

Abstract
Kinetochores are large multiprotein complexes that drive mitotic chromosome movements by mechanically coupling them to the growing and shortening tips of spindle microtubules. Kinetochores are also regulatory hubs, somehow sensing when they are erroneously attached and, in response, releasing their incorrect attachments and generating diffusible wait signals to delay anaphase until proper attachments can form. The remarkable ability of a kinetochore to sense and respond to its attachment status might stem from attachment- or tension-dependent changes in the structural arrangement of its core sub-complexes. However, direct tests of the relationship between attachment, tension, and core kinetochore structure have not previously been possible because of the difficulties of applying well-controlled forces and determining unambiguously the attachment status of individual kinetochores in vivo. The recent purification of native yeast kinetochores has enabled in vitro optical trapping-based assays of kinetochore tip-coupling and, in separate experiments, fluorescence imaging of single kinetochore particles. Here we introduce a dual instrument, combining optical trapping with multi-color total internal reflection fluorescence (TIRF) imaging, to allow kinetochore structure to be monitored directly with nanometer precision while mechanical tension is simultaneously applied. Our instrument incorporates differential interference contrast (DIC) imaging as well, to minimize the photo-bleaching of fluorescent tags during preparative bead and microtubule manipulations. A simple modification also allows the trapping laser to be easily converted into a real-time focus detection and correction system. Using this combined instrument, the distance between specific sub-complexes within a single kinetochore particle can be measured with 2-nm precision after 50 s observation time, or with 11-nm precision at 1 s temporal resolution. While our instrument was constructed specifically for studying kinetochores, it should also be useful for studying other filament-binding protein complexes, such as spindle poles, cortical microtubule attachments, focal adhesions, or other motor-cytoskeletal junctions.
Keywords: Total Internal Reflection Fluorescence (TIRF) Microscopy, Super-resolution, Centroid tracking, Differential Interference Contrast (DIC), Optical tweezers, Focus correction, Surface passivation, Kinetochore, Microtubule, Intra-molecular distance
1 Introduction
Optical tweezers are ideal tools for manipulating sub-micrometer objects such as colloidal particles or bacterial cells, and for generating and quantifying forces in the pico-Newton range [1-3]. They have become popular instruments for biophysical studies at the single molecule level in vitro, such as of motor proteins or protein complexes that are processive on linear cellular structures, including kinesin and dynein motors on microtubules [4-8], myosin on F-actin [9-11], and polymerases on DNA and RNA [12-16]. Typically, the optical tweezers are combined with wide-field microscopy or differential interference contrast (DIC) microscopy [17], which provides visual cues for the experimentalist to manipulate trapped particles into the desired spatial configuration. DIC microscopy is a popular choice, because the appearance of micrometer scale colloidal particles under DIC imaging is clearly distinctive from protein aggregates or bead clumps that, if trapped, would lead to errors in estimating the force generated by the optical tweezers. DIC imaging can also allow objects 10 times smaller than the illumination wavelength to be viewed. As an example, microtubules, which are 25 nm in diameter, are visible under DIC microscopy with video enhancements [18, 19]. This has made DIC imaging especially useful in single molecule tweezers studies of microtubule-associated motor proteins such as kinesins and cytoplasmic dynein. Another combination of optical tweezers that has become increasingly common is with fluorescence microscopy [5, 20, 21]. Simultaneously recording single-molecule fluorescence while also applying controlled mechanical perturbations can provide correlated information that is very valuable for uncovering how complex molecular machines function [22-24].
Most previous combined single-molecule fluorescence/optical tweezers instruments were designed for studying nucleic acid-interacting proteins rather than microtubule-interacting complexes. Studying protein machines that interact with microtubules, particularly dynamic microtubules, poses extra challenges. Microtubules are assembled in vitro and are never monodisperse in length, whereas DNAs can be made or purchased in highly monodisperse form. In experiments that must accommodate microtubule filaments of variable length, it is important to image the microtubules, whereas in DNA assays it is often feasible to leave the DNAs invisible. Video-enhanced differential interference contrast (VE-DIC) microscopy allows the use of unstained microtubules, avoiding possible interference from dye-labeled tubulin subunits, and enabling long-term imaging over hours without photobleaching [25-27].
In this chapter we introduce a combined instrument of optical tweezers, DIC and TIRF microscopy, each method working independently and simultaneously. To demonstrate its use, intra-kinetochore stretch distance is measured in vitro. Kinetochores are complex multi-protein machines that serve not only as mechanical couplers between mitotic chromosomes and dynamic microtubule tips [28, 29], but also as signaling hubs. Mechanical forces on kinetochores are widely believed to control the generation of wait signals (Spindle Assembly Checkpoint signals), which help to ensure the accuracy of mitosis by delaying anaphase chromosome separation until all kinetochores are properly tip-attached and under tension [30, 31]. Kinetochore tension is also believed to control an error avoidance mechanism that triggers detachment of erroneously attached kinetochores that lack tension, while selectively stabilizing proper kinetochore-microtubule attachments that bear tension [32-34]. Presumably, therefore, tension must affect kinetochore structure in such a way as to control the localization or activity of signal-generating and error-correcting factors. Recent work confirms the structural plasticity of kinetochores in vivo [35-38]. However, it remains unclear whether the rearrangements observed in vivo are direct responses to mechanical force, or whether they result indirectly from other signaling events or from differences in the attachment status of the kinetochores, which can be difficult to determine unambiguously in vivo. Our approach of observing the fluorescence from single kinetochore particles bound to individual microtubules in vitro (Fig. 1) provides a new and more direct means to uncover how mechanical force affects kinetochore architecture. Our combined fluorescence and trapping-based methods will also be useful for directly examining how attachment status affects the affinity of the kinetochore for key checkpoint signaling and error correction factors. Ultimately, the approach might also be useful for studying protein assemblies from other cytoskeletal junctions, such as spindle poles, cortical attachment sites, or focal adhesions.
Figure 1.
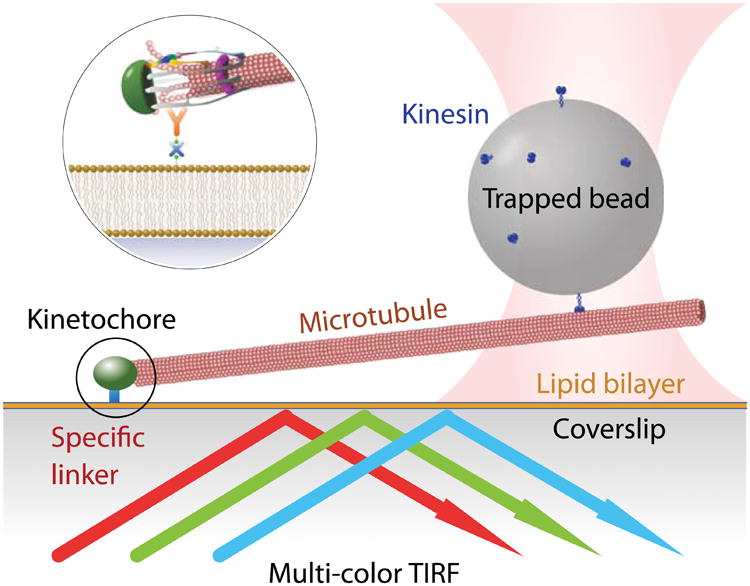
Schematic of the kinetochore stretching experiment. The glass surface is passivated by a supported lipid bilayer to provide specific binding of kinetochores. A small portion of the lipid is biotinylated, which allows avidin to bind, followed by biotinylated anti-His antibody. The kinetochores are His-tagged and are therefore diffusively anchored on the surface. Microtubules spontaneously form end-on attachments to the kinetochores, and a polystyrene bead that is functionalized by kinesin is trapped optically and then bound to the free end of the microtubule. The kinetochore is dragged along the lipid bilayer by the trapped bead, and time-lapse fluorescence images of the differentially labeled kinetochore are recorded.
This chapter describes the design of the three major optical components of our instrument, the surface preparation we use for single kinetochore imaging, and a novel kinetochore-stretching assay. We also describe the design of an extension to the instrument that converts the optical tweezers into a focus detection and correction system. Optical trap calibration and kinetochore purification are covered only very briefly, since more detailed information can be found in the cited references (and possibly in the other chapters of this volume).
2 Materials
2.1 Instrument Design and Optical Alignment
Many optical tweezer instruments are built by modifying commercial bright-field or DIC microscopes [39]. Whereas DIC and fluorescence imaging can be naturally combined with optical tweezers, the two imaging methods are not readily compatible by many standard microscope designs. The reason lies in the optical train of DIC, which has a pair of Wollaston or Nomarski prisms conjugated to the back-focal planes of the objective and condenser lenses. The prism on the objective side will split the emitted light based on its polarization, giving rise to two spatially displaced images corresponding to the two polarization directions. This artifact is undesirable in most applications, especially in single-molecule imaging, unless emission polarization is studied specifically. Wollaston or Nomarski prisms also affect the trapping laser in similar manner. For a circularly polarized incident trapping beam, the Nomarski prism will generate two slightly displaced traps, which can be approximated as one elliptically shaped trap. For a linearly polarized incident beam with polarization direction parallel with one of the axes of the prism, the beam will not be split, but the trap will still be anisotropic due to the polarization [40]. While anisotropy of the laser trap can be measured and accounted for [39], the artifact in fluorescence is not acceptable when imaging resolution is close to the diffraction limit. Here we overcome this limitation by relaying the back-focal plane of the objective lens via a 4f lens system (as detailed below, in Section 2.1.3). The trapping, fluorescence, and DIC imaging beam paths are then well separated, so that the Nomarski prism does not affect the other two optical paths. By implementing this modification, we show that DIC is perfectly compatible with fluorescence imaging and optical tweezers. In our combined system, we can manipulate an optically trapped object according to DIC images, and acquire fluorescence signals only when necessary to minimize photobleaching.
In addition to the incorporation of TIRF and DIC microscopy capability into optical tweezers, we also describe a practical modification that converts the optical tweezers setup into a real-time focus detection and correction system, or a focus lock, which compensates sample stage drift due to temperature fluctuations or slide creep. The principle is similar to many setups that use total internal reflection to convert sample movement into a translating optical signal [41]. Instead of using a separate light source, however, the trapping beam is converted from focusing at the imaging plane into a total-internal-reflection configuration by a few extra inexpensive optical elements, and is reflected back to a position-sensitive photodetector. This addition allows us to correct focal drift at video frequency, independent of DIC and TIRF functionalities.
Our instrument is built on a standard Nikon TE inverted microscope base, on which TIRF, optical tweezers, DIC, and focus lock are built sequentially. Three diode lasers at 488 nm, 561 nm, and 647 nm with a maximum power of 100 mW are used as excitation light sources that provide a good coverage of a wide range of fluorophore choices. Commercial DIC microscopes usually use green illumination, around 530 nm wavelength, which conflicts with one of the fluorescence channels. For this reason, we replaced the green DIC source with a 780-nm wavelength near-infrared LED (Thorlabs M780L3).
2.1.1 TIRF Microscopy and Image Channel Position Registration
The laser beams from three solid-state lasers at 488 nm (Coherent Sapphire), 561 nm (Coherent Sapphire) and 641 nm (Coherent Cube) pass three half-wave plates, three polarized cube beam splitters, and three mechanical shutters (Uniblitz VS 25S2ZMO). Then they are expanded individually each by a pair of telescope lenses L1 (f = 12mm) and L2 (f = 500mm), as shown in Fig. 2. The expanded beams then pass a 1:1 telescope with the first lens L3 (f = 100mm) on a translational stage so that the incident angle on the image plane is adjustable. Therefore, users can easily switch between TIR, epi, and oblique illumination. Three laser beams are combined by two dichroic mirrors, DM1 and DM2, placed between L3 and L4 (f = 100mm). The combined and steered beams that pass through lens L4 are focused by the tube lens L5 (f = 150mm) to the back-focal plane of the objective lens. A multi-band dichroic mirror DM3 between L5 and the objective lens reflects the incident excitation lasers while passing the emitted fluorescence and the bright-field (DIC) illumination. The focal points of lens pairs L1/L2, L3/L4, and L5/objective coincide. An illumination field diaphragm (FD) is placed downstream of L4 at a distance of 2f, and upstream of lens L5 at a distance of 1f, so that both the field iris and the translating lens L3 are conjugated to the image plane. The iris allows adjustment of the illuminated area to prevent unnecessary photobleaching outside the imaging field of view (FOV). With this design, when L3 moves, it only translates the focused beam at the back-focal plane of the objective, and thus changes the incident angle at the image plane without changing the illuminated area. The emission signals are collected by the objective lens, sent through dichroic mirror DM3, and imaged by the built-in tube lens of the microscope base, followed by L6 (f = 60mm) and L7 (f = 125mm) onto three EMCCD cameras (Andor iXon897+). The fluorescence is split by DM4 and DM5 and cleaned by emission filters BP2-4. The sample is mounted on a piezo-driven nano-positioning stage (Physik Instrumente P-571.3CL) that provides programmable movement and focusing correction in software.
Figure 2.
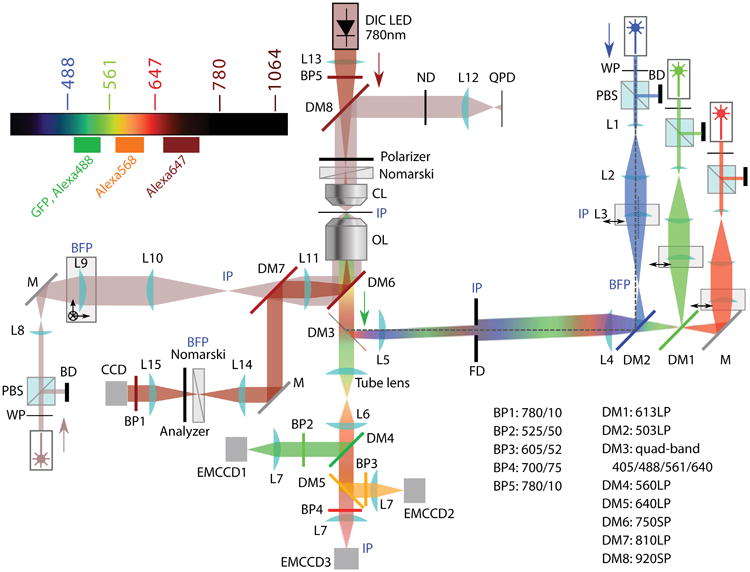
Diagram of the optical layout of the TIRF-Trap-DIC setup. BD beam dump; BFP back- focal plane; BP band-pass filter; CCD charge-coupled device (for bright-field detection); CL condenser/collection lens; DM dichroic mirror; EMCCD electron-multiplying CCD (for fluorescence detection); FD field diaphragm; IP image plane; L lens; LED light-emitting diode; M mirror; OL objective lens; PBS polarizing beam splitter; QPD quadrant photodiode; S shutter; WP half-wave plate.
To align the TIRF setup, a regular CCD camera is mounted at one of the view ports and aligned according to the images from the eyepiece. The camera is then used as a position indicator for laser beam alignment. First, the lasers are mounted and adjusted by several beam-folding mirrors. Beams of three colors are combined in this step by dichroic mirrors (DM1 and DM2). Then, the telescope assembly L4-iris-L5 is mounted to the backport of the microscope base and adjusted in position according to the beam pattern on the camera. Finally, lens L3 is installed, followed by the expansion telescope L2/L1 for each one of the three colors.
In applications where multiple fluorescence color channels are involved, accurate position registration across color channels is essential, especially when distances between components labeled in different colors need to be quantified. In the demonstration in this chapter, kinetochores are dragged along the lipid surface over micrometer-scale distances, while the nanometer-scale distance between two kinetochore components is measured by precisely tracking their locations in two different color channels. Accurate position registration is absolutely crucial. If, for example, the images in one channel were enlarged by just 1% relative to the other channel, then a 50 nm artificial displacement would arise from a 5 μm movement across the FOV.
There are many standard procedures that address the position registration problem. Typically, a fluorescent object, usually a bead that is visible in multiple color channels, is raster-scanned across the FOV [42, 43]. The bead positions are used to obtain the registration correction. Here, we describe a highly automated and quick procedure that takes advantage of the features of the instrument and the nature of the imaging data. Our kinetochore images contain sparsely distributed diffraction-limited spots at tunable density. The instrument also works in a “focus lock” mode (which will be introduced in Section 2.1.4), where the sample slide remains in focus while the slide is moved by the nano-positioner to explore other fields of view for imaging. A customized LabVIEW program automatically coordinates the stage movements, focus stabilization, and fluorescence imaging. This program adjusts the sample z-position into focus, sequentially takes fluorescence images in selected color channels, moves the stage lateral position to the next field, and repeats the cycle to fully explore the area accessible to the piezo stage. With this automated instrument and software, we can acquire hundreds of fields that contain 105 kinetochores within several minutes. Kinetochores in these images are randomly localized, and a fraction (typically between 10-50%) of the particles are dual-colored, containing fluorescent components in two color channels.
The transformation between corresponding coordinates in two channels is assumed to be T linear. That is, if one location at coordinate x = (x,y)T in channel 1 corresponds to a location at x′ = (x′,y′) T in channel 2, the two coordinates are related by x′ = Ax + b for all x and x′, where A is a 2 × 2 matrix and b = (xo,yo)T is a translation. This linear model covers rotation, scaling and shearing as four degrees of freedom in A. We find that this simple model is adequate for better than 5 nm precision. If more precision is needed, a more sophisticated, location-dependent nonlinear registration correction could also be obtained as described in [43].
To extract registration information A and b from the images of kinetochores, we implemented an unsupervised machine-learning algorithm that identifies the dual-colored particles. First, a spot detection routine finds the coordinates of bright spots in the two-color channels separately. Then the pair-wise relative positions of the detected bright spots are calculated within each FOV, and the relative positions are collected from all FOVs. The distribution of the pair-wise relative position is shown in Fig. 3. If the detected spots in the two channels are not co-localized at all and randomly distributed, the pair-wise relative position is uniformly distributed (not considering boundary effects). The co-localized spots, on the other hand, will generate a peaked distribution near the origin. The localization error and intrinsic physical distance between fluorophores broaden the distribution, while the registration error causes scaling, rotation, shearing, and nonlinear distortion of the distribution. Nevertheless, relative positions of separated particlesand co-localized particles have very distinctive features. The two clusters of spots are modeled by a mixture of two 2-D normal distributions (see Note 1), one of which has a very broad and isotropic spread centered at the origin, the other with a sharper but undetermined covariance matrix and center location. Because the widths of the two peaks are typically different by two orders of magnitude, they can be effectively separated following the standard expectation-maximization algorithm (see Note 2). The coordinates x and x′ of the co-localized pairs in both channels are then used to estimate A and b through a linear regression x′ = Ax + b. The registration information is immediately used to obtain the corrected coordinates, and a new pair-wise relative position is calculated, and the process iterates until A and b converge (Fig. 4).
Figure 3.
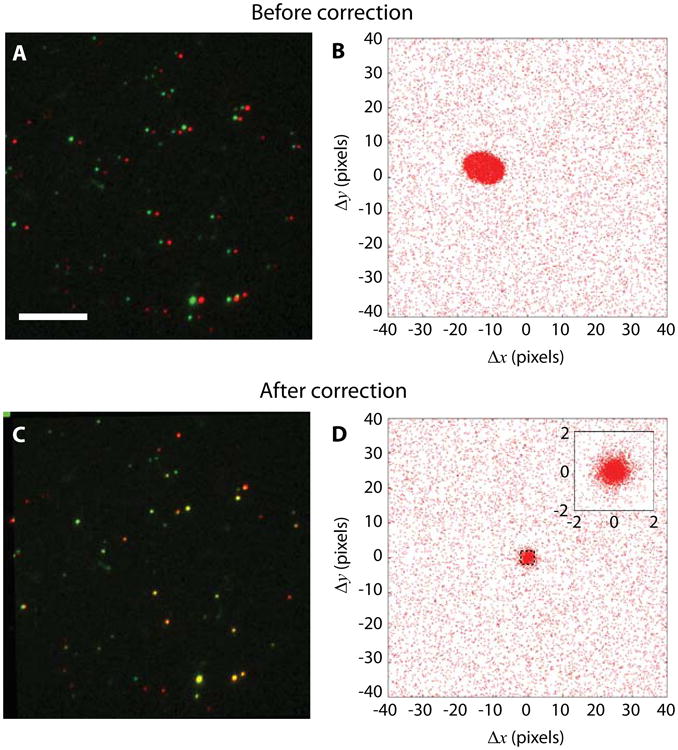
Color channel position registration can be obtained directly from kinetochore imaging data without need for a separate registration step. (A) Raw images of fluorescently labelled kinetochores with Alexa Fluor 546 (false-colored in green) and Alexa Fluor 647 (false-colored in red) on two kinetochore components. Scale bar, 5 μm. (B) Pair-wise distances between all detected spots in two-color channels before registration correction. A distinctive cluster is formed near the origin. (C) Kinetochores after registration correction appear overlapping. (D) Pair-wise relative distances form a tighter cluster at the origin after correction. The inset shows the zoomed-in distribution.
Figure 4.

Flow chart for the image registration algorithm. A spot detection procedure is first applied on the images in both channels. The coordinates are corrected using an initial guess of A and b, then the pair-wise relative position is calculated in each frame. An expectation-maximization routine identifies the co-localized pairs (distinguishing them from single-color spots lacking signal in the other channel), and the coordinates of these co-localized pairs are used to obtain registration correction through linear regression. The procedure is repeated until A and b converge.
The convergence is usually reached within a few iterations when the co-localization fraction is above 10%, so that the central co-localization peak is distinctive from the background. Figure 3 compares the pair-wise relative position before and after the linear registration correction. The distribution indicates that the distance of co-localized pairs after correction is tightly and isotropically centered around the origin. The cause of the residual spread is possibly due to residual localization inaccuracy, field-dependent image distortion, and intrinsic intra-molecular distance between the fluorescent markers.
2.1.2 Optical Tweezers
A 3W Nd:YAG laser at 1064 nm wavelength (Spectra-Physics J20-BL10-106C) is used for trapping. The beam passes a half-wave plate and a polarized cube beam splitter for manual power adjustment (see Note 3). The beam is then expanded by a pair of telescope lenses, L8 (f = 25mm) and L9 (f = 200mm), as shown in Fig. 2, with L9 mounted on a three-dimensional translation stage, and passes through a mechanical shutter (Uniblitz VS 25S2ZMO). The expanded trapping beam passes a pair of 1:1 telescope lenses, L10 and L11 (f = 175mm), and is directed to the objective lens by a dichroic mirror, DM6, mounted between the multi-band dichroic mirror for TIRF and the objective lens. The focal points of lens pairs L8/L9, L10/L11, and L11/objective coincide, and L9 is on the upstream focal plane of L10, so that telescope pair L10/L11 images L9 near the back aperture of the objective lens (see Note 4). When L9 is moved to adjust the optical trap position, the beam intensity does not change significantly. The transmitted light is collected by the condenser lens CL, collimated, and redirected by a dichroic mirror mounted inside the condenser turret through a cut hole. The transmitted beam is focused by lens L12 (f = 80mm) and sent to a quadrant photodiode (QPD). Lens L12 images the condenser back-focal plane onto the QPD, so that the position signal is insensitive to the specific location of the trap when L9 is moved (although it is not necessary to move the trap position in the application of this chapter, and L9 in practice is rarely moved). The QPD signal is low-passed filtered, amplified, and digitized by an A/D converter (National Instruments USB-6212), and recorded by a computer. The alignment process is similar to that used for the TIRF microscope. The fine alignment and calibration of the optical tweezers follows standard procedures as described in [7, 39, 44-46].
2.1.3 DIC Microscopy
Because visible bands are occupied by fluorescence channels, a 780 nm LED (Thorlabs M780L3) is used as the light source for the DIC imaging, fed in at the back port of the condenser turret, in place of the regular green illumination. The slightly longer wavelength compared to the usual green illumination makes it more challenging to image microtubules. Our Nikon microscope base offers DIC imaging by allowing users to insert two Wollaston prisms near the back apertures of the objective lens and the condenser lens. However, this setup would significantly degrade TIRF imaging, as the randomly polarized or un-polarized emission light will be split by the objective Wollaston prism so that the point-spread function for different polarizations are displaced. For this reason, the plane for placing the Wollaston prism under the objective lens is relayed downstream of the DIC beam path by a 4f lens pair.
The dichroic beam splitter DM6 for sending the trapping laser to the objective lens has a cutoff wavelength of 750 nm, and so the DIC illumination (780 nm) is also reflected along the trapping path, and goes through lens L11, the last telescope lens for the trapping beam. Past L11, the DIC beam is separated from the trapping beam by a NIR long-pass dichroic mirror DM7 with 810-nm cutoff wavelength. L11 and another lens with identical focal length, L14 (f = 175mm), form the 4f system that recreates the back aperture where the Wollaston prism is placed. The Wollaston prism is mounted on a stage with 3D-translational and rotational degrees of freedom. Finally, the DIC image is obtained by an imaging lens L15 (f = 125mm) and a CCD camera (Point Grey FL3-GE-03S1M-C). It is worth noting that in the Nikon DIC configuration, the direction of Wollaston prism axes are 45° from the x- and y-axes of the image, or the directions of the side ports and back port. The dichroic mirror that reflects both the trapping and DIC beams has a coating that complicates the phase relation between s-polarized and p-polarized waves at 780 nm. For this reason, the Wollaston prism axes are rotated to be parallel to the s- and p-directions of the dichroic mirror. The condenser turret is rotated 45° to achieve this configuration.
The optical tweezers should be aligned prior to setting up the DIC path. Lens L11 is shared by the tweezers and the DIC, so it should remain fixed in space. Using the auxiliary camera for TIRF alignment and the image of the field iris in the condenser turret, the condenser lens is aligned, so is the DIC illumination path. The second telescope lens L14 and the imaging lens L15 can be aligned similarly by centering the image on the DIC camera. Finally, the Wollaston prism is placed at the focal plane of L14 between L14 and L15. To adjust the x-y location of the Wollaston prism on the imaging side, move the prism until the image is the dimmest, and then move away from the dimmest position until the image is near saturation for the best contrast. To adjust the axial location of the Wollaston prism along the beam propagation direction, move the prism on both axial and perpendicular directions, so that the image changes in brightness uniformly, instead of having a wide dark stripe across the image.
Microtubules imaged by DIC have low contrast, and therefore the DIC images need to be processed for better visibility. Commercially available video enhancement devices can be found for analogue video signals (e.g., the Hamamatsu Argus). The same real-time image/video processing task can now be carried out by computers, with the advantage of better coordination with nano-positioner control, optical trapping laser control, and fluorescence cameras. To maximize microtubule contrast, the DIC camera works at near saturation so that its dynamic range is fully exploited. The images typically have an uneven illumination, and likely contain several dust particles in the field, both of which can have higher brightness variation than microtubule images. Fortunately, these static background contributions can be recorded at a clean field or by focusing inside the coverslip, and can be subtracted from the images with beads and microtubules. In case the background illumination is still uneven, a high-pass filter is applicable. For our particular setup, we use LabVIEW for image acquisition and processing. Filtering the images with a large kernel is slow and not suitable for real-time processing. Instead, we resize the images 32 times smaller, median filter them, and then resize them back to the original size using interpolation, which is significantly faster than filtering. The smoothed background is subtracted to approximate high-pass filtering.
2.1.4 Focus Detection
The 1064-nm trapping laser can be used for focal detection with simple modifications to the trapping beam path. The design is similar to the commercially available focus locks provided with most microscopes, but at far lower cost. In a TIR configuration, the incident beam is reflected by the glass-water interface and sent back to the objective. An axial shift in the position of the sample results in a translational movement of the reflected beam. This movement can be picked up by a position-sensitive detector (PSD; Pacific Silicon Sensors DL100-7PCBA3) as an indication of the sample axial location. Different from the trap configuration, the laser beam in TIR configuration is collimated on the imaging plane instead of focused. To convert the trapping beam to a TIR beam, a concave lens L16 (f = -150mm) is added between the 1:1 telescope L10-L11 (Fig. 5). With the concave lens, the laser beam will be collimated, centered and parallel to the objective axis. To create an incident angle greater than the critical angle, a pair of wedge prisms P, each having 2° beam deviation, is also inserted so that the incident angles can be adjusted to any value between 0° and 4° from any direction. The TIR beam is spatially separated from the incident beam, and is redirected onto the PSD by a D-shaped mirror (D) between L8 and L9 so that the incident beam is not blocked (Fig. 5).
Figure 5.
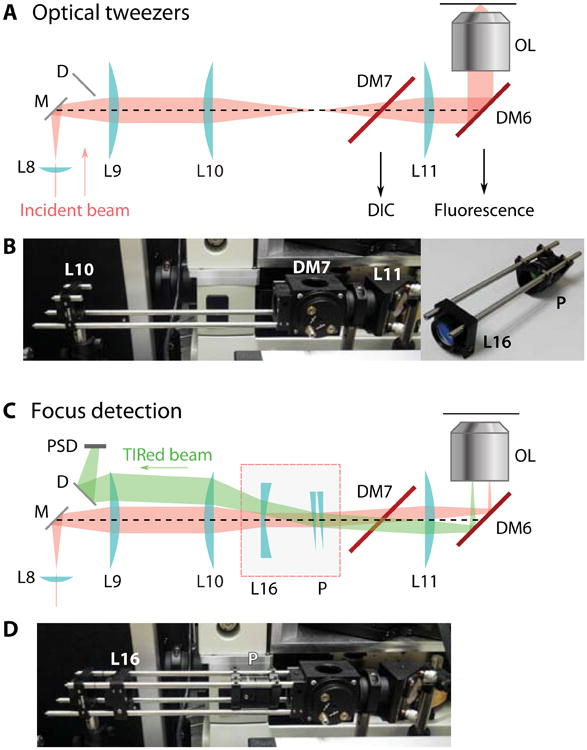
Focus lock design. (A) Diagram of the optical tweezers configuration. The incident 1064 nm beam is collimated at the back-focal point of the objective lens and focused on the image plane. (B) Photograph of the actual optical elements. The extra elements for converting to a focus lock are removed, and pictured separately at right. (C) Optical diagram of the focus detection configuration. An added convex lens L16 converts the beam collimation so that the laser is focused at the back-focal plane. A wedge glass pair P steers the incident angle beyond the critical angle. (D) Photograph showing the actual parts with focus detection components inserted.
For easy installation and removal of the three additional optical elements (one convex lens and two wedge prisms), lens L10 and L11 in the optical tweezers setup are mounted on a cage system with an open top, and the additions of the concave lens L16 and prism pair are mounted on complementary cage rails (Fig. 5). To adjust the prism pair, set the wedge of the prisms pointing in opposite directions initially, so that the beam out of the objective has zero incident angle about the axis. With a sample slide on the microscope stage, rotating the prisms in opposite directions will change the incident angle (the elevation angle), while rotating in the same direction rotates the collimated beam about the axis (the azimuthal angle). Adjust the prisms so that the incident beam reaches TIR condition. A bright reflection should be detectable along the beam path and on the PSD. The PSD signal is low-pass filtered, digitized by the acquisition board, and then monitored and controlled if needed by custom software written in LabVIEW.
2.2 Single Kinetochore Imaging and Manipulation Assay
2.2.1 Kinetochore, Tubulin and Kinesin Purification
For in vitro experiments on kinetochores, we follow the protocol in [47] to purify kinetochores from a yeast strain that carries genes encoding labeled kinetochore components. Tubulin is purified from calf brains following [48]. In order for the beads to attach to microtubules, we purify a His-tagged and biotinylated recombinant kinesin motor domain from E. coli cells [49, 50]. The biotinylation is achieved by an enzymatic ligation [51]. The materials can be found in the corresponding references.
2.2.2 Microbeads Functionalized with a Strong MT-Binding Protein
Streptavidin coated polystyrene beads, 440 nm in diameter, 1% weight/volume.
Biotinylated kinesin (see Note 6), 100 μg/ml.
Assay buffer, containing 25 mM HEPES-KOH at pH 7.6, 50 mM potassium glutamate, 1 mM magnesium acetate, 10% glycerol, 0.1 mM EDTA, 0.5 mM EGTA, 0.1% tergitol-type NP-40.
Oxygen-scavenging system for reducing photobleaching and photo-damage: mix glucose oxidase at 20 mg/ml, catalase at 4 mg/ml in BRB80 with 20% glycerol (100× stock), and 2 M glucose in water in a separate tube (100× stock).
5 mg/ml κ-casein in assay buffer.
2.2.3 Microtubules
BRB80 buffer: 80 mM PIPES, 1 mM MgCl2, 0.5 mM EGTA (see Note 7).
Microtubule polymerization buffer: 5% v/v DMSO, 1 mM GTP, 4 μM additional MgCl2 in BRB80 1×.
10 mM taxol stock solution in DMSO.
Purified bovine tubulin at approximately 1 mg/ml.
Fluorescently labeled bovine tubulin (optional).
Ultracentrifuge.
2.2.4 Small Unilamellar Vesicles (SUVs) for Lipid-Based Surface Passivation
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 25 mg/ml in chloroform.
1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(cap biotinyl) sodium salt (bio-cap-PE), 10 mg/ml.
Compressed nitrogen gas.
Lipid buffer: 25 mM HEPES-KOH and 150 mM NaCl at pH 7.6.
Tip sonifier.
Pasteur pipette, glass test tubes, desiccator and vacuum pump.
2.2.5 Disposable Channel-Slides
24×40 glass coverslips, glass slides and double-sided tape.
Bench top plasma cleaner with vacuum pump.
Hot plate or oven.
Nail polish.
2.2.6 Preparing Slides for the Single Kinetochore Imaging and Manipulation Assay
Assay buffer.
0.1 mg/ml avidin-DN in assay buffer.
20 nM penta-His antibody in assay buffer.
Vacuum source such as water aspirator with water trap.
Purified His-tagged kinetochore particles in assay buffer.
Functionalized beads prepared in section 3.1.2.
5 mg/ml κ-casein in assay buffer.
3 Methods
3.1 Single Kinetochore Imaging and Manipulation Assay
To demonstrate the application of our combined instrument, we use it to measure inter-molecular distances within a single kinetochore particle. Kinetochores are protein complexes that assemble on centromeric DNA during mitosis and meiosis and act as load-bearing linkers to the growing and shrinking tips of spindle microtubules in order to segregate chromosomes [52, 53]. Beyond their role as mechanical “tip-couplers”, kinetochores also facilitate crucial regulatory functions including modulation of microtubule tip dynamics [54, 47, 34] and monitoring the spindle-chromosome attachment configuration [30, 31]. The core kinetochore components are largely conserved in eukaryotes from yeast to human. The total molecular weight of a kinetochore is roughly in the megadalton range, but the total weight and the copy number of each component varies across species [55]. Kinetochores are organized into a layered structure, with inner kinetochore components bound to centromeric DNA and outer components forming attachments to spindle microtubules. Their architecture has been mapped with nanometer accuracy by fluorescently tagging particular pairs of kinetochore components and measuring their separation (the “delta measurement”) in fixed or live cells of both yeast and humans [36, 35, 38, 56, 57]. Electron microscopy has also provided high-resolution views of kinetochores. However, due to the difficulty of associating component identity with specific structures, only limited success has been achieved in resolving particular kinetochore proteins within electron micrographs [58, 59, 60].
Because of the mechanical role of kinetochores in spindle-chromosome attachment, optical tweezers have proven useful for probing the nature of these attachments [61, 54, 62, 47, 63, 64]. Similar to optical tweezer experiments on motor proteins, purified kinetochores (or kinetochore subcomplexes) were immobilized on dielectric beads, and were allowed to bind on surface-anchored microtubules. Mechanical measurements alone have revealed many interesting properties of kinetochores. For example, kinetochore-microtubule tip attachments exhibit catch bond-like behavior, where the lifetime of the attachment increases with increasing tension, until an optimal tension is reached, where the lifetime is maximal [47]. Optical tweezers experiments have also shown that meiotic kinetochores attach microtubule tips more strongly than mitotic kinetochores, suggesting that sister kinetochores in meiosis are mechanically fused together [64]. In the same work, TIRF microscopy was used, in addition to and separately from tweezers experiments, to show that the meiotic kinetochores contained more copies of the components than the mitotic ones, giving rise to brighter particles under fluorescence imaging. Thus, optical tweezers and quantitative TIRF microscopy have both provided independent means to quantify the differences between meiotic and mitotic kinetochores.
The structural arrangement of core kinetochore components changes dynamically in vivo depending on the cell cycle stage or after treatment with microtubule poisons. Because the separation distance between certain inner and outer kinetochore components increases at metaphase, when the kinetochores are likely to be supporting relatively high levels of tension, these rearrangements have sometimes been referred to as “intra-kinetochore stretch” [35, 37, 57]. In principle, such attachment- or tension-dependent changes in core kinetochore structure could control the key signaling events at kinetochores, including the generation of checkpoint wait signals and the selective destabilization of erroneous attachments [65]. However, whether the observed structural changes are caused directly by attachment or tension remains unproven and whether they play any role in modulating kinetochore function is also unclear [66, 67]. Resolving these issues will require observing nanometer-scale deformations under controlled forces in vitro. Optical tweezers can generate forces to recapitulate physiological levels of spindle tension while the movement of various kinetochore components is measured using TIRF microscopy and centroid tracking in real time. DIC imaging is particularly useful in this experiment as well, because the 25 nm thick microtubules are visible under DIC, so that fluorescence imaging is not required during the initial preparative manipulations before recording.
Single molecule TIRF experiments typically require surface engineering to achieve specificity for the molecules of interest and to passivate against all other components in the assay. This is often achieved by introducing non-specifically absorbed proteins (e.g., BSA or casein), or by chemical surface treatments that cover the glass surface. In less demanding situations, bovine serum albumin (BSA) or casein is sufficient to provide a layer of blocking protein to prevent nonspecific binding (see Note 5). When higher specificity is required, the glass surface can be silanized and polyethylene glycol (PEG) can be covalently attached at high density [68, 69]. In the third type of passivation strategy, a supported lipid bilayer self-assembles on the glass surface via vesicle fusion. Under this treatment, the ratio of specifically vs. nonspecifically immobilized molecules can be extremely high (Fig. 6). In these methods, a portion of the passivating molecules carry specific linkers, commonly biotin, so that molecules of interest can be immobilized via these linker molecules. Here, we use a lipid bilayer as our surface passivation. We find that supported lipid bilayers provide far superior blocking than simple protein passivation, and they are more reproducible in our hands than silanization followed by PEGylation.
Figure 6.
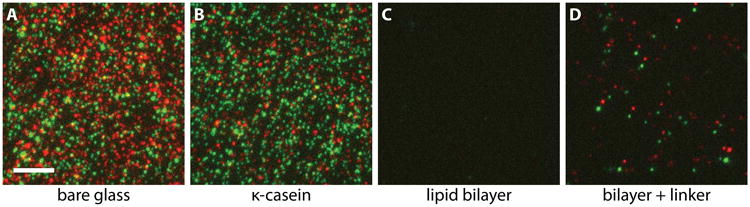
Lipid bilayer blocks nonspecific interactions while allowing kinetochores to be specifically anchored to the coverslip. (A) Kinetochores nonspecifically adsorbed on a clean glass surface at high density. Kinetochore components Mtw1 and Nuf2 that are fluorescently labeled are shown in red and green false colors, respectively. Image color channel registration is not corrected, so that the co-localized particles appear as red-green doublets. (B) The addition of 0.5 mg/ml κ-casein does not block nonspecific interaction to a workable level for single-molecule experiments. (C) A POPC lipid bilayer in coordination with 0.1 mg/ml κ-casein effectively blocks nonspecific adsorption of kinetochores onto the surface. (D) After specific linkers were provided to the same surface in (C), kinetochores were able to anchor specifically via their His-tags. Scale bar, 5 μm.
A unique property of supported lipid bilayers is their fluidity. Unlike linkers on a silanized surface that are covalently attached to the glass substrate, lipid molecules are diffusive within the bilayer, and thus the linked molecules are mobile. This property allows novel experimental designs such as DNA curtains [70], which are dense arrays of DNA molecules each with one end attached to the lipid head groups. Here we introduce a novel method to utilize the fluid nature of the lipid bilayer to study kinetochores structure. In our experimental setup, a kinetochore is dragged on the bilayer surface, thus remaining close to the surface and oriented in the direction of tension applied via an attached microtubule (Fig. 1). Under TIRF microscopy, the locations of multiple kinetochore components are tracked and the intra-kinetochore stretch is quantified (similarly to the “delta measurement”). To accommodate the finite movement range of our piezo-controlled specimen stage, each kinetochore is dragged in a circle instead of linearly. Following this routine, the intra-kinetochore stretch of an individual kinetochore is identified with better than 10 nm accuracy at a temporal resolution of seconds.
3.1.1 Kinetochore and Tubulin Purification
In brief, Saccharomyces cerevisiae (budding yeast) kinetochores are genetically tagged by 6×His and 3×Flag tags on the C-terminus of the kinetochore protein, Dsn1. In addition, SNAP and CLIP tags [71, 72] are fused onto selected kinetochore components. Yeast cells are grown, harvested, and lysed by either a blender, bead-beating, or a freezer mill. Flag tags on Dsn1 are used to purify the kinetochores using anti-Flag antibody coated magnetic beads. During the purification procedure, the fluorescently labeled SNAP- and CLIP-tag ligands are added to label the kinetochores. The kinetochores are then eluted by Flag peptides and stored at -80°C. Details of the purification procedure can be found in [47, 64].
Bovine tubulin can be obtained from commercial sources or purified from cow brains. We followed the protocol in [48] with minor modification to purify bovine tubulin. Calf brains were extracted, cleaned and homogenized at 4°C so that microtubules were forced to depolymerized, followed by ultracentrifugation to clarify the lysate. Then microtubule polymerization was promoted by the addition of glycerol and GTP at 37°C, and the filaments were then pelleted by ultracentrifugation. The resuspended microtubules were cold-treated again and ultracentrifuged. This cycle of polymerization-depolymerization was repeated one more time, and the harvested tubulin was aliquotted and stored at -80°C.
3.1.2 Microbeads Functionalized with a Strong MT-binding Protein
Optically trapped beads are used to apply force to a microtubule that has one end attached to a kinetochore. The beads are functionalized with kinesin so that they bind to the microtubule lattice when brought to the vicinity of the microtubule by the optical tweezers. To conjugate kinesin to the bead surface, we purchased streptavidin-functionalized polystyrene beads (Spherotech SVP-05-10), which were further functionalized by biotinylated kinesin. The kinesin was constructed based on the N-terminal 401-amino-acid motor domain (K401) of Drosophila kinesin [49, 50], and the biotinylation is achieved by an enzymatic ligation [51]. A 15-amino-acid AviTag recognized by biotin ligase BirA and a Flag-tag used for immunopurification are both fused to the K401 gene and transformed into E. coli BL21 (DE3) competent cells (New England BioLabs). A plasmid carrying the ligase gene, pBirA, was transformed into a different strain. Both strains were harvested separately, mixed and lysed with the addition of biotin so that the recombinant kinesin was biotinylated in vitro and purified to 100 mg/ml in BRB80. Note that the kinesin used in this chapter is purified via Flag-tag instead of the more common choice of His-tag because the kinetochores are immobilized on the surface via His-tags and surface bound anti-his antibody. Kinesin should not interact with the surface engineered this way. The following procedure describes how to make 100 μl functionalized beads, enough for approximately 10 tunnel slides.
Dilute 1% w/v streptavidin coated polystyrene beads in 25 mM HEPES buffer to 0.1%. Prepare at least 50 μl and sonicate (across the tube wall) by placing the tube near a flat-tip sonicator or in a bath sonicator immersed in a cold-water bath for 2 minutes at medium power setting. This step breaks most of the bead clumps into individual beads.
Take 2 μl of the sonicated beads, mix with 2 μl 100 μg/ml biotinylated kinesin. Place on rotor at 4°C for 1 hour.
Add 86 μl assay buffer with 10 μM taxol to the 4 μl bead-kinesin mix. Prior to use, briefly sonicate the diluted mixture for 5-10 seconds at medium to low power setting to break bead clumps. The diluted mixture can be kept on ice and remain active for a day. Sonicate when significant clumping occurs.
Dilute scavenger mix and glucose 5-fold in assay buffer separately. Take 8.8 μl diluted bead mix, add 0.2 μl 5 mg/ml κ-casein, 0.5 μl diluted glucose solution and 0.5 μl diluted scavenger mix. Avoid mixing scavenger and glucose much earlier before the slide is ready.
3.1.3 Microtubules
In live cells, kinetochores attach to dynamic spindle microtubules during mitosis and meiosis. In this in vitro experiment, microtubules are polymerized and stabilized by taxol before introduction into the flow chamber, where kinetochores have previously been anchored to the surface. Fluorescent tubulin is helpful to visualize attachment between a kinetochore and a microtubule, in addition to the DIC images.
Mix thawed 6.4 μl 1 mg/ml tubulin with 24.4 μl microtubule polymerization buffer. If fluorescence image is needed, mix 2% fluorescently-labeled tubulin.
Incubate at 37°C for 45 minutes.
Add 200 μl warm BRB80 with 10 μM taxol.
Spin the grown microtubule at 37°C at 100,000 g for 10 minutes in an ultracentrifuge.
Discard the supernatant, and resuspend the pelleted microtubule in BRB80 with 10 μM taxol by vigorous pipetting.
3.1.4 Small Unilamellar Vesicles (SUVs) for Lipid-Based Surface Passivation
Phospholipid small unilamellar vesicles (SUV) can spontaneously form supported bilayers on clean glass substrates by vesicle fusion onto the surface. A supported bilayer very effectively blocks soluble proteins and small molecules from adhering to the surface. A small fraction of the phospholipid can carry biotinylated head groups, so that molecules of interest can be specifically anchored to the surface via biotin-streptavidin interactions at controlled density. To prepare lipid SUVs, a lipid mixture dissolved in chloroform is thoroughly dried to form a lipid cake, followed by rehydration in lipid buffer. The suspension is sonicated with a micro-tip that is immersed directly into 300 μl of fluid to form SUVs. The following procedure describes how to make 300 μl lipid SUVs, enough for approximately 30 tunnel slides.
Add 12 μl 25 mg/ml POPC and 4 μl 0.1 mg/ml biotin-cap-PE to 84 μl chloroform in a glass test tube. The volume of the chloroform does not need to be exact. Mix well by brief vortexing. The mixture contains 0.1% biotinylated lipid. Never use plastic pipettes or syringes to transfer chloroform. Wash syringes 10 times with clean chloroform before and after each use.
Dry the lipid mixture in chloroform by blowing nitrogen over the mixture for 5-10 minutes. Rotate the test tube while drying the mixture until the lipid forms a thin semi-transparent film at the bottom of the tube.
Further dry the lipid cake in a vacuum desiccator without desiccant overnight. POPC lipid cake can be stored in a desiccator for weeks before passivation ability is compromised.
After the lipid cake is thoroughly dried, take the tube out of the desiccator. Add 300 μl lipid buffer, and rehydrate by rigorous vortexing. The lipid suspension should turn cloudy white.
Transfer the rehydrated lipid to a 1.5 ml tube, and mount the tube in a container filled with cooling water. Place the tube under the sonication tip so that the tip is immersed but not touching the tube wall.
Sonicate the lipid suspension for 5 minutes at 50% duty cycle and a low power setting, depending on the specific sonifier. The duty cycle setting prevents constant formation of cavitation bubbles, and helps heat dissipation. The lipid suspension should turn clear after sonication, which suggests that the vesicle size is well below the wavelength of visible light.
Keep the SUVs on ice and they are ready to use. SUVs can be stored at 4°C for days. A 5-minute sonication is recommended before use each day.
3.1.5 Disposable Channel-Slides
The channel slides used in this experiment are the common slides made of coverslips and double-sided tape as spacers. In order for the lipid SUVs to fuse on the surface, the coverslip needs to be cleaned thoroughly. A bench top plasma cleaner is a quick and convenient tool to provide sufficiently clean glass without additional washing.
Clean coverslip and glass slides on glass rack in plasma cleaner for 3 minutes.
Lay rows of double-sided tape perpendicular to the long edge of the cleaned glass slide with approximately 3 mm separation. Place the coverslip on top and briefly press. The volume of the tunnel at this width is approximately 5 ul.
Bake the tunnel slide on 60°C hot plate or oven for 5 minutes. Press to form tight seal between glass and coverslip. The baking step greatly reduces the chance that the coverslip lifts during the following steps.
If multiple tunnels are prepared on one slide, carefully brush a drop of nail polish between tunnels to prevent overflowing to adjacent tunnels in the following steps. Put the slide away until nail polish dries.
3.1.6 Preparing Slides for the Single Kinetochore Imaging and Manipulation Assay
In this step, the SUVs, linker molecules, kinetochores, microtubules and beads prepared above are flowed into the tunnel slide in turn to establish the designed experimental configuration illustrated in Fig. 1. The preparation steps are summarized in Fig. 7.
Feed 5 μl of lipid buffer into the tunnel slide.
Feed 10 μl SUVs into the tunnel slide with vacuum suction on the other opening of the tunnel. Incubate at room temperature for 5 minutes.
Wash the chamber with 35 μl lipid buffer. 5-10 times the volume of the chamber is sufficient to wash away the majority of the solution in the chamber.
Feed 10 μl 0.1 mg/ml avidin DN (see Note 9) into the tunnel slide. Incubate for 5 minutes.
Wash with 35 μl lipid buffer.
Feed 10 μl 20 nM anti-his antibody into the tunnel slide. Incubate for 10 minutes.
Wash with 35 μl lipid buffer.
Flow in tagged kinetochores in assay buffer at proper concentration. Incubate for 10 minutes. We found that 0.3 nM of kinetochore provides a good density of kinetochores on the surface. Incubate for 5 minutes.
Wash with 35 μl assay buffer.
Flow in taxol-stabilized microtubules in assay buffer with 10 μM taxol and 0.1 mg/ml κ-casein. Incubate for 5 minutes. To obtain ideal microtubule lengths, microtubules can be briefly vortexed for 5 seconds.
Without washing away the excess microtubules, very slowly feed in the kinesin-functionalized beads prepared in section 3.1.2. The flow speed should be slow because end-on attached microtubules can be washed away by the shear force. Seal the ends of the chamber with nail polish and mount on the microscope for tweezers manipulation and TIRF imaging.
Figure 7.
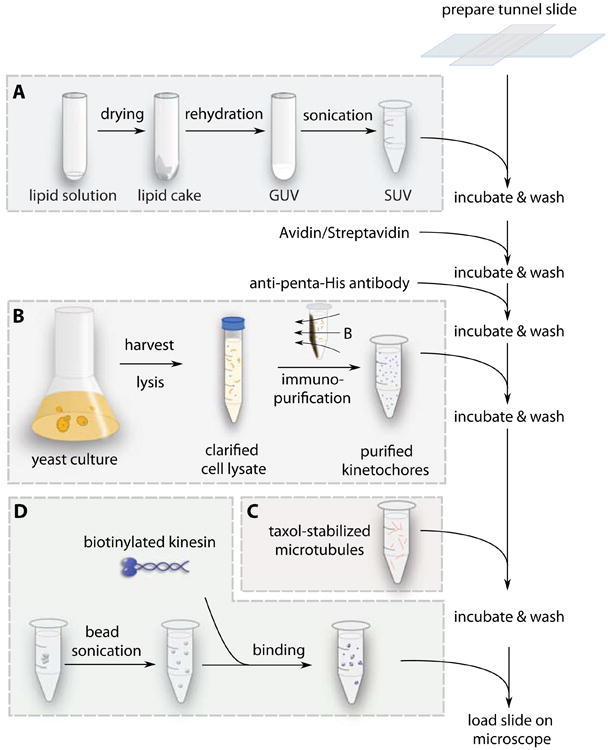
Procedure for sample slide preparation. The boxes summarize four separate procedures that prepare intermediate reagents used sequentially during slide preparation. (A) The lipid solution is dried, rehydrated, and sonicated into SUVs that will create a supported bilayer on a glass substrate. (B) Yeast cells are grown, harvested, and lysed. Kinetochore particles are labeled and immuno-purified from the cell lysate. (C) Microtubules are grown from purified tubulin, and stored in buffer with taxol. (D) Streptavidin coated polystyrene beads are briefly sonicated, and incubated with purified biotinylated kinesin. The functionalized beads are diluted in assay buffer. After the tunnel slides are prepared, lipid SUVs, avidin, anti-penta-His antibody, kinetochores, microtubules, and beads are flowed in sequentially with washes and incubations between each step.
3.1.7 Performing the Single Kinetochore Imaging and Manipulation Assay
Following the procedure described in the previous sections, kinetochores are anchored on the lipid bilayer via specific linkages. Microtubules are then flowed in and allowed to bind to the lipid-anchored kinetochores. The kinesin-functionalized beads are slowly introduced. The slide is mounted on the microscope. The slide is searched to find an end-on attached microtubule on a kinetochore that has fluorescent tags on both Mtw1 and Dam1 in two different color channels. Then the optical trap is switched on to trap a kinesin-functionalized bead nearby. The bead is brought close to the free end of the microtubule until a kinesin-microtubule attachment is established. During the bead manipulation, the fluorescence excitation is turned off and DIC is used to provide visual guidance. Because the transition temperature of the lipid mixture is below room temperature, the kinetochores anchored to the lipid heads remain diffusive on the surface. After the attachments on both ends of the microtubule are established, a customized LabVIEW program moves the piezo stage in a circle with radius larger than the distance between the kinetochore and the bead. The movement speed is set between 0.1 μm/s and 0.5 μm/s, which allows several revolutions of motion before photobleaching. Fluorescence images of the kinetochore components are recorded as it moves over the lipid surface. Because the lipid molecules to which the kinetochore is anchored are dragged through the bilayer, a tensile drag force is transmitted to the kinetochore.
In the laboratory frame of reference, the trapped bead remains nearly stationary, and the lipid surface moves in a circle driven by the piezo stage. The force balance and geometric relations are simpler to understand in the frame attached to the slide, in which the bead moves along a circle, and the kinetochore is dragged behind by the microtubule along a smaller circle (Fig. 8A). The bead velocity v′bead is along the tangential direction of the bead trajectory, but has an angle α with respect to the microtubule. The velocity component parallel to the microtubule v′‖ is equal to the kinetochore velocity v′KT, because the microtubule length l does not change. l and the radii of the two circular trajectories R and r form the three sides of a right triangle, with R2 = l2 + r2. The circular movement of the kinetochore has a phase lag α, behind the bead movement, that is determined by sin(α) = l⁄R. In the lab frame, the bead remains trapped, and all velocities in the slide frame are transformed by subtracting the instantaneous velocity of the bead in the slide frame v′bead = -vstage (Fig. 8B). The kinetochore velocity vkt equals the tangential component of the stage Cvelocity vstage, whereas the normal component of vstage creates an outward drag along the microtubule. Note that in the lab frame, the kinetochore moves with a phase lead π⁄2 - α, ahead of the stage movement. Figure 9A shows a snapshot of the bead, kinetochore and microtubule positions, overlaid by the trajectory of the kinetochore and the stage.
Figure 8.

Velocity diagrams of the trapped bead (gray sphere) and the dragged kinetochore (red dot) in the slide frame and the laboratory frame. (A) In the slide frame where the slide remains stationary, both the bead and the kinetochore move in circles with different radii R and r. The parallel component of the bead velocity v‖ is identical to the kinetochore velocity vKT. (B) In the lab frame, the bead is trapped and stationary. The stage moves in a circle of radius R and drives the kinetochore with it. The tangential component of the instantaneous stage velocity vstage is equal to the kinetochore velocity vKT, i.e. |vKT| = |vstage| cos(π⁄2 - α). The microtubule is depicted as a thick green line, and the bead-microtubule-kinetochore shown semi-transparently represents the configuration at an earlier time point.
Figure 9.
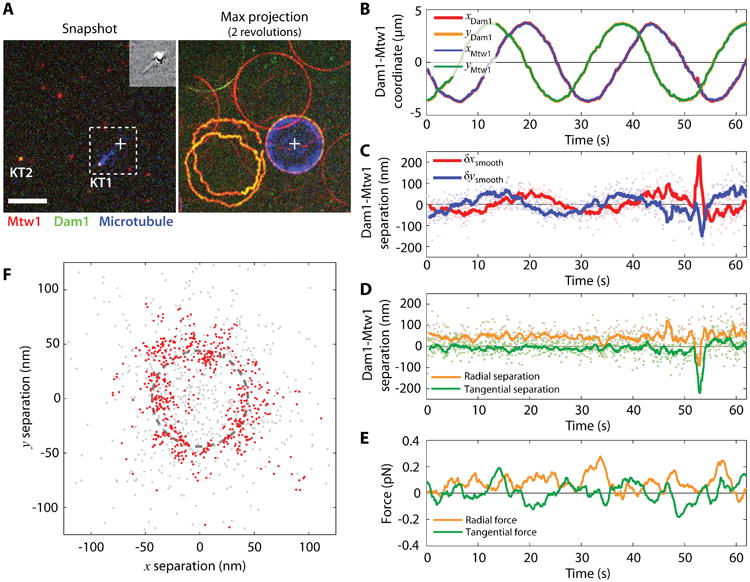
Intra-kinetochore stretch measured by tracking the centroids of kinetochore components as the kinetochore is dragged on the lipid bilayer. (A) One frame of the three-color fluorescence movie (left). Kinetochore components Mtw1, Dam1, and microtubules are pseudo-colored red, green, and blue, respectively. The cross marks the position of the trapped bead. Inset shows the simultaneous DIC image. The maximum projection of two revolutions is shown on the right. KT1 and KT2 mark a dragged kinetochore and a freely diffusive kinetochore, respectively. Note that the circular path of the dragged kinetochore is smaller than the circular path followed by the piezo, as indicated in Fig. 9. Scale bar, 5 μm. (B) The Cartesian coordinates of kinetochore 1 (KT1) Mtw1 and Dam1 indicate circular trajectories. (C) The position of Dam1 relative to Mtw1 also varies sinusoidally, in-phase with the orientation of the microtubule. (D) The parallel (radial) and perpendicular (tangential) components indicate that Dam1 and Mtw1 are separated by 44 ± 33 nm (mean ± standard deviation). (E) The parallel (radial) and perpendicular (tangential) components of the microtubule tension measured by the trapped bead. (F) The positions of Dam1 relative to Mtw1 follow a circle of radius 44 ± 33 nm. Gray points show raw positions and red points show the same data after smoothing with a 1-second window average. Dashed line marks a circle of 44 nm radius.
The centroids of the labeled kinetochore components are obtained from the time-lapse movies of both the Mtw1 and Dam1 channels. The coordinates are corrected according to the registration transformation. Figure 9B shows the kinetochore coordinates as functions of time. Because the kinetochores move along a circle, the coordinates are simple sinusoidal functions. The positions of Dam1 relative to Mtw1, which we chose as a fiducial marker, are calculated from the corrected coordinates, and plotted in Fig. 9C. The relative distance between Dam1 and Mtw1 also appears sinusoidal, and the phase follows the movement of the kinetochore shown in Fig. 9B. In rotating polar coordinates centered at the location of Mtw1 and following the direction of the microtubule, the radial and tangential coordinates of Dam1 are shown in Fig. 9D, with average values of 44 ± 33 nm and -7.5 ± 33 nm (mean ± SD), respectively. If smoothed by a 1 second moving window-average (as indicated by the solid lines), the standard deviation of the estimated mean location of the smoothed trace is 11 nm. Overall, the averaged separation in 45 seconds of data is 44.6 ± 1.8 nm. Force was measured by the bead displacement in the trap. Intra-kinetochore tension generated by the viscous lipid is 0.076 ± 0.003 pN along the radial direction and 0.008 ± 0.003 pN along the tangential direction (Fig. 9E). Note that at the typical speed of the bead moving in buffer, the viscous drag on the bead is below 10 fN (assuming a sphere of 0.5 μm diameter moving at 1 μm⁄s in water). Thus the major contribution to the tension measured by the optical trap is viscous drag associated with moving the kinetochore-lipid anchor through the bilayer. Figure 9F indicates the time projection of the position of Dam1 relative to Mtw1. Raw data (gray points), moving-window smoothed data (red points), and a circle of 44 nm radius are plotted together for comparison.
As a control, the position of Dam1 relative to Mtw1 on an unattached kinetochore is shown in Fig. 10. Because this kinetochore is not oriented to any particular direction, the separation is small and unbiased towards any direction. This negative control also proves that the position registration is accurate. The radial and tangential components of separation are -1.6 ± 1.1 nm and -0.4 ± 1.1 nm (mean ± SD of the mean).
Figure 10.

Un-stretched kinetochores showed no separation between components. (A) The Cartesian coordinates of Mtw1 and Dam1 of kinetochore 2 (KT2 in Fig. 9A). (B, C) The positions of Dam1 relative to Mtw1 in Cartesian (B) and polar (C) coordinates. (D) The positions of Dam1 relative to Mtw1 form a cluster. Gray points show raw positions and red points show the same data after smoothing with a 1-second window average. Dashed line marks a circle of 44 nm radius, as in Figure 9F.
In summary, we demonstrate the construction and application of a combined instrument of optical tweezers with TIRF and DIC microscopy. With this instrument, the distance between two protein components within a single kinetochore particle can be measured with 10 nm precision over one second of observation time, or with better than 2 nm precision over 50 seconds, internally controlled and bias-free. Fluorescence microscopy allowed us to observe the brightness and location of specifically tagged components, while the protein complex was simultaneously manipulated by optical tweezers at controllable and known force. DIC microscopy was used to avoid photobleaching when fluorescence images were not recorded, providing visual guidance to establish the correct experimental configuration and to monitor the progress of the dragging experiment.
The surface treatment selectively immobilized our protein complex of interest, while blocking all other components of the experiment such as microtubules, beads, and incomplete kinetochore assemblies. We adopted supported lipid bilayers as the surface passivation treatment, a less common method than silanization followed by PEGylation. Compared to PEGylation, we find that a lipid bilayer is considerably easier to prepare, and in our hands it provides a more reliable and cost-effective passivation. It also offers flexibility through the incorporation of different functionalized lipids. In the demonstration of this chapter, we utilized the fluidity property of the bilayer to mitigate possible errors due to color channel mis-registration. In principle, the viscosity of the lipid could be tuned by introducing various species of lipids to generate different forces. Alternatively, a lipid with transition temperature above room temperature could be used to create a gel-phase bilayer to more firmly immobilize the anchored molecules.
We have also introduced an addition to the combined instrument that incorporates focus detection into the existing setup. The addition shares the detection beam with the trapping laser, but converts it to a total-internal reflection configuration with a few extra, simple optical components. Axial position is converted to translational beam position, collected by a position-sensitive photodetector, and digitized. This position signal can be used to compensate focal drift during long-term imaging, or to execute a programmed axial scan for optical sectioning. Because a laser source, a data acquisition device, and a nano-positioning stage are usually standard components of an optical tweezer setup, the addition of focus detection is inexpensive and easy to implement.
The experimental platform we describe above is suitable for studying nanometer-scale structural changes in filament-binding complexes under tension. Our instrument was constructed with the goal of studying kinetochores, but we envision that it could also be useful for studying other cytoskeletal junctions as well. Kinetochores undergo conformational changes that are suspected to play important roles in spindle-assembly checkpoint signaling. It is not yet clear whether the changes in intra-kinetochore distance observed in vivo are caused by changes in kinetochore tension, by changes in kinetochore-microtubule attachment status, or by more complex signaling events [73]. Our single kinetochore imaging and manipulation assay represents a new, direct way of testing whether tension alone can induce structural changes in kinetochores, and whether the spindle assembly checkpoint system responds to such changes. The approach might also be generally useful for studying other protein complexes that associate with cytoskeletal filaments, such as the centrosomes and yeast spindle pole bodies that anchor spindle microtubules, the desmosomes that provide cell-to-cell adhesions, the LINC complexes that connect cytoskeletal to nucleoskeletal filaments, or the focal adhesion complexes that underlie cellular mechanosensation.
4 Notes
Strictly speaking, the distribution for the random relative position is the convolution of two uniform distributions with a boundary of the size of the FOV. In practice, it is approximated well by a two-dimensional normal distribution with a covariance matrix equal to the identity matrix multiplied by a constant proportional to the square of the FOV size.
Expectation-maximization is a powerful machine leaning algorithm that performs clustering in this case. The association of each pair of spots with the two clusters is unknown, and the shape and location of the clusters are also unknown. In the expectation-maximization algorithm, initial guesses of the cluster shape and location are given, followed by the clustering of pairs according to these guesses. The cluster shape and location are then updated using the clustering results. This process iterates until convergence is achieved.
Ideally, a Faraday optical isolator should be installed first to prevent back reflection from the surfaces.
In our setup, L9 is conjugated with the back-focal plane of the objective lens. But the back aperture is very close to the back-focal plane (in fact, the focal point is inside the objective lens), and the back aperture is slightly overfilled. The difference in beam profiles between the back aperture and the back-focal point is insignificant.
The role of casein in surface treatment is largely unclear. While casein is widely used as a passivation protein, whole casein is also frequently used in kinesin-microtubule gliding assays, and different casein components provide variable support. In our study, we found that κ-casein, a component of whole casein, provided the best passivation result. However, the presence of κ-casein severely impaired or even completely abolished the kinesin-microtubule interaction. While it is tempting to supplement the final assay buffer with a high amount of casein to prevent bead clumping, the bead-microtubule attachment tolerates the presence of only a small amount of κ-casein.
We used the K401 construct of drosophila kinesin, which will exhibit motility in the presence of ATP or GTP. This is not a concern in the demonstration, but one can implement the rigor mutation on kinesin when ATP or GTP will be present.
Microtubules are grown in PIPES based BRB80 buffer. The final dragging assay is performed in a different buffer based on HEPES. We found that PIPES buffer greatly reduced the interaction between the microtubules and kinetochores, as well as the kinetochore-associated motor protein, Cin8. The exact mechanism of this inhibition has not been further investigated. Microtubules cannot be stored stably in HEPES-based buffer over the time scale of hours, even in the presence of taxol. Therefore, prior to each experiment, microtubules were either freshly grown or stored in BRB80 with taxol (where they can remain stably assembled), pelleted and resuspended in the assay buffer.
The vacuum pump used for the plasma cleaner can also be used for drying lipid cake.
We find that streptavidin and avidin DN work indistinguishably. However, neutral avidin has a non-specific interaction with microtubules and should therefore be avoided.
References
- 1.Neuman KC, Nagy A. Single-molecule force spectroscopy: optical tweezers, magnetic tweezers and atomic force microscopy. Nat Methods. 2008;5:491–505. doi: 10.1038/nmeth.1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moffitt JR, Chemla YR, Smith SB, et al. Recent advances in optical tweezers. Annu Rev Biochem (Mosc) 2008;77:205–228. doi: 10.1146/annurev.biochem.77.043007.090225. [DOI] [PubMed] [Google Scholar]
- 3.Kapanidis AN, Strick T. Biology, one molecule at a time. Trends Biochem Sci. 2009;34:234–243. doi: 10.1016/j.tibs.2009.01.008. [DOI] [PubMed] [Google Scholar]
- 4.Lang MJ, Asbury CL, Shaevitz JW, et al. An automated two-dimensional optical force clamp for single molecule studies. Biophys J. 2002;83:491–501. doi: 10.1016/S0006-3495(02)75185-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blehm BH, Selvin PR. Single-molecule fluorescence and in vivo optical traps: How multiple dyneins and kinesins interact. Chem Rev. 2014;114:3335–3352. doi: 10.1021/cr4005555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Korten T, Nitzsche B, Gell C, et al. Fluorescence imaging of single kinesin motors on immobilized microtubules. Methods Mol Biol. 2011;783:121–137. doi: 10.1007/978-1-61779-282-3_7. [DOI] [PubMed] [Google Scholar]
- 7.Nicholas MP, Rao L, Gennerich A. An improved optical tweezers assay for measuring the force generation of single kinesin molecules. Methods Mol Biol. 2014;1136:171–246. doi: 10.1007/978-1-4939-0329-0_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gennerich A, Reck-Peterson SL. Probing the force generation and stepping behavior of cytoplasmic dynein. Methods Mol Biol. 2011;783:63–80. doi: 10.1007/978-1-61779-282-3_4. [DOI] [PubMed] [Google Scholar]
- 9.Vanzi F, Capitanio M, Sacconi L, et al. New techniques in linear and non-linear laser optics in muscle research. J Muscle Res Cell Motil. 2006;27:469–479. doi: 10.1007/s10974-006-9084-3. [DOI] [PubMed] [Google Scholar]
- 10.Ketelaar T, van der Honing HS, Emons AM. Probing cytoplasmic organization and the actin cytoskeleton of plant cells with optical tweezers. Biochem Soc Trans. 2010;38:823–828. doi: 10.1042/BST0380823. [DOI] [PubMed] [Google Scholar]
- 11.Sommese RF, Sung J, Nag S, et al. Molecular consequences of the R453C hypertrophic cardiomyopathy mutation on human β-cardiac myosin motor function. Proc Natl Acad Sci U S A. 2013;110:12607–12612. doi: 10.1073/pnas.1309493110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mehta AD, Rief M, Spudich JA, et al. Single-molecule biomechanics with optical methods. Science. 1999;283:1689–1695. doi: 10.1126/science.283.5408.1689. [DOI] [PubMed] [Google Scholar]
- 13.Abbondanzieri EA, Greenleaf WJ, Shaevitz JW, et al. Direct observation of base-pair stepping by RNA polymerase. Nature. 2005;438:460–465. doi: 10.1038/nature04268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Herbert KM, Greenleaf WJ, Block SM. Single-molecule studies of RNA polymerase: motoring along. Annu Rev Biochem. 2008;77:149–176. doi: 10.1146/annurev.biochem.77.073106.100741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Perkins TT. Ångström-precision optical traps and applications. Annu Rev Biophys. 2014;43:279–302. doi: 10.1146/annurev-biophys-042910-155223. [DOI] [PubMed] [Google Scholar]
- 16.Heller I, Hoekstra TP, King GA, et al. Optical tweezers analysis of DNA– protein complexes. Chem Rev. 2014;114:3087–3119. doi: 10.1021/cr4003006. [DOI] [PubMed] [Google Scholar]
- 17.Padawer J. The Nomarski interference-contrast microscope: an experimental basis for image interpretation. J R Microsc Soc. 1968;88:305–349. doi: 10.1111/j.1365-2818.1968.tb00616.x. [DOI] [PubMed] [Google Scholar]
- 18.Allen RD, Allen NS, Travis JL. Video-enhanced contrast, differential interference contrast (AVEC-DIC) microscopy: A new method capable of analyzing microtubule-related motility in the reticulopodial network of Allogromia laticollaris. Cell Motil. 1981;1:291–302. doi: 10.1002/cm.970010303. [DOI] [PubMed] [Google Scholar]
- 19.Walker RA, O'Brien ET, Pryer NK, et al. Dynamic instability of individual microtubules analyzed by video light microscopy: rate constants and transition frequencies. J Cell Biol. 1988;107:1437–1448. doi: 10.1083/jcb.107.4.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Elting MW, Spudich JA. Future challenges in single-molecule fluorescence and laser trap approaches to studies of molecular motors. Dev Cell. 2012;23:1084–1091. doi: 10.1016/j.devcel.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Joo C, Balci H, Ishitsuka Y, et al. Advances in single-molecule fluorescence methods for molecular biology. Annu Rev Biochem. 2008;77:51–76. doi: 10.1146/annurev.biochem.77.070606.101543. [DOI] [PubMed] [Google Scholar]
- 22.Swoboda M, Grieb MS, Hahn S, et al. Measuring two at the same time: combining magnetic tweezers with single-molecule FRET. EXS. 2014;105:253–276. doi: 10.1007/978-3-0348-0856-9_12. [DOI] [PubMed] [Google Scholar]
- 23.Savinov A, Perez CF, Block SM. Single-molecule studies of riboswitch folding. Biochim Biophys Acta. 2014;1839:1030–1045. doi: 10.1016/j.bbagrm.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hohng S, Lee S, Lee J, et al. Maximizing information content of single-molecule FRET experiments: multi-color FRET and FRET combined with force or torque. Chem Soc Rev. 2014;43:1007–1013. doi: 10.1039/c3cs60184f. [DOI] [PubMed] [Google Scholar]
- 25.Salmon E, Tran P. High-resolution video-enhanced differential interference contrast (VE-DIC) light microscopy. Methods Cell Biol. 1998:153–185. doi: 10.1016/s0091-679x(08)60426-4. [DOI] [PubMed] [Google Scholar]
- 26.Grishchuk EL, Molodtsov MI, Ataullakhanov FI, et al. Force production by disassembling microtubules. Nature. 2005;438:384–388. doi: 10.1038/nature04132. [DOI] [PubMed] [Google Scholar]
- 27.Driver JW, Powers AF, Sarangapani KK, et al. Measuring kinetochore-microtubule interaction in vitro. Methods Enzymol. 2014;540:321–337. doi: 10.1016/B978-0-12-397924-7.00018-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yamagishi Y, Sakuno T, Goto Y, et al. Kinetochore composition and its function: lessons from yeasts. FEMS Microbiol Rev. 2014;38:185–200. doi: 10.1111/1574-6976.12049. [DOI] [PubMed] [Google Scholar]
- 29.Cheerambathur DK, Desai A. Linked in: formation and regulation of microtubule attachments during chromosome segregation. Curr Opin Cell Biol. 2014;26:113–122. doi: 10.1016/j.ceb.2013.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.London N, Biggins S. Signalling dynamics in the spindle checkpoint response. Nat Rev Mol Cell Biol. 2014;15:736–748. doi: 10.1038/nrm3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gorbsky GJ. The spindle checkpoint and chromosome segregation in meiosis. FEBS J. 2015;282:2471–87. doi: 10.1111/febs.13166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nicklas RB. How cells get the right chromosomes. Science. 1997;275:632–637. doi: 10.1126/science.275.5300.632. [DOI] [PubMed] [Google Scholar]
- 33.Pinsky BA, Biggins S. The spindle checkpoint: tension versus attachment. Trends Cell Biol. 2005;15:486–493. doi: 10.1016/j.tcb.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 34.Joglekar AP, Bloom KS, Salmon E. Mechanisms of force generation by end-on kinetochore-microtubule attachments. Curr Opin Cell Biol. 2010;22:57–67. doi: 10.1016/j.ceb.2009.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wan X, O'Quinn RP, Pierce HL, et al. Protein architecture of the human kinetochore microtubule attachment site. Cell. 2009;137:672–684. doi: 10.1016/j.cell.2009.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Joglekar AP, Bloom K, Salmon ED. In vivo protein architecture of the eukaryotic kinetochore with nanometer scale accuracy. Curr Biol. 2009;19:694–699. doi: 10.1016/j.cub.2009.02.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maresca TJ, Salmon ED. Intrakinetochore stretch is associated with changes in kinetochore phosphorylation and spindle assembly checkpoint activity. J Cell Biol. 2009;184:373–381. doi: 10.1083/jcb.200808130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dumont S, Salmon ED, Mitchison TJ. Deformations within moving kinetochores reveal different sites of active and passive force generation. Science. 2012;337:355–358. doi: 10.1126/science.1221886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Neuman KC, Block SM. Optical trapping. Rev Sci Instrum. 2004;75:2787–2809. doi: 10.1063/1.1785844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rohrbach A. Stiffness of optical traps: quantitative agreement between experiment and electromagnetic theory. Phys Rev Lett. 2005;95:168102. doi: 10.1103/PhysRevLett.95.168102. [DOI] [PubMed] [Google Scholar]
- 41.Dempsey GT, Wang W, Zhuang X. Handbook of single-molecule biophysics. Springer; 2009. Fluorescence imaging at sub-diffraction-limit resolution with stochastic optical reconstruction microscopy; pp. 95–127. [Google Scholar]
- 42.Churchman LS, Okten Z, Rock RS, et al. Single molecule high-resolution colocalization of Cy3 and Cy5 attached to macromolecules measures intramolecular distances through time. Proc Natl Acad Sci U S A. 2005;102:1419–1423. doi: 10.1073/pnas.0409487102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pertsinidis A, Zhang Y, Chu S. Subnanometre single-molecule localization, registration and distance measurements. Nature. 2010;466:647–651. doi: 10.1038/nature09163. [DOI] [PubMed] [Google Scholar]
- 44.Berg-Sørensen K, Flyvbjerg H. Power spectrum analysis for optical tweezers. Rev Sci Instrum. 2004;75:594–612. [Google Scholar]
- 45.Shaevitz JW. A practical guide to optical trapping. [Accessed 11 January 2016];2006 http://genomics.princeton.edu/shaevitzlab/OT_Practicle_Guide.pdf.
- 46.Tolić-Nørrelykke SF, Schäffer E, Howard J, et al. Calibration of optical tweezers with positional detection in the back focal plane. Rev Sci Instrum. 2006;77:103101. [Google Scholar]
- 47.Akiyoshi B, Sarangapani KK, Powers AF, et al. Tension directly stabilizes reconstituted kinetochore-microtubule attachments. Nature. 2010;468:576–579. doi: 10.1038/nature09594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Castoldi M, Popov AV. Purification of brain tubulin through two cycles of polymerization–depolymerization in a high-molarity buffer. Protein Expr Purif. 2003;32:83–88. doi: 10.1016/S1046-5928(03)00218-3. [DOI] [PubMed] [Google Scholar]
- 49.Gilbert SP, Johnson KA. Expression, purification, and characterization of the Drosophila kinesin motor domain produced in Escherichia coli. Biochemistry (Mosc) 1993;32:4677–4684. doi: 10.1021/bi00068a028. [DOI] [PubMed] [Google Scholar]
- 50.Young EC, Berliner E, Mahtani HK, et al. Subunit interactions in dimeric kinesin heavy chain derivatives that lack the kinesin rod. J Biol Chem. 1995;270:3926–3931. doi: 10.1074/jbc.270.8.3926. [DOI] [PubMed] [Google Scholar]
- 51.de Boer E, Rodriguez P, Bonte E, et al. Efficient biotinylation and single-step purification of tagged transcription factors in mammalian cells and transgenic mice. Proc Natl Acad Sci U S A. 2003;100:7480–7485. doi: 10.1073/pnas.1332608100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cheeseman IM, Drubin DG, Barnes G. Simple centromere, complex kinetochore: linking spindle microtubules and centromeric DNA in budding yeast. J Cell Biol. 2002;157:199–203. doi: 10.1083/jcb.200201052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cheeseman IM. The kinetochore. Cold Spring Harb Perspect Biol. 2014;6:a015826. doi: 10.1101/cshperspect.a015826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Franck AD, Powers AF, Gestaut DR, et al. Tension applied through the Dam1 complex promotes microtubule elongation providing a direct mechanism for length control in mitosis. Nat Cell Biol. 2007;9:832–837. doi: 10.1038/ncb1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Joglekar AP, Salmon ED, Bloom KS. Counting kinetochore protein numbers in budding yeast using genetically encoded fluorescent proteins. Methods Cell Biol. 2008;85:127–151. doi: 10.1016/S0091-679X(08)85007-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Aravamudhan P, Felzer-Kim I, Gurunathan K, et al. Assembling the protein architecture of the budding yeast kinetochore-microtubule attachment using FRET. Curr Biol. 2014;24:1437–1446. doi: 10.1016/j.cub.2014.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Suzuki A, Badger BL, Wan X, et al. The architecture of CCAN proteins creates a structural integrity to resist spindle forces and achieve proper intrakinetochore stretch. Dev Cell. 2014;30:717–730. doi: 10.1016/j.devcel.2014.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brinkley B, Stubblefield E. The fine structure of the kinetochore of a mammalian cell in vitro. Chromosoma. 1966;19:28–43. doi: 10.1007/BF00332792. [DOI] [PubMed] [Google Scholar]
- 59.DeLuca JG, Dong Y, Hergert P, et al. Hec1 and Nuf2 are core components of the kinetochore outer plate essential for organizing microtubule attachment sites. Mol Biol Cell. 2005;16:519–531. doi: 10.1091/mbc.E04-09-0852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gonen S, Akiyoshi B, Iadanza MG, et al. The structure of purified kinetochores reveals multiple microtubule-attachment sites. Nat Struct Mol Biol. 2012;19:925–929. doi: 10.1038/nsmb.2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Asbury CL, Gestaut DR, Powers AF, et al. The Dam1 kinetochore complex harnesses microtubule dynamics to produce force and movement. Proc Natl Acad Sci U S A. 2006;103:9873–9878. doi: 10.1073/pnas.0602249103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Powers AF, Franck AD, Gestaut DR, et al. The Ndc80 kinetochore complex forms load-bearing attachments to dynamic microtubule tips via biased diffusion. Cell. 2009;136:865–875. doi: 10.1016/j.cell.2008.12.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sarangapani KK, Akiyoshi B, Duggan NM, et al. Phosphoregulation promotes release of kinetochores from dynamic microtubules via multiple mechanisms. Proc Natl Acad Sci U S A. 2013;110:7282–7287. doi: 10.1073/pnas.1220700110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sarangapani KK, Duro E, Deng Y, et al. Sister kinetochores are mechanically fused during meiosis I in yeast. Science. 2014;346:248–251. doi: 10.1126/science.1256729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Musacchio A. The molecular biology of spindle assembly checkpoint signaling dynamics. Curr Biol. 2015;25:R1002–R1018. doi: 10.1016/j.cub.2015.08.051. [DOI] [PubMed] [Google Scholar]
- 66.Maresca TJ, Salmon E. Welcome to a new kind of tension: translating kinetochore mechanics into a wait-anaphase signal. J Cell Sci. 2010;123:825–835. doi: 10.1242/jcs.064790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang Y, Jin F, Higgins R, et al. The current view for the silencing of the spindle assembly checkpoint. Cell Cycle. 2014;13:1694–1701. doi: 10.4161/cc.29027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sofia SJ, Premnath V, Merrill EW. Poly (ethylene oxide) grafted to silicon surfaces: grafting density and protein adsorption. Macromolecules. 1998;31:5059–5070. doi: 10.1021/ma971016l. [DOI] [PubMed] [Google Scholar]
- 69.Roy R, Hohng S, Ha T. A practical guide to single-molecule FRET. Nat Methods. 2008;5:507–516. doi: 10.1038/nmeth.1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Collins BE, Ye LF, Duzdevich D, et al. DNA curtains: Novel tools for imaging protein-nucleic acid interactions at the single-molecule level. Methods Cell Biol. 2013;123:217–234. doi: 10.1016/B978-0-12-420138-5.00012-4. [DOI] [PubMed] [Google Scholar]
- 71.Keppler A, Gendreizig S, Gronemeyer T, et al. A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat Biotechnol. 2003;21:86–89. doi: 10.1038/nbt765. [DOI] [PubMed] [Google Scholar]
- 72.Gautier A, Juillerat A, Heinis C, et al. An engineered protein tag for multiprotein labeling in living cells. Chem Biol. 2008;15:128–136. doi: 10.1016/j.chembiol.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 73.McEwen BF, Dong Y. Releasing the spindle assembly checkpoint without tension. J Cell Biol. 2009;184:355–356. doi: 10.1083/jcb.200812016. [DOI] [PMC free article] [PubMed] [Google Scholar]