

Abstract
Primary vesicoureteral reflux (VUR) is a common pediatric condition due to a developmental defect in the ureterovesical junction. The prevalence of VUR among individuals with connective tissue disorders, as well as the importance of the ureter and bladder wall musculature for the anti-reflux mechanism, suggest that defects in the extracellular matrix (ECM) within the ureterovesical junction may result in VUR. This review will discuss the function of the smooth muscle and its supporting ECM microenvironment with respect to VUR, and explore the association of VUR with mutations in ECM-related genes.
Keywords: Vesicoureteral reflux, Ureterovesical junction, Collagen, Elastin, Marfan syndrome, Williams syndrome, Cutis laxa, Ehlers–Danlos syndrome, Joint hypermobility, Tenascin-XB
Introduction
The ureterovesical junction (UVJ) is a critical structure in the urinary tract. It protects the low-pressure upper urinary tract from the intermittent high pressure in the bladder. The UVJ allows passage of urine into the bladder and prevents retrograde flow towards the kidneys when it is transiently occluded during micturition. Occlusion of the UVJ requires an adequate length to the distal end of the ureter-the intravesical ureter (IVU), an oblique angle of ureter entry into the bladder, and a properly developed smooth muscle and extracellular matrix microenvironment capable of compressing the ureteral orifice. Abnormalities in any of these elements, or in the three-dimensional structure of the bladder that encompasses these elements, results in backward flow of urine towards the upper urinary tract—vesicoureteral reflux (VUR). A specific example of the importance of the bladder can be seen in the Hutch bladder diverticulum that arises in the IVU and co-occurs with VUR.
Clinical spectrum of VUR and reflux nephropathy
VUR is a congenital urinary tract defect associated with the anomalous development of the UVJ, and is observed in ~1 % of general population, and in at least 30 % of children presenting with urinary tract infection [1–3]. However, the prevalence of VUR is likely underreported due to the invasive nature of the test used for diagnosis: the voiding cystourethrogram. VUR is highly heritable and a number of genes and susceptibility loci have been identified [3]. In addition to the genetic heterogeneity of the disorder, it is also phenotypically heterogeneous and loosely classified as primary and secondary VUR. Primary VUR is due to a defect in the intravesical ureter such that it is not occluded during voiding. Secondary VUR occurs as a result of elevated intravesical pressure that is typically seen in the presence of anatomical bladder obstruction (posterior urethral valves) or functional bladder obstruction (neurogenic bladder).
Both primary and secondary VUR can occur as an isolated finding, or as part of a syndrome. Syndromic VUR is the presence of VUR with other congenital and multiorgan defects including congenital anomalies of the kidney and the urinary tract (CAKUT) and hereditary connective tissue disorders (Table 1). The fact that VUR is observed in some connective tissue disorders suggests that derangements in extracellular matrix (ECM) may impair the function of the UVJ and/or the bladder. This is particularly of interest because of our recent findings that mutations in an ECM gene, tenascin XB (TNXB), result in familial and sporadic cases of VUR [4, 5].
Table 1.
Extracellular matrix syndromes with VUR as a urinary tract phenotype
| Syndrome | Inheritance | Gene | Characteristic features | Renal and urinary tract phenotypes |
|---|---|---|---|---|
| Cutis laxa | Autosomal dominant, Autosomal recessive | ELN FBLN4, FBLN5 ATP6V0A2, ATP7A, EFEMP2 | Lax and inelastic skin, vascular anomalies, gastrointestinal diverticula, abdominal hernia, genital prolapse | VUR, bladder diverticula |
| Ehlers–Danlos | Autosomal dominant, Autosomal recessive | ADAMTS2, COL-I-A1, COL-I-A2, COL-III-A1, COL-V-A1, COL-V-A2, PLOD1, TNXB | Hyperextensible skin, joint hypermobility, poor wound healing, easy bruising and scarring, molluscoid pseudotumors, subcutaneous spheroids, muscle hypotonia | VUR, bladder diverticula |
| Marfan | Autosomal dominant | FBN1 | Increased height, disproportionately long limbs and digits, anterior chest deformity, joint laxity, vertebral column deformity, highly arched palate | VUR, bladder diverticula |
| Williams | Autosomal dominant | ELN | Cardiovascular defects, mental retardation, joint, skin, and facial abnormalities | VUR, bladder diverticula, Renal artery stenosis, agenesis, ectopia |
VUR vesicoureteral reflux
In severe cases of VUR, recurrent pyelonephritis leads to parenchymal injury and fibrotic scarring of the kidney, known as reflux nephropathy (RN) [6–9]. Congenital reflux nephropathy has also been coined and refers to RN in the context of VUR with a congenital renal malformation, and is often observed in the absence of pyelonephritis. RN is a major cause of end-stage renal disease (ESRD), accounting for 10–15 % of all children who require long-term dialysis and renal transplantation [10, 11]. RN is defined histologically by the presence of interstitial infiltration with chronic inflammatory cells, tubular basement membrane thickening, tubular cell atrophy with dilation of tubules, medial and intimal thickening of arteries and arterioles, and fibrosis surrounding glomeruli and tubules [12–14]. The fibrosis is mediated by myofibroblasts that appear to arise from a diverse range of precursor cell types including epithelial cells, endothelial cells, smooth muscle cells, and pericytes, and in response to inflammatory cytokines such as transforming growth factor-β1 (TGF-β1) [15, 16]. It is worth mentioning that while a number of fate-mapping studies have been conducted to elucidate the source of myofibroblasts in renal fibrosis, this has not been done in a model of RN. TGF-β1 is a key mediator in the pathogenesis of renal fibrosis and is known to be released by myofibroblasts, tubular epithelial cells, as well as activated leukocytes in response to infection [15, 17–23]. TGF-β1 mediates the renal fibrotic response in many ways: it stimulates excessive production of ECM, it impairs degradation of ECM, and it induces epithelial-mesenchymal transition (EMT) to generate fibroblasts during progression of fibrosis. Interestingly, a recent study has shown that TNXB can promote EMT when its C-terminus binds to the TGF-β1 latency complex, resulting in the activation of TGF-β1 signaling (Fig. 1b) [22, 24, 25].
Fig. 1.
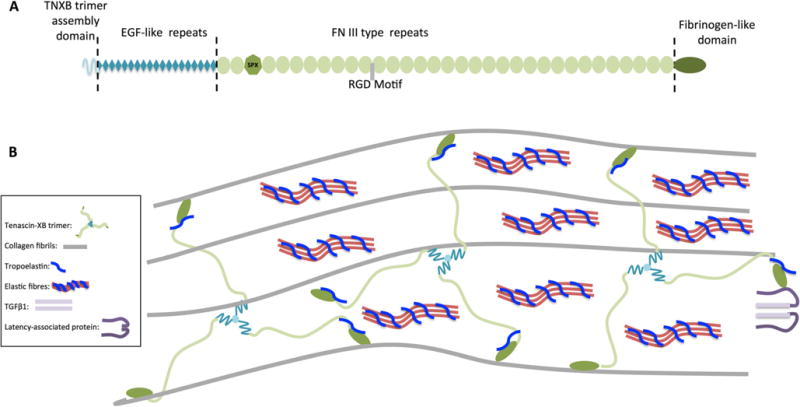
a Schematic representation of human tenascin-XB (TNXB) protein monomer and its major domains. SPX refers to a serine/prolinerich region in the FN III repeats thought to be important in phosphate homeostasis. RGD motif refers to the tri-peptide arginine, glycine, and asparagine sequence known to be a cell attachment site. b Model of interaction of TNXB with various ECM components. TNXB exists as a trimeric supramolecule and interacts with multiple components of the ECM. It has been shown to interact with fibrillary collagens through its EGF-like domains, FN III repeats, and FBG-like domain. At its C-terminus, TNXB interacts with tropoelastin, the major component of elastic fibers. TNXB modulates bioavailability of TGFβ1 (light purple) through interaction with the latency-associated peptide
Natural history of VUR
Longitudinal studies have shown that ~50–65 % of cases of non-syndromic primary VUR undergo spontaneous resolution with age [26–29]. Not much is known about the mechanism leading to this resolution, however the elongation of the intravesical ureter (0.5 cm at birth vs. 1.5–2.5 cm in adulthood) due to somatic growth has been offered as an explanation for the improved function of the UVJ [30–32]. Alternatively, morphological changes in the bladder and ureter smooth muscle layer and its supporting ECM microenvironment could be the basis for why VUR resolves over time.
Histology of the UVJ in VUR
The UVJ in humans has three distinct layers: the urothelium, the lamina propria, and the musculature layer. The urothelium is the innermost mucosal layer of the UVJ, and consists of a highly elastic and impermeable transitional epithelium. The urothelium includes the umbrella cell, the intermediate, and the basal cell layer [33, 34]. A single layer of multinucleated hexagonal-shaped cells, known as the umbrella cells, is directly exposed to urine and contributes to structural plaques seen by scanning electron microscopy. The umbrella cells express a specialized multi-protein complex of transmembrane glycoproteins known as uroplakins. Their function is not yet fully understood, however they maintain the permeability barrier of the uroepithelium, they regulate bladder contractions, and they play a role in closure of the UVJ [33, 34]. Glycosaminoglycans (GAGs) are ECM components that cover the apical surface of the urothelium and create a water barrier due to their high affinity to water. The GAG layer prevents the adherence of ions, proteins, microcrystals, and bacteria to the epithelium [35–38]. Below the urothelium is a thin layer of loose connective tissue known as the lamina propria. This layer is highly compressible and rich in afferent and efferent nerve endings as well as fibrillary collagens. Moving basally, smooth muscle fascicles are detected towards the outer edges of lamina propria; they then fuse to form a well-differentiated smooth muscle (SM) layer. The SM layer is uniform in thickness, and is longitudinal, unlike the rest of the ureter that contains both circular and longitudinal muscle bundles [39, 40]. The longitudinal muscles at the UVJ intercalate with the inner longitudinal muscle fibers of the detrusor. A combination of these fibers and the middle circular, and outer longitudinal muscle fibers of the detrusor form the roof of the UVJ. In contrast, the floor of the UVJ is made only from the inner circular and outer longitudinal layers of the detrusor. The specific arrangement of the detrusor and ureter muscle bundles is vital in providing firm support to the intravesical ureter and in preventing VUR [32, 41].
Histological studies of resected segments of UVJs from patients with reflux reveal the presence of severe atrophy and degeneration in the musculature with replacement by collagenous matrix. This perturbs the muscle: collagen ratio from the normal 1:0.3 to 1:3 [42–47]. The marked increase in the amount of collagen present is predicted to increase the stiffness of the SM layer and impair the occlusion of the UVJ. The ECM undergoes dynamic changes in its structure and composition as part of normal tissue homeostasis and repair, a process known as ECM turnover. As an example, collagen deposition is regulated by de novo synthesis and proteolytic degradation via a group of enzymes known as matrix metalloproteinases (MMPs). In states of health, the activity level of MMPs is low, while in disease and inflammatory states, increased activity is observed [48–50]. UVJs from patients with VUR show signs of pathological changes in the ECM as indicated by an increase in the expression of MMPs and the profibrotic cytokine TGF-β1 [45]. The same study noted that the expression levels of a number of growth factors critical to the growth and homeostasis of the SM layer (i.e., vascular endothelial growth factor (VEGF), insulin-like growth factor (IGF), and nerve growth factor (NGF)) were significantly lower in refluxing UVJs as shown by immunohistochemistry [45]. Other ECM changes that were noted included an increase in the expression of a tenascin, Tenascin C, fibronectin, and tetranectin within the smooth muscle layer of refluxing UVJs compared to control UVJs [45]. Refluxing UVJs show a decrease in density of capillaries due to a reported decrease in VEGF expression [44].
Surgical treatment of VUR
Ureteric re-implantation surgery and injection of a bulking compound in the submucosal layer at the orifice of the ureter are surgical techniques that correct VUR. The objective of ureteral re-implantation surgery is to lengthen the intravesical ureter. Introduction of bulking agents into the UVJ involves submucosal injection of biocompatible bulking agents including collagen is cross-linked with glutaraldehyde for injection, autologous fat, silicone, chondrocytes, and most commonly dextranomer/hyaluronic acid (Dex/HA). It is hypothesized that these compounds prevent VUR by elevating and narrowing the ureteral orifice. Post-injection, histological studies of human and animal UVJ segments show that both Dex/HA and collagen implants lead to histopathological changes in the ECM [51–57]. These changes include fibroblast invasion, collagen deposition, an increase in α-smooth muscle actin (α-SMA) surrounding the Dex/HA microspheres, and a foreign-body inflammatory response (granulation). The changes in the ECM microenvironment are predicted to increase the tensile forces at the UVJ and prevent VUR in addition to the structural narrowing of the ureteral orifice that is created by the presence of the bulking agent.
Molecular composition and biomechanical properties of the ECM
The biomechanical properties of a tissue dictate its response to internal or external forces and are specified by the composition of the extracellular matrix (ECM) microenvironment. Three important biomechanical properties are tensile strength, compliance, and recoil. Tensile strength refers to a tissue’s resistance to deformation, tearing or rupture when distended (i.e., stretched), and is defined by the amount, the organization, and the types of collagen that are present. Compliance is the ability of a tissue to distend with pressure, and is inversely related to a tissue’s tensile strength and stiffness. Recoil refers to a tissue’s ability to regain its original shape and dimension after an internal or external force is removed, and is defined by the quantity and the quality of elastic fibers. Aside from the collagens and elastin, the ECM consists of many other secreted glycoproteins (e.g., fibronectin, fibrillins, laminins), enzymes that post-translationally modify these components (e.g., lysyl oxidase), and proteinases that cleave peptide bonds (e.g., metalloproteinases) and facilitate ECM turnover [58]. The combination of these macromolecules within each tissue type is unique, and is determined during development through a dynamic biochemical and biophysical dialogue between various cellular components. A second category of ECM components consists of matricellular proteins such as thrombospondins and tenascins (i.e., Tenascin-C and Tenascin-XB) that regulate the formation or deposition of ECM components (i.e., collagen and elastin), and that modulate cell function through cell–matrix interactions. Of particular interest with respect to VUR is Tenascin-XB (TNXB), which is the largest member of the tenascin family of glycoproteins.
Collagen fibrils and tensile strength
Collagens are the best-characterized components of the ECM due to their great abundance in the body (~30 % of total protein). There are 28 types that are classified based on their ability to form fibrils into fibrillary and non-fibrillar collagens [58, 59]. Type I, II, III, V, XI, XXIV, and XXVII are all fibril-forming collagens with types I, III, and V most relevant to the urinary tract and the connective tissue disorders described in Table 1.
Each collagen molecule is composed of a triple helix of three polypeptide chains called α-chains. While the α-chains have some degree of structural similarity (i.e., they are all made of repeating Gly-Pro-X, where X can be any amino acid), their differences distinguish the types of collagen [59–62]. Collagen molecules can be homotrimers (i.e., triple helices made of the same α-chains), or heterotrimers. The trimers are organized into supramolecular structures that are tissue-specific and define each tissue’s biomechanical properties. Collagen fibrils are assembled from triple helices as mixtures of two or more types of fibril-forming collagens. The initiation of fibrillogenesis, as well as the thickness of the fibrils, is regulated by type V and type XI collagens [59–65]. As fibrillogenesis proceeds, two types of fibrils are formed: thick fibrils with larger cross-sectional area, and thin fibrils with smaller cross-section and better ability to form bonds with adjacent fibrils [59–61]. Thick fibrils have more tensile strength while thin fibrils provide higher resistance to distension.
Type I collagen, as the major contributor to tensile strength of tissues, forms thick fibrils and is expressed in most connective tissue. Type III collagen forms thinner fibrils than type I, has greater distensibility, and is therefore enriched in relatively elastic tissues such as lungs and blood vessels. Type III collagen forms heterotypic fibrils with type I collagen, and is crucial for type I fibril formation [59–62, 65–67]. Type V collagen is much less abundant, its major role is to regulate fibrillogenesis including the control of fibril size and diameter.
Elastic fibers and recoil response
Elastic fibers endow tissues with the ability to regain their original shape and dimension when undergoing repeated cycles of stretching [68–70]. Elastic fibers consist of an inner core of cross-linked elastin surrounded by microfibrils that are made of fibrillin. The deposition and cross-linking of elastin onto the fibrillin microfibrils is facilitated by members of the fibulin family (i.e., fibulin-4 and fibulin-5).
The protein tropoelastin, encoded by the elastin gene (ELN), is the fundamental component of elastin. The newly translated tropoelastin monomers are secreted into the extracellular environment where they are cross-linked to form elastin [70–72]. The ELN gene is expressed as early as 17 weeks of gestation, and is highly expressed in the first few years of life, allowing further development and growth of tissues in need of elastic recoil. At later ages, the expression of ELN is down-regulated, consistent with the observation that tissues lose their recoil properties over a lifespan [70, 73].
Fibrillins are a family of secreted glycoproteins with fibrillin-1 as their most abundant isoform [74, 75]. The precise mode of assembly of fibrillin microfibrils is unknown, but they are arranged as parallel bundles of four to eight fibrillin molecules that are joined in series in a head-to-tail manner [76]. The fibrillin microfibrils form a scaffold for the deposition of elastin; therefore perturbations in their synthesis will impair elastic fiber assembly [71, 76]. Fibrillin microfibrils play a key role in the regulation of TGF-β1 signaling in elastic tissues such as lungs and arteries. A number of proteins associated with fibrillin microfibrils bind and sequester TGF-β1 to prevent downstream signaling [70, 77, 78].
Fibulins are a family of seven secreted glycoproteins that interact with a variety of different ECM components and function in cell adhesion, migration, and proliferation. Fibulin-4 and -5 are crucial to elastic fiber assembly as they facilitate the deposition and cross-linking of elastin within the fibrillin microfibril scaffold [71, 79, 80]. Fibulin-5 is required for the development of the vasculature and the neural crest and down-regulates the transcription of TGF-β1 [80].
Tenascin-XB and tissue biomechanics
TNXB is a large protein with five distinct domains: a signaling domain, four heptad repeats, 18.5 epidermal growth factor (EGF)-like repeats, 32 fibronectin type III (FNIII) repeats, and a fibrinogen (FBG)-like globular domain at its C-terminus [81]. The heptad repeats facilitate the trimerization of TNXB, while the FNIII repeats endow it with conformational flexibility to physically contact other ECM components [82, 83] (Fig. 1). Like many other ECM components, TNXB has a cellular and molecular attachment site flanked by the arginine-glycine-asparagine sequence known as the RGD-motif. TNXB interacts with type I, III, V, XII, and XIV collagens via its EGF-like and FNIII repeats as well as its FBG-like domain [82–87]. Evidence that these interactions with collagens are functionally important has been revealed through the study of mutant mouse models and humans with mutations in TNXB. Tnxb−/− mice, and humans with heterozygous and homozygous mutations in TNXB, have hyperextensible skin [88–94]. Analysis of the skin of affected mice and humans has revealed that the collagen fibrils within the skin are not tightly packed, but spaced further apart, and this is associated with a decrease in the expression of type I collagen [92, 94–97]. In addition, there is a reduction in the quantity and quality of the elastic fibers that appear slender and fragmented. This is not surprising, given the fact that TNXB has been shown to interact with tropoelastin, the main component of elastic fibers, through its C-terminal region that includes the last few FNIII repeats and the FBG globular domain [98, 99]. In summary, TNXB influences the biomechanical properties of tissues by regulating the formation and stability of both collagen fibrils and elastic fibers.
In addition to its functions in tissue architecture, TNXB has regulatory roles in cell attachment, matrix turnover, and in the bioavailability of TGF-β1. In vitro studies have shown that TNXB, like other members of the tenascin family, has anti-adhesive properties and can cause cell detachment [100, 101]. Both in vivo and in vitro studies have shown that TNXB can negatively regulate matrix metalloproteinases (MMPs), and therefore modulate ECM turnover [102, 103]. TNXB interacts with vascular endothelial growth factor B (VEGF-B) through its FN III repeats, and this promotes endothelial cell proliferation [25, 104]. Finally, TNXB has been shown to modulate the bioavailability of TGF-β1 through activation of the latent form of TGF-β1 [25].
Biomechanical components of the ureter and the bladder
The ureter is a fibro-muscular tube that propels urine from the kidneys to the bladder through wave-like muscular contractions or peristalsis, the amplitude and frequency of which depends on the urine volume. As part of peristalsis, the ureteral wall exhibits multidirectional tensile strength and recoil. With every contractile wave, the urine bolus is pushed forward, distending the ureteral wall, while simultaneously, proximal to the bolus, the wall recoils back to its original shape and dimension. The smooth muscle layer drives the contractile wave and provides structural support, while collagen fibrils within the lamina propria and the connective sheath surrounding the smooth muscle regulate the tension within the ureter wall through repeated cycles of uncoiling and recoiling of the collagen fibrils. The limited data available on the collagen composition of the human ureter indicates the presence of both type I and type III collagen fibrils [105]. Biomechanical studies have shown that the ureter has higher tensile strength along its longitudinal axis perhaps due to the presence of more longitudinally oriented collagen fibrils within the lamina propria layer [106–108].
Recoil is required by the ureter in two contexts: to restore the distended portion of ureter that contains the urine to its original shape and to reopen the tubular lumen after it has been occluded during a peristaltic wave. The integrity and abundance of elastic fibers facilitates the recoil of the ureter wall. Elastic fibers are detected within the urothelium, the lamina propria, and the smooth muscle layer [109].
The intravesical portion of the ureter (IVU) lacks circular muscles, and therefore is unable to generate peristaltic waves. The passage of urine through the IVU depends on the pressure gradient between the extravesical portion of the ureter and the IVU. Because the IVU is narrower than the extravesical ureter, maintaining low pressure within the IVU is dependent on low resistance, which is determined by the composition and amount of collagens.
Tensile and recoil properties are also critical to the function of the bladder that undergoes repeated cycles of filling and emptying. The continuous distension of the bladder during filling (i.e., storage phase), and its recoil during emptying (i.e., voiding phase) depends on the integrity of the detrusor muscle bundles as well as the tensile properties of the fibrillary collagens and the recoil properties of the elastic fibers. Indeed, biomechanical studies have shown that lamina propria, which is rich with collagen fibrils and elastic fibers, is responsible for bearing most of the mechanical load of the bladder (i.e., resistance, compliance, and recoil) [110–112]. Biochemical and histological studies have shown that in the normal human bladder, the collagen composition is approximately 75 % type I collagen fibrils and 25 % type III collagen fibrils [113]. The thick type I collagen fibrils provide tensile strength and resistance against deformation, while the thinner type III collagen fibrils facilitate compliance and distension. As with the ureter, the elastic fibers of the bladder facilitate its recoil post-micturition.
For urine to flow freely across the IVU and into the bladder, the pressure of the bladder during the storage phase must remain low. The occlusion of the IVU upon completion of the storage phase and initiation of the voiding phase depends on the contraction of the detrusor muscle and the transiently higher pressure inside the bladder.
Ehlers–Danlos syndrome and mutations in fibrillary collagens
Ehlers–Danlos syndrome (EDS) encompasses a spectrum of monogenic disorders primarily affecting the skin, the ligaments, the joints, blood vessels, and internal organs [114, 115]. EDS is currently classified into six subtypes, most of which have been associated with mutations in genes that encode fibrillary collagens or the enzymes involved in post-translational modification of these proteins [115].
Classical types of EDS are associated with mutations in genes encoding either the α1- or α2-chains of type V collagen. Vascular type EDS is caused by mutations in the gene encoding the α1-chain of type III collagen. The arthrochalasia type, characterized by hip dislocation, is caused by mutations in the genes encoding either the α1- or α2-chain of type I collagen [115].
Much of what is known about the ECM derangements in connective tissue disorders including EDS has been gathered through skin biopsies and fibroblast cultures. These studies have revealed that most EDS subtypes exhibit disrupted collagen fibrillogenesis, as indicated by irregular and loosely packed collagen fibrils [116–119].
The most commonly reported urinary tract phenotype among individuals with various types of EDS is bladder diverticula; there are at least 30 reported cases [118, 120–136]. Some reports describe cases of recurrent upper urinary tract infections (UTIs), urinary incontinence, VUR, and reflux nephropathy [118, 120–123, 125–127, 134, 136]. No large-scale studies have been conducted to investigate the incidence of bladder diverticula and VUR amongst patients with EDS.
Joint hypermobility and fibrillary collagen perturbations due to mutations in TNXB
Generalized joint hypermobility is seen in~ 40 % of school-age children depending on race, ethnicity, and gender, and is diagnosed using criteria established by Beighton (1998) [137–142]. Generalized joint hypermobility is also seen in many heritable connective tissue disorders including Marfan syndrome and joint hypermobility type EDS (HT-EDS). HTEDS is characterized by joint laxity, recurrent joint dislocations, musculoskeletal pain, as well as soft velvety skin that is slightly hyperextensible [143]. There are no specific clinical criteria to distinguish between generalized and EDS type hypermobility, therefore, many clinicians believe they arise from the same mechanism [144, 145]. This is of particular interest because of the high prevalence of individuals who have both joint hypermobility and VUR. Van Eerde et al. studied 50 children with VUR and found that they had a fourfold greater likelihood of having joint hypermobility than age-matched controls without VUR [146]. These results have been echoed by Pournasiri et al., Beiraghdar et al., and Adib et al., who demonstrated that patients with joint hypermobility have an increased likelihood of having VUR compared to the general population [147–149].
HT-EDS is reported to have an autosomal dominant mode of inheritance, with variable penetrance and incomplete expressivity influenced by sex [150]. While the major fibrillary collagens (i.e., type I, III, and V) have been ruled out as candidate genes by linkage studies, a subset of patients with HTEDS have been found to harbor heterozygous or homozygous mutations in the ECM glycoprotein, Tenascin-XB [89, 97]. We recently identified a large extended pedigree with autosomal dominant inheritance of VUR and joint hypermobility due to missense mutations in TNXB [4]. We then screened children with familial and non-familial primary VUR and identified rare variants in TNXB in 9 % of familial cases (5/57) and 4 % of sporadic cases [5]. These results suggest that VUR may commonly occur in the presence of joint hypermobility and that Tenascin-XB and other ECM genes that are regulated by TNXB are all candidate genes that could explain these combined phenotypes.
Cutis laxa, Marfan syndrome, and Williams syndrome, and mutations in elastic fiber components
Cutis laxa is a genetically heterogeneous group of rare connective tissue disorders (incidence of ~1:400,000) characterized by the presence of sagging and inelastic skin [151]. The autosomal dominant form is caused by mutations in elastin (ELN) or more rarely in fibulin 5 (FBLN5) and presents later in life, typically with the following features: inelastic and redundant skin, aged appearance, high forehead, large ear lobes, cardiac disease, and pulmonary disease including bronchiectasis and emphysema [151]. The autosomal recessive form of cutis laxa is caused by mutations in fibulins, namely fibulin-4 and -5 genes, and presents in early childhood with a number of features including: severe cardiac disease (i.e., aortic aneurysm), pulmonary emphysema, inguinal and umbilical hernias, gastro-esophageal reflux, and diverticula formation in the intestines [151]. Most children with the recessive form of cutis laxa die in early childhood or before puberty from cardiac or pulmonary failure [151]. Urinary tract defects are described in a number of case series and include VUR and bladder diverticula [152–155]. Pathogenic mutations of FBLN4 and FBLN5 result in abnormal protein folding that prevents secretion of the protein so that is not available to cross-link elastic fibers. This leads to a shortage of elastic fibers and a reduction in tissue recoil [151, 152].
Marfan syndrome is a common (prevalence of ~1:10,000) multisystem disorder characterized by musculoskeletal (e.g., hypermobility), cardiovascular, and ocular abnormalities. Dominantly inherited mutations in fibrillin-1 are the cause of this syndrome and alter the stability of the protein, resulting in its enhanced degradation [75, 156, 157]. The reduction in fibrillin-1 protein impairs the formation of the fibrillin microfibrils that form the scaffold to assemble the elastic fibers [75, 156, 157]. From case series, urinary tract phenotypes have been reported in Marfan syndrome and include: voiding dysfunction, bladder diverticula, and VUR [158–160].
Williams syndrome is a common (prevalence of ~1:10,000) disorder caused by the hemizygous deletion of a specific region of chromosome 7 that removes 26 to 28 genes, depending on the size of the deletion [161, 162]. The large deletion typically includes the elastin gene (ELN), which accounts for the connective tissue and cardiovascular phenotypes noted with the disorder. The haploinsufficiency for the elastin gene results in 50 % less elastin protein, a reduction in the formation of elastic fibers, and impaired recoil for all tissues. Affected individuals exhibit varying ranges of intellectual disability, unique personality characteristics, distinctive facial features, cardiovascular and joint problems, as well as fragile and sagging skin. The urinary tract phenotypes reported for this syndrome are bladder diverticula and VUR. Amongst the connective tissue disorders discussed in this review, Williams syndrome is the only one with larger-scale case series that have systematically screened patients for urinary tract phenotypes. A combined total of 213 patients with a variety of urinary tract phenotypes have been evaluated of which 20 % have bladder diverticula and 7 % have VUR [163–167].
Making connections: how biomechanical anomalies of the ureter and the bladder can cause VUR
The normal physiological function of the urinary tract requires a delicate balance of tensile strength and recoil that is mediated by the composition and abundance of collagen fibrils and elastic fibers. In the setting of a genetic predisposition or an acquired injury that perturbs the balance, urinary tract defects will arise. An increase in tensile strength could arise from an increase in the production and deposition of fibrillary collagens, particularly type I fibrils. This could arise in the setting of fibrosis with activation of the TGFβ1 signaling pathway. An increase in tensile strength in the ureter is predicted to result in high resistance and an increase in the intraureteral pressure that could impede the flow of urine into the IVU by diminishing the amplitude of the contractile wave. In the IVU, the increased stiffness would impair its closure and promote VUR. Within the bladder, the decrease in bladder wall compliance would result in higher bladder pressures with filling that would diminish the pressure gradient between the IVU and the bladder, thereby promoting VUR. High resistance in the bladder would also result in high-pressure voiding that could predispose to the formation of diverticula.
A decrease in tensile strength could arise from a decrease or derangement in type I collagen fibrils as seen in classic, arthrochalasia, and hypermobility type EDS. In the ureter, a decrease in tensile strength is predicted to result in less resistance to distension and a reduction in intraureteral pressure. Because type III collagen forms heterotypic fibrils with type I, a reduction in type I collagen could result in an adaptive increase in production of type III fibrils. An increase in type III fibrils could increase the distensibility of the ureter and impede its ability to withstand repeated cycles of distension and recoil as part of the contractile wave, giving rise to megaureter [105]. In the IVU, a decrease in tensile strength coupled with an increase in distensibility would lead to an over-stretched lumen that would not be completely occluded and therefore prone to VUR. In the bladder, the loss of tensile strength and the increase in distensibility could promote the formation of bladder diverticula, like the Hutch diverticulum, and could also promote VUR secondary to ineffective closure of the IVU.
A decrease in recoil could arise from a deficiency in elastic fibers due to a genetic origin from loss-of-function mutations in the genes encoding tropoelastin, fibrillin, and fibulin, as seen in cutis laxa, Marfan, and Williams syndrome or secondary to injury. A decrease in recoil is predicted to impair contractile waves along the ureter and predispose to megaureter. In the IVU, the loss of recoil is expected to maintain a patent IVU even during voiding and promote VUR. In the bladder, impaired recoil would result in a distended and deformed bladder and a predisposition to the formation of diverticula and potentially VUR.
The high prevalence of joint hypermobility in children with VUR could reflect a decrease in tensile strength and/or a decrease in recoil in both the joints and the UVJ. Indeed, mutations in TNXB result in a decrease in tensile strength as well as perturbed recoil and explain some cases of VUR with joint hypermobility. We would speculate that mutations in other ECM genes that affect tensile strength or recoil are also candidate genes that warrant investigation in affected families.
Conclusions
Our current understanding of urinary tract phenotypes among individuals with connective tissue disorders is limited by the few published case reports and series that describe small numbers of patients. Based on what is known about the ECM composition and biomechanical properties of the IVU and the bladder, we would predict that the association of VUR and bladder diverticula with hereditary connective tissue disorders is much more prevalent than reported. We would recommend that future research needs to focus on systematic assessment of patients with hereditary connective tissue disorders to determine the frequency of urinary tract defects in this population. Future research should also identify and evaluate patients with VUR and joint hypermobility so that this phenotype can be better defined in terms of prevalence, genetic risk factors, and long-term prognosis.
Footnotes
Conflict of interest The authors declare no conflicts of interest.
References
- 1.Williams G, Fletcher JT, Alexander SI, Craig JC. Vesicoureteral reflux. J Am Soc Nephrol. 2008;19:847–862. doi: 10.1681/ASN.2007020245. [DOI] [PubMed] [Google Scholar]
- 2.Tullus K. Vesicoureteric reflux in children. Lancet. 2015;385:371–379. doi: 10.1016/S0140-6736(14)60383-4. [DOI] [PubMed] [Google Scholar]
- 3.Fillion ML, Watt CL, Gupta IR. Vesicoureteric reflux and reflux nephropathy: from mouse models to childhood disease. Pediatr Nephrol. 2014;29:757–766. doi: 10.1007/s00467-014-2761-3. [DOI] [PubMed] [Google Scholar]
- 4.Gbadegesin RA, Brophy PD, Adeyemo A, Hall G, Gupta IR, Hains D, Bartkowiak B, Rabinovich CE, Chandrasekharappa S, Homstad A, Westreich K, Wu G, Liu Y, Holanda D, Clarke J, Lavin P, Selim A, Miller S, Wiener JS, Ross SS, Foreman J, Rotimi C, Winn MP. TNXB mutations can cause vesicoureteral reflux. J Am Soc Nephrol. 2013;24:1313–1322. doi: 10.1681/ASN.2012121148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Elahi S, Homstad A, Vaidya H, Stout J, Hall G, Wu G, Conlon P, Jr, Routh JC, Wiener JS, Ross SS, Nagaraj S, Wigfall D, Foreman J, Adeyemo A, Gupta IR, Brophy PD, Rabinovich CE, Gbadegesin RA. Rare variants in tenascin genes in a cohort of children with primary vesicoureteric reflux. Pediatr Nephrol. 2015;31:247–253. doi: 10.1007/s00467-015-3203-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arze RS, Ramos JM, Owen JP, Morley AR, Elliott RW, Wilkinson R, Ward MK, Kerr DN. The natural history of chronic pyelonephritis in the adult. Q J Med. 1982;51:396–410. [PubMed] [Google Scholar]
- 7.el-Khatib MT, Becker GJ, Kincaid-Smith PS. Morphometric aspects of reflux nephropathy. Kidney Int. 1987;32:261–266. doi: 10.1038/ki.1987.201. [DOI] [PubMed] [Google Scholar]
- 8.el-Khatib MT, Becker GJ, Kincaid-Smith PS. Reflux nephropathy and primary vesicoureteric reflux in adults. Q J Med. 1990;77:1241–1253. doi: 10.1093/qjmed/77.3.1241. [DOI] [PubMed] [Google Scholar]
- 9.Nakashima Y, Matsuoka H, Oshima K, Sakamoto K. Progression of renal disease in patients with reflux nephropathy. Follow-up study. Nihon Hinyokika Gakkai Zasshi. 1997;88:557–565. doi: 10.5980/jpnjurol1989.88.557. [DOI] [PubMed] [Google Scholar]
- 10.Bailey RR, Lynn KL, Robson RA. End-stage reflux nephropathy. Ren Fail. 1994;16:27–35. doi: 10.3109/08860229409044845. [DOI] [PubMed] [Google Scholar]
- 11.Smellie JM, Barratt TM, Chantler C, Gordon I, Prescod NP, Ransley PG, Woolf AS. Medical versus surgical treatment in children with severe bilateral vesicoureteric reflux and bilateral nephropathy: a randomised trial. Lancet. 2001;357:1329–1333. doi: 10.1016/S0140-6736(00)04520-7. [DOI] [PubMed] [Google Scholar]
- 12.Becker GJ, Kincaid-Smith P. Reflux nephropathy: the glomerular lesion and progression of renal failure. Pediatr Nephrol. 1993;7:365–369. doi: 10.1007/BF00857540. [DOI] [PubMed] [Google Scholar]
- 13.Dillon MJ, Goonasekera CD. Reflux nephropathy. J Am Soc Nephrol. 1998;9:2377–2383. doi: 10.1681/ASN.V9122377. [DOI] [PubMed] [Google Scholar]
- 14.Goonasekera CD, Gordon I, Dillon MJ. 15-year follow-up of reflux nephropathy by imaging. Clin Nephrol. 1998;50:224–231. [PubMed] [Google Scholar]
- 15.Duffield JS. Cellular and molecular mechanisms in kidney fibrosis. J Clin Invest. 2014;124:2299–2306. doi: 10.1172/JCI72267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu Y. Cellular and molecular mechanisms of renal fibrosis. Nat Rev Nephrol. 2011;7:684–696. doi: 10.1038/nrneph.2011.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schnaper HW, Hayashida T, Hubchak SC, Poncelet AC. TGF-beta signal transduction and mesangial cell fibrogenesis. Am J Physiol Renal Physiol. 2003;284:F243–F252. doi: 10.1152/ajprenal.00300.2002. [DOI] [PubMed] [Google Scholar]
- 18.Schnaper HW, Kopp JB. Renal fibrosis. Front Biosci. 2003;8:e68–e86. doi: 10.2741/925. [DOI] [PubMed] [Google Scholar]
- 19.Solari V, Owen D, Puri P. Association of transforming growth factor-beta1 gene polymorphism with reflux nephropathy. J Urol. 2005;174:1609–1611. doi: 10.1097/01.ju.0000179385.64585.dc. discussion 1611. [DOI] [PubMed] [Google Scholar]
- 20.Bottinger EP. TGF-beta in renal injury and disease. Semin Nephrol. 2007;27:309–320. doi: 10.1016/j.semnephrol.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 21.Sabasinska A, Zoch-Zwierz W, Wasilewska A, Porowski T. Laminin and transforming growth factor beta-1 in children with vesicoureteric reflux. Pediatr Nephrol. 2008;23:769–774. doi: 10.1007/s00467-007-0723-8. [DOI] [PubMed] [Google Scholar]
- 22.Lan HY. Diverse roles of TGF-beta/Smads in renal fibrosis and inflammation. Int J Biol Sci. 2011;7:1056–1067. doi: 10.7150/ijbs.7.1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lopez-Hernandez FJ, Lopez-Novoa JM. Role of TGF-beta in chronic kidney disease: an integration of tubular, glomerular and vascular effects. Cell Tissue Res. 2012;347:141–154. doi: 10.1007/s00441-011-1275-6. [DOI] [PubMed] [Google Scholar]
- 24.Leask A, Denton CP, Abraham DJ. Insights into the molecular mechanism of chronic fibrosis: the role of connective tissue growth factor in scleroderma. J Invest Dermatol. 2004;122:1–6. doi: 10.1046/j.0022-202X.2003.22133.x. [DOI] [PubMed] [Google Scholar]
- 25.Alcaraz LB, Exposito JY, Chuvin N, Pommier RM, Cluzel C, Martel S, Sentis S, Bartholin L, Lethias C, Valcourt U. Tenascin-X promotes epithelial-to-mesenchymal transition by activating latent TGF-beta. J Cell Biol. 2014;205:409–428. doi: 10.1083/jcb.201308031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smellie JM, Prescod NP, Shaw PJ, Risdon RA, Bryant TN. Childhood reflux and urinary infection: a follow-up of 10–41 years in 226 adults. Pediatr Nephrol. 1998;12:727–736. doi: 10.1007/s004670050535. [DOI] [PubMed] [Google Scholar]
- 27.Smellie JM, Jodal U, Lax H, Mobius TT, Hirche H, Olbing H, Writing Committee IRSiC Outcome at 10 years of severe vesicoureteric reflux managed medically: Report of the International Reflux Study in Children. J Pediatr. 2001;139:656–663. doi: 10.1067/mpd.2001.117583. [DOI] [PubMed] [Google Scholar]
- 28.Sjostrom S, Sillen U, Bachelard M, Hansson S, Stokland E. Spontaneous resolution of high grade infantile vesicoureteral reflux. J Urol. 2004;172:694–698. doi: 10.1097/01.ju.0000130747.89561.cf. discussion 699. [DOI] [PubMed] [Google Scholar]
- 29.Arlen AM, Garcia-Roig M, Weiss AD, Leong T, Cooper CS, Kirsch AJ. Vesicoureteral reflux index: 2-institution analysis and validation. J Urol. 2015 doi: 10.1016/j.juro.2015.03.094. [DOI] [PubMed] [Google Scholar]
- 30.Agostiniani R, Mariotti P. The natural history of vesicoureteral reflux. J Matern Fetal Neonatal Med. 2011;24(Suppl 1):2–3. doi: 10.3109/14767058.2011.607557. [DOI] [PubMed] [Google Scholar]
- 31.Roshani H, Dabhoiwala NF, Verbeek FJ, Kurth KH, Lamers WH. Anatomy of ureterovesical junction and distal ureter studied by endoluminal ultrasonography in vitro. J Urol. 1999;161:1614–1619. [PubMed] [Google Scholar]
- 32.Tanagho EA, Pugh RC. The anatomy and function of the ureterovesical junction. Br J Urol. 1963;35:151–165. doi: 10.1111/j.1464-410x.1963.tb02610.x. [DOI] [PubMed] [Google Scholar]
- 33.Yu W, Hill WG. Defining protein expression in the urothelium: a problem of more than transitional interest. Am J Physiol Renal Physiol. 2011;301:F932–F942. doi: 10.1152/ajprenal.00334.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu XR, Kong XP, Pellicer A, Kreibich G, Sun TT. Uroplakins in urothelial biology, function, and disease. Kidney Int. 2009;75:1153–1165. doi: 10.1038/ki.2009.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lazzeri M. The physiological function of the urothelium—more than a simple barrier. Urol Int. 2006;76:289–295. doi: 10.1159/000092049. [DOI] [PubMed] [Google Scholar]
- 36.Lilly JD, Parsons CL. Bladder surface glycosaminoglycans is a human epithelial permeability barrier. Surg Gynecol Obstet. 1990;171:493–496. [PubMed] [Google Scholar]
- 37.Parsons CL, Boychuk D, Jones S, Hurst R, Callahan H. Bladder surface glycosaminoglycans: an epithelial permeability barrier. J Urol. 1990;143:139–142. doi: 10.1016/s0022-5347(17)39897-x. [DOI] [PubMed] [Google Scholar]
- 38.Parsons CL, Stauffer C, Schmidt JD. Bladder-surface glycosaminoglycans: an efficient mechanism of environmental adaptation. Science. 1980;208:605–607. doi: 10.1126/science.6154316. [DOI] [PubMed] [Google Scholar]
- 39.Noordzij JW, Dabhoiwala NF. A view on the anatomy of the ureterovesical junction. Scand J Urol Nephrol. 1993;27:371–380. doi: 10.3109/00365599309180449. [DOI] [PubMed] [Google Scholar]
- 40.Gearhart JP, Canning DA, Gilpin SA, Lam EE, Gosling JA. Histological and histochemical study of the vesicoureteric junction in infancy and childhood. Br J Urol. 1993;72:648–654. doi: 10.1111/j.1464-410x.1993.tb16226.x. [DOI] [PubMed] [Google Scholar]
- 41.Hutch JA. Theory of maturation of the intravesical ureter. J Urol. 1961;86:534–538. doi: 10.1016/S0022-5347(17)65215-7. [DOI] [PubMed] [Google Scholar]
- 42.Oswald J, Brenner E, Schwentner C, Deibl M, Bartsch G, Fritsch H, Radmayr C. The intravesical ureter in children with vesicoureteral reflux: a morphological and immunohistochemical characterization. J Urol. 2003;170:2423–2427. doi: 10.1097/01.ju.0000097146.26432.9a. [DOI] [PubMed] [Google Scholar]
- 43.Oswald J, Schwentner C, Brenner E, Deibl M, Fritsch H, Bartsch G, Radmayr C. Extracellular matrix degradation and reduced nerve supply in refluxing ureteral endings. J Urol. 2004;172:1099–1102. doi: 10.1097/01.ju.0000135673.28496.70. [DOI] [PubMed] [Google Scholar]
- 44.Schwentner C, Oswald J, Lunacek A, Schlenck B, Berger AP, Deibl M, Fritsch H, Bartsch G, Radmayr C. Structural changes of the intravesical ureter in children with vesicoureteral reflux-does ischemia have a role? J Urol. 2006;176:2212–2218. doi: 10.1016/j.juro.2006.07.062. [DOI] [PubMed] [Google Scholar]
- 45.Schwentner C, Oswald J, Lunacek A, Pelzer AE, Fritsch H, Schlenck B, Karatzas A, Bartsch G, Radmayr C. Extracellular microenvironment and cytokine profile of the ureterovesical junction in children with vesicoureteral reflux. J Urol. 2008;180:694–700. doi: 10.1016/j.juro.2008.04.048. [DOI] [PubMed] [Google Scholar]
- 46.Radmayr C, Fritsch H, Schwentner C, Lunacek A, Deibl M, Bartsch G, Oswald J. Fetal development of the vesicoureteric junction, and immunohistochemistry of the ends of refluxing ureters. J Pediatr Urol. 2005;1:53–59. doi: 10.1016/j.jpurol.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 47.Radmayr C, Schwentner C, Lunacek A, Karatzas A, Oswald J. Embryology and anatomy of the vesicoureteric junction with special reference to the etiology of vesicoureteral reflux. Ther Adv Urol. 2009;1:243–250. doi: 10.1177/1756287209348985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stetler-Stevenson WG. Dynamics of matrix turnover during pathologic remodeling of the extracellular matrix. Am J Pathol. 1996;148:1345–1350. [PMC free article] [PubMed] [Google Scholar]
- 49.Bonnans C, Chou J, Werb Z. Remodelling the extracellular matrix in development and disease. Nat Rev Mol Cell Biol. 2014;15:786–801. doi: 10.1038/nrm3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lu P, Takai K, Weaver VM, Werb Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb Perspect Biol. 2011;3(12):a005058. doi: 10.1101/cshperspect.a005058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Leonard MP, Canning DA, Epstein JI, Gearhart JP, Jeffs RD. Local tissue reaction to the subureteral injection of glutaraldehyde cross-linked bovine collagen in humans. J Urol. 1990;143:1209–1212. doi: 10.1016/s0022-5347(17)40227-8. [DOI] [PubMed] [Google Scholar]
- 52.Frey P, Lutz N, Berger D, Herzog B. Histological behavior of glutaraldehyde cross-linked bovine collagen injected into the human bladder for the treatment of vesicoureteral reflux. J Urol. 1994;152:632–635. doi: 10.1016/s0022-5347(17)32669-1. [DOI] [PubMed] [Google Scholar]
- 53.Cerwinka WH, Scherz HC, Kirsch AJ. Endoscopic treatment of vesicoureteral reflux with dextranomer/hyaluronic acid in children. Adv Urol. 2008;2008:513854. doi: 10.1155/2008/513854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stenberg A, Larsson E, Lackgren G. Endoscopic treatment with dextranomer-hyaluronic acid for vesicoureteral reflux: histological findings. J Urol. 2003;169:1109–1113. doi: 10.1097/01.ju.0000053013.49676.89. [DOI] [PubMed] [Google Scholar]
- 55.Alkan M, Talim B, Ciftci AO, Senocak ME, Caglar M, Buyukpamukcu N. Histological response to injected gluteraldehyde cross-linked bovine collagen based implant in a rat model. BMC Urol. 2006;6:3. doi: 10.1186/1471-2490-6-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Routh JC, Ashley RA, Sebo TJ, Vandersteen DR, Slezak J, Reinberg Y. Histopathological changes associated with dextranomer/hyaluronic acid injection for pediatric vesicoureteral reflux. J Urol. 2007;178:1707–1710. doi: 10.1016/j.juro.2007.03.165. [DOI] [PubMed] [Google Scholar]
- 57.Arena S, Fazzari C, Implatini A, Torre S, Villari D, Arena F, Di Benedetto V. Dextranomer/hyaluronic Acid copolymer implant for vesicoureteral reflux: role of myofibroblast differentiation. J Urol. 2009;181:2695–2701. doi: 10.1016/j.juro.2009.02.059. [DOI] [PubMed] [Google Scholar]
- 58.Jarvelainen H, Sainio A, Koulu M, Wight TN, Penttinen R. Extracellular matrix molecules: potential targets in pharmacotherapy. Pharmacol Rev. 2009;61:198–223. doi: 10.1124/pr.109.001289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gordon MK, Hahn RA. Collagens. Cell Tissue Res. 2010;339:247–257. doi: 10.1007/s00441-009-0844-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ottani V, Raspanti M, Ruggeri A. Collagen structure and functional implications. Micron. 2001;32:251–260. doi: 10.1016/s0968-4328(00)00042-1. [DOI] [PubMed] [Google Scholar]
- 61.Gelse K, Poschl E, Aigner T. Collagens—structure, function, and biosynthesis. Adv Drug Deliv Rev. 2003;55:1531–1546. doi: 10.1016/j.addr.2003.08.002. [DOI] [PubMed] [Google Scholar]
- 62.Martin R, Waldmann L, Kaplan DL. Supramolecular assembly of collagen triblock peptides. Biopolymers. 2003;70:435–444. doi: 10.1002/bip.10464. [DOI] [PubMed] [Google Scholar]
- 63.Birk DE, Fitch JM, Babiarz JP, Doane KJ, Linsenmayer TF. Collagen fibrillogenesis in vitro: interaction of types I and V collagen regulates fibril diameter. J Cell Sci. 1990;95(Pt 4):649–657. doi: 10.1242/jcs.95.4.649. [DOI] [PubMed] [Google Scholar]
- 64.Wenstrup RJ, Florer JB, Brunskill EW, Bell SM, Chervoneva I, Birk DE. Type V collagen controls the initiation of collagen fibril assembly. J Biol Chem. 2004;279:53331–53337. doi: 10.1074/jbc.M409622200. [DOI] [PubMed] [Google Scholar]
- 65.Mouw JK, Ou G, Weaver VM. Extracellular matrix assembly: a multiscale deconstruction. Nat Rev Mol Cell Biol. 2014;15:771–785. doi: 10.1038/nrm3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liu X, Wu H, Byrne M, Krane S, Jaenisch R. Type III collagen is crucial for collagen I fibrillogenesis and for normal cardiovascular development. Proc Natl Acad Sci U S A. 1997;94:1852–1856. doi: 10.1073/pnas.94.5.1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Henkel W, Glanville RW. Covalent crosslinking between molecules of type I and type III collagen. The involvement of the N-terminal, nonhelical regions of the alpha 1 (I) and alpha 1 (III) chains in the formation of intermolecular crosslinks. Eur J Biochem. 1982;122:205–213. doi: 10.1111/j.1432-1033.1982.tb05868.x. [DOI] [PubMed] [Google Scholar]
- 68.Sherratt MJ. Tissue elasticity and the ageing elastic fibre. Age (Dordr) 2009;31:305–325. doi: 10.1007/s11357-009-9103-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gosline J, Lillie M, Carrington E, Guerette P, Ortlepp C, Savage K. Elastic proteins: biological roles and mechanical properties. Philos Trans R Soc Lond B Biol Sci. 2002;357:121–132. doi: 10.1098/rstb.2001.1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kielty CM, Sherratt MJ, Shuttleworth CA. Elastic fibres. J Cell Sci. 2002;115:2817–2828. doi: 10.1242/jcs.115.14.2817. [DOI] [PubMed] [Google Scholar]
- 71.Liu X, Zhao Y, Gao J, Pawlyk B, Starcher B, Spencer JA, Yanagisawa H, Zuo J, Li T. Elastic fiber homeostasis requires lysyl oxidase-like 1 protein. Nat Genet. 2004;36:178–182. doi: 10.1038/ng1297. [DOI] [PubMed] [Google Scholar]
- 72.Kielty CM. Elastic fibres in health and disease. Expert Rev Mol Med. 2006;8:1–23. doi: 10.1017/S146239940600007X. [DOI] [PubMed] [Google Scholar]
- 73.Shapiro SD, Endicott SK, Province MA, Pierce JA, Campbell EJ. Marked longevity of human lung parenchymal elastic fibers deduced from prevalence of D-aspartate and nuclear weapons-related radiocarbon. J Clin Invest. 1991;87:1828–1834. doi: 10.1172/JCI115204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kielty CM, Shuttleworth CA. Fibrillin-containing microfibrils: structure and function in health and disease. Int J Biochem Cell Biol. 1995;27:747–760. doi: 10.1016/1357-2725(95)00028-n. [DOI] [PubMed] [Google Scholar]
- 75.Milewicz DM, Grossfield J, Cao SN, Kielty C, Covitz W, Jewett T. A mutation in FBN1 disrupts profibrillin processing and results in isolated skeletal features of the Marfan syndrome. J Clin Invest. 1995;95:2373–2378. doi: 10.1172/JCI117930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kielty CM, Wess TJ, Haston L, Ashworth JL, Sherratt MJ, Shuttleworth CA. Fibrillin-rich microfibrils: elastic biopolymers of the extracellular matrix. J Muscle Res Cell Motil. 2002;23:581–596. [PubMed] [Google Scholar]
- 77.Hyytiainen M, Penttinen C, Keski-Oja J. Latent TGF-beta binding proteins: extracellular matrix association and roles in TGF-beta activation. Crit Rev Clin Lab Sci. 2004;41:233–264. doi: 10.1080/10408360490460933. [DOI] [PubMed] [Google Scholar]
- 78.Rifkin DB. Latent transforming growth factor-beta (TGF-beta) binding proteins: orchestrators of TGF-beta availability. J Biol Chem. 2005;280:7409–7412. doi: 10.1074/jbc.R400029200. [DOI] [PubMed] [Google Scholar]
- 79.Huang J, Davis EC, Chapman SL, Budatha M, Marmorstein LY, Word RA, Yanagisawa H. Fibulin-4 deficiency results in ascending aortic aneurysms: a potential link between abnormal smooth muscle cell phenotype and aneurysm progression. Circ Res. 2010;106:583–592. doi: 10.1161/CIRCRESAHA.109.207852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yanagisawa H, Schluterman MK, Brekken RA. Fibulin-5, an integrin-binding matricellular protein: its function in development and disease. J Cell Commun Signal. 2009;3:337–347. doi: 10.1007/s12079-009-0065-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bristow J, Tee MK, Gitelman SE, Mellon SH, Miller WL. Tenascin-X: a novel extracellular matrix protein encoded by the human XB gene overlapping P450c21B. J Cell Biol. 1993;122:265–278. doi: 10.1083/jcb.122.1.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lethias C, Descollonges Y, Boutillon MM, Garrone R. Flexilin: a new extracellular matrix glycoprotein localized on collagen fibrils. Matrix Biol. 1996;15:11–19. doi: 10.1016/s0945-053x(96)90122-5. [DOI] [PubMed] [Google Scholar]
- 83.Lethias C, Carisey A, Comte J, Cluzel C, Exposito JY. A model of tenascin-X integration within the collagenous network. FEBS Lett. 2006;580:6281–6285. doi: 10.1016/j.febslet.2006.10.037. [DOI] [PubMed] [Google Scholar]
- 84.Bristow J, Carey W, Egging D, Schalkwijk J. Tenascin-X, collagen, elastin, and the Ehlers–Danlos syndrome. Am J Med Genet C: Semin Med Genet. 2005;139C:24–30. doi: 10.1002/ajmg.c.30071. [DOI] [PubMed] [Google Scholar]
- 85.Veit G, Hansen U, Keene DR, Bruckner P, Chiquet-Ehrismann R, Chiquet M, Koch M. Collagen XII interacts with avian tenascin-X through its NC3 domain. J Biol Chem. 2006;281:27461–27470. doi: 10.1074/jbc.M603147200. [DOI] [PubMed] [Google Scholar]
- 86.Egging D, van den Berkmortel F, Taylor G, Bristow J, Schalkwijk J. Interactions of human tenascin-X domains with dermal extracellular matrix molecules. Arch Dermatol Res. 2007;298:389–396. doi: 10.1007/s00403-006-0706-9. [DOI] [PubMed] [Google Scholar]
- 87.Minamitani T, Ikuta T, Saito Y, Takebe G, Sato M, Sawa H, Nishimura T, Nakamura F, Takahashi K, Ariga H, Matsumoto K. Modulation of collagen fibrillogenesis by tenascin-X and type VI collagen. Exp Cell Res. 2004;298:305–315. doi: 10.1016/j.yexcr.2004.04.030. [DOI] [PubMed] [Google Scholar]
- 88.Burch GH, Gong Y, Liu W, Dettman RW, Curry CJ, Smith L, Miller WL, Bristow J. Tenascin-X deficiency is associated with Ehlers–Danlos syndrome. Nat Genet. 1997;17:104–108. doi: 10.1038/ng0997-104. [DOI] [PubMed] [Google Scholar]
- 89.Schalkwijk J, Zweers MC, Steijlen PM, Dean WB, Taylor G, van Vlijmen IM, van Haren B, Miller WL, Bristow J. A recessive form of the Ehlers–Danlos syndrome caused by tenascin-X deficiency. N Engl J Med. 2001;345:1167–1175. doi: 10.1056/NEJMoa002939. [DOI] [PubMed] [Google Scholar]
- 90.Voermans NC, Jenniskens GJ, Hamel BC, Schalkwijk J, Guicheney P, van Engelen BG. Ehlers–Danlos syndrome due to tenascin-X deficiency: muscle weakness and contractures support overlap with collagen VI myopathies. Am J Med Genet Part A. 2007;143A:2215–2219. doi: 10.1002/ajmg.a.31899. [DOI] [PubMed] [Google Scholar]
- 91.Voermans NC, Altenburg TM, Hamel BC, de Haan A, van Engelen BG. Reduced quantitative muscle function in tenascin-X deficient Ehlers–Danlos patients. Neuromuscul Disord. 2007;17:597–602. doi: 10.1016/j.nmd.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 92.Voermans NC, Verrijp K, Eshuis L, Balemans MC, Egging D, Sterrenburg E, van Rooij IA, van der Laak JA, Schalkwijk J, van der Maarel SM, Lammens M, van Engelen BG. Mild muscular features in tenascin-X knockout mice, a model of Ehlers–Danlos syndrome. Connect Tissue Res. 2011;52:422–432. doi: 10.3109/03008207.2010.551616. [DOI] [PubMed] [Google Scholar]
- 93.Lindor NM, Bristow J. Tenascin-X deficiency in autosomal recessive Ehlers–Danlos syndrome. Am J Med Genet Part A. 2005;135:75–80. doi: 10.1002/ajmg.a.30671. [DOI] [PubMed] [Google Scholar]
- 94.Mao JR, Taylor G, Dean WB, Wagner DR, Afzal V, Lotz JC, Rubin EM, Bristow J. Tenascin-X deficiency mimics Ehlers–Danlos syndrome in mice through alteration of collagen deposition. Nat Genet. 2002;30:421–425. doi: 10.1038/ng850. [DOI] [PubMed] [Google Scholar]
- 95.Egging DF, van Vlijmen I, Starcher B, Gijsen Y, Zweers MC, Blankevoort L, Bristow J, Schalkwijk J. Dermal connective tissue development in mice: an essential role for tenascin-X. Cell Tissue Res. 2006;323:465–474. doi: 10.1007/s00441-005-0100-5. [DOI] [PubMed] [Google Scholar]
- 96.Zweers MC, Hakim AJ, Grahame R, Schalkwijk J. Joint hypermobility syndromes: the pathophysiologic role of tenascin-X gene defects. Arthritis Rheum. 2004;50:2742–2749. doi: 10.1002/art.20488. [DOI] [PubMed] [Google Scholar]
- 97.Zweers MC, Bristow J, Steijlen PM, Dean WB, Hamel BC, Otero M, Kucharekova M, Boezeman JB, Schalkwijk J. Haploinsufficiency of TNXB is associated with hypermobility type of Ehlers–Danlos syndrome. Am J Hum Genet. 2003;73:214–217. doi: 10.1086/376564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zweers MC, van Vlijmen-Willems IM, van Kuppevelt TH, Mecham RP, Steijlen PM, Bristow J, Schalkwijk J. Deficiency of tenascin-X causes abnormalities in dermal elastic fiber morphology. J Invest Dermatol. 2004;122:885–891. doi: 10.1111/j.0022-202X.2004.22401.x. [DOI] [PubMed] [Google Scholar]
- 99.Zweers MC, Schalkwijk J, van Kuppevelt TH, van Vlijmen-Willems IM, Bergers M, Lethias C, Lamme EN. Transplantation of reconstructed human skin on nude mice: a model system to study expression of human tenascin-X and elastic fiber components. Cell Tissue Res. 2005;319:279–287. doi: 10.1007/s00441-004-1011-6. [DOI] [PubMed] [Google Scholar]
- 100.Chiquet-Ehrismann R, Kalla P, Pearson CA, Beck K, Chiquet M. Tenascin interferes with fibronectin action. Cell. 1988;53:383–390. doi: 10.1016/0092-8674(88)90158-4. [DOI] [PubMed] [Google Scholar]
- 101.Fujie S, Maita H, Ariga H, Matsumoto K. Tenascin-X induces cell detachment through p38 mitogen-activated protein kinase activation. Biol Pharm Bull. 2009;32:1795–1799. doi: 10.1248/bpb.32.1795. [DOI] [PubMed] [Google Scholar]
- 102.Matsumoto K, Takayama N, Ohnishi J, Ohnishi E, Shirayoshi Y, Nakatsuji N, Ariga H. Tumour invasion and metastasis are promoted in mice deficient in tenascin-X. Genes Cells. 2001;6:1101–1111. doi: 10.1046/j.1365-2443.2001.00482.x. [DOI] [PubMed] [Google Scholar]
- 103.Matsumoto K, Minamitani T, Orba Y, Sato M, Sawa H, Ariga H. Induction of matrix metalloproteinase-2 by tenascin-X deficiency is mediated through the c-Jun N-terminal kinase and protein tyrosine kinase phosphorylation pathway. Exp Cell Res. 2004;297:404–414. doi: 10.1016/j.yexcr.2004.03.041. [DOI] [PubMed] [Google Scholar]
- 104.Ikuta T, Ariga H, Matsumoto KI. Effect of tenascin-X together with vascular endothelial growth factor A on cell proliferation in cultured embryonic hearts. Biol Pharm Bull. 2001;24:1320–1323. doi: 10.1248/bpb.24.1320. [DOI] [PubMed] [Google Scholar]
- 105.Lee BR, Silver RI, Partin AW, Epstein JI, Gearhart JP. A quantitative histologic analysis of collagen subtypes: the primary obstructed and refluxing megaureter of childhood. Urology. 1998;51:820–823. doi: 10.1016/s0090-4295(98)00013-2. [DOI] [PubMed] [Google Scholar]
- 106.Sokolis DP. Multiaxial mechanical behaviour of the passive ureteral wall: experimental study and mathematical characterisation. Comput Methods Biomech Biomed Engin. 2012;15:1145–1156. doi: 10.1080/10255842.2011.581237. [DOI] [PubMed] [Google Scholar]
- 107.Rassoli A, Shafigh M, Seddighi A, Seddighi A, Daneshparvar H, Fatouraee N. Biaxial mechanical properties of human ureter under tension. Urol J. 2014;11:1678–1686. [PubMed] [Google Scholar]
- 108.Shilo Y, Pichamuthu JE, Averch TD, Vorp DA. Evaluation of the tensile strength of the human ureter—preliminary results. J Endourol. 2014;28:1470–1473. doi: 10.1089/end.2014.0226. [DOI] [PubMed] [Google Scholar]
- 109.Escala JM, Keating MA, Boyd G, Pierce A, Hutton JL, Lister J. Development of elastic fibres in the upper urinary tract. J Urol. 1989;141:969–973. doi: 10.1016/s0022-5347(17)41080-9. [DOI] [PubMed] [Google Scholar]
- 110.Andersson KE, McCloskey KD. Lamina propria: the functional center of the bladder? Neurourol Urodyn. 2014;33:9–16. doi: 10.1002/nau.22465. [DOI] [PubMed] [Google Scholar]
- 111.Chang SL, Howard PS, Koo HP, Macarak EJ. Role of type III collagen in bladder filling. Neurourol Urodyn. 1998;17:135–145. doi: 10.1002/(sici)1520-6777(1998)17:2<135::aid-nau7>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 112.Aitken KJ, Bagli DJ. The bladder extracellular matrix. Part I: architecture, development and disease. Nat Rev Urol. 2009;6:596–611. doi: 10.1038/nrurol.2009.201. [DOI] [PubMed] [Google Scholar]
- 113.Macarak EJ, Howard PS. The role of collagen in bladder filling. Adv Exp Med Biol. 1999;462:215–223. doi: 10.1007/978-1-4615-4737-2_17. discussion 225–233. [DOI] [PubMed] [Google Scholar]
- 114.Callewaert B, Malfait F, Loeys B, De Paepe A. Ehlers–Danlos syndromes and Marfan syndrome. Best Pract Res Clin Rheumatol. 2008;22:165–189. doi: 10.1016/j.berh.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 115.De Paepe A, Malfait F. The Ehlers–Danlos syndrome, a disorder with many faces. Clin Genet. 2012;82:1–11. doi: 10.1111/j.1399-0004.2012.01858.x. [DOI] [PubMed] [Google Scholar]
- 116.Wenstrup RJ, Florer JB, Cole WG, Willing MC, Birk DE. Reduced type I collagen utilization: a pathogenic mechanism in COL5A1 haplo-insufficient Ehlers–Danlos syndrome. J Cell Biochem. 2004;92:113–124. doi: 10.1002/jcb.20024. [DOI] [PubMed] [Google Scholar]
- 117.Malfait F, Wenstrup RJ, De Paepe A. Clinical and genetic aspects of Ehlers–Danlos syndrome, classic type. Genet Med. 2010;12:597–605. doi: 10.1097/GIM.0b013e3181eed412. [DOI] [PubMed] [Google Scholar]
- 118.Malfait F, De Coster P, Hausser I, van Essen AJ, Franck P, Colige A, Nusgens B, Martens L, De Paepe A. The natural history, including orofacial features of three patients with Ehlers–Danlos syndrome, dermatosparaxis type (EDS type VIIC) Am H Med Genet Part A. 2004;131:18–28. doi: 10.1002/ajmg.a.30299. [DOI] [PubMed] [Google Scholar]
- 119.Symoens S, Malfait F, Renard M, Andre J, Hausser I, Loeys B, Coucke P, De Paepe A. COL5A1 signal peptide mutations interfere with protein secretion and cause classic Ehlers–Danlos syndrome. Hum Mutat. 2009;30:E395–E403. doi: 10.1002/humu.20887. [DOI] [PubMed] [Google Scholar]
- 120.Zalis EG, Roberts DC. Ehlers–Danlos syndrome with a hypoplastic kidney, bladder diverticulum, and diaphragmatic hernia. Arch Dermatol. 1967;96:540–544. doi: 10.1001/archderm.96.5.540. [DOI] [PubMed] [Google Scholar]
- 121.Burrows NP, Monk BE, Harrison JB, Pope FM. Giant bladder diverticulum in Ehlers–Danlos syndrome type I causing outflow obstruction. Clin Exp Dermatol. 1998;23:109–112. doi: 10.1046/j.1365-2230.1998.00338.x. [DOI] [PubMed] [Google Scholar]
- 122.Cuckow PM, Blackhall RJ, Mouriquand PD. Huge bladder diverticula associated with Ehlers–Danlos syndrome. J R Soc Med. 1994;87:290–291. doi: 10.1177/014107689408700517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Levard G, Aigrain Y, Ferkadji L, Elghoneimi A, Pichon J, Boureau M. Urinary bladder diverticula and the Ehlers–Danlos syndrome in children. J Pediatr Surg. 1989;24:1184–1186. doi: 10.1016/s0022-3468(89)80115-0. [DOI] [PubMed] [Google Scholar]
- 124.Berger J, Lang E, Schaeffer EM. Urological radiographic manifestations of the Ehlers–Danlos syndrome. J Urol. 2007;178:1490. doi: 10.1016/j.juro.2007.06.037. [DOI] [PubMed] [Google Scholar]
- 125.Jorion JL, Michel M. Spontaneous rupture of bladder diverticula in a girl with Ehlers–Danlos syndrome. J Pediatr Surg. 1999;34:483–484. doi: 10.1016/s0022-3468(99)90506-7. [DOI] [PubMed] [Google Scholar]
- 126.Handa S, Sethuraman G, Mohan A, Sharma VK. Ehlers–Danlos syndrome with bladder diverticula. Br J Dermatol. 2001;144:1084–1085. doi: 10.1046/j.1365-2133.2001.04205.x. [DOI] [PubMed] [Google Scholar]
- 127.Shukla AR, Bellah RA, Canning DA, Carr MC, Snyder HM, Zderic SA. Giant bladder diverticula causing bladder outlet obstruction in children. J Urol. 2004;172:1977–1979. doi: 10.1097/01.ju.0000140450.50242.50. [DOI] [PubMed] [Google Scholar]
- 128.Eadie DG, Wilkins JL. Bladder-neck obstruction and the Ehlers–Danlos syndrome. Br J Urol. 1967;39:353–358. doi: 10.1111/j.1464-410x.1967.tb09815.x. [DOI] [PubMed] [Google Scholar]
- 129.Kuivaniemi H, Peltonen L, Palotie A, Kaitila I, Kivirikko KI. Abnormal copper metabolism and deficient lysyl oxidase activity in a heritable connective tissue disorder. J Clin Invest. 1982;69:730–733. doi: 10.1172/JCI110503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Breivik N, Refsum S, Jr, Oppedal BR, Vesterhus P. Ehlers–Danlos syndrome and diverticula of the bladder. Z Kinderchir. 1985;40:243–246. doi: 10.1055/s-2008-1059784. [DOI] [PubMed] [Google Scholar]
- 131.Schippers E, Dittler HJ. Multiple hollow organ dysplasia in Ehlers–Danlos syndrome. J Pediatr Surg. 1989;24:1181–1183. doi: 10.1016/s0022-3468(89)80114-9. [DOI] [PubMed] [Google Scholar]
- 132.Kahn T, Reiser M, Gmeinwieser J, Heuck A. The Ehlers–Danlos syndrome, type IV, with an unusual combination of organ malformations. Cardiovasc Intervent Radiol. 1988;11:288–291. doi: 10.1007/BF02577038. [DOI] [PubMed] [Google Scholar]
- 133.Bade JJ, Ypma AF, van Elk P, Mensink HJ. A pelvic mass: bladder diverticulum with haemorrhage in Ehlers–Danlos patient. Scand J Urol Nephrol. 1994;28:319–321. [PubMed] [Google Scholar]
- 134.Stage KH, Tank ES. Primary congenital bladder diverticula in boys. Urology. 1992;40:536–538. doi: 10.1016/0090-4295(92)90410-x. [DOI] [PubMed] [Google Scholar]
- 135.Kumar S, Jayant K, Barapatra Y, Rani J, Agrawal S. Giant Urinary Bladder Diverticula presenting as Epigastric Mass and Dyspepsia. Nephrourol Mon. 2014;6:e18918. doi: 10.5812/numonthly.18918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ghosh AK, O’Bryan T. Ehlers–Danlos syndrome with reflux nephropathy. Nephron. 1995;70:266. doi: 10.1159/000188598. [DOI] [PubMed] [Google Scholar]
- 137.Beighton P, De Paepe A, Steinmann B, Tsipouras P, Wenstrup RJ. Ehlers–Danlos syndromes: revised nosology, Villefranche, 1997. Ehlers–Danlos National Foundation (USA) and Ehlers–Danlos Support Group (UK) Am J Med Genet. 1998;77:31–37. doi: 10.1002/(sici)1096-8628(19980428)77:1<31::aid-ajmg8>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 138.Forleo LH, Hilario MO, Peixoto AL, Naspitz C, Goldenberg J. Articular hypermobility in school children in Sao Paulo, Brazil. J Rheumatol. 1993;20:916–917. [PubMed] [Google Scholar]
- 139.Larsson LG, Baum J, Mudholkar GS, Srivastava DK. Hypermobility: prevalence and features in a Swedish population. Br J Rheumatol. 1993;32:116–119. doi: 10.1093/rheumatology/32.2.116. [DOI] [PubMed] [Google Scholar]
- 140.Decoster LC, Vailas JC, Lindsay RH, Williams GR. Prevalence and features of joint hypermobility among adolescent athletes. Arch Pediatr Adolesc Med. 1997;151:989–992. doi: 10.1001/archpedi.1997.02170470023005. [DOI] [PubMed] [Google Scholar]
- 141.Rikken-Bultman DG, Wellink L, van Dongen PW. Hypermobility in two Dutch school populations. Eur J Obstet Gynecol Reprod Biol. 1997;73:189–192. doi: 10.1016/s0301-2115(97)02745-0. [DOI] [PubMed] [Google Scholar]
- 142.Remvig L, Jensen DV, Ward RC. Epidemiology of general joint hypermobility and basis for the proposed criteria for benign joint hypermobility syndrome: review of the literature. J Rheumatol. 2007;34:804–809. [PubMed] [Google Scholar]
- 143.De Wandele I, Rombaut L, Malfait F, De Backer T, De Paepe A, Calders P. Clinical heterogeneity in patients with the hypermobility type of Ehlers–Danlos syndrome. Res Dev Disabil. 2013;34:873–881. doi: 10.1016/j.ridd.2012.11.018. [DOI] [PubMed] [Google Scholar]
- 144.Remvig L, Jensen DV, Ward RC. Are diagnostic criteria for general joint hypermobility and benign joint hypermobility syndrome based on reproducible and valid tests? A review of the literature. J Rheumatol. 2007;34:798–803. [PubMed] [Google Scholar]
- 145.Tinkle BT, Bird HA, Grahame R, Lavallee M, Levy HP, Sillence D. The lack of clinical distinction between the hypermobility type of Ehlers–Danlos syndrome and the joint hypermobility syndrome (a.k.a. hypermobility syndrome) Am J Med Genet Part A. 2009;149A:2368–2370. doi: 10.1002/ajmg.a.33070. [DOI] [PubMed] [Google Scholar]
- 146.van Eerde AM, Verhoeven VJ, de Jong TP, van de Putte EM, Giltay JC, Engelbert RH. Is joint hypermobility associated with vesico-ureteral reflux? An assessment of 50 patients. BJU Int. 2012;109:1243–1248. doi: 10.1111/j.1464-410X.2011.10469.x. [DOI] [PubMed] [Google Scholar]
- 147.Pournasiri Z, Madani A, Zandi H, Salehpour S, Abdollah Gorji F, Ahmadzahe A. Relationship of generalized joint hypermobility with vesicoureteral reflux and urinary tract infection. Iran J Kidney Dis. 2014;8:189–193. [PubMed] [Google Scholar]
- 148.Beiraghdar F, Rostami Z, Panahi Y, Einollahi B, Teimoori M. Vesicourethral reflux in pediatrics with hypermobility syndrome. Nephrourol Mon. 2013;5:924–927. doi: 10.5812/numonthly.10770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Adib N, Davies K, Grahame R, Woo P, Murray KJ. Joint hypermobility syndrome in childhood. A not so benign multisystem disorder? Rheumatology (Oxford) 2005;44:744–750. doi: 10.1093/rheumatology/keh557. [DOI] [PubMed] [Google Scholar]
- 150.Malfait F, Hakim AJ, De Paepe A, Grahame R. The genetic basis of the joint hypermobility syndromes. Rheumatology (Oxford) 2006;45:502–507. doi: 10.1093/rheumatology/kei268. [DOI] [PubMed] [Google Scholar]
- 151.Mohamed M, Voet M, Gardeitchik T, Morava E. Cutis Laxa. Adv Exp Med Biol. 2014;802:161–184. doi: 10.1007/978-94-007-7893-1_11. [DOI] [PubMed] [Google Scholar]
- 152.Morava E, Guillard M, Lefeber DJ, Wevers RA. Autosomal recessive cutis laxa syndrome revisited. Eur J Hum Genet. 2009;17:1099–1110. doi: 10.1038/ejhg.2009.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Alehossein M, Pourgholami M, Kamrani K, Soltani M, Yazdi A, Salamati P. Radiologic findings in cutis laxa syndrome and unusual association with hypertrophic pyloric stenosis. Iran J Radiol. 2013;10:94–98. doi: 10.5812/iranjradiol.4064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Agha A, Sakati NO, Higginbottom MC, Jones KL, Jr, Bay C, Nyhan WL. Two forms of cutis laxa presenting in the newborn period. Acta Paediatr Scand. 1978;67:775–780. doi: 10.1111/j.1651-2227.1978.tb16260.x. [DOI] [PubMed] [Google Scholar]
- 155.Robertson SP, Bankier A. Sotos syndrome and cutis laxa. J Med Genet. 1999;36:51–56. [PMC free article] [PubMed] [Google Scholar]
- 156.Kielty CM, Davies SJ, Phillips JE, Jones CJ, Shuttleworth CA, Charles SJ. Marfan syndrome: fibrillin expression and microfibrillar abnormalities in a family with predominant ocular defects. J Med Genet. 1995;32:1–6. doi: 10.1136/jmg.32.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Robinson PN, Godfrey M. The molecular genetics of Marfan syndrome and related microfibrillopathies. J Med Genet. 2000;37:9–25. doi: 10.1136/jmg.37.1.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Clunie GJ, Mason JM. Visceral diverticula and the Marfan syndrome. Br J Surg. 1962;50:51–52. doi: 10.1002/bjs.18005021913. [DOI] [PubMed] [Google Scholar]
- 159.de Silva DG, Gunawardena TP, Law FM. Unusual complications in siblings with marfanoid phenotype. Arch Dis Child. 1996;75:247–248. doi: 10.1136/adc.75.3.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Al-Hakim W, Goldsmith DJ. Bilateral popliteal aneurysms complicating adult polycystic kidney disease in a patient with a marfanoid habitus. Postgrad Med J. 2003;79:474–475. doi: 10.1136/pmj.79.934.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Stromme P, Bjornstad PG, Ramstad K. Prevalence estimation of Williams syndrome. J Child Neurol. 2002;17:269–271. doi: 10.1177/088307380201700406. [DOI] [PubMed] [Google Scholar]
- 162.Pober BR. Williams-Beuren syndrome. N Engl J Med. 2010;362:239–252. doi: 10.1056/NEJMra0903074. [DOI] [PubMed] [Google Scholar]
- 163.Stoermer J, Olbing H, Hentrich F, Even K, Galal O, Bachmann J. Syndrome of supravalvular aortic stenosis (Williams-Beuren syndrome) in association with changes in the kidney and efferent urinary tract. Monatsschr Kinderheilkd. 1984;132:110–112. [PubMed] [Google Scholar]
- 164.Pober BR, Lacro RV, Rice C, Mandell V, Teele RL. Renal findings in 40 individuals with Williams syndrome. Am J Med Genet. 1993;46:271–274. doi: 10.1002/ajmg.1320460306. [DOI] [PubMed] [Google Scholar]
- 165.Pankau R, Partsch CJ, Winter M, Gosch A, Wessel A. Incidence and spectrum of renal abnormalities in Williams-Beuren syndrome. Am J Med Genet. 1996;63:301–304. doi: 10.1002/(SICI)1096-8628(19960503)63:1<301::AID-AJMG49>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 166.Sugayama SM, Koch VH, Furusawa EA, Leone C, Kim CA. Renal and urinary findings in 20 patients with Williams-Beuren syndrome diagnosed by fluorescence in situ hybridization (FISH) Rev Hosp Clin Fac Med Sao Paulo. 2004;59:266–272. doi: 10.1590/s0041-87812004000500008. [DOI] [PubMed] [Google Scholar]
- 167.Morris CA, Leonard CO, Dilts C, Demsey SA. Adults with Williams syndrome. Am J Med Genet Suppl. 1990;6:102–107. doi: 10.1002/ajmg.1320370619. [DOI] [PubMed] [Google Scholar]