

Abstract
c‐MYC controls more than 15% of genes responsible for proliferation, differentiation, and cellular metabolism in pancreatic as well as other cancers making this transcription factor a prime target for treating patients. The transcriptome of 55 patient‐derived xenografts show that 30% of them share an exacerbated expression profile of MYC transcriptional targets (MYC‐high). This cohort is characterized by a high level of Ki67 staining, a lower differentiation state, and a shorter survival time compared to the MYC‐low subgroup. To define classifier expression signature, we selected a group of 10 MYC target transcripts which expression is increased in the MYC‐high group and six transcripts increased in the MYC‐low group. We validated the ability of these markers panel to identify MYC‐high patient‐derived xenografts from both: discovery and validation cohorts as well as primary cell cultures from the same patients. We then showed that cells from MYC‐high patients are more sensitive to JQ1 treatment compared to MYC‐low cells, in monolayer, 3D cultured spheroids and in vivo xenografted tumors, due to cell cycle arrest followed by apoptosis. Therefore, these results provide new markers and potentially novel therapeutic modalities for distinct subgroups of pancreatic tumors and may find application to the future management of these patients within the setting of individualized medicine clinics.
Keywords: bromodomains, c‐MYC, JQ1, pancreatic adenocarcinoma, transcriptomic signature
Subject Categories: Cancer; Chromatin, Epigenetics, Genomics & Functional Genomics
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is one of the most lethal cancers and a major public health issue since there are approximately 230,000 new PDAC cases per year worldwide with approximately the same number of deaths (Jemal et al, 2005). Like others malignant diseases, PDAC results from a complex combination of genetic, epigenetic, and environmental factors which gives rise to a particularly heterogeneous disease, with patients having different set of symptoms, predisposition to early metastasis, and therapeutic responses (Yachida & Iacobuzio‐Donahue, 2013; Dunne & Hezel, 2015; Waddell et al, 2015). This heterogeneity highlights the necessity to stratify patients with the goal of predicting better responses to therapies (Heller et al, 2015; Koay et al, 2016; Noll et al, 2016). One strategy to discover potential markers for patient stratification is to focus on identifying pathways that are deregulated in tumors, particularly when tumor cells absolutely depend of keeping these alterations (e.g., oncogene “dependence” to survive and grow; Mancias & Kimmelman, 2011; Cohen et al, 2015). Consequently, it is logical to assume that blockage of these pathways with specific inhibitors should lead to cell growth arrest, death, and tumor regression. Using this rational, it would be possible to select, by means of a few markers, a particular subgroup of patients “addicted” to a distinct pathways, a major goal of modern individualized medicine.
A frequently deregulated, although insufficiently therapeutically exploited pathway in PDAC involves the “dependence” to c‐MYC oncogene (Mertz et al, 2011). This transcription factor influences the expression of a significant number of genes involved in cell growth, proliferation, and apoptosis (Dang, 1999, 2012; Prendergast, 1999; Schmidt, 1999). In fact, this oncogene has been implicated in the pathogenesis of one‐third of all human malignancies. As it relates to pancreatic cancer, the disease focus of the current study, c‐MYC was found to be originally amplified in more than 30% of PDAC (Schleger et al, 2002) by using interphase fluorescence in situ hybridization, as well as overexpressed in more than 40% of tumors (Schleger et al, 2002). However, more recently, whole‐exome sequencing of microdissected PDAC revealed that the percentage of PDAC with amplified c‐MYC gene is approximately 12% (Witkiewicz et al, 2015). Early studies confirmed the oncogenic role of c‐MYC in PDAC using genetically engineered mouse models, which upon overexpression of this gene display increased pancreatic tumorigenesis (Morton & Sansom, 2013). In addition, using a variety of experimental models, it has been later shown that upregulation of c‐MYC is sufficient to induce the formation of PDAC without additional genetic manipulation of any cell survival pathway (Lin et al, 2013), deletion of one c‐MYC allele decelerates tumor development in vivo (Walz et al, 2014), MYC targeted by an RNAi approach in vivo blocks PDAC development (Saborowski et al, 2014), and the subsequent increase in PGC‐1α is a key determinant for the OXPHOS dependency of cancer stem cells (Sancho et al, 2015). Interestingly, in a more recent work, Wirth and Schneider propose to use c‐MYC as a stratification marker of PDAC (Wirth & Schneider, 2016). All these features indicate that c‐MYC behaves as a cancer driver gene for PDAC. Consequently, many efforts have been dedicated to identify potent MYC inhibitors as new therapeutic options (Soucek et al, 2008; Annibali et al, 2014; McKeown & Bradner, 2014; Fletcher & Prochownik, 2015). Key to these efforts have been the discovery that the bromodomain and extraterminal family of proteins (BET), which are efficiently inhibited by the JQ1 compound, are necessary for c‐MYC activity (Nesbit et al, 1999; Delmore et al, 2011; Kandela et al, 2015). Notably, JQ1 suppresses PDAC development in mice by inhibiting both c‐MYC activity and inflammatory signals (Mazur et al, 2015). Conversely, inhibition of c‐MYC expression is thought to be also an essential mechanism by which BET inhibitors suppress tumor progression in hematological malignancies (Knoechel et al, 2014; Roderick et al, 2014; Trabucco et al, 2015). Thus, identifying the subgroup of pancreatic patients based on their MYC‐high status and testing their response to JQ1 is timely and of paramount medical importance.
Several studies have focused on the discovery of predictive markers of response to BET inhibitors. Puissant et al (2013) reported that amplification of MYCN in medulloblastoma was the most robust marker for predicting the sensitivity of those tumors to JQ1. Moreover, certain rare tumors called NUT midline carcinomas carrying tandem fusion of BRD4 and NUT genes (nuclear protein in testis) show an important sensitivity to BET inhibitors (Stathis et al, 2016). However, outside to these relatively rare examples it is very difficult to predict an efficient response to the BET inhibitors by genomic approaches. To overcome this issue, the use of tumoral transcriptional program can be an effective way to develop and characterize robust predictive signatures notably in terms of chemosensitivity.
In this work, we define a transcriptomic signature that classifies PDAC, whose growth appears to depend on c‐MYC. This result was confirmed in a prospective validation cohort of 16 independent PDAC. We determined that a third of patient‐derived PDAC xenograft bear this signature and thus were likely to respond to JQ1 treatment. Indeed, experimental therapeutic studies using matching patient‐derived cells in monolayer and 3D cultures confirm this prediction. We conclude that having tools to determine tumors with high c‐MYC activity is of clinical interest to select patients sensitive to BET inhibitors suggesting that a similar strategy may be useful in the setting of individualized medicine efforts aimed at stratifying patients to novel treatments.
Results
Selection of PDAC patients with MYC‐high or MYC‐low activity by using a gene expression profile signature
In order to stratify a cohort of 55 PDAC patients, 30 primary tumors obtained from surgery and 25 biopsy samples taken by EUS‐FNA were implanted subcutaneously into mice and preserved as patient‐derived xenografts (PDX). The histopathologic and clinical characteristics of patients from the learning cohort are displayed in Table 1. The main anatomopathological characteristics of patient primary tumors (e.g., nuclear shape and staining intensity, nucleocytoplasmic ratio, eosinophilia, mucins production, and differentiation degree) were preserved in xenografts after at least six successive passages (Duconseil et al, 2015). Growth rates to reach a tumor volume of 1 cm3 ranged from 2 to 6 months in most of the PDX. Total RNA was obtained from the 55 PDX, and gene expression profiling was performed using Affymetrix platform. Subsequently, we selected a panel of 239 RNAs regulated by c‐MYC in accordance with the MYC targets v1 and v2 list from Molecular Signatures Database (MSigDB). Figure 1A represents the hierarchical clustering and heatmap for the top significantly high‐expressed genes in MYC‐high patients. The dendrogram showing the genetic distance between patients indicates the presence of two major subgroups that we define as MYC‐high and MYC‐low in red and blue colors, respectively. Interestingly, we observe that 17/55 (30.9%) patients are characterized by an increase in the expression of 134/239 c‐MYC target RNAs (P‐value = 0.01996 and q‐value (FDR) = 0.044). The rank‐listed transcripts are available in Appendix Table S1. In order to gain insight into the potential biological processes enriched in MYC‐high vs. MYC‐low subgroups, we performed a Gene Set Enrichment Analysis (GSEA). As shown in Fig 1B, the MYC‐high subgroup is characterized by a low differentiated phenotype and their two most significant associated biological processes are cell cycle process (Normalized Enrichment Score = 3.60 and FDR = 0.00) and DNA replication and genome maintenance (Normalized Enrichment Score = 3.24 and FDR = 0.00). In contrast, the MYC‐low subgroup is characterized by biological processes that reflect a more differentiated state of pancreatic tumors such as digestion (Normalized Enrichment Score = −2.23 and FDR = 0.00) and glycoprotein metabolism (Normalized Enrichment Score = −1.76 and FDR = 0.00). In addition, a complete list of statistically significant enriched signatures using Biological Process, Curated Geneset Enriched, and Hallmarks Enriched tools is presented in Datasets EV1, EV2, EV3, EV4, EV5, EV6. To confirm that MYC‐high patients give rise to PDX with high proliferative index, we performed an IHC‐based Ki67 staining scoring on the epithelial compartment of the 55 PDX. As shown in Fig 1C (left part), this semi‐quantitative scoring reveals that MYC‐high patient‐derived PDX proliferate more than the MYC‐low subgroup (Ki67 mean score 2.88 ± 0.25 [n = 17] vs. 2.06 ± 0.12 [n = 38], P = 0.0018). In addition, we determine the degree of differentiation for both subgroups on H&E staining on paraffin‐embedded tissues sections. As shown in Fig 1C (right part), MYC‐high subgroup shows lower differentiation state than MYC‐low subgroup (differentiation mean score 0.77 ± 0.2 [n = 17] vs. 1.82 ± 0.08 [n = 38], P = 0.0002). The Ki67 and differentiation scores are provided in Appendix Fig S1A and B, respectively. Moreover, we analyzed the clinical outcome of both MYC‐high and MYC‐low patients using a Kaplan–Meier analysis and considering both the overall and the relapsing free survival time for the 55 patient cohort. As shown in Fig 1D, the overall survival median is 9.2 months for the MYC‐high vs. 18.8 months for the MYC‐low subgroup (HR = 2.43 [1.1–5.1]). The relapse‐free survival median is 5.6 and 11.5 months for MYC‐high and MYC‐low subgroup, respectively (HR = 2.7 [1.3–5.8]). Altogether, these observations indicate that we can identify patients with MYC‐high and MYC‐low activity. Moreover, PDAC with MYC‐high activity is characterized by increased proliferation, lower differentiation status and they have poor survival expectancy. Combined, these observations constitute a solid characterization of the molecular, biological, and medical features of the c‐MYC status in patient‐derived xenografts, which is necessary to build the trajectory toward the testing of novel therapies aimed at treating this distinct subgroup of tumors.
Table 1.
Clinicopathological parameters from the learning cohort of patients
| Patient distribution (learning cohort) | |||
|---|---|---|---|
| All (%) | Resectable | Unresectable | |
| n | 55 | 30 | 25 |
| Sex | |||
| Male | 34 (62) | 18 | 16 |
| Female | 21 (38) | 12 | 9 |
| Age | |||
| Mean | 64 | 66 | 61 |
| Min–Max | 41–86 | 45–86 | 41–83 |
| Other cancers | |||
| No | 44 (80) | 20 | 24 |
| Yes | 11 (20) | 10 | 1 |
| Tumor location | |||
| Head | 32 (58) | 19 | 13 |
| Undefined | 6 (11) | 0 | 6 |
| Body | 3 (5.5) | 2 | 1 |
| Tail | 14 (25.5) | 9 | 5 |
| Specimen type | |||
| Primary tumor | 46 (84) | 30 | 16 |
| Hepatic metastasis | 5 (9) | 0 | 5 |
| Carcinomatosis | 4 (7) | 0 | 4 |
| Tumor status at diagnosis | |||
| Localized | 29 (53) | 28 | 1 |
| Locally advanced | 8 (14.5) | 2 | 6 |
| Metastasis | 13 (23.5) | 0 | 13 |
| Carcinomatosis | 5 (9) | 0 | 5 |
Figure 1. Identification of a c‐MYC transcriptional signature in 55 pancreatic cancer‐derived xenografts.
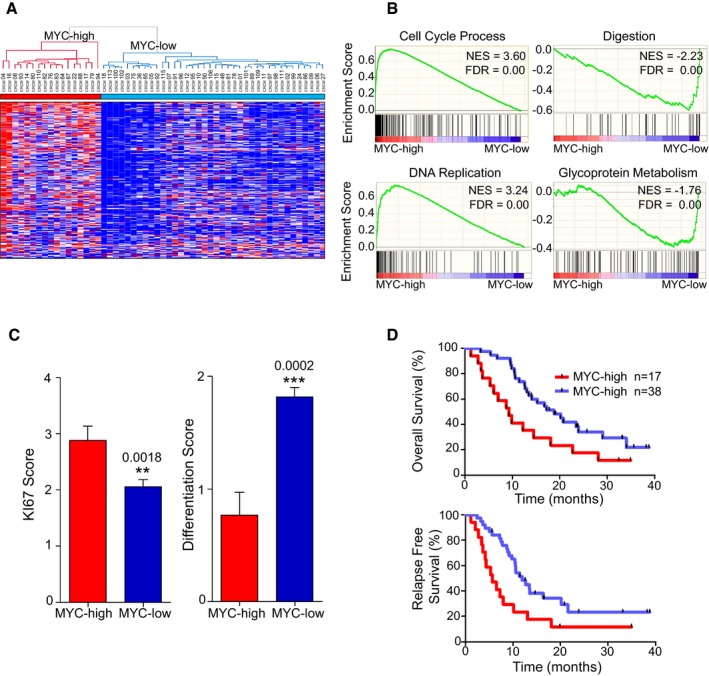
- Hierarchical clustering and expression heatmap analyzed by a non‐supervised method. MYC‐high and MYC‐low subgroups present different expression patterns based on the selected 239 probe sets corresponding to MYC target genes (Hugene 2.0 ST Array, Affymetrix Genechips). MYC‐high (n = 17 patients) and MYC‐low (n = 38 patients). RMA normalized gene expression is represented in color to indicate relative gene expression (high in red, low in blue).
- GSEA analysis of RMA normalized gene expression. Top score biological process significantly different between both groups (MYC‐high and MYC‐low) are represented; 825 gene sets from MSigDB collections were used. NES is the normalized enrichment score, and FDR corresponds to the false discovery rate.
- Ki67 expression level and differentiation degree: Samples were determined by IHC and scored from 0 to 4 for Ki67 staining (0 corresponding to negative staining and 4 to maximal staining) and from 0 to 2 for differentiation state (0 corresponding to the lowest and 2 to the maximal differentiation). Pictures representing the different scores are provided in the Appendix. **P = 0.0018; ***P = 0.0002 (mean ± SEM, n = 17 vs. 38, unpaired t‐test two‐tailed).
- Kaplan–Meier curves showing the overall (upper graph) and relapse‐free survival (lower graph) for MYC‐high and MYC‐low subgroups. The P‐values were calculated using log‐rank test.
Source data are available online for this figure.
MYC‐dependent RNA signatures can be used for classifying distinct PDAC subtypes
To define a specific MYC signature that can be used to classify tumor subtypes, we selected a total of 16 genes. The first 10 (Figs 1A and 2C) were identified from the gene set corresponding to the upregulated genes in the MYC‐high group of patients. To obtain the genes downregulated in the MYC‐high subgroup, we identified the six top‐score downregulated genes in the MYC‐high patients by a t‐test analysis from the whole gene expression profiles (Fig 2A and B). The list of these 16 markers is shown in the Table 2. For each patient, 60 ratios were computed after mean centered normalization of the 16 markers (see Materials and Methods for the normalization method) revealing the MYC‐high or MYC‐low profiles. As shown in Fig 2D, we were able to detect the 17 MYC‐high patients with medians of expression ratios up to 1 with an excellent specificity and accuracy. Affymetrix data were then confirmed by RT–qPCR on four putative MYC‐high and four MYC‐low patients. Each transcript was normalized to the 28S ribosomal RNA, the relative quantity was calculated by the ∆∆C t method and the ratios were calculated after normalization. The signature was able to definitively detect all MYC‐high and MYC‐low profiles by RT–qPCR as shown in Fig 3A and B. We then assessed the MYC signature on primary cultures derived from the same eight xenografts (Fig 3C) and found that the corresponding MYC‐high or MYC‐low profiles were correctly detected. Therefore, we conclude that the signature based on these transcripts is a reliable for identifying tumor subtypes based on their c‐MYC status.
Figure 2. Determination of a set of 16 genes specific for MYC .
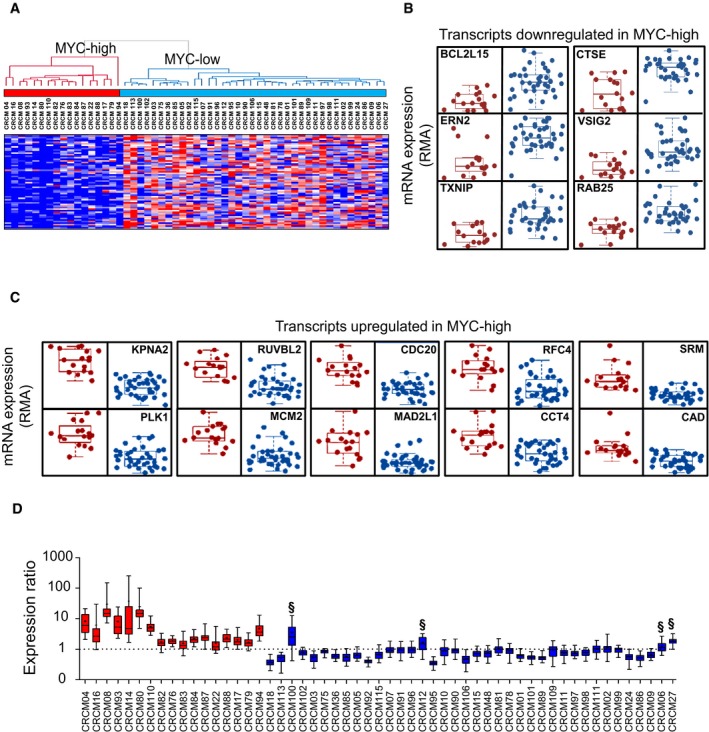
-
AHierarchical clustering and expression heatmap for the top significantly low‐expressed genes in MYC‐high patients. Sixty transcripts were ranked (P = 0.01996; Wilcoxon t‐test). Data correspond to RMA normalized expression values. Red and blue colors represent relative gene expression as in Fig 1A.
-
B, CBox plots of the sixteen selected markers for the MYC‐associated signature. In (B) are plotted the six selected transcripts that are downregulated in the MYC‐high patient group, this set was selected from the 60 transcripts indicated in (A) (P = 0.01996 and FDR ≤ 0.044). In (C) are plotted the set of 10 MYC target transcripts upregulated in MYC‐high patients which was selected from the 239 transcripts indicated in Fig 1A (P = 0.01996 and FDR ≤ 0.044).
-
DBox plots representing the normalized expression ratios (see Materials and Methods) for the sixteen selected transcripts in the MYC‐associated signature. Ratios were done with transcriptomic data obtained from the 55 patients used to select the MYC signature (training cohort). Ratios > 1 indicate a MYC‐high profile, and ratios < 1 correspond to MYC‐low profile. § symbols indicate the four false positives detected with the signature (duplicates [2 chips/PDX]).
Table 2.
List of biomarkers used in the transcriptomic signature
| Gene symbol | Affymetrix ID | RefSeq | mRNA_assignment | |
|---|---|---|---|---|
| Upregulated transcripts in MYC‐high cohort | CDC20 | 16663514 | NM_001255 | Homo sapiens cell division cycle 20 |
| KPNA2 | 16837270 | NM_002266 | Homo sapiens karyopherin alpha 2 | |
| PLK1 | 16817017 | ENST00000300093 | Homo sapiens polo‐like kinase 1 | |
| SRM | 16681611 | NM_003132 | Homo sapiens spermidine synthase | |
| RFC4 | 16962493 | NM_002916 | Homo sapiens replication factor C (activator 1) 4 | |
| MCM2 | 16945101 | ENST00000265056 | Homo sapiens minichromosome maintenance complex component 2 | |
| RUVBL2 | 16863946 | NM_006666 | Homo sapiens RuvB‐like 2 (E. coli) | |
| MAD2L1 | 16979389 | ENST00000296509 | Homo sapiens MAD2 mitotic arrest deficient‐like 1 | |
| CCT4 | 16898175 | NM_006430 | Homo sapiens chaperonin containing TCP1, subunit 4 (delta) | |
| CAD | 16878137 | NM_004341 | Homo sapiens carbamoyl‐phosphate synthetase 2 | |
| Downregulated transcripts in MYC‐high cohort | VSIG2 | 16745683 | NM_014312 | Homo sapiens V‐set and immunoglobulin domain containing 2 |
| BCL2L15 | 16691121 | NM_001010922 | Homo sapiens BCL2‐like 15 (BCL2L15) | |
| RAB25 | 16671901 | NM_020387 | Homo sapiens RAB25, member RAS oncogene family | |
| TXNIP | 16669796 | NM_006472 | Homo sapiens thioredoxin interacting protein | |
| CTSE | 16676547 | NM_001910 | Homo sapiens cathepsin E, transcript variant 1 | |
| ERN2 | 16825120 | NM_033266 | Homo sapiens endoplasmic reticulum to nucleus signaling 2 |
Figure 3. Validation of the MYC signature.
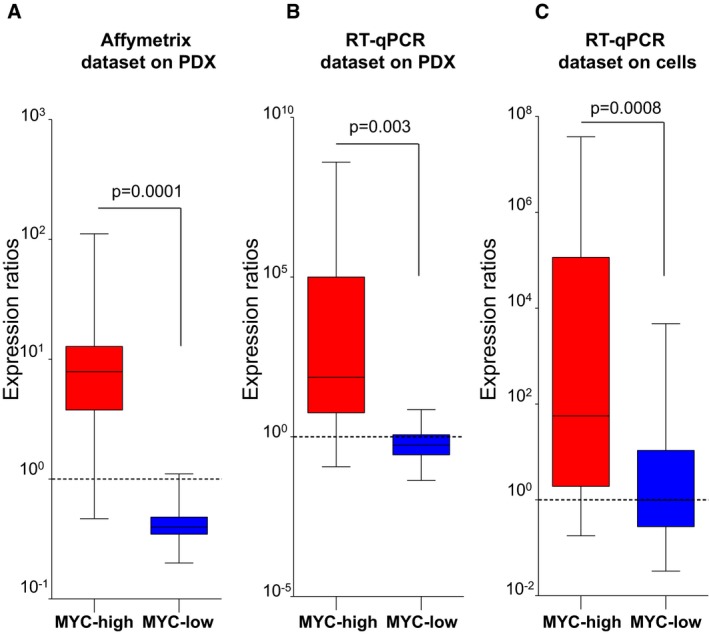
-
A–CBox plots representing normalized expression ratios (A) from transcriptomic data as in Fig 2D for four PDX of each group (MYC‐high and MYC‐low), (B) by RT–qPCR for the same PDX as in (A), and (C) by RT–qPCR for the same eight PDX‐derived primary cell cultures. The dotted line indicates the threshold 1 that differentiates between MYC‐high and MYC‐low profiles. The line in the box‐plot representation shows the median value of mRNA expression ratios, the lower and upper limits of each box represents the first and third quartiles, respectively. Whiskers represent the limits of extreme measurements (Mann–Whitney t‐test).
Source data are available online for this figure.
We also analyzed whether or not the genetic alterations of the c‐MYC gene in PDAC tumors are predictive of the response to BET inhibitors. To this end, we examined the gain (one or more alleles) of c‐MYC gene in the PDX collection. We provide the c‐MYC gain status for both MYC‐high and MYC‐low samples in Appendix Fig S2. This analysis reveals that 15 of 17 PDX samples with a MYC‐high phenotype show a gain in the c‐Myc gene copy number, but also 20 of 38 PDX samples from the MYC‐low group. Thus, in the MYC‐low samples, which are not good responders to JQ1, approximately half of patients also display increases in c‐MYC copy number. This phenomenon suggests that potential epigenetic mechanisms are deployed by cells to compensate for the increase in c‐MYC copy number in this tumor. Based on this observation, we conclude that c‐MYC CNV alterations, although more frequent in MYC‐high tumors, are an unsuitable prediction method to estimate the higher BET inhibitors sensitivity.
MYC‐high PDX are sensitive to growth inhibition by the BET inhibitor JQ1 in vitro
We hypothesized that the subgroup of PDX belonging to the MYC‐high phenotype should be more sensitives to pharmacological inhibition of MYC activity, which currently cannot be targeted directly but instead through the inactivation of BET proteins. To test our hypothesis, we treated a panel of pancreatic PDX‐derived primary cultures with the well‐characterized BET inhibitor JQ1. According to their MYC signature, we selected four MYC‐high PDX (CRCM16, CRCM17, CRCM04, and CRCM08) and four MYC‐low patients (CRCM05, CRCM11, CRCM10, and CRCM109) (see Fig 2D). We assessed the viability of cells with increasing dose of drug (chemograms) for 72 h. As shown in Fig 4A and B, MYC‐high cells exhibit higher sensitivity to JQ1 treatment compared to the MYC‐low ones. The mean of IC50 for the MYC‐high cells is 2.3 μM ± 0.8, whereas the one corresponding to MYC‐low primary cells was of 39.22 μM ± 16 (Fig 4C). We also evaluated the effect of JQ1 in patient‐derived cells grown in 3D culture conditions. As shown in Fig 4D, MYC‐high spheroids were more sensitive to the JQ1 treatment for 72 h (50% reduction in volume) than their MYC‐low counterpart (25% reduction in volume). As a positive control, we analyzed the effect of JQ1 treatment on the MYC‐high primary cell CRCM16 and found a significant decrease of MYC protein level (Fig 4E). Importantly, MYC depletion is accompanied with an increase in p27Kip1 level and an increase in the cleavage of caspase‐3 which may explain the antitumor effect of the compound. Thus, these experimental therapeutic experiments suggest that inhibition of BET proteins by small drugs like JQ will be beneficial to antagonize the growth of pancreatic cells which carry the MYC‐high status.
Figure 4. MYC‐high PDX were sensitive to the JQ1 compounds.
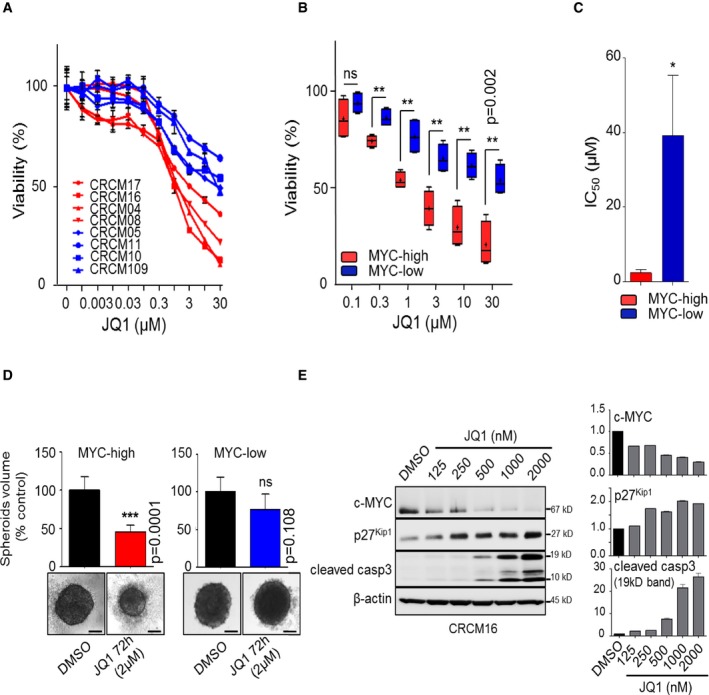
- Chemograms for eight PDX‐derived cell lines. Dose–response curves after 72 h of JQ1 treatment. Cell viability is indicated in % to the control (vehicle treated). Error bars represent SEM; n = 3.
- Box plots representing JQ1 sensitivity for the six highest concentrations used in chemograms. The line in the box‐plot representation shows the median value of mRNA expression ratios; the lower and upper limits of each box represent the first and third quartiles, respectively. Whiskers represent the limits of extreme measurements (**P = 0.002, unpaired t‐test with Welch's correction, triplicates).
- Histograms representing IC50 for JQ1 for the four MYC‐high and the four MYC‐low cell lines (*P = 0.05, unpaired t‐test with Welch's correction).
- Histograms representing spheroid volumes from three derived cell lines in each group treated with 2 μM JQ1 for 72 h or with DMSO (0.05%). Data are representative of three independent experiments made in triplicate. Phase contrast pictures were taken under a 4×/NA 0.45 μm objective lens. Black scale bars represent 100 μm (***P = 0.0001, unpaired t‐test with Welch's correction).
- Expression of c‐MYC, p27kip1 and cleaved caspase‐3 of CRCM16 in primary cells treated with increasing concentrations of JQ1 or vehicle (DMSO). The graphs represent the densitometry of each protein normalized on the β‐actin levels. Densitometries were made with ImageJ software (ImageJ, National Institutes of Health, Bethesda, Maryland, USA) (duplicates).
Source data are available online for this figure.
MYC‐dependent RNA signature identify MYC‐high patients on an independent validation cohort
Sixteen new PDAC patients were included in the study as an independent validation cohort. The histopathologic and clinical characteristics of patients from the test cohort are displayed in Table 3. We obtained 16 PDX and from them six PDX‐derived cells. We measured the expression of 16 MYC‐associated markers by RT–qPCR and found that eight patients present a MYC‐high profile (CRCM43, CRCM26, CRCM50, CRCM19, CRCM30, CRCM114, CRCM116, and CRCM34) and eight show a MYC‐low profile (CRCM23, CRCM21, CRCM108, CRCM25, CRCM28, CRCM112, CRCM29, CRCM42) as described in Fig 5A. Of the six primary cultures available, three presented a MYC‐high (CRCM116, CRCM114, and CRCM34) and three a MYC‐low profile (CRCM112, CRCM21, and CRCM28). To test their sensitivity to BET inhibitors, cells were treated with increasing concentrations of JQ1 and as expected the three MYC‐high cell cultures showed to be more sensitive than the MYC‐low cells as shown in Fig 5B and C. The mean of IC50 for the MYC‐high cells is 5.65 μM ± 2.4, whereas it is 223.71 μM ± 191.5 for the MYC‐low primary cultures (Fig 5D) which are close to the study cohort presented in Fig 5C. These results confirm that cells from PDAC presenting a MYC‐high profile are more sensitive to JQ1 treatment compared to the cells presenting a MYC‐low profile.
Table 3.
Clinicopathological parameters from the validation cohort of patients
| Patient distribution (validation cohort) | |||
|---|---|---|---|
| All (%) | Resectable | Unresectable | |
| n | 16 | 7 | 9 |
| Sex | |||
| Male | 10 (62.5) | 3 | 7 |
| Female | 6 (37.5) | 4 | 2 |
| Age | |||
| Mean | 69 | 67 | 70 |
| Min–Max | 53–83 | 58–71 | 53–83 |
| Other cancers | |||
| No | 11 (68.75) | 4 | 7 |
| Yes | 5 (31.25) | 3 | 2 |
| Tumor location | |||
| Head | 5 (31.25) | 3 | 2 |
| Undefined | 2 (12.5) | 0 | 2 |
| Body | 2 (12.5) | 1 | 1 |
| Tail | 7 (43.75) | 3 | 4 |
| Specimen type | |||
| Primary tumor | 13 (81.25) | 7 | 6 |
| Hepatic metastasis | 2 (12.5) | 0 | 2 |
| Carcinomatosis | 1 (6.25) | 0 | 1 |
| Tumor status at diagnosis | |||
| Localized | 7 (43.75) | 7 | 0 |
| Locally advanced | 1 (6.25) | 0 | 1 |
| Metastasis | 6 (37.5) | 0 | 6 |
| Carcinomatosis | 2 (12.5) | 0 | 2 |
Figure 5. Verification in a validation cohort.
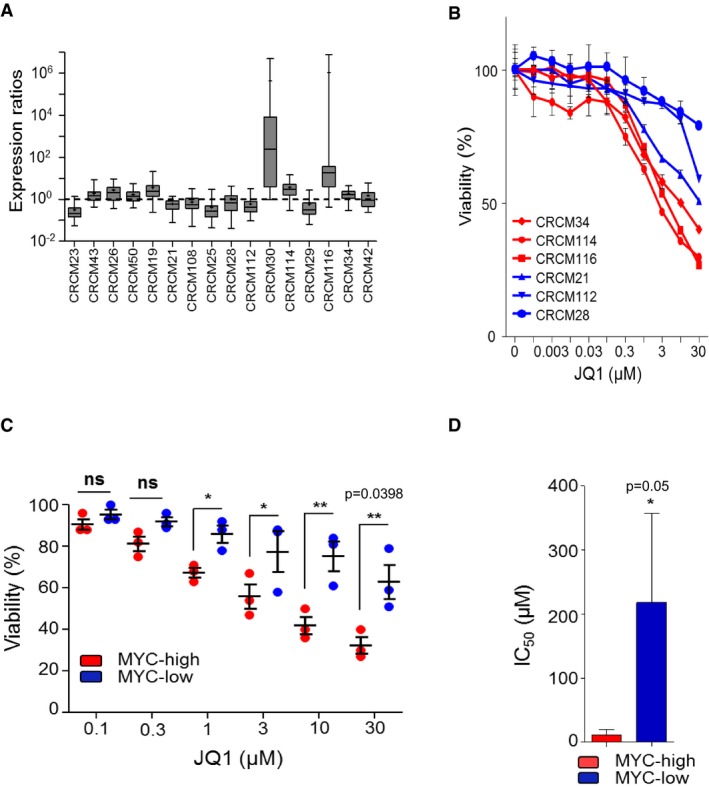
- Box plots representing normalized expression ratios for the MYC signature in 16 new PDX used as validation or test cohort. The line in the box‐plot representation shows the median value of mRNA expression ratios; the lower and upper limits of each box represent the first and third quartiles, respectively. Whiskers represent the limits of extreme measurements. qPCR in duplicate for each PDX.
- Chemograms for eight PDX‐derived cell lines with MYC‐high (red) or MYC‐low (blue) profiles were subjected to JQ1 treatment as in Fig 4A. Cell viability is indicated as % of the control (vehicle treated). Error bars represent SEM; n = 3.
- Graph representing JQ1 sensitivity for the six highest concentrations used in chemograms. Horizontal lines represent the median ± SEM (*P = 0.014; **P = 0.0398, Welch's t‐test).
- Histograms representing IC50 for JQ1 for the three MYC‐high and the three MYC‐low cell lines taken from the validation cohort (mean ± SEM, unpaired t‐test with Welch's correction).
Source data are available online for this figure.
MYC‐high PDX are sensitive to the BET inhibitor JQ1 in nude mice model
Finally, we performed a preclinical analysis by treating four PDX with MYC‐high and four with MYC‐low phenotypes with the JQ1 compound (50 mg/kg/day) to validate the obtained in vitro results. As shown in Fig 6, CRCM16, CRCM04, CRCM114, and CRCM116 (MYC‐high) samples efficiently responded to treatment. On the contrary, samples with the MYC‐low phenotype (CRCM05, CRCM10, CRCM109, and CRCM112) were more resistant. Altogether, from the in vitro and in vivo results, we can assume that MYC‐high tumors are more sensitive to the BET inhibitors.
Figure 6. JQ1 is a more potent inhibitor in MYC‐high PDX in vivo .
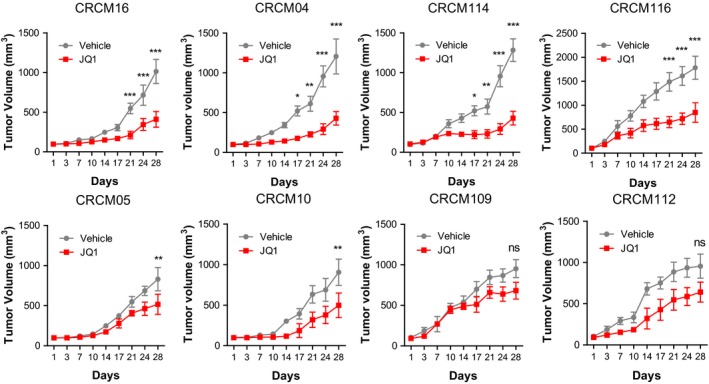
Four PDX with MYC‐high (CRCM16, CRCM04, CRCM114, and CRCM116) and four with MYC‐low (CRCM05, CRCM10, CRCM109, and CRCM112) phenotypes were treated with 50 mg/kg/day of JQ1 or with vehicle for 28 days by i.p. injections (n = 3 per group). ***P < 0.001, **P < 0.01 and *P < 0.05 in two‐way ANOVA with Bonferroni post‐test (mean ± SEM).Source data are available online for this figure.
Discussion
Current treatments for patients with a PDAC are not highly effective primarily due to the recently discovered fact that these tumors are both molecularly and clinically heterogeneous. For example, the response of these tumors to gemcitabine and Folfirinox, the two gold standard therapies against PDAC, is only 10% (Burris et al, 1997) and 31% (Conroy et al, 2011), respectively. The variability in this response seems to be due, on one hand, to the difficulty for the drugs to reach the transformed cells because of the compact PDAC stroma (resulting in hypovascularization) and, on the other hand, to the marked differences in cellular susceptibility to drugs into the tumors due to their molecular heterogeneity. Therefore, it has become important to develop methods to stratify patients in a manner that allows predicting their susceptibility to the treatments so as to increase their therapeutic responses which will result in increased survival expectancies. Consequently, in this work, we focused our attention on a subgroup of PDAC, which are characterized by a deregulation of the c‐MYC pathway, established a molecular signature that allows us to identify those tumors of the MYC‐high subtype and showed that they are highly sensitive to BET inhibitors. Based on this observation, we conclude that the development of tools to determine tumors with high deregulation in the c‐MYC pathway is of clinical interest to select patients sensitive to BET inhibitors, a goal that can help with the design and administration of individualized medicine protocols.
The oncogene c‐MYC is the “corner stone” of several proliferative pathways involved in PDAC development such as RAS‐RAF‐MEK‐ERK and PI3K‐AKT pathways that stabilize c‐MYC by preventing its degradation by the proteasome (Sears et al, 2000; Dai et al, 2006; Zhu et al, 2008). Consistent with these observations, we chose to define c‐MYC transcriptional program as a restricted signature that allowed the selection of distinct subtype of PDAC tumors. This MYC‐associated signature was established by using selected c‐MYC target genes available from the molecular signature database and who are reported to be directly regulated by this transcription factor (MYC target v1 and v2 from MsigDB). From the 239 putative c‐MYC‐dependent genes, 16 targets were selected according to the fold change between MYC‐high and MYC‐low PDAC. Those targets usefully identify highly proliferative PDAC with low degree of differentiation and patients with poor clinical outcome as shown in Fig 1C and D. An efficient signature, like the one reported here, which is easily applicable and low cost, for detecting patients having a MYC‐high PDAC is of clear clinical interest, particularly in non‐operable patients which represent about 85% of PDAC. Currently, in these patients, a biopsy is systematically taken by EUS‐FNA before starting the antitumor treatment as a diagnosis confirmation procedure. These biopsies represent a valuable source of cancer cells which may serve as the source of tumor RNA. In turn, this RNA may be used for measuring the expression of a set of RNAs of interest. We envision that in the near future treatments of cancer patients will be preceded by the molecular characterization of their tumor in order to select the more appropriate treatments toward an individualized medicine approach. PDAC is undoubtedly one of the malignant diseases that most urgently need this type of approaches since the treatment with standard available drugs are almost inefficient. Conceptually, targeting on the pathways, which are dominantly deregulated such as those driven by oncogenes such as Kras and c‐MYC by specific drugs, could be of major interest for patients and we presume that it may have repercussions in terms of survival. This work was focused on c‐MYC, but a similar approach can be applied to other pathways for PDAC as well as for other cancer types. Our work was also benefited by the existence of well‐characterized drugs that reliably inhibit the c‐MYC pathway such as JQ1, a thienotriazolodiazepine, and a potent inhibitor of the BET family of proteins which includes BRD2, BRD3, BRD4, and BRDT (Filippakopoulos et al, 2010; Asangani et al, 2014). Mechanistically, JQ1 competitively inhibits the BET proteins from binding to acetylated lysine residues of histones (Jung et al, 2014; Korb et al, 2015). This process prevents the association of transcriptional complexes with chromatin and thus decreases expression of RNA species that are dependent of this mechanism of transcription (Smith & Zhou, 2016). Many studies suggest that the main mechanism by which BET inhibitors affect tumoral growth is by their effects on c‐MYC expression and activity (Knoechel et al, 2014; Roderick et al, 2014; Trabucco et al, 2015). However, a recent paper suggests an additional antitumor effect of JQ1 on PDAC via the inhibition of CDC25B, a regulator of cell cycle progression. Whether this effect is specific to JQ1 or common to BET inhibitors remains to be demonstrated (Garcia et al, 2016). Lastly, in hepatocellular carcinoma, MYC expression does not seem to be predictive of the response of these tumors to JQ1 (Huang et al, 2014). Therefore, we can assume that MYC level is not indicative of the MYC activity or, alternatively, that the response to JQ1 could be specific in certain tissues, although testing both of these hypotheses remains the topic of future investigations.
In conclusion, this study is the first to report a strategy, which begun by molecularly characterizing patient‐derived PDX as well as to define a molecular signature that can help to select PDAC patients with deregulation in the c‐MYC pathway and showing that these tumors are more sensitive to the BET inhibitor treatment. We presume that a therapeutic strategy against PDAC by using BET inhibitors, in combination with standard anticancer drugs, could be a promising strategy in well‐selected patients. A similar approach can be applied to other pathways for PDAC or other cancer types, highlighting its potential to contribute to the development of novel individualized therapies for malignant diseases, which currently remain incurable.
Materials and Methods
Ethics statements
The study was performed according to the principles set out in the WMA Declaration of Helsinki and to the protocols approved by the Department of Health and Human Services Belmont Report. Patients were included in this project under the Paoli Calmettes Institute clinical trial NCT01692873 (https://clinicaltrials.gov/show/NCT01692873). Three expert clinical centers collaborated on this project after receiving ethics review board approval. Patient informed consent forms were collected and registered in a central database.
For animal studies, all mice, housed under specific pathogen‐free conditions, were 4‐week‐old males on a Swiss nude background (Crl: Nu(lco)‐Foxn1nu, Charles River, Wilmington, MA). Mice were used at 6 weeks of age, and all animal studies were approved by the Animal Facility and Experimental Platform (Scientific and Technological Park of Luminy, Marseille, France) and the French Ministry of National Education and Research under the reference number: 02859.01. All animal experiments were conducted in accordance with the Guides for the Use and Care of Laboratory Animals (ARRIVE guidelines). The animal house is run by professional employees fully equipped with state‐of‐the‐art instrumentation in order to maintain the standard of animal welfare at the maximum levels. All mice were housed in individual, ventilated cages (IVCs) with 12‐h light/dark cycles with food and water ad libitum.
PDAC samples and cell culture
Two types of samples were obtained: endoscopic ultrasound‐guided fine‐needle aspiration (EUS‐FNA) biopsies from patients with unresectable tumors and tumor tissue samples from patients undergoing surgery. All the samples were anonymized, and postsurgical anatomopathology reports were provided. Histopathologic evaluation was performed on 5‐μm hematoxylin and eosin stained sections of patient tumors and xenografts and examined under a light microscope. These sections were compared with the original human tumor when available. Samples from EUS‐FNA were mixed with 100 μl of Matrigel™ (BD Biosciences, Franklin Lakes, NJ) and injected in the upper right flank of a nude mouse (Swiss Nude Mouse Crl: NU(lco)‐Foxn1nu; Charles River Laboratories, Wilmington, MA). Samples from surgery were fragmented, mixed with 100 μl of Matrigel™, and implanted with a trocar (10 gauge; Innovative Research of America, Sarasota, FL) in the subcutaneous right upper flank of an anesthetized and disinfected mouse. When the tumors reached 1 cm3, the mice were sacrificed, and the tumors were removed. Xenografts that failed to develop within 6 months were discontinued. The study on animals was approved by the Animal Facility and Experimental Platform (Scientific and Technological Park of Luminy, Marseille, France).
Primary cell cultures were obtained from xenografts. Tissues were split into several small pieces and processed in a biosafety chamber. After a fine mincing, they were treated with collagenase type V (ref C9263; Sigma) and trypsin/EDTA (ref 25200‐056; Gibco, Life Technologies) and suspended in DMEM supplemented with 1% w/w penicillin/streptomycin (Gibco, Life Technologies) and 10% fetal bovine serum (Lonza). After centrifugation, cells were re‐suspended in Serum Free Ductal Media (SFDM) adapted from Schreiber et al (2004) without antibiotic and incubated at 37°C in a 5% CO2 incubator.
Gene expression microarrays
Total RNA was purified from xenograft using TRIzol® Reagent (Gibco, Life Technologies) according to the manufacturer. Briefly, 50–100 mg of fresh frozen tissue per ml of TRIzol® was disrupted using a homogenizer followed by a single step of phenol/chloroform purification. Total RNA was quantified using the Nanodrop spectrophotometer (NanoDrop Technologies, Inc), and RNA Integrity Number (RIN) was calculated using the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). RNA samples that reached a RIN between 8 and 10 were used for microarray hybridization (GeneChip; Affymetrix Inc., Santa Clara, CA). The Genechip® Human Gene 2.0 ST Arrays were washed and stained using the Affymetrix GeneChip fluidic station 450 (protocol EukGE‐WS2v5_450) and were scanned using a GeneChip scanner 3000G7 (Affymetrix Inc., Santa Clara, CA). GeneChip operating software version 1.4 (Affymetrix Inc., Santa Clara, CA) was used to obtain chip images and for quality control. Background subtraction and normalization of probe set intensities were performed using the method of Robust Multi‐array Analysis (RMA; Irizarry et al, 2003). Microarray analysis was performed by the CHU de Québec Research Center Gene Expression Platform (Quebec City, Quebec, Canada). Seventeen samples of the cohort were previously published (Duconseil et al, 2015) as GEO accession numbers GSE55513 and GSE89792.
Bioinformatics analysis
Robust Multi‐array Analysis normalized data from microarrays were imported into GENE‐E (version 3.0.204; Broad Institute, Cambridge, MA, USA) to generate heatmaps. The color intensity on the heatmap reflects global expression within a minimum (25%) in blue and a maximum (75%) in red. Cluster analysis, using Euclidean distance correlation of samples only, and distance for the clustering were calculated using a complete linkage algorithm. Differentially expressed genes were identified using t‐test ratio, and false discovery rate was estimated using Benjamini & Hochberg method (Benjamini et al, 2001). Gene set enrichment analysis (GSEA) was performed using the Broad Institute platform, and statistical significance (false discovery rate) was seated at 0.05.
GSEA analysis
Two categories of pre‐defined gene sets in the Molecular Signatures Database (MSigDB, Broad Institute, Cambridge, MA, USA) were selected for analysis named the Hallmarks set, and the C5 set, a Gene Ontology (GO) molecular function gene set derived from the Molecular Function Ontology database (Subramanian et al, 2005). The gene sets included in the analysis were limited to those that contained between 15 and 500 genes. Permutation was conducted 1,000 times according to default‐weighted enrichment statistics and by using a t‐test ratio metric to rank genes according to their differential expression levels across the MYC‐high and MYC‐low groups. Significant gene sets were defined as those with a nominal P < 0.05. Calculation of the false discovery rate (FDR) was used to correct for multiple comparisons and gene set sizes (Benjamini et al, 2001).
SNP arrays analysis
DNA was extracted for 55 PDX samples using the Blood & Cell culture DNA mini kit (Qiagen) following the manufacturer's instructions. Illumina Infinium HumanCode‐24 BeadChip SNP arrays were used to analyze the DNA samples. Integragen SA (Evry, France) carried out hybridization, according to the manufacturer's recommendations. The BeadStudio software (Illumina) was used to normalize raw fluorescent signals and to obtain log R ratio (LRR) and B allele frequency (BAF) values. Asymmetry in BAF signals due to bias between the two dyes used in Illumina assays was corrected using the tQN normalization procedure (Staaf et al, 2008). We used the circular binary segmentation algorithm (Venkatraman & Olshen, 2007) to segment genomic profiles and assign corresponding smoothed values of log R ratio and B allele frequency. The Genome Alteration Print (GAP) method was used to determine the ploidy of each sample, the level of contamination with normal cells, and the allele‐specific copy number of each segment (Popova et al, 2009). Chromosomal instability index (CIN) was estimated by the mean number of SNP probes with a loss or gained status normalized by chromosomes length. SNP data are available through ArrayExpress (http://www.ebi.ac.uk/arrayexpress) under accession E‐MTAB‐5006.
Chemograms
Cells were screened for their chemosensitivity to JQ1 compound (Sigma‐Aldrich, St Louis, MO, USA). Five thousand cells per well were plated in 96‐well plates in SFDM medium. Twenty‐four hours later, the media was supplemented with increasing concentrations of JQ1 and incubated for an additional 72‐h period. Each experiment was done in triplicate and repeated at least three times. Ten increasing concentrations of JQ1 were used ranging from 0 to 30 μM.
Spheroids outgrowth assay
Fifteen thousand cells per well were seeded in 96‐well round bottom plates with medium containing 20% methylcellulose (Sigma‐Aldrich, St Louis, MO, USA). After 48‐h incubation, cells with spheroids of uniform size and shape were incubated with 2 μM JQ1 during 72 h. Images were captured every day with an Evos microscope, equipped with a 4×/N.A. (0.13) objective lens (Thermo Fisher Scientifics, Waltham, MA, USA). Spheroids volumes were determined by the following equation: Vspheroids = 4/3 π*r3. Results were expressed as a percentage of spheroid growth compared with no treatment condition (DMSO 0.05%).
Viability assays
Cell viability was estimated after addition of PrestoBlue™ reagent (Life Technologies, Carlsbad, CA, USA) for 3 h, following the supplier protocol. For spheroid viability assays, the viability was estimated after addition of CellTiter‐Glo® 3D for 1 h, following the supplier protocol (Promega, Madison, USA).
Proteins extraction and Western blotting
Cells were washed with ice‐cold PBS and lysed in Laemmli sodium dodecyl sulfate‐sample buffer (90 mM Tris–HCl [pH 6.8], 2.5% sodium dodecyl sulfate, 15% glycerol). Samples were then boiled and sonicated, and protein concentrations were determined using the bicinchoninic acid (BCA) assay (Bio‐Rad, Hercules, CA, USA) with bovine serum albumin as standard. β‐mercaptoethanol and bromophenol blue were then added to a final concentration of 1% and 0.005%, respectively. Proteins (20 μg) were separated by SDS–PAGE in 10% or 12.5% gels and were detected immunologically following electro‐transfer onto equilibrated PVDF (Imobilon‐P membranes, Millipore, Billerica, MA, USA). PVDF membranes were stained with Ponceau Red to assure a correct transfer of proteins and molecular weight markers. Membranes were blocked in PBS containing 5% powdered milk and 0.05% Tween‐20 for 1 h at 25°C. Membranes were then incubated overnight at 4°C with primary antibodies in blocking solution and thereafter with horseradish peroxidase‐conjugated IgG for 1 h. Blots were visualized using the Amersham ECL system. The c‐MYC antibody was purified from hybridomas clone 9E10 and used at 1/500 (ATCC® CRL‐1729, ATCC France). The p27kip1 antibody (C19) was purchased from Santa Cruz and used at 1/1,000. The cleaved caspase‐3 (Asp175) antibodies was purchased from Cell Signaling (#9661) and used at 1/500. The β‐actin antibody (AC‐74) was purchased from Sigma‐Aldrich and used at 1/10,000.
Real‐time quantitative PCR
Total RNA (1 μg) was used as a template for cDNA synthesis, using the GoScript™ reverse transcription kit (Promega, Madison, WI, USA). The GoTaq® qPCR 2X Master Mix (Promega, Madison, WI, USA) that include all components for quantitative PCR, except sample DNA, primers and water, was used to quantify the sixteen MYC‐high signature markers. Primer lists for each transcript are available in Appendix Table S2. Reaction conditions were denaturation at 95°C for 2 min; 40 cycles of 15 s at 95°C, 45 s at 60°C. Reactions were carried out using the AriaMx real‐time PCR system and analyzed using the AriaMx software v1.1 (Agilent Technologies, Santa Clara, CA, USA).
Ki67 staining and quantification
Full‐thickness, 5‐μm sections were cut from formalin‐fixed, paraffin‐embedded blocks from all 55 PDX. The samples were then stained with the Ki67 antibody (MIB‐1 clone, 1:160; Dako, France) using tonsillar tissue as a positive control. Negative controls were run simultaneously with the primary antibody replaced with a buffer. Antigen retrieval was conducted in citrate buffer at pH 6.0 under pressure for 3 min. Envision Dual Link Kit (Dako) was used for detection, with diaminobenzidine (DAB) as the chromogen and hematoxylin as the counterstain. Staining was considered positive when brown nuclear labeling was observed in epithelial compartment. A standard Olympus BX41 microscope (Olympus Corp., Tokyo, Japan) was used to identify tumor hot spots in each case. The percentage of tumor cell staining was independently counted by three reviewers (BB, MB, and ND) and with “eyeballing” methodology.
Differentiation scoring
Formalin‐fixed tumors were submitted to the Hôpital Nord histology core facility, and paraffin‐embedded sections were cut for hematoxylin and eosin (H&E) staining. For histopathologic scoring, H&E‐stained slides were scored for the penetrance of each histological hallmark on a scale of 0–2. The predominant tumor phenotype gave the pathological score for the whole tumor (0 = poorly differentiated, 1 = moderately differentiated, 2 = well differentiated).
Method for normalizing expression ratios
Considering the expression of upregulated gene i in patient α as u iα and the expression of downregulated gene j in patient α as d jα. We calculate the sum of expression of each marker in all patients:
The mean centered normalized expression was then calculated for the two set of markers:
The normalized ratio between upregulated gene U i and downregulated gene D j is:
The median of the 60 normalized ratios is then calculated:
show a MYC‐high profile and show a MYC‐low profile.
Treatment with JQ1 of PDX with MYC‐high and MYC‐low phenotype in vivo
We transplanted four PDX samples with the MYC‐high phenotype (CRCM16, CRCM04, CRCM114, and CRCM116) and four with the MYC‐low phenotype (CRCM05, CRCM10, CRCM109, and CRCM112) subcutaneously into 6‐week‐old male Swiss nude mice (Crl: Nu(lco)‐Foxn1nu, Charles River, Wilmington, MA). Each PDX sample was inoculated into six nude NMRI mice who were randomized for treatment (n = 3) and vehicle (n = 3). When the PDX reached 100 mm3, we started the treatment with JQ1 or vehicle and followed their growth for 28 days. JQ1 was prepared according to published procedures (Filippakopoulos et al, 2010) and administered intraperitoneally every day at 50 mg/kg. Tumor size was measured with a Vernier caliper twice weekly, and the tumor volume was calculated with the equation v = (length/width2)/2.
Statistical analysis
Overall survival and relapse‐free survival were analyzed using the Kaplan–Meier log‐rank test to assess differences in survival. Box‐and‐whisker plots show the medians, quartiles, and range of continuous data to demonstrate the variability of data and the degree of normality. For continuous variables, non‐parametric unpaired two‐tailed t‐test was performed under the assumption of equal variance.
Author contributions
BB, MB, OG, CL, AM, RN, YB, LM, and AM performed experiments. MGil, VM, OT, J‐RD, MGio, PG, MGas, MO, JEB, and JQ provided material. SG, VS, MR, and FP performed the histochemical stainings and immunohistochemical scoring. J‐LR, GL, RU, AS, ND, and JI performed or coordinated the bioinformatic and biostatistical analysis. RU, ND, and JI contributed to the experimental design, data analysis, and discussion. ND and JI directed the project. BB, RU, ND, and JI wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
The paper explained.
Problem
One strategy to discover potential markers for patients stratification is to focus on identifying pathways that are deregulated in tumors, particularly when tumor cells absolutely depend of keeping these alterations (e.g., oncogene “dependence” to survive and grow). Consequently, it is logical to assume that blockage of these pathways with specific inhibitors should lead to cell growth arrest, death, and tumor regression. Using this rational, it would be possible to select, by means of a few markers, a particular subgroup of patients “addicted” to a distinct pathways, a major goal of modern individualized medicine.
Results
To define classifier expression signature, we selected a group of 10 c‐MYC target transcripts whose expression is increased in the MYC‐high group and six transcripts increased in the MYC‐low group. We validated the ability of these markers panel to identify MYC‐high patient‐derived xenografts from both: discovery and validation cohorts as well as primary cell cultures from the same patients. We then showed that cells from MYC‐high patients are more sensitive to BET inhibitor treatment compared to MYC‐low cells, in both monolayers, 3D cultured spheroids and in xenografted tumors, due to cell cycle arrest followed by apoptosis.
Impact
The results provide new markers and potentially novel therapeutic modalities for distinct subgroups of pancreatic tumors and may find application to the future management of these patients within the setting of individualized medicine clinics.
Supporting information
Appendix
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Dataset EV6
Source Data for Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
We thank Emilie Nouguerede, Jihane Pakradouni and members of the cell culture platform (CRCM, Unité 1068) for their technical assistance. This work was supported by La Ligue Contre le Cancer, INCa, Canceropole PACA, DGOS (labellisation SIRIC) and INSERM to JI.
EMBO Mol Med (2017) 9: 482–497
Contributor Information
Nelson Dusetti, Email: nelson.dusetti@inserm.fr.
Juan Iovanna, Email: juan.iovanna@inserm.fr.
References
- Annibali D, Whitfield JR, Favuzzi E, Jauset T, Serrano E, Cuartas I, Redondo‐Campos S, Folch G, Gonzalez‐Junca A, Sodir NM et al (2014) Myc inhibition is effective against glioma and reveals a role for Myc in proficient mitosis. Nat Commun 5: 4632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asangani IA, Dommeti VL, Wang X, Malik R, Cieslik M, Yang R, Escara‐Wilke J, Wilder‐Romans K, Dhanireddy S, Engelke C et al (2014) Therapeutic targeting of BET bromodomain proteins in castration‐resistant prostate cancer. Nature 510: 278–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I (2001) Controlling the false discovery rate in behavior genetics research. Behav Brain Res 125: 279–284 [DOI] [PubMed] [Google Scholar]
- Burris HA III, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo AM, Tarassoff P et al (1997) Improvements in survival and clinical benefit with gemcitabine as first‐line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol 15: 2403–2413 [DOI] [PubMed] [Google Scholar]
- Cohen R, Neuzillet C, Tijeras‐Raballand A, Faivre S, de Gramont A, Raymond E (2015) Targeting cancer cell metabolism in pancreatic adenocarcinoma. Oncotarget 6: 16832–16847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y, Adenis A, Raoul JL, Gourgou‐Bourgade S, de la Fouchardiere C et al (2011) FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 364: 1817–1825 [DOI] [PubMed] [Google Scholar]
- Dai MS, Jin Y, Gallegos JR, Lu H (2006) Balance of Yin and Yang: ubiquitylation‐mediated regulation of p53 and c‐Myc. Neoplasia 8: 630–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV (1999) c‐Myc target genes involved in cell growth, apoptosis, and metabolism. Mol Cell Biol 19: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV (2012) MYC on the path to cancer. Cell 149: 22–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi JW, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J et al (2011) BET bromodomain inhibition as a therapeutic strategy to target c‐Myc. Cell 146: 903–916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duconseil P, Gilabert M, Gayet O, Loncle C, Moutardier V, Turrini O, Calvo E, Ewald J, Giovannini M, Gasmi M et al (2015) Transcriptomic analysis predicts survival and sensitivity to anticancer drugs of patients with a pancreatic adenocarcinoma. Am J Pathol 185: 1022–1032 [DOI] [PubMed] [Google Scholar]
- Dunne RF, Hezel AF (2015) Genetics and biology of pancreatic ductal adenocarcinoma. Hematol Oncol Clin North Am 29: 595–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I et al (2010) Selective inhibition of BET bromodomains. Nature 468: 1067–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher S, Prochownik EV (2015) Small‐molecule inhibitors of the Myc oncoprotein. Biochim Biophys Acta 1849: 525–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia PL, Miller AL, Kreitzburg KM, Council LN, Gamblin TL, Christein JD, Heslin MJ, Arnoletti JP, Richardson JH, Chen D et al (2016) The BET bromodomain inhibitor JQ1 suppresses growth of pancreatic ductal adenocarcinoma in patient‐derived xenograft models. Oncogene 35: 833–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heller A, Gaida MM, Mannle D, Giese T, Scarpa A, Neoptolemos JP, Hackert T, Strobel O, Hoheisel JD, Giese NA et al (2015) Stratification of pancreatic tissue samples for molecular studies: RNA‐based cellular annotation procedure. Pancreatology 15: 423–431 [DOI] [PubMed] [Google Scholar]
- Huang CH, Lujambio A, Zuber J, Tschaharganeh DF, Doran MG, Evans MJ, Kitzing T, Zhu N, de Stanchina E, Sawyers CL et al (2014) CDK9‐mediated transcription elongation is required for MYC addiction in hepatocellular carcinoma. Genes Dev 28: 1800–1814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry RA, Hobbs B, Collin F, Beazer‐Barclay YD, Antonellis KJ, Scherf U, Speed TP (2003) Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4: 249–264 [DOI] [PubMed] [Google Scholar]
- Jemal A, Murray T, Ward E, Samuels A, Tiwari RC, Ghafoor A, Feuer EJ, Thun MJ (2005) Cancer statistics, 2005. CA Cancer J Clin 55: 10–30 [DOI] [PubMed] [Google Scholar]
- Jung M, Philpott M, Muller S, Schulze J, Badock V, Eberspacher U, Moosmayer D, Bader B, Schmees N, Fernandez‐Montalvan A et al (2014) Affinity map of bromodomain protein 4 (BRD4) interactions with the histone H4 tail and the small molecule inhibitor JQ1. J Biol Chem 289: 9304–9319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandela I, Jin HY, Owen K, Biol RPC (2015) Registered report: BET bromodomain inhibition as a therapeutic strategy to target c‐Myc. eLife 4: e07072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoechel B, Roderick JE, Williamson KE, Zhu J, Lohr JG, Cotton MJ, Gillespie SM, Fernandez D, Ku M, Wang H et al (2014) An epigenetic mechanism of resistance to targeted therapy in T cell acute lymphoblastic leukemia. Nat Genet 46: 364–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koay EJ, Amer AM, Baio FE, Ondari AO, Fleming JB (2016) Toward stratification of patients with pancreatic cancer: past lessons from traditional approaches and future applications with physical biomarkers. Cancer Lett 381: 237–243 [DOI] [PubMed] [Google Scholar]
- Korb E, Herre M, Zucker‐Scharff I, Darnell RB, Allis CD (2015) BET protein Brd4 activates transcription in neurons and BET inhibitor Jq1 blocks memory in mice. Nat Neurosci 18: 1464–1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin WC, Rajbhandari N, Liu C, Sakamoto K, Zhang Q, Triplett AA, Batra SK, Opavsky R, Felsher DW, DiMaio DJ et al (2013) Dormant cancer cells contribute to residual disease in a model of reversible pancreatic cancer. Cancer Res 73: 1821–1830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancias JD, Kimmelman AC (2011) Targeting autophagy addiction in cancer. Oncotarget 2: 1302–1306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazur PK, Herner A, Mello SS, Wirth M, Hausmann S, Sanchez‐Rivera FJ, Lofgren SM, Kuschma T, Hahn SA, Vangala D et al (2015) Combined inhibition of BET family proteins and histone deacetylases as a potential epigenetics‐based therapy for pancreatic ductal adenocarcinoma. Nat Med 21: 1163–1171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeown MR, Bradner JE (2014) Therapeutic strategies to inhibit MYC. Cold Spring Harb Perspect Med 4: a014266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, Mele DA, Bergeron L, Sims RJ (2011) Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc Natl Acad Sci USA 108: 16669–16674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton JP, Sansom OJ (2013) MYC‐y mice: from tumour initiation to therapeutic targeting of endogenous MYC. Mol Oncol 7: 248–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesbit CE, Tersak JM, Prochownik EV (1999) MYC oncogenes and human neoplastic disease. Oncogene 18: 3004–3016 [DOI] [PubMed] [Google Scholar]
- Noll EM, Eisen C, Stenzinger A, Espinet E, Muckenhuber A, Klein C, Vogel V, Klaus B, Nadler W, Rosli C et al (2016) CYP3A5 mediates basal and acquired therapy resistance in different subtypes of pancreatic ductal adenocarcinoma. Nat Med 22: 278–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popova T, Manie E, Stoppa‐Lyonnet D, Rigaill G, Barillot E, Stern MH (2009) Genome Alteration Print (GAP): a tool to visualize and mine complex cancer genomic profiles obtained by SNP arrays. Genome Biol 10: R128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prendergast GC (1999) Mechanisms of apoptosis by c‐Myc. Oncogene 18: 2967–2987 [DOI] [PubMed] [Google Scholar]
- Puissant A, Frumm SM, Alexe G, Bassil CF, Qi J, Chanthery YH, Nekritz EA, Zeid R, Gustafson WC, Greninger P et al (2013) Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer Discov 3: 308–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roderick JE, Tesell J, Shultz LD, Brehm MA, Greiner DL, Harris MH, Silverman LB, Sallan SE, Gutierrez A, Look AT et al (2014) c‐Myc inhibition prevents leukemia initiation in mice and impairs the growth of relapsed and induction failure pediatric T‐ALL cells. Blood 123: 1040–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saborowski M, Saborowski A, Morris JPT, Bosbach B, Dow LE, Pelletier J, Klimstra DS, Lowe SW (2014) A modular and flexible ESC‐based mouse model of pancreatic cancer. Genes Dev 28: 85–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancho P, Burgos‐Ramos E, Tavera A, Bou Kheir T, Jagust P, Schoenhals M, Barneda D, Sellers K, Campos‐Olivas R, Grana O et al (2015) MYC/PGC‐1alpha balance determines the metabolic phenotype and plasticity of pancreatic cancer stem cells. Cell Metab 22: 590–605 [DOI] [PubMed] [Google Scholar]
- Schleger C, Verbeke C, Hildenbrand R, Zentgraf H, Bleyl U (2002) c‐MYC activation in primary and metastatic ductal adenocarcinoma of the pancreas: incidence, mechanisms, and clinical significance. Mod Pathol 15: 462–469 [DOI] [PubMed] [Google Scholar]
- Schmidt EV (1999) The role of c‐myc in cellular growth control. Oncogene 18: 2988–2996 [DOI] [PubMed] [Google Scholar]
- Schreiber FS, Deramaudt TB, Brunner TB, Boretti MI, Gooch KJ, Stoffers DA, Bernhard EJ, Rustgi AK (2004) Successful growth and characterization of mouse pancreatic ductal cells: functional properties of the Ki‐RAS(G12V) oncogene. Gastroenterology 127: 250–260 [DOI] [PubMed] [Google Scholar]
- Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR (2000) Multiple Ras‐dependent phosphorylation pathways regulate Myc protein stability. Genes Dev 14: 2501–2514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SG, Zhou MM (2016) The bromodomain: a new target in emerging epigenetic medicine. ACS Chem Biol 11: 598–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soucek L, Whitfield J, Martins CP, Finch AJ, Murphy DJ, Sodir NM, Karnezis AN, Swigart LB, Nasi S, Evan GI (2008) Modelling Myc inhibition as a cancer therapy. Nature 455: 679–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staaf J, Vallon‐Christersson J, Lindgren D, Juliusson G, Rosenquist R, Hoglund M, Borg A, Ringner M (2008) Normalization of Illumina Infinium whole‐genome SNP data improves copy number estimates and allelic intensity ratios. BMC Bioinformatics 9: 409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stathis A, Zucca E, Bekradda M, Gomez‐Roca C, Delord JP, de La Motte Rouge T, Uro‐Coste E, de Braud F, Pelosi G, French CA (2016) Clinical response of carcinomas harboring the BRD4‐NUT oncoprotein to the targeted bromodomain inhibitor OTX015/MK‐8628. Cancer Discov 6: 492–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES et al (2005) Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA 102: 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trabucco SE, Gerstein RM, Evens AM, Bradner JE, Shultz LD, Greiner DL, Zhang H (2015) Inhibition of bromodomain proteins for the treatment of human diffuse large B‐cell lymphoma. Clin Cancer Res 21: 113–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatraman ES, Olshen AB (2007) A faster circular binary segmentation algorithm for the analysis of array CGH data. Bioinformatics 23: 657–663 [DOI] [PubMed] [Google Scholar]
- Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, Johns AL, Miller D, Nones K, Quek K et al (2015) Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 518: 495–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walz S, Lorenzin F, Morton J, Wiese KE, von Eyss B, Herold S, Rycak L, Dumay‐Odelot H, Karim S, Bartkuhn M et al (2014) Activation and repression by oncogenic MYC shape tumour‐specific gene expression profiles. Nature 511: 483–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth M, Schneider G (2016) MYC: a stratification marker for pancreatic cancer therapy. Trends Cancer 2: 1–3 [DOI] [PubMed] [Google Scholar]
- Witkiewicz AK, McMillan EA, Balaji U, Baek G, Lin WC, Mansour J, Mollaee M, Wagner KU, Koduru P, Yopp A et al (2015) Whole‐exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun 6: 6744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yachida S, Iacobuzio‐Donahue CA (2013) Evolution and dynamics of pancreatic cancer progression. Oncogene 32: 5253–5260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Blenis J, Yuan J (2008) Activation of PI3K/Akt and MAPK pathways regulates Myc‐mediated transcription by phosphorylating and promoting the degradation of Mad1. Proc Natl Acad Sci USA 105: 6584–6589 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Dataset EV6
Source Data for Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6