

This study shows that the anion transporter antagonists furosemide and DIDS cause a marked decrease of axonal excitability in rat hippocampal CA1 region and prevent the induction of activity-dependent hyperexcitability in Schaffer collateral axons. The data are consistent with direct effects on axonal membrane properties. We also find that activity-dependent enhancement and depression of axonal excitability can be modified independently, suggesting that these events are governed by different underlying processes.
Keywords: furosemide, anion transport, axon, excitability, fiber volley
Abstract
The loop diuretic furosemide is known to have anticonvulsant effects, believed to be exerted through blockade of glial Na+-K+-2Cl− cotransport causing altered volume regulation in brain tissue. The possibility that direct effects of furosemide on neuronal properties could also be involved is supported by previous observations, but such effects have not been thoroughly investigated. In the present study we show that furosemide has two opposing effects on stimulus-induced postsynaptic excitation in the nonepileptic rat hippocampal slice: 1) an enhancement of e-s coupling, which depended on intact GABAA transmission and was partially mimicked by selective blockade of K+-2Cl− cotransport, and 2) a decrement of field excitatory postsynaptic potentials. The balance between these effects varied, depending on the amount of synaptic drive. In addition, the compound action potential (fiber volley) recorded from the stimulated Schaffer collateral axons in stratum radiatum showed a progressive decrease during perfusion of furosemide. This effect was activity-independent, was mimicked by the stilbene derivative DIDS, and could be reproduced on fiber volleys in the alveus. Furosemide also reduced the initial enhancement of the fiber volley observed during trains of high-frequency stimulation (HFS). Results of hyperosmotic expansion of the extracellular volume, with 30 mM sucrose, indicated that both the induction and antagonism of the HFS-induced enhancement were independent of signaling via the extracellular space. Furosemide caused an increased decay of paired-pulse-induced supranormal axonal excitability, which was antagonized by ZD7288. We conclude that furosemide decreases axonal excitability and prevents HFS-induced hyperexcitability via mechanisms downstream of blockage of anion transport, which could include hyperpolarization of axonal membranes.
NEW & NOTEWORTHY This study shows that the anion transporter antagonists furosemide and DIDS cause a marked decrease of axonal excitability in rat hippocampal CA1 region and prevent the induction of activity-dependent hyperexcitability in Schaffer collateral axons. The data are consistent with direct effects on axonal membrane properties. We also find that activity-dependent enhancement and depression of axonal excitability can be modified independently, suggesting that these events are governed by different underlying processes.
evidence from a number of experimental and clinical studies suggests that loop diuretics such as furosemide and bumetanide have anticonvulsant effect (Ahmad et al. 1976; Dzhala et al. 2005; Eftekhari et al. 2013; Gutschmidt et al. 1999; Haglund and Hochman 2005; Hesdorffer et al. 2001; Hochman et al. 1995; Holtkamp et al. 2003; Kahle et al. 2008; Kilb et al. 2007; Löscher et al. 2013; Maa et al. 2011; Margineanu and Klitgaard 2006; Uwera et al. 2015). Understanding the precise cause of this effect could potentially be helpful in the search for new strategies in seizure management. However, with respect to furosemide, the literature offers several different explanations for the anticonvulsant mechanism, some of which are conflicting. These difficulties mainly result from the fact that furosemide acts on multiple targets including the Na+-K+-2Cl− cotransporter (NKCC1), the K+-2Cl− cotransporter (KCC2), the Na+-independent Cl−/ exchanger (AE3), some subtypes of GABAA receptors, and the carbonic anhydrase (Maa et al. 2011; Staley 2002). From several studies using different experimental approaches, it appears that in adult brain tissue blockade of the glial NKCC1 is a critical step in the anticonvulsant effect of furosemide by causing antagonism of activity-dependent shrinkage of the extracellular volume during seizures (Haglund and Hochman 2005; Hochman 2012; Hochman et al. 1995). Furthermore, antagonism of the NKCC1 with furosemide or low extracellular Cl− concentration has been shown to induce desynchronization of postsynaptic pyramidal neurons, which is proposed to be due to action potential propagation failure in distal axonal arbors or varicosities (Hochman and Schwartzkroin 2000). It is unclear whether the latter effect depends on conditions evolving during epileptiform activity (changes in extracellular volume, ion concentrations, pH, etc.) or whether it is inducible in nonepileptic tissue. Studies in brain slices have shown that the selective blockade of NKCC1 with bumetanide is ineffective in blocking epileptiform activity (Dzhala et al. 2005; Margineanu and Klitgaard 2006; Uwera et al. 2015; Wahab et al. 2011), suggesting that other mechanisms contribute to the anticonvulsant effect of furosemide. Recent evidence from our laboratory has indicated a contributory effect from blockade of AE3, which could involve a depression of neuronal excitation due to interference with pH homeostasis during hyperactivity (Uwera et al. 2015). In addition, studies in nonepileptic brain slices have suggested that basic electrophysiological parameters could be changed during perfusion of furosemide. Such observations include a decrement of the orthodromically evoked population spikes (Gutschmidt et al. 1999) and field excitatory postsynaptic potentials (fEPSPs) (Müller 2000) in the hippocampal CA1 area. These findings would seem to indicate that furosemide has a primary effect of reducing excitability, which might significantly contribute to (or even explain) its anticonvulsant property (Gutschmidt et al. 1999). These inhibitory effects have not been studied in detail, and their mechanisms remain unsettled. On the other hand, several reports have shown that furosemide increases neuronal excitation in response to afferent stimulation, presumably resulting from its effects on inhibition (Hochman et al. 1995; MacVicar and Hochman 1991; Müller 2000; Thompson et al. 1988; Thompson and Gähwiler 1989). Thus available data give no clear picture of how furosemide affects excitability under normal basic conditions, even less whether any such effect could play a role in the anticonvulsant property of the drug. In the present study we attempted to address this issue by investigating, in a quantitative fashion, the effect of furosemide and related agents on the coupling of excitatory inputs and firing efficiency (e-s coupling) and on presynaptic axon excitability in the CA1 area of the nonepileptic hippocampal slice.
METHODS
Animal care and protocol for euthanasia were in accordance with Danish and European law and approved by the Animal Experimentation Board under the Danish Ministry of Justice. Male Wistar rats (4–5 wk old) were anesthetized with isoflurane and decapitated. After removal, the brain was placed in dissection medium at 4°C (see below), and horizontal slices containing the hippocampus (400 μm) were cut on a vibratome. One slice was immediately transferred to the recording chamber, where it was placed on a nylon mesh grid at the interface between warm (31–32°C) standard perfusion medium (see below) and warm humidified carbogen (95% O2-5% CO2). Perfusion flow rate was 1 ml/min. The slice rested for at least 1 h before electrophysiological recordings began. The remaining slices were stored in dissection medium bubbled with carbogen at room temperature until use.
Electrophysiology and analyses.
Extracellular recordings were obtained with borosilicate glass electrodes (1.2-mm OD; Clark Electromedical, Pangbourne, UK) filled with 1 M NaCl (tip resistance 10–20 MΩ). A bipolar Teflon-insulated platinum electrode (tip diameter: 50 μm, intertip distance: 25 μm), placed within the stratum (str.) radiatum of CA1, was used for orthodromic stimulation of Schaffer collateral commissural fibers with constant-current pulses (0.5 μs). Field potentials in CA1 were recorded with an electrode placed in str. pyramidale to record the population spike (PS) and another electrode placed in str. radiatum perpendicular to str. pyramidale in line with the other electrode to record the fEPSP. Both electrodes were placed ~200 µm away from the stimulation site. Conventional recording techniques were employed, using a high-input impedance amplifier (Axoclamp2A, Molecular Devices) with bridge balance and current injection facilities. Signals were digitized online with a Digidata 1440 interface and transferred to a computer for analysis employing pCLAMP (version 10, Molecular Devices). The fEPSP slope was measured within the first millisecond after onset, before the start of the PS. The PS was measured as the area below a fictive line connecting the start and end point of the PS. During washin of drugs, single stimuli were delivered at 0.2 Hz. The stimulus intensity was adjusted to produce a PS of ~70% of maximal amplitude, which was typically 1.10–1.25 times the intensity at PS threshold. A 15-min baseline was recorded before drug application. Measurements were done on averaged traces from 12 consecutive stimuli (1 min). For statistical evaluation of drug effects, averaged values from the last 5 min of drug application were compared with the corresponding values from the last 5 min of the baseline (control). Drugs were applied only once per slice. Before experiments using GABAA receptor blockers, a knife cut was made at the border between CA1 and CA3, separating the two areas.
Plots of PS area vs. fEPSP slope (e-s coupling plots) were obtained by stimulation at incrementing steps covering the intensity range from no evoked response to maximal PS size. In each slice, identical stimulus protocols were applied in a control period >15 min before drug application and at the end of the drug application. Measurements were done on averaged traces from five consecutive stimuli at the same intensity. In each experiment, fEPSP slopes were normalized to the slope that induced a PS area of 95% of the maximal (= 1.0). This value was chosen instead of 100% to avoid contribution from random “jitter” to the measurement. The PS areas were normalized to the maximal value. e-s coupling plots were constructed in each experiment and fitted to a sigmoidal curve with an built-in function in SigmaPlot 13 (Alfasoft). The fEPSP slope that gave 50% of maximal PS size (E50) was read from the curve data. To construct e-s coupling plots from multiple experiments, the normalized fEPSP values from individual experiments were binned (bin width 0.1–0.2) and the averaged values of PS and fEPSP from each bin were plotted.
To record fiber volleys in str. radiatum and the alveus, the stimulus electrode and recording electrode were separated by 500–1,000 µm. Stimulation was set to induce a fiber volley of ≥1.5 mV. Alvear recording and stimulation were done in a section of the alveus that had been physically isolated from the str. oriens with a knife cut. High-frequency stimulation (HFS) was generated in trains of 100 stimuli at 20 Hz or 100 Hz. The interval between trains was set to 50 s, which allowed full recovery of the fiber volley amplitude, as confirmed in each experiment. Fiber volley amplitudes were measured as the voltage difference between the trough of the negative deflection and the peak of the following positive deflection. Fiber volley areas were taken as the integral of the negative voltage trajectory under a fictive line between the peaks of the positive deflections on either side. HFS signals were low-pass filtered at 2 kHz before measurement.
For statistical comparisons, paired or unpaired Student’s t-test was used unless otherwise stated.
Drugs and solutions.
The composition of the dissection medium was (in mM) 120 NaCl, 2 KCl, 1.25 KH2PO4, 6.6 HEPES acid, 2.6 NaHEPES, 20 NaHCO3, 2 CaCl2, 2 MgSO4, and 10 d-glucose, bubbled with carbogen. The perfusion medium used during recordings contained (in mM) 124 NaCl, 3.25 KCl, 1.25 NaH2PO4, 20 NaHCO3, 2 CaCl2, 2 MgSO4, and 10 d-glucose, bubbled with carbogen (pH = 7.3). 6-Cyano-7-nitroquinoxaline-2,3-dione (CNQX), (5S,10R)-(+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d] cyclohepten-5,10-imine-maleate (MK 801), (2S)-3-[[(1S)-1-(3,4-dichlorophenyl)ethyl]amino-2-hydroxypropyl](phenylmethyl)phosphinic acid hydrochloride (CGP55845), 2-chloro-N-[[2′-[(cyanoamino)sulfonyl][1,1′-biphenyl]-4-yl]methyl]-N-[(4-methylphenyl)methyl]-benzamide (S0859), 4-ethylphenylamino-1,2-dimethyl-6-methylaminopyrimidinium chloride (ZD7288), bicuculline methbromide, and 2-(3-carboxypropyl)-3-amino-6-(4 methoxyphenyl)pyridazinium bromide (SR-95531, gabazine) were made up in aqueous stock solutions at 100–1,000 times the required final concentration and diluted in the perfusion medium as appropriate. N-(4-methyl-2-thiazolyl)-2-[(6-phenyl-3-pyridazinyl)thio]acetamide (VU0240551) was dissolved in DMSO at 1,000 times final concentration. Furosemide, bumetanide, and 4,4′-diisothiocyanostilbene-2,2′-disulfonic acid (DIDS) were added directly to the standard perfusion medium followed by repetitive cycles of desiccation for 30–60 min to ensure that the substances were thoroughly dissolved before use. VU0240551, ZD7288, and CGP55845 were purchased from Tocris. All other drugs were purchased from Sigma.
RESULTS
e-s coupling.
We monitored the fEPSP and PS during 30-min perfusion of furosemide (2.5 mM). Furosemide caused a gradual reduction of the fEPSP slope (Fig. 1A1), which began within 10 min and reached 82 ± 6% of control at 30 min (n = 9, P < 0.05). The PS area showed little change (100 ± 5% of control after 30 min, P = 1.00; Fig. 1A2). As a result, the e-s ratio increased in response to furosemide, reaching 124 ± 5% of control (at 30 min, P < 0.01; Fig. 1A3). Furosemide also caused a leftward shift in the e-s coupling curve (Fig. 1A4). The fEPSP slope (normalized) that evoked a population spike of 50% of the maximal size (E50) was significantly reduced from an average of 0.60 ± 0.06 in the control period to 0.43 ± 0.04 in the presence of furosemide (P < 0.05, n = 5). An isolated reduction of the fEPSP is expected to be accompanied by an increased stimulus threshold for exciting the postsynaptic neurons. In line with this expectation, we observed that stimulus intensities that induced a near-minimal PS in the control condition gave either a smaller PS or no PS in the presence of furosemide (7 of 9 experiments; Fig. 2). Conversely, strong stimuli (>1.7–2.4 × threshold for the PS) induced additional spiking in the presence of furosemide (6 of 9 experiments; Fig. 2), which was not observed in control or during the lower-intensity stimulation used during washin of furosemide. To examine whether the above effects could be attributed to blockade of either NKCC1 or KCC2, we replaced furosemide with the selective antagonists of NKCC1 (bumetanide; Fig. 1B) or KCC2 (VU0240551; Fig. 1C). Perfusion of bumetanide (20 µM) for 30 min induced no significant change in the fEPSP, the PS, or the e-s ratio (Fig. 1, B1–B3; n = 7, P > 0.05 for each parameter). The e-s coupling curve obtained with bumetanide showed little deviation from the control curve (Fig. 1B4), and the E50 was not significantly altered (control: 0.61 ± 0.10, bumetanide: 0.68 ± 0.18, n = 7, P = 0.28). VU0240551 (20 µM, 30 min) had no overall effect on the fEPSP (slope: 97 ± 4% of control, P = 0.53; Fig. 1C1) but it significantly increased the PS (area: 109 ± 3% of control, P < 0.05; Fig. 1C2) and the e-s ratio (114 ± 3% of control, P < 0.01, n = 8; Fig. 1C3) and caused a leftward shift in e-s coupling (Fig. 1C4). The E50 was reduced from 0.60 ± 0.03 to 51 ± 0.02 in the presence of VU0240551, which was statistically significant (P < 0.05, n = 8). These data suggested that the enhanced e-s coupling induced by furosemide could, at least partly, be related to blockade of KCC2 and likely result from a diminished GABAA receptor-mediated inhibition. To determine whether furosemide’s effects were altogether dependent on GABAA transmission, we blocked the GABAA receptor with gabazine. Perfusion of furosemide in the presence of gabazine (15 µM) resulted in a significant decrease of both the fEPSP and PS (Fig. 1, D1 and D2; fEPSP slope: 58 ± 8% of control, P < 0.01; PS area: 59 ± 12%, P < 0.05, n = 7). The time course of the e-s ratio showed high variability among experiments, and no significant change was detected after 30 min (ratio: 103 ± 20% of control, P = 0.90; Fig. 1D3). In the presence of gabazine, furosemide induced little change in e-s coupling (Fig. 1D4), and the E50 was not significantly altered [gabazine alone: 47 ± 4%, gabazine + furosemide (30 min): 53 ± 9%, n = 6, P = 0.47].
Fig. 1.

Effects of furosemide, bumetanide, and VU0240551 on e-s coupling in the CA1 area. A1–A3: normalized fEPSP slope, PS area, and e-s ratio vs. time in a control period 0–15 min before and 0–30 min after switch to a perfusion medium containing either furosemide (FURO; 2.5 mM) or no added drug (ACSF). Each point represents the average value ± SE from 9 experiments. A1 and A2, top: example records of fEPSPs and PSs averaged over the last 5 min in control (solid trace) and the last 5 min in furosemide (dashed trace). A4: e-s coupling curves showing averaged results from the same experiments. Data were obtained in a control period >15 min before (control) and after 30-min exposure to furosemide (FURO). B–D: plots and traces as in A, but instead of furosemide bumetanide (BUM; 20 µM) was applied in B, VU0240551 (VU; 20 µM) was applied in C, and furosemide (2.5 mM) was applied in the presence of gabazine (15 µM; perfused for >20 min before recordings began) in D. Scale bars: 2 ms, 5 mV throughout.
Fig. 2.
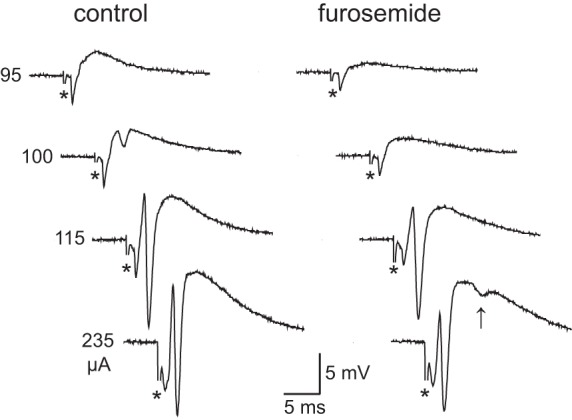
Effects of furosemide on stimulus-induced population activity: field potentials in str. pyramidale in response to afferent stimuli at different intensities (95–235 µA) before (control) and after 30-min perfusion of furosemide (2.5 mM). Note that in the presence of furosemide the 100-µA stimulus becomes subthreshold (with respect to the PS) and an extra PS is apparent with the 235-µA stimulus (indicated by arrow). Asterisks denote stimulus artifacts.
Fiber volley in str. radiatum.
The above results suggest that furosemide reduces the fEPSP independent of blockage of either KCC2 or NKCC1. We hypothesized that decrement of the fEPSP slope was due to a decreased excitation of the presynaptic axon. We therefore isolated the electrical stimulus-induced afferent fiber volley in str. radiatum by recording in the presence of CNQX (20–40 µM), MK 801 (10 µM), and CGP55845 (2 µM) to block ionotropic glutamate receptors and GABAB receptors.
Perfusion of furosemide (2.5 mM) for 30 min resulted in a monophasic reduction of the fiber volley amplitude discernible after ~10 min and reaching 79 ± 6% of control (n = 9, P < 0.01; Fig. 3A1, Table 1). The decrement of the amplitude was accompanied by an increase in the latency from the time of stimulation to the peak of the fiber volley, which was significant (Fig. 3A1, Table 1). Inclusion of gabazine (15 µM) before furosemide did not alter the magnitude or the time course of the effect (n = 6; Table 1). These changes could reflect a desynchronization of otherwise unaltered action potential discharge in individual axons, in which case the fiber volley area should be constant. However, the area of the fiber volley also declined significantly in the presence of furosemide (to 84 ± 6% of control, P < 0.05; Table 1), indicating that effects other than desynchronization contributed to the response. In these experiments, test stimuli were delivered at 0.2 Hz to enable continuous monitoring of the fiber volley. To examine whether the effect of furosemide depended on stimulus-induced activity, we made a parallel series of experiments in which stimulation was halted during 0–25 min of perfusion (Fig. 3A1). The fiber volley amplitude measured after resumption of the 0.2-Hz stimulation (25–30 min) was significantly reduced compared with control before furosemide (84 ± 4%, n = 8, P < 0.01). The magnitude of the reduction did not deviate significantly from that observed with continuous stimulation (P = 0.22, unpaired t-test), suggesting that the decrement of the fiber volley occurred independently of stimulus-induced axonal activity. HFS at 10–100 Hz is known to produce a biphasic change in fiber volley amplitude consisting of an initial enhancement followed by a depression (Kim et al. 2012). We tested whether furosemide could influence this response by delivering trains of 100 pulses at 100 Hz and 20 Hz. We measured the fiber volley amplitude at every fifth stimulus (normalized to the amplitude at the first stimulus) and expressed it as a function of the position of the stimulus within the train. During 100-Hz stimulation in the control condition, the fiber volley amplitude showed a bell-shaped increase during the first 20 stimuli, reaching a maximum at stimulus 10, followed by a decay (Fig. 3A2). Repetition of the 100-Hz train after 30-min perfusion of furosemide revealed a reduction of the initial enhancement (stimulus 10: amplitude 1.22 ± 0.04 in control, 1.02 ± 0.02 in furosemide, n = 17, P < 0.01). The decay observed at the end of the train was unaltered (stimulus 100: amplitude in control 0.27 ± 0.02, furosemide 0.31 ± 0.04, P = 0.35, Wilcoxon signed-rank test; Fig. 3, A2 and A3). In the control condition, a 20-Hz train gave an enhancement during the first 40 stimuli (the peak being close to stimulus 20) followed by a reduction that was less than that produced by the 100-Hz train. Furosemide reduced the initial enhancement (stimulus 20: amplitude 1.21 ± 0.03 in control, 1.02 ± 0.04 in furosemide, n = 16, P < 0.01, Wilcoxon signed-rank test), and it also reduced the amplitude of fiber volleys evoked late in the 20-Hz train (stimulus 100: amplitude 0.84 ± 0.02 in control, 0.71 ± 0.02 in furosemide, n = 16, P < 0.01; Fig. 3, A2 and A3). The normalized fiber volley area measured during HFS showed changes qualitatively similar to the amplitude in response to furosemide (summarized in Fig. 3A3). These results were obtained in absence of GABAA receptor blockade, as adding gabazine (15 µM) or bicuculline (10 µM) to the blocker-containing medium led to the induction of aberrant synchronous discharge during HFS trains, which occluded the directly evoked fiber volleys (data not shown).
Fig. 3.

Effects of furosemide, bumetanide, and DIDS on the fiber volley in CA1 str. radiatum. A1: plots of the normalized amplitude and latency to peak of the fiber volley (fv) vs. time, 0–10 min before and 0–30 min after starting perfusion of furosemide. Symbols represent the means ± SE of 12 consecutive responses (1 min) evoked by continuous orthodromic stimulation (filled circles) or paused (0–25 min) stimulation (open circles). Top: example records of averaged fiber volleys taken 0–1 min before (solid trace) and 29–30 min after starting perfusion of furosemide (dashed trace). A2: averaged fiber volley amplitude as function of stimulus number in HFS trains of 100 Hz and 20 Hz (100 stimuli in both). Results from every 5th stimulus are shown. Measurements were done >10 min before (control) and after exposure to furosemide for >30 min (FURO). Data from each experiment were normalized to the amplitude at stimulus 1. Means ± SE from 17 experiments are shown. Top: example records of fiber volleys evoked at stimuli 1, 10, and 100 during a 100-Hz train. A3: summary histograms comparing the normalized amplitudes (top) and areas (bottom) of near-maximally enhanced fiber volleys (stimulus 10 at 100 Hz; stimulus 20 at 20 Hz) and of the volleys evoked at stimulus 100 in control (filled bars) and in the presence of furosemide (open bars). B and C: plots as in A, but instead of furosemide bumetanide (20 µM, 30 min) was applied in B and DIDS (0.5 mM, 45 min) was applied in C. Data in A–C were all obtained in the presence of CNQX (20 µM), MK 801 (10 µM), and CGP55845 (2 µM). Scale bars: 1 ms, 2 mV. *P < 0.05; **P < 0.01.
Table 1.
Effects of anion transport inhibitors, sucrose, and ZD7288 on evoked fiber volleys in stratum radiatum and alveus of CA1
| Drug Effect on Fiber Volley |
||||
|---|---|---|---|---|
| n | Amplitude | Area | Latency | |
| Furosemide (2.5 mM, 30 min) | 9 | 79 ± 6%** | 84 ± 6%* | 110 ± 2%** |
| Furosemide (2.5 mM, 30 min) with gabazine | 6 | 81 ± 4%** | 84 ± 4%* | 109 ± 2%* |
| Bumetanide (20 µM, 30 min) | 7 | 96 ± 2% | 98 ± 4% | 100 ± 2% |
| VU0240551(20 µM, 30 min) | 5 | 108 ± 6% | 99 ± 2% | 98 ± 2% |
| DIDS (0.5 mM, 45 min) | 9 | 80 ± 5%** | 90 ± 6% | 113 ± 3%** |
| DIDS + furosemide (45 min) | 5 | 55 ± 6%* | 74 ± 7%* | 124 ± 2%** |
| S0859 (30 µM, 45 min) | 7 | 102 ± 1% | 102 ± 5% | 101 ± 2% |
| Sucrose (30 mM, 30 min) | 6 | 89 ± 4% | 93 ± 4% | 97 ± 1%* |
| ZD7288 (20 µM, 30 min) | 11 | 100 ± 4% | 101 ± 4% | 101 ± 2% |
| Furosemide (2.5 mM, 30 min) with ZD7288 | 7 | 76 ± 4%** | 83 ± 5%* | 113 ± 2%** |
| Alveus, furosemide (2.5 mM, 30 min) | 11 | 66 ± 4%** | 74 ± 4%** | 128 ± 3%** |
| Alveus, bumetanide (20–50 µM, 30 min) | 9 | 100 ± 1% | 103 ± 4% | 103 ± 1%** |
Percentages (means ± SE) are relative to control before drug application. All data were collected in the presence of CNQX (20–40 µM), MK 801 (10 µM), and CGP55845 (2 µM).
P < 0.05;
P < 0.01.
Perfusion of bumetanide (20 µM, 30 min) induced no overall change in the fiber volley amplitude or latency (Fig. 3B1, Table 1) and had minor and insignificant effects on HFS-induced enhancement/depression at either 100 Hz or 20 Hz (n = 7; Fig. 3, B2 and B3). A similar lack of effects was recorded in five experiments with VU0240551 (20 µM; Table 1). These findings indicated that the effect of furosemide on the fiber volley was not mediated through blockade of NKCC1 or KCC2 transporters. In search for an alternative cause of this effect, we used the stilbene derivative DIDS (500 µM), which is an antagonist of several anion transporters, some of which are also antagonized by furosemide. Initial experiments using 30-min application of DIDS indicated a decrement of the fiber volley that began late in this period. We therefore extended the perfusion of DIDS to 45 min. Results from nine experiments showed a slight increase in the fiber volley amplitude during the first 20 min, which subsequently reversed into a decrease. The fiber volley latency increased along with the decrease in amplitude (Fig. 3C1). Both changes were significant after 45 min (P < 0.01; Table 1). The fiber volley area also showed an average decrease, which, however, did not reach statistical significance (P = 0.11; Table 1). As seen with furosemide, DIDS reduced the early enhancement of the fiber volley during HFS trains at 100 Hz (Fig. 3, C2 and C3). In contrast to furosemide, DIDS also reduced the late depression: the fiber volley at stimulus 100 was significantly larger in the presence of DIDS (both amplitude and area) compared with control (P < 0.01). The effects of DIDS with 20-Hz trains was similar to furosemide, consisting of a significant reduction of the fiber volley (amplitude and area) in both the early and late parts of the train (Fig. 3, C2 and C3).
Figure 4A compares the effect on the fiber volley of furosemide and DIDS applied alone and in combination (45 min). The data showed that combined application beyond 30 min had larger effects than either drug alone, but the washin curve did not reach a stable level within 45 min, precluding an assessment of whether the effects were additive or occlusive. The effects of coapplied furosemide and DIDS on HFS trains were similar to furosemide with respect to late depression of the fiber volley, which was unaltered. The effect on the early enhancement was larger than with either drug alone (Fig. 4, A2 and A3). DIDS is an antagonist of the Na+- transporter (NBC), which is present in both neurons and glial cells and is not reported to be sensitive to furosemide. We tested the possible influence of NBC by perfusion of the selective antagonist S0859 (30 µM). The fiber volley evoked at 0.2 Hz during washin of S0859 was unaltered (Fig. 4B1, Table 1), and the responses evoked during 100-Hz and 20-Hz HFS trains were not significantly changed after 45-min exposure to S0859 (Fig. 4, B2 and B3).
Fig. 4.

Effects of coapplied DIDS and furosemide and of S0859 on the fiber volley. A1: normalized fiber volley amplitude and latency recorded during washin of furosemide (2.5 mM, n = 5), DIDS (0.5 mM, n = 9), or the 2 drugs combined (n = 5). A2: fiber volley amplitude as function of stimulus number (normalized to stimulus 1) in HFS trains of 100 Hz before (control) and after 45-min perfusion of DIDS + furosemide. A3: summary histograms comparing amplitude (top) and area (bottom) of near-maximally enhanced fiber volleys (stimulus 10 at 100 Hz; stimulus 20 at 20 Hz) and of the volleys evoked at stimulus 100 from the control period (filled bars) and in the presence of DIDS + furosemide (open bars) B: plots as in A but recorded with perfusion of S0859 (30 µM). *P < 0.05; **P < 0.01.
Effects of sucrose.
The nature and relationship of the mechanisms underlying the early increase and late decrease of the fiber volley during sustained HFS are unclear, but both events might be governed by extracellular ion concentration changes (Owen and Grover 2015). However, the observed differential sensitivity to furosemide with 100-Hz HFS is indicative of different underlying mechanisms. We sought to verify that changes in extracellular ion composition could explain the observed changes of the volley size during HFS trains. The magnitude of activity-dependent changes in ion concentrations of the extracellular space would be expected to depend on the volume of the space. To induce a hyperosmotic increase in the extracellular volume we included 30 mM sucrose in the perfusion medium. Perfusion of sucrose (for 30 min) gave insignificant reduction of the fiber volley evoked at 0.2 Hz, which was accompanied by a modest, but statistically significant, decrease in the latency (Fig. 5A1, Table 1). Sucrose had no overall effect on fiber volleys evoked in the early part of the HFS train at 100 Hz but caused a significant increase in the normalized fiber volley area recorded at stimulus 100. The amplitude also increased, but this was not significant (Fig. 5, A2 and A3). In the same experiments, we subsequently increased the rate of perfusion of the recording chamber from 1 to 3 ml/min (with sucrose still present) in order to increase the rate of fluid exchange within the slice. After 5 min of increased perfusion, the normalized amplitude (Fig. 5, A2 and A3) and area (Fig. 5A3) of the fiber volleys evoked late in the train showed a further increase. Both parameters were significantly changed compared with control (P < 0.01). Even with a high perfusion rate there was no detectable effect of sucrose on the fiber volleys evoked early in the train, nor was there any significant effect of sucrose on fiber volleys (early or late) evoked by 20-Hz trains (Fig. 5A3). To test whether expansion of the extracellular volume could counteract the effect of furosemide, we perfused sucrose and furosemide together. The inhibitory effect of furosemide on fiber volley amplitude (Fig. 5B1) and on HFS-induced enhancement of the fiber volley during 100-Hz and 20-Hz trains (Fig. 5, B2 and B3) persisted during coapplication of sucrose (n = 6), and the average magnitude of these effects did not deviate significantly from those induced by furosemide or sucrose alone (P > 0.05, unpaired t-test).
Fig. 5.

Effects of sucrose on the fiber volley and modification of paired-pulse enhancement of excitability by furosemide and ZD7288. A1: normalized fiber volley amplitude and latency before (10 min) and during perfusion of sucrose (30 min). A2: fiber volley amplitude as function of stimulus number in 100-Hz trains evoked before sucrose (control), during perfusion of sucrose at normal rate (1 ml/min; sucrose), and after 5 min of increased perfusion rate (3 ml/min; sucrose + ↑perf). A3: comparison of amplitudes (top) and areas (bottom) of near-maximally enhanced fiber volleys (stimulus 10 at 100 Hz; stimulus 20 at 20 Hz) and of volleys evoked at stimulus 100 in the control period (filled bars) and in the presence of sucrose with normal (gray bars) and increased (open bars) perfusion rate. B: plots as in A, but with coapplication of sucrose (30 mM) and furosemide (2.5 mM). In B2 and B3 only control data and data with high perfusion rate (with sucrose and furosemide) are included. C: recordings of fiber volleys evoked by equal test stimuli (t) in the absence (tu) and presence (tc) of a preceding conditioning stimulus (c) of larger intensity. Stimulation frequency = 0.1 Hz. D: amplitude ratio (tc/tu) plotted as a function of delay from the conditioning stimulus in the control condition and during perfusion of furosemide (2.5 mM). E: fiber volley amplification [(tc − tu)/tu] at 200-ms delay normalized to the amplification at 50-ms delay (= 1.0) in the control condition and during perfusion of furosemide (2.5 mM), ZD7288 (20 µM), and the 2 drugs combined. F: plots as in A, showing effects of ZD7288 (20 µM) applied alone and furosemide (2.5 mM) applied during continuous perfusion of ZD7288. *P < 0.05; **P < 0.01.
Paired-pulse modification of axonal excitability.
Action potentials in Schaffer collaterals induce a period of supranormal excitability lasting ≥200 ms (Soleng et al. 2004; Wigström and Gustafsson 1981). To investigate whether furosemide alters this type of response, we measured the fiber volley amplitude in the presence and absence of a strong conditioning stimulus (3 × duration of the test stimulus) delivered 10–200 ms before the test stimulus (Fig. 5C). In control recordings the fiber volley was amplified after the conditioning stimulus, reaching a maximum at short delays (10 or 50 ms) followed by decay. Furosemide did not alter the magnitude of the response between 10 and 100 ms but caused a significant decrease at 150 ms and 200 ms delay (Fig. 5D; n = 7). The decay of supranormal excitability is reported to be promoted by activation of the hyperpolarization-activated cation current, Ih (Soleng et al. 2004). To test whether the effect of furosemide depended on Ih, we blocked Ih with ZD7288 (20 µM, 30 min). We obtained an estimate of the decay at 200-ms delay by measuring the residual amplification of the fiber volley at this time point normalized to the amplification at 50-ms delay. ZD7288 by itself caused a significant increase of the residual amplification, consistent with a decreased rate of decay as reported by Soleng et al. (2004). Subsequent addition of furosemide induced, on average, some reversal, but this was not statistically significant (Fig. 5E; n = 5, P = 0.28). These findings indicated that furosemide directly or indirectly enhances Ih in the axons. It therefore seemed plausible that Ih activation could be behind furosemide’s effect on the fiber volley size and on HFS-induced enhancement of the fiber volley. We therefore tested the effects on these parameters of ZD7288 (20 µM) applied alone and furosemide (2.5 mM) applied in the presence of ZD7288. We found no change in the fiber volley dimension or latency during 30-min perfusion of ZD7288 (n = 11; Fig. 5F1, Table 1) and no overall change in HFS-induced effects on the fiber volley (Fig. 5, F2 and F3). In the presence of ZD7288, furosemide still caused significant reduction of the fiber volley (n = 7; Fig. 5F1, Table 1) and the HFS-induced early enhancement (Fig. 5, F2 and F3). These changes were not significantly different from those obtained with furosemide alone [see Fig. 3A and Table 1; fiber volley amplitude: P = 0.75, amplitude at stimulus 10 (100 Hz): P = 0.11].
Fiber volley in the alveus.
It is possible that the effect of furosemide on the fiber volley in str. radiatum depends on properties that are specific for axons of the Schaffer collateral pathway. To clarify, we recorded fiber volleys originating from axons of CA1 pyramidal neurons by stimulating and recording locally within a part of the alveus that had been physically isolated from the str. oriens of the CA1 region. Furosemide (2.5 mM, 30 min) significantly depressed the fiber volley in the alveus (both amplitude and area) and increased the latency to peak (Fig. 6A1, Table 1). The onset of the effect occurred after 5 min, which was faster than in str. radiatum, and it reached a maximum within ~25 min. HFS trains at 100 Hz resulted in a reduction of the fiber volley that began after a few stimuli and showed steady progression in proportion to the length of the train. Compared with control, furosemide caused no significant change in the normalized amplitude or area of fiber volleys occurring either early or late in the train (100 Hz or 20 Hz; Fig. 6, A2 and A3). As observed in the str. radiatum, bumetanide (20 µM) had no effects on the baseline fiber volley in the alveus (n = 9; Fig. 6B1, Table 1), nor did it alter the depression occurring during HFS trains (Fig. 6, B2 and B3).
Fig. 6.

Effects of furosemide and bumetanide on fiber volleys in the alveus. A1: time course of the amplitude and latency to peak (normalized) of the fiber volley measured in the alveus before and during perfusion of furosemide. Top: example records show fiber volley before (solid trace) and after 29–30 min in furosemide (dashed trace). A2: fiber volley amplitude as function of stimulus number in HFS trains of 100 Hz before (control) and after exposure to furosemide for >30 min (FURO). Top: records of fiber volleys evoked at stimuli 1, 10, and 100 (100 Hz). A3: comparison of the normalized amplitudes (top) and areas (bottom) of fiber volleys evoked at stimuli 10 (100 Hz), 20 (20 Hz), and 100 (100 Hz and 20 Hz) taken from control period (filled bars) and in the presence of furosemide (open bars). B: plots as in A, but instead of furosemide bumetanide (20 µM, 30 min) was perfused. All recordings were done with CNQX (20 µM), MK 801 (10 µM), and CGP55845 (2 µM) present. Scale bars: 1 ms, 2 mV.
DISCUSSION
e-s relationship.
The present results show that furosemide has several effects on normal neuronal signaling in the hippocampal CA1 region. First, furosemide increases the stimulus-induced excitation of pyramidal neurons as revealed by an increased e-s coupling. This effect is abolished by gabazine, suggesting that it depends on a reduction of GABAA receptor-mediated inhibition. The results with VU0240551 indicate that blockade of the neuronal KCC2 transporter (Delpire et al. 2009) contributes to this effect of furosemide, which likely results from a decreased Cl− extrusion leading to intraneuronal accumulation of Cl− and a reduced gradient for Cl− influx through the GABAA channel (Deisz et al. 2014; Deisz and Lux 1982; Misgeld et al. 1986; Thompson et al. 1988; Thompson and Gähwiler 1989). The potentiation of e-s coupling was considerably larger with furosemide than with VU0240551, which possibly can be explained by an additional direct antagonism of certain subtypes of GABAA receptors by furosemide (Korpi et al. 1995; Pearce 1993).
Despite the enhancement of the e-s relationship, the PS remained constant during washin of furosemide because of a concomitant decrease of excitatory input (reduced fEPSP). The impact of the latter change on the ability to activate the postsynaptic neurons will, as expected, depend on the amount of presynaptic activity, which can explain why furosemide caused decreased excitation at low stimulus intensities and increased excitation at high intensities. These findings are therefore both consistent with reports showing “hyperexcited” synaptic response with high-intensity stimulation (Hochman et al. 1995; MacVicar and Hochman 1991) and with reports showing insignificant or inhibitory effects of furosemide (Gutschmidt et al. 1999). It should be noted that such dependency on the synaptic drive makes it difficult to assess how furosemide influences excitability during spontaneous epileptic activity. The results here indicate that effects that are measured with electrical stimulation cannot immediately be extrapolated onto spontaneous discharges unless the amount of synaptic drive is equalized.
Müller (2000) reported that the fEPSP slope is reduced by furosemide or DIDS. Our data confirm the result with furosemide, including the magnitude of the effect. In addition, we found that bumetanide and VU0240551 in relevant doses for blocking NKCC1 and KCC2, respectively, in brain slices (Dzhala et al. 2012) were inefficacious, suggesting that blockade of these transporters was not causing the decrement of the fEPSP.
Fiber volley.
Furosemide significantly reduced the amplitude of the isolated fiber volley in CA1. The concomitant increase in latency and decrement of the area of the fiber volley indicate a lowered excitability of individual axons but do not exclude other influences, such as conduction block, desynchronization, or altered action potential waveform. As either of such changes could cause a decreased or delayed release of vesicles from synaptic terminals, they are likely causally related to the decrease in fEPSP slope. In keeping with this idea, the effect of furosemide on the fiber volley was insensitive to GABAA receptor blockade, similar to the fEPSP, and the fiber volley was sensitive to DIDS but insensitive to bumetanide or VU0240551, as was also found for the fEPSP (see Müller 2000 for the effect of DIDS on the fEPSP). Other possible factors that could affect the fEPSP include changes in vesicular glutamate uptake, which is known to be influenced by cytosolic Cl− concentration, and might therefore be sensitive to furosemide (Staley 2002).
The stilbene derivative DIDS (500 µM) reproduced the effect of furosemide on the fiber volley, indicating that specific interference with anion transport systems, some of which are known to be common targets for DIDS and furosemide, is the primary cause of this effect. DIDS is an antagonist of several proteins involved in transport of Cl− and , including the NBC. Blockade of the NBC seems to have no major role, as S0859 was unable to reproduce the effect. Transporters that are sensitive to both furosemide and DIDS include the KCC2 and the Na+-independent Cl−/ exchanger AE3, present in both glial cells and neurons (Deitmer and Rose 2010), the blockade of which will likely result in altered gradients for Cl−, , and/or H+. It is unclear to what extent (and by which mechanism) such changes could specifically impact the conduction properties of axons. VU240551 had no effect on the fiber volley, questioning a role of the KCC2. Previous studies on epileptic brain slice tissue have suggested that furosemide can induce failure of action potential invasion into axonal arborizations and varicosities (Hochman and Schwartzkroin 2000). On the basis of indirect observations it was hypothesized that this effect was induced by an increased extracellular potassium concentration ([K+]o) resulting from interference with Na+-K+-2Cl− cotransport. While mechanisms involving elevated [K+]o can significantly influence axonal fidelity under hyperexcitable conditions (Meeks et al. 2005), it seems unlikely that a rise in [K+]o can explain the present results with furosemide or DIDS. First, furosemide (2–5 mM) applied to hippocampal slices has been shown to cause either a small rise (0.6 mM on average; Jauch et al. 2002) or no change (Xiong and Stringer 2000) in baseline [K+]o, and DIDS (1 mM) is reported to have no effect on baseline [K+]o (Jauch et al. 2002). Second, even though axonal excitability is sensitive to changes in [K+]o, it requires a relatively large increase to reduce the fiber volley, and moderate elevations (up to 6 mM) increase the fiber volley amplitude and decrease its latency (Poolos et al. 1987). During washin of furosemide we could detect no initial increase in fiber volley amplitude, as would be expected if [K+]o was building up. A transient increase was seen during washin of DIDS, but this was not accompanied by a decreased latency. Alternatively, furosemide’s effect on the fiber volley could be due to hyperpolarization of the axons, which would increase the latency of action potentials and reduce the number of fibers that were depolarized to threshold by the electrical stimulus. One finding here seems to support this proposal: furosemide was seen to affect the decay of paired-pulse enhancement of excitability. Increased activation of the Ih, presumably resulting from axonal hyperpolarization, has been shown to increase the rate of decay of paired-pulse enhancement without changing the magnitude of the response at its peak (Soleng et al. 2004). A similar combination of effect was observed here with furosemide and was subsequently antagonized by Ih blockade (Fig. 5E). These findings therefore seem consistent with the hypothesis that furosemide induces hyperpolarization of the axons. It should be noted that nonspecific effects of ZD7288, including blockade of sodium currents (Wu et al. 2012) and T-type Ca2+ channels (Sánchez-Alonso et al. 2008), could have contributed to the observed response. The present data do not exclude that action potentials of individual fibers were modified to some extent, but the lack of effect of ZD7288 on the fiber volley amplitude and the HFS-induced dynamics (Fig. 5F) indicates that such nonspecific effects had no major impact on excitability or conduction properties of the stimulated axons. The lack of effect of ZD7288 on the baseline amplitude of the fiber volley is consistent with previous findings (Soleng et al. 2003) and suggests little activation of the Ih at axonal resting membrane potential. The reason why ZD7288 could influence the amplitude of fiber volleys evoked by paired stimuli but not by HFS trains is unclear, but this finding indicates that the relative impact of the Ih on axonal excitability decreases during prolonged high-frequency firing. Whether this results from a progressive depolarization of the baseline membrane potential (deactivating the HCN channels) and/or is due to alteration of other membrane currents during HFS trains remains to be investigated. It is interesting to note that at lower frequencies (2–5 Hz) blockade of the Ih is reported to cause a marked decrease in the fiber volley amplitude (Soleng et al. 2003). We conclude that furosemide and DIDS decrease axonal excitability by a mechanism downstream of blockade of anion transport, which is unrelated to an increase in [K+]o, and which likely involves hyperpolarization of the axons.
HFS effect on the fiber volley.
During HFS trains, the observed changes in the fiber volley, consisting of an initial increase in amplitude followed by a decrease, are largely in accordance with recent studies (Kim et al. 2012; Owen and Grover 2015). It should be noted that whereas we found a parallel change in fiber volley amplitude and area, Owen and Grover (2015) found no depression when response area was measured. The reason for this discrepancy is uncertain but may relate to different recording conditions. Evidence suggests that the decrement of the fiber volley during prolonged HFS stimulation reflects specific changes in individual axons, including conduction failure and decreased conduction velocity (Kim et al. 2012). Such changes are presumably caused by progressive modification of ion channel properties (Kress and Mennerick 2009) and/or changes in extracellular concentration of ions, such as decrease in extracellular Na+ concentration and increase in [K+]o (He et al. 2002; Heinemann et al. 1990; Kim et al. 2012; Meeks and Mennerick 2004). In addition, as noted above, a hyperexcitable period is known to follow individual spikes in Schaffer collateral axons (Wigström and Gustafsson 1981), which is likely controlled by mechanisms intrinsic to the activated axon (Palani et al. 2010; Soleng et al. 2004). The relative importance of these individual factors for axonal conduction during the different phases of high-frequency activity is not clarified, making it difficult to identify the exact cause of the effect of furosemide. Interestingly, the early increase in the fiber volley reflects a state of hyperexcitability that is selectively expressed in the distal Schaffer collateral axons as recently shown by Owen and Grover (2015). These authors proposed that this state, which seems to have a function of allowing successful propagation of short-duration, high-frequency firing, might be caused by an initial small increase in [K+]o leading to a moderate depolarization in the distal part of the axons. The present results with sucrose suggest that mechanisms sensitive to hyperosmotic expansion of the extracellular volume (e.g., activity-dependent increase in [K+]o) make a significant contribution to the large depression of the fiber volley induced with very high stimulation frequency (100 Hz). However, the same data gave no support that such mechanisms were behind moderate depressions (20 Hz) or the initial enhancement. Furosemide was seen to induce the opposite combination of changes as sucrose; it reduced the initial enhancement, amplified moderate depressions (20 Hz), but had no effect on large depressions (100 Hz). The ability to differentially manipulate the enhancement and the depression is highly indicative that the two phenomena are governed by different mechanisms. The lack of sensitivity to sucrose indicates that furosemide’s effect on HFS-induced enhancement of the fiber volley was exerted locally on individual axons rather than being dependent on signaling from surrounding cells (glia or other axons) via the extracellular space. Such local effect may involve membrane hyperpolarization, as discussed above, consistent with the hypothesis that voltage-dependent currents in the stimulated axons contribute to HFS-induced enhancement of the fiber volley. The observed resistance to ZD7288 suggests that currents other than the Ih are involved in this response. Gutschmidt et al. (1999) showed that furosemide reduces activity-evoked rises in [K+]o, most likely due to interference with excitability. Insofar as a rise in [K+]o is a major cause of activity-dependent depression of the fiber volley (Kim et al. 2012; Owen and Grover 2015), and furosemide (or DIDS) decreases axonal excitability (present study), one would expect less depression in the presence of these drugs. Indeed, DIDS was seen to reduce the large depressions of the fiber volley induced during 100-Hz HFS, consistent with a primary effect on axonal excitability. On the other hand, furosemide had no effect on the large depressions. This difference seems to be due to an additional effect of furosemide, as the copresence of furosemide and DIDS also failed to reduce the large depressions. Interestingly, moderate depression of the fiber volley seen with 20-Hz HFS was not reduced but enhanced by furosemide and DIDS. The reason for the latter effect is unclear, but a simple explanation would be that it reflects the continuation of the antagonism of the early enhancement. This interpretation implies that the processes underlying the enhancement and depression of the fiber volley coexist during prolonged HFS, at least at lower frequencies, and could be representing variable states of excitability in segregated areas of the axonal arbor.
The sensitivity of the fiber volley to furosemide is not specific for axons running in str. radiatum of the CA1. The induced decrement of the fiber volley in the alveus was larger and occurred faster than in the str. radiatum, which may be ascribed to a better penetration of the drug, possibly favored by the physical isolation of the tissue. Fiber volleys in the alveus exhibited a monophasic depression in response to HFS trains, and the relative magnitude of the depression was unaltered by furosemide. This result seems equivalent to the findings on the large depressions in str. radiatum discussed above.
Relevance for epilepsy.
This study shows that furosemide applied in doses required for inhibition of epileptiform activity in slices has both enhancing and depressing effects on stimulus-induced excitation in normal slice tissue. As the balance between these opposing effects appears to depend on the level of excitation (at least in the CA1 region), it is difficult to ascertain whether the resultant effect would be one that contributes to the antiepileptic properties of the drug. It seems reasonable to assume that a general decrease in axonal excitability will tend to inhibit the spread of hyperactivity from an epileptic focus via efferent projections into normal tissue surrounding the focus. In the Schaffer collaterals, high-frequency bursting patterns, characteristic of epileptiform activity, would seem to be particularly prone to filtering in the presence of furosemide due to antagonism of activity-dependent enhancement of axonal excitation. In addition, changes in axonal fidelity would likely influence epileptogenesis even within a focal area. To investigate, studies should be designed to isolate the depressant effects for direct testing in epileptic tissue.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
M.A. and S.N. conceived and designed research; M.A. and S.N. interpreted results of experiments; M.A. and S.N. edited and revised manuscript; M.A. and S.N. approved final version of manuscript; S.N. performed experiments; S.N. analyzed data; S.N. prepared figures; S.N. drafted manuscript.
ACKNOWLEDGMENTS
The authors thank Bertha Beck Mortensen for excellent technical assistance.
REFERENCES
- Ahmad S, Clarke L, Hewett AJ, Richens A. Controlled trial of frusemide as an antiepileptic drug in focal epilepsy. Br J Clin Pharmacol 3: 621–625, 1976. doi: 10.1111/j.1365-2125.1976.tb04885.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deisz RA, Lux HD. The role of intracellular chloride in hyperpolarizing post-synaptic inhibition of crayfish stretch receptor neurones. J Physiol 326: 123–138, 1982. doi: 10.1113/jphysiol.1982.sp014181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deisz RA, Wierschke S, Schneider UC, Dehnicke C. Effects of VU0240551, a novel KCC2 antagonist, and DIDS on chloride homeostasis of neocortical neurons from rats and humans. Neuroscience 277: 831–841, 2014. doi: 10.1016/j.neuroscience.2014.07.037. [DOI] [PubMed] [Google Scholar]
- Deitmer JW, Rose CR. Ion changes and signalling in perisynaptic glia. Brain Res Brain Res Rev 63: 113–129, 2010. doi: 10.1016/j.brainresrev.2009.10.006. [DOI] [PubMed] [Google Scholar]
- Delpire E, Days E, Lewis LM, Mi D, Kim K, Lindsley CW, Weaver CD. Small-molecule screen identifies inhibitors of the neuronal K-Cl cotransporter KCC2. Proc Natl Acad Sci USA 106: 5383–5388, 2009. doi: 10.1073/pnas.0812756106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzhala V, Valeeva G, Glykys J, Khazipov R, Staley K. Traumatic alterations in GABA signaling disrupt hippocampal network activity in the developing brain. J Neurosci 32: 4017–4031, 2012. doi: 10.1523/JNEUROSCI.5139-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzhala VI, Talos DM, Sdrulla DA, Brumback AC, Mathews GC, Benke TA, Delpire E, Jensen FE, Staley KJ. NKCC1 transporter facilitates seizures in the developing brain. Nat Med 11: 1205–1213, 2005. doi: 10.1038/nm1301. [DOI] [PubMed] [Google Scholar]
- Eftekhari S, Mehvari Habibabadi J, Najafi Ziarani M, Hashemi Fesharaki SS, Gharakhani M, Mostafavi H, Joghataei MT, Beladimoghadam N, Rahimian E, Hadjighassem MR. Bumetanide reduces seizure frequency in patients with temporal lobe epilepsy. Epilepsia 54: e9–e12, 2013. doi: 10.1111/j.1528-1167.2012.03654.x. [DOI] [PubMed] [Google Scholar]
- Gutschmidt KU, Stenkamp K, Buchheim K, Heinemann U, Meierkord H. Anticonvulsant actions of furosemide in vitro. Neuroscience 91: 1471–1481, 1999. doi: 10.1016/S0306-4522(98)00700-3. [DOI] [PubMed] [Google Scholar]
- Haglund MM, Hochman DW. Furosemide and mannitol suppression of epileptic activity in the human brain. J Neurophysiol 94: 907–918, 2005. doi: 10.1152/jn.00944.2004. [DOI] [PubMed] [Google Scholar]
- He Y, Zorumski CF, Mennerick S. Contribution of presynaptic Na+ channel inactivation to paired-pulse synaptic depression in cultured hippocampal neurons. J Neurophysiol 87: 925–936, 2002. [DOI] [PubMed] [Google Scholar]
- Heinemann U, Stabel J, Rausche G. Activity-dependent ionic changes and neuronal plasticity in rat hippocampus. Prog Brain Res 83: 197–214, 1990. doi: 10.1016/S0079-6123(08)61250-9. [DOI] [PubMed] [Google Scholar]
- Hesdorffer DC, Stables JP, Hauser WA, Annegers JF, Cascino G. Are certain diuretics also anticonvulsants? Ann Neurol 50: 458–462, 2001. doi: 10.1002/ana.1136. [DOI] [PubMed] [Google Scholar]
- Hochman DW. The extracellular space and epileptic activity in the adult brain: explaining the antiepileptic effects of furosemide and bumetanide. Epilepsia 53, Suppl 1: 18–25, 2012. doi: 10.1111/j.1528-1167.2012.03471.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochman DW, Baraban SC, Owens JW, Schwartzkroin PA. Dissociation of synchronization and excitability in furosemide blockade of epileptiform activity. Science 270: 99–102, 1995. doi: 10.1126/science.270.5233.99. [DOI] [PubMed] [Google Scholar]
- Hochman DW, Schwartzkroin PA. Chloride-cotransport blockade desynchronizes neuronal discharge in the “epileptic” hippocampal slice. J Neurophysiol 83: 406–417, 2000. [DOI] [PubMed] [Google Scholar]
- Holtkamp M, Matzen J, Buchheim K, Walker MC, Meierkord H. Furosemide terminates limbic status epilepticus in freely moving rats. Epilepsia 44: 1141–1144, 2003. doi: 10.1046/j.1528-1157.2003.14003.x. [DOI] [PubMed] [Google Scholar]
- Jauch R, Windmüller O, Lehmann T-N, Heinemann U, Gabriel S. Effects of barium, furosemide, ouabaine and 4,4′-diisothiocyanatostilbene-2,2′-disulfonic acid (DIDS) on ionophoretically-induced changes in extracellular potassium concentration in hippocampal slices from rats and from patients with epilepsy. Brain Res 925: 18–27, 2002. doi: 10.1016/S0006-8993(01)03254-1. [DOI] [PubMed] [Google Scholar]
- Kahle KT, Staley KJ, Nahed BV, Gamba G, Hebert SC, Lifton RP, Mount DB. Roles of the cation-chloride cotransporters in neurological disease. Nat Clin Pract Neurol 4: 490–503, 2008. doi: 10.1038/ncpneuro0883. [DOI] [PubMed] [Google Scholar]
- Kilb W, Sinning A, Luhmann HJ. Model-specific effects of bumetanide on epileptiform activity in the in-vitro intact hippocampus of the newborn mouse. Neuropharmacology 53: 524–533, 2007. doi: 10.1016/j.neuropharm.2007.06.015. [DOI] [PubMed] [Google Scholar]
- Kim E, Owen B, Holmes WR, Grover LM. Decreased afferent excitability contributes to synaptic depression during high-frequency stimulation in hippocampal area CA1. J Neurophysiol 108: 1965–1976, 2012. doi: 10.1152/jn.00276.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korpi ER, Kuner T, Seeburg PH, Lüddens H. Selective antagonist for the cerebellar granule cell-specific γ-aminobutyric acid type A receptor. Mol Pharmacol 47: 283–289, 1995. [PubMed] [Google Scholar]
- Kress GJ, Mennerick S. Action potential initiation and propagation: upstream influences on neurotransmission. Neuroscience 158: 211–222, 2009. doi: 10.1016/j.neuroscience.2008.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löscher W, Puskarjov M, Kaila K. Cation-chloride cotransporters NKCC1 and KCC2 as potential targets for novel antiepileptic and antiepileptogenic treatments. Neuropharmacology 69: 62–74, 2013. doi: 10.1016/j.neuropharm.2012.05.045. [DOI] [PubMed] [Google Scholar]
- Maa EH, Kahle KT, Walcott BP, Spitz MC, Staley KJ. Diuretics and epilepsy: will the past and present meet? Epilepsia 52: 1559–1569, 2011. doi: 10.1111/j.1528-1167.2011.03203.x. [DOI] [PubMed] [Google Scholar]
- MacVicar BA, Hochman D. Imaging of synaptically evoked intrinsic optical signals in hippocampal slices. J Neurosci 11: 1458–1469, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margineanu DG, Klitgaard H. Differential effects of cation-chloride co-transport-blocking diuretics in a rat hippocampal slice model of epilepsy. Epilepsy Res 69: 93–99, 2006. doi: 10.1016/j.eplepsyres.2006.01.005. [DOI] [PubMed] [Google Scholar]
- Meeks JP, Jiang X, Mennerick S. Action potential fidelity during normal and epileptiform activity in paired soma-axon recordings from rat hippocampus. J Physiol 566: 425–441, 2005. doi: 10.1113/jphysiol.2005.089086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeks JP, Mennerick S. Selective effects of potassium elevations on glutamate signaling and action potential conduction in hippocampus. J Neurosci 24: 197–206, 2004. doi: 10.1523/JNEUROSCI.4845-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misgeld U, Deisz RA, Dodt HU, Lux HD. The role of chloride transport in postsynaptic inhibition of hippocampal neurons. Science 232: 1413–1415, 1986. doi: 10.1126/science.2424084. [DOI] [PubMed] [Google Scholar]
- Müller M. Effects of chloride transport inhibition and chloride substitution on neuron function and on hypoxic spreading-depression-like depolarization in rat hippocampal slices. Neuroscience 97: 33–45, 2000. doi: 10.1016/S0306-4522(00)00025-7. [DOI] [PubMed] [Google Scholar]
- Owen B, Grover LM. Activity-dependent differences in function between proximal and distal Schaffer collaterals. J Neurophysiol 113: 3646–3662, 2015. doi: 10.1152/jn.00446.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palani D, Baginskas A, Raastad M. Bursts and hyperexcitability in non-myelinated axons of the rat hippocampus. Neuroscience 167: 1004–1013, 2010. doi: 10.1016/j.neuroscience.2010.03.021. [DOI] [PubMed] [Google Scholar]
- Pearce RA. Physiological evidence for two distinct GABAA responses in rat hippocampus. Neuron 10: 189–200, 1993. doi: 10.1016/0896-6273(93)90310-N. [DOI] [PubMed] [Google Scholar]
- Poolos NP, Mauk MD, Kocsis JD. Activity-evoked increases in extracellular potassium modulate presynaptic excitability in the CA1 region of the hippocampus. J Neurophysiol 58: 404–416, 1987. [DOI] [PubMed] [Google Scholar]
- Sánchez-Alonso JL, Halliwell JV, Colino A. ZD 7288 inhibits T-type calcium current in rat hippocampal pyramidal cells. Neurosci Lett 439: 275–280, 2008. doi: 10.1016/j.neulet.2008.05.016. [DOI] [PubMed] [Google Scholar]
- Soleng AF, Baginskas A, Andersen P, Raastad M. Activity-dependent excitability changes in hippocampal CA3 cell Schaffer axons. J Physiol 560: 491–503, 2004. doi: 10.1113/jphysiol.2004.071225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soleng AF, Chiu K, Raastad M. Unmyelinated axons in the rat hippocampus hyperpolarize and activate an H current when spike frequency exceeds 1 Hz. J Physiol 552: 459–470, 2003. doi: 10.1113/jphysiol.2003.048058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley KJ. Diuretics as antiepileptic drugs: Should we go with the flow? Epilepsy Curr 2: 35–38, 2002. doi: 10.1046/j.1535-7597.2002.00018.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson SM, Deisz RA, Prince DA. Relative contributions of passive equilibrium and active transport to the distribution of chloride in mammalian cortical neurons. J Neurophysiol 60: 105–124, 1988. [DOI] [PubMed] [Google Scholar]
- Thompson SM, Gähwiler BH. Activity-dependent disinhibition. II. Effects of extracellular potassium, furosemide, and membrane potential on ECl- in hippocampal CA3 neurons. J Neurophysiol 61: 512–523, 1989. [DOI] [PubMed] [Google Scholar]
- Uwera J, Nedergaard S, Andreasen M. A novel mechanism for the anticonvulsant effect of furosemide in rat hippocampus in vitro. Brain Res 1625: 1–8, 2015. doi: 10.1016/j.brainres.2015.08.014. [DOI] [PubMed] [Google Scholar]
- Wahab A, Albus K, Heinemann U. Age- and region-specific effects of anticonvulsants and bumetanide on 4-aminopyridine-induced seizure-like events in immature rat hippocampal-entorhinal cortex slices. Epilepsia 52: 94–103, 2011. doi: 10.1111/j.1528-1167.2010.02722.x. [DOI] [PubMed] [Google Scholar]
- Wigström H, Gustafsson B. Increased excitability of hippocampal unmyelinated fibres following conditioning stimulation. Brain Res 229: 507–513, 1981. doi: 10.1016/0006-8993(81)91013-1. [DOI] [PubMed] [Google Scholar]
- Wu X, Liao L, Liu X, Luo F, Yang T, Li C. Is ZD7288 a selective blocker of hyperpolarization-activated cyclic nucleotide-gated channel currents? Channels (Austin) 6: 438–442, 2012. doi: 10.4161/chan.22209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong ZQ, Stringer JL. Sodium pump activity, not glial spatial buffering, clears potassium after epileptiform activity induced in the dentate gyrus. J Neurophysiol 83: 1443–1451, 2000. [DOI] [PubMed] [Google Scholar]