

Abstract
Engineered stem cell (SC)-based therapy holds enormous promise for treating the incurable brain cancer glioblastoma (GBM). Retaining the cytotoxic SCs in the surgical cavity after GBM resection is one of the greatest challenges to this approach. Here, we describe a biocompatible electrospun nanofibrous scaffold (bENS) implant capable of delivering and retaining tumor-homing cytotoxic stem cells that suppress recurrence of post-surgical GBM. As a new approach to GBM therapy, we created poly(l-lactic acid) (PLA) bENS bearing drug-releasing human mesenchymal stem cells (hMSCs). We discovered that bENS-based implant increased hMSC retention in the surgical cavity 5-fold and prolonged persistence 3-fold compared to standard direct injection using our mouse model of GBM surgical resection/recurrence. Time-lapse imaging showed cytotoxic hMSC/bENS treatment killed co-cultured human GBM cells, and allowed hMSCs to rapidly migrate off the scaffolds as they homed to GBMs. In vivo, bENS loaded with hMSCs releasing the anti-tumor protein TRAIL (bENSsTR) reduced the volume of established GBM xenografts 3-fold. Mimicking clinical GBM patient therapy, lining the post-operative GBM surgical cavity with bENSsTR implants inhibited the re-growth of residual GBM foci 2.3-fold and prolonged post-surgical median survival from 13.5 to 31 days in mice. These results suggest that nanofibrous-based SC therapies could be an innovative new approach to improve the outcomes of patients suffering from terminal brain cancer.
Keywords: Cytotoxic stem cell, Electrospun nanofibrous scaffolds, Surgical resection, Cancer, Molecular imaging
1. Introduction
Glioblastoma (GBM) is the most common primary brain tumor and a devastating disease [1]. 9000 new patients are diagnosed annually in the United States with GBM. The standard of care for these patients remains surgical tumor resection followed by radio-and chemo-therapy. Yet, these treatments are ineffective and median survival of GBM patients remains only 12–15 months [2,3]. In contrast to traditional stem cell therapies for regenerative medicine, engineered stem cells (SCs) are emerging as an efficacious treatment strategy for GBM [4]. SCs have a unique tumor-homing capacity that allows them to migrate to both local and invasive GBM foci [5]. This makes SCs an ideal drug carrier, and SCs have been engineered with a variety of different anti-tumor payloads. Using pre-clinical models, SC-based therapy has been shown to regress solid GBM [6,7] and significantly extend the survival of GBM-bearing animals [4,6–8]. SC-based therapy for GBM recently entered patient trials based on these exciting pre-clinical studies, [9]. In this first-in-human study, therapeutic SCs in solution were injected into the walls of the GBM surgical cavity and shown to be safe [9,10].
Although the initial Phase I testing of SC therapies for GBM is conducted in surgically resected patients, solid tumor models remain the mainstay of pre-clinical GBM studies. Recently, we used a unique model of GBM surgical resection in mice [11] to investigate the persistence and efficacy of SCs delivered into the walls of the GBM resection cavity [8]. Unlike the long-term persistence that is observed with traditional intraparenchymal SC injection [12,13], we found that >90% of cells injected into the walls of the GBM resection cavity are lost within 7 days. This resulted in an insufficient number of cytotoxic SCs in the brain, and post-surgical GBM regrowth was rapid with treated animal only surviving several days longer than control-treated animals. The rapid cell clearance was slowed and tumor killing restored by delivering therapeutic SCs in a hydrogel scaffold. This suggests polymer scaffolds will significantly improve the efficacy of SC therapy for surgically resected GBM in patients. The hydrogel scaffold remains the only approach tested, yet major obstacles to the clinical utility of this approach include the potential for significant mass caused by filling the resection cavity with scaffold material, substantial wash-out of therapeutic SCs during the long gelation time, reduced drug release due to shielding of the encapsulated cells, and the increased migratory distance for SCs encapsulated in the interior of the matrix. Therefore, it is critical to develop scaffolds suitable for intracavity SC-based GBM therapy that are compliant with human patient testing.
Interestingly, the properties of biocompatible electrospun nanofibrous scaffolds (bENS) are ideally suited for SC transplant into the GBM resection cavity [14]. bENS are known to improve the survival of transplanted SCs in the injured brain and improve the capacity of cells to repair the brain [15,16]. The biocompatibility of bENS minimizes toxicity to SCs attached on their surface and to the local brain tissue surrounding the implants. SCs migrate on the matrices, and SC migration as well as differentiation can be controlled by tuning fiber composition and structure [17]. The ability to seed cytotoxic SCs on the surface of bENS should maximize the levels of cytotoxic agents that engage GBM cells by minimizing shielding. The minimal cross-sectional thickness of nanofiber matrices could line the walls of the GBM resection cavity with a scaffold that is only nano-to micrometers thick. This would permit post-operative fluid to fill the cavity as normal and minimize intracranial pressure and mass effect of the scaffold. Despite the potential of this technology, the use of electrospun materials to improve the transplant and tumor killing effects of therapeutic SCs in the GBM surgical cavity has not been explored.
Here, we describe the generation and characterization of a new composite therapy for surgically resected GBM comprised of bENS and cytotoxic human mesenchymal SCs (hMSCs). We used novel surgical resection models of GBM in mice and non-invasive kinetic imaging to reveal the retention, survival, and tumor-selective homing of engineered hMSCs delivered into the GBM surgical cavity after tumor resection. Additionally, we investigated the anti-cancer efficacy of tumoricidal SCs transplanted on bENS as a novel therapy for both solid and surgically resected human GBM xenografts in mice. To clearly assess therapeutic efficacy, we employed hMSCs delivering a secreted form of tumor-necrosis factor-related apoptosis-inducing ligand (TRAIL), a potent cytotoxic agent proven to induce robust anti-tumor effects when delivered from hMSCs [18–20]. These studies are the first of their kind, and begin to define a novel and essential new therapeutic strategy to eradicate post-surgical minimal GBM using a unique biomaterials-based approach.
2. Material and methods
All experimental protocols were approved by the Animal Care and Use Committees at The University of North Carolina at Chapel Hill and care of the mice was in accordance with the standards set forth by the National Institutes of Health Guide for the Care and Use of Laboratory Animals, USDA regulations, and the American Veterinary Medical Association.
2.1. Cell lines and viral vectors
U87, LN229 and U251 human glioma cells (American Type Culture Collection) were cultured in DMEM (Invitrogen) supplemented with 10% heat-inactivated fetal bovine serum (Sigma-Aldrich), 100 μg/mL penicillin (GIBCO), 100 μg/mL streptomycin (GIBCO) [12]. hMSCs were purchased from the Texas A&M MSC Distribution Center (Darwin Prokopf, University of Texas A&M). The cells were isolated and characterized according to standard protocols. Briefly, hMSCs were isolated from a bone marrow aspirate. Each batch was tested by flow cytometry for various cell markers to ensure they are positive for CD105, CD166, CD29, and CD44 but absent for expression of CD14, CD34, and CD45.
Several different lentiviral vectors were used in these studies: 1) fluorescent and bioluminescent reporters: GFP-firefly luciferase (GFP-FL), mCherry-firefly luciferase (mC-FL), GFP-Renilla luciferase (GFP-RL), Gaussia Luciferase-TRAIL fusion (LV-diTR); 2) therapeutic vector: LV-sTR contains secreted TRAIL driven by the CMV promoter and contains and IRES-GFP cassette. All vectors were generated using custom vector synthesis services from Invitrogen. All LV constructs were packaged as LV vectors in 293T/17 cells using a helper virus-free packaging system as described previously [21]. GBMs and hMSCs were transduced with LVs at varying multiplicity of infection (MOI) by incubating virions in a culture medium containing 5 μg/ml protamine sulfate (Sigma) and cells were visualized for fluorescent protein expression by fluorescence microscopy.
2.2. Scaffold fabrication
PLA scaffolds were developed as previously described [22,23]. Briefly, PLA (MW = 70,000) was dissolved in chloroform and dimethylformamide (3:1) (Sigma) at 80 °C to create a 11 wt% solution. A freshly prepared solution was used in a custom electro-spinning system with a grounded collector using 15 kV voltage, feed rate of 0.7 μl/h and spinning distance of 13–15 cm.
2.3. Seeding of scaffolds
3-mm diameter and 0.5-mm diameter scaffolds were immersed in 70% ethanol for 10 min, rinsed several times with PBS, incubated in culture medium over night. The scaffolds were then seeded from 100,000 (0.5 mm scaffolds) to 500,000 (3-mm scaffold) cells.
2.4. Scanning electron microscopy (SEM)
SEM images were captured using field emission scanning electron microscopy (FESEM JEOL 6400 F) at 15 kV and 200 kV accelerating voltage, respectively. Cell seeded samples were fixed in 10% buffered formalin for 30 min and dehydrated using a graded concentration (50–100% v/v) of ethanol. Drying of PLA scaffolds was accomplished by immersion in hexamethyldisilazane for 15 min followed by air-drying overnight under a fume hood. Dried samples were then sputter coated with gold to observe the morphology of fibers and the attached hMSCs using FESEM.
2.5. Cell viability
To define the viability of hMSCs on scaffolds, hMSC-GFP-FL (1 × 105 cells/scaffold) were seeded on bENS. A separate group of hMSC-GFP-FL (1 × 105 cells) was directly seeded in culture dishes without bENS. 12 hrs and 3, 6, 9 and 14 days later, viability was measured by luciferase assay as described. bENS-induced apoptosis was determine by seeding hMSC-GFP-FL (1 × 105 cells) either on bENS or directly in culture wells and reading caspase 3/7 activity 48 h later using a caspase 3/7-activateable DEVD-aminoluciferin (Caspase-Glo 3/7, Promega, Madison, WI, USA) as described previously [12].
2.6. Real-time imaging and motion analysis
A 0.6% agarose was prepared to mimic brain tissue as described previously [24]. 3 ml of the agarose solution was added to each well of 6-well culture plates and allowed to solidify. A 2 ml aspirating pipette attached to a vacuum was used to create cavities in the agarose ∼500 μm apart. bENS containing hMSC-GFP-FL were placed in one of the agarose cavities. Human U87-mC-FL cells were seeded in the adjacent hole or the cavity was left empty and the wells were filled with media. 24 hrs after plating, the cell/agarose system was placed in a VivaView incubator-microscope live cell imaging system (Olympus) and allowed to equilibrate. Fluorescent images were captured at 20× magnification every 20 min for 44 h in 6 locations per well (to monitor sufficient cell numbers) in three independent experiments. NIH Image was then used to generate movies and to define the migratory path of hMSCs, the directionality of migration, and the velocity of hMSC migration using the “Manual Tracking” and “Chemotaxis Tool” plug-ins.
2.7. In vitro imaging of TRAIL secretion
hMSCs engineered with GFP-FL and the diagnostic TRAIL variant diTR were seeded on bENS. A subset of cells were place directly in culture dishes in solution. On days 3, 6, and 9 after seeding, cell growth was determined using FLuc imaging. Simultaneously, cell culture medium containing secreted fusion proteins was collected and diTR levels determined by Gaussia luciferase imaging as described [6,12]. The diTR photon emission was normalized to the FLuc photon emission to correct for changes in cell number.
2.8. Co-culture viability assays
The effects of biocompatible electrospun nanofiber scaffolds (bENS) bearing hMSC-sTR (bENSsTR) therapy on human GBM cells in vitro were determined by seeding bENSsTR or control bENS with hMSC-GFP (bENSGFP) (2 × 105 cells) adjacent to Human U87, LN229, and U251 GBM cells expressing mCherry-FLuc (2 × 105 cells). A previous 3-D levitation culture strategy was performed in GBM cells, using the bio-assembler kit (Nano3D Biosciences, Houston, TX), in order to mimic in vivo characteristics. Briefly, GBM cells in a 6 well plate with an 80% of confluence, were treated overnight with 72 μl of nanoshuttle magnetic particles. The next day, cells were detached with trypsin and plated in an ultra-low attachment 6-well plate. A magnetic driver of 6 neodymium magnets (field strength = 50 G) designed for 6-well plates were placed atop the well plate to levitate the cells to the air–liquid interface and cultured for an additional 18–24 h to form spheroids. Finally, the spheroids were plated adjacent to bENS and viability was measured at different time points (0, 1, 2, 3, 4, and 6 days) by quantitative in vitro fluorescent imaging.
2.9. In vivo study, fluorescence-guided tumor resection
To perform image-guided GBM resection in mice, we modified our previously reported strategy [11]. Nude mice (6–8 weeks of age; Charles River Laboratories) 25–30 g in weight were used for the intracranial xenograft GBM model. U87-mC-FL were harvested at 80% confluency and implanted stereotactically (5 × 105 cells) in the right frontal lobe 2 mm lateral to the bregma and 0.5 mm from the dura. Following immobilization on a stereotactic frame mice were placed under an Olympus MVX-10 microscope connected to a Hamamatsu ORCA 03G CCD camera. Intraoperative microscopic white light, GFP, and mCherry images were captured throughout the procedure. A midline incision was made in the skin above the skull exposing the cranium of the mouse. The intracranial xenograft was identified using mCherry fluorescence. A small portion of the skull covering the tumor was surgically removed using a bone drill and forceps and the overlying dura was gently peeled back from the cortical surface to expose the tumor. Under mCherry fluorescence, the U87-mC-FL tumor was surgically excised using a combination of surgical dissection and aspiration, and images of mCherry fluorescence were continuously captured to assess accuracy of mCherry-guided surgical resection. Following tumor removal, the resulting resection cavity was copiously irrigated and the skin closed with 7-0 Vicryl suture. No procedure-related mortality was observed.
2.10. In vivo study, bENS transplant of hMSCs
To study the retention and survival of hMSCs on bENS and non-bENS hMSCs transplanted into the surgical resection cavity, U87mC-FL tumors were resected as described above. hMSC-GFP-FL (5 × 105 cells) were seeded on bENS and transplanted onto the walls of the tumor cavity or suspended in PBS and directly injected into the boarders of surgical cavity. The skin was then closed with 7-0 vicryl suture. hMSC retention and survival were measured using bioluminescence imaging 2–21 days after seeding as described below. In a subset of mice, brains were harvested 14 days after transplant, and ex vivo immunofluorescent imaging was performed using the MVX microscope. The bENS was removed from the brain, and hMSC content and GBM cells were imaged in the bENS and the surgical cavity using immunofluorescence microscopy. The co-localization of GFP+ hMSCs and mCherry+ U87 in the brain was determined using intensity scans plots that consist of a graph of pixel intensity values measured at each position along a line through a RGB image. For this purpose a line was drawn over the RGB image and intensity values plotted in a graph using the Analyze Measure RGB plug-in from NIH Image.
2.11. In vivo study, bENS-based therapy for solid xenografts
To study the ability of bENS-based hMSC therapy for solid tumors, nude mice (6–8 weeks of age; Charles River Laboratories, Wilmington, Massachusetts) 25–30 g were anesthetized by isoflurane. 3 × 106 U87-mC-FL were mixed with 100 μl of PBS and injected into the paraspinal space (2 independent injection sites per animal, 4 injected animals) using a 27G size needle [12]. One week later, the skin covering the developing tumor was lifted. bENS bearing hMSC-sTR or control hMSC-GFP-RL were implanted over the established tumors (4 tumors/group). Tumor volumes were monitored using luciferase imaging performed 1, 2, 5, 7, 9 and 15 days post-transplant as described below.
2.12. In vivo study, bENS-based therapy of surgically resected GBMs
To study the therapeutic efficacy of bENS-based hMSC transplant into the resection cavity, established orthotopic U87-mC-FL tumors were resected as described above. Immediately following resection, hMSC-sTRor hMSC-GFP-RL (5 × 105 cells) on bENS were transplanted into the post-operative GBM cavity. GBM volumes were visualized in the bENSsTR and control bENSGFP treatment groups using FLuc imaging as described below. Images were captured 1, 2, 5, 6, 7, 11, 15, and 18 days after bENS-based therapy.
2.13. Tissue processing
Mice with resected tumors or mice with resected tumors and implanted with bENS carrying hMSCs were perfused with formalin and brains were removed and sectioned. The tissue was immediately immersed in formalin. 30 μm coronal sections were generated using a vibrating microtome (Fisher). Sections were washed three times with PBS and nuclei were counterstained with Hoechst 33342 (Life Technologies) and visualized using a confocal microscope (Olympus). In a subset of mice, brains were isolated and fluorescent imaging was performed using an Olympus MVX-10 microscope.
2.14. Statistical analysis
Data were analyzed by Student t-test when comparing 2 groups and by ANOVA followed by Dunnetts post-test when comparing greater than 2 groups. Data were expressed as mean ± SEM and differences were considered significant at P < 0.05. Survival times of mice groups (n = 5/group) were compared using log-rank test.
3. Results
3.1. Generation and in vitro characterization of bENS bearing engineered SCs
To create an electrospun scaffold system for effective transplant of therapeutic SCs into the surgical resection cavity following GBM debulking (strategy outlined in Fig. 1a), we created biocompatible electrospun nanofibrous scaffolds (bENS) bearing human mesenchymal SCs (hMSCs) engineered with optical reporters or tumoricidal gene products using lentiviral vectors. To initially create diagnostic systems, bENS were synthesized from poly-l-lactic acid (PLA; Fig. 1b) and seeded with hMSCs expressing GFP and firefly luciferase fusion proteins (bENSGFPFL; Fig. 1c). Scanning electron microscopy (SEM) cross-sectional images showed the minimal thickness of the scaffolds (Fig. 1d). While, 3-D reconstruction of the fluorescent images revealed the minimal cross-sectional thickness of bENSGFPFL composites. SEM images revealed the efficient attachment and spreading of the engineered hMSCs along the bENS fibers (Fig. 1e). We also used the SEM images to evaluate the physical characteristics of our nanofibrous materials. The average fiber diameter and pore size of each scaffold was calculated from at least 50 measurements per scaffold. Our results indicate that the fibers had average fiber diameters of 450 ± 72 nm and the average pore size of 0.45 ± 0.44 mm. Our previous findings suggest the PLA nanofibers should degrade 25% after 36 days [22].
Fig. 1.
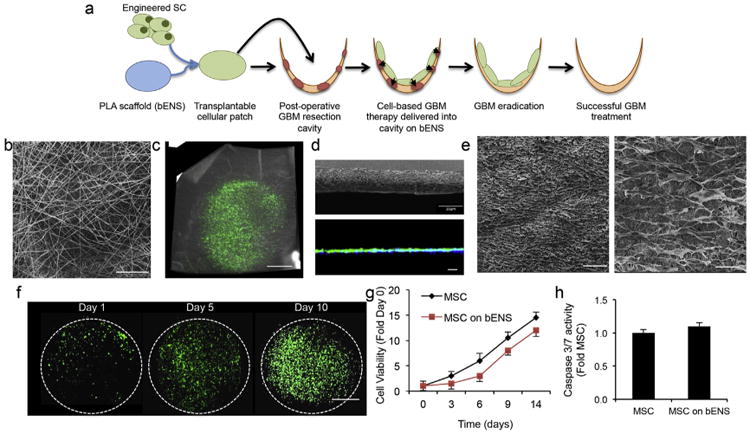
Characterization of engineered hMSCs on bENS. (a) Schematic representation of our new approach to GBM therapy using bENS to deliver drug releasing SCs into the GBM resection cavity. Once implanted, the bENS are designed to allow the therapeutic SCs to migrate into the GBM foci and deliver tumoricidal agents that will eradicate residual tumor. (b) Representative SEM image of the bENS. (c) Photomicrograph showing hMSCs expressing GFP and firefly luciferase grown on bENS. (d) SEM image showing cross-sectional thickness of bENS and 3-D cross-sectional z-stack of hMSCs seeded on bENS created using fluorescence confocal microscopy. (e) SEM images of bENS bearing engineered hMSCs at 100× and 500× magnification. (f) Representative fluorescent images depicting the growth of hMSC-GFP-FL on bENS. (g) Summary graph comparing the growth rates of hMSC-GFP-FL cultured on or off bENS. p > 0.05 by Two-way ANOVA. (h). Apoptosis assay depicting the caspase 3/7 activation in hMSCs and hMSCs on bENS. p > 0.05 by Students t-Test. Data in f and g are mean ± SEM (error bars are from 3 to 4 independent experiments). Scale bars in b, e are 50 μm, 40 μm in d, 200 μm in e, and 1.6 mm in c, f.
To determine the compatibility of bENS with the engineered hMSCs, we tracked the growth, viability, and apoptosis of hMSC-GFP-FL in vitro. Serial fluorescent imaging showed hMSC-GFP-FL proliferated on the bENS in vitro (Fig. 1f). Quantitative bioluminescent cell viability assays revealed the growth rate of hMSC-GFP-FL on bENS was initially slightly slower than hMSCs grown without scaffolds at early time points, but were comparable 1 week after seeding (Fig. 1g). The bENS did not increase apoptosis, as the levels of activated caspase 3/7 were equivalent between hMSC-GFP-FL grown on and off bENS (Fig. 1h). These studies show the bENS platform promotes the attachment and growth of engineered hMSCs.
3.2. bENS-based implant increase the retention and persistence ofSCs in the GBM resection cavity
Scaffolds must provide long-term persistence of engineered hMSCs in the GBM resection cavity to improve SC-based therapy for GBM. This would overcome the rapid loss of SCs suspensions delivered via traditional direct injection methods. To determine if bENS improves retention of engineered SCs in the surgical GBM resection cavity, we measured the delivery efficiency and persistence of hMSCs transplanted into the GBM cavity with or without bENS using our novel mouse model of GBM resection. mCherry-expressing human U87 glioblastoma tumor cells (U87-mC) were implanted into the parenchyma of nude mice (Fig. 2a).10 days later, an incision in the scalp was made to expose the developing GBM. Fluorescence guided surgical techniques were used to de-bulk the tumor (Fig. 2a). This created a resection cavity that is surrounded by minimal residual deposits of GBM foci. The tumor resection cavity was then lined with bENSGFPFL. Additionally, hMSC-GFP-FL were directly injected into the wall of the surgical cavity in a subset of animals to mimic the scaffold-free transplant that is currently used in clinical patient trials. Serial bioluminescence imaging showed the directly injected hMSC-GFP-FL were rapidly cleared from the tumor cavity with <10% remaining 10 days post-injection (Fig. 2b). In contrast, the retention of hMSC-GFP-FL transplanted on bENS was 11-fold greater than direct injection at day 10, and bENS extended the persistence of hMSC-GFP-FL in the resection cavity to more than 3-fold. The high retention and persistence by bENS observed in vivo was verified by ex vivo analysis. Fluorescence imaging of ex vivo brains confirmed showed GFP+ hMSCs were still present on the bENS in the resection cavity 2 weeks post-transplant (Fig. 2c–d). After the bENS was removed from the brain, whole-brain fluorescence imaging revealed hMSC-GFP-FL were present in the underlying boarders of the resection cavity (Fig. 2e). Interestingly, the hMSC-GFP-FL signal extensively co-localized with residual U87-GFP-mC foci (Fig. 2e–f). This could suggest that hMSC-GFP-FL migrate off the bENS in response to local GBM cells. The presence of hMSCs in the borders of the surgical resection cavity was confirmed byhigh-resolution fluorescence microscopy of postmortem tissue sections (Fig. 2g). GFP+ hMSC-GFP-FL were found to co-localize with residual mCherry+ U87-mC-FL in resection cavity 10 days post-implantation. Together, these data demonstrate that bENS improves the transplant of engineered SCs into the GBM resection cavity.
Fig. 2.
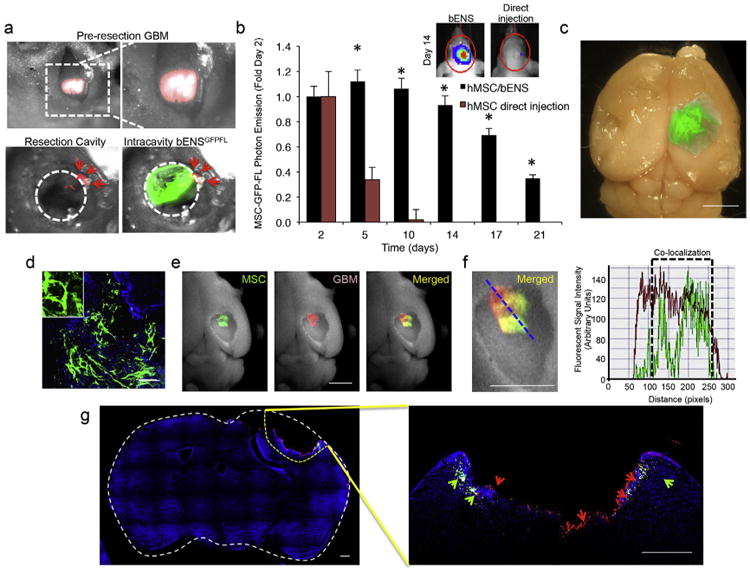
The survival and retention of engineered SCs delivered into the GBM resection cavity on bENS. (a) Photomicrographs showing the image-guided GBM resection in mice and intracavity seeding of bENSGFPFL. mCherry+ human U87 GBMs were de-bulked using imaged guided microsurgery. bENS bearing hMSC-GFP-FL were seeded into the resection cavity. The circular dotted line indicates the resection cavity boarder. Arrowheads indicate residual GBM. (b) Summary graph and representative bioluminescence images showing the levels and persistence of hMSCs transplanted into the resection cavity on bENS or directly injected into the cavity walls in suspension. *p < 0.01 vs. direct injection by Repeated Measures. (c) Ex vivo fluorescent image of bENSGFPFL in the resection cavity 14 days post-transplant. (d) High resolution ex vivo image showing GFP+ hMSCs on bENS after residing in the GBM surgical cavity for 2 weeks. (e) Fluorescent and bright field images of the resection cavity after removal of the bENSGFPFL revealing the presence of hMSCs (green) in the walls of the resection cavity, the presence of residual GBM (red), and their co-localization (merged). (f) Intensity analysis showing the co-localization of the mCherry signal (residual GBM) and GFP signal (hMSCs) in the boarder of the resection cavity. (g) High resolution fluorescence imaging of post-mortem tissue sections demonstrating the presence of GFP+ hMSCs and mCherry+ U87-mC-FL along the borders of the GBM surgical resection cavity. Arrows indicate the presence of hMSCs (green) and U87 tumors cells (red) in the brain tissue adjacent to the resection cavity. Data in b are mean ± SEM. Scale bars in c, e, f are 2.5 mm, 200 μm in d, and 400 μm in g.
3.3. Cytotoxic SCs migrate off bENS and home to GBM cells
The tumor-homing migration of SCs is a unique and vital component of their anti-GBM efficacy [4]. Thus, it is essential that scaffold platforms permit engineered SCs to migrate on the matrix as well as move off the scaffolds and localize with cancer foci. To investigate the migration and tumoritropic homing of hMSCs on bENS, we developed a novel strategy that combines time-lapse motion analysis with 3-dimensional (3-D) under-agarose migration systems (outlined in Fig. 3a). A tissue culture dish is filled with a 0.6% agarose solution to mimic the composition of the brain [24]. A cavity is created in the agarose and lined with bENSGFPFL. Human U87-mC-FL are seeded into a second cavity 500 mm away to establish a chemotactic signaling gradient. The system is placed in an incubator microscope and kinetic images are captured every 20 min for 44 h to define the migration of hMSCs as they move off the bENS, through the agarose matrix, and towards the mCherry+ GBM cells.
Fig. 3.
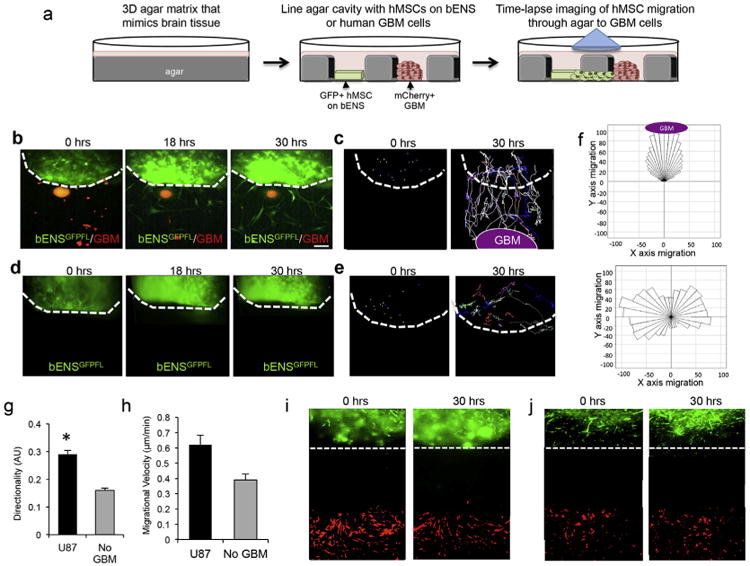
hMSCs tumoritropic migration on bENS. (a) Schematic representation of studies exploring tumor-directed migration of hMSC on bENS using time-lapse motion analysis and 3-dimensional (3-D) under-agarose migration system. hMSC-GFP-RL were seeded on bENS and placed in the well of a 3-D agarose gel in culture. U87-mC-FL were plated in an adjacent well. Time-lapse imaging was performed to define the tumoritropic migration of hMSCs on bENS through/under the agarose to GBM cells. (b) Representative fluorescent images showing the kinetics of hMSCs (green) migration off bENS and homing to co-cultured GBM cells (red). The dotted line indicates the boarder of the bENS. (c) Single-cell analysis showing the migratory path of hMSCs as they exit bENS and home to GBM cells over time. Each line depicts the path of one cell. The dotted line indicates the boarder of the bENS. (d–e) Fluorescent images (d) and single-cell analysis (e) showing the migration of hMSCs on bENS cultured in the absence of GBM cells. (f) Rosetta graphs demonstrating the hMSCs off bENS is directed towards GBM cells and when cancer cells are not present. (g–h) Summary graphs showing the directionality (g) and velocity (h) of hMSCs on bENS in the presence and absence of human GBM cells. (i–j) Representative images showing hMSCs do not migrate off bENS we co-cultured with astrocytes (i) or fibroblasts (j). *p < 0.05 vs. no GBM by Students t-Test. Data in g and h are mean ± SEM (error bars are from 3 independent experiments). Scale bars in b, d are 200 mm.
Using this 3-D culture strategy, time-lapse imaging revealed that hMSCs rapidly migrated off the bENS to co-localize with co-cultured GBM cells. hMSCs had exited the bENS by 18 h post-seeding (Fig. 3b, Supplemental Fig. S1). Single cell migratory path analysis showed the hMSCs directly migrated to the GBM cells as they exited the bENS and migrated through the agar (Fig. 3c, Supplemental Fig. S2). In contrast, bENSGFPFL cultured without GBM cells did not exit the scaffold but only moved on the scaffold fibers (Fig. 3d, Supplemental Fig. S4). Single cell migration analysis showed the migratory pattern of the bENSGFPFL was random in the absence of GBM cells and verified that few hMSCs migrated off the scaffolds (Fig. 3e, Supplemental Fig. S5). Quantitative analysis clearly demonstrated the high directional migration of bENSGFPFL in the presence of GBM cells, with all cells moving in the direction of the GBM and few cells moving in opposite directions (Fig. 3f, Supplemental Fig. S3, 6). Alternatively, the migration of hMSCs was distributed in a 360° pattern in the absence of GBM cells. To quantify the directionality of movement, we determined the ratio of Euclidean distance to the accumulated distance. Linear motion was then given a value of 1 and non-linear motion a value of 0. The presence of GBM cells increased the directionality of hMSC migration 23% compared to cells cultured without GBM (Fig. 3g). Velocity analysis revealed that hMSCs was no significant different in the migratory rate of hMSCs on bENS in the presence or absence of GBM cells (Fig. 3h). Migration was specific to GBM cells, as hMSCs did not migrate to astrocytes or fibroblasts (Fig. 3i–j). Together, these studies demonstrated that hMSCs are highly migratory on bENS, easily migrate off bENS, and retain the tumoritropic homing capacity to selectively target GBM cells.
Supplementary data related to this article can be found online at http://dx.doi.org/10.1016/j.biomaterials.2016.03.008.
3.4. Cytotoxic SCs/bENS therapy decreases the viability of GBM cells in vitro
To investigate the efficacy of engineered hMSCs delivered on bENS, we genetically engineered hMSCs with a secreted variant of the pro-apoptotic protein TRAIL (hMSC-sTR). We then created bENS seeded with the hMSC-sTR (bENSsTR; Fig. 4a) or control hMSCs expressing GFP and renilla luciferase fusions (bENSGFPRL). To investigate the impact of bENS on hMSC drug release, we developed bENS with hMSCs engineered with both firefly luciferase (FLuc) to track cell growth and a diagnostic fusion between Gaussia luciferase and TRAIL (diTR) to monitor protein secretion. We have previously shown diTR can be used to track TRAIL release from different SC lines in vitro and in vivo [8,12]. diTR imaging of media samples collected over 9 days revealed hMSCs on bENS stably release therapeutic proteins (Fig. 4b). Importantly, bENS did not interfere with drug release as the levels of the diTR fusion were not statistically different between hMSC-diTR cultured with or without bENS. Additionally, diTR was found to increase in a dose-dependent manner as MSC-diTR number increased (Fig. 4c).
Fig. 4.
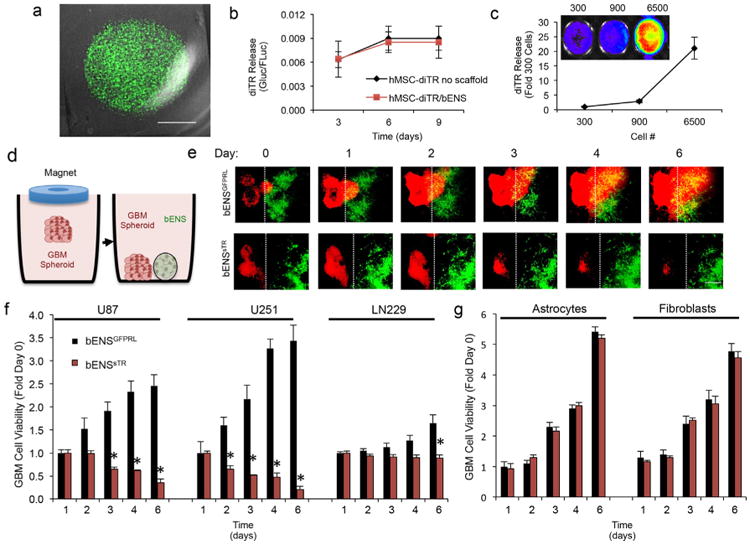
Anti-tumor effects of therapeutic hMSCs delivered on bENS on GBM spheroids in culture. (a) Representative merged fluorescent/DIC images of therapeutic bENSsTR. (b) In vitro luciferase assay revealing the levels of diTR secreted over time from hMSCs grown on bENS or without a scaffold. *p > 0.05 by Students t-Test. (c) Representative images and summary data showing levels of secreted diTR increase as increasing numbers of hMSC-diTR are seeded on bENS. (d–f) To assess the efficacy of hMSC-sTR/bENS therapy against different GBM cell lines, 3-dimensional spheroid assays were performed (described in d). Human GBM cells were loaded with magnetic nanoparticles and seeded in 6-well plates. A magnetic driver was placed over the dish to levitate the GBM cells and create tumor spheroids. Once formed, the GBM spheroids were co-cultured with bENSsTR or control. Real-time fluorescent images showed bENSsTR therapy (green) induced a time-dependent decrease in the viability of U87 GBM spheroids (red, e). This was not observed in U87 GBM spheroids treated with control bENSGFPRL (e). Dotted line indicated boarder of the bENS. Summary data (f) showed that bENSsTR therapy significantly reduced the viability of U87, U251, and LN229 human GBM spheroids compared to control-treated spheroids, although treatment response varied between tumor lines. *p < 0.01 vs. bENSGFPRL in all panels by Repeated Measures. Data in b, c, and f are mean ± SEM (error bars are from 3 to 4 independent experiments). Scale bars are 1.6 mm in a and 400 μm in e.
To determine the anti-tumor effects of bENS-based SC therapy in vitro, we performed co-culture studies of mCherry+ human GBM cells and GFP+ bENSsTR or control bENSGFPRL. We used a novel 3-D levitation cultured strategy to more accurately mimic therapy for solid GBM foci (Fig. 4d). GBM cells were labeled with magnetic material and cultured in levitation by a magnet placed overt the culture dish to form 3-D human GBM spheroids. The GBM spheroids were co-cultured with bENSsTR or control bENSGFPRL and tumor killing was determined by kinetic optical imaging. We found that bENSsTR induced a time dependent decrease in GBM cell viability (Fig. 4e), reducing solid GBM spheroids by 8.3-fold (U87), 16.3-fold (U251), and 1.9-fold (LN229) compared to control-treated spheroids (Fig. 4f). These findings suggest bENSsTR therapy can reduce the viability of different solid GBM spheroids in vitro.
3.5. bENS-based SC therapy inhibits progression of established GBM xenografts
To determine the effects of bENSsTR therapy on established solid GBM, we xenografted U87-mC-FL GBMs into mice. After tumors were established, bENSsTR or control bENSGFPRL was surgically implanted over the solid tumors (Fig. 5a). Serial bioluminescence imaging showed bENSsTR-treated xenografts were significantly smaller than control-treated tumors by day 7, and over 3-fold small by 2 weeks post-treatment (Fig. 5b–c). These findings begin to establish the anti-tumor efficacy of hMSC therapy delivered on bENS in vivo.
Fig. 5.
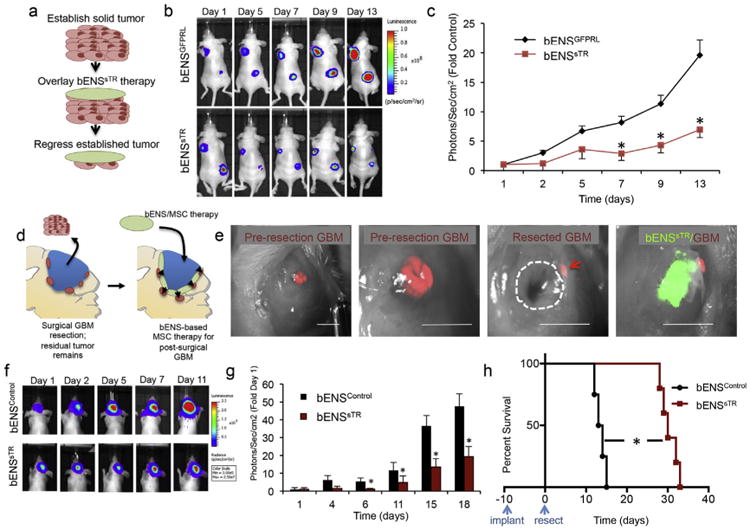
In Vivo efficacy of cytotoxic MSC/bENS therapy for solid and surgically resected GBM. (a) To determine the efficacy of bENS-based hMSC therapy on established tumors, U87-mC-FL xenografts were created in mice. bENSsTR or control bENSGFPRL were surgically implanted over each established tumor (detailed in a). Serial bioluminescence imaging was used to follow the growth tumors in each treatment group. Significantly smaller tumor volumes were detected in bENSsTR–treated tumors across multiple time points as demonstrated in the representative images (b) and summary graph (c). *p < 0.01 vs. bENSGFPRL by Repeated Measures. (d) Schematic representation of bENS-based cytotoxic SC therapy for surgically resected GBM. Following microsurgical resection of human GBMs in mice, drug-releasing hMSCs are delivered into the resection cavity on bENS. (e) Representative images showing pre-resection GBMs, the post-surgical GBM cavity, and bENSsTR seeded in the resection cavity. The dotted line indicates the resection cavity. The arrowheads indicate residual GBM foci. (f–g) Representative serial bioluminescence images (f) and summary graph (g) showed a significant reduction in GBM regrowth in bENSsTR-treated animals compared to bENSControl-treated animals. *p < 0.01 vs. bENSGFPRL by Repeated Measures. (h) Kaplan-Meyer survival analysis and summary table showing bENSsTR therapy prolongs the survival of animals with resected GBMs compared to animals with unresected GBMs or post-surgical GBMs treated with control bENSGFPRL. *p < 0.01 vs. bENSControl log-rank test. Scale bar is 2.5 mm in e.
3.6. Intracavity SC/bENS therapy inhibits recurrence of surgically resected GBM
Lastly, we investigated the effect of bENSsTR therapy on post-surgical minimal GBM in mice to mimic clinical GBM treatment for post-operative human patients (Fig. 5d). Human U87-mC-FL were implanted orthotopically into the parenchyma of Nude mice. 10 days later, the GBMs were surgically resected using image-guided microsurgery as described above (Fig. 5e). The walls of the surgical resection cavity were lined with either bENSsTR or control bENSGFPRL. Serial bioluminescence imaging showed GBM recurrence was markedly delayed by bENSsTR therapy (Fig. 5f–g). This reduction in GBM volumes was accompanied by a significant extension in survival of tumor-bearing animals, as bENSsTR-treated mice survived longer than 31 days post-therapy compared to only 13.5 days for control-treated animals (Fig. 5h). When taken together, our results strongly suggest that bENS-based transplant is a highly efficacious strategy for intracavity hMSC therapy of surgically resected GBM.
4. Discussion
As SC-based therapy from GBM enters patient testing [9], it is essential that the cytotoxic SCs persist in the surgical resection cavity to enable tumor-trophic migration and suppression of post-surgical GBM foci. To improve SC therapy for post-surgical GBM, we created bENS/hMSC composites that increase the persistence and efficacy of SC therapy for residual minimal GBM. bENS have been used in regenerative medicine to control SC differentiation, migration, and survival [14], but have never been used to transplant cytotoxic SCs for cancer therapy. We discovered that bENS-based transplant increased both the initial retention and long-term persistence of hMSCs in the GBM resection cavity. We show that SCs can rapidly migrate off the bENS, homing to GBM cells. In vivo, we found bENS loaded with cytotoxic hMSCs regressed solid GBM xenografts and both suppressed post-surgical GBM recurrence while extending the survival of mice surgically resected GBM. bENS are a highly adaptable platform and were created from material that is approved from clinical use by the FDA [25]. Therefore, these findings suggest that bENS-based transplant of human cytotoxic SCs is a new approach to effectively treat post-surgical GBM that could eventually be used in clinical patient testing.
We found that bENS-based transplant significantly increases the retention of hMSCs in the surgical resection cavity compared to hMSCs directly injected into the walls of the surgical cavity in solution. Ex vivo analysis showed hMSCs were still present in the scaffolds 14 days after transplant. This technique utilized the bENS to line the walls of the resection cavity, and did not rely on cavity-filling cell-encapsulation strategies used in previous studies. Although these previous studies showed the hydrogel scaffolds improved human SC therapy for GBM, no studies have investigated the impact of scaffolds on human SC persistence in the post-resection cavity. We found that nearly 40% of hMSCs on bENS remains in the GBM resection cavity 21 days after seeding. The scaffolds were also able to retain the hMSCs at the site of the GBM foci, as fluorescent whole-brain images showed hMSCs in the brain localized with residual GBM foci. Here, we only monitored one time point after transplant, therefore further studies will be required to fully define the rate of hMSC exit from bENS in vivo. Few studies have explored the use of scaffolds to improve the retention and efficacy of stem cell-mediated therapy for post-surgical GBM. Therefore, the ideal scaffold material and architecture remains unknown. In contrast to previous studies using hydrogel, the minimal cross-sectional thickness of bENS is advantageous because it should minimize the risk of mass effect and associated morbidity and mortality that cavity-filling strategies could cause if the surgical cavity of human patients is filled with polymer/stem cell mixtures. Unlike previous strategies [8,26], bENS delivers therapeutic hMSCs seeded on the surface of the scaffold rather than encapsulated. This raises the potential to explore new parameters in scaffold architecture, topography, and composition as we work towards defining the optimal scaffold properties for stem cell-based GBM therapy. Finally, the great tunability of bENS compared to hydrogels should enable fabrication and testing of scaffolds with a wider architecture and composition. This will be valuable in identifying the optimal scaffold design that concurrently maximizes intra-cavity retention, tumor-directed migration, and anti-GBM efficacy as the impact of parameters such as fiber alignment, fiber diameter, composition, fiber coating, and degradation rates are explored in future studies. Such evaluations will be critical to maximize the efficacy of cytotoxic stem cell/bENS therapy in eventual human patient testing.
The ability of SCs to home to GBM cells is an essential benefit of this approach. Unlike regenerative medicine applications, it is essential for GBM therapy that cytotoxic SCs exit the bENS matrix and migrate to GBM cells. We found that hMSCs migrated off the bENS within 18 h of seeding and homed to co-cultured GBM cells. Interestingly, hMSCs cultured without GBMs did not exit the bENS suggesting the migration off the bENS was induced in part by the presence of GBM cells. We used time-lapse imaging to create movies that could be used for motion analysis to evaluate hMSC migration. This unique approach provided kinetic and quantitative data that is not achievable with traditional Boyden chamber assays as well as time-lapse video of hMSC movement over 44 h. We generated single-cell line tracing which depicted the specific migratory path as hMSCs homed to GBM cells. We were also able to determine the directionality and velocity of hMSC movement. We found both parameters were increased by the presence of GBM cells. SCs are known to move faster on aligned fibers. Therefore, it is possible that adjusting the design of bENS matrix could increase the migratory velocity of cytotoxic hMSCs on scaffolds as well as their exit off the matrix.
We employed hMSCs releasing the cytotoxic agent TRAIL. Our goal was to explore the impact of bENS matrix on SC therapy. The therapeutic benefits of this unexplored transplant system could be most clearly assessed using a therapeutic molecule with proven cytotoxic effects. We purposefully chose TRAIL because its proven anti-GBM effects make it ideal to accomplish these aims [7,18–20] Through interactions with death receptors on the surface of cancer cells, TRAIL elicits a well-established apoptotic response in tumor cells [27]. A variety of studies have demonstrated the potential of stem cell-mediated delivery of TRAIL as a robust therapeutic for GBM. In vitro, cell-mediated secretion of TRAIL markedly reduces the viability of both established and primary GBM cells. In vivo, the sustained delivery of TRAIL by cellular delivery vehicles leads to dramatic reductions in established tumor volumes, suppression of post-surgical GBM recurrence, and correlated extensions in survival compared to control-treated animals [8,19,20]. Importantly, these effects have been demonstrated for both NSC and hMSC cell carriers [7,18–20]. Our study is the first to characterize the therapeutic efficacy of stem cell-mediated TRAIL therapy delivered into the GBM surgical cavity on bENS. We used a diagnostic variant, and first showed that hMSCs on bENS stably release cytotoxic gene products for 9 days. Importantly, quantitative bioluminescence imaging showed there was no difference in secretion levels between hMSCs cultured on or off bENS. This suggests that bENS do not interfere with SC drug release. hMSCs-sTR/bENS therapy markedly reduced the viability of GBM co-cultures in a time-dependent and dose-dependent manner. We observed 3-D GBM cell killing by 6 days. This bENSsTR therapy was effective against multiple 3-D GBM lines. This suggests bENSsTR should be effective against a wide range of GBM cells.
In vivo, we found that bENSsTR therapy reduced the volumes of solid GBM and delayed the regrowth of post-surgical GBMs. We found a statistically significant inhibition in GBM growth within 1 week post-treatment. Clinical bENS therapy will likely be used to treat post-operative GBM as surgical resection is part of the clinical standard-of-care for GBM patients. We used our novel mouse model of GBM resection/recurrence and found that bENSsTR induced statistically significant reductions in GBM regrowth 6 days after treatment. Importantly, this inhibition in tumor regrowth allowed bENSsTR-treated animals to survive over 2-fold longer than control animals. We did not investigate the impact of transplanting multiple therapeutic bENS into the resection cavity due to the size constraints of the mouse model. Despite the potent anti-GBM effects of bENSsTR-therapy, robust tumor suppression was not maintained and animals eventually succumbed to tumor re-growth. Identifying the molecular mechanisms mediating GBM escape from cytotoxic hMSC therapy will enable tuning of bENS to convert the initial robust tumor kill into sustained tumor suppression. Although the precise cause of tumor escape remains unknown, we found that hMSCs on bENS were still eventually lost from the resection cavity, suggesting carrier clearance may play a role. Alternatively, inadequate or sub-optimal hMSC migration from the bENS may limit accumulation in distant GBM foci. Lastly, it is possible that sub-populations of GBM cells adapt to single agent therapy and are mediate tumor regrowth. In efforts to address these limitations, we are testing the ability of bENS with different architectures and compositions to improve hMSC persistence within the surgical cavity as well as maximize the migration of hMSCs off bENS and into neighboring GBM cells. We are also exploring the potential of hMSCs engineered with multiple cytotoxic agents to kill tumor populations that are resistant to single-agent hMSC therapy when delivered into the surgical cavity on bENS.
In this study, we utilized lentiviral vectors to modify the hMSCs. This approach has been widely utilized in the past to engineer a variety of cell therapies for cancer [12,20,28]. Yet, aberrant virus production or tumorigenicity caused by insertional mutagenesis are concerns that must be screened for prior to delivery of engineered cells into human patients [29]. Transfection of non-integrating vectors is an alternative that eliminates the potentially harmful risks associated with virus transduction but reduces the duration of drug delivery as the plasmids are typically lost form the cells after only several days. An intermediate strategy is transduction of cells with non-integrating virus, such as adenovirus. Although the gene is not stably integrated, the transgene persists significantly longer than plasmid transfection but eliminates the risk of insertional mutagenesis associated with lentiviral vectors [29]. Future studies will be required to define the most effective and safest approach for introducing therapeutic transgenes into hMSCs for cancer therapy.
5. Conclusion
In conclusion, these data suggest bENS are an effective new approach for cytotoxic SC therapy of post-surgical GBM. We envision an eventual clinical approach wherein cytotoxic SCs are seeded on bENS and neurosurgeons line the walls of the resection cavity immediately following GBM debulking. Future studies will focus on determining the scaffold composition and architecture that maximizes killing of residual GBM.
Supplementary Material
Acknowledgments
This work was supported by the UNC Lineberger Comprehensive Cancer Center's University Cancer Research Fund and the UNC Translational and Clinical Sciences Institute (KL2TR001109, UL1TR001111).
We thank the UNC-Olympus Imaging Research Center for microscope usage.
References
- 1.Adamson C, Kanu OO, Mehta AI, et al. Glioblastoma multiforme: a review of where we have been and where we are going. Expert Opin Investig Drugs. 2009;18(8):1061–1083. doi: 10.1517/13543780903052764. [DOI] [PubMed] [Google Scholar]
- 2.Erpolat OP, Akmansu M, Goksel F, Bora H, Yaman E, Buyukberber S. Outcome of newly diagnosed glioblastoma patients treated by radiotherapy plus concomitant and adjuvant temozolomide: a long-term analysis. Tumori. 2009;95(2):191–197. doi: 10.1177/030089160909500210. [DOI] [PubMed] [Google Scholar]
- 3.Stupp R, Hegi ME, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10(5):459–466. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 4.Aboody KS, Najbauer J, Danks MK. Stem and progenitor cell-mediated tumor selective gene therapy. Gene Ther. 2008;15(10):739–752. doi: 10.1038/gt.2008.41. [DOI] [PubMed] [Google Scholar]
- 5.Shah K, Hingtgen S, Kasmieh R, et al. Bimodal viral vectors and in vivo imaging reveal the fate of human neural stem cells in experimental glioma model. J Neurosci. 2008;28(17):4406–4413. doi: 10.1523/JNEUROSCI.0296-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hingtgen S, Kasmieh R, Elbayly E, et al. A first-generation multi-functional cytokine for simultaneous optical tracking and tumor therapy. PLoS One. 2012;7(7):e40234. doi: 10.1371/journal.pone.0040234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hingtgen S, Ren X, Terwilliger E, Classon M, Weissleder R, Shah K. Targeting multiple pathways in gliomas with stem cell and viral delivered S-TRAIL and temozolomide. Mol Cancer Ther. 2008;7(11):3575–3585. doi: 10.1158/1535-7163.MCT-08-0640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kauer TM, Figueiredo JL, Hingtgen S, Shah K. Encapsulated therapeutic stem cells implanted in the tumor resection cavity induce cell death in gliomas. Nat Neurosci. 2012;15(2):197–204. doi: 10.1038/nn.3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aboody KS, Najbauer J, Metz MZ, et al. Neural stem cell-mediated enzyme/prodrug therapy for glioma: preclinical studies. Sci Transl Med. 2013;5(184):184ra59. doi: 10.1126/scitranslmed.3005365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aboody K, Capela A, Niazi N, Stern JH, Temple S. Translating stem cell studies to the clinic for CNS repair: current state of the art and the need for a Rosetta stone. Neuron. 2011;70(4):597–613. doi: 10.1016/j.neuron.2011.05.007. [DOI] [PubMed] [Google Scholar]
- 11.Hingtgen S, Figueiredo JL, Farrar C, et al. Real-time multi-modality imaging of glioblastoma tumor resection and recurrence. J Neuro-oncol. 2013;111(2):153–161. doi: 10.1007/s11060-012-1008-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hingtgen SD, Kasmieh R, van de Water J, Weissleder R, Shah K. A novel molecule integrating therapeutic and diagnostic activities reveals multiple aspects of stem cell-based therapy. Stem Cells. 2010;28(4):832–841. doi: 10.1002/stem.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ahmed AU, Alexiades NG, Lesniak MS. The use of neural stem cells in cancer gene therapy: predicting the path to the clinic. Curr Opin Mol Ther. 2010;12(5):546–552. [PMC free article] [PubMed] [Google Scholar]
- 14.Pettikiriarachchi JTS, Parish CL, Shoichet MS, Forsythe JS, Nisbet DR. Biomaterials for brain tissue engineering. Aust J Chem. 2010;63(8):1143–1154. [Google Scholar]
- 15.Guan J, Zhu Z, Zhao RC, et al. Transplantation of human mesenchymal stem cells loaded on collagen scaffolds for the treatment of traumatic brain injury in rats. Biomaterials. 2013;34(24):5937–5946. doi: 10.1016/j.biomaterials.2013.04.047. [DOI] [PubMed] [Google Scholar]
- 16.Bible E, Chau DY, Alexander MR, Price J, Shakesheff KM, Modo M. The support of neural stem cells transplanted into stroke-induced brain cavities by PLGA particles. Biomaterials. 2009;30(16):2985–2994. [Google Scholar]
- 17.Christopherson GT, Song H, Mao HQ. The influence of fiber diameter of electrospun substrates on neural stem cell differentiation and proliferation. Biomaterials. 2009;30(4):556–564. doi: 10.1016/j.biomaterials.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 18.Menon LG, Kelly K, Yang HW, Kim SK, Black PM, Carroll RS. Human bone marrow-derived mesenchymal stromal cells expressing S-TRAIL as a cellular delivery vehicle for human glioma therapy. Stem Cells. 2009;27(9):2320–2330. doi: 10.1002/stem.136. [DOI] [PubMed] [Google Scholar]
- 19.Ehtesham M, Kabos P, Gutierrez MA, et al. Induction of glioblastoma apoptosis using neural stem cell-mediated delivery of tumor necrosis factor-related apoptosis-inducing ligand. Cancer Res. 2002;62(24):7170–7174. [PubMed] [Google Scholar]
- 20.Sasportas LS, Kasmieh R, Wakimoto H, et al. Assessment of therapeutic efficacy and fate of engineered human mesenchymal stem cells for cancer therapy. Proc Natl Acad Sci U S A. 2009;106(12):4822–4827. doi: 10.1073/pnas.0806647106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sena-Esteves M, Tebbets JC, Steffens S, Crombleholme T, Flake AW. Optimized large-scale production of high titer lentivirus vector pseudotypes. J Virol Methods. 2004;122(2):131–139. doi: 10.1016/j.jviromet.2004.08.017. [DOI] [PubMed] [Google Scholar]
- 22.Asli MM, Pourdeyhimi B, Loboa EG. Release profiles of tricalcium phosphate nanoparticles from poly(L-lactic acid) electrospun scaffolds with single component, core-sheath, or porous fiber morphologies: effects on hASC viability and osteogenic differentiation. Macromol Biosci. 2012;12(7):893–900. doi: 10.1002/mabi.201100470. [DOI] [PubMed] [Google Scholar]
- 23.Mohiti-Asli M, Pourdeyhimi B, Loboa EG. Skin tissue engineering for the infected wound site: biodegradable PLA nanofibers and a novel approach for silver ion release evaluated in a 3D coculture system of keratinocytes and Staphylococcus aureus. Tissue Eng C Methods. 2014;20(10):790–797. doi: 10.1089/ten.tec.2013.0458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen ZJ, Gillies GT, Broaddus WC, et al. A realistic brain tissue phantom for intraparenchymal infusion studies. J Neurosurg. 2004;101(2):314–322. doi: 10.3171/jns.2004.101.2.0314. [DOI] [PubMed] [Google Scholar]
- 25.Kulkarni RK, Pani KC, Neuman C, Leonard F. Polylactic acid for surgical implants. Arch Surg. 1966;93(5):839–843. doi: 10.1001/archsurg.1966.01330050143023. [DOI] [PubMed] [Google Scholar]
- 26.Bago JR, Pegna GJ, Okolie O, Hingtgen SD. Fibrin matrices enhance the transplant and efficacy of cytotoxic stem cell therapy for post-surgical cancer. Biomaterials. 2016;84:42–53. doi: 10.1016/j.biomaterials.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lim B, Allen JE, Prabhu VV, Talekar MK, Finnberg NK, El-Deiry WS. Targeting TRAIL in the treatment of cancer: new developments. Expert Opin Ther Targets. 2015;19(9):1171–1185. doi: 10.1517/14728222.2015.1049838. [DOI] [PubMed] [Google Scholar]
- 28.Balyasnikova IV, Prasol MS, Ferguson SD, et al. Intranasal delivery of mesenchymal stem cells significantly extends survival of irradiated mice with experimental brain tumors. Mol Ther. 2014;22(1):140–148. doi: 10.1038/mt.2013.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karra D, Dahm R. Transfection techniques for neuronal cells. J Neurosci. 2010;30(18):6171–6177. doi: 10.1523/JNEUROSCI.0183-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.