

Abstract
Heart failure (HF) is the inability of the heart to provide sufficient cardiac output for the energy demands of the body. Over the last decades, our understanding of the role of microRNAs (miRNAs), a class of small non-coding RNA regulators of gene expression at the post-transcriptional level, in cardiovascular diseases has expanded at a rapid rate. Importantly, multiple miRNAs have been specifically implicated in the progression of HF. Growing evidence suggests that miRNAs regulate central metabolic pathways and thus are highly implicated in the maintenance of energy homeostasis. In this review, we highlight recent discoveries of the mechanistic role of miRNAs in regulating metabolic functions in HF, with specific focus on the implication of miRNAs in metabolic rearrangements, discuss the potential value of miRNA profiles as novel HF biomarkers, and summarize the recent investigations on therapeutic approaches using miRNAs in heart disease.
This article is part of a Special Issue entitled: The role of post-translational protein modifications on heart and vascular metabolism edited by Jason R.B. Dyck & Jan F.C. Glatz.
Keywords: Heart failure, MicroRNA, Metabolism
1. Introduction
Heart failure (HF) is a disease of increasing incidence and prevalence with high morbidity and mortality. It includes derangements in metabolic pathways and disturbed energy homeostasis. MicroRNAs (miRNAs) are highly expressed in the heart and the significance of miRNAs in cardiac development, physiology and disease was elucidated over the last years through various key studies. Cardiac-specific deletion of the miRNA biogenesis key enzyme Dicer in mice has been shown to result in embryonic lethality due to HF [1]. In the past decades, miRNAs have been increasingly described to regulate a diverse spectrum of processes in the heart, and are a vital part of normal cardiac function and cardiac development. Interestingly, a growing body of evidence indicates that miRNAs are implicated as sensors of environmental changes in the settings of heart disease and HF, suggesting their up- or downregulation plays either a causative or protective role by modulating key signaling elements and enzymes in the metabolic pathways. The diagnostic roles of miRNAs in the assessment of heart disease and the modulation of miRNAs as a new therapeutic option have been widely explored, and new findings are continuously emerging. In the present review, we summarize the dysregulation of miRNAs, their role as biomarkers in the failing myocardium, highlight the role of miRNA-mediated metabolic regulation in the cardiac function and discuss the strategy of targeting miRNAs for cardiac disease therapy.
2. Role of miRNAs in regulating gene expression
miRNAs are small single-stranded non-coding RNAs (approximately 22 nucleotides in length), and their best characterized function is to regulate gene expression at the post-transcriptional level [2]. Indeed, most mammalian protein coding genes are under the control of miRNAs via their near-perfect base-pairing with the 3’UTR of the target mRNAs through their 5′-proximal “seeding” region [3,4].
miRNAs are encoded within the genomes of species ranging from protozoans to plants to mammals. miRNAs are first transcribed by RNA polymerase II enzyme into long (~1000 nucleotides in length) precursor molecules called primary miRNAs (pri-miRNAs), and are further transformed into smaller (< 100 nucleotides) hairpin-shaped precursor miRNAs (pre-miRNAs) by the nuclear microprocessor complex consisting of RNase III enzyme Drosha. Once cleaved by Drosha, pre-miRNAs are transported from the nucleus into the cytoplasm, where pre-miRNAs are further cleaved by RNase III-type endonuclease Dicer, generating small double-stranded RNAs containing the mature miRNAs. After being processed into single-stranded miRNAs, the mature miRNAs are incorporated into miRNA-induced silencing complex (miRISC) with the Argonaute (Ago) proteins. This is the functional unit that delivers miRNAs to their mRNA targets [5–7] (Fig. 1).
Fig. 1.
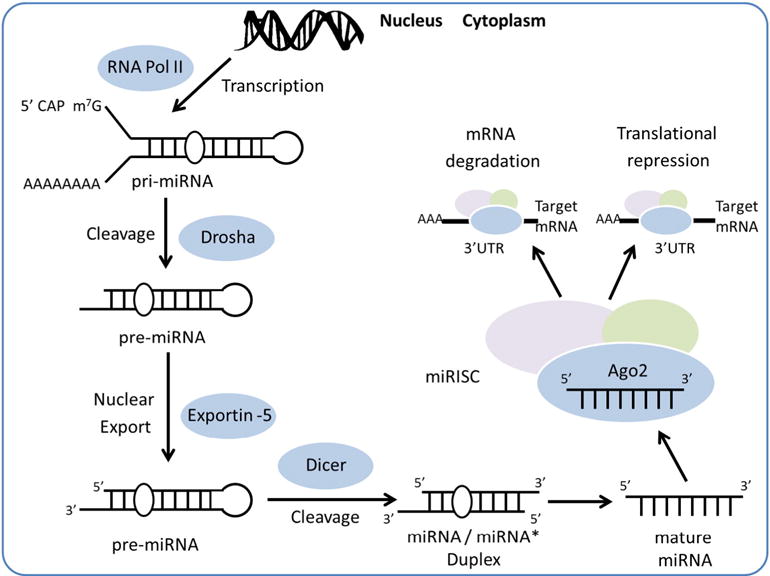
Schematic representation of miRNAs biogenesis and function in gene regulation. RNA Pol II: RNA polymerase II; pri-miRNA: primary miRNA; pre-miRNA: precursor miRNA; miRISC: miRNA-induced silencing complex; Ago2: Argonaute2.
miRNAs regulate the gene expression either through translational repression and/or through deadenylation activation, which subsequently results in transcript degradation. These two mechanisms have been shown to be two distinct independent pathways [8–10]. Interestingly, it has been shown that each mRNA could be targeted by multiple miRNAs, whereas each miRNA could base-pair with multiple target mRNAs, providing another layer of complexity into miRNA-mediated regulation. Thus, these molecules are powerful regulators with the potential to intercept a wide network of cardiovascular diseases.
3. HF-related miRNA profiling in myocardium
miRNAs are newly emerging key regulators of cardiac diseases. miRNA expression varies widely by cell type and cell maturation, under normal and pathological conditions. In recent years, the specific cardiovascular profiling of miRNAs has been studied extensively in the context of HF. Numerous studies have demonstrated that a signature of miRNA alteration is implicated in the preceding pathologies under various cardiac disease settings [11,12–23].
3.1. miRNA signature of HF pathogenesis
Numerous studies have been conducted in the human failing heart using large-scale screening. Matkovich et al. measured the expression of 467 miRNAs in the end-stage congestive failing myocardium, and identified 28 upregulated miRNAs. Among these miRNAs, eight of them (miR-21, -23a, -24, -26, -27, -125, -195 and -199-3p) were demonstrated to be directly associated with HF pathophysiology [12]. In another recent study, the analysis of human failing left ventricles using next generation sequencing method detected significant changes in more than 250 miRNAs of the 800 known human miRNAs, in end-stage heart failure patients diagnosed with dilated cardiomyopathy (DCM) or hypertrophic cardiomyopathy (HCM) in an etiology-dependent manner [13]. Other studies focused on specific miRNAs have also shown the upregulation of miR-24, miR-100, miR-125b, miR-195, miR-199a, miR-214, and downregulation of miR-18, miR-19 family and miR-133 in different types of HF, such as idiopathic dilated cardiomyopathy (IDC), ischemic cardiomyopathy (ISC) and in age-associated HF [14–16]. Interestingly, the altered expression of another set of miRNAs, such as miR-25 [19], miR-199b [24] and miR-204 [25,26], which are regulated by or associated with the critical transcription factor Nuclear Factor of Activated T cells (NFAT) and NFAT signaling, has also been observed in patients with end-stage HF secondary to ischemic heart disease and in pulmonary arterial hypertension (PAH) associated heart disease.
Besides investigations in human HF, a series of studies using animal models have also provided valuable insights into miRNA expression contributing to the pathogenesis of hypertrophy and HF. In mice undergoing transverse aortic constriction (TAC)-induced hypertrophy, a broad variety of miRNAs were upregulated, such as miR-23a, miR-23b, miR-24, miR-125b, miR-195, miR-199a and miR-214, while other specific miRNAs were downregulated, such as miR-93, miR-133a, miR-150 and miR-181b [15]. Similar miRNA expression changes were observed in another study, using left ventricles of a TAC rat model during early hypertrophic growth, showing that 4 miRNAs, miR-23a, miR-27b, miR-125b and miR-195 were upregulated [21].
Controversy exists regarding cardiac expression of miR-25 which seemed to be decreased in both patients with end-stage HF secondary to ischemic heart disease and in hearts from TAC-operated mouse model of HF [19], while another group showed that miR-25 was upregulated in both human myocardial samples from patients with severe HF and in cardiomyocytes of TAC-induced failing mouse hearts [20]. These discrepancies may reflect the existence of distinct miRNA signatures in the failing hearts of different etiologies, or to different stages of disease progression.
3.2. Dynamic changes of miRNA expression in cardiac disease progression
Numerous miRNAs display characteristic changes in expression during pathogenesis of cardiac diseases, in response to acute cardiac stress and in some cases during long-term compensatory response to a chronic injury [21 ]. Their dynamic profile alteration may represent the stage of cardiovascular disease progression.
During the development of hypertrophy, miR-1 and miR-133a were shown to be downregulated. However, both miRNAs displayed overexpression in the study of hearts from canines with chronic HF [22]. Another interesting study investigated the miRNA expression shift during transition from right ventricular hypertrophy (RVH) to HF induced by pulmonary artery constriction (PAC) [23]. At the time points corresponding to early compensated hypertrophy, no significant changes were detected, while 32 and 49 miRNAs were identified as deregulated at the time point associated with early decompensated hypertrophy and HF in mice. This finding further supports the notion that different sets of miRNAs are involved in early-phase of hypertrophic growth and in end-stage HF when HF progression has ceased.
Right ventricular (RV) failure is a complex clinical syndrome resulting from functional cardiac disorder that impairs the ability of the RV to fill or to eject blood. RV failure has mortality as high as left ventricular (LV) failure, and has emerged as an important research priority in the cardiopulmonary field. Interestingly, impairment of miRNA’s expression has been implicated in RV dysfunction [27]. By comparing the miRNA profile, Reddy et al. also pointed out the differences between PAC-induced RVH/HF model and TAC-induced LVH/HF model. 4 particular miRNAs, miR-34a, miR-28, miR-148a, and miR-93, were identified upregulated in the RVH/HF but downregulated in the LVH/HF, suggesting the distinct signature of miRNA expression in response to different pressure overload stresses [23]. miR-126 expression has been shown to be decreased in RV tissues from patients with decompensated RV failure when compared with patients with compensated RV hypertrophy, whereas LV tissues were not affected [28]. Another study showed that the myocardium-specific miR-208 was progressively downregulated as RV failure progressed; while its expression was maintained in the progression of LV failure [29], further suggesting a chamber-specific regulatory mechanism of miRNAs. These studies illustrate that the HF-related aberrant shift in miRNA expression pattern contributes to various aspects of cardiovascular disease, thus suggesting their use as novel diagnostic tools and potential therapeutic targets of HF.
4. Circulating miRNAs as biomarkers of HF
Detection of early diagnostic biomarkers is important for effective management of cardiac disease. Even though the levels of cardiac miRNAs have been assayed for correlation to a wide variety of heart diseases, the miRNA profiling in cardiac tissue has a limited diagnostic value due to the requirement of a cardiac biopsy. Therefore, there is a strong demand for exploring more simple and novel circulating biomarkers for clinical management of HF.
4.1. Properties of circulating miRNAs
miRNAs detected in serum or plasma are collectively called circulating miRNAs. The recent discovery showed that circulating miRNAs remain in a stable form in bodily fluids, even after exposure to severe conditions, such as high temperatures, extreme pH, and prolonged storage [30]. The encapsulated circulating miRNAs protect themselves through several mechanisms, such as packing in membrane vesicles, bounding to transporter proteins, and inclusion in macromolecular complexes, making their degradation particularly difficult [31–35]. The signature of circulating miRNAs has also been shown to be associated with various heart diseases with more sensitivity and specificity. These essential characteristics of circulating miRNAs as excellent blood-based biomarkers for noninvasive molecular diagnosis have attracted more and more interest in the scientific community for their potential applications in cardiovascular diseases, such as early disease detection, monitoring disease proceeding, and response to therapeutic interventions.
4.2. Signature of circulating miRNAs in HF
The expression profiles of miRNAs have been systematically assessed in blood from subjects with HF, which showed significantly altered miRNA expression in HF patients compared to healthy individuals, and during the disease progression in HF (Table 1). One study showed that circulating miRNAs displayed less than 5-fold differences compared with normal patients in stable HF, while the levels of circulating miRNAs increased up to 140-fold in advanced HF. Interestingly, These extracellular changes nearly completely reverted 3 months after initiation of left ventricular assist device (LVAD) support [36]. In a study recruiting 39 healthy controls and 50 cases with symptoms of dyspnea (30 patients were diagnosed with HF, while 20 had non-HF-related disease), 6 miRNAs, miR-18b, miR-129-5p, miR-423-5p, miR-622, miR-675, and miR-1254 were elevated in patients with HF. Among these miRNAs, miR-423-5p was specifically enriched and strongly correlated with the clinical diagnosis of HF [37]. By meticulous screening of 186 miRNAs, the expression of 4 prominent miRNAs, miR-22, miR-92b, miR-320a and miR-423-5p, was found to be significantly elevated in the serum of HF patients. The miRNA-score was also closely related to several important prognostic parameters [38]. Another study reported 4 miRNAs, miR-30b, miR-103, miR-142-3p and miR-342-3p, differentially expressed between HF patients and controls [39]. In a study with 10 HF patient and 17 controls, miR-126 was reported to inversely correlate with age and severity of HF [40]. A different study compared miRNA expression profiles in peripheral blood mononuclear cells (PBMCs) from HF patients either with ischemic (ICM) or non-ischemic dilated cardiomyopathy (NIDCM). 3 miRNAs, miR-107, miR-139 and miR-142-5p, displayed decreased levels in both HF conditions. Additionally, miR-125b and miR-497 levels decreased in ICM fractions only, while miR-142-3p and miR-29b levels increased in NIDCM fractions only [41].
Table 1.
Circulating microRNAs as potential biomarkers in heart failure.
| Study design | Sources | miRNAs expression alteration | miRNA detection | References |
|---|---|---|---|---|
| 39 Healthy subjects | Plasma | miR-18b ↑ | Microarray | [37] |
| 30 HF | miR-129-5p ↑ | qRT-PCR | ||
| 20 Non-HF with dyspnea | miR-423-5p ↑ | |||
| miR-622 ↑ | ||||
| miR-675 ↑ | ||||
| miR-1254 ↑ | ||||
| 30 Healthy subjects | Serum | miR-22 ↑ | qRT-PCR | [38] |
| 30 CHF | miR-92b ↑ | |||
| miR-320a ↑ | ||||
| miR-423-5p ↑ | ||||
| 15 Healthy subjects | Plasma | miR-30b ↓ | Microarray | [39] |
| 44 HF | miR-103 ↓ | qRT-PCR | ||
| 32 COPD | miR-142-3p ↓ | |||
| miR-342-3p ↓ | ||||
| 17 Healthy subjects (Asymptomatic) | Plasma | miR-126 ↓ | Microarray | [40] |
| 10HF | qRT-PCR | |||
| 19 Healthy subjects | PBMCs | ICM and NIDCM: | qRT-PCR | [41] |
| 15 Ischemic cardiomyopathy ICM | miR-107 ↓ | |||
| 19 Non-ischemic dilated cardiomyopathy NIDCM | miR-139 ↓ | |||
| miR-142-5p ↓ | ||||
| ICM only: | ||||
| miR-125b ↓ | ||||
| miR-497 ↓ | ||||
| NIDCM only: | ||||
| miR-142-3p ↑ | ||||
| miR-29b ↑ | ||||
| 10 Healthy subjects | Plasma | miR-423-5p unchanged | qRT-PCR | [42] |
| 41 Right ventricular HF | ||||
| 34 Healthy subjects | Plasma | miR-122 ↑ | qRT-PCR | [43] |
| 33 AHF | miR-499 ↑ | |||
| 10 Healthy subjects | Plasma | miR-126 ↓ | qRT-PCR | [44] |
| 9 MI | miR-122 unchanged | |||
| 15 HF | miR-499 unchanged |
HF: heart failure; CHF: congestive heart failure; COPD: chronic obstructive pulmonary disease; ICM: ischemic cardiomyopathy; NIDCM: non-ischemic dilated cardiomyopathy; AHF: acute heart failure; MI: myocardial infarction.
However, conflicts in the alteration levels of circulating miRNA expression detected by different groups also occur. For example, in contrast to previous results showing miR-423-5p elevated in HF, Tutarel et al. failed to find changes of miR-423-5p levels in patients with right ventricular HF [42]. This discrepancy might be caused by differences in study population, in particular the importance of right versus LV failure, and the impact of right or LV overload versus ischemia in the study.
Another study reported circulating levels of miR-122 and miR-499 elevated in patients with acute heart failure (AHF) by comparing 33 AHF patients and 34 healthy subjects [43]. However, Adachi et al. showed increased plasma miR-499 levels in acute myocardial infarction only, but the concentration of miR-499 was below the limit of detection for all individuals in congestive heart failure (CHF) group [44]. These discrepancies, to some extent, indicate a specific miRNA response to different pathological conditions. At the same time, they also reflect the demand of a reliable technique for circulating miRNAs detection in CHF.
4.3. Circulating miRNAs as new cell-to-cell regulatory species
Emerging evidence has shown that miRNAs in circulation may be involved in cell-to-cell communication by “shuttling” between intracellular compartments. In fact, several studies have suggested that circulating miRNAs can be transferred from a donor cell into a distant recipient cell via exosomes and micro-particles, and regulate gene expression by mediating the repression of critical mRNA targets in recipient cells. Interestingly, extracellular vesicles contain specific miRNAs rather than the complete spectrum of miRNAs of a cell, indicating specific mechanisms for their recognition, packaging, and secretion [45]. The disease-based selection in miRNAs packaging highlights the possibility that administration of miRNAs through the peripheral circulation might potentially result in successful miRNA-based therapeutics.
Several studies have shown how circulating miRNAs work as functional regulators in recipient cells by transferred from donor cells via exosomes during heart diseases. Peripartum cardiomyopathy (PPCM), a life-threatening pregnancy-associated HF in previously healthy women, has been associated with transferred circulating miRNAs [46]. miR-146a overexpression in endothelial cells resulted in the packing and releasing of miR-146a-loaded exosomes, which were further taken up by cardiomyocytes. This process led to subsequent deleterious effects on the recipient cell, by reducing metabolic activity through repression of mRNA targets involved in glucose uptake. Another study showed that miR-21-3p was packed into exosome vesicle in cardiac fibroblasts, and transferred to myocytes as a functional regulator of cardiomyocyte hypertrophy [47].
4.4. Biomarker limitations
The distinct profiling of miRNAs in circulation enables monitoring physiological status changes associated with cardiac disease. However there are still several major limitations to the biomarker value of circulating miRNAs. Firstly, the majority of studies aimed to identify circulating miRNAs as biomarkers of cardiovascular diseases thus far included relatively small patient cohorts; therefore, the conclusions should be validated and confirmed in independent and larger cohort studies. Second, no standardized method for the isolation and quantification of circulating miRNAs has been established so far. Although the main technique for miRNA detection is qRT-PCR, the various choices of normalization make it difficult to compare miRNA expression profiles identified by different laboratories. Thus the generation of standard measurement protocols is still required.
5. The role of miRNAs in cardiac metabolic homeostasis
Cardiac metabolic pathways are governed by a regulatory network that depends on the coordinated action of multiple metabolic enzymes involved in substrate utilization and oxidative phosphorylation for generating ATP supply. A growing body of evidence suggests that numerous miRNAs are implicated in the regulation of energy balance and the maintenance of metabolic homeostasis, by regulating the expression of important components of multiple pathways (Table 2). Hence, to understand the mechanistic basis of miRNAs in the heart, it will be important to decipher the functional targets to investigate the miRNA-metabolism interplay. Glucose and lipid metabolism, which are extensively modulated by the miRNA machinery, will be discussed respectively.
Table 2.
miRNAs involved in metabolic homeostasis.
| miRNAs | Targets | Function | References |
|---|---|---|---|
| Let-7 | Insulin-PI3K-mTOR pathway | Leads to insulin resistance and impaired glucose tolerance | [50,51] |
| miR-1 | G6PD, 6PGD, TKT, FABP3 | Inhibits fatty acid uptake | [60,67] |
| miR-15a | UCP-2 | Reduces ATP generation | [76] |
| miR-15b | Arl2 | Reduces ATP generation | [77] |
| miR-26a | PDHX | Increases glucose uptake, but reduces production of acetyl-CoA | [62] |
| miR-30c | MTP, LPGAT1 | Reduces plasma lipids | [73] |
| miR-33a, miR-33b | CROT, CPT1A, HADHB, SREBP | Down-regulates fatty acid β-oxidation rate | [64] |
| miR-34a | HK, GPI, PDK | Represses glycolysis | [61] |
| miR-93 | GLUT4 | Inhibits glucose uptake | [59] |
| miR-103/107 | CAV1 | Reduces insulin sensitivity and glucose homeostasis | [53] |
| miR-122 | HMGCR, HMGCS1, DHCR7 | Increases cholesterol and triglyceride plasma level | [70,71] |
| miR-132 | CACT | Reduces the transporting rate of fatty-acyl-carnitines into the mitochondria | [66] |
| miR-143 | ORP8 | Impairs insulin-stimulated AKT activation and glucose homeostasis | [55] |
| miR-155 | GLUT1, HK2, PFK2, PKM2, LDHA | Increases glucose consumption rate and lactate production | [63] |
| miR-181a | IDH | Modulate TCA cycle | [74] |
| miR-183 | IDH | Modulate TCA cycle | [75] |
| miR-195 | INSR | Impairs insulin signaling and glycogen synthesis | [54] |
| miR-199a | CAV1 | Down-regulates fatty acid β-oxidation rate | [68] |
| miR-199a/214 | PPAR | Metabolic switch from fatty acid oxidation towards glucose utilization | [49] |
| miR-208 | MED 13 | Reduces insulin sensitivity and glucose tolerance | [48] |
| miR-210 | COX 10 | Reduces ATP generation and up-regulates glycolysis | [79] |
| miR-212 | CACT | Reduces the transporting rate of fatty-acyl-carnitines into the mitochondria | [66] |
| miR-223 | GLUT4 | Inhibits glucose uptake | [58] |
| miR-338 | COX IV | Decreases oxidative phosphorylation and reduces ATP generation | [78] |
| miR-370 | DGAT2, FAS, ACC1 | Reduces the transporting rate of fatty-acyl-CoA into the mitochondria | [65] |
| miR-375 | CAV1, RGS16, AIFM1, ID3, CADM1 etc | Maintains normal glucose homeostasis | [52] |
| miR-378 | ERRϒ, GABPA | Reduces TCA cycle and oxygen consumption, but increases lactate production | [56] |
| miR-696 | PGC-1α | Down-regulates fatty acid β-oxidation rate and up-regulates aerobic metabolism | [69] |
| miR-758 | ABCA1 | Regulates cholesterol efflux | [72] |
| miR-802 | Hnf1b | Leads to impaired glucose tolerance and attenuates insulin sensitivity | [57] |
mTOR: mammalian target of rapamycin; G6PD: glucose-6-phosphate dehydrogenase; 6PGD: 6-phophogluconate dehydrogenase; TKT: transketolase; FABP3: fatty acid binding protein 3; UCP-2: uncoupling protein-2; Arl2: ADP-ribosylation factor-like 2; PDHX: pyruvate dehydrogenase protein X component; MTP: microsomal triglyceride transfer protein; LPGAT1: lysophosphatidylglycerol acyltransferase 1; CROT: carnitine O-octanoyltransferase; CPT1A: carnitine palmitoyltransferase 1A; HADHB: hydroxyacyl-coenzyme A-dehydrogenase; SREBP: sterol-regulatory element-binding protein; HK: hexokinase; GPI: glucose-phosphate isomerase; PDK: phosphoinositide dependent kinase; GLUT: glucose transporter; CAV1: caveolin 1; HMGCR: 3-hydroxy-3-methylglutaryl-coenzyme A reductase; HMGCS: 3-hydroxy-3-methylglutaryl-coenzyme A synthase; DHCR7: dehydrocholesterol reductase; CACT: carnitine acyl-carnitine translocase; ORP8: oxysterol-binding-protein-related protein 8; PFK2: phosphofructokinase 2; PKM2: pyruvate dehydrogenase isoform M2; LDHA: lactate dehydrogenase isoform A; IDH: isocitrate dehydrogenase; INSR: insulin receptor; PPAR: peroxisome proliferator-activated receptor; MED13: mediator complex subunit 13; COX: cytochrome c oxidase; DGAT2: diacylglycerolacyltransferase-2; FAS: fatty acid synthase; ACC1: acyl-CoA carboxylase 1; RGS16: regulator of G protein signaling 16; AIFM1: apoptosis-inducing factor, mitochondrion-associated 1; ID3: inhibitor of DNA binding 3; CADM1: cell adhesion molecule 1; ERRϒ: estrogen-related receptor T; GABPA: GA-binding protein alpha chain; PGC-1α: PPAR coactivator 1α; ABCA1: ATP-binding cassette transporter A1; Hnf1b: hepatocyte nuclear factor 1 homeobox B.
5.1. Glucose metabolism
A recent study revealed an interesting role of miR-208 in the regulation of cardiac metabolism. Heart-specific miR-208 negatively regulates MED13, a regulator of energy expenditure and numerous genes involved in energy balance in the heart. Indeed, pharmacologic inhibition of miR-208 in mice conferred resistance to obesity improving systemic insulin sensitivity and glucose tolerance [48]. The miR-199a/214 cluster has been shown to play a regulatory role in the metabolic switch from mitochondrial fatty acid oxidation in the healthy myocardium towards glucose utilization at the onset of HF, by actively repressing its cardiac target genes PPARβ/δ [49].
Additional miRNAs have been implicated in glucose metabolism in other organs as well. By repression of multiple key components in the insulin-PI3K-mTOR pathway, global and muscle-specific and pancreas-specific overexpression of let-7 induced insulin resistance and impaired glucose tolerance in mice, resulting in reduced fat mass and body weight, as well as decreased body size [50,51]. Another study showed that miR-375 was essentially required for normal glucose homeostasis. miR-375 knockout mice suffered from hyperglycemia and glucose intolerance associated with increased gluconeogenesis [52]. A critical insulin receptor regulator, caveolin 1 (CAV1), is a direct target of miR-103/107. By modulating the expression of CAV1, silencing of miR-103/107 in mice resulted in improved insulin sensitivity and glucose homeostasis, whereas forced expression of miR-103/107 in either liver or adipose tissue was sufficient to induce impaired glucose homeostasis coupled to increased gluconeogenesis [53]. Further, miR-195 suppressed the expression of insulin receptor (INSR), a direct target of miR-195, thereby impairing the insulin signaling cascade and glycogen synthesis [54]). miR-143 has also been shown to associate with insulin-stimulated AKT activation and glucose homeostasis in the liver through regulating its target oxysterol-binding-protein-related protein (ORP) 8 [55]. Another study showed that miR-378* reduced tricarboxylic acid cycle and oxygen consumption but increased lactate production by inhibiting the expression of two PGC-1β partners, ERRγ and GABPA [56]. miR-802 has also been demonstrated to be involved in development of obesity-associated impairment of glucose metabolism through targeting of Hnf1b in mice [57].
miRNAs also modulate fluctuations of metabolic substrates by directly targeting at metabolic enzymes in various glucose metabolism pathways. miR-223 and miR-93 have been reported to inhibit cardiomyocyte glucose uptake through downregulation of the glucose transporter GLUT4 [58,59]. Another group showed that miR-1 regulated glucose flux through the pentose phosphate pathway (PPP) by modulating multiple enzymes, such as glucose-6-phosphate dehydrogenase (G6PD), 6-phophogluconate dehydrogenase (6PGD), and transketolase (TKT) [60]. By directly targeting several glycolytic enzymes, hexokinase (HK) 1 and 2, glucose-6-phosphate isomerase (GPI) and pyruvate dehydrogenase kinase (PDK)-1, miR-34a repressed cellular glycolysis and regulated glucose metabolism [61]. miR-26a has been shown to repress the expression of pyruvate dehydrogenase protein X component (PDHX), and modulates production of acetyl coenzyme A (acetyl-CoA) [62]. Another interesting finding is that numerous enzymes involved in glucose transport, glycolysis, and lactate production were regulated in concert by one miRNA. They demonstrated that miR-155 promoted the glucose consumption rate and lactate production through targeting glucose transporter 1 (GLUT1), hexokinase 2 (HK2), phosphofructokinase 2 (PFK2), pyruvate dehydrogenase isoform M2 (PKM2), and lactate dehydrogenase isoform A (LDHA) [63]. Collectively, these studies highlight the critical roles of miRNAs in glucose metabolism.
5.2. Fatty acid and lipid metabolism
Mitochondrial fatty acid (β-oxidation is the process in which fatty acids are degraded to produce acetyl-CoA, for citric acid cycle activity and ATP/energy generation. Numerous mitochondrial enzymes involved in this process have been shown to be under the control of miRNAs. miR-33a and miR-33b played crucial role in controlling fatty acid β-oxidation by inhibiting the expression of several metabolic enzymes: carnitine O-octanoyltransferase (CROT), which breaks down very long-chain fatty acids, carnitine palmitoyltransferase 1A (CPT1A), the rate-limiting transporter of fatty acids into mitochondria, and hydroxyacyl-coenzyme A-dehydrogenase (HADHB), an enzyme directly involved in mitochondrial fatty acid (β-oxidation [64]. miR-370 was another miRNA revealed to target CPT1A to regulate the rate of β-oxidation. Besides CPT1A, miR-370 was also been shown to functionally participate in fatty acid and triglyceride biosynthesis by regulating diacylglycerol acyltransferase-2 (DGAT2), fatty acid synthase (FAS),and acyl-CoA carboxylase 1 (ACC1) [65]. In another study, miR-132 and miR-212 inhibited the expression of carnitine acyl-carnitine translocase (CACT), and modulated the transporting rate of long-chain acyl-carnitines into the mitochondria for β-oxidation [66].
Additional studies have identified other fatty acid metabolism-associated miRNAs. miR-1 is associated with fatty acid uptake in cardiomyocytes by negatively regulating the expression of heart-type fatty acid binding protein 3 (FABP3) [67]. miR-199a participated in mitochondrial activity by inhibiting the expression of mitochondrial fatty acid β-oxidation related genes through CAV1 suppression [68]. In cultured myocytes, overexpression of miR-696 resulted in decreased fatty acid oxidation compared with normal control myocytes, through direct targeting PGC-1α [69].
The first miRNA linked to lipid metabolic control is miR-122. Two earlier studies showed that miR-122 inhibition resulted in decreased plasma cholesterol and triglyceride levels, probably due to the regulation of its targets: 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR), 3-hydroxy-3-methylglutaryl-coenzyme A synthase 1 (HMGCS1), and 7-dehydrocholesterol reductase (DHCR7) [70,71]. Besides miR-122, a number of other miRNAs are also implicated in the regulation of lipid metabolism. miR-33a and miR-33b have been shown to regulate cholesterol and lipid homeostasis in concert with their host genes, the sterol-regulatory element-binding protein (SREBP) [64]. miR-758 was shown to regulate cholesterol efflux through repression of ATP-binding cassette transporter A1 (ABCA1) [72]. In another study, miR-30c reduced plasma lipids without causing steatosis, by repression of two genes involved in lipid synthesis, microsomal triglyceride transfer protein (MTP) and lysophosphatidylglycerol acyltransferase 1 (LPGAT1) [73].
5.3. Energy metabolism in mitochondria
Mitochondria maintain homeostasis through ATP generation and play a vital role in cardiac function. Both miR-181a and miR-183 have been shown to directly target isocitrate dehydrogenase (IDH) to modulate tricarboxylic acid (TCA) cycle [74,75]. miR-15a inhibited the expression of uncoupling protein-2 (UCP-2) and reduced ATP generation [76]. Another study showed miR-15b decreased ATP synthesis and mitochondrial integrity by targeting ADP-ribosylation factor-like 2 (Arl2) in rat cardiac myocytes [77]. miR-338 and miR-210 respectively modulated ATP production by depressing the expression of cytochrome c oxidase IV (COX IV) and cytochrome c oxidase assembly protein (COX 10), which are the main components in the electron transfer chain [78,79].
5.4. miRNA expression modulated by metabolic stimuli
Interestingly, the relationship between miRNAs and metabolism is a bidirectional link. Multiple studies have shown that miRNA expression alters in response to various environmental stimuli, indicating a complex regulation of homeostasis. Recent studies have shown that high glucose significantly reduced miR-126 and miR-375 expression respectively [80–82]. Prolonged exposure of the beta-cell line M1N6B1 and pancreatic islets to palmitate has induced miR-34a and miR-146 expression in a time and dose dependent manner. In response to a high-fat diet, miR-378 and miR-378* were upregulated in the liver of mice [83]. Two other interesting studies have shown that under metabolic stress observed in pulmonary arterial hypertension (PAH), activation of hypoxia-inducible factor 1α (HlF-1α) repressed miR-223 expression. Interestingly, it has been shown that the downregulation of miR-223 was further implicated in RV dysfunction and DNA damage in HF by subsequently inducing insulin-like growth factor-I receptor (IGF-IR) and 1GF-I downstream signaling [84], and poly(ADP-ribose) polymerase 1 (PARP-1)/DNA damage signaling exacerbation [85].
All the above studies suggest that miRNAs could act as energetic sensors of environmental changes in the regulation metabolic pathways, to adapt to the energetic demands of cells, tissues and organs.
6. Sirtuin-mediated deacetylation in metabolism
Protein acetylation on lysine residues has been discovered as a widespread and evolutionarily conserved post-translational modification, participating in a great number of biological functions, including heart diseases [86]. Altered acetylation states of metabolic enzymes involved in metabolism pathways or signaling cascades may lead to the development of HF, due to long-term metabolic demand not being met [87]. The balance between acetylation/deacetylation is tightly regulated by a group of deacetylases named sirtuins (SIRTs). Interestingly, the expression of SIRTs is also under the control of miRNAs, pointing to a complex regulatory axis involving miRNA function, acetylation/deacetylation rearrangement, metabolic alteration and cardiac dysfunction. In this regard, understanding how SIRTs mediate metabolic alteration through post-translational acetylation, and how SIRTs are regulated by miRNAs is important for the development of therapeutic targets for treating heart diseases associated with SIRTs dysfunction.
6.1. SIRT family
ln mammalian cells, seven SIRT (SIRT 1–7) homologs of the yeast SIRT2 gene function in enzyme-driven protein deacetylation, modulating distinct metabolic pathways. The dependence of SIRTs on NAD + makes them energetic sensors with their enzymatic activities adaptable in response to dynamic energy fluctuations in metabolism [88]. SIRT1 and SIRT2 shuttle between the nucleus and cytoplasm, while SIRT6 and SIRT7 are predominately located in the nucleus [89]. SIRT3, SIRT4 and SIRT5 are found in mitochondria [86], and are not equally effective as deacetylases. Of the three mitochondrial SIRTs, SIRT3 has been reported as a major mitochondrial deacetylase with broadest activities [90], while SIRT4 functions as an efficient ADP-ribosyltransferase and SIRT5 is more widely recognized as mediating malonylation and succinylation [91,92].
SIRT1 plays a critical role in regulating cell death/survival and has been implicated in the pathogenesis of HF. The studies in transgenic mice with cardiac-specific overexpression of SIRT1 identified the beneficial effects of SIRT1 against oxidative stress in the heart [93]. SIRT2 moderates cellular stress-tolerance and the knockdown or inhibition of SIRT2 is protective against ischemia-reperfusion (I/R) injury [94]. The cardiac-protective role of SIRT3 has been demonstrated by several studies showing that the loss of SIRT3, in cardiomyocytes or in mouse models, contributed to the development of cardiac hypertrophy and HF [95–98]. SIRT6 has been shown to be a crucial regulator of glucose metabolism and stress resistance and to attenuate cardiac hypertrophy [99–102]. Finally SIRT7 has been shown to regulate apoptosis and stress responses in the heart [103]. Among the SIRT family, SIRT1 and SIRT3 have been most extensively investigated in the cardiovascular system, playing protective roles in failing hearts.
6.2. SIRTs regulated by miRNAs
Expression of SIRT1 is regulated by several miRNAs to maintain energy homeostasis and metabolic adaption (Table 3). miR-34a, which is highly expressed in high fat diet-induced obese mice, has been demonstrated to directly target SIRT1 and reduce its expression [104,105]. Interestingly, miR-34a itself was shown to be regulated by SIRT1 through several mechanisms, suggesting a regulatory feedback loop [106]. miR-181a displayed an elevated expression in diabetes patients, and directly associated with SIRT1 mRNA 3′UTR, inversely regulating SIRT1 expression and affecting insulin resistance [107]. Furthermore, modulation of miR-181a levels in a diet induced mouse model was sufficient to impair insulin signaling or improve glucose homeostasis. miR-143 suppressed SIRT1 expression and resulted in decreased glucose uptake and glucose intolerance [108]. miR-132 has also been shown to reduce SIRT1 expression, leading to increased acetylation levels of SIRT1 targets [109]. In the heart, miR-217 has been shown to be involved in the development of atherosclerosis by targeting SIRT1 and thus affecting Forkhead Box O1 (FOXO1) acetylation status [110]. Excess glucose has been shown to trigger upregulation of miR-486-5p, modulate SIRT1 expression and activity and affect metabolic processes [111]. Furthermore, both miR-9 and miR-146 have been shown to inhibit SIRT1, and therefore repress SIRT1-mediated deacetylation, leading to attenuated insulin secretion [112–114]. Interestingly, miR-195, which is upregulated in early and late stage hypertrophy and HF, has also been shown to directly bind to the 3’UTR of SIRT1 and inhibit its expression in cardiomyocytes [21,115,116].
Table 3.
SIRTs regulated by miRNAs.
| SIRTs | Location | Enzymatic activity | miRNAs that targets SIRTs | References |
|---|---|---|---|---|
| SIRT1 | Nucleus, cytosol | Lysine deacetyalase | miR-9 | [21,104–116] |
| miR-34a | ||||
| miR-132 | ||||
| miR-143 | ||||
| miR-146 | ||||
| miR-181a | ||||
| miR-195 | ||||
| miR-217 | ||||
| miR-486-5p | ||||
| SIRT2 | Nucleus, cytosol | Lysine deacetyalase | miR-339 | [117] |
| SIRT3 | Mitochondria, nucleus | Lysine deacetyalase | miR-28-5p | [118] |
| SIRT4 | Mitochondria | ADP-ribosyltransferase | ||
| SIRT5 | Mitochondria | Succinyltransferase, malonyltransferase | ||
| SIRT6 | Nucleus | Demyristoylase, depalmitoylase, ADP-ribosyltransferase, deacetylase | miR-33a | [119–121] |
| miR-34a | ||||
| miR-766 | ||||
| SIRT7 | Nucleus | ADP-ribosyltransferase, deacetylase | miR-93 | [122–123] |
| miR-125a | ||||
| miR-125b |
While miRNA-mediated SIRT1 regulation is well known, the regulation of other SIRT family members by miRNAs has just begun to be explored (Table 3). 1t has been identified by luciferase assay that, miR-339 directly targets SIRT2. Moreover, overexpression of miR-339 suppressed SIRT2 expression, while miR-339 knockdown upregulated SIRT2 in human SH-SY5Y neuroblastoma cells and rat PC12 pheochromocytoma cells [117]. miR-28-5p, which is elevated due to high glucose coupled oxidative stress, inhibited SIRT3 expression and led to increased p53 acetylation [118]. 1n another study, miR-33a was shown to target SIRT6, resulting in increased acetylation of several lipid metabolic target genes [119]. miR-34a and miR-766 have also been reported to directly associate with SIRT6 mRNA 3’UTR and downregulate SIRT6 expression [120,121]. Two recent studies demonstrated that miR-93, miR-125a and miR-125b targeted SIRT7 and suppressed its expression level [122,123]. To our knowledge, so far no miRNAs has been reported to directly target SIRT4 or SIRT5.
6.3. SIRT deacetylase-mediated metabolism rearrangement
SIRT1-deficient mice have been shown to exhibit notable developmental defects in the heart [124]. Other studies showed that the expression of SIRT1 was upregulated in failing hearts [125], suggesting a cardioprotective role in the early stage of HF. By determining the acetylation status, SIRT1 regulates numerous targets, key transcription factors and metabolic enzymes, involved in a wide range of signaling pathways and metabolic processes in the heart (Table 4). SIRT1 induced gluconeogenesis by deacetylating and activating the PGC-1α. 1n addition, SIRT1 has been reported to repress glycolytic genes through modulating the activity of PGC-1α in response to pyruvate [126]. Besides directly targeting PGC-1α, SIRT1 has also been shown to deacetylate serine/threonine kinase liver kinase B1 (LKB1) to activate AMPK, which further induced the expression of PGC-1 α, and thus promoted fat oxidation [127,128]. Another study showed that SIRT1 modulated the production of energy via glycolysis by deacetylating and repressing glycolytic enzyme phosphoglycerate mutase-1 (PGAM-1) [129]. SIRT1 can stimulate fatty acid oxidation by deacetylating and activating another target, PPAR-α, to increase energy production and protect heart from hypertrophy [130,131]. For regulating cholesterol homeostasis, SIRT1 deacetylated sterol regulatory element binding protein (SREBP), the major transcription factors for lipogenesis and cholesterol synthesis, farnesoid X receptor (FXR), which is important for cholesterol catabolic pathways, and oxysterol receptor respectively to regulate cholesterol metabolism [132–134]. Another study showed SIRT1’s protective role against metabolic stress by deacetylating FOXO and p53, upregulating expression of antioxidants and inhibiting myocyte apoptosis in the heart [93,135–138]. In another study, SIRT1 also activated Akt/PI3K signaling, by deacetylating Akt, and resulted in enhancement of cardiac hypertrophic response in cardiomyocytes [139]. In summary, SIRT1 mediates a sophisticated switch in metabolism to adapt the energy demanding in the heart.
Table 4.
SIRTs targets involved in metabolism alteration.
| SIRTs | Deacetylation targets | Signaling/metabolic pathways | References |
|---|---|---|---|
| SIRT1 | PGC-1α | Fatty acid oxidation | [93,126–139] |
| LKB1 | Gluconeogenesis | ||
| PGAM-1 | Glycolysis | ||
| PPAR-α | Cholesterol metabolism | ||
| SREBP | AMPK pathway | ||
| FXR | Apoptosis | ||
| Oxysterol receptor | Akt/PI3K signaling | ||
| FOXO | |||
| P53 | |||
| Akt | |||
| SIRT2 | PGC-1α | Fatty acid oxidation | [153–154] |
| SIRT3 | HMGCS2 | Lipid utilization | [140–152] |
| LCAD | Fatty acid oxidation | ||
| AceCS2 | Glucose oxidation | ||
| PDH | TCA cycle | ||
| IDH2 | Oxidation phosphorylation | ||
| GDH | |||
| NDUFA9 | |||
| SDH | |||
| OSCP | |||
| ATP synthase β subunit | |||
| SIRT4 | GDH | Fatty acid oxidation | [92] |
| SIRT5 | HMGCS2 | Lipid utilization | [155] |
| SIRT6 | c-jun | IGF/Akt signaling | [101–102,156–161] |
| PGC-1α | Glycolysis | ||
| H1F1-α | Lipogenesis | ||
| ACC1 | Triglyceride metabolism | ||
| SCD1 | Cholesterol metabolism | ||
| Bcl2 | AMPK pathway | ||
| NFkB | |||
| SIRT7 | TR4/TAK1 | Lipid metabolism | [103,162] |
PGC-1α: PPAR coactivator 1α; LKB1: liver kinase B1; PGAM-1: phosphoglycerate mutase-1; PPAR: peroxisome proliferator-activated receptor; SREBP: sterol regulatory element binding protein; FXR: farnesoid X receptor; FOXO: Forkhead Box Class O; LCAD: long-chain acyl CoA dehydrogenase; HMGCS: 3-hydroxy-3-methylglutaryl-coenzyme A synthase; AceCS2: acetyl-CoA synthase 2; PDH: pyruvate dehydrogenases; IDH2: Isocitrate dehydrogenase 2; GDH: glutamate dehydrogenase; SDH: succinate dehydrogenase; OSCP: oligomycin sensitivity conferring protein; HIF1-α: hypoxia-inducible factor 1α; ACC1: acetyl-CoA carboxylase 1; SCD1: stearoyl-CoA desaturase 1; Bcl2: B-cell lymphoma 2; NFkB: nuclear factor kappa-light-chain-enhancer of activated B cells; TR4/TAK1: testicular receptor 4/transforming growth factor-β-activated kinase.
Interestingly, while SIRT1 regulates metabolic function by mostly deacetylating nuclear transcription factors, SIRT3 does so by directly targeting mitochondrial metabolic enzymes (Table 4). Mitochondria are the main cellular organelles responsible for energy balance and metabolism homeostasis in the heart. Thus the mitochondrial deacetylase SIRT3 plays a pivotal role in the maintenance of appropriate mitochondrial function in the heart, by mediating deacetylation of a series of key modulators and their relevant pathways. SIRT3 has been reported to modulate the process of lipid utilization and fatty acid oxidation, by targeting 3-hydroxy-3-methylglutaryl CoA synthase 2 (HMGCS2) and long-chain acyl CoA dehydrogenase (LCAD) respectively [140,141]. SIRT3 was shown to target a cardiac-enriched mitochondrial enzyme acetyl-CoA synthase 2 (AceCS2), which promotes the entry of acetate into the TCA cycle in the form of acetyl-CoA [142,143], and increase its activity. SIRT3 has also been reported to regulate the TCA cycle and glycolysis, which connects glucose oxidation with oxidative phosphorylation. In another study, pyruvate dehydrogenases (PDH), isocitrate dehydrogenase 2 (IDH2) and glutamate dehydrogenase (GDH) have been identified as targets of SIRT3, which deacetylated and activated their enzymatic activities [144–146]. SIRT3 has also been demonstrated to be important for the mitochondrial energy maintenance, by deacetylating multiple proteins participating in electron transport chain (ETC) and oxidative phosphorylation, such as NDUFA9 subunit of complex I of ETC [147], succinate dehydrogenase (SDH, complex II) [148,149], oligomycin sensitivity conferring protein (OSCP) [150], and ATP synthase β subunit [151] of complex V. Since SIRT3 has been shown to involve in every aspect of metabolic associated scenarios, its function in the development of HF has also been elucidated. SIRT3 deficient mice produced a severe cardiac hypertrophic response to hypertrophic stimuli, while the overexpression of SIRT3 prevented cardiac hypertrophy and dysfunction in primary cultures of cardiomyocytes [152]. Therefore, SIRT3 plays a beneficial role in the heart and the regulation of its function could be used as a novel cardiac-protective strategy.
Besides SIRT1 and SIRT3, the role of other SIRTs has also been investigated in maintaining energy homeostasis and cardiac health (Table 4). SIRT2 has been reported to affect fatty acid oxidation in adipocytes through PGC-1α deacetylation [153], and to interact with Akt in insulin-responsive cell lines [154]. Inhibition of SIRT4 promoted fatty acid oxidation and mitochondrial function, probably due to its regulation on glutamate dehydrogenase, while overexpression of SIRT5 increased oxygen consumption and ATP synthesis [92]. In addition, it has been shown that SIRT5 regulated succinylation of the rate-limiting ketogenic enzyme HMGCS2 in the mitochondria [155]. The expression level of SIRT6, another nuclear SIRT, decreases during cardiac dysfunction in humans. The beneficial role of SIRT6 has been demonstrated that this SIRT attenuated IGF-Akt signaling and the development of cardiac hypertrophy by targeting c-jun [102]. Interestingly, recent studies of SIRT6 knockout mice showed increased glycolysis, lipogenesis and triglyceride production, probably due to targeting PGC-1α [156,157]. SIRT6 has also been reported to regulate glucose homeostasis via HIF1-α [101]. Moreover, SIRT6 modulated cholesterol and fatty acid metabolism, by regulating SREBP-1 target genes implicated in fatty acid production such as acetyl-CoA carboxylase 1 (ACC1), and stearoyl-CoA desaturase 1 (SCD1) [156,158–160]. Other protective mechanisms of SIRT6 have been also well studied, including the regulation of AMPKα pathway, B-cell lymphoma 2 (Bcl2), nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB), and Akt pathway during metabolic stress [161]. SIRT7 has been reported to regulate lipid metabolism, fatty acid uptake and synthesis/storage of triglycerides, probably by targeting nuclear receptor testicular receptor 4/transforming growth factor-(β-activated kinase (TR4/TAK1) [162]. In a study with animal models, SIRT7-deficient mice exhibited inflammatory cardiomyopathy and cardiac hypertrophy [103].
Together, these findings indicate that SIRTs are not only sensors of energy status, but can also dynamically and intrinsically regulate cellular metabolism, protecting myocardium against metabolic stresses. The beneficial net effects of SIRT family on cardiac health point out the collaborative roles of SIRTs with coordinated nuclear and mitochondrial programs. The complex regulatory effects of miRNAs on the energy homeostasis in the failing myocardium through SIRTs-mediated alteration of metabolic protein acetylation highlight a promising strategy to develop new miRNA implicated therapeutic protocols to contrast metabolism-associated cardiac dysfunction.
7. MiRNA-based therapeutic strategies
miRNA-mediated intervention in cardiovascular disease is a promising avenue for the development of therapeutics in the preservation of cardiac function. In recent years, numerous studies have shown the role of miRNAs in the maladaptive processes involved in HF, and how the aberrant expressed miRNAs can be therapeutically manipulated by the administration of extracellular miRNAs. There are two approaches to normalize the dysregulated miRNAs, using antimiRs to suppress the level of miRNAs or using miRNA mimics to elevate the level of miRNAs. Their mode of action is described in Fig. 2.
Fig. 2.
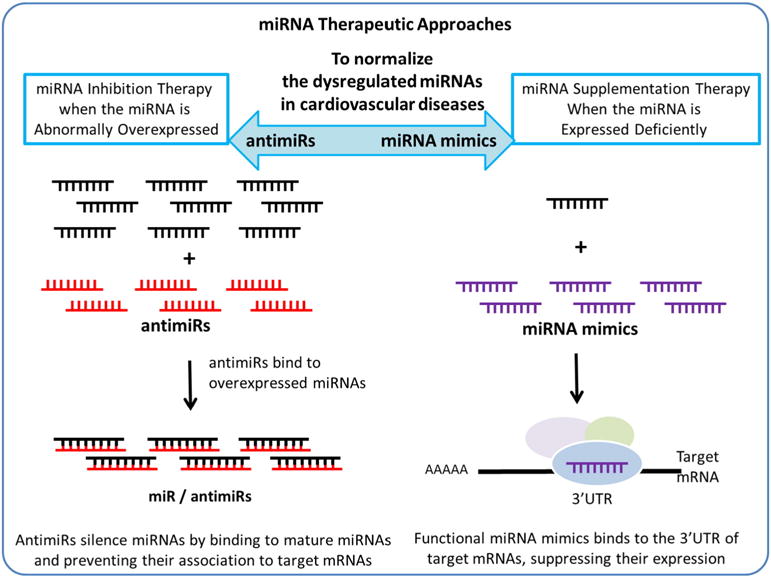
Schematic representation of miRNA-based therapies to compensate the aberrant expressed miRNAs in heart diseases.
7.1. Loss of function - miRNA silencing
AntimiRs are single-stranded antisense oligonucleotide molecules which could directly silence miRNAs by binding to the mature miRNAs and consequently prevent their association to target mRNAs. Varying chemical modifications are designed to enhance their stability and uptake efficiency by the recipient cells. Functional antimiRs, such as antagomiRs and locked nucleic acids (LNAs), could effectively silence miRNAs that are overexpressed in HF. Moreover, it has also been demonstrated that in preclinical animal models, the long-term inhibition of miRNAs by antimiRs is capable of attenuating cardiovascular disease, with no evidence of toxicity.
In a cardiac hypertrophy mouse model, miR-21 showed elevated expression levels in cardiac fibroblasts. The use of an antagomiR, which could functionally inhibit miR-21, significantly reduced cardiac hypertrophy and improved cardiac function [163]. In addition, inhibition of the miR-15 family by LNA-modified antimiRs protected cardiomyocytes and resulted in an improvement in cardiac function [164]. Studies in a Dahl salt-sensitive rat HF model have shown that the delivery of antimiR-208a not only prevents cardiac remodeling, but also improves cardiac function and survival [165]. Injection of antagomiR-25 resulted in a remarkable slowdown in the development of HF in a mouse model, improved cardiac function and survival [20]. Global knockdown of Let-7 with antimiRs was sufficient to prevent and treat impaired glucose tolerance in mice with diet-induced obesity, demonstrating the role of Let-7 in the regulation of glucose metabolism and suggesting the antimiR-mediated Let-7 silencing as a potential treatment of type 2 diabetes [51]. In another study, the LNA-modified antimiR-mediated inhibition of miR-33 family, key regulators of cholesterol and lipid homeostasis, increased high-density lipoprotein cholesterol and led to cardiovascular protection [166]. In a mouse model of peripartum cardiomyopathy, knockdown of miR-146a with LNA-modified antagomiR-146a reduced HF phenotype, suggesting miR-146a as a therapeutic target in HF [46]. The cardiac expression level of miR-199b was detected to be increased in HF. The antagomiR-mediated inhibition of miR-199b successfully caused a marked reversal of hypertrophy and prevented the establishment of HF in a mouse model of cardiac pressure-overload hypertrophy, by reducing the critical transcription factor NFAT activity [24].
7.2. Gain of function - miRNA overexpression
miRNA mimics, a class of synthetic double-stranded oligonucleotides miRNA analogs, are widely used to restore the expression levels of dysregulated “protective” miRNAs implicated in HF. In target cells, they are further processed into single-stranded miRNAs. The functional miRNA mimics are capable of binding to the 3’UTR of target mRNAs, and consequently suppress their expression. So far, adenovirus, lentivirus, and adeno-associated viruses (AAV) are widely used for exogenous administration. Since the miRNA mimics have to undergo the miRNA maturation processes, the technologies for miRNA functional overexpression and the optimization for their better delivery and uptake are not as well developed as the inhibition of miRNAs via antimiRs.
In a cardiac-specific miR-133 inducible transgenic mouse model, miR-133 overexpression decreased cardiomyocyte apoptosis and promoted cardiac function following pressure-overload [167]. Another group has also shown that in vitro, ultrasound-mediated miR-133 mimic-overexpression could reverse hypertrophy on cardiomyocytes [168], confirming the cardiac-protective role of miR-133. Overexpression of miR-590 and miR-199a via an AAV system induced cardiac regeneration and preserved cardiac function after myocardial infarction, suggesting a novel therapy that can restore the proliferative capacity of the damaged heart by miRNA overexpression [169]. AAV-mediated long-term overexpression of miR-669a reduced cardiac hypertrophy and improves survival in mice [170]. In a myocardial ischemia/reperfusion rat model, adenovirus-mediated miR-22 overexpression improved cardiac function by significantly reducing infarct size and cardiomyocyte apoptosis [171].
To date, most studies are based on manipulating single miRNAs. Since multiple miRNAs are likely to be involved in the development and progression of HF, the therapeutic combination to modulate several miRNAs could be a potential future medical intervention. However, to our knowledge, so far no human clinical HF studies with antimiRs or miRNA mimics are ongoing. Many problems need to be resolved to use in clinical setting, and the design and efficiency of these methods need to be thoroughly tested for further validation.
In summary, cardiovascular diseases, including HF, are the predominant cause of death in developed countries. Recent discoveries on the implication of miRNAs in HF have improved our understanding of miRNA-mediated regulatory network associated to metabolic abnormalities and energy homeostasis perturbations in the setting of heart disease or HF. Therefore, miRNAs are emerging as important biomolecular candidates for their potential value in the development of novel diagnostic tools and therapeutic approaches. However, the contrasting findings in some studies emphasize the gaps in our knowledge and indicate that our understanding of the molecular mechanisms underlying the regulation of HF progress by miRNAs remains limited. Further investigation is required for a deeper understanding to fully explain miRNA function in cardiac biology.
Footnotes
Transparency Document
The Transparency document associated with this article can be found, in the online version.
References
- 1.Zhao Y, et al. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1-2. Cell. 2007;129(2):303–317. doi: 10.1016/j.cell.2007.03.030. [DOI] [PubMed] [Google Scholar]
- 2.Ghildiyal M, Zamore PD. Small silencing RNAs: an expanding universe. Nat Rev Genet. 2009;10(2):94–108. doi: 10.1038/nrg2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kozomara A, Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014;42:D68–D73. doi: 10.1093/nar/gkt1181. (Database issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Friedman RC, et al. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19(1):92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 6.Gregory RI, et al. The microprocessor complex mediates the genesis of microRNAs. Nature. 2004;432(7014):235–240. doi: 10.1038/nature03120. [DOI] [PubMed] [Google Scholar]
- 7.Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol. 2014;15(8):509–524. doi: 10.1038/nrm3838. [DOI] [PubMed] [Google Scholar]
- 8.Zhang X, Virtanen A, Kleiman FE. To polyadenylate or to deadenylate: that is the question. Cell Cycle. 2010;9(22):4437–4449. doi: 10.4161/cc.9.22.13887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of posttranscriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet. 2008;9(2):102–114. doi: 10.1038/nrg2290. [DOI] [PubMed] [Google Scholar]
- 10.Eulalio A, et al. Deadenylation is a widespread effect of miRNA regulation. RNA. 2009;15(1):21–32. doi: 10.1261/rna.1399509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kalozoumi G, Yacoub M, Sanoudou D. MicroRNAs in heart failure: small molecules with major impact. Glob Cardiol Sci Pract. 2014;2014(2):79–102. doi: 10.5339/gcsp.2014.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matkovich SJ, et al. Reciprocal regulation of myocardial microRNAs and messenger RNA in human cardiomyopathy and reversal of the microRNA signature by biomechanical support. Circulation. 2009;119(9):1263–1271. doi: 10.1161/CIRCULATIONAHA.108.813576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leptidis S, et al. A deep sequencing approach to uncover the miRNOME in the human heart. PLoS One. 2013;8(2):e57800. doi: 10.1371/journal.pone.0057800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sucharov C, Bristow MR, Port JD. miRNA expression in the failing human heart: functional correlates. J Mol Cell Cardiol. 2008;45(2):185–192. doi: 10.1016/j.yjmcc.2008.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van Rooij E, et al. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc Natl Acad Sci U S A. 2006;103(48):18255–18260. doi: 10.1073/pnas.0608791103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van Almen GC, et al. MicroRNA-18 and microRNA-19 regulate CTGF and TSP-1 expression in age-related heart failure. Aging Cell. 2011;10(5):769–779. doi: 10.1111/j.1474-9726.2011.00714.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elia L, et al. Reciprocal regulation of microRNA-1 and insulin-like growth factor-1 signal transduction cascade in cardiac and skeletal muscle in physiological and pathological conditions. Circulation. 2009;120(23):2377–2385. doi: 10.1161/CIRCULATIONAHA.109.879429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takasu N, Yoshimura Noh J. Hashimoto’s thyroiditis: TGAb, TPOAb, TRAb and recovery from hypothyroidism. Expert Rev Clin Immunol. 2008;4(2):221–237. doi: 10.1586/1744666X.4.2.221. [DOI] [PubMed] [Google Scholar]
- 19.Dirkx E, et al. Nfat and miR-25 cooperate to reactivate the transcription factor Hand2 in heart failure. Nat Cell Biol. 2013;15(11):1282–1293. doi: 10.1038/ncb2866. [DOI] [PubMed] [Google Scholar]
- 20.Wahlquist C, et al. Inhibition of miR-25 improves cardiac contractility in the failing heart. Nature. 2014;508(7497):531–535. doi: 10.1038/nature13073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Busk PK, Cirera S. MicroRNA profiling in early hypertrophic growth of the left ventricle in rats. Biochem Biophys Res Commun. 2010;396(4):989–993. doi: 10.1016/j.bbrc.2010.05.039. [DOI] [PubMed] [Google Scholar]
- 22.Belevych AE, et al. MicroRNA-1 and -133 increase arrhythmogenesis in heart failure by dissociating phosphatase activity from RyR2 complex. PLoS One. 2011;6(12):e28324. doi: 10.1371/journal.pone.0028324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reddy S, et al. Dynamic microRNA expression during the transition from right ventricular hypertrophy to failure. Physiol Genomics. 2012;44(10):562–575. doi: 10.1152/physiolgenomics.00163.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.da Costa Martins PA, et al. MicroRNA-199b targets the nuclear kinase Dyrk1a in an auto-amplification loop promoting calcineurin/NFAT signalling. Nat Cell Biol. 2010;12(12):1220–1227. doi: 10.1038/ncb2126. [DOI] [PubMed] [Google Scholar]
- 25.Potus F, et al. Vascular remodeling process in pulmonary arterial hypertension, with focus on miR-204 and miR-126 (2013 Grover conference series) Pulm Circ. 2014;4(2):175–184. doi: 10.1086/675980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Courboulin A, et al. Role for miR-204 in human pulmonary arterial hypertension. J Exp Med. 2011;208(3):535–548. doi: 10.1084/jem.20101812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boucherat O, Potus F, Bonnet S. microRNA and pulmonary hypertension. Adv Exp Med Biol. 888(2015):237–252. doi: 10.1007/978-3-319-22671-2_12. [DOI] [PubMed] [Google Scholar]
- 28.Potus F, et al. Downregulation of microRNA-126 contributes to the failing right ventricle in pulmonary arterial hypertension. Circulation. 2015;132(10):932–943. doi: 10.1161/CIRCULATIONAHA.115.016382. [DOI] [PubMed] [Google Scholar]
- 29.Paulin R, et al. A miR-208-Mef2 axis drives the decompensation of right ventricular function in pulmonary hypertension. Circ Res. 2015;116(1):56–69. doi: 10.1161/CIRCRESAHA.115.303910. [DOI] [PubMed] [Google Scholar]
- 30.Chen X, et al. Characterization of microRNAs in serum: a novel class of biomarkers for diagnosis of cancer and other diseases. Cell Res. 2008;18(10):997–1006. doi: 10.1038/cr.2008.282. [DOI] [PubMed] [Google Scholar]
- 31.Hunter MP, et al. Detection of microRNA expression in human peripheral blood microvesicles. PLoS One. 2008;3(11):e3694. doi: 10.1371/journal.pone.0003694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Skog J, et al. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol. 2008;10(12):1470–1476. doi: 10.1038/ncb1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Valadi H, et al. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9(6):654–659. doi: 10.1038/ncb1596. [DOI] [PubMed] [Google Scholar]
- 34.Wang K, et al. Export of microRNAs and microRNA-protective protein by mammalian cells. Nucleic Acids Res. 2010;38(20):7248–7259. doi: 10.1093/nar/gkq601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Janowska-Wieczorek A, et al. Platelet-derived microparticles bind to hematopoietic stem/progenitor cells and enhance their engraftment. Blood. 2001;98(10):3143–3149. doi: 10.1182/blood.v98.10.3143. [DOI] [PubMed] [Google Scholar]
- 36.Akat KM, et al. Comparative RNA-sequencing analysis of myocardial and circulating small RNAs in human heart failure and their utility as biomarkers. Proc Natl Acad Sci USA. 2014;111(30):11151–11156. doi: 10.1073/pnas.1401724111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tijsen AJ, et al. MiR423-5p as a circulating biomarker for heart failure. Circ Res. 2010;106(6):1035–1039. doi: 10.1161/CIRCRESAHA.110.218297. [DOI] [PubMed] [Google Scholar]
- 38.Goren Y, et al. Serum levels of microRNAs in patients with heart failure. Eur J Heart Fail. 2012;14(2):147–154. doi: 10.1093/eurjhf/hfr155. [DOI] [PubMed] [Google Scholar]
- 39.Ellis KL, et al. Circulating microRNAs as candidate markers to distinguish heart failure in breathless patients. Eur J Heart Fail. 2013;15(10):1138–1147. doi: 10.1093/eurjhf/hft078. [DOI] [PubMed] [Google Scholar]
- 40.Fukushima Y, et al. Assessment of plasma miRNAs in congestive heart failure. Circ J. 2011;75(2):336–340. doi: 10.1253/circj.cj-10-0457. [DOI] [PubMed] [Google Scholar]
- 41.Voellenkle C, et al. MicroRNA signatures in peripheral blood mononuclear cells of chronic heart failure patients. Physiol Genomics. 2010;42(3):420–426. doi: 10.1152/physiolgenomics.00211.2009. [DOI] [PubMed] [Google Scholar]
- 42.Tutarel O, et al. Circulating miR-423_5p fails as a biomarker for systemic ventricular function in adults after atrial repair for transposition of the great arteries. Int J Cardiol. 2013;167(1):63–66. doi: 10.1016/j.ijcard.2011.11.082. [DOI] [PubMed] [Google Scholar]
- 43.Corsten MF, et al. Circulating MicroRNA-208b and MicroRNA-499 reflect myocardial damage in cardiovascular disease. Circ Cardiovasc Genet. 2010;3(6):499–506. doi: 10.1161/CIRCGENETICS.110.957415. [DOI] [PubMed] [Google Scholar]
- 44.Adachi T, et al. Plasma microRNA 499 as a biomarker of acute myocardial infarction. Clin Chem. 2010;56(7):1183–1185. doi: 10.1373/clinchem.2010.144121. [DOI] [PubMed] [Google Scholar]
- 45.Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013;200(4):373–383. doi: 10.1083/jcb.201211138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Halkein J, et al. MicroRNA-146a is a therapeutic target and biomarker for peripartum cardiomyopathy. J Clin Invest. 2013;123(5):2143–2154. doi: 10.1172/JCI64365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bang C, et al. Cardiac fibroblast-derived microRNA passenger strand-enriched exosomes mediate cardiomyocyte hypertrophy. J Clin Invest. 2014;124(5):2136–2146. doi: 10.1172/JCI70577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grueter CE, et al. A cardiac microRNA governs systemic energy homeostasis by regulation of MED13. Cell. 2012;149(3):671–683. doi: 10.1016/j.cell.2012.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.el Azzouzi H, et al. The hypoxia-inducible microRNA cluster miR-199a approximately 214 targets myocardial PPARdelta and impairs mitochondrial fatty acid oxidation. Cell Metab. 2013;18(3):341–354. doi: 10.1016/j.cmet.2013.08.009. [DOI] [PubMed] [Google Scholar]
- 50.Zhu H, et al. The Lin28/let-7 axis regulates glucose metabolism. Cell. 2011;147(1):81–94. doi: 10.1016/j.cell.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Frost RJ, Olson EN. Control of glucose homeostasis and insulin sensitivity by the Let-7 family of microRNAs. Proc Natl Acad Sci U S A. 2011;108(52):21075–21080. doi: 10.1073/pnas.1118922109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Poy MN, et al. miR-375 maintains normal pancreatic alpha- and beta-cell mass. Proc Natl Acad Sci U S A. 2009;106(14):5813–5818. doi: 10.1073/pnas.0810550106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Trajkovski M, et al. MicroRNAs 103 and 107 regulate insulin sensitivity. Nature. 2011;474(7353):649–653. doi: 10.1038/nature10112. [DOI] [PubMed] [Google Scholar]
- 54.Yang WM, et al. Saturated fatty acid-induced miR-195 impairs insulin signaling and glycogen metabolism in HepG2 cells. FEBS Lett. 2014;588(21):3939–3946. doi: 10.1016/j.febslet.2014.09.006. [DOI] [PubMed] [Google Scholar]
- 55.Jordan SD, et al. Obesity-induced overexpression of miRNA-143 inhibits insulin-stimulated AKT activation and impairs glucose metabolism. Nat Cell Biol. 2011;13(4):434–446. doi: 10.1038/ncb2211. [DOI] [PubMed] [Google Scholar]
- 56.Eichner LJ, et al. miR-378(*) mediates metabolic shift in breast cancer cells via the PGC-1beta/ERRgamma transcriptional pathway. Cell Metab. 2010;12(4):352–361. doi: 10.1016/j.cmet.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 57.Kornfeld JW, et al. Obesity-induced overexpression of miR-802 impairs glucose metabolism through silencing of Hnf1b. Nature. 2013;494(7435):111–115. doi: 10.1038/nature11793. [DOI] [PubMed] [Google Scholar]
- 58.Lu H, Buchan RJ, Cook SA. MicroRNA-223 regulates Glut4 expression and cardiomyocyte glucose metabolism. Cardiovasc Res. 2010;86(3):410–420. doi: 10.1093/cvr/cvq010. [DOI] [PubMed] [Google Scholar]
- 59.Chen YH, et al. miRNA-93 inhibits GLUT4 and is overexpressed in adipose tissue of polycystic ovary syndrome patients and women with insulin resistance. Diabetes. 2013;62(7):2278–2286. doi: 10.2337/db12-0963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Singh A, et al. Transcription factor NRF2 regulates miR-1 and miR-206 to drive tumorigenesis. J Clin Invest. 2013;123(7):2921–2934. doi: 10.1172/JCI66353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim HR, et al. p53 regulates glucose metabolism by miR-34a, Biochem. Biophys Res Commun. 2013;437(2):225–231. doi: 10.1016/j.bbrc.2013.06.043. [DOI] [PubMed] [Google Scholar]
- 62.Chen B, et al. MicroRNA-26a regulates glucose metabolism by direct targeting PDHX in colorectal cancer cells. BMC Cancer. 2014;14:443. doi: 10.1186/1471-2407-14-443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jiang S, et al. A novel miR-155/miR-143 cascade controls glycolysis by regulating hexokinase 2 in breast cancer cells. EMBO J. 2012;31(8):1985–1998. doi: 10.1038/emboj.2012.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rottiers V, et al. MicroRNAs in metabolism and metabolic diseases. Cold Spring Harb Symp Quant Biol. 2011;76:225–233. doi: 10.1101/sqb.2011.76.011049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Iliopoulos D, et al. MicroRNA-370 controls the expression of microRNA-122 and Cpt1alpha and affects lipid metabolism. J Lipid Res. 2010;51(6):1513–1523. doi: 10.1194/jlr.M004812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Soni MS, et al. Downregulation of carnitine acyl-carnitine translocase by miRNAs 132 and 212 amplifies glucose-stimulated insulin secretion. Diabetes. 2014;63(11):3805–3814. doi: 10.2337/db13-1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Varrone F, et al. The circulating level of FABP3 is an indirect biomarker of microRNA-1. J Am Coll Cardiol. 2013;61(1):88–95. doi: 10.1016/j.jacc.2012.08.1003. [DOI] [PubMed] [Google Scholar]
- 68.Li B, et al. Aberrant miR199a-5p/caveolin1/PPARalpha axis in hepatic steatosis. J Mol Endocrinol. 2014;53(3):393–403. doi: 10.1530/JME-14-0127. [DOI] [PubMed] [Google Scholar]
- 69.Aoi W, et al. The microRNA miR-696 regulates PGC-1{alpha} in mouse skeletal muscle in response to physical activity. Am J Physiol Endocrinol. 2010;298(4):E799–E806. doi: 10.1152/ajpendo.00448.2009. [DOI] [PubMed] [Google Scholar]
- 70.Esau C, et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006;3(2):87–98. doi: 10.1016/j.cmet.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 71.Krutzfeldt J, et al. Silencing of microRNAs in vivo with ‘antagomirs’. Nature. 2005;438(7068):685–689. doi: 10.1038/nature04303. [DOI] [PubMed] [Google Scholar]
- 72.Ramirez CM, et al. MicroRNA-758 regulates cholesterol efflux through posttranscriptional repression of ATP-binding cassette transporter A1. Arterioscler Thromb Vasc Biol. 2011;31(11):2707–2714. doi: 10.1161/ATVBAHA.111.232066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Soh J, et al. MicroRNA-30c reduces hyperlipidemia and atherosclerosis in mice by decreasing lipid synthesis and lipoprotein secretion. Nat Med. 2013;19(7):892–900. doi: 10.1038/nm.3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chu B, et al. MiR-181a regulates lipid metabolism via IDH1. Sci Rep. 2015;5:8801. doi: 10.1038/srep08801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tanaka H, et al. MicroRNA-183 upregulates HIF-1 alpha by targeting isocitrate dehydrogenase 2 (IDH2) in glioma cells. J Neuro-Oncol. 2013;111(3):273–283. doi: 10.1007/s11060-012-1027-9. [DOI] [PubMed] [Google Scholar]
- 76.Sun LL, et al. MicroRNA-15a positively regulates insulin synthesis by inhibiting uncoupling protein-2 expression. Diabetes Res Clin Pract. 2011;91(1):94–100. doi: 10.1016/j.diabres.2010.11.006. [DOI] [PubMed] [Google Scholar]
- 77.Nishi H, et al. MicroRNA-15b modulates cellular ATP levels and degenerates mitochondria via Arl2 in neonatal rat cardiac myocytes. J Biol Chem. 2010;285(7):4920–4930. doi: 10.1074/jbc.M109.082610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Aschrafi A, et al. MicroRNA-338 regulates local cytochrome c oxidase IV mRNA levels and oxidative phosphorylation in the axons of sympathetic neurons. J Neurosci. 2008;28(47):12581–12590. doi: 10.1523/JNEUROSCI.3338-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chen Z, et al. Hypoxia-regulated microRNA-210 modulates mitochondrial function and decreases ISCU and COX10 expression. Oncogene. 2010;29(30):4362–4368. doi: 10.1038/onc.2010.193. [DOI] [PubMed] [Google Scholar]
- 80.Zampetaki A, et al. Plasma microRNA profiling reveals loss of endothelial miR-126 and other microRNAs in type 2 diabetes. Circ Res. 2010;107(6):810–817. doi: 10.1161/CIRCRESAHA.110.226357. [DOI] [PubMed] [Google Scholar]
- 81.El Ouaamari A, et al. miR-375 targets 3′-phosphoinositide-dependent protein kinase-1 and regulates glucose-induced biological responses in pancreatic beta-cells. Diabetes. 2008;57(10):2708–2717. doi: 10.2337/db07-1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Keller DM, Clark EA, Goodman RH. Regulation of microRNA-375 by cAMP in pancreatic beta-cells. Mol Endocrinol. 2012;26(6):989–999. doi: 10.1210/me.2011-1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Carrer M, et al. Control of mitochondrial metabolism and systemic energy homeostasis by microRNAs 378 and 378*. Proc Natl Acad Sci U S A. 2012;109(38):15330–15335. doi: 10.1073/pnas.1207605109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shi L, et al. miR-223-IGF-IR signalling in hypoxia- and load-induced right-ventricular failure: a novel therapeutic approach. Cardiovasc Res. 2016 doi: 10.1093/cvr/cvw065. [DOI] [PubMed] [Google Scholar]
- 85.Meloche J, et al. miR-223 reverses experimental pulmonary arterial hypertension, Am. J. Physiol. Cell Physiol. 2015;309(6):C363–C372. doi: 10.1152/ajpcell.00149.2015. [DOI] [PubMed] [Google Scholar]
- 86.Tanno M, et al. Emerging beneficial roles of sirtuins in heart failure. Basic Res Cardiol. 2012;107(4):273. doi: 10.1007/s00395-012-0273-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wagner GR, Payne RM. Mitochondrial acetylation and diseases of aging. J Aging Res. 2011(2011):234875. doi: 10.4061/2011/234875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Imai S, et al. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403(6771):795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- 89.Finkel T, Deng CX, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature. 2009;460(7255):587–591. doi: 10.1038/nature08197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Onyango P, et al. SIRT3, a human SIR2 homologue, is an NAD-dependent deacetylase localized to mitochondria. Proc Natl Acad Sci U S A. 2002;99(21):13653–13658. doi: 10.1073/pnas.222538099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Michishita E, et al. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol Biol Cell. 2005;16(10):4623–4635. doi: 10.1091/mbc.E05-01-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Haigis MC, et al. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell. 2006;126(5):941–954. doi: 10.1016/j.cell.2006.06.057. [DOI] [PubMed] [Google Scholar]
- 93.Alcendor RR, et al. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ Res. 2007;100(10):1512–1521. doi: 10.1161/01.RES.0000267723.65696.4a. [DOI] [PubMed] [Google Scholar]
- 94.Lynn EG, et al. SIRT2 is a negative regulator of anoxia-reoxygenation tolerance via regulation of 14-3-3 zeta and BAD in H9c2 cells. FEBS Lett. 2008;582(19):2857–2862. doi: 10.1016/j.febslet.2008.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hafner AV, et al. Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging (Albany NY) 2010;2(12):14–923. doi: 10.18632/aging.100252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chen T, et al. Mouse SIRT3 attenuates hypertrophy-related lipid accumulation in the heart through the deacetylation of LCAD. PLoS One. 2015;10(3):e0118909. doi: 10.1371/journal.pone.0118909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zeng H, et al. Apelin gene therapy increases myocardial vascular density and ameliorates diabetic cardiomyopathy via upregulation of sirtuin 3. Am J Physiol Heart Circ Physiol. 2014;306(4):H585–H597. doi: 10.1152/ajpheart.00821.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sundaresan NR, et al. SIRT3 is a stress-responsive deacetylase in cardiomyocytes that protects cells from stress-mediated cell death by deacetylation of Ku70. Mol Cell Biol. 2008;28(20):6384–6401. doi: 10.1128/MCB.00426-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mostoslavsky R, et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell. 2006;124(2):315–329. doi: 10.1016/j.cell.2005.11.044. [DOI] [PubMed] [Google Scholar]
- 100.Michishita E, et al. SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature. 2008;452(7186):492–496. doi: 10.1038/nature06736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhong L, et al. The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1alpha. Cell. 2010;140(2):280–293. doi: 10.1016/j.cell.2009.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sundaresan NR, et al. The sirtuin SIRT6 blocks IGF-Akt signaling and development of cardiac hypertrophy by targeting c-Jun. Nat Med. 2012;18(11):1643–1650. doi: 10.1038/nm.2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Vakhrusheva O, et al. Sirt7 increases stress resistance of cardiomyocytes and prevents apoptosis and inflammatory cardiomyopathy in mice. Circ Res. 2008;102(6):703–710. doi: 10.1161/CIRCRESAHA.107.164558. [DOI] [PubMed] [Google Scholar]
- 104.Lee J, et al. A pathway involving farnesoid X receptor and small heterodimer partner positively regulates hepatic sirtuin 1 levels via microRNA-34a inhibition. J Biol Chem. 2010;285(17):12604–12611. doi: 10.1074/jbc.M109.094524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yamakuchi M, Ferlito M, Lowenstein CJ. miR-34a repression of SIRT1 regulates apoptosis. Proc Natl Acad Sci U S A. 2008;105(36):13421–13426. doi: 10.1073/pnas.0801613105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rottiers V, Naar AM. MicroRNAs in metabolism and metabolic disorders. Nat Rev Mol Cell Biol. 2012;13(4):239–250. doi: 10.1038/nrm3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhou B, et al. Downregulation of miR-181a upregulates sirtuin-1 (SIRT1) and improves hepatic insulin sensitivity. Diabetologia. 2012;55(7):2032–2043. doi: 10.1007/s00125-012-2539-8. [DOI] [PubMed] [Google Scholar]
- 108.Pramanik D, et al. Restitution of tumor suppressor microRNAs using a systemic nanovector inhibits pancreatic cancer growth in mice. Mol Cancer Ther. 2011;10(8):1470–1480. doi: 10.1158/1535-7163.MCT-11-0152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Strum JC, et al. MicroRNA 132 regulates nutritional stress-induced chemokine production through repression of SirT1. Mol Endocrinol. 2009;23(11):1876–1884. doi: 10.1210/me.2009-0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Menghini R, et al. MicroRNA 217 modulates endothelial cell senescence via silent information regulator 1. Circulation. 2009;120(15):1524–1532. doi: 10.1161/CIRCULATIONAHA.109.864629. [DOI] [PubMed] [Google Scholar]
- 111.Kim YJ, et al. miR-486-5p induces replicative senescence of human adipose tissue-derived mesenchymal stem cells and its expression is controlled by high glucose. Stem Cells Dev. 2012;21(10):1749–1760. doi: 10.1089/scd.2011.0429. [DOI] [PubMed] [Google Scholar]
- 112.Lovis P, et al. Alterations in microRNA expression contribute to fatty acid-induced pancreatic beta-cell dysfunction. Diabetes. 2008;57(10):2728–2736. doi: 10.2337/db07-1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ramachandran D, et al. Sirt1 and mir-9 expression is regulated during glucose-stimulated insulin secretion in pancreatic beta-islets. FEBS J. 2011;278(7):1167–1174. doi: 10.1111/j.1742-4658.2011.08042.x. [DOI] [PubMed] [Google Scholar]
- 114.Ahn J, et al. MicroRNA-146b promotes adipogenesis by suppressing the SIRT1-FOXO1 cascade. EMBO Mol Med. 2013;5(10):1602–1612. doi: 10.1002/emmm.201302647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mortuza R, Feng B, Chakrabarti S. miR-195 regulates SIRT1-mediated changes in diabetic retinopathy. Diabetologia. 2014;57(5):1037–1046. doi: 10.1007/s00125-014-3197-9. [DOI] [PubMed] [Google Scholar]
- 116.Zhu H, et al. MicroRNA-195 promotes palmitate-induced apoptosis in cardiomyocytes by down-regulating Sirt1. Cardiovasc Res. 2011;92(1):75–84. doi: 10.1093/cvr/cvr145. [DOI] [PubMed] [Google Scholar]
- 117.Wang JY, et al. Acupuncture may exert its therapeutic effect through microRNA-339/Sirt2/NFkappaB/FOXO1 axis. Biomed Res Int. 2015;2015:249013. doi: 10.1155/2015/249013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Poulsen RC, et al. Cell differentiation versus cell death: extracellular glucose is a key determinant of cell fate following oxidative stress exposure. Cell Death Dis. 5(2014):e1074. doi: 10.1038/cddis.2014.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Davalos A, et al. miR-33a/b contribute to the regulation of fatty acid metabolism and insulin signaling. Proc Natl Acad Sci U S A. 2011;108(22):9232–9237. doi: 10.1073/pnas.1102281108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lefort K, et al. A miR-34a-SIRT6 axis in the squamous cell differentiation network. EMBO J. 2013;32(16):2248–2263. doi: 10.1038/emboj.2013.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Sharma A, et al. The role of SIRT6 protein in aging and reprogramming of human induced pluripotent stem cells. J Biol Chem. 2013;288(25):18439–18447. doi: 10.1074/jbc.M112.405928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Cioffi M, et al. MiR-93 controls adiposity via inhibition of Sirt7 and Tbx3. Cell Rep. 2015;12(10):1594–1605. doi: 10.1016/j.celrep.2015.08.006. [DOI] [PubMed] [Google Scholar]
- 123.Kim JK, et al. Sirtuin7 oncogenic potential in human hepatocellular carcinoma and its regulation by the tumor suppressors MiR-125a-5p and MiR-125b. Hepatology. 2013;57(3):1055–1067. doi: 10.1002/hep.26101. [DOI] [PubMed] [Google Scholar]
- 124.Cheng HL, et al. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc Natl Acad Sci U S A. 2003;100(19):10794–10799. doi: 10.1073/pnas.1934713100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Alcendor RR, et al. Silent information regulator 2alpha, a longevity factor and class III histone deacetylase, is an essential endogenous apoptosis inhibitor in cardiac myocytes. Circ Res. 2004;95(10):971–980. doi: 10.1161/01.RES.0000147557.75257.ff. [DOI] [PubMed] [Google Scholar]
- 126.Rodgers JT, et al. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434(7029):113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 127.Lan F, et al. SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. J Biol Chem. 2008;283(41):27628–27635. doi: 10.1074/jbc.M805711200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Gerhart-Hines Z, et al. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1alpha. EMBO J. 2007;26(7):1913–1923. doi: 10.1038/sj.emboj.7601633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hallows WC, Yu W, Denu JM. Regulation of glycolytic enzyme phosphoglycerate mutase-1 by Sirt1 protein-mediated deacetylation. J Biol Chem. 2012;287(6):3850–3858. doi: 10.1074/jbc.M111.317404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Purushotham A, et al. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab. 2009;9(4):327–338. doi: 10.1016/j.cmet.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Planavila A, et al. Sirt1 acts in association with PPARalpha to protect the heart from hypertrophy, metabolic dysregulation, and inflammation. Cardiovasc Res. 2011;90(2):276–284. doi: 10.1093/cvr/cvq376. [DOI] [PubMed] [Google Scholar]
- 132.Walker AK, et al. Conserved role of SIRT1 orthologs in fasting-dependent inhibition of the lipid/cholesterol regulator SREBP. Genes Dev. 2010;24(13):1403–1417. doi: 10.1101/gad.1901210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kemper JK, et al. FXR acetylation is normally dynamically regulated by p300 and SIRT1 but constitutively elevated in metabolic disease states. Cell Metab. 2009;10(5):392–404. doi: 10.1016/j.cmet.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Li X, et al. SIRT1 deacetylates and positively regulates the nuclear receptor LXR. Mol Cell. 2007;28(1):91–106. doi: 10.1016/j.molcel.2007.07.032. [DOI] [PubMed] [Google Scholar]
- 135.Zhang C, et al. Resveratrol attenuates doxorubicin-induced cardiomyocyte apoptosis in mice through SIRT1-mediated deacetylation of p53. Cardiovasc Res. 2011;90(3):538–545. doi: 10.1093/cvr/cvr022. [DOI] [PubMed] [Google Scholar]
- 136.Brunet A, et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303(5666):2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 137.Daitoku H, et al. Silent information regulator 2 potentiates Foxo1-mediated transcription through its deacetylase activity. Proc Natl Acad Sci U S A. 2004;101(27):10042–10047. doi: 10.1073/pnas.0400593101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Hsu CP, et al. Silent information regulator 1 protects the heart from ischemia/reperfusion. Circulation. 2010;122(21):2170–2182. doi: 10.1161/CIRCULATIONAHA.110.958033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Sundaresan NR, et al. The deacetylase SIRT1 promotes membrane localization and activation of Akt and PDK1 during tumorigenesis and cardiac hypertrophy. Sci Signal. 2011;4(182):ra46. doi: 10.1126/scisignal.2001465. [DOI] [PubMed] [Google Scholar]
- 140.Shimazu T, et al. SIRT3 deacetylates mitochondrial 3-hydroxy-3-methylglutaryl CoA synthase 2 and regulates ketone body production. Cell Metab. 2010;12(6):654–661. doi: 10.1016/j.cmet.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Hirschey MD, et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature. 2010;464(7285):121–125. doi: 10.1038/nature08778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Schwer B, et al. Reversible lysine acetylation controls the activity of the mitochondrial enzyme acetyl-CoA synthetase 2. Proc Natl Acad Sci U S A. 2006;103(27):10224–10229. doi: 10.1073/pnas.0603968103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Hallows WC, Lee S, Denu JM. Sirtuins deacetylate and activate mammalian acetyl-CoA synthetases. Proc Natl Acad Sci U S A. 2006;103(27):10230–10235. doi: 10.1073/pnas.0604392103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Jing E, et al. Sirt3 regulates metabolic flexibility of skeletal muscle through reversible enzymatic deacetylation. Diabetes. 2013;62(10):3404–3417. doi: 10.2337/db12-1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Stobbe MD, et al. Improving the description of metabolic networks: the TCA cycle as example. FASEB J. 2012;26(9):3625–3636. doi: 10.1096/fj.11-203091. [DOI] [PubMed] [Google Scholar]
- 146.Falcon AA, et al. Acetyl-coenzyme A synthetase 2 is a nuclear protein required for replicative longevity in. Saccharomyces cerevisiae, Mol Cell Biochem. 2010;333(1–2):99–108. doi: 10.1007/s11010-009-0209-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ahn BH, et al. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc Natl Acad Sci U S A. 2008;105(38):14447–14452. doi: 10.1073/pnas.0803790105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Cimen H, et al. Regulation of succinate dehydrogenase activity by SIRT3 in mammalian mitochondria. Biochemistry. 2010;49(2):304–311. doi: 10.1021/bi901627u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Finley LW, et al. Succinate dehydrogenase is a direct target of sirtuin 3 deacetylase activity. PLoS One. 2011;6(8):e23295. doi: 10.1371/journal.pone.0023295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Vassilopoulos A, et al. SIRT3 deacetylates ATP synthase F1 complex proteins in response to nutrient- and exercise-induced stress. Antioxid Redox Signal. 2014;21(4):551–564. doi: 10.1089/ars.2013.5420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Rahman M, et al. Drosophila Sirt2/mammalian SIRT3 deacetylates ATP synthase beta and regulates complex V activity. J Cell Biol. 2014;206(2):289–305. doi: 10.1083/jcb.201404118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Sundaresan NR, et al. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J Clin Invest. 2009;119(9):2758–2771. doi: 10.1172/JCI39162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Krishnan J, et al. Dietary obesity-associated Hif1alpha activation in adipocytes restricts fatty acid oxidation and energy expenditure via suppression of the Sirt2-NAD+ system. Genes Dev. 2012;26(3):259–270. doi: 10.1101/gad.180406.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Ramakrishnan G, et al. Sirt2 deacetylase is a novel AKT binding partner critical for AKT activation by insulin. J Biol Chem. 2014;289(9):6054–6066. doi: 10.1074/jbc.M113.537266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Rardin MJ, et al. SIRT5 regulates the mitochondrial lysine succinylome and metabolic networks. Cell Metab. 2013;18(6):920–933. doi: 10.1016/j.cmet.2013.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kim HS, et al. Hepatic-specific disruption of SIRT6 in mice results in fatty liver formation due to enhanced glycolysis and triglyceride synthesis. Cell Metab. 2010;12(3):224–236. doi: 10.1016/j.cmet.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Dominy JE, Jr, et al. The deacetylase Sirt6 activates the acetyltransferase GCN5 and suppresses hepatic gluconeogenesis. Mol Cell. 2012;48(6):900–913. doi: 10.1016/j.molcel.2012.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Masri S, et al. Partitioning circadian transcription by SIRT6 leads to segregated control of cellular metabolism. Cell. 2014;158(3):659–672. doi: 10.1016/j.cell.2014.06.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Tao R, et al. Hepatic SREBP-2 and cholesterol biosynthesis are regulated by FoxO3 and Sirt6. J Lipid Res. 2013;54(10):2745–2753. doi: 10.1194/jlr.M039339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Elhanati S, et al. Multiple regulatory layers of SREBP1/2 by SIRT6. Cell Rep. 2013;4(5):905–912. doi: 10.1016/j.celrep.2013.08.006. [DOI] [PubMed] [Google Scholar]
- 161.Maksin-Matveev A, et al. Sirtuin 6 protects the heart from hypoxic damage. Exp Cell Res. 2015;330(1):81–90. doi: 10.1016/j.yexcr.2014.07.013. [DOI] [PubMed] [Google Scholar]
- 162.Yoshizawa T, et al. SIRT7 controls hepatic lipid metabolism by regulating the ubiquitin-proteasome pathway. Cell Metab. 2014;19(4):712–721. doi: 10.1016/j.cmet.2014.03.006. [DOI] [PubMed] [Google Scholar]
- 163.Thum T, et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature. 2008;456(7224):980–984. doi: 10.1038/nature07511. [DOI] [PubMed] [Google Scholar]
- 164.Hullinger TG, et al. Inhibition of miR-15 protects against cardiac ischemic injury. Circ Res. 2012;110(1):71–81. doi: 10.1161/CIRCRESAHA.111.244442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Montgomery RL, et al. Therapeutic inhibition ofmiR-208a improves cardiac function and survival during heart failure. Circulation. 2011;124(14):1537–1547. doi: 10.1161/CIRCULATIONAHA.111.030932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Rottiers V, et al. Pharmacological inhibition of a microRNA family in nonhuman primates by a seed-targeting 8-mer antimiR. Sci Transl Med. 2013;5(212):212ra162. doi: 10.1126/scitranslmed.3006840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Castaldi A, et al. MicroRNA-133 modulates the beta1-adrenergic receptor transduction cascade. Circ Res. 2014;115(2):273–283. doi: 10.1161/CIRCRESAHA.115.303252. [DOI] [PubMed] [Google Scholar]
- 168.Gill SL, et al. Enhanced delivery of microRNA mimics to cardiomyocytes using ultrasound responsive microbubbles reverses hypertrophy in an in-vitro model. Technol Health Care. 2014;22(1):37–51. doi: 10.3233/THC-130772. [DOI] [PubMed] [Google Scholar]
- 169.Eulalio A, et al. Functional screening identifies miRNAs inducing cardiac regeneration. Nature. 2012;492(7429):376–381. doi: 10.1038/nature11739. [DOI] [PubMed] [Google Scholar]
- 170.Quattrocelli M, et al. Long-term miR-669a therapy alleviates chronic dilated cardiomyopathy in dystrophic mice. J Am Heart Assoc. 2013;2(4):e000284. doi: 10.1161/JAHA.113.000284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Yang J, et al. MicroRNA-22 targeting CBP protects against myocardial ischemia-reperfusion injury through anti-apoptosis in rats. Mol Biol Rep. 2014;41(1):555–561. doi: 10.1007/s11033-013-2891-x. [DOI] [PubMed] [Google Scholar]