. 2014;21:14. doi: 10.1051/parasite/2014014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attia H, Sghaier RM, Gelanew T, Bali A, Schweynoch C, Guerfali FZ, Mkannez G, Chlif S, Belhaj-Hamida S, Dellagi K, Schonian GDL. Genetic micro-heterogeneity of Leishmania major in emerging foci of zoonotic cutaneous leishmaniasis in Tunisia. Infect Genet Evol. 2016;43:179–85. doi: 10.1016/j.meegid.2016.04.032. [DOI] [PubMed] [Google Scholar]
- Banuls AL, Bastien P, Pomares C, Arevalo J, Fisa R, Hide M. Clinical pleiomorphism in human leishmaniases, with special mention of asymptomatic infection. Clin Microbiol Infect. 2011;17:1451–1461. doi: 10.1111/j.1469-0691.2011.03640.x. [DOI] [PubMed] [Google Scholar]
- Beetham JK, Donelson JE, Dahlin RR. Surface glycoprotein PSA (GP46) expression during short- and long-term culture of Leishmania chagasi. Mol Biochem Parasitol. 2003;131:109–117. doi: 10.1016/s0166-6851(03)00197-x. [DOI] [PubMed] [Google Scholar]
- Bettaieb J, Toumi A, Chlif S, Chelghaf B, Boukthir A, Gharbi A, Ben Salah A. Prevalence and determinants of Leishmania major infection in emerging and old foci in Tunisia. Parasit Vectors. 2014;7:386. doi: 10.1186/1756-3305-7-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brotherton MC, Bourassa S, Leprohon P, Legare D, Poirier GG, Droit A, Ouellette M. Proteomic and genomic analyses of antimony resistant Leishmania infantum mutant. PLoS One. 2013;8:e81899. doi: 10.1371/journal.pone.0081899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantacessi C, Dantas-Torres F, Nolan MJ, Otranto D. The past, present, and future of Leishmania genomics and transcriptomics. Trends Parasitol. 2015;31:100–108. doi: 10.1016/j.pt.2014.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun S, Fay JC. Identification of deleterious mutations within three human genomes. Genome Res. 2009;19:1553–1561. doi: 10.1101/gr.092619.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coderre JA, Beverley SM, Schimke RT, Santi DV. Overproduction of a bifunctional thymidylate synthetase-dihydrofolate reductase and DNA amplification in methotrexate-resistant Leishmania tropica. Proc Natl Acad Sci U S A. 1983;80:2132–2136. doi: 10.1073/pnas.80.8.2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coelho AC, Boisvert S, Mukherjee A, Leprohon P, Corbeil J, Ouellette M. Multiple mutations in heterogeneous miltefosine-resistant Leishmania major population as determined by whole genome sequencing. PLoS Negl Trop Dis. 2012;6:e1512. doi: 10.1371/journal.pntd.0001512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copin R, Coscolla M, Seiffert SN, Bothamley G, Sutherland J, Mbayo G, Gagneux S, Ernst JD. Sequence diversity in the pe_pgrs genes of Mycobacterium tuberculosis is independent of human T cell recognition. MBio. 2014;5:e00960–00913. doi: 10.1128/mBio.00960-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz AK, Titus R, Beverley SM. Plasticity in chromosome number and testing of essential genes in Leishmania by targeting. Proc Natl Acad Sci U S A. 1993;90:1599–1603. doi: 10.1073/pnas.90.4.1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David M, Gabdank I, Ben-David M, Zilka A, Orr I, Barash D, Shapira M. Preferential translation of Hsp83 in Leishmania requires a thermosensitive polypyrimidine-rich element in the 3′ UTR and involves scanning of the 5′ UTR. RNA. 2010;16:364–374. doi: 10.1261/rna.1874710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devault A, Banuls AL. The promastigote surface antigen gene family of the Leishmania parasite: differential evolution by positive selection and recombination. BMC Evol Biol. 2008;8:292. doi: 10.1186/1471-2148-8-292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downing T, Imamura H, Decuypere S, Clark TG, Coombs GH, Cotton JA, Hilley JD, de Doncker S, Maes I, Mottram JC, Quail MA, Rijal S, Sanders M, Schonian G, Stark O, Sundar S, Vanaerschot M, Hertz-Fowler C, Dujardin JC, Berriman M. Whole genome sequencing of multiple Leishmania donovani clinical isolates provides insights into population structure and mechanisms of drug resistance. Genome Res. 2011;21:2143–2156. doi: 10.1101/gr.123430.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folgueira C, Requena JM. A postgenomic view of the heat shock proteins in kinetoplastids. FEMS Microbiol Rev. 2007;31:359–377. doi: 10.1111/j.1574-6976.2007.00069.x. [DOI] [PubMed] [Google Scholar]
- Haraksingh RR, Snyder MP. Impacts of variation in the human genome on gene regulation. J Mol Biol. 2013;425:3970–7. doi: 10.1016/j.jmb.2013.07.015. [DOI] [PubMed] [Google Scholar]
- Hunt R, Sauna ZE, Ambudkar SV, Gottesman MM, Kimchi-Sarfaty C. Silent (synonymous) SNPs: should we care about them? Methods Mol Biol. 2009;578:23–39. doi: 10.1007/978-1-60327-411-1_2. [DOI] [PubMed] [Google Scholar]
- Imamura H, Downing T, Van den Broeck F, Sanders MJ, Rijal S, Sundar S, Mannaert A, Vanaerschot M, Berg M, De Muylder G, Dumetz F, Cuypers B, Maes I, Domagalska M, Decuypere S, Rai K, Uranw S, Bhattarai NR, Khanal B, Prajapati VK, Sharma S, Stark O, Schonian G, De Koning HP, Settimo L, Vanhollebeke B, Roy S, Ostyn B, Boelaert M, Maes L, Berriman M, Dujardin JC, Cotton JA. Evolutionary genomics of epidemic visceral leishmaniasis in the Indian subcontinent. Elife. 2016;22:5. doi: 10.7554/eLife.12613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isnard A, Shio MT, Olivier M. Impact of Leishmania metalloprotease GP63 on macrophage signaling. Front Cell Infect Microbiol. 2012;2:72. doi: 10.3389/fcimb.2012.00072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivens AC, Peacock CS, Worthey EA, Murphy L, Aggarwal G, Berriman M, Sisk E, Rajandream MA, Adlem E, Aert R, Anupama A, Apostolou Z, Attipoe P, Bason N, Bauser C, Beck A, Beverley SM, Bianchettin G, Borzym K, Bothe G, Bruschi CV, Collins M, Cadag E, Ciarloni L, Clayton C, Coulson RM, Cronin A, Cruz AK, Davies RM, De Gaudenzi J, Dobson DE, Duesterhoeft A, Fazelina G, Fosker N, Frasch AC, Fraser A, Fuchs M, Gabel C, Goble A, Goffeau A, Harris D, Hertz-Fowler C, Hilbert H, Horn D, Huang Y, Klages S, Knights A, Kube M, Larke N, Litvin L, Lord A, Louie T, Marra M, Masuy D, Matthews K, Michaeli S, Mottram JC, Muller-Auer S, Munden H, Nelson S, Norbertczak H, Oliver K, O’Neil S, Pentony M, Pohl TM, Price C, Purnelle B, Quail MA, Rabbinowitsch E, Reinhardt R, Rieger M, Rinta J, Robben J, Robertson L, Ruiz JC, Rutter S, Saunders D, Schafer M, Schein J, Schwartz DC, Seeger K, Seyler A, Sharp S, Shin H, Sivam D, Squares R, Squares S, Tosato V, Vogt C, Volckaert G, Wambutt R, Warren T, Wedler H, Woodward J, Zhou S, Zimmermann W, Smith DF, Blackwell JM, Stuart KD, Barrell B, Myler PJ. The genome of the kinetoplastid parasite, Leishmania major. Science. 2005;309:436–442. doi: 10.1126/science.1112680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer JP, Kaprakkaden A, Choudhary ML, Shaha C. Crucial role of cytosolic tryparedoxin peroxidase in Leishmania donovani survival, drug response and virulence. Mol Microbiol. 2008;68:372–391. doi: 10.1111/j.1365-2958.2008.06154.x. [DOI] [PubMed] [Google Scholar]
- Kane MM, Mosser DM. Leishmania parasites and their ploys to disrupt macrophage activation. Curr Opin Hematol. 2000;7:26–31. doi: 10.1097/00062752-200001000-00006. [DOI] [PubMed] [Google Scholar]
- Kebaier C, Louzir H, Chenik M, Ben Salah A, Dellagi K. Heterogeneity of wild Leishmania major isolates in experimental murine pathogenicity and specific immune response. Infect Immun. 2001;69:4906–4915. doi: 10.1128/IAI.69.8.4906-4915.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leprohon P, Legare D, Girard I, Papadopoulou B, Ouellette M. Modulation of Leishmania ABC protein gene expression through life stages and among drug-resistant parasites. Eukaryot Cell. 2006;5:1713–1725. doi: 10.1128/EC.00152-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leprohon P, Legare D, Raymond F, Madore E, Hardiman G, Corbeil J, Ouellette M. Gene expression modulation is associated with gene amplification, supernumerary chromosomes and chromosome loss in antimony-resistant Leishmania infantum. Nucleic Acids Res. 2009;37:1387–1399. doi: 10.1093/nar/gkn1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Calvillo S, Stuart K, Myler PJ. Ploidy changes associated with disruption of two adjacent genes on Leishmania major chromosome 1. Int J Parasitol. 2005;35:419–429. doi: 10.1016/j.ijpara.2004.12.014. [DOI] [PubMed] [Google Scholar]
- McGwire BS, Satoskar AR. Leishmaniasis: clinical syndromes and treatment. QJM. 2014;107:7–14. doi: 10.1093/qjmed/hct116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta D, Iwamoto K, Ueda J, Bundo M, Adati N, Kojima T, Kato T. Comprehensive survey of CNVs influencing gene expression in the human brain and its implications for pathophysiology. Neurosci Res. 2014;79:22–33. doi: 10.1016/j.neures.2013.10.009. [DOI] [PubMed] [Google Scholar]
- Morales MA, Watanabe R, Dacher M, Chafey P, Osorio y Fortea J, Scott DA, Beverley SM, Ommen G, Clos J, Hem S, Lenormand P, Rousselle JC, Namane A, Spath GF. Phosphoproteome dynamics reveal heat-shock protein complexes specific to the Leishmania donovani infectious stage. Proc Natl Acad Sci U S A. 2010;107:8381–8386. doi: 10.1073/pnas.0914768107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee A, Boisvert S, Monte-Neto RL, Coelho AC, Raymond F, Mukhopadhyay R, Corbeil J, Ouellette M. Telomeric gene deletion and intrachromosomal amplification in antimony-resistant Leishmania. Mol Microbiol. 2013;88:189–202. doi: 10.1111/mmi.12178. [DOI] [PubMed] [Google Scholar]
- Murray HW, Berman JD, Davies CR, Saravia NG. Advances in leishmaniasis. Lancet. 2005;366:1561–1577. doi: 10.1016/S0140-6736(05)67629-5. [DOI] [PubMed] [Google Scholar]
- Naderer T, McConville MJ. Intracellular growth and pathogenesis of Leishmania parasites. Essays Biochem. 2011;51:81–95. doi: 10.1042/bse0510081. [DOI] [PubMed] [Google Scholar]
- Nandan D, Cherkasov A, Sabouti R, Yi T, Reiner NE. Molecular cloning, biochemical and structural analysis of elongation factor-1 alpha from Leishmania donovani: comparison with the mammalian homologue. Biochem Biophys Res Commun. 2003;302:646–652. doi: 10.1016/s0006-291x(03)00216-x. [DOI] [PubMed] [Google Scholar]
- Nandan D, Yi T, Lopez M, Lai C, Reiner NE. Leishmania EF-1alpha activates the Src homology 2 domain containing tyrosine phosphatase SHP-1 leading to macrophage deactivation. J Biol Chem. 2002;277:50190–50197. doi: 10.1074/jbc.M209210200. [DOI] [PubMed] [Google Scholar]
- Nollen EA, Morimoto RI. Chaperoning signaling pathways: molecular chaperones as stress-sensing ‘heat shock’ proteins. J Cell Sci. 2002;115:2809–2816. doi: 10.1242/jcs.115.14.2809. [DOI] [PubMed] [Google Scholar]
- Olivier M, Gregory DJ, Forget G. Subversion mechanisms by which Leishmania parasites can escape the host immune response: a signaling point of view. Clin Microbiol Rev. 2005;18:293–305. doi: 10.1128/CMR.18.2.293-305.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olliaro P, Vaillant M, Arana B, Grogl M, Modabber F, Magill A, Lapujade O, Buffet P, Alvar J. Methodology of clinical trials aimed at assessing interventions for cutaneous leishmaniasis. PLoS Negl Trop Dis. 2013;7:e2130. doi: 10.1371/journal.pntd.0002130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlopoulos GA, Oulas A, Iacucci E, Sifrim A, Moreau Y, Schneider R, Aerts J, Iliopoulos I. Unraveling genomic variation from next generation sequencing data. BioData Min. 2013;6:13. doi: 10.1186/1756-0381-6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR. BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Curr Protoc Bioinformatics. 2014;47:11.12.1–34. doi: 10.1002/0471250953.bi1112s47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravel C, Dubessay P, Bastien P, Blackwell JM, Ivens AC. The complete chromosomal organization of the reference strain of the Leishmania Genome Project, L. major; Friedlin. Parasitol Today. 1998;14:301–303. doi: 10.1016/s0169-4758(98)01275-7. [DOI] [PubMed] [Google Scholar]
- Ritt JF, Raymond F, Leprohon P, Legare D, Corbeil J, Ouellette M. Gene amplification and point mutations in pyrimidine metabolic genes in 5-fluorouracil resistant Leishmania infantum. PLoS Negl Trop Dis. 2013;7:e2564. doi: 10.1371/journal.pntd.0002564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers MB, Hilley JD, Dickens NJ, Wilkes J, Bates PA, Depledge DP, Harris D, Her Y, Herzyk P, Imamura H, Otto TD, Sanders M, Seeger K, Dujardin JC, Berriman M, Smith DF, Hertz-Fowler C, Mottram JC. Chromosome and gene copy number variation allow major structural change between species and strains of Leishmania. Genome Res. 2011;21:2129–2142. doi: 10.1101/gr.122945.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohousova I, Volf P. Sand fly saliva: effects on host immune response and Leishmania transmission. Folia Parasitol (Praha) 2006;53:161–171. [PubMed] [Google Scholar]
- Sakthianandeswaren A, Foote SJ, Handman E. The role of host genetics in leishmaniasis. Trends Parasitol. 2009;25:383–391. doi: 10.1016/j.pt.2009.05.004. [DOI] [PubMed] [Google Scholar]
- Sauna ZE, Kimchi-Sarfaty C. Understanding the contribution of synonymous mutations to human disease. Nat Rev Genet. 2011;12:683–691. doi: 10.1038/nrg3051. [DOI] [PubMed] [Google Scholar]
- Schonian G, Nasereddin A, Dinse N, Schweynoch C, Schallig HD, Presber W, Jaffe CL. PCR diagnosis and characterization of Leishmania in local and imported clinical samples. Diagn Microbiol Infect Dis. 2003;47:349–358. doi: 10.1016/s0732-8893(03)00093-2. [DOI] [PubMed] [Google Scholar]
- Schriefer A, Wilson ME, Carvalho EM. Recent developments leading toward a paradigm switch in the diagnostic and therapeutic approach to human leishmaniasis. Curr Opin Infect Dis. 2008;21:483–488. doi: 10.1097/QCO.0b013e32830d0ee8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapira M, McEwen JG, Jaffe CL. Temperature effects on molecular processes which lead to stage differentiation in Leishmania. EMBO J. 1988;7:2895–2901. doi: 10.1002/j.1460-2075.1988.tb03147.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapira M, Zilka A, Garlapati S, Dahan E, Dahan I, Yavesky V. Post transcriptional control of gene expression in Leishmania. Med Microbiol Immunol. 2001;190:23–26. doi: 10.1007/s004300100073. [DOI] [PubMed] [Google Scholar]
- Sterkers Y, Lachaud L, Bourgeois N, Crobu L, Bastien P, Pages M. Novel insights into genome plasticity in Eukaryotes: mosaic aneuploidy in Leishmania. Mol Microbiol. 2012;86:15–23. doi: 10.1111/j.1365-2958.2012.08185.x. [DOI] [PubMed] [Google Scholar]
- Stuart K, Brun R, Croft S, Fairlamb A, Gurtler RE, McKerrow J, Reed S, Tarleton R. Kinetoplastids: related protozoan pathogens, different diseases. J Clin Invest. 2008;118:1301–1310. doi: 10.1172/JCI33945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira PC, Velasquez LG, Lepique AP, de Rezende E, Bonatto JM, Barcinski MA, Cunha-Neto E, Stolf BS. Regulation of Leishmania (L.) amazonensis protein expression by host T cell dependent responses: differential expression of oligopeptidase B, tryparedoxin peroxidase and HSP70 isoforms in amastigotes isolated from BALB/c and BALB/c nude mice. PLoS Negl Trop Dis. 2015;9:e0003411. doi: 10.1371/journal.pntd.0003411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdivia HO, Reis-Cunha JL, Rodrigues-Luiz GF, Baptista RP, Baldeviano GC, Gerbasi RV, Dobson DE, Pratlong F, Bastien P, Lescano AG, Beverley SM, Bartholomeu DC. Comparative genomic analysis of Leishmania (Viannia) peruviana and Leishmania (Viannia) braziliensis. BMC Genomics. 2015;16:715. doi: 10.1186/s12864-015-1928-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinkenoog R, Speranca MA, van Breemen O, Ramesar J, Williamson DH, Ross-MacDonald PB, Thomas AW, Janse CJ, del Portillo HA, Waters AP. Malaria parasites contain two identical copies of an elongation factor 1 alpha gene. Mol Biochem Parasitol. 1998;94:1–12. doi: 10.1016/s0166-6851(98)00035-8. [DOI] [PubMed] [Google Scholar]
- Vrijenhoek T, Kraaijeveld K, Elferink M, de Ligt J, Kranendonk E, Santen G, Nijman IJ, Butler D, Claes G, Costessi A, Dorlijn W, van Eyndhoven W, Halley DJ, van den Hout MC, van Hove S, Johansson LF, Jongbloed JD, Kamps R, Kockx CE, de Koning B, Kriek M, Deprez RL, Lunstroo H, Mannens M, Mook OR, Nelen M, Ploem C, Rijnen M, Saris JJ, Sinke R, Sistermans E, van Slegtenhorst M, Sleutels F, van der Stoep N, van Tienhoven M, Vermaat M, Vogel M, Waisfisz Q, Weiss JM, van den Wijngaard A, van Workum W, Ijntema H, van der Zwaag B, van IWF, den Dunnen JT, Veltman JA, Hennekam R, Cuppen E. Next-generation sequencing-based genome diagnostics across clinical genetics centers: implementation choices and their effects. Eur J Hum Genet. 2015;23:1270. doi: 10.1038/ejhg.2015.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO. Control of the leishmaniasis: Report of a meeting of the WHO Expert Committee on the Control of Leishmaniases. World Health Organization; Geneva, Switzerland: 2010. [Google Scholar]
- Yamage M, Debrabant A, Dwyer DM. Molecular characterization of a hyperinducible, surface membrane-anchored, class I nuclease of a trypanosomatid parasite. J Biol Chem. 2000;275:36369–36379. doi: 10.1074/jbc.M004036200. [DOI] [PubMed] [Google Scholar]
- Zhang WW, Peacock CS, Matlashewski G. A genomic-based approach combining in vivo selection in mice to identify a novel virulence gene in Leishmania. PLoS Negl Trop Dis. 2008;2:e248. doi: 10.1371/journal.pntd.0000248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang WW, Ramasamy G, McCall LI, Haydock A, Ranasinghe S, Abeygunasekara P, Sirimanna G, Wickremasinghe R, Myler P, Matlashewski G. Genetic analysis of Leishmania donovani tropism using a naturally attenuated cutaneous strain. PLoS Pathog. 2014;10:e1004244. doi: 10.1371/journal.ppat.1004244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Utsunomiya YT, Xu L, Hay el HA, Bickhart DM, Alexandre PA, Rosen BD, Schroeder SG, Carvalheiro R, de Rezende Neves HH, Sonstegard TS, Van Tassell CP, Ferraz JB, Fukumasu H, Garcia JF, Liu GE. Genome-wide CNV analysis reveals variants associated with growth traits in Bos indicus. BMC Genomics. 2016;17:419. doi: 10.1186/s12864-016-2461-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.