

ABSTRACT
Cyclin E, in conjunction with its catalytic partner cyclin-dependent kinase 2 (CDK2), regulates cell cycle progression as cells exit quiescence and enter S-phase. Multiple mechanisms control cyclin E periodicity during the cell cycle, including phosphorylation-dependent cyclin E ubiquitylation by the SCFFbw7 ubiquitin ligase. Serine 384 (S384) is the critical cyclin E phosphorylation site that stimulates Fbw7 binding and cyclin E ubiquitylation and degradation. Because S384 is autophosphorylated by bound CDK2, this presents a paradox as to how cyclin E can evade autocatalytically induced degradation in order to phosphorylate its other substrates. We found that S384 phosphorylation is dynamically regulated in cells and that cyclin E is specifically dephosphorylated at S384 by the PP2A-B56 phosphatase, thereby uncoupling cyclin E degradation from cyclin E-CDK2 activity. Furthermore, the rate of S384 dephosphorylation is high in interphase but low in mitosis. This provides a mechanism whereby interphase cells can oppose autocatalytic cyclin E degradation and maintain cyclin E-CDK2 activity while also enabling cyclin E destruction in mitosis, when inappropriate cyclin E expression is genotoxic.
KEYWORDS: cell cycle, cyclin-dependent kinases, cyclins, serine/threonine phosphatases, ubiquitination
INTRODUCTION
The mammalian cell cycle is regulated by cyclin-dependent kinases (CDKs) and their associated cyclin regulatory subunits. Cyclin E-CDK2 has essential roles as cells exit quiescence and during endoreduplication. It also regulates numerous processes during the G1 and S-phases of the cell cycle, including S-phase entry, DNA replication, and centrosome duplication (1–5).
Cells must tightly regulate cyclin E-CDK2 activity, and this is accomplished through a variety of mechanisms, including E2F-dependent cyclin E transcription, binding of CDK inhibitor proteins (p27Kip1 and p21Cip1), activating and inhibitory phosphorylation of CDK2, and cyclin E degradation by the ubiquitin-proteasome system (1, 2). Abnormal cyclin E-CDK2 activity disrupts normal G1/S control and causes genomic instability (6–12). Importantly, cyclin E is oncogenic in multiple contexts: the cyclin E gene is amplified in human serous ovarian and basal breast cancers, and its deregulated activity promotes tumorigenesis in mice (13–16).
The F-box protein Fbw7 is the substrate recognition component of an SCF ubiquitin ligase that targets cyclin E for degradation after cyclin E becomes phosphorylated within conserved motifs called Cdc4 phosphodegrons (CPDs) (17–22). Fbw7 substrate CPDs contain two negative charges: a phosphorylated-Thr/Ser (pT/S) in the 0 position, and a second pT/S or an acidic amino acid in the +4 position (23, 24). Cyclin E contains two CPDs: a high-affinity C-terminal degron that is phosphorylated at T380 and S384 and is the primary determinant of Fbw7 binding and a low-affinity N-terminal degron that contains only one phosphorylation site, at T62 (Fig. 1A). Phosphorylation-dependent cyclin E ubiquitylation by SCFFbw7 is essential for cyclin E periodicity and normal cell division. Accordingly, inhibition of cyclin E degradation via ablation of CPD phosphorylation sites or removal of Fbw7 results in constitutive cyclin E-CDK2 activity throughout the cell cycle, abnormal S-phase and mitotic progression, hyperproliferation in epithelial and hematopoietic cells, and accelerated tumorigenesis (11, 15, 25–29).
FIG 1.

Cyclin E is dephosphorylated specifically at S384 both in vivo and in vitro. (A) Schematic depicting cyclin E CPDs and the relevant kinases phosphorylating each residue. Cyclin E contains two CPDs: a low-affinity N-terminal degron centered at T62 that contains only one phosphosite (pT62) and a high-affinity C-terminal degron centered at T380 that contains two phosphosites (pT380 and pS384). All three cyclin E CPD phosphosites can be phosphorylated by CDK2. T62 can also be phosphorylated by an unknown kinase, while T380 can also phosphorylated by glycogen synthase kinase 3-β (GSK3-β). Importantly, S384 can only be phosphorylated by CDK2 in cis (by the bound CDK2 molecule), as depicted. (B) HeLa cells stably expressing an shRNA against FBXW7 (HeLa-shFBXW7) were treated with the serine/threonine phosphatase inhibitor calyculin A for 5 min. Endogenous cyclin E was immunoprecipitated from whole-cell lysates, and changes in phosphorylation of T62, T380, and S384 (all three cyclin E CPD phosphosites) were determined by Western blotting (IB). Efficiency of Fbw7 knockdown was determined by analyzing Fbw7 protein depletion in HeLa cells transduced with either nontargeting control shRNA (NT control) or FBXW7-specific shRNA. (C) Hct116 Fbw7−/− cells were treated with calyculin A for up to 12 min and processed as for panel B. (D) HeLa-shFBXW7 cells were treated with the CDK1/2 inhibitor roscovitine for 5 min and processed as for panel B. (E) Hct116 Fbw7−/− cells were treated with roscovitine for up to 20 min and processed as for panel B. (F) 293A cells were cotransfected with Myc-cyclin E and HA-CDK2 and then treated with roscovitine for up to 12 min. Myc-cyclin E was immunoprecipitated from whole-cell lysates, and changes in S384 phosphorylation were assayed by Western blotting. (G) 293A cells were lysed in buffer lacking phosphatase inhibitors and incubated with recombinant cyclin E for 0 to 60 min in an in vitro dephosphorylation assay. Changes in CPD dephosphorylation over time were measured by Western blotting.
While T380 is phosphorylated by multiple kinases, and its phosphorylation is largely constitutive, S384 can be autophosphorylated only by the bound molecule of CDK2 (in cis) (Fig. 1A) (20). Additionally, Fbw7's affinity for the doubly phosphorylated (pT380/pS384) C-terminal CPD is over 1,000-fold greater than for the singly phosphorylated (pT380) form (30). S384 phosphorylation is the critical switch that initiates cyclin E degradation; therefore, Fbw7 binding to cyclin E is stimulated by a phosphorylation that is predicted to occur almost simultaneously with the formation of an active cyclin E-CDK2 complex. Autocatalytically stimulated cyclin E degradation thus presents a paradox: how can an active cyclin E-CDK2 complex persist in cells long enough to phosphorylate its substrates before catalyzing its recognition and ubiquitylation by SCFFbw7? We hypothesized that this could be counteracted by a phosphatase that dephosphorylates cyclin E's CPDs, thus preventing Fbw7 binding. Indeed, degron dephosphorylation regulates the stability of c-Myc, an Fbw7 substrate with a CPD highly related to the cyclin E T380 region (31, 32), as well as that of Cln2, the cyclin E ortholog of budding yeast (33).
PP2A is a member of the phosphoprotein phosphatase (PPP) family of serine/threonine phosphatases. These enzymes share a conserved catalytic core domain and affect much of the total cellular serine/threonine phosphatase activity (34). PP2A regulates diverse cellular processes, including cell division (35, 36). The active PP2A holoenzyme is a heterotrimer consisting of a catalytic (C) subunit, a scaffold (A) subunit, and a regulatory (B) subunit that dictates substrate specificity. The PP2A catalytic and scaffold subunits are each encoded by two genes that produce nearly identical α and β isoforms of the proteins, which are thought to be largely functionally redundant. In contrast, PP2A regulatory subunits are divided into at least four families: B/B55, B′/B56, B″, and B‴/striatins. The structural and functional distinctions among these families alter how PP2A holoenzymes interact with substrates and confer substrate specificity (37, 38).
In this study, we determined the role of cyclin E CPD dephosphorylation in opposing cyclin E degradation. We found that S384 phosphorylation is highly labile in interphase cells and that its dephosphorylation is catalyzed by PP2A. While both PP2A-B55 and PP2A-B56 holoenzymes can dephosphorylate S384 in vitro, only PP2A-B56 complexes regulate S384 phosphorylation in vivo. By opposing cyclin E-CDK2 autophosphorylation at S384, PP2A-B56 promotes cyclin E-CDK2 activity and cyclin E stability. In contrast with interphase cells, mitotic cells exhibit very low S384 phosphatase activity, which may reinforce cyclin E periodicity. PP2A-mediated S384 dephosphorylation thus uncouples cyclin E activity from its degradation during the portion of the cell cycle in which its activity is required to coordinate G1/S progression.
RESULTS
Cyclin E pS384 is rapidly dephosphorylated.
To determine the role of phosphatases in regulating cyclin E phosphorylation, we treated cells with the serine/threonine phosphatase inhibitor calyculin A and examined the three major regulatory cyclin E CPD phosphorylation sites (pT62, pT380, and pS384) using phospho-specific antibodies. Because endogenous cyclin E is rapidly degraded following S384 phosphorylation, pS384 cyclin E is readily detectable only when cyclin E turnover is disabled (27). We studied two cell systems in which Fbw7 function was inactivated: HeLa cells stably expressing a short hairpin RNA (shRNA) against FBXW7 (the gene name for Fbw7; Fbw7 knockdown is shown in Fig. 1B) and Hct116 cells in which FBXW7 was deleted by gene targeting (27). Calyculin A treatment rapidly increased S384 phosphorylation in both cell types, whereas neither T62 nor T380 phosphorylation changed appreciably (Fig. 1B and C). The kinetics of S384 dephosphorylation was also evaluated by treating cells with the CDK1/2 inhibitor roscovitine to prevent ongoing S384 autophosphorylation. CDK2 inhibition led to the near-complete loss of S384 phosphorylation within minutes, whereas pT62 and pT380 were unaffected, although this may partially reflect their continued phosphorylation by kinases not inhibited by rosocovitine (Fig. 1D and E). To ensure that these effects were not an indirect consequence of Fbw7 inactivation, we also overexpressed cyclin E-CDK2 in 293A cells, which exceeds the ubiquitylation capacity of endogenous SCFFbw7 and enables detection of S384-phosphorylated cyclin E. Again, S384 was rapidly dephosphorylated within minutes of roscovitine addition (Fig. 1F).
To study cyclin E CPD dephosphorylation without using broadly acting phosphatase inhibitors, we incubated recombinant glutathione S-transferase (GST)–cyclin E-CDK2 with whole-cell lysates and examined the rate at which pT62, pT380, and pS384 were dephosphorylated (recombinant cyclin E-CDK2 isolated from baculovirus-infected SF9 cells is highly phosphorylated at each site). Cell lysates contained robust phosphatase activity against pS384 but had little activity against either pT62 or pT380 (Fig. 1G). Together, these experiments indicate that in contrast to the case with T62 and T380, S384 phosphorylation is highly labile in vivo and robustly dephosphorylated in vitro.
PP2A dephosphorylates cyclin E at S384.
Calyculin A inhibits several PPP family phosphatases, including PP1, PP2A, PP4, PP5, and PP6 (39). We used pharmacological and genetic methods to identify which of these enzymes target S384. First, we performed in vitro dephosphorylation assays using lysates pretreated with either okadaic acid (which targets several PPP family members), or the PP1-specific inhibitor tautomycetin (40). While okadaic acid prevented pS384 dephosphorylation, tautomycetin had no effect, suggesting that PP1 does not dephosphorylate pS384 (Fig. 2A). Next, we used RNA interference (RNAi) to identify which PPP catalytic subunit(s) targets pS384 in vitro. Lysates from 293A cells transfected with small interfering RNA (siRNA) pools targeting each PPP catalytic subunit were used to dephosphorylate recombinant cyclin E-CDK2. While each targeted phosphatase was efficiently and specifically depleted by the corresponding siRNA pool, only lysates depleted of PP2A catalytic subunit had decreased pS384 phosphatase activity (Fig. 2B). To exclude possible off-target effects of the PP2A siRNA pool, we confirmed this result with two independent siRNA pools targeting the PP2A catalytic subunit (siPP2A-C_1 and siPP2A-C_2 [Fig. 2C]). These data implicate PP2A as the calyculin A- and okadaic acid-sensitive phosphatase that dephosphorylates cyclin E at S384.
FIG 2.

The serine/threonine phosphatase PP2A dephosphorylates cyclin E at S384. (A) In vitro dephosphorylation assay of recombinant cyclin E-CDK2 using cell lysates pretreated with either dimethyl sulfoxide (DMSO), the PPP family phosphatase inhibitor okadaic acid (10 nM), or the PP1-specific inhibitor tautomycetin (10 nM). (B) 293A cells were transfected with siRNA pools, each consisting of four distinct siRNAs, against the PPP family phosphatases PP1α (PPP1CA), PP1β (PPP1CB), PP1γ (PPP1CC), PP2A (PPP2CA/B), PP4 (PPP4C), PP5 (PPP5C), and PP6 (PPP6C) or a negative-control siRNA (siAllstar control). Lysates collected from these cells were incubated with recombinant cyclin E-CDK2 in an in vitro dephosphorylation assay. The efficacy of catalytic subunit depletion was measured by Western blotting using antibodies specific for each subunit. (C) Same as panel B but performed with two distinct pools of siRNAs targeting both PP2A catalytic subunits (PPP2CA and PPP2CB), siPP2A-C_1 and siPP2A-C_2, in comparison with a negative-control siRNA (siAllstar).
PP2A-B56 dephosphorylates S384 in vivo.
PP2A substrate specificity is in large part determined by the B subunit in the PP2A holoenzyme. To identify specific PP2A complexes that regulate pS384, we screened lysates depleted of individual PP2A B subunits for reduced S384 phosphatase activity. 293A cells were transfected with Qiagen siRNA screening pools, each containing four unique siRNAs, targeting each of 12 different PP2A regulatory subunits. These lysates were then used to dephosphorylate recombinant cyclin E-CDK2 in vitro. Depletion of three B subunits—PPP2R2A (B55α), PPP2R5C (B56γ), and PPP2R5D (B56δ)—partially decreased S384 phosphatase activity (Fig. 3A). However, depleting individual regulatory subunits had a smaller effect on S384 dephosphorylation than did PP2A catalytic subunit depletion (depicted in the last lane of Fig. 3A), consistent with functional redundancies among the individual B subunits (41–43).
FIG 3.
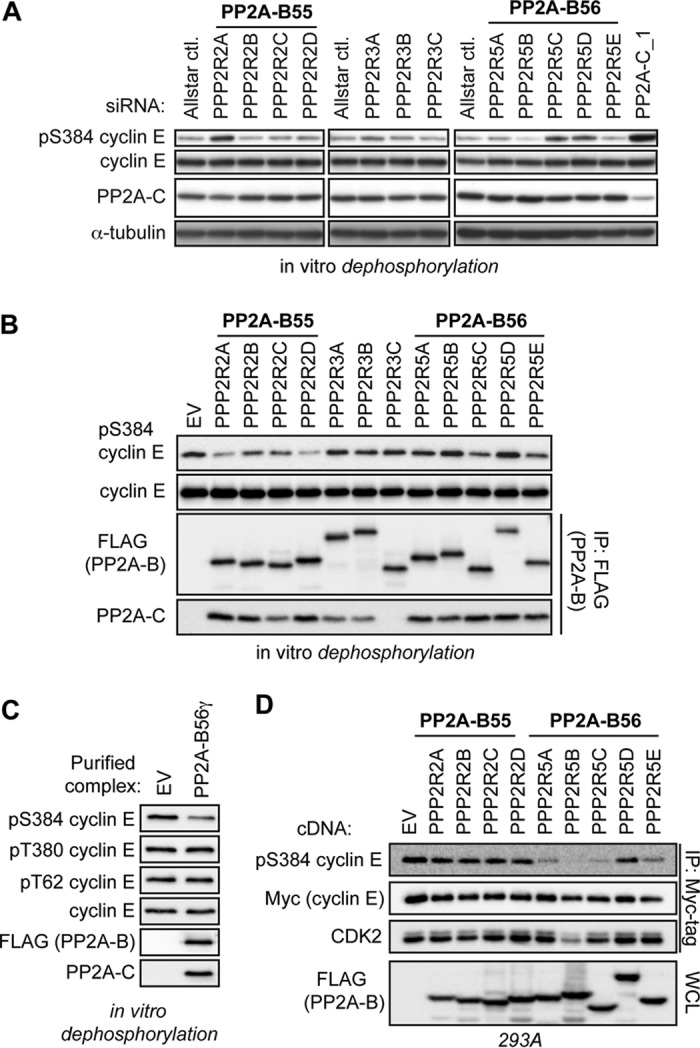
PP2A-B56 complexes regulate cyclin E S384 phosphorylation in vitro and in vivo. (A) Specific PP2A B subunits were depleted by transfecting 293A cells with pools of siRNAs, as done for Fig. 2B. Lysates harvested from these cells were incubated with purified cyclin E-CDK2 complexes in an in vitro dephosphorylation assay. Lysates depleted of the PP2A catalytic subunit were used as a positive control (siPP2A-C_1, far right lane). (B) 293A cells were transfected with FLAG-B subunits and then lysed in Tween 20 lysis buffer without phosphatase inhibitors. PP2A holoenzymes were purified via anti-FLAG immunoprecipitation and then incubated with recombinant cyclin E-CDK2 complexes to test for direct dephosphorylation of S384 in vitro. IPs from cells transfected with an empty vector (EV) served to control for nonspecific coprecipitating phosphatase activity. The relative abundance of PP2A-C served to approximate the efficiency of purification of intact PP2A heterotrimers. (C) 293A cells were transfected with either an EV or FLAG-tagged B56γ and lysed in Tween 20 lysis buffer without phosphatase inhibitors. Next, anti-FLAG immunoprecipitations were used to purify PP2A-B56γ complexes; immunoprecipitations of EV lysate served as a control for nonspecific background phosphatase activity. These complexes were then used for in vitro dephosphorylation reactions of recombinant cyclin E-CDK2. The specificity of PP2A-B56γ for each cyclin E CPD phosphosite was determined by Western blotting. (D) 293A cells were cotransfected with FLAG-B subunits, Myc-cyclin E, and HA-CDK2. Cells were lysed, and exogenous cyclin E was immunoprecipitated and analyzed for pS384 and bound CDK2 by Western blotting.
To determine if purified PP2A complexes can directly dephosphorylate S384, FLAG-tagged B subunits were expressed in 293A cells and PP2A holoenzymes were isolated via anti-FLAG immunoprecipitation (IP). These purified PP2A complexes were then used to dephosphorylate recombinant cyclin E-CDK2. Intact holoenzymes were obtained for each B subunit except PPP2R3C (B″γ), as evidenced by the coprecipitating PP2A catalytic subunit (Fig. 3B). Multiple PP2A complexes dephosphorylated S384 in vitro, with two B55 subunits (B55α and B55δ) and two B56 subunits (B56γ and B56ε) exhibiting the most activity (Fig. 3B). Both the in vivo inhibitor data and in vitro dephosphorylation experiments using whole-cell lysates suggested that phosphatases specifically target cyclin E at pS384 and have little effect on either pT62 or pT380 (Fig. 1B, D, and G). To test the specificity of PP2A for specific cyclin E CPD phosphosites, we performed anti-FLAG immunoprecipitations from cells transfected with either empty vector or FLAG-B56γ and assayed these purified complexes for in vitro phosphatase activity toward all three cyclin E CPD phosphosites. As we observed previously, PP2A-B56γ had significant S384 phosphatase activity but little effect on either pT62 or pT380 (Fig. 3C). Together, these data suggest that S384 is the crucial cyclin E phosphosite regulated by dephosphorylation.
Because key factors influencing cellular phosphatase regulation are not recapitulated in vitro (e.g., subcellular localization), we used gain-of-function and loss-of-function approaches to study the role of PP2A-B55 and PP2A-B56 complexes in S384 phosphorylation in vivo. First, we cotransfected Myc-cyclin E and hemagglutinin (HA)-CDK2 with FLAG-tagged B55 and B56 family subunits and found that most of the overexpressed B56 subunits (B56α, B56β, B56γ, and B56ε), but none of the overexpressed B55 proteins, affected the amount of pS384 (Fig. 3D). Because reduced S384 phosphorylation could reflect the amount cyclin E-CDK2 activity, rather than pS384 dephosphorylation, we confirmed that B56 overexpression did not impair cyclin E-CDK2 binding (with the exception of B56β) or cyclin H-CDK7 (CAK)-mediated activating phosphorylation of CDK2 (visualized as the faster-migrating CDK2 band).
To study how endogenous PP2A complexes regulate pS384, we used siRNA pools to knock down the B55 and B56 families in their entirety to help overcome functional redundancies between regulatory subunits, as suggested by our results (Fig. 3A to C) and previous studies (41–43). Both PP2A-B55 and PP2A-B56 complexes are required for mitotic exit, and their inhibition causes mitotic arrest (44–46). We therefore synchronized HeLa-shFBXW7 cells at the G1/S transition using the double-thymidine protocol shown in Fig. 4A to specifically study pS384 regulation by PP2A in interphase cells. Strikingly, depletion of endogenous B56 subunits (siB56 pool 1) increased endogenous cyclin E S384 phosphorylation, whereas B55 family knockdown (siB55 pool) had no effect (Fig. 4B). Each pool effectively silenced their respective subunits, as shown by representative B subunits in Fig. 4B and D and also by FLAG-tagged overexpressed subunits (data not shown). siB56 pool 1 resulted in a small fraction (10%) of G2/M-arrested cells (Fig. 4E). To ensure that these effects were not due to off-target effects of the siRNA pool, we also used an independent pool of siRNAs targeting each of the PP2A-B56 subunits (siB56 pool 2) and again observed an increase in endogenous cyclin E S384 phosphorylation in HeLa-shFBXW7 cells (Fig. 4C). Thus, while multiple PP2A complexes exhibit S384 phosphatase activity in vitro, only B56-containing holoenzymes regulate pS384 in vivo.
FIG 4.

PP2A-B56 controls cyclin E kinase activity and protein stability. (A) Schematic of the experimental protocol used for the other panels. Cells were seeded and transfected with siRNAs on the next day (time zero). Cells were then synchronized at the G1/S transition using a double-thymidine block, and endpoint assays were conducted approximately 51 h after siRNA transfection. (B) HeLa-shFBXW7 cells were transfected with siRNAs targeting all four PP2A-B55 regulatory subunits (siB55 pool), all five PP2A-B56 regulatory subunits (siB56 pool 1), or a negative-control siRNA (siAllstar control). Following transfection, the cells were synchronized as depicted in panel A, and changes in S384 phosphorylation were assayed by Western blotting. To measure siRNA efficacy, the abundances of endogenous B55α and B56γ isoforms were analyzed by Western blotting. The asterisk indicates a nonspecific band that cross-reacts with the B56γ antibody. (C) HeLa-shFBXW7 cells were transfected with an independent set of siRNAs targeting all five PP2A-B56 regulatory subunits (siB56 pool 2) or a negative-control siRNA (siAllstar control) and processed as described for panel B. Numbers correspond to relative cyclin E S384 phosphorylation normalized to total immunoprecipitated cyclin E. (D) HeLa cells were transfected with siRNAs targeting all four PP2A-B55 regulatory subunits (siB55 pool), all five PP2A-B56 regulatory subunits (siB56 pool 1), or a negative-control siRNA (siAllstar control). Following transfection, cells were synchronized as depicted in panel A, released for 2 h, and assayed for cyclin E protein abundance and kinase activity. To assay for changes in the amount of bound CDK2, CDK2 inhibitory phosphorylation, and bound p27, we performed cyclin E immunoprecipitations after normalizing the input for the amount of cyclin E present in the whole-cell extract. To measure siRNA efficacy, the abundances of multiple B55 (B55α and B55δ) and B56 (B56α and B56γ) isoforms were analyzed by Western blotting. An asterisk indicates a nonspecific band that cross-reacts with the B56γ antibody. (E) Cell cycle fluorescence-activated cell sorter (FACS) analysis of samples treated as for panel D. DNA was stained using propidium iodide (PI). (F) Analysis of CCNE1 transcript levels in cells treated as for panel D. Transcripts were normalized to ACTB expression. Data are means ± SEMs of two independent biological replicates. (G) Endogenous cyclin E half-life was measured using a [35S]Met pulse-chase in cells synchronized at G1/S, as depicted in panel A.
PP2A-B56 regulates cyclin E kinase activity and stability.
A large fraction of cellular cyclin E is sequestered in inactive complexes with CDK2 due to the binding of Cip/Kip CDK inhibitor proteins or CDK2 inhibitory phosphorylation. Because cyclin E cannot be phosphorylated at S384 within these inactive complexes, only cyclin E bound to active CDK2 is regulated by SCFFbw7. Thus, modulating Fbw7-dependent cyclin E degradation has a substantial effect on cyclin E-associated kinase activity, which is a readout of only the active pool of cyclin E. In contrast, steady-state cyclin E abundance and half-life, which reflect both the active and inactive pools, are changed less substantially. We used the synchronization protocol (Fig. 4A) and siRNA pools described above to determine whether PP2A-B55 and PP2A-B56 regulate cyclin E activity and stability at the G1/S transition. PP2A-B56 knockdown greatly decreased cyclin E-associated kinase activity and modestly decreased cyclin E abundance, while knockdown of the B55 subunits slightly increased kinase activity and did not affect cyclin E abundance (Fig. 4D). B55 and B56 family knockdown each modestly decreased cyclin E mRNA abundance (Fig. 4F), while neither affected the amount of CDK2 bound to cyclin E, the amount of CDK2-inhibitory phosphorylation (pY15), or the amount of bound p27 (in cyclin E immunoprecipitates that were normalized for the different amounts of cyclin E in the input) (Fig. 4D). Finally, we used a [35S]Met metabolic pulse-chase to determine the effect of B56 depletion on endogenous cyclin E stability. As predicted, inhibition of PP2A-B56 decreased cyclin E half-life (from 6.7 h to 3.6 h) in siB56 pool cells (Fig. 4G). However, because this half-life reflects the stability of both active and inactive cyclin E complexes, it underrepresents the full impact of PP2A-B56 on Fbw7-mediated cyclin E degradation. In summary, PP2A-B56 regulates cyclin E S384 phosphorylation, cyclin E-CDK2 activity, and cyclin E stability at the G1/S transition.
S384 phosphatase activity varies throughout the cell cycle.
PP2A-B56 complexes are inhibited at the start of mitosis before being reactivated by the PP1 phosphatase to initiate mitotic exit in fission yeast (46), and mitotic inhibition of PP2A-B56 complexes in human cells has also been reported (47). To test for differential S384 phosphatase activity at different points of the cell cycle, we treated either asynchronous or nocodazole-arrested Hct116 Fbw7−/− cells with roscovitine and analyzed the kinetics of S384 dephosphorylation in asynchronously proliferating versus prometaphase-arrested cells (Fig. 5A). As we observed previously, asynchronous cells rapidly dephosphorylated S384 upon inhibition of CDK2. However, we observed almost no dephosphorylation of S384 in the nocodazole-arrested cells, indicating that phosphatase activity toward S384 is markedly lower in prometaphase than in interphase. Similar results were obtained when we repeated this experiment in nocadazole-arrested HeLa-shFBXW7 cells (Fig. 5B). Finally, to ensure that this was not an artifact of activating the spindle checkpoint via nocodazole treatment, we also analyzed cyclin E S384 dephosphorylation in Hct116 Fbw7−/− cells that were allowed to enter mitosis unperturbed following release from a double-thymidine block. After the cells were treated with roscovitine for 10 min, mitotic cells were harvested by shake-off (Fig. 5B). Again, we observed markedly less S384 dephosphorylation in mitotic cells than in asynchronous cultures. Thus, S384 dephosphorylation—which protects cyclin E from autocatalytically stimulated degradation—is highest in the portions of the cell cycle in which cyclin E-CDK2 activity is needed and lowest in mitosis.
FIG 5.
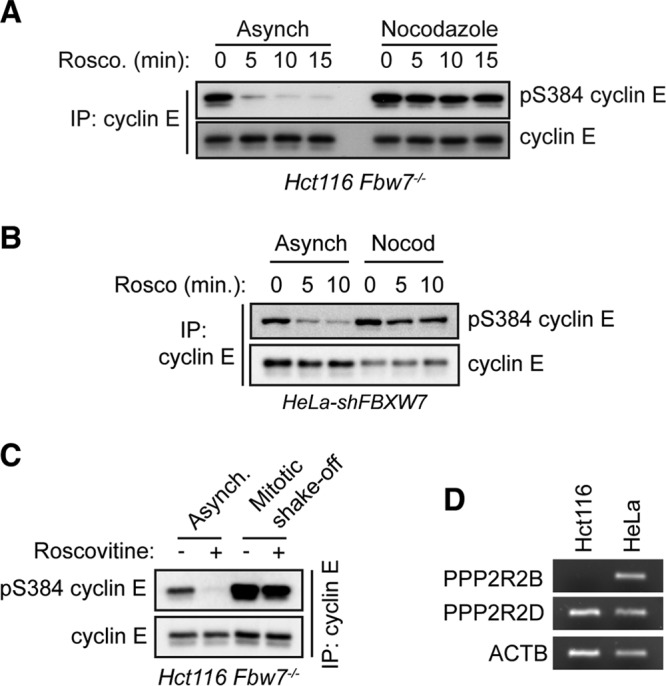
Phosphatase activity toward S384 is decreased in mitosis. (A) Hct116 Fbw7−/− cells were grown as asynchronous cultures or arrested in prometaphase by adding nocodazole for 18 h. The CDK1/2 inhibitor roscovitine was then added for up to 15 min before harvesting the cells. Endogenous cyclin E was immunoprecipitated, and changes in pS384 were analyzed by Western blotting. (B) HeLa-shFBXW7 cells were treated and processed as described for panel A. (C) Hct116 Fbw7−/− cells were either grown asynchronously or harvested by mitotic shake-off, following a double-thymidine block and release. Prior to harvesting, cells were treated for 10 min with roscovitine. Changes in endogenous cyclin E S384 phosphorylation were measured by immunoprecipitation followed by Western blotting. (D) Semiquantitative RT-PCR analysis of PPP2R2B (B55β), PPP2R2D (B55δ), and ACTB mRNA expression in Hct116 and HeLa cells.
DISCUSSION
In this study, we have shown that PP2A-B56 specifically dephosphorylates cyclin E at S384, which is the phosphorylation site that links cyclin E activity to its degradation. Preventing S384 dephosphorylation by inactivating PP2A-B56 decreased cyclin E-associated kinase activity and shortened its half-life, which is consistent with the increased degradation of catalytically active cyclin E. Dephosphorylation of S384 thus provides a solution to the paradox of how cyclin E evades autocatalytically mediated destruction and can phosphorylate its substrates.
Our finding that mitotic cells have low S384 phosphatase activity is consistent with other reports showing that PP2A-B56 phosphatases are repressed during mitosis (46, 47). Additionally, we have previously shown that Fbw7-null cells have high cyclin E-CDK2 specific activity in mitosis due to decreased inhibitory phosphorylation of CDK2 (27). Together, the combined effects of maximal S384 autophosphorylation by CDK2 and decreased S384 dephosphorylation by PP2A-B56 may synergistically protect mitotic cells against inappropriate cyclin E-CDK2 activity, which causes chromosome segregation failures and aneuploidy (11).
In contrast with our work, a previous study found that PP2A-B55β dephosphorylates all three cyclin E CPD phosphosites (pT62, pT380, and pS384) and stabilizes cyclin E (48). While we detected phosphatase activity of PP2A-B55 complexes toward S384 in vitro, we found that only B56 regulatory subunits affected S384 phosphorylation and cyclin E-CDK2 kinase activity in vivo (Fig. 3B and D and 4B and C). Moreover, Hct116 cells contain robust S384 phosphatase activity (Fig. 1C and D) but have been reported not to express the B55β isoform due to promoter hypermethylation at the PPP2R2B gene (49). We also confirmed the lack of B55β mRNA in Hct116 cells experimentally (Fig. 5D). Thus, a phosphatase other than PP2A-B55β must regulate cyclin E dephosphorylation in Hct116 cells. However, because the reasons underlying the discrepancies between our studies remain unclear, we cannot preclude roles for PP2A-B55β in cyclin E regulation in other contexts. Finally, while S384 is autophosphorylated only by CDK2, T62 and T380 are phosphorylated by multiple cellular kinases, even when cyclin E is in an inactive CDK2 complex (20). The difference between general cyclin E CPD dephosphorylation and specific S384 dephosphorylation suggests that PP2A-B56 functions to specifically stabilize cyclin E within active CDK2 complexes. Cell cycle-regulated cyclin E S384 dephosphorylation, in turn, provides a mechanism to protect cyclin E from autocatalytic degradation specifically when its activity is required, and we speculate that this is an important mechanism to reinforce cyclin E periodicity during the cell cycle.
The cyclin E gene (CCNE1) is commonly amplified in human cancers, including serous ovarian adenocarcinomas and basal breast cancers (13, 14, 50). Because overexpression of B56 regulatory subunits might be expected to phenocopy cyclin E overexpression in tumorigenesis, we used cBioportal to examine TCGA ovarian and breast cancer data sets for amplifications of CCNE1 and the B56 subunits (13, 14, 51, 52). As shown in Fig. 6, PP2A-B56 subunits are amplified in cancers from both organ sites, and the amplifications of the various B56 subunits are nearly mutually exclusive of one another. This suggests that they may, in part, function redundantly. Intriguingly, there is also a strong tendency toward mutual exclusivity with CCNE1 amplifications. While merely correlative, these data raise the possibility that amplifications of B56 subunits contribute to tumorigenesis and that this may, in part, involve cyclin E deregulation. While overexpression of PPP2R5D did not drive S384 dephosphorylation in 293A cells (Fig. 3D), it is possible that this PP2A subunit regulates cyclin E phosphorylation in other cell types and/or in tumors with PPP2R5D amplifications. Several lines of evidence have implicated PP2A as a tumor suppressor, including its opposition of many progrowth signaling pathways, inactivation by the simian virus 40 (SV40) small T viral oncoprotein, and the frequent mutation of the A scaffold subunit in cancer (53–58). The finding that PP2A-B56 complexes promote cyclin E activity at the G1/S transition and are amplified in primary human tumors suggests that in some contexts, a subset of PP2A complexes may alternatively function as oncogenes. Thus, to fully understand the multifaceted roles of PP2A in tumorigenesis, it will be necessary to continue to characterize the molecular pathways regulated by the large number of unique PP2A complexes and the cellular contexts of their regulation.
FIG 6.

CCNE1 and B56 subunits are amplified in breast and ovarian cancers. (A) The cBioPortal online interface (51, 52) was used to analyze amplifications of CCNE1 and PP2A-B56 subunits in the TCGA ovarian cancer data set (14). Individual patient samples are presented in columns. Percentages represent fraction of patients in study that had an alteration in each gene. (B) Same analysis as panel A, except for the TCGA breast cancer data set (13).
MATERIALS AND METHODS
Cell lines, plasmids, and drug treatments.
All cells were maintained in Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and penicillin-streptomycin. Myc-cyclin E and HA-CDK2 have been described previously (59). All PP2A B subunits were PCR amplified from either a collection of green fluorescent protein (GFP)-tagged expression constructs (a gift from T. Kapoor, Rockefeller University) as previously described (41) or HEK293A cDNA, cloned into pCS2-3×FLAG vectors, and sequence verified. Transient transfections of plasmids were performed using the calcium phosphate precipitation method as described previously (60). Tautomycetin (Tocris) and okadaic acid (Santa Cruz Biotechnology) were used at 10 nM, roscovitine (Sigma) was used at 25 μM or 10 μM (Fig. 5), and calyculin A (Cell Signaling Technology) was used at 50 nM.
RNAi.
FBXW7 shRNA was obtained from Open Biosystems (pGIPZ backbone; sense strand; 5′-CAGAGAAATTGCTTGCTTT-3′), and lentiviral particles were produced in HEK293T cells by cotransfection of pGIPZ, psPAX, and pMD2.G (obtained from Addgene). The following siRNAs were purchased from Qiagen: Allstar negative control, PPP2CA_1 (sense strand; 5′-GGAACUUGACGAUACUCUATT-3′), PPP2CA_2 (sense strand; 5′-CAAACAAUCAUUGGAGCUUAATT-3′), PPP2CB_1 (sense strand; 5′-GGAAUUAGAUGACACUUUATT-3′), and PPP2CB_2 (sense strand; 5′-CCGACAAAUUACCCAAGUAUATT-3′). siRNAs used in the siB55 pool were purchased from Qiagen: PPP2R2A (sense strand; 5′-CUGCAGAUGAUUUGCGGAUUATT-3′), PPP2R2B (sense strand; 5′-CCGGAAGAUCCAAGCAACAGATT-3′), PPP2R2C (sense strand; 5′-CGCUCAUUCUUCUCGGAAATT-3′), and PPP2R2D (sense strand; 5′-UUCAUCCAUAUCCGAUGUAAATT-3′). siRNAs used in siB56 pool 1 have been described previously (41) and were ordered from IDT. siB56 pool 2 siRNAs were ordered from Qiagen and targeted the following sequences: PPP2R5A (5′-CTGTATCATGGCCATAGTATA-3′), PPP2R5B (5′-CCGCATGATCTCAGTGAATAT-3′), PPP2R5C (5′-AACGAGCTGCTTTAAGTGAAA-3′), PPP2R5D (5′-ACGGGCCGAGATGCCCTATAA-3′), and PPP2R5E (5′-CACCGGGATTGCAAATCTAAT-3′). siRNAs used for PPP family catalytic subunit and PP2A-B subunit in vitro screens were comprised of pools of four independent siRNAs targeting each gene (Qiagen). Sequences are provided in Table S1 in the supplemental material. All siRNA transfections were performed using Lipofectamine RNAiMax (Life Technologies) by following the manufacturer's protocol.
Antibodies.
Antibodies were purchased from suppliers as follows: Santa Cruz Biotechnology, cyclin E IP (HE111; mouse monoclonal), cyclin E WB (HE12; mouse monoclonal; 1:1,000), CDK2 (M2; rabbit polyclonal; 1:1,000), p27 (C-19; rabbit polyclonal; 1:500), and γ-tubulin (C-20; goat polyclonal; 1:1,000); BD Biosciences, PP2A-C (610555; mouse monoclonal; 1:20,000); Calbiochem, CDK1/2 pY15 (219440; rabbit polyclonal; 1:1,000); Cell Signaling Technology, B55α (5689; mouse monoclonal; 1:1,000); Thermo Fisher, B55δ (PA5-30763; rabbit polyclonal; 1:500); Sigma, FLAG (M2; mouse monoclonal; 1:4,000) and α-tubulin (DM1A; mouse monoclonal, 1:1,000); and Bethyl Antibodies, PP1α (A300-904A; rabbit polyclonal; 1:1,000), PP1β (A300-905A; rabbit polyclonal; 1:1,000), PP1γ (A300-906A; rabbit polyclonal; 1:1,000), PP4 (A300-835A; rabbit polyclonal; 1:1,000), PP5C (A300-909A; rabbit polyclonal; 1:1,000), PP6C (A300-844A; rabbit polyclonal; 1:1,000), B56α (A300-967A; rabbit polyclonal; 1:1,000), B56γ (A303-814A; rabbit polyclonal; 1:1,000), Fbw7 IP (A301-721A; rabbit polyclonal), and Fbw7 WB (A301-702A; rabbit polyclonal; 1:1,000). Cyclin E phospho-specific rabbit polyclonal antibodies for T62, T380, and S384 were developed by PhosphoSolutions as described previously (20, 27, 59). 9E10 hybridoma supernatant was used at 1:5.
Western blotting.
Cells were harvested in NP-40 lysis buffer (50 mM Tris [pH 8.0], 150 mM NaCl, 0.5% NP-40, 1 mM dithiothreitol [DTT]) supplemented with protease and phosphatase inhibitor cocktail, with the following exceptions. Lysates collected for in vitro dephosphorylation assays were harvested in lysis buffer containing only protease inhibitors. Cells harvested for PP2A complex purification were lysed in Tween 20 lysis buffer (50 mM Tris [pH 7.5], 150 mM NaCl, 0.1% Tween 20, 10% glycerol, 1 mM EDTA, 2.5 mM EGTA, and 1 mM DTT) supplemented with a protease inhibitor cocktail. All lysates for Western blotting were prepared in Laemmli sample buffer, boiled, and resolved on polyacrylamide gels before semidry transfer to polyvinylidene difluoride (PVDF) membranes. All membranes were blocked in 5% milk–Tris-buffered saline with Tween 20 (TBST) and probed with primary antibodies from 1 h at room temperature to overnight at 4°C. Horseradish peroxidase (HRP)-conjugated secondary antibodies were prepared in blocking solution at 1:10,000.
In vitro dephosphorylation assays.
In vitro dephosphorylation assays using whole-cell lysates were performed by incubating cell extracts with either recombinant glutathione S-transferase (GST)–cyclin E-CDK2 (Fig. 1F, 2A to C, and 3B) or immunoprecipitated Myc-cyclin E/HA-CDK2 complexes (Fig. 3A) at 30°C for up to 60 min, with agitation. Assays testing the activity of specific PP2A complexes were performed by immunoprecipitating PP2A holoenzymes from cell lysates transfected with FLAG-tagged B subunits. These immunoprecipitates were washed twice in lysis buffer and once in phosphatase reaction buffer (50 mM Tris [pH 7.4], 150 mM NaCl, 1 mM MnCl2, and 5 mM MgCl2, supplemented with a protease inhibitor cocktail) and then incubated with recombinant GST-cyclin E-CDK2 in phosphatase reaction buffer at 30°C for up to 60 min. All reactions were quenched by the addition of 4× Laemmli sample buffer, boiled, and assayed by Western blotting.
qRT-PCR.
Total RNA was extracted from cells using TRIzol and purified with an RNA miniprep kit (Zymo Research). RNA was quantified and equal amounts were DNased before reverse transcription using a high-capacity cDNA reverse transcription kit (Applied Biosystems). Quantitative reverse transcription-PCRs (qRT-PCRs) were performed using transcript-specific primer probe sets (Applied Biosystems) (Fig. 4D) or transcript-specific SYBR primer sets (IDT) (Fig. 5D) and 2× Taq universal PCR master mix (Applied Biosystems) on a QuantStudio 5 instrument. Transcripts were normalized using ACTB as an endogenous control. Primer sequences for Fig. 5D are as follows: for PPP2R2B, 5′-AGGACATTGATACCCGCAAA (forward [Fwd]) and 5′-AATTCTCCCGTGTGGTTGAA (reverse [Rev]); for PPP2R2D, 5′-TGCGACAGACACTCCAAGTT (Fwd) and 5′-CGCCCACTATGACTGAATTT (Rev); and for ACTB, 5′-GCACAGAGCCTCGCCTT (Fwd) and 5′-GTTGTCGACGACGAGCG (Rev).
Cyclin E kinase assays and [35S]Met pulse-chase.
Kinase assays were performed by immunoprecipitating endogenous cyclin E from cell lysates for 2 h at 4°C. IPs were washed twice in lysis buffer and once in kinase assay buffer (50 mM HEPES [pH 7.4], 10 mM MgCl2, 1 mM DTT) and incubated at 30°C for 30 min in a 20-μl reaction mixture containing 30 μM ATP, 2 μCi of [γ-32P]ATP, and histone H1 as a substrate. Reactions were quenched by the addition of sample buffer, boiled, resolved on polyacrylamide gels, and exposed to film. Endogenous cyclin E half-life was measured by [35S]Met pulse-chase as previously described (61).
Flow cytometry.
For cell cycle flow cytometry analysis, cells were fixed in 90% ethanol before staining with a propidium iodide-RNase A solution and processed on a CANTO II flow cytometer (Becton Dickinson). Data were analyzed using FlowJo.
Supplementary Material
ACKNOWLEDGMENTS
We thank members of the Clurman Lab for helpful discussions and feedback on the work.
This work was supported by NIH grants R01 CA193808-02 and R21 CA187324-02 (B.E.C.) and by the National Science Foundation Graduate Research Fellowship Program under grant numbers DGE-0718124 and DGE-1256082 (R.J.D.).
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
We declare that we have no conflicts of interest regarding the contents of this article.
R.J.D. contributed to research direction, designed and performed most of the experiments, and wrote the manuscript. J.S. performed experiments and contributed to the manuscript. B.T.H. performed experiments and contributed to the research direction. B.E.C. led the research, designed experiments, and contributed to the manuscript.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/MCB.00657-16.
REFERENCES
- 1.Siu KT, Rosner MR, Minella AC. 2012. An integrated view of cyclin E function and regulation. Cell Cycle 11:57–64. doi: 10.4161/cc.11.1.18775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hwang HC, Clurman BE. 2005. Cyclin E in normal and neoplastic cell cycles. Oncogene 24:2776–2786. doi: 10.1038/sj.onc.1208613. [DOI] [PubMed] [Google Scholar]
- 3.Chi Y, Welcker M, Hizli AA, Posakony JJ, Aebersold R, Clurman BE. 2008. Identification of CDK2 substrates in human cell lysates. Genome Biol 9:R149. doi: 10.1186/gb-2008-9-10-r149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Geng Y, Lee YM, Welcker M, Swanger J, Zagozdzon A, Winer JD, Roberts JM, Kaldis P, Clurman BE, Sicinski P. 2007. Kinase-independent function of cyclin E. Mol Cell 25:127–139. doi: 10.1016/j.molcel.2006.11.029. [DOI] [PubMed] [Google Scholar]
- 5.Matsumoto Y, Maller JL. 2004. A centrosomal localization signal in cyclin E required for Cdk2-independent S phase entry. Science 306:885–888. doi: 10.1126/science.1103544. [DOI] [PubMed] [Google Scholar]
- 6.Spruck CH, Won KA, Reed SI. 1999. Deregulated cyclin E induces chromosome instability. Nature 401:297–300. doi: 10.1038/45836. [DOI] [PubMed] [Google Scholar]
- 7.Ohtsubo M, Roberts JM. 1993. Cyclin-dependent regulation of G1 in mammalian fibroblasts. Science 259:1908–1912. doi: 10.1126/science.8384376. [DOI] [PubMed] [Google Scholar]
- 8.Minella AC, Grim JE, Welcker M, Clurman BE. 2007. p53 and SCFFbw7 cooperatively restrain cyclin E-associated genome instability. Oncogene 26:6948–6953. doi: 10.1038/sj.onc.1210518. [DOI] [PubMed] [Google Scholar]
- 9.Bester AC, Roniger M, Oren YS, Im MM, Sarni D, Chaoat M, Bensimon A, Zamir G, Shewach DS, Kerem B. 2011. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 145:435–446. doi: 10.1016/j.cell.2011.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Costantino L, Sotiriou SK, Rantala JK, Magin S, Mladenov E, Helleday T, Haber JE, Iliakis G, Kallioniemi OP, Halazonetis TD. 2014. Break-induced replication repair of damaged forks induces genomic duplications in human cells. Science 343:88–91. doi: 10.1126/science.1243211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Keck JM, Summers MK, Tedesco D, Ekholm-Reed S, Chuang LC, Jackson PK, Reed SI. 2007. Cyclin E overexpression impairs progression through mitosis by inhibiting APC(Cdh1). J Cell Biol 178:371–385. doi: 10.1083/jcb.200703202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Minella AC, Swanger J, Bryant E, Welcker M, Hwang H, Clurman BE. 2002. p53 and p21 form an inducible barrier that protects cells against cyclin E-cdk2 deregulation. Curr Biol 12:1817–1827. doi: 10.1016/S0960-9822(02)01225-3. [DOI] [PubMed] [Google Scholar]
- 13.Cancer Genome Atlas Network. 2012. Comprehensive molecular portraits of human breast tumours. Nature 490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cancer Genome Atlas Research Network. 2011. Integrated genomic analyses of ovarian carcinoma. Nature 474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Loeb KR, Kostner H, Firpo E, Norwood T, Tsuchiya DK, Clurman BE, Roberts JM. 2005. A mouse model for cyclin E-dependent genetic instability and tumorigenesis. Cancer Cell 8:35–47. doi: 10.1016/j.ccr.2005.06.010. [DOI] [PubMed] [Google Scholar]
- 16.Smith AP, Henze M, Lee JA, Osborn KG, Keck JM, Tedesco D, Bortner DM, Rosenberg MP, Reed SI. 2006. Deregulated cyclin E promotes p53 loss of heterozygosity and tumorigenesis in the mouse mammary gland. Oncogene 25:7245–7259. doi: 10.1038/sj.onc.1209713. [DOI] [PubMed] [Google Scholar]
- 17.Koepp DM, Schaefer LK, Ye X, Keyomarsi K, Chu C, Harper JW, Elledge SJ. 2001. Phosphorylation-dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science 294:173–177. doi: 10.1126/science.1065203. [DOI] [PubMed] [Google Scholar]
- 18.Strohmaier H, Spruck CH, Kaiser P, Won KA, Sangfelt O, Reed SI. 2001. Human F-box protein hCdc4 targets cyclin E for proteolysis and is mutated in a breast cancer cell line. Nature 413:316–322. doi: 10.1038/35095076. [DOI] [PubMed] [Google Scholar]
- 19.Clurman BE, Sheaff RJ, Thress K, Groudine M, Roberts JM. 1996. Turnover of cyclin E by the ubiquitin-proteasome pathway is regulated by cdk2 binding and cyclin phosphorylation. Genes Dev 10:1979–1990. doi: 10.1101/gad.10.16.1979. [DOI] [PubMed] [Google Scholar]
- 20.Welcker M, Singer J, Loeb KR, Grim J, Bloecher A, Gurien-West M, Clurman BE, Roberts JM. 2003. Multisite phosphorylation by Cdk2 and GSK3 controls cyclin E degradation. Mol Cell 12:381–392. doi: 10.1016/S1097-2765(03)00287-9. [DOI] [PubMed] [Google Scholar]
- 21.Moberg KH, Bell DW, Wahrer DC, Haber DA, Hariharan IK. 2001. Archipelago regulates cyclin E levels in Drosophila and is mutated in human cancer cell lines. Nature 413:311–316. doi: 10.1038/35095068. [DOI] [PubMed] [Google Scholar]
- 22.Won KA, Reed SI. 1996. Activation of cyclin E/CDK2 is coupled to site-specific autophosphorylation and ubiquitin-dependent degradation of cyclin E EMBO J 15:4182–4193. [PMC free article] [PubMed] [Google Scholar]
- 23.Nash P, Tang X, Orlicky S, Chen Q, Gertler FB, Mendenhall MD, Sicheri F, Pawson T, Tyers M. 2001. Multisite phosphorylation of a CDK inhibitor sets a threshold for the onset of DNA replication. Nature 414:514–521. doi: 10.1038/35107009. [DOI] [PubMed] [Google Scholar]
- 24.Davis RJ, Welcker M, Clurman BE. 2014. Tumor suppression by the Fbw7 ubiquitin ligase: mechanisms and opportunities. Cancer Cell 26:455–464. doi: 10.1016/j.ccell.2014.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Siu KT, Xu Y, Swartz KL, Bhattacharyya M, Gurbuxani S, Hua Y, Minella AC. 2014. Chromosome instability underlies hematopoietic stem cell dysfunction and lymphoid neoplasia associated with impaired Fbw7-mediated cyclin E regulation. Mol Cell Biol 34:3244–3258. doi: 10.1128/MCB.01528-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu Y, Swartz KL, Siu KT, Bhattacharyya M, Minella AC. 2014. Fbw7-dependent cyclin E regulation ensures terminal maturation of bone marrow erythroid cells by restraining oxidative metabolism. Oncogene 33:3161–3171. doi: 10.1038/onc.2013.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grim JE, Gustafson MP, Hirata RK, Hagar AC, Swanger J, Welcker M, Hwang HC, Ericsson J, Russell DW, Clurman BE. 2008. Isoform- and cell cycle-dependent substrate degradation by the Fbw7 ubiquitin ligase. J Cell Biol 181:913–920. doi: 10.1083/jcb.200802076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ekholm-Reed S, Spruck CH, Sangfelt O, van Drogen F, Mueller-Holzner E, Widschwendter M, Zetterberg A, Reed SI, Reed SE. 2004. Mutation of hCDC4 leads to cell cycle deregulation of cyclin E in cancer. Cancer Res 64:795–800. doi: 10.1158/0008-5472.CAN-03-3417. [DOI] [PubMed] [Google Scholar]
- 29.Minella AC, Loeb KR, Knecht A, Welcker M, Varnum-Finney BJ, Bernstein ID, Roberts JM, Clurman BE. 2008. Cyclin E phosphorylation regulates cell proliferation in hematopoietic and epithelial lineages in vivo. Genes Dev 22:1677–1689. doi: 10.1101/gad.1650208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hao B, Oehlmann S, Sowa ME, Harper JW, Pavletich NP. 2007. Structure of a Fbw7-Skp1-cyclin E complex: multisite-phosphorylated substrate recognition by SCF ubiquitin ligases. Mol Cell 26:131–143. doi: 10.1016/j.molcel.2007.02.022. [DOI] [PubMed] [Google Scholar]
- 31.Arnold HK, Sears RC. 2006. Protein phosphatase 2A regulatory subunit B56alpha associates with c-myc and negatively regulates c-myc accumulation. Mol Cell Biol 26:2832–2844. doi: 10.1128/MCB.26.7.2832-2844.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yeh E, Cunningham M, Arnold H, Chasse D, Monteith T, Ivaldi G, Hahn WC, Stukenberg PT, Shenolikar S, Uchida T, Counter CM, Nevins JR, Means AR, Sears R. 2004. A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat Cell Biol 6:308–318. doi: 10.1038/ncb1110. [DOI] [PubMed] [Google Scholar]
- 33.McCourt P, Gallo-Ebert C, Gonghong Y, Jiang Y, Nickels JT. 2013. PP2A(Cdc55) regulates G1 cyclin stability. Cell Cycle 12:1201–1210. doi: 10.4161/cc.24231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shi Y. 2009. Serine/threonine phosphatases: mechanism through structure. Cell 139:468–484. doi: 10.1016/j.cell.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 35.Wlodarchak N, Xing Y. 2016. PP2A as a master regulator of the cell cycle. Crit Rev Biochem Mol Biol 51:162–184. doi: 10.3109/10409238.2016.1143913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wurzenberger C, Gerlich DW. 2011. Phosphatases: providing safe passage through mitotic exit. Nat Rev Mol Cell Biol 12:469–482. doi: 10.1038/nrm3149. [DOI] [PubMed] [Google Scholar]
- 37.Xu Y, Chen Y, Zhang P, Jeffrey PD, Shi Y. 2008. Structure of a protein phosphatase 2A holoenzyme: insights into B55-mediated Tau dephosphorylation. Mol Cell 31:873–885. doi: 10.1016/j.molcel.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu Y, Xing Y, Chen Y, Chao Y, Lin Z, Fan E, Yu JW, Strack S, Jeffrey PD, Shi Y. 2006. Structure of the protein phosphatase 2A holoenzyme. Cell 127:1239–1251. doi: 10.1016/j.cell.2006.11.033. [DOI] [PubMed] [Google Scholar]
- 39.Swingle M, Ni L, Honkanen RE. 2007. Small-molecule inhibitors of ser/thr protein phosphatases: specificity, use and common forms of abuse. Methods Mol Biol 365:23–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mitsuhashi S, Matsuura N, Ubukata M, Oikawa H, Shima H, Kikuchi K. 2001. Tautomycetin is a novel and specific inhibitor of serine/threonine protein phosphatase type 1, PP1. Biochem Biophys Res Commun 287:328–331. doi: 10.1006/bbrc.2001.5596. [DOI] [PubMed] [Google Scholar]
- 41.Foley EA, Maldonado M, Kapoor TM. 2011. Formation of stable attachments between kinetochores and microtubules depends on the B56-PP2A phosphatase. Nat Cell Biol 13:1265–1271. doi: 10.1038/ncb2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Espert A, Uluocak P, Bastos RN, Mangat D, Graab P, Gruneberg U. 2014. PP2A-B56 opposes Mps1 phosphorylation of Knl1 and thereby promotes spindle assembly checkpoint silencing. J Cell Biol 206:833–842. doi: 10.1083/jcb.201406109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu P, Raetz EA, Kitagawa M, Virshup DM, Lee SH. 2013. BUBR1 recruits PP2A via the B56 family of targeting subunits to promote chromosome congression. Biol Open 2:479–486. doi: 10.1242/bio.20134051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schmitz MH, Held M, Janssens V, Hutchins JR, Hudecz O, Ivanova E, Goris J, Trinkle-Mulcahy L, Lamond AI, Poser I, Hyman AA, Mechtler K, Peters JM, Gerlich DW. 2010. Live-cell imaging RNAi screen identifies PP2A-B55alpha and importin-beta1 as key mitotic exit regulators in human cells. Nat Cell Biol 12:886–893. doi: 10.1038/ncb2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cundell MJ, Bastos RN, Zhang T, Holder J, Gruneberg U, Novak B, Barr FA. 2013. The BEG (PP2A-B55/ENSA/Greatwall) pathway ensures cytokinesis follows chromosome separation. Mol Cell 52:393–405. doi: 10.1016/j.molcel.2013.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grallert A, Boke E, Hagting A, Hodgson B, Connolly Y, Griffiths JR, Smith DL, Pines J, Hagan IM. 2015. A PP1-PP2A phosphatase relay controls mitotic progression. Nature 517:94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Porter IM, Schleicher K, Porter M, Swedlow JR. 2013. Bod1 regulates protein phosphatase 2A at mitotic kinetochores. Nat Commun 4:2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tan Y, Sun D, Jiang W, Klotz-Noack K, Vashisht AA, Wohlschlegel J, Widschwendter M, Spruck C. 2014. PP2A-B55β antagonizes cyclin E1 proteolysis and promotes its dysregulation in cancer. Cancer Res 74:2006–2014. doi: 10.1158/0008-5472.CAN-13-1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tan J, Lee PL, Li Z, Jiang X, Lim YC, Hooi SC, Yu Q. 2010. B55β-associated PP2A complex controls PDK1-directed myc signaling and modulates rapamycin sensitivity in colorectal cancer. Cancer Cell 18:459–471. doi: 10.1016/j.ccr.2010.10.021. [DOI] [PubMed] [Google Scholar]
- 50.Mayr D, Kanitz V, Anderegg B, Luthardt B, Engel J, Löhrs U, Amann G, Diebold J. 2006. Analysis of gene amplification and prognostic markers in ovarian cancer using comparative genomic hybridization for microarrays and immunohistochemical analysis for tissue microarrays. Am J Clin Pathol 126:101–109. doi: 10.1309/N6X5MB24BP42KP20. [DOI] [PubMed] [Google Scholar]
- 51.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C, Schultz N. 2012. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N. 2013. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6:pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kuo YC, Huang KY, Yang CH, Yang YS, Lee WY, Chiang CW. 2008. Regulation of phosphorylation of Thr-308 of Akt, cell proliferation, and survival by the B55alpha regulatory subunit targeting of the protein phosphatase 2A holoenzyme to Akt. J Biol Chem 283:1882–1892. doi: 10.1074/jbc.M709585200. [DOI] [PubMed] [Google Scholar]
- 54.Rundell K, Parakati R. 2001. The role of the SV40 ST antigen in cell growth promotion and transformation. Semin Cancer Biol 11:5–13. doi: 10.1006/scbi.2000.0341. [DOI] [PubMed] [Google Scholar]
- 55.Pallas DC, Shahrik LK, Martin BL, Jaspers S, Miller TB, Brautigan DL, Roberts TM. 1990. Polyoma small and middle T antigens and SV40 small t antigen form stable complexes with protein phosphatase 2A. Cell 60:167–176. [DOI] [PubMed] [Google Scholar]
- 56.Calin GA, di Iasio MG, Caprini E, Vorechovsky I, Natali PG, Sozzi G, Croce CM, Barbanti-Brodano G, Russo G, Negrini M. 2000. Low frequency of alterations of the alpha (PPP2R1A) and beta (PPP2R1B) isoforms of the subunit A of the serine-threonine phosphatase 2A in human neoplasms. Oncogene 19:1191–1195. doi: 10.1038/sj.onc.1203389. [DOI] [PubMed] [Google Scholar]
- 57.Wang SS, Esplin ED, Li JL, Huang L, Gazdar A, Minna J, Evans GA. 1998. Alterations of the PPP2R1B gene in human lung and colon cancer. Science 282:284–287. [DOI] [PubMed] [Google Scholar]
- 58.Chen W, Arroyo JD, Timmons JC, Possemato R, Hahn WC. 2005. Cancer-associated PP2A Aalpha subunits induce functional haploinsufficiency and tumorigenicity. Cancer Res 65:8183–8192. doi: 10.1158/0008-5472.CAN-05-1103. [DOI] [PubMed] [Google Scholar]
- 59.Ye X, Nalepa G, Welcker M, Kessler BM, Spooner E, Qin J, Elledge SJ, Clurman BE, Harper JW. 2004. Recognition of phosphodegron motifs in human cyclin E by the SCF(Fbw7) ubiquitin ligase. J Biol Chem 279:50110–50119. doi: 10.1074/jbc.M409226200. [DOI] [PubMed] [Google Scholar]
- 60.Graham FL, van der Eb AJ. 1973. A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology 52:456–467. [DOI] [PubMed] [Google Scholar]
- 61.Hughes BT, Sidorova J, Swanger J, Monnat RJ, Clurman BE. 2013. Essential role for Cdk2 inhibitory phosphorylation during replication stress revealed by a human Cdk2 knockin mutation. Proc Natl Acad Sci U S A 110:8954–8959. doi: 10.1073/pnas.1302927110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.