

Abstract
Chloroquine, long the default first-line treatment against malaria, is now abandoned in large parts of the world because of widespread drug-resistance in Plasmodium falciparum. In spite of its importance as a cost-effective and efficient drug, a coherent understanding of the cellular mechanisms affected by chloroquine and how they influence the fitness and survival of the parasite remains elusive. Here, we used a systems biology approach to integrate genome-scale transcriptomics to map out the effects of chloroquine, identify targeted metabolic pathways, and translate these findings into mechanistic insights. Specifically, we first developed a method that integrates transcriptomic and metabolomic data, which we independently validated against a recently published set of such data for Krebs-cycle mutants of P. falciparum. We then used the method to calculate the effect of chloroquine treatment on the metabolic flux profiles of P. falciparum during the intraerythrocytic developmental cycle. The model predicted dose-dependent inhibition of DNA replication, in agreement with earlier experimental results for both drug-sensitive and drug-resistant P. falciparum strains. Our simulations also corroborated experimental findings that suggest differences in chloroquine sensitivity between ring- and schizont-stage P. falciparum. Our analysis also suggests that metabolic fluxes that govern reduced thioredoxin and phosphoenolpyruvate synthesis are significantly decreased and are pivotal to chloroquine-based inhibition of P. falciparum DNA replication. The consequences of impaired phosphoenolpyruvate synthesis and redox metabolism are reduced carbon fixation and increased oxidative stress, respectively, both of which eventually facilitate killing of the parasite. Our analysis suggests that a combination of chloroquine (or an analogue) and another drug, which inhibits carbon fixation and/or increases oxidative stress, should increase the clearance of P. falciparum from the host system.
Keywords: Plasmodium, Chloroquine, Metabolic network modeling, Redox metabolism, Carbon fixation
Graphical abstract
1. Introduction
Malaria, which is caused by a protozoan parasite of the Plasmodium genus, represents a serious global health concern given that nearly half of the world population is at risk of infection (WHO, 2014). Although several anti-malarial drugs for treating the symptomatic stage (or blood stage) of malarial infection are commercially available (Antony and Parija, 2016), their efficacy has declined appreciably in the last few decades owing to widespread drug resistance developed by the parasite (Breman et al., 2004).
Plasmodium falciparum—the most lethal species of malaria—causes approximately 50% of all malarial infections. Chloroquine was the first-line malaria treatment for many decades until drug-resistant P. falciparum strains became common. The drug causes a dose-dependent decrease in hemozoin formation (Chou and Fitch, 1992, Slater and Cerami, 1992) and an associated increase in toxic free heme in the food vacuole of the parasite (Combrinck et al., 2013, Loria et al., 1999). Over the past few decades, researchers have proposed many different mechanisms for chloroquine action, including 1) DNA intercalation (Meshnick, 1990), 2) alteration of digestive food vacuole pH (Yayon et al., 1985), 3) inhibition of heme polymerase (Yayon et al., 1985), and 4) formation of a toxic chloroquine-ferriprotoporphyrin IX complex (Sugioka et al., 1987). Yet, there is no consensus on the exact mechanisms by which chloroquine kills the parasite and under what circumstances they operate. Shedding light on the possible actions of chloroquine is critical for developing more potent drugs or combination drug treatments against the parasite and for understanding how the parasite can develop resistance against them.
Here we used systems biology model and transcriptional profiles of P. falciparum to obtain information about metabolic and biological precursors that determine the physiological or pathophysiological state of the parasite. Specifically, we used the P. falciparum transcriptomic data obtained by Hu and colleagues during the intraerythrocytic development cycle (IDC) at different concentrations of chloroquine (Hu et al., 2010). Our primary goal was to use these data to investigate the effect of chloroquine on P. falciparum DNA replication. We chose to study this effect because it has been proposed as a mechanism of chloroquine action in different Plasmodium species (Gutteridge et al., 1972, Meshnick, 1990, Polet and Barr, 1968a, Polet and Barr, 1968b), in bacteria (Ciak and Hahn, 1966), and in bioassays examining the effect of chloroquine on RNA and DNA polymerases (Cohen and Yielding, 1965, O'Brien et al., 1966b, Whichard et al., 1972).
We first developed a method that integrates the information embedded in the transcriptome data with the whole-genome metabolic network model for P. falciparum (Fang et al., 2014) to predict the metabolic phenotype corresponding to those transcription data. We validated the developed method by using a data set from an independent study that jointly capture transcriptomic and metabolomic data from P. falciparum, to correctly predict the metabolic phenotype tied to genetic perturbation of two Krebs cycle enzymes (Ke et al., 2015). We then focused on the inhibition of DNA replication in P. falciparum (as a consequence of chloroquine treatment) a critical metabolic phenotype (Ciak and Hahn, 1966, Cohen and Yielding, 1965, Polet and Barr, 1968a), and used our method to simulate the effect of chloroquine on the metabolic fluxes of P. falciparum during the IDC. We identified genes that were substantially altered in response to chloroquine treatment and linked to the inhibition of P. falciparum DNA replication. The cohort of identified genes suggests that DNA replication is inhibited by the downstream effect of heme accumulation. Specifically, our analysis suggests that continuous accumulation of heme inhibits redox metabolism, carbon fixation, and pyrimidine metabolism, which leads to inhibition of DNA replication and facilitates death of the parasite. Our results provide a mechanistic explanation for why parasites with an efficient redox metabolism may have a lower sensitivity to chloroquine (Kasozi et al., 2013, Lehane et al., 2012, Meierjohann et al., 2002).
2. Materials and methods
2.1. Transcriptome data used for simulating metabolic flux profile during IDC
Metabolic flux profiles of P. falciparum strains 3D7 (Pf3D7) and Dd2 (PfDd2) during the IDC were estimated by using transcriptome data previously reported by Llinas and colleagues (Llinas et al., 2006), who extracted cDNA from parasites cultured in erythrocytes and hybridized them to DNA microarrays to yield hourly levels of gene expression for the Pf3D7 (or PfDd2) strain during the IDC. The temporal resolution of these hourly gene expression profiles allowed us to make high-resolution time-dependent metabolic flux predictions during the IDC.
We used previously reported gene expression data (Hu et al., 2010) to simulate the effect of chloroquine on the time-dependent metabolic fluxes of Pf3D7 and PfDd2 during the IDC. Briefly, Hu and colleagues (Hu et al., 2010) administered chloroquine 18 h after erythrocyte invasion at concentrations of 41, 72, and 144 nM for Pf3D7 and at 43 nM for PfDd2, and obtained gene expression profiles every hour for 8 h during the IDC following the chloroquine treatment.
2.2. Metabolic network model and data processing
The metabolic network model used involved 1045 reactions and 376 genes. The model was identical to a previously published model (Fang et al., 2014), with one minor modification: we added reactions explicitly carried out by Pdx2 (PF11_0169) and Pdx1 (MAL6P1.215). This was done by 1) modifying an old reaction that incorrectly assumed that pyridoxal 5-phosphate (PLP) was synthesized from ribulose 5-phosphate and 2) adding a new (and separate) reaction for Pdx1 because experiments suggest that it can synthesize PLP independent of the ammonia generated by Pdx2 (Hanes et al., 2008). In the updated model, Pdx2 hydrolyzes L-glutamine to yield glutamate and ammonia while Pdx1 incorporates the ammonia generated by Pdx2 to yield PLP from the substrates ribose 5-phosphate and glyceraldehyde 3-phosphate (Gengenbacher et al., 2006), with the isomerization of ribose 5-phosphate to ribulose 5-phosphate as a side reaction.
We used gene-to-reaction mappings available in the metabolic network model to obtain reaction expressions , which depend on the expression of a gene (or genes) catalyzing a particular metabolic reaction. For a reaction catalyzed by a single enzyme, was equal to the expression of the gene catalyzing that reaction. However, if a reaction was associated with more than one gene, then their gene-to-reaction mappings were defined by using the Boolean operators “AND” and “OR.” We implemented these operations by taking the minimal (for the AND operator) and maximal (for the OR operator) values of the associated gene expression data (Song et al., 2014).
2.3. Estimating metabolic flux changes in response to external stress
We recently developed a method (Fang et al., 2014) that integrates hourly gene expression data with a whole-genome metabolic network model to estimate the hourly metabolic flux profiles of P. falciparum during the 48-h long IDC period. To take advantage of additional studies that collect gene expression data as a function of genetic or chemical perturbations, we modified the method because such studies usually do not provide the same granularity and density of gene expression data to fully cover the entire IDC. Therefore, we aimed to develop a method that could utilize a reduced transcriptomic data set to predict alterations in metabolic flux profiles during the IDC. Briefly, We used the following steps to estimate metabolic fluxes under no-stress conditions:
Step 1: Estimate nutrient fluxes sufficient for the nominal growth rate (40% of the maximum growth rate).
Step 2: Estimate reaction fluxes for metabolite i corresponding to the nominal growth rate constrained by the nutrient fluxes obtained in Step 1.
Step 3: Estimate reaction fluxes for reaction i at any time point t during the IDC by incorporating time-dependent transcriptomic profiles under stoichiometric and nutrient uptake constraints.
The last step was achieved by globally minimizing for all reactions and time points the value of t that minimized the weighted absolute difference, , where G represents the set of unidirectional reactions with known gene-to-reaction mapping. The factor ; where, represents reaction expression of ith reaction at time t obtained from the gene-to-reaction mapping of the P. falciparum metabolic network, according to the approach outlined in Section 2.2. This factor, , ensured that the mean of the ith reaction during the IDC was equal to . The complete procedure has been detailed elsewhere (Fang et al., 2014).
In this work, we estimated the organism-level impact of a drug on metabolism by using transcriptomic data obtained after drug treatment. The original formulation outlined above (Steps 1–3) cannot be directly translated to stress conditions unless the complete transcriptomic profile for each stress condition during the entire IDC is available (Fang et al., 2014). Therefore, we introduced a parameter to account for up- or down-regulation of individual reactions in response to stress to simulate the effect of exogenous stress on metabolic flux profiles during the IDC. We modified the factor to , where denotes the median relative change in the reaction expression of the ith reaction following chloroquine treatment, as observed in previous experiments (Hu et al., 2010). Note that the mean of each individual modifying factor , i.e., , obtained by summing the values across all time points in the IDC, tracks with ; that is, when the mean is less than one, greater than one, or equal to one, , , or , respectively. In other words, increases or decreases the nominal flux of the ith reaction during the entire IDC depending on the relative increase or decrease in the ith reaction expression. In principle, a drug may differentially affect gene transcription at different time points during the IDC. Here, we assumed that the median change in gene transcription after a stress treatment was representative of the transcriptional response throughout the IDC.
2.4. Validating whether reaction flux estimates capture the effect of external stress?
We used a previously published data set (Ke et al., 2015) to validate our methodology to simulate the effect of external stress on metabolic fluxes during the IDC. Ke and colleagues (Ke et al., 2015) constructed a P. falciparum mutant with two tricarboxylic acid (TCA) cycle enzymes deleted (ΔIDH/ΔKDH), cultured wild-type and mutant parasites in erythrocytes, and obtained matched transcriptomic and metabolomic data. We used this data set because it describes changes in gene expression (transcriptomics) and phenotype (metabolomics) under the same experimental stress conditions.
If the reaction fluxes estimated from previously reported transcriptome data (Ke et al., 2015) adequately represent the reaction fluxes in vitro, then the predicted changes in reaction fluxes should be sufficient to predict the changes observed in the corresponding metabolomic data. We used a computer model of mitochondrial oxidative phosphorylation and the TCA cycle (Wu et al., 2007) to predict changes in TCA cycle metabolites in response to changes in these estimated reaction fluxes (Ke et al., 2015). The kinetic parameters of this computer model were determined and validated by using extensive experimental data. We simulated wild-type reactions by running this model, with its default parameters, for 2000 s (time required to reach steady-state condition) and stored the concentrations of TCA cycle metabolites. We performed simulations for the mutant parasite in the same way, albeit after scaling the model parameters that govern TCA cycle enzyme activities by the estimated increase or decrease in reaction fluxes of those enzymes. We used a freely available software package called BISEN (Vanlier et al., 2009) to implement the TCA cycle computer model (Wu et al., 2007).
2.5. Prediction of time-dependent P. falciparum DNA synthesis
P. falciparum takes up nutrients to synthesize DNA, RNA, proteins, and phospholipids. These processes are not constitutively activated, because the parasite will synthesize them as needed throughout the IDC. We used a validated in-house developed method (Fang et al., 2014) to simulate time-dependent DNA synthesis during the IDC. For comprehensiveness, we introduce the constraint governing the rate of DNA synthesis in the metabolic network model:
Here, dATP, dCTP, dGTP, and dTTP refer to deoxyadenosine, deoxycytidine, deoxyguanosine, and deoxythymidine triphosphates, respectively. We refer the reader to the original article (Fang et al., 2014) for further details on this method for simulating time-dependent P. falciparum macro-molecules during the IDC.
2.6. Identifying genes critical for P. falciparum DNA synthesis
The criticality of a gene is determined by optimizing the following objective function: , subjected to: and . Here, is the gene set containing the metabolites that contribute to DNA synthesis (introduced above) and is the contribution of the ith metabolite (. In addition, S is the stoichiometry matrix, is a vector containing all metabolic reactions of the P. falciparum metabolic network model, m is the number of metabolites in the network, n is the number of reactions in the network, and and are the upper and lower bounds, respectively, of metabolic reactions. We deemed a reaction as critical if there was no DNA synthesis after knocking out the reaction in silico. We denoted any gene encoding such a reaction as a critical gene.
2.7. Identifying genes underlying chloroquine-induced DNA synthesis inhibition
Chloroquine administration inhibits DNA replication in malaria parasites (Cohen and Yielding, 1965, Polet and Barr, 1968a). Likewise, existing transcriptome data (Hu et al., 2010) also show that metabolic genes critical for P. falciparum DNA synthesis are suppressed by chloroquine treatment. We sought to identify the genes that are substantially altered in response to chloroquine and cause inhibition of P. falciparum DNA synthesis. Obvious candidates are genes that are down-regulated and critical for P. falciparum DNA synthesis. However, not all such genes will have a pronounced effect on DNA synthesis, which depends on the extent to which an individual gene is down-regulated and/or the individual contribution of that gene to the DNA synthesis of the parasite. This suggests that many combinations of such genes may underlie the metabolic response associated with chloroquine treatment. Following Occam's razor (Blumer et al., 1990), we aimed at identifying the minimal number of genes (hereinafter referred to as the core genes) that are sufficient to completely account for the effect of chloroquine on DNA synthesis by the parasite.
We identified the core genes based on the Bayesian information criteria (BIC) score (Schwarz, 1978), which is given by, , where and denotes the number of time points, k the number of genes being tested, the estimated rate of DNA synthesis under control conditions, and the estimated rate of DNA synthesis after chloroquine treatment (with for the genes being tested). We implemented an algorithm that starts with a set of size and increases the size of the test set as long as a given combination of genes for the new set (size ) has a BIC score lower than the minimal BIC score of the previous size. We exhaustively repeated this process at each chloroquine dose (41, 72, and 144 nM for Pf3D7, and 43 nM for PfDd2).
3. Results
3.1. Validating the phenotype of reaction flux estimates
We modified our previous approach (Fang et al., 2014) to simulate the effect of external stress, as depicted in Fig. 1 and described in the Materials and methods Section. We validated the methodology by demonstrating that reaction fluxes obtained in response to stress were sufficient to predict metabolic phenotypes corresponding to that stress. Fig. 2A shows the biochemical pathways incorporated into the computer model used to predict the functional consequences of altered fluxes of TCA cycle enzymes in response to previously studied genetic perturbations (Ke et al., 2015). Fig. 2B shows the predicted alterations in TCA cycle metabolites in response to genetic perturbations of TCA cycle enzymes (Fig. 2A, red circles). We calculated the altered metabolite concentrations by scaling the control parameters in the TCA cycle model (Wu et al., 2007) by the median of the predicted changes in TCA cycle enzyme activities (Fig. 2C). Our predictions of TCA cycle metabolites (Fig. 2B, filled bars) qualitatively captured the trends of relative change in experimentally measured TCA cycle metabolites (Fig. 2B, open bars). However, our prediction of the citrate level (Fig. 2B, filled bars) was not within the margin of error of the experimental measurements (Fig. 2B, open bars). We examined all possible enzymes/transporters (e.g., citrate synthase, pyruvate dehydrogenase, aspartate aminotransferase, citrate-malate antiporter) whose perturbation could yield mitochondrial citrate levels higher than those in the wild-type system. In each scenario, either the predicted citrate levels did not increase or the predicted oxaloacetate levels increased. Therefore, based on our model analysis, the source of this increase in the experimentally reported citrate level appears to be non-mitochondrial in nature.
Fig. 1.

Schematic representation of the method developed to simulate the effect of chloroquine treatment on metabolic flux profiles of P. falciparum during the intraerythrocytic development cycle. The method shown above with ‘’ is identical to a previously proposed method (Fang et al., 2014) and is used to estimate reaction fluxes under control conditions (black curves in the bottom panel). We modified the previous approach (Fang et al., 2014) to integrate the effect of stress (such as chloroquine treatment). In the metabolic network shown above, are intracellular fluxes; is the input; is the output; and is the flux representing the growth rate. We simulated the effect of external stress by scaling the baseline flux of a reaction (), using the relative change in the transcriptome data (α) of that reaction in response to stress. The objective was to find a new intracellular flux distribution (red curves in the bottom panel) based on the modified . These new intracellular fluxes were sufficient to capture the phenotypic alterations that occur in response to external stress. In the dotted box, w represents the weight of individual reactions, S represents the stoichiometry of reactions, and lb and ub represent lower and upper bounds of reactions, respectively. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Fig. 2.

Reaction fluxes based on changes in gene transcription obtained in response to a certain stress condition are sufficient to mimic the metabolic phenotype corresponding to that condition. A) Schematic representation of the thermodynamically balanced computer model of mitochondrial oxidative phosphorylation used to predict relative changes in metabolites on the basis of relative changes in estimated reaction fluxes under wild-type (WT) and knockout (KO) conditions; figure modified and reproduced from (Wu et al., 2007). The two red circles represent the two genes that were deleted in a previous study (Ke et al., 2015). B) Alterations in the concentrations of TCA cycle metabolites after the deletion of two TCA cycle enzymes. Open bars show experimentally observed values and closed bars the computationally predicted values. Metabolite concentrations were computed by using the model shown in A and reaction flux ratios shown in C. C) Median relative changes in TCA cycle enzyme fluxes, estimated by using the approach shown in Fig. 1, during the intraerythrocytic development cycle under two conditions (WT and KO). The effect of the stress condition, i.e., knockout of two TCA cycle enzymes, is captured by the transcriptome data obtained under WT and KO conditions (Ke et al., 2015). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.2. Chloroquine inhibits P. falciparum DNA synthesis
In agreement with previous experimental observations (Cohen and Yielding, 1965, Polet and Barr, 1968a), our simulations suggested that chloroquine treatment results in dose-dependent inhibition of DNA replication (Fig. 3A). At chloroquine concentrations of 41 nM (Pf3D7, panel A, blue diamonds) and 43 nM (PfDd2, panel B, blue diamonds), the rate of DNA synthesis during the ring stage (the first 18 h after infection) was less inhibited than it was during the late trophozoite and early schizont phases (beyond 30 h after infection). These results are in agreement with previous observations (Yayon et al., 1983).
Fig. 3.

Simulated effect of chloroquine on rates of DNA synthesis by the parasite during the intraerythrocytic development cycle (IDC). In the legend, CQN denotes chloroquine. A) Dose-dependent decrease in the rate of DNA synthesis for Pf3D7. The DNA synthesis rate at a CQN dose of 0 nM was estimated from transcriptome data of Pf3D7 (Llinas et al., 2006). The effect of CQN dose on the DNA synthesis rate was simulated by the approach described in the Materials and methods Section, using transcriptome data of Pf3D7 (Hu et al., 2010). B) The DNA synthesis rate at a CQN dose of 0 nM was estimated from transcriptome data of PfDd2 (Llinas et al., 2006). The effect of CQN dose on the DNA synthesis rate was estimated by the same approach as that in A, using transcriptome data of PfDd2 (Hu et al., 2010). For both strains, the decrease in DNA synthesis occurring later in the IDC (beyond 30 h after infection) was more pronounced than that occurring early in the IDC (the first 18 h after infection) in response to CQN treatment. To account for variability of gene expression in our simulations, we added 10% Gaussian noise to the expression level of each gene. The results were averaged across twenty independent simulations. Error bars represent the standard error associated with each prediction.
Because many studies suggest that PfDd2 is less sensitive to chloroquine than Pf3D7 (Moneriz et al., 2011), DNA synthesis might have been expected to be more inhibited in Pf3D7 than in PfDd2. In our simulations, however, a similar concentration of chloroquine resulted in greater DNA synthesis inhibition in PfDd2 (Fig. 3B; blue diamonds versus black circles, paired-sample t-test; p < 0.0001) than in Pf3D7 (Fig. 3A; blue diamonds versus black circles, paired-sample t-test; p = 0.15). These differences likely stem from differences in the relative gene expression ratios of PfDd2 and Pf3D7. Notably, the transcriptional profile for PfDd2 was more down-regulated than that for Pf3D7 at a similar concentration of chloroquine. In other words, the percentage of down-regulated genes critical for P. falciparum DNA synthesis was greater in PfDd2 than in Pf3D7 (Table S1). Together with the highly similar IC50 values reported previously (Hu et al., 2010) for Pf3D7 (41 nM) and PfDd2 (43 nM), these results suggest that the PfDd2 strain used here may have diverged from its true (chloroquine-resistant) phenotype.
3.3. Genes critical for P. falciparum DNA synthesis
We found that 81 metabolic reactions in our network model are critical for P. falciparum DNA synthesis. We denoted these reactions as critical because their elimination resulted in abolished DNA synthesis (see Section 2.6). Of these 81 reactions, 64 had a known gene association whereas 17 did not. These 64 metabolic reactions were encoded by a total of 44 unique genes. These genes belonged to pathways involved in glycolysis, pentose phosphate synthesis, pyruvate metabolism, purine metabolism, pyrimidine metabolism, redox metabolism, and mitochondrial metabolism. In the online supplement (see spreadsheet), we provide detailed information, including the reaction name, chemical reaction, gene association, and pathway, of all reactions critical for P. falciparum DNA synthesis.
3.4. Genes significantly affected by chloroquine treatment
Experimental observations (Cohen and Yielding, 1965, Polet and Barr, 1968a) and the present simulations (Fig. 3) indicate that chloroquine treatment inhibits replication of P. falciparum DNA, which in turn suggests that pathways essential for DNA synthesis are inhibited. Fig. 4 shows the relative change in the amount of DNA synthesized by the parasite during the IDC period in response to chloroquine treatment. Table S2 lists the actual amounts of synthesized DNA for both strains under each simulated condition. We observed a dose-dependent decrease in the amount of DNA synthesized by the parasite during the IDC (Fig. 4, red markers). We used BIC scores (Schwarz, 1978) and employed the algorithm described in Section 2.7 to identify core genes that caused this net decrease of synthesized DNA. Preventing only the core genes from being down-regulated by chloroquine was sufficient to restore the amount of DNA synthesized to control values (Fig. 4, green markers); for these simulations, the reaction expressions of the core genes were set to be equal for control and chloroquine-treated conditions. A list of the core genes identified for each strain and chloroquine dose (Table S3) suggests that thioredoxin reductase and ribonucleotide reductase substantially affect the amount of synthesized DNA. For PfDd2, only two genes (thioredoxin reductase and ribonucleotide reductase) caused the nearly 50% reduction in the net amount of synthesized DNA (Table S3 and Fig. 4). These genes were not mentioned in the original transcriptome analysis of Hu and colleagues, who used a fundamentally different approach to interpret their transcriptome data (Hu et al., 2010). These authors focused on genes that were either co-regulated or showed strongly differential expression (≥8 log2-fold change) in response to a drug. In contrast, our methodology of coupling transcriptomic changes with a metabolic network model can predict physiological outcomes as they relate to parasite metabolism, regardless of the magnitude of transcriptional change for an individual gene. Transcriptional changes are not proportional to changes in metabolic reaction fluxes (Moxley et al., 2009). Our analysis suggests that low-amplitude alterations in these identified genes (Table S3) were sufficient to cause a significant reduction in parasite DNA synthesis.
Fig. 4.

Simulated effect of chloroquine on net synthesized DNA, obtained by integrating the rate of DNA synthesis (Fig. 3) over the intraerythrocytic development cycle. The ordinate shows values of net synthesized DNA (•) relative to control values as a function of chloroquine dose. The net amount of synthesized DNA decreases as the dose increases (red symbols). The amount of synthesized DNA returns back to control levels (green symbols) when is set to 1 for the core genes in Table S3, while simulating the effect of chloroquine on metabolic fluxes. Control values of net synthesized DNA (•) were obtained from transcriptome data of Pf3D7 or PfDd2 (Llinas et al., 2006) (Table S2). In the legend, CTL denotes DNA synthesized under control conditions after chloroquine treatment, whereas -CG denotes that after chloroquine treatment with set to 1 for core genes. The results were averaged across twenty independent simulations. Error bars represent the standard error associated with each prediction. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
4. Discussion
4.1. Simulating effect of exogenous stress
Several existing methods integrate whole-genome metabolic networks and transcriptome data (Colijn et al., 2009, Song et al., 2014, Wallqvist et al., 2016, Zur et al., 2010). Recently, we developed a method that predicts metabolic flux profiles of P. falciparum during the IDC (Fang et al., 2014). The transcriptome data of P. falciparum obtained under different stress conditions (e.g., drug exposure, genetic mutation, etc.) contain information about its functional state and can be used to predict qualitative alterations, if any, in metabolic flux profiles during the IDC. However, such data are often collected at a limited number of time points; for instance, Hu and colleagues (Hu et al., 2010) obtained P. falciparum transcriptome data at only 8 selected time points during the IDC. Currently, there is no approach to integrate limited data, such as those reported previously (Hu et al., 2010), with a whole-genome network to predict metabolic flux profiles during the IDC. This is mainly because such an approach requires gene expression data throughout the IDC to predict the corresponding metabolic flux profiles. Therefore, we developed an approach that uses gene expression data obtained under a stress condition to predict alterations of metabolic fluxes that correspond to that condition. We validated the developed method by integrating transcriptome data obtained under a given stress condition and predicting metabolomic data corresponding to that stress (Ke et al., 2015). Using the developed method, we simulated the effect of chloroquine on the metabolic flux profiles of P. falciparum during the IDC.
4.2. Chloroquine: mechanism of action and resistance
Chloroquine inhibits the formation of hemozoin and leads to heme accumulation, which is toxic to the parasite (Fitch, 1998). However, the specific mechanism underlying this toxicity is not known (Ginsburg et al., 1998, Lehane et al., 2012). Some experiments suggest inhibition of DNA replication as a potential mechanism of chloroquine action (Cohen and Yielding, 1965, Kwakye-Berko and Meshnick, 1989, Polet and Barr, 1968a). We developed a method that successfully predicted a reduction in the ability of the parasite to synthesize DNA in response to chloroquine treatment (see Fig. 3, Fig. 4). We identified twelve distinct genes that were substantially affected in Pf3D7 and PfDd2 after chloroquine treatment; importantly, preventing their down-regulation in response to chloroquine treatment was sufficient for restoring the P. falciparum DNA synthesis inhibited by chloroquine treatment (see Table 1). These genes belonged to pathways involved in glycolysis, redox metabolism, and pyrimidine metabolism.
Table 1.
Core genes significantly affected by chloroquine.
| EC Numbera | Gene ID | Enzyme name | Gene abbreviation | Pathway |
|---|---|---|---|---|
| 1.2.1.12 | PF14_0598 | Glyceraldehyde 3P dehydrogenase | GAPDH | Glycolysis |
| 2.7.2.3 | PFI1105w | Phosphoglycerate kinase | PGK | Glycolysis |
| 5.4.2.1 | PF11_0208 | Phosphoglycerate mutase | PGM | Glycolysis |
| 4.2.1.11 | PF10_0155 | Enolase | Enolase | Glycolysis |
| 1.11.1.15 | PF14_0368 | Thioredoxin peroxidase 1 | Tpx | Redox |
| 1.11.1.15 | PFL0725w | Thioredoxin peroxidase 2 | Tpx | Redox |
| 1.17.4.1 | PF10_0154 | Ribonucleotide reductase | RNR | Redox |
| 1.8.1.9 | PFI1170c | Thioredoxin reductase | TrxR | Redox |
| 1.10.2.2 | PF14_0248 | Ubiquinol-cytochrome c reductase | UQCR | Electron transport |
| 1.3.3.1 | PFF0160c | Dihydroorotate dehydrogenase | DHODH | Electron transport |
| 4.2.1.2 | PFI1340w | Fumarate hydratase | FUM | TCA cycleb |
| 2.7.4.6 | PF13_0349 | Nucleoside diphosphokinase | NDPK | Pyrimidine |
Enzyme Commission number.
Tricarboxylic acid cycle.
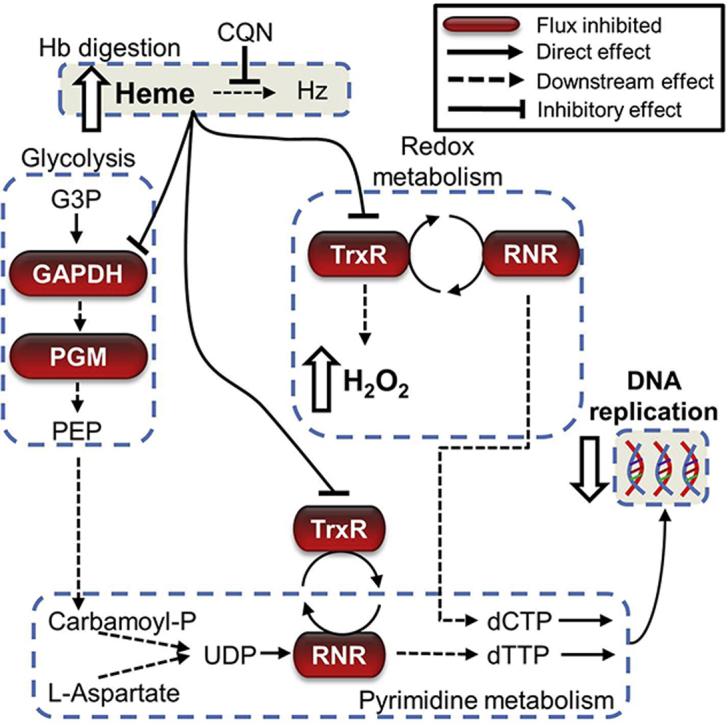
A closer look at these genes reveals a potential mechanism of chloroquine action that unifies previous theories and observations involving heme (Fitch, 1998), DNA replication (Polet and Barr, 1968a), and oxidative stress (Lehane et al., 2012, Radfar et al., 2008). Our proposed mechanism, which is summarized in Fig. 5, suggests that administration of chloroquine has two major metabolic effects:
-
i.
It interferes with the function of glyceraldehyde 3-phosphate dehydrogenase (GAPDH), which reduces the synthesis of phosphoenolpyruvate, whose carbon fixation activity is essential for anaplerotic TCA metabolism and redox balance (Storm et al., 2014).
-
ii.
Its interference of thioredoxin reductase (TrxR) reduces conversion of oxidized thioredoxin Trx (S2) to reduced thioredoxin Trx (SH2), which is essential for donating electrons to reactions catalyzed by thioredoxin peroxidase (Tpx) and ribonucleotide reductase (RNR). Tpx is essential for converting H2O2 (reactive) to H2O (harmless) (Pannala and Dash, 2015), and RNR for converting ribonucleoside 5′-diphosphates species (ADP, UDP, GDP and CDP), to deoxy-species (dADP, dUDP, dGDP and dCDP) which are used for DNA synthesis (Munro and Silva, 2012).
Fig. 5.

Predicted mechanism by which chloroquine kills the malaria parasite. Chloroquine (CQN) leads to the accumulation of heme. Although the downstream consequences of this effect are unknown, our analysis suggests that the accumulated heme leads to inhibition of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and thioredoxin reductase (TrxR), and that the downstream effects of these inhibitory events are the major determinants of chloroquine efficacy. This hypothesis also provides a mechanistic explanation of how chloroquine inhibits DNA synthesis (Cohen and Yielding, 1965, Polet and Barr, 1968a). Abbreviations: CDP, cytidine diphosphate; CO2, carbon dioxide; dCDP, deoxycytidine diphosphate; dCTP, deoxycytidine triphosphate; DHODH, dihydroorotate dehydrogenase; dTDP, deoxythymidine diphosphate; dTTP, deoxythymidine triphosphate; G3P, glyceraldehyde 3-phosphate; H2O, water; H2O2, hydrogen peroxide; Hb, hemoglobin; Hz, hemozoin; NADPH, reduced nicotinamide adenine dinucleotide phosphate; NADP+, nicotinamide adenine dinucleotide phosphate; NDPK, nucleoside diphosphokinase; PEP, phosphoenolpyruvate; PGK, phosphoglycerate kinase; PGM, phosphoglycerate mutase; PPP, pentose phosphate pathway; RNR, ribonucleotide reductase; TCA, tricarboxylic acid; Tpx, thioredoxin peroxidase; Trx (S2), oxidized thioredoxin; Trx (SH2), reduced thioredoxin; UDP, uridine diphosphate.
The proposed mechanism reveals candidate pathways that alter the efficacy of chloroquine. For example, our analysis points to TrxR and GAPDH as candidate molecules that determine the efficacy of chloroquine. Here, we focused on candidates susceptible to a low dose of chloroquine because several pathways will be inhibited at higher concentrations (see Table S3; chloroquine = 144 nM). In addition, proteins that transport chloroquine out of food vacuoles should decrease the effective concentration of chloroquine. Targeting these pathways while administering chloroquine treatment should enhance the efficacy of chloroquine at a given effective concentration.
4.3. Chloroquine as DNA intercalator?
The hypothesis that chloroquine kills the malarial parasite by DNA intercalation is decades old (Meshnick, 1990). Although it was the prime candidate mechanism of chloroquine from the 1950s to the 1970s, it has now largely been abandoned for the past two decades. Our model analyses do not identify a drug target or targets; however, they suggest that the effect of chloroquine administration leads to inhibition of DNA synthesis. According to our model, the parasite responds to chloroquine by reducing the expression of genes critical for DNA synthesis. Although chloroquine could specifically intercalate with the identified core gene set in accord with the DNA intercalation theory of chloroquine (Meshnick, 1990, O'Brien et al., 1966a), this is a highly unlikely scenario. Heme is a more plausible molecular inhibitor, given that it accumulates in large amounts within the parasite's food vacuole and is capable of causing DNA damage (Ishikawa et al., 2010, Rahman et al., 1997), protein oxidation (Kitatsuji et al., 2016), NADPH oxidation (Bodaness, 1983), and apoptosis (Chiabrando et al., 2014). However, there is no direct evidence suggesting that heme causes any of these damages in P. falciparum.
4.4. Implications of model analyses
Chloroquine, which was once a highly effective drug, is now abandoned in many malaria endemic regions because of widespread resistance. We identified a potential mechanism by which chloroquine inhibits parasite DNA synthesis. Knowledge of this mechanism allowed us to identify metabolic pathways that could be exploited to increase chloroquine efficacy. For example, our model analyses suggest that administration of H2O2 would potentiate parasite killing by chloroquine, consistent with in vitro experiments (Malhotra et al., 1990). Similarly, our analyses suggest the hypothesis that targeting parasite-specific pathways that synthesize phosphoenolpyruvate and reduce thioredoxin would potentiate parasite killing. Targeting these pathways, while administering amodiaquine (a chloroquine analogue), should result in higher efficacy than administering chloroquine, especially because amodiaquine accumulates more than chloroquine in the parasite’s food vacuole and has a mechanism similar to that of chloroquine (Hawley et al., 1996).
5. Limitations of the current study
Our aim was to identify and investigate enzymes that contribute to decreased DNA replication in response to chloroquine treatment. Using a transcriptomics-based approach, we can identify cellular responses that are specific to chloroquine by expression data taken from chloroquine-treated parasites. Although other antimalarial drugs may activate similar stress pathways, the details of the response will be specific to the particular conditions. The approach makes no assumptions on the underlying molecular actions that cause or affect the response. The identified genes are not likely to be affected by chloroquine directly. The effect of chloroquine on DNA replication is likely to be indirect, being mediated by heme accumulation, which evokes multiple toxic effects in the cell.
We simulated the effect of chloroquine over the entire IDC based on the available transcriptome data taken from an eight-hour window during the trophozoite stage. Ideally, for any specific inhibitor, it is desirable to have hourly transcriptional readouts to predict the effect of a systemic perturbation throughout the entire IDC. In the case presented here, the largest effect of chloroquine occurs during the trophozoite stage and the protocol for collecting experimental transcriptomic data was designed to characterize transcriptional changes during this stage. Our use of these data to identify changes in metabolite fluxes throughout the IDC relative to untreated controls rests on the assumption that altering the expression of genes not associated with any biological function specific to a particular stage will have minimal physiological effects at other stages. This is an assumption that is specific to P. falciparum, because malaria relies on the execution of a highly regimented transcriptional program during the IDC, which initiates specific stage-dependent cellular functions. These assumptions were ultimately corroborated because the observed metabolite phenotype changes are mainly associated with the trophozoite stage and metabolic alterations during previous time points were non-existent or immaterial to DNA synthesis.
6. Conclusion
Herein, we investigated the potential mechanism by which chloroquine inhibits DNA synthesis in P. falciparum and kills the parasite. We first developed an approach (that requires limited transcriptome data) to simulate the effect of exogenous stress on P. falciparum metabolic fluxes during the IDC. We then developed an algorithm employing Bayesian information theory to identify potential pathways that inhibit DNA synthesis in response to chloroquine treatment. Our analysis suggests that 1) chloroquine inhibits DNA synthesis via inhibition of redox metabolism, carbon fixation, and anaplerotic TCA cycle metabolism; 2) P. falciparum may maintain DNA synthesis after chloroquine treatment by preserving a reduced pool of thioredoxin and by invoking alternate mechanisms for synthesizing phosphoenolpyruvate, and 3) a drug or combination of drugs that target redox metabolism, carbon fixation, and heme accumulation could be an effective way to target the parasite.
The method developed here used limited transcriptome data to identify pathways that underlie the inhibition of DNA synthesis of P. falciparum in response to chloroquine treatment. This suggests that the developed method could also be suitable to simulate and capture the effect of other drugs (or stress conditions) on complex metabolic and physiological responses of P. falciparum, based solely on transcriptome data.
Acknowledgments
The authors thank Drs. Francisco Vital-Lopez and Tatsuya Oyama for valuable discussions and comments on a previous version of the manuscript. The authors were supported by the US Army Medical Research and Materiel Command as part of the US Army’s Network Science Initiative. The opinions and assertions contained herein are the private views of the authors and are not to be construed as official or as reflecting the views of the US Army or the US Department of Defense. This paper has been approved for public release with unlimited distribution.
Footnotes
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.ijpddr.2017.03.004.
Contributor Information
Shivendra G. Tewari, Email: stewari@bhsai.org.
Sean T. Prigge, Email: sprigge2@jhu.edu.
Jaques Reifman, Email: jaques.reifman.civ@mail.mil.
Anders Wallqvist, Email: sven.a.wallqvist.civ@mail.mil.
Appendix A. Supplementary data
The following are the supplementary data related to this article:
References
- Antony H.A., Parija S.C. Antimalarial drug resistance: an overview. Trop. Parasitol. 2016;6:30–41. doi: 10.4103/2229-5070.175081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumer A., Ehrenfeucht A., Haussler D., Warmuth M.K. Occam's razor. In: Shavlik J.W., Dietterich, Thomas G., editors. Readings in Machine Learning. Morgan Kaufmann Publishers, Inc.; San Matero, CA: 1990. pp. 201–204. [Google Scholar]
- Bodaness R.S. The H2O2-mediated oxidation of NADPH to NADP+ catalyzed by the heme-undecapeptide from cytochrome C. Biochem. Biophys. Res. Commun. 1983;113:710–716. doi: 10.1016/0006-291x(83)91784-9. [DOI] [PubMed] [Google Scholar]
- Breman J.G., Alilio M.S., Mills A. Conquering the intolerable burden of malaria: what's new, what's needed: a summary. Am. J. Trop. Med. Hyg. 2004;71:1–15. [PubMed] [Google Scholar]
- Chiabrando D., Vinchi F., Fiorito V., Mercurio S., Tolosano E. Heme in pathophysiology: a matter of scavenging, metabolism and trafficking across cell membranes. Front. Pharmacol. 2014;5:61. doi: 10.3389/fphar.2014.00061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou A.C., Fitch C.D. Heme polymerase: modulation by chloroquine treatment of a rodent malaria. Life Sci. 1992;51:2073–2078. doi: 10.1016/0024-3205(92)90158-l. [DOI] [PubMed] [Google Scholar]
- Ciak J., Hahn F.E. Chloroquine: mode of action. Science. 1966;151:347–349. doi: 10.1126/science.151.3708.347. [DOI] [PubMed] [Google Scholar]
- Cohen S.N., Yielding K.L. Inhibition of DNA and RNA polymerase reactions by chloroquine. Proc. Natl. Acad. Sci. U. S. A. 1965;54:521–527. doi: 10.1073/pnas.54.2.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colijn C., Brandes A., Zucker J., Lun D.S., Weiner B., Farhat M.R., Cheng T.Y., Moody D.B., Murray M., Galagan J.E. Interpreting expression data with metabolic flux models: predicting Mycobacterium tuberculosis mycolic acid production. PLoS Comput. Biol. 2009;5:e1000489. doi: 10.1371/journal.pcbi.1000489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combrinck J.M., Mabotha T.E., Ncokazi K.K., Ambele M.A., Taylor D., Smith P.J., Hoppe H.C., Egan T.J. Insights into the role of heme in the mechanism of action of antimalarials. ACS Chem. Biol. 2013;8:133–137. doi: 10.1021/cb300454t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang X., Reifman J., Wallqvist A. Modeling metabolism and stage-specific growth of Plasmodium falciparum HB3 during the intraerythrocytic developmental cycle. Mol. Biosyst. 2014;10:2526–2537. doi: 10.1039/c4mb00115j. [DOI] [PubMed] [Google Scholar]
- Fitch C.D. Involvement of heme in the antimalarial action of chloroquine. Trans. Am. Clin. Climatol. Assoc. 1998;109:97–105. discussion 105–106. [PMC free article] [PubMed] [Google Scholar]
- Gengenbacher M., Fitzpatrick T.B., Raschle T., Flicker K., Sinning I., Muller S., Macheroux P., Tews I., Kappes B. Vitamin B6 biosynthesis by the malaria parasite Plasmodium falciparum: biochemical and structural insights. J. Biol. Chem. 2006;281:3633–3641. doi: 10.1074/jbc.M508696200. [DOI] [PubMed] [Google Scholar]
- Ginsburg H., Famin O., Zhang J., Krugliak M. Inhibition of glutathione-dependent degradation of heme by chloroquine and amodiaquine as a possible basis for their antimalarial mode of action. Biochem. Pharmacol. 1998;56:1305–1313. doi: 10.1016/s0006-2952(98)00184-1. [DOI] [PubMed] [Google Scholar]
- Gutteridge W.E., Trigg P.I., Bayley P.M. Effects of chloroquine on Plasmodium knowlesi in vitro. Parasitology. 1972;64:37–45. doi: 10.1017/s0031182000044620. [DOI] [PubMed] [Google Scholar]
- Hanes J.W., Burns K.E., Hilmey D.G., Chatterjee A., Dorrestein P.C., Begley T.P. Mechanistic studies on pyridoxal phosphate synthase: the reaction pathway leading to a chromophoric intermediate. J. Am. Chem. Soc. 2008;130:3043–3052. doi: 10.1021/ja076604l. [DOI] [PubMed] [Google Scholar]
- Hawley S.R., Bray P.G., Park B.K., Ward S.A. Amodiaquine accumulation in Plasmodium falciparum as a possible explanation for its superior antimalarial activity over chloroquine. Mol. Biochem. Parasitol. 1996 Sep;80(1):15–25. doi: 10.1016/0166-6851(96)02655-2. [DOI] [PubMed] [Google Scholar]
- Hu G., Cabrera A., Kono M., Mok S., Chaal B.K., Haase S., Engelberg K., Cheemadan S., Spielmann T., Preiser P.R., Gilberger T.W., Bozdech Z. Transcriptional profiling of growth perturbations of the human malaria parasite Plasmodium falciparum. Nat. Biotechnol. 2010;28:91–98. doi: 10.1038/nbt.1597. [DOI] [PubMed] [Google Scholar]
- Ishikawa S., Tamaki S., Ohata M., Arihara K., Itoh M. Heme induces DNA damage and hyperproliferation of colonic epithelial cells via hydrogen peroxide produced by heme oxygenase: a possible mechanism of heme-induced colon cancer. Mol. Nutr. Food Res. 2010;54:1182–1191. doi: 10.1002/mnfr.200900348. [DOI] [PubMed] [Google Scholar]
- Kasozi D., Mohring F., Rahlfs S., Meyer A.J., Becker K. Real-time imaging of the intracellular glutathione redox potential in the malaria parasite Plasmodium falciparum. PLoS Pathog. 2013;9:e1003782. doi: 10.1371/journal.ppat.1003782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke H., Lewis I.A., Morrisey J.M., McLean K.J., Ganesan S.M., Painter H.J., Mather M.W., Jacobs-Lorena M., Llinas M., Vaidya A.B. Genetic investigation of tricarboxylic acid metabolism during the Plasmodium falciparum life cycle. Cell Rep. 2015;11:164–174. doi: 10.1016/j.celrep.2015.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitatsuji C., Izumi K., Nambu S., Kurogochi M., Uchida T., Nishimura S., Iwai K., O'Brian M.R., Ikeda-Saito M., Ishimori K. Protein oxidation mediated by heme-induced active site conversion specific for heme-regulated transcription factor, iron response regulator. Sci. Rep. 2016;6:18703. doi: 10.1038/srep18703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwakye-Berko F., Meshnick S.R. Binding of chloroquine to DNA. Mol. Biochem. Parasitol. 1989;35:51–55. doi: 10.1016/0166-6851(89)90141-2. [DOI] [PubMed] [Google Scholar]
- Lehane A.M., McDevitt C.A., Kirk K., Fidock D.A. Degrees of chloroquine resistance in Plasmodium - is the redox system involved? Int. J. Parasitol. Drugs Drug Resist. 2012;2:47–57. doi: 10.1016/j.ijpddr.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinas M., Bozdech Z., Wong E.D., Adai A.T., DeRisi J.L. Comparative whole genome transcriptome analysis of three Plasmodium falciparum strains. Nucleic Acids Res. 2006;34:1166–1173. doi: 10.1093/nar/gkj517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loria P., Miller S., Foley M., Tilley L. Inhibition of the peroxidative degradation of haem as the basis of action of chloroquine and other quinoline antimalarials. Biochem. J. 1999;339(Pt 2):363–370. [PMC free article] [PubMed] [Google Scholar]
- Malhotra K., Salmon D., Le Bras J., Vilde J.L. Potentiation of chloroquine activity against Plasmodium falciparum by the peroxidase-hydrogen peroxide system. Antimicrob. Agents Chemother. 1990 Oct;34(10):1981–1985. doi: 10.1128/aac.34.10.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meierjohann S., Walter R.D., Muller S. Regulation of intracellular glutathione levels in erythrocytes infected with chloroquine-sensitive and chloroquine-resistant Plasmodium falciparum. Biochem. J. 2002;368:761–768. doi: 10.1042/BJ20020962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meshnick S.R. Chloroquine as intercalator: a hypothesis revived. Parasitol. Today. 1990;6:77–79. doi: 10.1016/0169-4758(90)90215-p. [DOI] [PubMed] [Google Scholar]
- Moneriz C., Marin-Garcia P., Garcia-Granados A., Bautista J.M., Diez A., Puyet A. Parasitostatic effect of maslinic acid. I. Growth arrest of Plasmodium falciparum intraerythrocytic stages. Malar. J. 2011;10:82. doi: 10.1186/1475-2875-10-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moxley J.F., Jewett M.C., Antoniewicz M.R., Villas-Boas S.G., Alper H., Wheeler R.T., Tong L., Hinnebusch A.G., Ideker T., Nielsen J., Stephanopoulos G. Linking high-resolution metabolic flux phenotypes and transcriptional regulation in yeast modulated by the global regulator Gcn4p. Proc. Natl. Acad. Sci. U. S. A. 2009;106:6477–6482. doi: 10.1073/pnas.0811091106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro J.B., Silva J.C. Ribonucleotide reductase as a target to control apicomplexan diseases. Curr. Issues Mol. Biol. 2012;14:9–26. [PubMed] [Google Scholar]
- O'Brien R.L., Allison J.L., Hahn F.E. Evidence for intercalation of chloroquine into DNA. Biochim. Biophys. Acta. 1966;129:622–624. doi: 10.1016/0005-2787(66)90078-5. [DOI] [PubMed] [Google Scholar]
- O'Brien R.L., Olenick J.G., Hahn F.E. Reactions of quinine, chloroquine, and quinacrine with DNA and their effects on the DNA and RNA polymerase reactions. Proc. Natl. Acad. Sci. U. S. A. 1966;55:1511–1517. doi: 10.1073/pnas.55.6.1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannala V.R., Dash R.K. Mechanistic characterization of the thioredoxin system in the removal of hydrogen peroxide. Free Radic. Biol. Med. 2015;78:42–55. doi: 10.1016/j.freeradbiomed.2014.10.508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polet H., Barr C.F. Chloroquine and dihydroquinine. In vitro studies by their antimalarial effect upon Plasmodium knowlesi. J. Pharmacol. Exp. Ther. 1968;164:380–386. [PubMed] [Google Scholar]
- Polet H., Barr C.F. DNA, RNA, and protein synthesis in erythrocytic forms of Plasmodium knowlesi. Am. J. Trop. Med. Hyg. 1968;17:672–679. doi: 10.4269/ajtmh.1968.17.672. [DOI] [PubMed] [Google Scholar]
- Radfar A., Diez A., Bautista J.M. Chloroquine mediates specific proteome oxidative damage across the erythrocytic cycle of resistant Plasmodium falciparum. Free Radic. Biol. Med. 2008;44:2034–2042. doi: 10.1016/j.freeradbiomed.2008.03.010. [DOI] [PubMed] [Google Scholar]
- Rahman Q., Mahmood N., Khan S.G., Arif J.M., Athar M. Mechanism of asbestos-mediated DNA damage: role of heme and heme proteins. Environ. Health Perspect. 1997;105(Suppl. 5):1109–1112. doi: 10.1289/ehp.97105s51109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz G. Estimating the dimension of a model. Ann. Stat. 1978;6:461–464. [Google Scholar]
- Slater A.F., Cerami A. Inhibition by chloroquine of a novel haem polymerase enzyme activity in malaria trophozoites. Nature. 1992;355:167–169. doi: 10.1038/355167a0. [DOI] [PubMed] [Google Scholar]
- Song H.S., Reifman J., Wallqvist A. Prediction of metabolic flux distribution from gene expression data based on the flux minimization principle. PLoS One. 2014;9:e112524. doi: 10.1371/journal.pone.0112524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storm J., Sethia S., Blackburn G.J., Chokkathukalam A., Watson D.G., Breitling R., Coombs G.H., Muller S. Phosphoenolpyruvate carboxylase identified as a key enzyme in erythrocytic Plasmodium falciparum carbon metabolism. PLoS Pathog. 2014;10:e1003876. doi: 10.1371/journal.ppat.1003876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugioka Y., Suzuki M., Sugioka K., Nakano M. A ferriprotoporphyrin IX-chloroquine complex promotes membrane phospholipid peroxidation. A possible mechanism for antimalarial action. FEBS Lett. 1987;223:251–254. doi: 10.1016/0014-5793(87)80299-5. [DOI] [PubMed] [Google Scholar]
- Vanlier J., Wu F., Qi F., Vinnakota K.C., Han Y., Dash R.K., Yang F., Beard D.A. BISEN: biochemical simulation environment. Bioinformatics. 2009;25:836–837. doi: 10.1093/bioinformatics/btp069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallqvist A., Fang X., Tewari S.G., Ye P., Reifman J. Metabolic host responses to malarial infection during the intraerythrocytic developmental cycle. BMC Syst. Biol. 2016;10:58. doi: 10.1186/s12918-016-0291-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whichard L.P., Washington M.E., Holbrook D.J., Jr. The inhibition in vitro of bacterial DNA polymerases and RNA polymerase by antimalarial 8-aminoquinolines and by chloroquine. Biochim. Biophys. Acta. 1972;287:52–67. doi: 10.1016/0005-2787(72)90329-2. [DOI] [PubMed] [Google Scholar]
- WHO . World Health Organization; Geneva: 2014. World Malaria Report 2014. [Google Scholar]
- Wu F., Yang F., Vinnakota K.C., Beard D.A. Computer modeling of mitochondrial tricarboxylic acid cycle, oxidative phosphorylation, metabolite transport, and electrophysiology. J. Biol. Chem. 2007;282:24525–24537. doi: 10.1074/jbc.M701024200. [DOI] [PubMed] [Google Scholar]
- Yayon A., Cabantchik Z.I., Ginsburg H. Susceptibility of human malaria parasites to chloroquine is pH dependent. Proc. Natl. Acad. Sci. U. S. A. 1985;82:2784–2788. doi: 10.1073/pnas.82.9.2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yayon A., Vande Waa J.A., Yayon M., Geary T.G., Jensen J.B. Stage-dependent effects of chloroquine on Plasmodium falciparum in vitro. J. Protozool. 1983;30:642–647. doi: 10.1111/j.1550-7408.1983.tb05336.x. [DOI] [PubMed] [Google Scholar]
- Zur H., Ruppin E., Shlomi T. iMAT: an integrative metabolic analysis tool. Bioinformatics. 2010;26:3140–3142. doi: 10.1093/bioinformatics/btq602. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.