

Abstract
Heterozygous inactivating mutations in ribosomal protein genes (RPGs) are associated with hematopoietic and developmental abnormalities, activation of p53, and altered risk of cancer in humans and model organisms. Here we performed a large‐scale analysis of cancer genome data to examine the frequency and selective pressure of RPG lesions across human cancers. We found that hemizygous RPG deletions are common, occurring in about 43% of 10,744 cancer specimens and cell lines. Consistent with p53‐dependent negative selection, such lesions are underrepresented in TP53‐intact tumors (P ≪ 10−10), and shRNA‐mediated knockdown of RPGs activated p53 in TP53‐wild‐type cells. In contrast, we did not see negative selection of RPG deletions in TP53‐mutant tumors. RPGs are conserved with respect to homozygous deletions, and shRNA screening data from 174 cell lines demonstrate that further suppression of hemizygously deleted RPGs inhibits cell growth. Our results establish RPG haploinsufficiency as a strikingly common vulnerability of human cancers that associates with TP53 mutations and could be targetable therapeutically.
Keywords: cancer, ribosomal gene haploinsufficiency, ribosome function
Subject Categories: Cancer; Genetics, Gene Therapy & Genetic Disease
Introduction
The human ribosome consists of an rRNA scaffold and about 80 proteins, divided into two subunits. Ribosomal protein genes (RPGs) are denoted RPS1, RPS2, etc. for the small (40S) subunit; RPL1, RPL2, etc. for the large (60S) subunit.
Several recent lines of evidence hold that mutation of RPGs leads to specific clinical and cellular phenotypes. Germline heterozygous inactivating mutations or deletions in RPS19 and at least eight other RPGs cause Diamond‐Blackfan anemia (DBA), a disorder characterized by macrocytic anemia and cancer predisposition, and the founding member of a class of disorders known as ribosomopathies (Draptchinskaia et al, 1999; Gazda et al, 2006, 2008, 2012; Farrar et al, 2008; Narla & Ebert, 2010; Vlachos et al, 2012; Raiser et al, 2014). Acquired somatic mutations in specific RPGs associate with certain malignancies, including the 5q‐ subtype of myelodysplastic syndrome (hemizygous deletions of RPS14; Ebert et al, 2008), T‐cell acute lymphoblastic leukemia (mutations in RPL5, RPL10, and RPL22; Rao et al, 2012; De Keersmaecker et al, 2013), and microsatellite‐unstable endometrial and gastric cancer (mutations in RPL22; Nagarajan et al, 2012; Novetsky et al, 2013). Eleven RPGs are tumor suppressor genes in zebrafish, where hemizygous inactivation of these genes causes malignant peripheral nerve sheath tumors with high penetrance (Amsterdam et al, 2004; MacInnes et al, 2008; Lai et al, 2009). In mouse models, RPG haploinsufficiency alters stem cell quiescence (Signer et al, 2014), homeobox gene translation, and tissue patterning (Kondrashov et al, 2011; Xue et al, 2015).
Despite these observations, the occurrence of RPG lesions in human cancers has not been investigated systematically. We therefore carried out a large‐scale analysis of cancer genome data to determine the frequency and selective pressure of RPG lesions across human cancers. We first looked for chromosomal deletions and amplifications and point mutations in RPGs using pre‐existing DNA copy number microarray and whole‐exome sequencing data from a total of 10,744 cancer specimens and cell lines. Because recent studies have shown that RPG haploinsufficiency activates p53 in ribosomopathies (Narla & Ebert, 2010; Raiser et al, 2014) and the pathobiology of ribosomopathies can be alleviated by p53 inhibition (Volarevic et al, 2000; Sulic et al, 2005; Fumagalli et al, 2009; Barlow et al, 2010; Dutt et al, 2011; Jaako et al, 2011), we hypothesized that inactivation of RPGs could lead to negative selection unless the cells have mutated TP53, and, accordingly, looked for associations between inactivating RPG lesions and TP53 mutation. While we observed few point mutations and homozygous deletions in RPGs, we detected hemizygous RPG deletions in a large proportion (43%) of samples. Consistent with negative selection, further analyses revealed an underrepresentation of RPG deletions in TP53‐intact tumors, whereas we did not see any signs of negative selection in TP53‐mutant tumors. Furthermore, functional experiments showed that deficiency of frequently deleted RPGs increases p53 activity in TP53‐intact cell lines and perturbs rRNA maturation both in cell lines cultured ex vivo and in primary acute leukemia cells with specific RPG deletions and expanded in vivo in xenograft models. Finally, consistent with the low frequency of homozygous deletion, analysis of genomewide shRNA screening data showed that further suppression of hemizygously deleted RPGs inhibits the growth of RPG‐haploinsufficient cancer cells. We conclude that RPG haploinsufficiency as a common feature of human cancers that associates with TP53 mutations and could be targetable therapeutically.
Results
Haploinsufficiency for RPGs on the basis of hemizygous regional deletions is a common feature of cancer genomes
We first analyzed DNA copy number profiles of 7,225 cancer specimens belonging to 24 tumor types using data from The Cancer Genome Atlas, TCGA (Cancer Genome Atlas Research Network et al, 2013). Using log2 copy number ratio thresholds between −0.3 and −0.7 (median −0.5), we detected deletions affecting RPGs in 67–22% of specimens (median 43%), and in all tumor types analyzed (Fig 1A and Table 1). In 58–12% (median 32%) of tumors, we detected multiple RPG deletions (Fig 1C). Consistent with negative selection, we observed lower deletion frequencies for RPG than for other genes (Fig 1C and D). Validation in two independent sets of copy number profiles of 2,476 specimens belonging to 13 tumor types from Tumorscape (Beroukhim et al, 2010) and 1,043 cancer cell lines from the Cancer Cell Line Encyclopedia (CCLE; Barretina et al, 2012) identified the same RPGs as frequently deleted (Figs 1B and EV1, and Table 1). In all three data sets, almost all (97–100%) of RPG deletions had copy numbers consistent with hemizygous loss (Fig 2A), and few RPGs showed recurrent homozygous deletions (Table EV1). Throughout, RPGs were hit by regional deletions covering multiple genes, and all frequently deleted RPGs were co‐deleted with well‐known tumor suppressor genes, including RPL26 on chromosome 17p, which is always co‐deleted with TP53, and RPS6 on chromosome 9p, which is always co‐deleted with CDKN2A. In addition to copy number changes, we examined the spectrum of point mutations in RPGs using whole‐exome sequencing data from 4,655 TCGA samples. In contrast to the high frequency of hemizygous deletions, we found a low frequency of point mutations in RPGs, although we noted recurrent mutations in RPL5, RPL10, and RPL22 in several tumor types as reported previously (Nagarajan et al, 2012; Rao et al, 2012; De Keersmaecker et al, 2013; Novetsky et al, 2013; Table EV2). In summary, these data show that RPG haploinsufficiency on the basis of regional deletions occurs in a large proportion of human cancers.
Figure 1. Hemizygous deletion of RPGs is a common feature of cancer genomes.
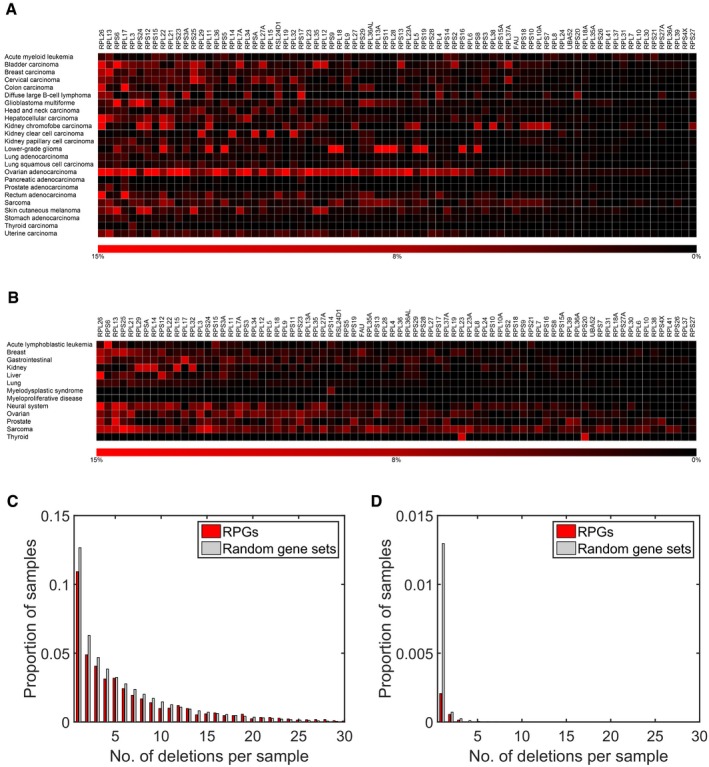
- Deletion frequencies for different RPGs estimated from DNA copy number profiles of 7,275 primary cancer specimens belonging to 24 different tumor types (red).
- Analysis of DNA copy number profiles of 2,476 primary cancer specimens belonging to 13 tumor types (including four not represented in the TCGA) identified the same RPGs as frequently deleted. Similar results were also calculated with DNA copy number profiles of 1,043 cancer cell lines (Fig EV1). The results shown were obtained using log2 ratio −0.5 as threshold. Similar results were obtained with other reasonable thresholds.
- Histogram of number of deletions per tumor for RPGs and for 1,000 equally sized random gene sets in the TCGA samples. Tumors with multiple RPG deletions were common. Consistent with negative selection, we also detected fewer deletions in RPGs than in 1,000 random gene sets of the same size. This plot was obtained with a log2 threshold of −0.5, detecting both hemizygous and homozygous deletions.
- Corresponding plot obtained with the more conservative log2 threshold −2.0, primarily detecting homozygous deletions.
Table 1.
Ten most frequently deleted RPGs
| Gene | Chr | Start (bp) | End (bp) | TCGA | CCLE | Tumorscape | |||
|---|---|---|---|---|---|---|---|---|---|
| n | % | n | % | n | % | ||||
| RPL26 | 17 | 8,280,833 | 8,286,565 | 1,752 | 24.2 | 340 | 32.6 | 310 | 12.5 |
| RPL13 | 16 | 89,627,064 | 89,633,237 | 1,430 | 19.8 | 204 | 19.6 | 222 | 9.0 |
| RPS6 | 9 | 19,376,253 | 19,380,235 | 1,411 | 19.5 | 373 | 35.8 | 269 | 10.9 |
| RPL17 | 18 | 47,014,850 | 47,018,935 | 1,335 | 18.5 | 393 | 37.7 | 173 | 7.0 |
| RPL29 | 3 | 52,027,643 | 52,029,958 | 1,283 | 17.8 | 340 | 32.6 | 229 | 9.2 |
| RPL3 | 22 | 39,708,886 | 39,715,670 | 1,205 | 16.7 | 263 | 25.2 | 152 | 6.1 |
| RPL14 | 3 | 40,498,782 | 40,503,863 | 1,133 | 15.7 | 292 | 28.0 | 209 | 8.4 |
| RPSA | 3 | 39,448,203 | 39,454,032 | 1,129 | 15.6 | 284 | 27.2 | 202 | 8.2 |
| RPL21 | 13 | 27,825,691 | 27,830,702 | 1,121 | 15.5 | 328 | 31.4 | 224 | 9.0 |
| RPS15 | 19 | 1,438,362 | 1,440,492 | 1,108 | 15.3 | 264 | 25.3 | 170 | 6.9 |
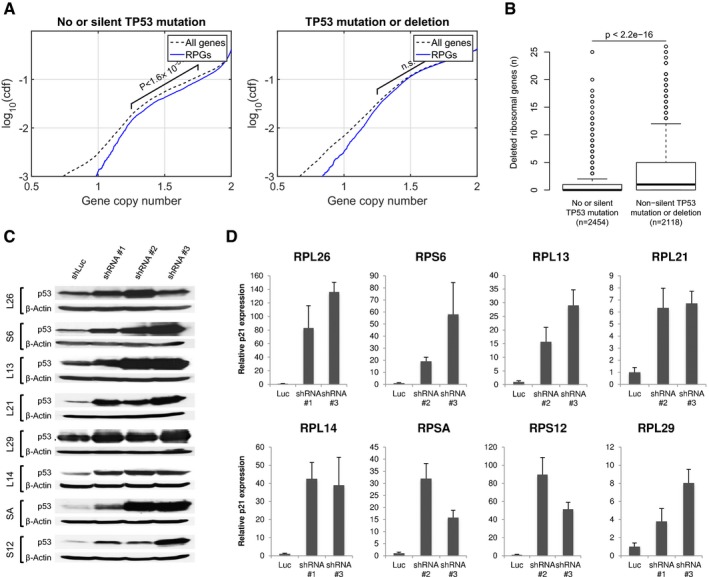
Figure EV1. RPG deletion frequencies in the CCLE data.
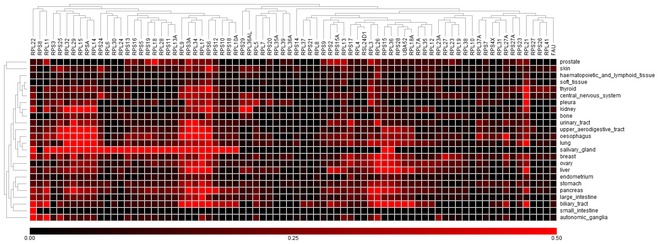
Figure 2. RPG haploinsufficiency associates with TP53 mutation status and p53 activation.
-
ARPG deletions are underrepresented in TP53‐intact tumors, but not in TP53‐mutant tumors. Plots show cumulative copy number distributions for RPGs (blue) versus all genes (dashed black) for 4,675 TCGA samples having both copy number microarray and whole‐exome sequence data. However, cases without TP53 mutation/deletion show fewer copy numbers consistent with heterozygous loss of RPGs compared to other genes (brackets), whereas cases harboring TP53 mutations/deletions show no corresponding difference. These plots also show that, regardless of TP53 mutation status, homozygous loss of RPGs is extremely rare.
-
BNumber of hemizygous RPG deletions in TP53‐mutant and ‐wild‐type tumors. Boxes indicate medians and the first and third quartiles. Whiskers indicate first and third quartiles ±1.5 times the interquartile range. Notches indicate confidence intervals around the median. P‐values indicate significance by Wilcoxon rank‐sum test.
-
C, DTo obtain further support for p53‐negative selection, we used shRNA‐mediated knockdown of RPGs in A549 cells, which are TP53‐wild type. Panels show p53 protein levels as analyzed by Western blot (β‐actin loading control), and P21 transcript levels as analyzed by qPCR (ACTB normalization control) after 4–6 days of expression of two shRNAs per tested RPG. Error bars indicate standard deviation between triplicates. Throughout, we observed increased p53 levels and P21 expression. These data support that RPG deletions lead to p53 activation and negative selection in cancer cells.
Deletion of RPGs in cancer cells is restricted by p53‐dependent negative selection
Recently, studies aimed at understanding the molecular mechanisms of ribosomopathies have identified p53 as a central mediator of the clinical features of these diseases (Narla & Ebert, 2010; Raiser et al, 2014). In both DBA and the 5q‐ syndrome, the hematopoietic phenotype is at least partly linked to p53 activation, and animal models have confirmed p53 as a sensor of ribosome dysfunction (Volarevic et al, 2000; Sulic et al, 2005; Fumagalli et al, 2009; Barlow et al, 2010; Dutt et al, 2011; Jaako et al, 2011). In normal cells, the expression of RPGs is tightly coordinated. In DBA and the 5q‐ syndrome, however, the RPG haploinsufficiency is thought to perturb the stoichiometry of ribosomal proteins, leading to inefficient ribosome assembly and increased concentrations of free ribosomal proteins, some of which (RPL5 and RPL11) regulate key components of the 5S ribonucleoprotein particle (Macias et al, 2010; Donati et al, 2013; Sloan et al, 2013; Goudarzi & Lindstrom, 2016). When ribosome biogenesis is blocked, the 5S SNP pre‐ribosomal complex is re‐directed from assembly into 60S ribosomes to MDM2 E3 ubiquitin ligase inhibition, thereby inhibiting the ability of MDM2 to target p53 for proteasomal degradation (Fumagalli et al, 2009; Zhang & Lu, 2009; Deisenroth & Zhang, 2010; Miliani de Marval & Zhang, 2011; Zhou et al, 2013; Goudarzi & Lindstrom, 2016). Moreover, in vitro and in vivo studies support that the phenotypic effects of RPG haploinsufficiency can be alleviated by genetic or pharmacological inhibition of p53 (Volarevic et al, 2000; Sulic et al, 2005; Fumagalli et al, 2009; Barlow et al, 2010; Dutt et al, 2011; Jaako et al, 2011).
Because of these reports, we hypothesized that acquisition of RPG deletions in cancer cells could lead to p53 activation and thereby negative selection, unless the p53 pathway has been inactivated. To test this hypothesis, we compared the copy number distributions for RPGs versus other genes across 4,675 TCGA samples having both copy number microarray and whole‐exome sequence data, enabling the detection of RPG deletions as well as deletions and point mutations in TP53. We found an underrepresentation of hemizygous RPG deletions in TP53‐wild‐type tumors (Wilcoxon rank‐sum P ≪ 10−10), whereas RPGs and other genes showed identical copy number distributions in TP53‐mutant tumors (Fig 2A). Similarly, among TP53‐wild‐type tumors, but not among TP53‐mutant tumors, we found fewer cases with hemizygous deletions affecting RPGs than with hemizygous deletions affecting random gene sets of the same size (permutation testing P = 0.04 with 1,000 random sets of genes located on other autosomes than chromosome 17), whereas no difference was detected in TP53‐mutant tumors. Additionally, TP53‐mutant tumors showed higher numbers of RPG deletions in total (Fig 2B), RPG deletions that could be detected in TP53‐wild‐type tumors were enriched in tumors harboring alternative p53 pathway‐inactivating lesions, including MDM2 amplifications and CDKN2A deletions (Fig EV2). Taken together, these results indicate that the p53 pathway restricts the acquisition of RPG deletions in cancer cells.
Figure EV2. Deletions of key genes of the p53 pathway in TP53‐wild‐type cases.
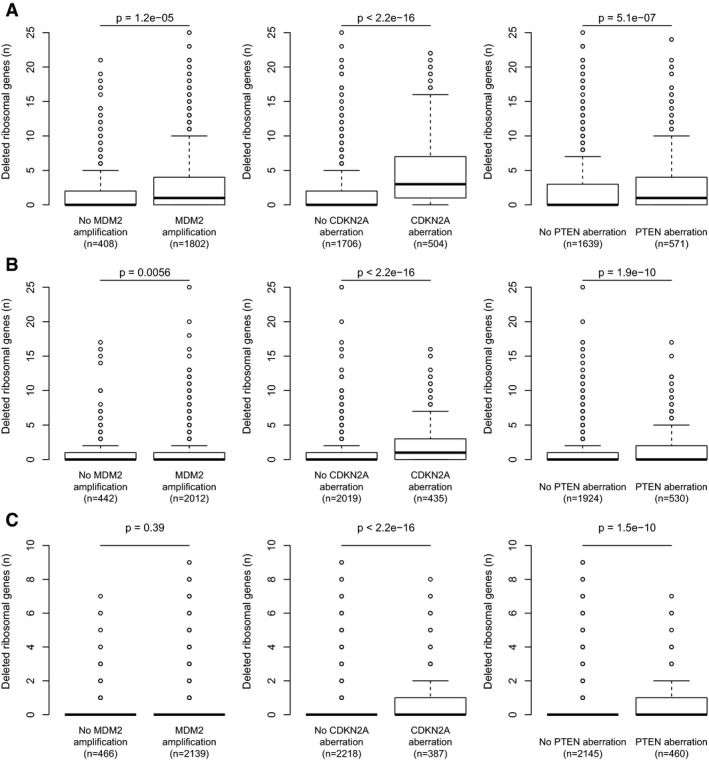
-
A–CAmong tumors without detectable TP53 deletion/mutation, the RPG deletions that could be detected were enriched in tumors harboring alternative p53‐inactivating lesions, including MDM2 amplifications, CDKN2A deletions, and PTEN deletions. Results obtained using log2 ratio thresholds (A) −0.3; (B) −0.5; (C) −0.7. To avoid biases introduced by co‐deletion, RPGs on chromosome 12, 9, or 10 were excluded from the MDM2, CDKN2A, and PTEN analyses, respectively. Boxes indicate medians and the first and third quartiles. Whiskers indicate first and third quartiles ±1.5 times the interquartile range. Notches indicate confidence intervals around the median. P‐values indicate significance by Wilcoxon rank‐sum test.
Deletion of RPGs leads to loss of coordinated RPG expression and p53 activation
Previous studies aimed at understanding the molecular mechanisms of DBA and the 5q‐ syndrome have shown that decreased expression of specific RPGs (RPS6, RPS14, and RPS19) leads to p53 activation (Narla & Ebert, 2010; Raiser et al, 2014). However, the RPGs we found to be deleted in human cancers have not been studied functionally in this regard. To examine whether p53 senses decreased expression of the frequently deleted RPGs, we used shRNA‐mediated knockdown in the TP53‐wild‐type lung adenocarcinoma cell line A549 (Fumagalli et al, 2009; Dutt et al, 2011). For all eight RPGs tested, multiple shRNAs caused elevated p53 protein levels and increased P21 transcript levels (Figs 2C and D, and EV3, and Table EV3). Similarly, knockdown of three of the most frequently deleted RPGs in TP53‐wild‐type MOLM13 leukemia cells caused elevated expression of p53 and four p53 target genes P21, BAX, PUMA, and NOXA (Figs EV4 and EV5). Taken together, these data indicate that p53 activation results not only from decreased expression of the RPGs that are mutated in ribosomopathies, but also from decreased expression of the RPGs that are frequently deleted in cancer cells.
Figure EV3. Validation of shRNAs targeting frequently deleted RPGs.
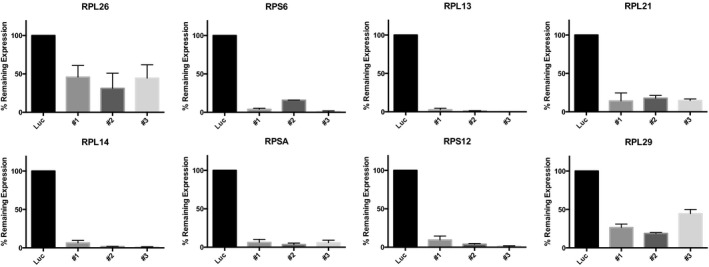
mRNA expression levels of the indicated ribosomal protein gene were measured by quantitative real‐time PCR. Ribosomal protein gene knockdown is shown as percentage of the gene expression in cells with control (luciferase) knockdown. Error bars are min‐max of duplicates.
Figure EV4. Knockdown of identified RPGs induces p53 target genes.
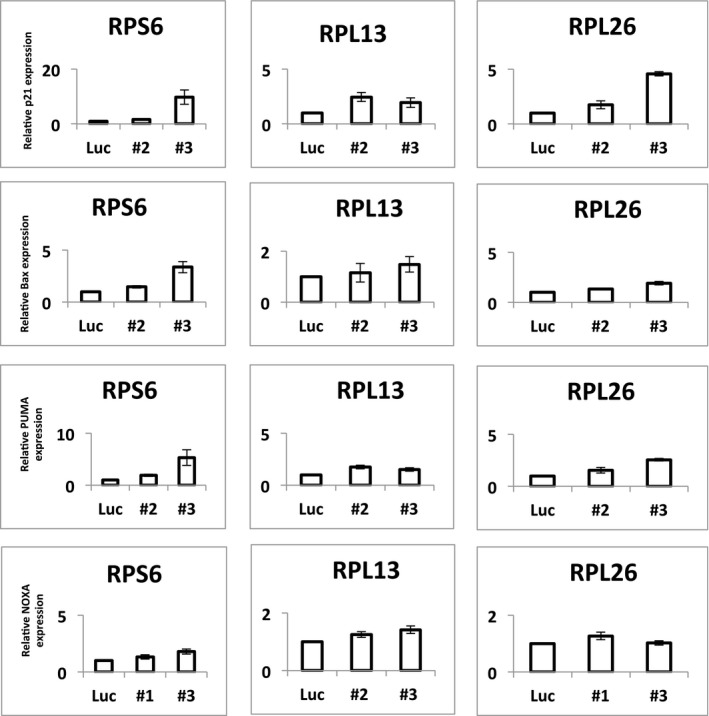
Expression of the p53 target genes P21, BAX, PUMA, and NOXA in MOLM13 cells, as measured by qPCR after knockdown with shRNAs toward RPS6, RPL13, RPL26 (two per gene; as in Fig 2), or luciferase (control). As shown, knockdown of these three RPGs induces the p53 target genes. Error bars are min‐max. Duplicates.
Figure EV5. Knockdown of identified RPGs induces p53.
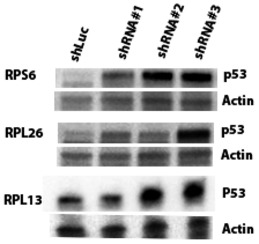
Western blots showing p53 protein levels in MOLM13 leukemia cells after knockdown with shRNAs toward RPS6, RPL26, RPL13 (three per gene), or luciferase (control).
Since RPG haploinsufficiency is thought to activate p53 by altering ribosomal protein stoichiometry (Fumagalli et al, 2009), we tested for associations between RPG copy number anomalies (RPG‐CNAs) and altered RPG expression patterns using 4,919 TCGA samples having both DNA copy number microarray data and global mRNA‐sequencing data. We found a correlation between copy number and expression for most individual RPGs (Fig EV6A). Quantifying RPG co‐expression by calculating correlation coefficients for all pairs of RPGs, we detected less correlated RPG expression in tumors with a high RPG‐CNA burden (Fig EV6B and C), whereas tumors with few RPG‐CNAs showed RPG co‐expression patterns comparable to those observed in non‐cancerous tissues (Fig EV6D). These data indicate that RPG‐CNAs lead to uncoordinated RPG expression, which, together with the shRNA knockdown data, provides a possible explanation for the negative selection of RPG deletion observed in TP53‐wild‐type tumors (Fig 2).
Figure EV6. While RPG are tightly co‐expressed in normal tissues, copy number aberrations affecting RPGs lead to uncoordinated expression.
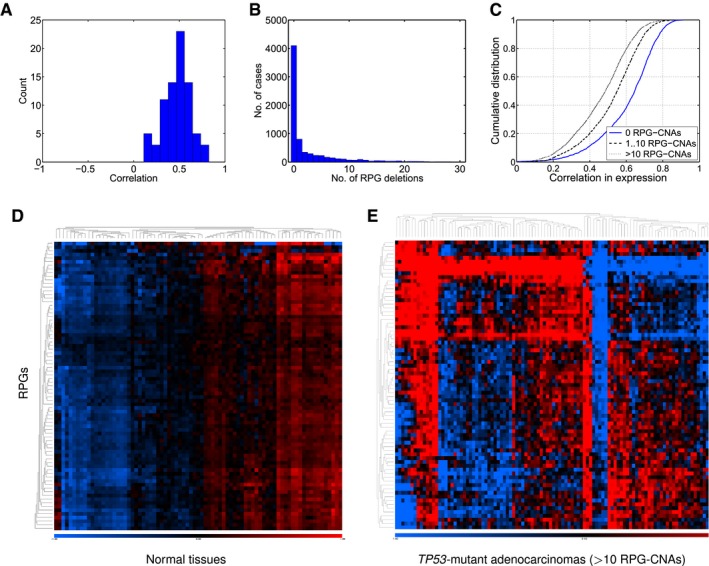
- Correlations between RPG copy number and expression across 4,919 TCGA samples.
- Numbers of RPG deletions per sample. Multiple RPG deletions are common.
- Cumulative distributions of Pearson correlation coefficient for all pairs of RPGs across the TCGA RNA‐sequencing data. Tumors with more RPG copy number aberrations show less coordinated RPG expression.
- Expression of RPGs in normal tissues (SymAtlas; NCBI Gene Expression Omnibus GSE96).
- Expression of RPG in colorectal adenocarcinomas harboring 10 or more RPG copy number aberrations (TCGA samples).
Deletion of RPGs in cancer cells with intact p53 function
We asked which RPG deletions are permissible in cancer cells with intact p53 function. To this end, we specifically analyzed the 30 cell lines from CCLE that are both TP53‐wild‐type and sensitive to the p53‐activating compound Nutlin‐3 (IC50 < 8 μmol; Barretina et al, 2012; Sonkin et al, 2013), where the latter supports that p53 has not been inactivated by alternative mutations in other genes. Interestingly, the most recurrently deleted RPG in these cell lines was RPL22 (Table EV4). Firstly, Rpl22−/− knockout mice have only subtle phenotypes with no significant translation defects, probably because these mice show increased expression of the paralog Rpl22‐like1 (Rpl22 l1) which is incorporated in the ribosome instead of Rps22 (O'Leary et al, 2013). Secondly, RPL22 has been identified as a potential tumor suppressor gene that is mutated or deleted in T‐ALL and several epithelial tumor types (Rao et al, 2012; Novetsky et al, 2013; Goudarzi & Lindstrom, 2016). Another interesting observation was recurrent deletion of RPS6, which (like in the primary tumors) was always associated with co‐deletion of CDKN2A. These data indicate that some RPG deletions are less likely to cause negative selection, either because of gene redundancy, because they do not activate p53, or because they are associated with a pro‐proliferative effect that allows the cells to escape the negative effect of p53 activation.
Further suppression of deleted RPGs inhibits the proliferation of cancer cells
The almost complete lack of homozygous RPG deletions in the three copy number data sets supports that such lesions are not tolerated. These observations are consistent with the notion that ribosomal proteins are essential for generation of functional ribosomes and cell survival. To investigate whether RPG haploinsufficiency renders cancer cells vulnerable to further suppression of ribosome function, we analyzed two sets of shRNA screening data from 102 and 72 genomically annotated cancer cell lines (Cheung et al, 2011; Marcotte et al, 2012). These pooled screens targeted 11,194 and 16,056 genes, including 26 and 55 RPGs, respectively. For each targeted gene, the effect of knockdown on proliferation in cell lines carrying one gene copy, relative to the effect in cell lines carrying at least two gene copies, was scored (Marcotte et al, 2012; Shao et al, 2013). In both data sets, we observed strongly significant enrichments of negative gene scores among RPGs (P = 1.0 × 10−8 and P = 1.7 × 10−4, respectively; Fig 3A and B), indicating that further suppression of RPGs preferentially inhibits the growth of RPG‐haploinsufficient cells.
Figure 3. Consequences of hemizygous RPG deletions.
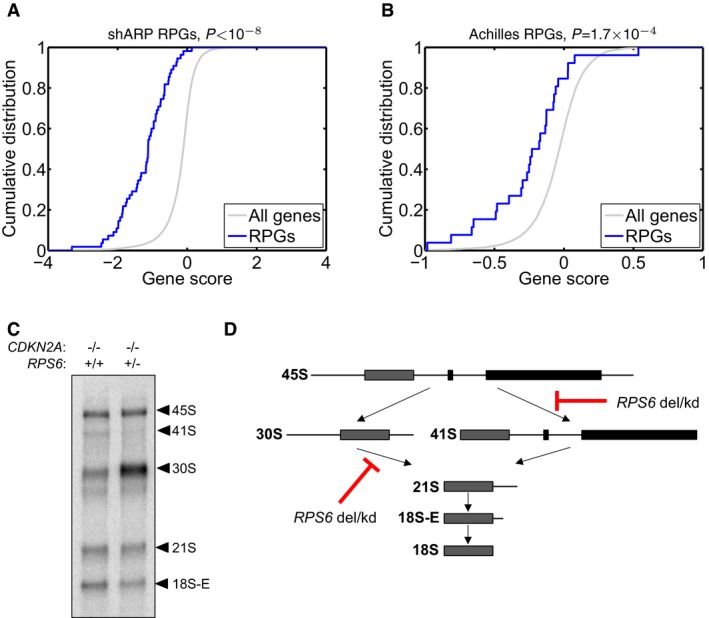
-
A, BTo investigate whether RPG haploinsufficiency renders cancer cells susceptible to further suppression of ribosome function, we analyzed genomewide pooled shRNA screening data from the (A) shARP and (B) Achilles studies (data for 72 and 102 genomically annotated cancer cell lines, respectively). For each targeted gene, the effect of knockdown on proliferation in cell lines carrying one gene copy, relative to those carrying at least two gene copies, was scored. We observed strongly significant enrichments of negative gene scores for RPGs (solid blue; P = 1.0 × 10−8 and P = 1.7 × 10−4, respectively) compared to other genes (gray), demonstrating that further suppression of hemizygously deleted RPGs inhibits cell growth.
-
C, D(C) To explore whether RPG haploinsufficiency causes rRNA maturation defects in cancer cells, we focused on pediatric ALL with 9p deletions targeting CDKN2A. From two previously banked samples (one with homozygous CDKN2A deletion but intact RPS6 and one with homozygous deletion of CDKN2A and hemizygous deletion of RPS6), we obtained viable cells, which were xenografted into NOD scid gamma immunodeficient mice and expanded in vivo. RNA from sorted tumor cells was assessed by Northern blot to analyze pre‐ribosomal RNA processing. The RPS6‐haploinsufficient case exhibited rRNA maturation defects similar to those seen previously in shRNA‐targeted RPS6‐deficient cells, namely (D) a reduction in 41S species (18S‐E, 21S and 41S) and an accumulation of 30S species (30S and 45S). These observations provide the first evidence indicating that the rRNA maturation defect that is observed in ribosomopathies may also be present in RPG‐haploinsufficient cancer cells.
Deletion of RPGs influences rRNA maturation in cancer cells
Previously, in vitro studies have shown that knockdown of RPS6 and other 40S RPGs impairs the processing of pre‐ribosomal RNA species into mature rRNA in ribosomopathies(Narla & Ebert, 2010; O'Donohue et al, 2010; Raiser et al, 2014). To explore whether RPG haploinsufficiency leads to rRNA maturation defects also in cancer cells, we decided to analyze rRNA maturation across pediatric acute lymphoblastic leukemia (ALL) samples harboring CDKN2A (9p) deletions with and without concurrent deletion of RPS6. Deletions targeting CDKN2A are present in 20–30% of B‐cell ALL and 95% of T‐cell ALL. We first genotyped 47 previously banked pediatric ALL samples using copy number microarrays. For two samples (one with homozygous deletion of CDKN2A but intact RPS6 and one with homozygous deletion of CDKN2A and hemizygous deletion of RPS6), we were able to obtain viable cells, which were xenografted into NOD scid gamma immunodeficient mice and expanded in vivo. RNA from sorted tumor cells was assessed by Northern blot to analyze pre‐ribosomal RNA processing. Interestingly, the RPS6‐haploinsufficient case exhibited rRNA maturation defects similar to those seen previously in shRNA‐targeted RPG‐deficient cells (cf. Narla & Ebert, 2010; Raiser et al, 2014 and references therein), namely a reduction in 41S species (18S‐E, 21S, and 41S) and an accumulation of 30S species (30S and 45S), indicating that heterozygous deletion of RPS6 impacts on ribosomal RNA biogenesis in ALL cells (Fig 3C and D). The difference in rRNA pattern observed in ALL cells with and without deletion of RPS6 was comparable to the difference in rRNA pattern observed in TP53‐wild‐type MOLM13 leukemia cells with and without shRNA knockdown of RPS6 (Fig EV7). Collectively, these observations provide the first evidence that the rRNA maturation defect that is observed in ribosomopathies on the basis of heterozygous inactivating mutations in other RPGs (Narla & Ebert, 2010; Raiser et al, 2014) may also be present in cancer cells with hemizygous RPG deletions.
Figure EV7. Effects of RPS6 knockdown on rRNA pattern.
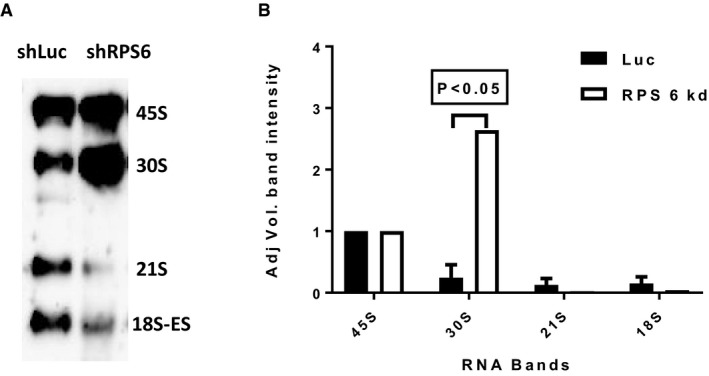
- Representative Northern blot showing rRNA patterns in MOLM13 leukemia cells after knockdown with shRNA against RPS6 (shRPS6) or luciferase control (shLuc).
- Quantification of rRNA specifies from three Northern blot gels (error bars show standard deviation; Student's t‐test P‐value). As shown, knockdown of RPS6 yielded increased 30S rRNA levels and decreased 21S and 18S rRNA levels, which is congruent with the results obtained for primary ALL cells shown in Fig 3.
Discussion
Here we report the novel finding that ribosomal protein genes are routinely deleted across human cancers, particularly in concert with TP53 mutation. Such a finding could lead to new possibilities for cancer therapy in TP53‐mutant patients. Previously, mutation of RPGs (without concurrent TP53 mutation) has primarily been associated with rare ribosomopathies, specific tumor subtypes, and cancer development in zebrafish models.
The analyses in this study are based on a large number of samples from primary samples, belonging to a broad range of different cancer types. The results indicate that RPG deletions are enriched among the samples that have concurrent TP53 mutation. This finding is in accordance with previous studies on ribosomopathies (particularly DBA and the 5q‐syndrome) showing that RPG haploinsufficiency leads to activation of the p53 pathway, most likely through the 5S RNP‐MDM2 pathway. One of the current main hypotheses is that the ribosomal assembly intermediate 5S ribonucleoprotein particle, containing RPL5, RPL11, and the 5S rRNA, accumulates when ribosome biogenesis is blocked; the excess 5S RNP binds to murine double minute 2 (MDM2), the main p53‐suppressor in the cell, inhibiting its function and leading to p53 activation. Our data indicate that targeted degradation of the mRNAs for RPGs that are frequently deleted in cancer leads to p53 activation in A549 and MOLM13 cells, implicating p53 activation and (secondary to that) negative selection as a probable cause of the lower frequencies of RPG deletions seen among TP53‐wild‐type tumors. Moving forward, follow‐up studies will be needed to examine whether this activation is mediated through the 5S RNP‐MDM2 pathway. Additionally, analyses of clonal heterogeneity (e.g., using single‐cell experiments or mathematical modeling of allele ratios) could illuminate whether p53 mutation precedes RPG deletion during oncogenesis.
Further, a central question about the biochemical consequences of RPG haploinsufficiency will be to determine precisely how these alterations affect rRNA maturation in cancer cells. Our data, though limited in sample size, indicate that rRNA patterns in acute lymphoblastic leukemia cells harboring RPS6 deletion are perturbed in the same way as in cell lines with shRPS6 knockdown. These observations motivate further studies with larger numbers of samples to determine how variation in rRNA patterns associates with genetics lesions in RPGs.
Finally, genes that are hemizygously deleted are not necessarily drivers but can confer susceptibility for therapies (Muller et al, 2012; Nijhawan et al, 2012; Liu et al, 2015). Our study represents the first detailed examination of vulnerabilities in a specific cellular component. Our data show that RPG haploinsufficiency is a strikingly common vulnerability that is enriched among p53‐deficient tumors, which are often hard to treat. Our results raise the question whether it could be possible to exploit RPG haploinsufficiency to selectively kill cancer cells. A number of drugs that modulate ribosomal function exist, including rapamycin and other inhibitors (“rapalogs”) of the mammalian target of rapamycin (mTOR; Easton & Houghton, 2006), and compounds that inhibit ribosome biogenesis by inhibiting rRNA synthesis (Drygin et al, 2011; Bywater et al, 2012; Peltonen et al, 2014; Colis et al, 2014). These drugs are active against subsets of human tumors, but their therapeutic scope is unknown and could depend on the presence of ribosome defects. Future studies will inform how RPG deletions can be used for the treatment of a large subset of human cancer.
Materials and Methods
Cancer genome data sets
Firstly, we obtained normalized, segmented Affymetrix and Agilent DNA copy number data (downloadable.seg files) representing 7,225 primary tumor samples belonging to 24 tumor types from TCGA and with matched whole‐exome sequencing data (somatic mutations; 4,675 samples) and RNA‐sequencing data (4,919 samples; Cancer Genome Atlas Research Network et al, 2013; http://cancergenome.nih.gov/). Gene‐wise copy numbers by averaging the copy number signal across genes (from annotated gene start to gene end) in each sample. Secondly, we obtained corresponding data Affymetrix DNA copy number data representing 2,476 primary tumor samples belonging to 13 tumor types from Tumorscape (Beroukhim et al, 2010; http://www.broadinstitute.org/tumorscape), and for 1,043 cancer cell lines from the CCLE (Barretina et al, 2012; http://www.broadinstitute.org/ccle). To determine whether a gene was deleted, we applied log2 copy number thresholds between −0.3 and −0.7, corresponding to linear scale copy numbers 1.6 and 1.2, respectively. A log2 ratio of −0.7 corresponds to the theoretical copy number amplitude for a hemizygous deletion that present in all tumor cells in a sample with 80% tumor cell fraction (a TCGA inclusion criterion). This threshold is conservative as it does not leave any room for clonal heterogeneity, technical underestimation, or noise. The log2 ratio −0.3 corresponds to the midpoint between the theoretical copy number and the euploid, and is less conservative. To call homozygous deletions, we used a log2 ratio threshold of −2.0, corresponding to copy number 0.5 in linear scale. For completeness, we repeated our analyses with other reasonable thresholds, yielding results in broad agreement with those shown. The file processing and statistical analyses were done with Matlab (http://www.mathworks.com), R (http://www.r-project.org), Ultrasome (Nilsson et al, 2009), RenderCat (Nilsson et al, 2007), and various scripts. The set of RPGs was defined by the 78 annotated genes encoding the known proteins of the small and large ribosomal subunits (Fig 1).
Lentiviral shRNA vectors and infection
Lentiviral shRNAs targeting frequently RPGs in the pLKO.1 or pLKO_TRC005 vector were obtained from the The RNAi Consortium (TRC) at the Broad Institute (Table EV3). Lentivirus was produced in 293TL cells. A549 cells were infected with 1 day after plating in the presence of 8 μg/ml polybrene (Sigma‐Aldrich) and selected 24 h later with 2 μg/ml puromycin (Sigma‐Aldrich) for at least 48 h before collection and processing for protein lysates or mRNA. A549 cells were maintained at 37°C and 5% CO2 in F‐12K medium (ATCC) supplemented with 10% fetal bovine serum and 1% pen/strep/glutamine (Gibco). MOLM13 cells were transduced by spinfection with 8 μg/ml polybrene (Sigma‐Aldrich). The cells were selected at 24 h after transduction with 0.3 μg/ml puromycin for 48 h. Puromycin‐selected cells were harvested and divided as per experimental need for processing RNA or protein lysate. MOLM13 cells were maintained in RPMI 1640 supplemented with 10% fetal bovine serum and 1% PEST (Gibco).
Western blot
Cells for protein analysis were lysed with Pierce® IP Lysis Buffer (Thermo Scientific). Western blots were performed using 25–50 μg of protein and primary antibodies against p53 (DO‐1; Santa Cruz Biotechnology) at 1:500 dilution and β‐actin (C4; Santa Cruz Biotechnology) at 1:3,000 dilution. HRP‐conjugated anti‐mouse secondary antibody (GE Healthcare) was used at 1:10,000 dilution. Immunoreactive proteins were visualized using SuperSignal® West Pico Chemiluminescence Substrate (Thermo Scientific). MOLM13 cells were lysed with Biorupter Pico (Diagenode) using 30/30 s on/off for 10‐min program in Laemmli sample buffer (Bio‐Rad). Blots were probed with primary antibody against p53 (Sc‐126, DO‐1, Santa Cruz Biotechnology) at 1:1,000 dilution and β‐actin (Sc‐8432, C‐1; Santa Cruz Biotechnology) at 1:3,000 dilution.
Quantitative real‐time PCR
RNA was purified from A549 and MOLM13 cells using TRIzol (Invitrogen) and RNeasy mini kit (cat no 74104), respectively. First‐strand cDNA was generated using 100–200 ng of total RNA and oligo(dT) primers with the Superscript III reverse transcription kit (Invitrogen), and Omniscript RT kit (cat no 205113) was used for cDNA synthesis from MOLM13 cells. Quantitative RT–PCR was performed using TaqMan® Gene Expression Master Mix (Applied Biosystems) or SYBR® Green PCR Master Mix (Applied Biosystems) on an ABI Prism 7900HT system and StepOnePlus Real‐Time PCR system (Applied Biosystems). We used TaqMan® assays to assess the knockdown effect for RPS6 (Hs01058685_g1), RPL26 (Hs00864008_m1), RPL13 (Hs00744303_s1), RPL21 (Hs03003806_g1), RPL29 (Hs00426490_g1), RPL14 (Hs03004339_g1), RPSA (Hs00347791_s1), and RPS12 (Hs04184906_g1) using ACTB (ABI #401846) as control, and P21 (Hs00355782_m1), NOXA (Hs00560402_m1), PUMA (Hs00248075_m1), BAX (Hs00180269_m1) were assessed using GAPDH (Hs02758991_g1) as endogenous control for MOLM13 cells. Quantitative PCR for P21 and ACTB was performed using SYBR® Green PCR Master Mix (Applied Biosystems) and the following primers: P21 forward 5′‐GCTCTGCTGCAGGGGACAGC‐3′; P21 reverse 5′‐GCCGCCGTTTTCGACCCTGA‐3′; ACTB forward: 5′‐AGCGAGCATCCCCCAAAGTT‐3′; and ACTB forward: 5′‐GGGCACGAAGGCTCATCATT‐3′ for A549 cells.
Analysis of leukemia samples
We obtained DNA extracted from 47 blood and bone marrow samples taken at diagnosis from patients with pediatric ALL. The samples were collected and banked at the Dane Farber Cancer Institute subject to informed consent (Dana Farber Cancer Institute, institutional review board protocol no. 05‐001). The experiments conformed to the principles set out in the WMA Declaration of Helsinki [http://www.wma.net/en/30publications/10policies/b3/] and the NIH Belmont Report [https://www.hhs.gov/ohrp/regulations-and-policy/belmont-report/]. The samples were analyzed on Affymetrix 6.0 arrays (Affymetrix Inc.; data available for download from the ArrayExpress repository accession no. E‐MTAB‐5450). Copy number changes were delineated using the program Ultrasome (Nilsson et al, 2009). We obtained banked tumor cells and expanded them in vivo in NOD scid gamma immunodeficient mice for about 6 months. When the mice appeared moribund, they were sacrificed. Cells harvested from the spleens and bone marrow were stained with anti‐human CD45 (Miltenyi Biotec) and sorted by fluorescence‐activated cell sorting to purify tumor cells, from which RNA was isolated for rRNA maturation analysis by Northern blot (Qiagen kits). For Northern blot analysis, gel‐fractionated RNA was transferred to zeta‐probe membranes (Bio‐Rad). An oligonucleotide probe 5′‐CCTCGCCCTCCGGGCTCCGTTAATGATC‐3′ (complementary to sequences 5520–5547 spanning the boundary between 18S rRNA and ITS1) was labeled with 32P using T4 polynucleotide kinase and hybridized overnight with membrane‐bound RNA at 37°C in ULTRAHyb‐Oligonucleotide hybridization buffer (Ambion). Membranes were washed at 37°C with 6× SSC and subjected to phosphorimager analysis. MOLM13 RPS6 knockdown RNA samples were electrophoretically fractioned in MOPS‐gel running buffer (cat no AM8671, Ambion) and transferred to Biodyne nylon membrane (cat no 77016, Thermo Scientific) using semi‐dry blot method. Hybridization and washing steps were followed as recommended in Northern Max kit (AM1940, Thermo Scientific). The biotinylated cDNA probe hybridization was visualized following Chemiluminescent Nucleic Acid Detection Module (cat no 89880, Thermo Scientific).
Analysis of shRNA screening data
To test whether further suppression of hemizygously deleted RPGs inhibits cell growth, we used genomewide, pooled shRNA screening data from the Achilles and shARP studies (Cheung et al, 2011; Marcotte et al, 2012). The Achilles study provided data on 102 cell lines infected with a pool of lentivirally delivered shRNAs, composed of 54,020 shRNAs targeting 11,194 genes. The shARP study provided data on 72 cancer cell lines infected with a pool of 78,432 shRNAs targeting 16,056 genes. Gene scores reflecting the effect on knockdown on cell growth in cell lines carrying one copy versus at least two copies were provided with the original publications (Cheung et al, 2011; Marcotte et al, 2012; Shao et al, 2013). To test for enrichment of negative scores (indicating depletion) among RPGs compared to other genes, we used RenderCat (Nilsson et al, 2007).
Author contributions
BN and BLE designed and supervised the project, performed computational analyses, and wrote the manuscript with input from DMW, SA, and SRE. Experiments were done by DR, RA, MM, BB and BM. Additional computational analyses were done by MJ and GS. All authors contributed to the final manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
For more information
The Cancer Genome Atlas (http://cancergenome.nih.gov/)
Tumorscape (http://www.broadinstitute.org/tumorscape)
Cancer Cell Line Encyclopedia (http://www.broadinstitute.org/ccle)
Ultrasome (http://www.broadinstitute.org/ultrasome)
Matlab (http://www.mathworks.com)
R project (http://www.r-project.org)
The paper explained.
Problem
The human ribosome consists of an rRNA scaffold and about 80 proteins, divided into two subunits. Ribosomal protein genes (RPGs) are denoted RPS1, RPS2, etc. for the small (40S) subunit; RPL1, RPL2, etc. for the large (60S) subunit. Increasing evidence holds that mutation of RPGs leads to specific clinical and cellular phenotypes, including Diamond‐Blackfan anemia, the 5q‐ subtype of myelodysplastic syndrome, and specific tumor types. Furthermore, many RPGs are tumor suppressor genes in animal models_ENREF_13. Despite these observations, which support a link between RPG lesions and cancer, the occurrence of RPG lesions in human cancers has not been investigated systematically.
Results
We carried out a large‐scale analysis of cancer genome data to determine the frequency and selective pressure of RPG lesions across human cancers. We first looked for chromosomal deletions and amplifications and point mutations in RPGs using pre‐existing DNA copy number microarray and whole‐exome sequencing data from a total of 10,744 cancer specimens and cell lines. Because recent studies have shown that RPG haploinsufficiency activates p53 in ribosomopathies, and the pathobiology of ribosomopathies can be alleviated by p53 inhibition, we hypothesized inactivation of RPGs could lead to negative selection unless the cells have mutated TP53, and, accordingly, looked for associations between inactivating RPG lesions and TP53 mutation. While we observed few point mutations and homozygous deletions in RPGs, we detected hemizygous RPG deletions in about 43% of samples. Consistent with negative selection, further analyses revealed an underrepresentation of RPG deletions in TP53‐intact tumors, whereas we did not see any signs of negative selection in TP53‐mutant tumors. Furthermore, functional experiments showed that deficiency of frequently deleted RPGs increases p53 activity in TP53‐intact cell lines and perturbs rRNA maturation both in cell lines cultured ex vivo and in primary acute leukemia cells with specific RPG deletions and expanded in vivo in xenograft models. Finally, consistent with the low frequency of homozygous deletion, analysis of genomewide shRNA screening data showed that further suppression of hemizygously deleted RPGs inhibits the growth of RPG‐haploinsufficient cancer cells.
Impact
Genes that are hemizygously deleted are not necessarily drivers but can confer susceptibility for therapies. Our data show that RPG haploinsufficiency is a strikingly common vulnerability that is enriched among p53‐deficient tumors, which are often hard to treat. Our results raise the question whether it could be possible to exploit RPG haploinsufficiency to selectively kill cancer cells. A number of drugs that modulate ribosomal function exist. These drugs are active against subsets of human tumors, but their therapeutic scope is unknown and could depend on the presence of ribosome defects. Future studies will inform how RPG deletions can be used for the treatment of a large subset of human cancer.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Review Process File
Acknowledgements
The project was supported by research grants from the Swedish Foundation for Strategic Research (ICA08‐0057), the Marianne and Marcus Wallenberg Foundation (2010.0112), the Swedish Children's Cancer Fund (PR2012‐0084), and a Wallenberg Academy Fellows Award to B.N. (2012.0193). The project was also supported by the National Institute of Health (R01 HL082945 and P01 CA108631) and a Leukemia and Lymphoma Society Scholar Award to B.L.E.
EMBO Mol Med (2017) 9: 498–507
Contributor Information
Benjamin L Ebert, Email: benjamin_ebert@dfci.harvard.edu.
Björn Nilsson, Email: bjorn.nilsson@med.lu.se.
References
- Amsterdam A, Sadler KC, Lai K, Farrington S, Bronson RT, Lees JA, Hopkins N (2004) Many ribosomal protein genes are cancer genes in zebrafish. PLoS Biol 2: E139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow JL, Drynan LF, Hewett DR, Holmes LR, Lorenzo‐Abalde S, Lane AL, Jolin HE, Pannell R, Middleton AJ, Wong SH et al (2010) A p53‐dependent mechanism underlies macrocytic anemia in a mouse model of human 5q‐ syndrome. Nat Med 16: 59–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D et al (2012) The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483: 603–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M et al (2010) The landscape of somatic copy‐number alteration across human cancers. Nature 463: 899–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bywater MJ, Poortinga G, Sanij E, Hein N, Peck A, Cullinane C, Wall M, Cluse L, Drygin D, Anderes K et al (2012) Inhibition of RNA polymerase I as a therapeutic strategy to promote cancer‐specific activation of p53. Cancer Cell 22: 51–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network , Weinstein JN, Collisson EA, Mills GB, Mills Shaw KR, Ozenberger BA, Ellrott K, Shmulevich I, Sander C, Stuart JM (2013) The Cancer Genome Atlas Pan‐Cancer analysis project. Nat Genet 45: 1113–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung HW, Cowley GS, Weir BA, Boehm JS, Rusin S, Scott JA, East A, Ali LD, Lizotte PH, Wong TC et al (2011) Systematic investigation of genetic vulnerabilities across cancer cell lines reveals lineage‐specific dependencies in ovarian cancer. Proc Natl Acad Sci USA 108: 12372–12377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colis L, Peltonen K, Sirajuddin P, Liu H, Sanders S, Ernst G, Barrow JC, Laiho M (2014) DNA intercalator BMH‐21 inhibits RNA polymerase I independent of DNA damage response. Oncotarget 5: 4361–4369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Keersmaecker K, Atak ZK, Li N, Vicente C, Patchett S, Girardi T, Gianfelici V, Geerdens E, Clappier E, Porcu M et al (2013) Exome sequencing identifies mutation in CNOT3 and ribosomal genes RPL5 and RPL10 in T‐cell acute lymphoblastic leukemia. Nat Genet 45: 186–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deisenroth C, Zhang Y (2010) Ribosome biogenesis surveillance: probing the ribosomal protein‐Mdm2‐p53 pathway. Oncogene 29: 4253–4260 [DOI] [PubMed] [Google Scholar]
- Donati G, Peddigari S, Mercer CA, Thomas G (2013) 5S ribosomal RNA is an essential component of a nascent ribosomal precursor complex that regulates the Hdm2‐p53 checkpoint. Cell Rep 4: 87–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draptchinskaia N, Gustavsson P, Andersson B, Pettersson M, Willig TN, Dianzani I, Ball S, Tchernia G, Klar J, Matsson H et al (1999) The gene encoding ribosomal protein S19 is mutated in Diamond‐Blackfan anaemia. Nat Genet 21: 169–175 [DOI] [PubMed] [Google Scholar]
- Drygin D, Lin A, Bliesath J, Ho CB, O'Brien SE, Proffitt C, Omori M, Haddach M, Schwaebe MK, Siddiqui‐Jain A et al (2011) Targeting RNA polymerase I with an oral small molecule CX‐5461 inhibits ribosomal RNA synthesis and solid tumor growth. Cancer Res 71: 1418–1430 [DOI] [PubMed] [Google Scholar]
- Dutt S, Narla A, Lin K, Mullally A, Abayasekara N, Megerdichian C, Wilson FH, Currie T, Khanna‐Gupta A, Berliner N et al (2011) Haploinsufficiency for ribosomal protein genes causes selective activation of p53 in human erythroid progenitor cells. Blood 117: 2567–2576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easton JB, Houghton PJ (2006) mTOR and cancer therapy. Oncogene 25: 6436–6446 [DOI] [PubMed] [Google Scholar]
- Ebert BL, Pretz J, Bosco J, Chang CY, Tamayo P, Galili N, Raza A, Root DE, Attar E, Ellis SR et al (2008) Identification of RPS14 as a 5q‐ syndrome gene by RNA interference screen. Nature 451: 335–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrar JE, Nater M, Caywood E, McDevitt MA, Kowalski J, Takemoto CM, Talbot CC Jr, Meltzer P, Esposito D, Beggs AH et al (2008) Abnormalities of the large ribosomal subunit protein, Rpl35a, in Diamond‐Blackfan anemia. Blood 112: 1582–1592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli S, Di Cara A, Neb‐Gulati A, Natt F, Schwemberger S, Hall J, Babcock GF, Bernardi R, Pandolfi PP, Thomas G (2009) Absence of nucleolar disruption after impairment of 40S ribosome biogenesis reveals an rpL11‐translation‐dependent mechanism of p53 induction. Nat Cell Biol 11: 501–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazda HT, Grabowska A, Merida‐Long LB, Latawiec E, Schneider HE, Lipton JM, Vlachos A, Atsidaftos E, Ball SE, Orfali KA et al (2006) Ribosomal protein S24 gene is mutated in Diamond‐Blackfan anemia. Am J Hum Genet 79: 1110–1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazda HT, Sheen MR, Vlachos A, Choesmel V, O'Donohue MF, Schneider H, Darras N, Hasman C, Sieff CA, Newburger PE et al (2008) Ribosomal protein L5 and L11 mutations are associated with cleft palate and abnormal thumbs in Diamond‐Blackfan anemia patients. Am J Hum Genet 83: 769–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazda HT, Preti M, Sheen MR, O'Donohue MF, Vlachos A, Davies SM, Kattamis A, Doherty L, Landowski M, Buros C et al (2012) Frameshift mutation in p53 regulator RPL26 is associated with multiple physical abnormalities and a specific pre‐ribosomal RNA processing defect in diamond‐blackfan anemia. Hum Mutat 33: 1037–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goudarzi KM, Lindstrom MS (2016) Role of ribosomal protein mutations in tumor development (Review). Int J Oncol 48: 1313–1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaako P, Flygare J, Olsson K, Quere R, Ehinger M, Henson A, Ellis S, Schambach A, Baum C, Richter J et al (2011) Mice with ribosomal protein S19 deficiency develop bone marrow failure and symptoms like patients with Diamond‐Blackfan anemia. Blood 118: 6087–6096 [DOI] [PubMed] [Google Scholar]
- Kondrashov N, Pusic A, Stumpf CR, Shimizu K, Hsieh AC, Xue S, Ishijima J, Shiroishi T, Barna M (2011) Ribosome‐mediated specificity in Hox mRNA translation and vertebrate tissue patterning. Cell 145: 383–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai K, Amsterdam A, Farrington S, Bronson RT, Hopkins N, Lees JA (2009) Many ribosomal protein mutations are associated with growth impairment and tumor predisposition in zebrafish. Dev Dyn 238: 76–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Zhang X, Han C, Wan G, Huang X, Ivan C, Jiang D, Rodriguez‐Aguayo C, Lopez‐Berestein G, Rao PH et al (2015) TP53 loss creates therapeutic vulnerability in colorectal cancer. Nature 520: 697–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macias E, Jin A, Deisenroth C, Bhat K, Mao H, Lindstrom MS, Zhang Y (2010) An ARF‐independent c‐MYC‐activated tumor suppression pathway mediated by ribosomal protein‐Mdm2 Interaction. Cancer Cell 18: 231–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacInnes AW, Amsterdam A, Whittaker CA, Hopkins N, Lees JA (2008) Loss of p53 synthesis in zebrafish tumors with ribosomal protein gene mutations. Proc Natl Acad Sci USA 105: 10408–10413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotte R, Brown KR, Suarez F, Sayad A, Karamboulas K, Krzyzanowski PM, Sircoulomb F, Medrano M, Fedyshyn Y, Koh JL et al (2012) Essential gene profiles in breast, pancreatic, and ovarian cancer cells. Cancer Discov 2: 172–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miliani de Marval PL, Zhang Y (2011) The RP‐Mdm2‐p53 pathway and tumorigenesis. Oncotarget 2: 234–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller FL, Colla S, Aquilanti E, Manzo VE, Genovese G, Lee J, Eisenson D, Narurkar R, Deng P, Nezi L et al (2012) Passenger deletions generate therapeutic vulnerabilities in cancer. Nature 488: 337–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajan N, Bertrand D, Hillmer AM, Zang ZJ, Yao F, Jacques PE, Teo AS, Cutcutache I, Zhang Z, Lee WH et al (2012) Whole‐genome reconstruction and mutational signatures in gastric cancer. Genome Biol 13: R115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narla A, Ebert BL (2010) Ribosomopathies: human disorders of ribosome dysfunction. Blood 115: 3196–3205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijhawan D, Zack TI, Ren Y, Strickland MR, Lamothe R, Schumacher SE, Tsherniak A, Besche HC, Rosenbluh J, Shehata S et al (2012) Cancer vulnerabilities unveiled by genomic loss. Cell 150: 842–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson B, Hakansson P, Johansson M, Nelander S, Fioretos T (2007) Threshold‐free high‐power methods for the ontological analysis of genome‐wide gene‐expression studies. Genome Biol 8: R74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson B, Johansson M, Al‐Shahrour F, Carpenter AE, Ebert BL (2009) Ultrasome: efficient aberration caller for copy number studies of ultra‐high resolution. Bioinformatics 25: 1078–1079 [DOI] [PubMed] [Google Scholar]
- Novetsky AP, Zighelboim I, Thompson DM Jr, Powell MA, Mutch DG, Goodfellow PJ (2013) Frequent mutations in the RPL22 gene and its clinical and functional implications. Gynecol Oncol 128: 470–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donohue MF, Choesmel V, Faubladier M, Fichant G, Gleizes PE (2010) Functional dichotomy of ribosomal proteins during the synthesis of mammalian 40S ribosomal subunits. J Cell Biol 190: 853–866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Leary MN, Schreiber KH, Zhang Y, Duc AC, Rao S, Hale JS, Academia EC, Shah SR, Morton JF, Holstein CA et al (2013) The ribosomal protein Rpl22 controls ribosome composition by directly repressing expression of its own paralog, Rpl22l1. PLoS Genet 9: e1003708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peltonen K, Colis L, Liu H, Jaamaa S, Zhang Z, Af Hallstrom T, Moore HM, Sirajuddin P, Laiho M (2014) Small molecule BMH‐compounds that inhibit RNA polymerase I and cause nucleolar stress. Mol Cancer Ther 13: 2537–2546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raiser D, Narla A, Ebert B (2014) The emerging importance of ribosomal dysfunction in the pathogenesis of hemaotologic disorders. Leuk Lymphoma 55: 491–500 [DOI] [PubMed] [Google Scholar]
- Rao S, Lee SY, Gutierrez A, Perrigoue J, Thapa RJ, Tu Z, Jeffers JR, Rhodes M, Anderson S, Oravecz T et al (2012) Inactivation of ribosomal protein L22 promotes transformation by induction of the stemness factor, Lin28B. Blood 120: 3764–3773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao DD, Tsherniak A, Gopal S, Weir BA, Tamayo P, Stransky N, Schumacher SE, Zack TI, Beroukhim R, Garraway LA et al (2013) ATARiS: computational quantification of gene suppression phenotypes from multisample RNAi screens. Genome Res 23: 665–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Signer RA, Magee JA, Salic A, Morrison SJ (2014) Haematopoietic stem cells require a highly regulated protein synthesis rate. Nature 509: 49–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan KE, Bohnsack MT, Watkins NJ (2013) The 5S RNP couples p53 homeostasis to ribosome biogenesis and nucleolar stress. Cell Rep 5: 237–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonkin D, Hassan M, Murphy DJ, Tatarinova TV (2013) Tumor suppressors status in cancer cell line Encyclopedia. Mol Oncol 7: 791–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulic S, Panic L, Barkic M, Mercep M, Uzelac M, Volarevic S (2005) Inactivation of S6 ribosomal protein gene in T lymphocytes activates a p53‐dependent checkpoint response. Genes Dev 19: 3070–3082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlachos A, Rosenberg PS, Atsidaftos E, Alter BP, Lipton JM (2012) Incidence of neoplasia in Diamond Blackfan anemia: a report from the Diamond Blackfan anemia registry. Blood 119: 3815–3819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volarevic S, Stewart MJ, Ledermann B, Zilberman F, Terracciano L, Montini E, Grompe M, Kozma SC, Thomas G (2000) Proliferation, but not growth, blocked by conditional deletion of 40S ribosomal protein S6. Science 288: 2045–2047 [DOI] [PubMed] [Google Scholar]
- Xue S, Tian S, Fujii K, Kladwang W, Das R, Barna M (2015) RNA regulons in Hox 5′ UTRs confer ribosome specificity to gene regulation. Nature 517: 33–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Lu H (2009) Signaling to p53: ribosomal proteins find their way. Cancer Cell 16: 369–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Hao Q, Liao J, Zhang Q, Lu H (2013) Ribosomal protein S14 unties the MDM2‐p53 loop upon ribosomal stress. Oncogene 32: 388–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Review Process File