

Abstract
How MYC reprograms metabolism in primary tumors remains poorly understood. Using integrated gene expression and metabolite profiling, we identify six pathways that are coordinately deregulated in primary MYC‐driven liver tumors: glutathione metabolism; glycine, serine, and threonine metabolism; aminoacyl‐tRNA biosynthesis; cysteine and methionine metabolism; ABC transporters; and mineral absorption. We then focus our attention on glutathione (GSH) and glutathione disulfide (GSSG), as they are markedly decreased in MYC‐driven tumors. We find that fewer glutamine‐derived carbons are incorporated into GSH in tumor tissue relative to non‐tumor tissue. Expression of GCLC, the rate‐limiting enzyme of GSH synthesis, is attenuated by the MYC‐induced microRNA miR‐18a. Inhibition of miR‐18a in vivo leads to increased GCLC protein expression and GSH abundance in tumor tissue. Finally, MYC‐driven liver tumors exhibit increased sensitivity to acute oxidative stress. In summary, MYC‐dependent attenuation of GCLC by miR‐18a contributes to GSH depletion in vivo, and low GSH corresponds with increased sensitivity to oxidative stress in tumors. Our results identify new metabolic pathways deregulated in primary MYC tumors and implicate a role for MYC in regulating a major antioxidant pathway downstream of glutamine.
Keywords: cancer, glutathione, metabolism, miRNA, MYC
Subject Categories: Cancer, Metabolism
Introduction
Metabolic reprogramming is a hallmark of MYC‐driven tumor growth and maintenance 1. Most notably, MYC orchestrates tumor cell dependence on glucose and glutamine for biomass accumulation 1, 2, 3, 4. MYC regulates many points in glycolysis through transcriptional regulation of glycolytic genes. For example, MYC regulates the GLUT1 transporter 5 and glycolytic enzymes such as LDHA 6 and PKM2 7. Expression of PKM2 in cancer has been suggested to enhance proliferative metabolism by causing a “backflow” of metabolites into anabolic pathways such as the serine biosynthesis pathway 8.
MYC also regulates glutaminolysis at multiple nodes. Many MYC‐overexpressing cell lines are addicted to glutamine 9, 10 and primary MYC‐driven murine lung and liver tumors display increased glutamine metabolism 11. Expression of the glutamine transporter Slc1a5 is regulated by MYC and is elevated in MYC‐driven liver tumors 11, 12. MYC also activates expression of glutamine synthetase through promoter demethylation in some tumors 13. Glutaminase (GLS), the enzyme that catalyzes the conversion of glutamine to glutamate, is upregulated in lymphoma cells due to MYC‐dependent inhibition of miR‐23a/b expression 14. GLS inhibition in cell lines or xenograft tumors causes MYC‐dependent cell death or reduced proliferation 11, 12, 15. Elevated glutamine uptake and metabolism can contribute to TCA cycle flux and glutathione synthesis in MYC‐expressing cells 12, 14, 15. However, whether MYC regulates specific pathways downstream of glutamine conversion to glutamate in tumor cells remains unknown.
Most work to uncover MYC's role in metabolism to date has been conducted using cultured tumor cells that are removed from the native tumor environment 3, 4. However, cell culture conditions may not accurately reflect nutrient availability and uptake in the host tissue. In this study, we sought to identify novel metabolic pathways that are altered in MYC‐driven primary tumors in vivo. MYC drives human hepatocarcinogenesis 16, is frequently amplified or overexpressed in human hepatocellular carcinoma (HCC), and is associated with poorly differentiated tumors and poor liver cancer prognosis 17, 18, 19, 20, 21. We thus sought to characterize altered metabolism in a mouse model of MYC‐driven liver cancer. We performed mRNA gene expression and mass spectrometry‐based biochemical profiling of tumor samples from the LAP‐tTA × TetO‐MYC (LT2‐MYC) bi‐transgenic mouse, a conditional model that drives MYC expression and tumorigenesis specifically in hepatocytes 22, 23 (Fig 1A). We recently showed that this model has gene expression changes consistent with aggressive, poorly differentiated human liver cancers 24. Use of this model allows us to observe MYC‐mediated metabolic reprogramming in the native tumor environment 23. Additionally, we can take advantage of its conditional nature to identify changes that are a direct effect of MYC signaling in vivo (Fig 1A). Our work identifies a novel role for MYC in regulating the synthesis of glutathione, a major cellular antioxidant, via miR‐18a in primary tumors. This finding has implications for the use of oxidative stress‐inducing drugs for therapy of MYC liver tumors.
Figure 1. Integrated metabolic analysis of MYC‐driven liver tumors.
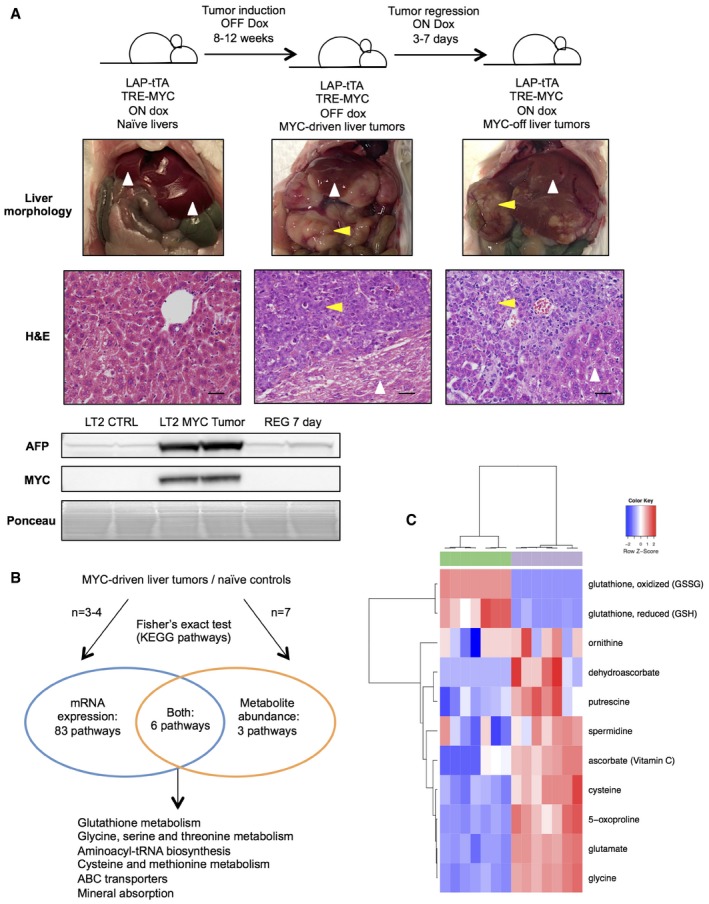
- Summary of LT2‐MYC conditional transgenic mouse model of MYC‐induced hepatocarcinogenesis. Prolonged MYC overexpression induces tumor nodules that are morphologically and histologically distinct from non‐tumor tissue. MYC protein expression can be turned off in established tumors and correlates with alpha‐fetoprotein (AFP) expression, a marker of aggressive liver cancer (see REG 7 day Western blot). In images, white arrows indicate non‐tumor liver tissue and yellow arrows indicate liver tumor tissue. Scale bars in hematoxylin and eosin‐stained (H&E) sections represent 20 μm.
- Transcriptional and biochemical profiling analyses identify six pathways that are significantly altered in LT2‐MYC tumors versus control livers (n = 3 LT2 control and n = 4 LT2‐MYC for transcriptional profiling, n = 7 in each group for biochemical profiling, Fisher's exact test, P < 0.05).
- Glutathione pathway (KEGG #ko00480) metabolite abundances segregate LT2‐MYC tumors from control livers by unsupervised hierarchical clustering (n = 7 in each group, LT2 control liver samples in green, LT2‐MYC tumor samples in gray).
Source data are available online for this figure.
Results
Integrated metabolic analysis of MYC‐driven liver tumors
To identify novel metabolic pathways that are altered in primary liver tumors with high MYC expression (Fig 1A), we performed mRNA expression and mass spectrometry‐based metabolite profiling of LT2‐MYC tumor samples and naïve LT2 liver controls (Fig 1B and Dataset EV1). Of 333 detected metabolites with KEGG PATHWAY database identifiers 25, 188 were significantly altered in LT2‐MYC tumors versus control liver tissue (FDR < 0.05). Likewise, 3,706 genes with KEGG identifiers exhibited significant deregulation in LT2‐MYC tumors versus controls (FDR < 0.05). We performed pathway enrichment analysis, referencing all metabolic pathways defined by KEGG, of the significantly altered transcripts and metabolites. We identified six KEGG pathways that were significantly altered, both transcriptionally and biochemically, in LT2‐MYC tumors compared to control liver tissues: glutathione metabolism; glycine, serine, and threonine metabolism; aminoacyl‐tRNA biosynthesis; cysteine and methionine metabolism; ABC transporters; and mineral absorption (Fig 1B and Appendix Figs S1–S6).
Of the six KEGG pathways that were coordinately deregulated, glutathione metabolism exhibited the most significantly altered gene expression and the second highest rate of metabolite abundance change (Appendix Table S1). Glutathione (GSH) is an important cellular antioxidant synthesized from glutamine carbons 26. In accordance with our integrated analyses, altered glutathione pathway metabolites and transcripts readily segregate LT2‐MYC tumors from LT2 control livers by unsupervised hierarchical clustering (Figs 1C and EV1A, respectively). Glutathione (GSH) and glutathione disulfide (GSSG) were among the most dramatically depleted metabolites profiled in the MYC‐driven tumors; we independently confirmed the depletion of total glutathione (GSH + GSSG) in the same MYC liver tumor samples using an enzymatic assay (Fig 2A). Interestingly, the depletion of GSH and GSSG was observed in murine liver tumors driven by MYC but not in those driven by RAS (LT2‐RAS model described in 24) (Fig EV1B and C). This indicates that GSH depletion is not due to liver tumorigenesis in general, but instead is MYC oncogene‐specific.
Figure EV1. Glutathione metabolism is altered in MYC‐driven liver tumors.
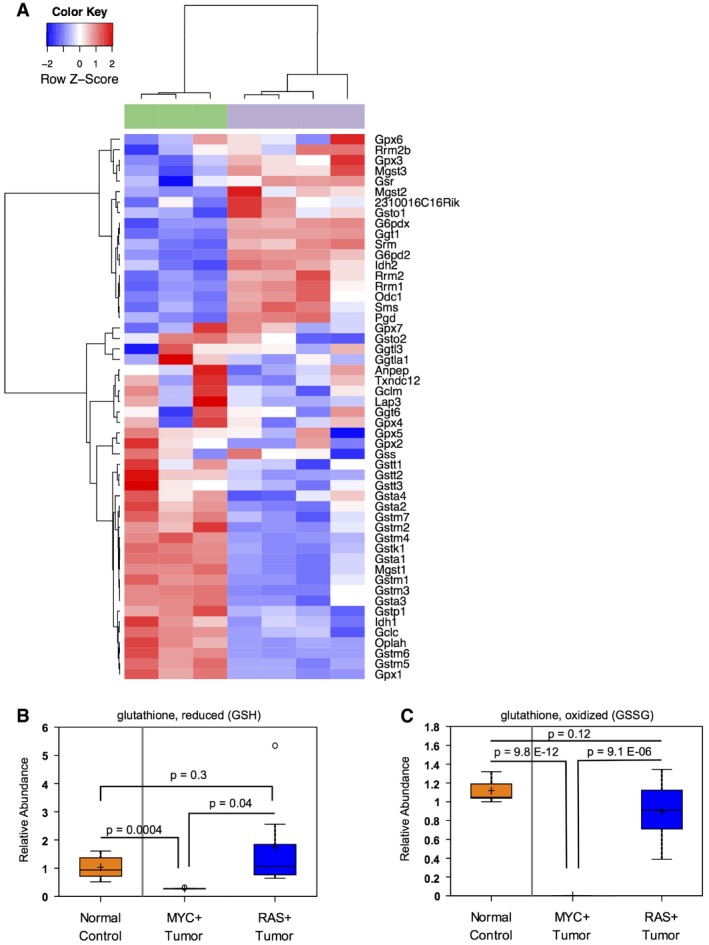
-
AGlutathione pathway transcript expression segregates LT2‐MYC tumors from control livers by unsupervised hierarchical clustering (n = 3 control livers in green, n = 4 tumors in gray).
-
B, CRelative metabolite abundance of GSH (B) or GSSG (C) in tissue samples from murine liver tumors driven by MYC or RAS, as compared to normal liver controls (n = 7 control liver, n = 7 MYC tumor, n = 7 RAS tumor, data represented as box plots with horizontal bar representing the median, box ranges representing the first (bottom) and third (top) quartiles, and vertical bars representing the standard error, unpaired two‐tailed t‐test, Bonferroni adjusted P‐value for multiple comparisons is 0.02).
Figure 2. Characterization of aberrant glutathione metabolism in MYC‐driven liver tumors.
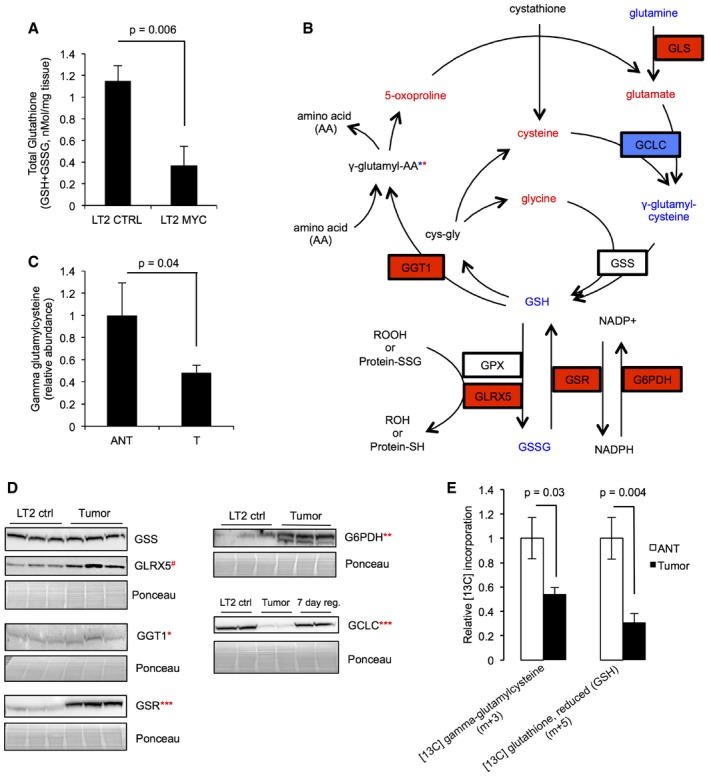
- Total glutathione (GSH + GSSG) measured by enzymatic assay in LT2‐MYC tumors versus control livers (n = 5 LT2 control samples, n = 6 LT2‐MYC tumor samples, data represented as mean ± SEM, unpaired two‐tailed t‐test, P = 0.006).
- Multiple metabolites and enzymes in the glutathione metabolism pathway are significantly altered in LT2‐MYC tumors versus control livers (unpaired two‐tailed t‐test, P < 0.1). Red = significantly elevated at P < 0.1, blue = significantly depleted at P < 0.05, and red and blue asterisks indicate that individual gamma‐glutamyl amino acids are significantly increased or decreased at P < 0.05. Increased protein expression of the GLS1 isoform of glutaminase was previously reported for LT2‐MYC tumors 11.
- Gamma‐glutamylcysteine abundance in MYC‐driven tumors as compared to adjacent non‐tumor tissue (n = 6 each group, data represented as normalized mean ± SEM, paired one‐tailed t‐test, P = 0.04).
- Western blot analysis of key enzymes involved in the glutathione metabolism pathway in LT2‐MYC tumors versus non‐tumor LT2 controls (n = 2–3 each as indicated in images, unpaired two‐tailed t‐test on normalized expression, GSS P = 0.7, GLRX5 # P = 0.09, GGT1 *P = 0.05, GSR ***P = 0.0004, G6PDH **P = 0.001, GCLC ***P = 0.0004). For GCLC, LT2‐MYC tumors regressed for 7 days by feeding doxycycline chow are also shown.
- Relative incorporation of [U‐13C]‐glutamine into gamma‐glutamylcysteine and GSH in MYC‐driven tumors compared to adjacent non‐tumor liver tissue (n = 6 each group, data represented as normalized mean ± SEM, unpaired two‐tailed t‐test, gamma‐glutamylcysteine P = 0.03, GSH P = 0.004).
Source data are available online for this figure.
Characterization of aberrant glutathione metabolism in MYC‐driven liver tumors
We next sought to identify likely reasons for GSH depletion in MYC‐driven liver tumors. Decreased GSH synthesis, increased gamma‐glutamyl cycling, and increased protein S‐glutathionylation may all contribute to low tissue GSH 26.
Our metabolic profiling found that abundance of glutamine, GSH, and GSSG decreased while abundance of glutamate, cysteine, and glycine increased in MYC tumors relative to non‐tumors (Fig 2B and Appendix Fig S1B). Decreased glutamine concomitant with elevated glutamate is consistent with elevated GLS expression and activity and has been previously described in LT2‐MYC tumors 11. We performed a separate metabolic analysis and found that gamma‐glutamylcysteine, the direct precursor of GSH, is depleted in MYC‐driven tumors relative to non‐tumor tissue (Fig 2B and C). Taken together, these data indicate a bottleneck in GSH production, particularly at the step catalyzed by glutamate‐cysteine ligase, catalytic subunit (GCLC) (Fig 2B).
Our metabolic profiling did not strongly suggest that gamma‐glutamyl cycling was elevated in MYC tumors relative to non‐tumor controls. Although 5‐oxoproline was significantly elevated and multiple individual gamma‐glutamyl amino acids were significantly altered in MYC tumors relative to non‐tumors, the gamma‐glutamyl amino acids were not uniformly altered—some were elevated and some were depleted (Figs 2B and EV2A). Further, we did not find evidence for elevated S‐glutathionylation in the tumors. Our metabolic profiling indicated that both cysteine–glutathione disulfide and S‐methylglutathione were dramatically depleted in MYC‐driven liver tumors relative to non‐tumor controls (Fig EV2B). Taken together, our biochemical profiling data strongly suggest that impairment of GSH production, rather than elevated gamma‐glutamyl cycling or S‐glutathionylation, contributes to GSH loss in the MYC‐driven tumors.
Figure EV2. Characterization of gamma‐glutamyl amino acid and S‐glutathionylation abundance in MYC‐driven liver tumors.
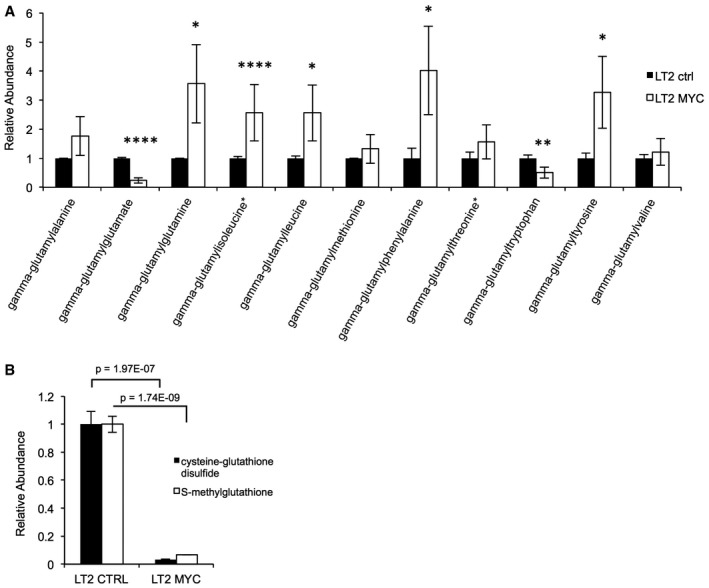
- Metabolite profiling of gamma‐glutamyl amino acids in LT2‐MYC tumors versus control livers (n = 7 control liver, n = 7 MYC tumor, data represented as normalized mean ± SEM, unpaired two‐tailed t‐test, *P < 0.05, **P < 0.01, ****P < 0.00001).
- Metabolite profiling of cysteine–glutathione disulfide and S‐methylglutathione in LT2‐MYC tumors versus control livers (n = 7 control liver, n = 7 MYC tumor, data represented as normalized mean ± SEM, unpaired two‐tailed t‐test, cysteine–glutathione disulfide P = 1.97305E‐07, S‐methylglutathione P = 1.73948E‐09).
We next sought to characterize the expression of enzymes that regulate GSH metabolism (Fig 2B). We performed Western blot analysis to determine the protein expression of several key GSH pathway enzymes, including GCLC; glutathione synthetase (GSS); gamma‐glutamyltransferase 1 (GGT1); glutaredoxin 5 (GLRX5); glutathione reductase (GSR); and glucose‐6‐phosphate dehydrogenase (G6PDH) (Fig 2D).
Our Western blot analysis indicated that protein expression of GLRX5, GGT1, GSR, and G6PDH increased (P ≤ 0.05 for GGT1, GSR, G6PDH; 0.05 < P < 0.10 for GLRX5), expression of GCLC markedly decreased (P < 0.001), and expression of GSS did not change in MYC‐driven liver tumors compared to naïve liver tissue (Fig 2B and D). Although significant, the increase in GGT1 expression in tumors was very small. On the other hand, substantial downregulation of GCLC, the rate‐limiting enzyme of GSH synthesis, is consistent with our hypothesis that GSH synthesis is impaired in tumors. Taken together, our metabolomic and Western blot data strongly suggest that decreased GSH synthesis contributes to depletion of free GSH in LT2‐MYC tumors. However, we could not entirely rule out contributions from elevated gamma‐glutamyl cycling and glutaredoxin activity.
Isotopic tracing of glutamine in MYC‐driven liver tumors
MYC activates expression of the glutamine transporter Slc1a5, which increases cellular uptake of glutamine 11, 12. GSH is synthesized downstream of the conversion of glutamine to glutamate by GLS, which is elevated in a MYC‐dependent manner 27 (Fig 2B). Previous studies suggest that elevated glutamine uptake contributes to GSH synthesis in MYC‐overexpressing cells 12, 14, 15. Because we saw depleted GSH concomitant with decreased GCLC expression and increased abundance of several GSH precursors in LT2‐MYC tumors, we sought to trace the flow of glutamine‐derived carbons in MYC‐driven liver tumors to confirm whether GSH synthesis is impaired. We used mice from a somatic transgenic model of MYC‐driven liver tumorigenesis 28, which also have elevated MYC and depleted GCLC protein expression (Fig EV3A). Tumor‐bearing mice were injected with fully labeled [U‐13C]‐glutamine and mass spectrometry‐based isotopic tracing of liver tumors was performed and compared to adjacent non‐tumor liver tissue (Fig EV3B and C). We observed decreased incorporation of [U‐13C]‐glutamine carbons into GSH and γ‐glutamyl‐cysteine in tumors relative to adjacent non‐tumor liver tissue (Figs 2E and EV3C). This is consistent with the diminished steady state abundances of GCLC protein (Figs 2D and EV3A) and GSH and GSSG metabolites we observed (Fig 2A). These results, together with the unchanged expression of GSS in tumors (Fig 2B and D), indicate that GSH synthesis via GCLC is impaired in MYC‐driven liver tumors.
Figure EV3. Isotopic tracing of glutamine in MYC‐driven liver tumors.
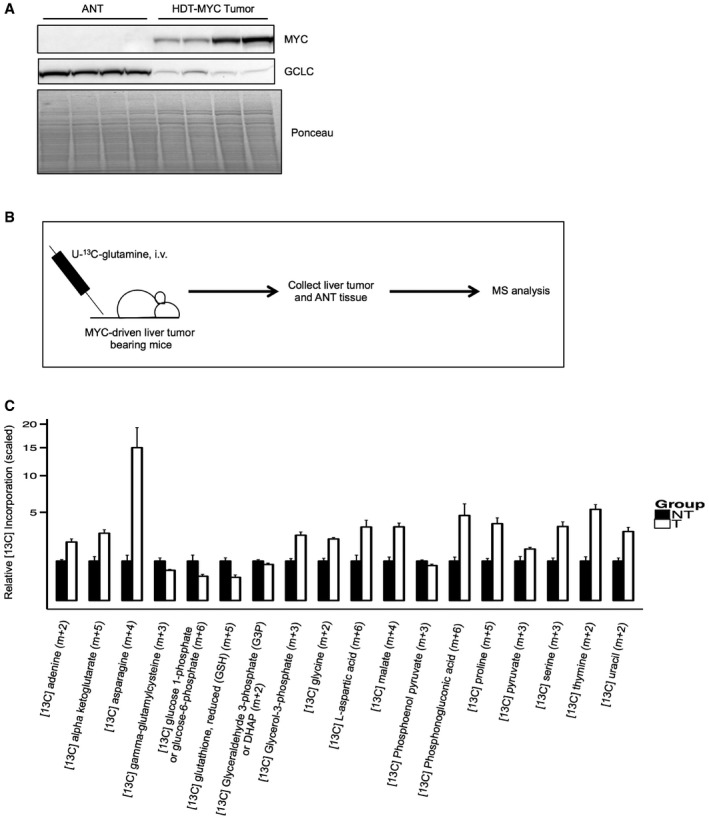
- Western blot analysis of MYC and GCLC protein expression in MYC‐driven murine liver tumors established by hydrodynamic transfection 28 (n = 4 adjacent non‐tumor (ANT), n = 4 MYC tumor).
- Schematic summary of U‐[13C]‐glutamine tracing experiment in MYC‐driven liver tumor‐bearing mice.
- Relative incorporation of [13C]‐glutamine into metabolites in MYC‐driven tumors compared to adjacent non‐tumor liver tissue (n = 6 adjacent non‐tumor (NT), n = 6 MYC tumor (T), all data represented as normalized mean ± SEM, unpaired two‐tailed t‐test, all data significant at P < 0.05).
Source data are available online for this figure.
GCLC expression is attenuated by miRNA‐18a in MYC‐driven liver tumors
Because MYC regulates numerous genes involved in tumor metabolism, we reasoned that MYC might also regulate GCLC expression to control GSH synthesis. In support of this hypothesis, we found that GCLC protein and mRNA exhibit an inverse correlation with MYC signaling in vivo. Both GCLC protein (Fig 2D) and transcript (Fig EV4A) are low in tumor tissues relative to non‐tumor tissue, and return to baseline when MYC is turned off in tumors by ad libitum doxy chow feeding (Fig 1A). Additionally, we find that total GSH levels increase in some tumors upon 72 h tumor regression, relative to tumor tissue (Fig EV4B). We further observed MYC‐dependent changes in GCLC protein expression in a murine liver tumor cell line derived from the LT2‐MYC model 29. When cells are grown in the presence of 8 ng/ml doxycycline, MYC expression is rapidly inhibited (Fig 3A). Using this conditional system, we found that GCLC protein increases when MYC is conditionally turned off over several days (Fig 3A).
Figure EV4. GCLC expression and activity are altered in MYC‐driven liver tumors.
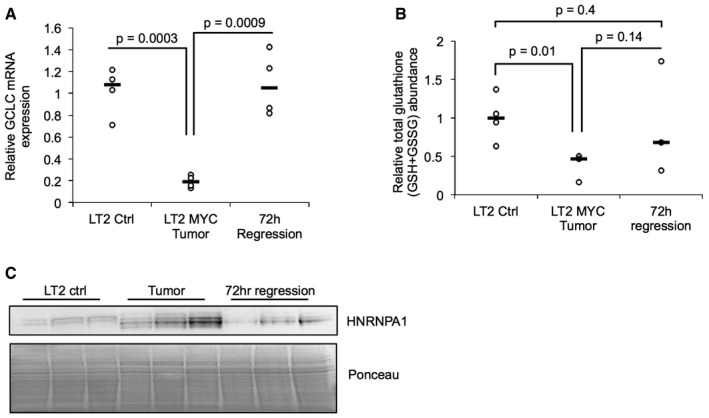
- Quantitative PCR analysis of Gclc mRNA expression in LT2‐MYC tumors, control livers, and liver tumors regressed for 72 h (n = 4 each group, data represented as univariate scatter plot with median, unpaired two‐tailed t‐test, LT2 ctrl versus LT2‐MYC P = 0.0003, LT2‐MYC versus 72 h regression P = 0.0009).
- Total glutathione (GSH + GSSG) abundance measured by enzymatic analysis in LT2 control livers, LT2‐MYC tumors, and tumors regressed for 72 h (n = 4 LT2 ctrl, n = 3 LT2‐MYC tumor, n = 3 72 h reg tumor, data represented as univariate scatter plot with median, unpaired one‐tailed t‐test, LT2 ctrl versus LT2‐MYC P = 0.01, LT2‐MYC versus 72 h reg P = 0.14, LT2 ctrl versus 72 h reg P = 0.4, Bonferroni adjusted P‐value is 0.02).
- Western blot analysis of HNRNPA1 in LT2‐MYC tumors, control livers, and tumors regressed for 72 h (n = 3 each group).
Source data are available online for this figure.
Figure 3. GCLC is attenuated by miRNA‐18a in MYC‐driven liver tumors.
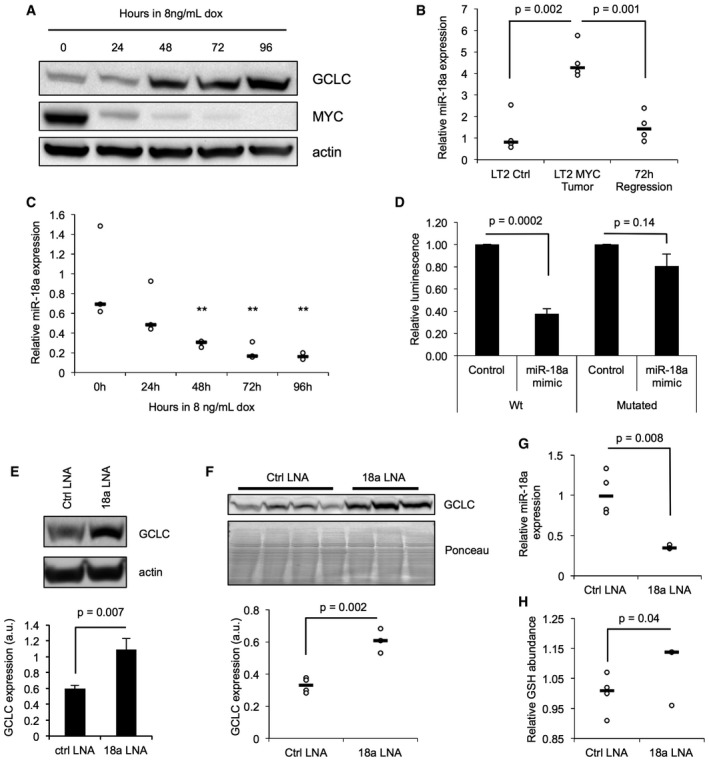
-
AWestern blot analysis of GCLC and MYC protein expression in conditional liver tumor cells derived from an LT2‐MYC tumor (Western blot is representative of a minimum of four experimental replicates).
-
BQuantitative PCR (qPCR) analysis of miR‐18a expression in LT2‐MYC tumors, control liver tissues, and tumors regressed for 3 days (n = 4 each group, data represented as univariate scatter plot with median, unpaired two‐tailed t‐test, LT2 ctrl versus LT2‐MYC tumor P = 0.002, LT2‐MYC tumor versus 72 h regression tumors P = 0.001).
-
CqPCR analysis of miR‐18a expression in conditional liver tumor cells treated with doxycycline (data represented as univariate scatter plot with median, data points represent three experimental replicates comprised of three technical replicates each, unpaired two‐tailed t‐test; compared to 0 h: 24 h, P = 0.14; 48 h, P = 0.008; 72 h, P = 0.005; 96 h, P = 0.003, **P‐values for 48 h, 72 h, and 96 h fall below Bonferroni adjusted P‐value of 0.01).
-
DLuciferase reporter expression in cultured murine liver tumor cells treated with a miR‐18a mimic or control. Wt, wild‐type Gclc 3′ UTR; mutated, Gclc 3′ UTR with four base pairs of the putative miR‐18a binding site mutated (data represented as normalized mean ± SEM of three experimental replicates with three technical replicates each, unpaired two‐tailed t‐test, Wt UTR ctrl versus 18a mimic P = 0.0002, mutated UTR control versus 18a mimic P = 0.14).
-
EWestern blot (WB) analysis of GCLC protein expression following treatment of cultured LT2‐MYC liver tumor cells with locked nucleic acid (LNA) inhibitors of miR‐18a (WB representative of two experimental replicates with three technical replicates each, pooled data represented as mean ± SEM, unpaired two‐tailed t‐test, P = 0.007).
-
F–HWB of GCLC protein expression (F), qPCR of miR‐18a expression (G), and enzymatic quantitation of GSH abundance (H) in liver tissue samples following treatment of LT2‐MYC tumor‐bearing mice with LNA inhibitors of miR‐18a or control LNA (n = 4 control LNA, n = 3 18a LNA, data represented as univariate scatter plots with median, unpaired two‐tailed t‐test, WB P = 0.002; qPCR P = 0.008; GSH assay P = 0.04).
Source data are available online for this figure.
In prior work it was found that MYC indirectly regulates glutamine metabolism via suppression of miR‐23a/b, which target the 3′ UTR of GLS 14. Because we observed MYC‐dependent suppression of GCLC protein and transcript, we hypothesized that MYC may regulate GCLC expression, at least in part, via miRNA. Using the Targetscan database (v6.2), we identified miRNAs whose seed sequences are predicted to bind the 3′ UTR of the GCLC transcript. We found that miR‐18a, a MYC‐regulated miRNA that is part of the oncogenic miR‐17‐92 miRNA cluster 30, 31, 32, had the lowest predicted Total Context Score (Appendix Table S2), indicating a strong probability of binding the GCLC 3′ UTR. Additionally, of the miRNAs predicted to bind the GCLC 3′ UTR, miR‐18a was one of the most highly upregulated in LT2‐MYC tumors compared to LT2 control liver (miRNA profiling dataset described in 24). Elevated expression of miR‐18a was confirmed by multiple probes in the array (Appendix Table S3).
Using qRT–PCR, we confirmed that miR‐18a is elevated in LT2‐MYC tumors and that its expression is MYC‐dependent in vivo (Fig 3B). We also observed downregulation of miR‐18a when MYC expression is inhibited in cultured LT2‐MYC tumor cells (Fig 3C). Previous work suggests that the MYC‐regulated splicing factor HNRNPA1 7 directs processing of mature miR‐18a 33. Accordingly, we observe that HNRNPA1 protein is elevated in liver tumors in a MYC‐dependent manner (Fig EV4C). Thus, both miR‐18a transcription and processing may be coordinately increased by MYC in the LT2‐MYC tumor model.
To determine whether miR‐18a directly targets the Gclc transcript, we generated a luciferase reporter fusion containing the Gclc 3′ UTR downstream of firefly luciferase. Reporter expression was diminished following transfection of a miR‐18a mimic into murine liver tumor cells but was unchanged when four bases of the predicted miR‐18a seed sequence binding site were mutated in the Gclc 3′ UTR (Fig 3D). We next asked whether miR‐18a antagonists could regulate GCLC expression in MYC‐driven liver tumor cells. When tumor cells derived from the LT2‐MYC model are transfected with locked nucleic acid (LNA) antagonists against miR‐18a, we observe that GCLC protein expression increases relative to cells treated with control LNA (Fig 3E).
We next sought to determine whether miR‐18a regulates GCLC expression in vivo in MYC‐driven liver tumors. We treated LT2‐MYC tumor‐bearing mice with miR‐18a antagonist LNA or control LNA twice weekly for 3 weeks. Tumor tissues were collected and Western blotting for GCLC protein was performed. We found that specific inhibition of miR‐18a in vivo increases GCLC protein expression (Fig 3F). Elevated GCLC protein corresponded with diminished miR‐18a expression in tumor tissue (Fig 3G). We next asked whether this change in GCLC protein expression corresponded to a change in GSH abundance in MYC tumors. Enzymatic analysis of GSH activity confirmed a small but significant increase of GSH in MYC‐driven liver tumors treated with miR‐18a LNA, compared to those treated with control LNA (Fig 3H). Thus, our results demonstrate that MYC‐dependent miR‐18a regulates GCLC, and subsequently GSH synthesis, in primary liver tumors.
miR‐18a is elevated in human HCC and correlates with altered glutathione pathway gene expression
Elevated MYC expression is associated with aggressive human liver cancer. MiR‐18a is elevated in a subset of HCCs 34 and may serve as a serum biomarker for HBV‐associated HCC 35. Because of the striking correlation between MYC and miR‐18a expression we observed in murine liver tumors, along with the regulation of GSH synthesis by MYC and miR‐18a described above, we next sought to determine whether there is a link between miR‐18a expression and altered GSH metabolism in human liver cancers. Using a previously published dataset 36, we confirmed that miR‐18a expression is significantly elevated in human HCC (Fig 4A) and inversely correlates with GCLC mRNA expression (Fig 4B). When the same HCCs are stratified by miR‐18a expression, the top tertile exhibits a GSH pathway gene expression pattern that is similar to that of the MYC‐driven liver tumor model (Fig 4C). This indicates that GSH pathway expression is similarly regulated in mouse and human tumors with elevated miR‐18a.
Figure 4. miR‐18a is elevated in human HCC and correlates with altered GSH pathway gene expression.
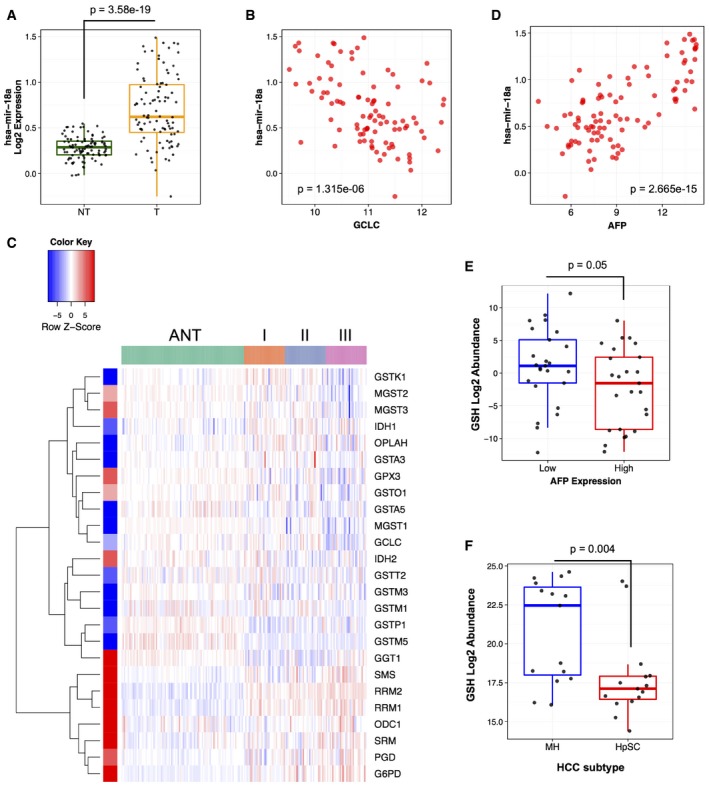
- miR‐18a expression in human HCC versus adjacent non‐tumor tissue (n = 96 each group, data represented as box plot with horizontal bar representing the median, box ranges representing the first (bottom) and third (top) quartiles, and vertical bars representing the standard error, Mann–Whitney U‐test, P = 3.58E‐19).
- miR‐18a expression inversely correlates with GCLC mRNA expression in human HCC (n = 96, Pearson correlation, Rp = −0.47, two‐tailed t‐test P = 1.315E‐06).
- Human HCCs with high miR‐18a expression exhibit a GSH pathway gene expression pattern similar to LT2‐MYC tumors. ANT, adjacent non‐tumor. Roman numerals represent ranked tertiles of increasing miR‐18a expression. Colored bar on left indicates relative gene expression in LT2‐MYC tumors for reference (n = 96 ANT, n = 96 total tumor samples).
- miR‐18a expression correlates with AFP mRNA in human HCC (n = 96, Pearson correlation, Rp = 0.70, two‐tailed t‐test P = 2.665E‐15).
- Tumor GSH abundance, normalized to matched NT liver tissue, in human HCCs with low (left) versus high (right) serum AFP status (n = 25 each group, data represented as box plot with horizontal bar representing the median, box ranges representing the first (bottom) and third (top) quartiles, and vertical bars representing the standard error, Mann–Whitney U‐test, P = 0.05).
- Tumor GSH abundance, normalized to matched NT liver tissue, in hepatic stem cell‐like (HpSC) human HCC versus mature hepatocyte‐like (MH) HCC (n = 15 each group, data represented as box plot with horizontal bar representing the median, box ranges representing the first (bottom) and third (top) quartiles, and vertical bars representing the standard error, Mann–Whitney U‐test, P = 0.004).
MYC is associated with a variety of poorly differentiated human tumors 37. In human liver cancer, elevated tumor alpha‐fetoprotein (AFP) expression is a clinical marker of poorly differentiated and aggressive disease. Previous studies have indicated a correlation between MYC overexpression in HCC tissue and increased serum AFP 21, 38. Accordingly, we find that MYC and AFP protein expression is correlated in MYC‐driven murine liver tumors (Fig 1A); we also find that tumor AFP transcript strongly correlates with miR‐18a in human HCC (Fig 4D). We thus sought to determine whether poorly differentiated human liver tumors characterized by elevated AFP expression have alterations in GSH abundance. Metabolite profiling of primary human HCCs has recently been reported 39, 40. Examining these datasets, we find that HCC patients with high serum AFP levels (Fig 4E) or high tissue AFP expression (HpSC subtype in Fig 4F) exhibit lower tumor GSH abundance than those with low serum or tissue AFP (correlation between GSH abundance and MYC expression in MH and HpSC subtypes in Fig EV5A and B). Taken together, our results indicate that miR‐18a strongly correlates with AFP expression, a marker of aggressive tumors, in human HCC. Further, elevated miR‐18a expression and AFP abundance correlate with diminished GCLC expression and GSH abundance, respectively, in human liver cancer.
Figure EV5. Evaluation of relationship between GSH abundance and MYC expression in human liver cancer.
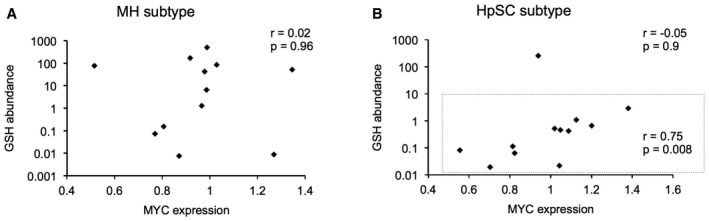
-
A, BCorrelation plots of GSH abundance versus MYC mRNA expression in mature hepatocyte (MH) subtype (A) or hepatic stem cell (HpSC) subtype (B) of HCC tumors (samples from 40) (MH: n = 12, Pearson coefficient = 0.02, two‐tailed P = 0.96; HpSC: n = 12, Pearson coefficient = −0.05, two‐tailed P = 0.9; HpSC inset: r = 0.75 and two‐tailed P = 0.008 when an outlier is removed).
MYC‐driven liver tumors are sensitive to exogenous oxidative stress
Because GSH is an important cellular antioxidant, we next sought to understand how depleted GSH affects MYC‐driven liver tumor survival. To assess how MYC‐driven liver tumors respond to acute oxidative stress, tumor‐bearing LT2‐MYC mice were treated via i.p. injection with the redox‐active compound diquat, which induces superoxide generation and ROS damage 41, 42. Tissues were collected at 6 and 24 h post‐treatment. We hypothesized that diquat treatment would specifically target tumor tissues and spare adjacent non‐tumor tissues.
Diquat treatment of tumor‐bearing LT2‐MYC mice led to decreased cellularity in tumors following treatment, which was not observed in adjacent non‐tumor tissue (Fig 5A, top panel). Additionally, diquat‐treated LT2‐MYC tumors showed a small but significant increase in TUNEL staining, a marker of cell death, as compared to saline‐treated tumors. This elevation was not observed in adjacent non‐tumor tissue; instead, we observed a small, but significant, decrease in TUNEL staining in adjacent non‐tumor treated with diquat, as compared to saline‐treated tissue (Fig 5A, middle panel). Finally, we observed fewer MYC‐positive cells in tumor tissues following diquat treatment (Fig 5A, lower panel). This observation was supported by decreased expression of MYC, as indicated by Western blot, in tumor tissues at both 6 and 24 h post‐treatment compared to saline controls (Fig 5B and C). These data suggest that depletion of GSH in MYC‐driven liver tumors leads to increased sensitivity to exogenous oxidative stress, and that residual tumor tissues have diminished MYC expression.
Figure 5. MYC‐driven liver tumors are sensitive to exogenous oxidative stress.
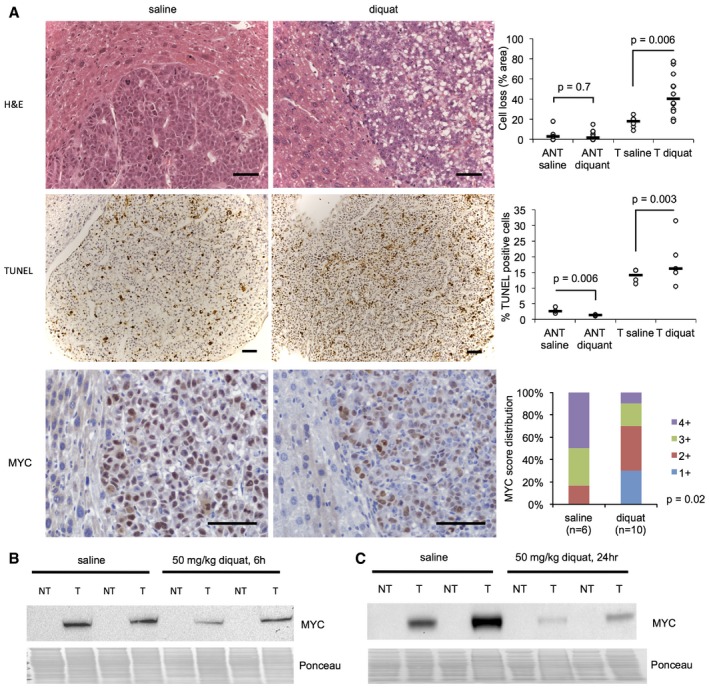
-
AHistological evaluation of MYC‐driven tumors following acute diquat treatment. Upper panel: Representative histology of H&E sections of LT2‐MYC tumors (T) and adjacent non‐tumor (ANT) tissue 24 h after saline (control) or 50 mg/kg diquat treatment. Quantitation is percent area of cell loss per sample (n = 6 saline ANT and T, n = 12 diquat ANT and T, data represented as univariate scatter plot with median, unpaired two‐tailed t‐test, ANT saline versus diquat P = 0.7, T saline versus diquat P = 0.006). Middle panel: representative TUNEL (cell death marker) staining of LT2‐MYC tumors 24 h after saline or 50 mg/kg diquat treatment. Quantitation is average percentage of TUNEL‐positive cells in 10 high‐powered fields for ANT or T samples 24 h after treatment (n = 3 ANT saline, n = 3 ANT diquat, n = 4 T saline, n = 5 T diquat, data represented as univariate scatter plot with median, unpaired two‐tailed t‐test, ANT saline versus diquat P = 0.006, T saline versus diquat P = 0.003). Lower panel: Representative MYC staining of LT2‐MYC tumors 24 h after saline or 50 mg/kg diquat treatment. Quantitation is percent of tumor samples with MYC‐positive score 1+ to 4+ after diquat treatment (n = 6 saline, n = 10 diquat, 6 h and 24 h combined, 1+ corresponds to 1–50% of tumor cells staining positive for MYC, 2+ = 51–74%, 3+ = 75–84%, 4+ = 85–100%, unpaired two‐tailed t‐test, P = 0.02). For all images, scale bar is 50 μm.
-
B, CWestern blot analysis of MYC protein expression in LT2‐MYC tumors (T) and adjacent non‐tumor (NT) 6 h (B) and 24 h (C) after 50 mg/kg diquat treatment (n = 2 samples/condition).
Source data are available online for this figure.
Discussion
In this study, we sought to identify novel metabolic pathways that are dysregulated in MYC‐driven tumors. Our biochemical and transcriptional profiling of murine liver tumors identified six pathways that are coordinately altered in vivo (Fig 1B and Appendix Figs S1–S6). At least two of these pathways, serine metabolism and ABC transporters, have been described in connection with MYC signaling previously 43, 44, while to our knowledge, the others have yet to be associated with MYC overexpression in cancer.
We describe an inverse relationship between MYC and GCLC, the rate‐limiting enzyme of GSH biogenesis, in a mouse model of MYC‐driven liver cancer. We show evidence for a novel regulatory axis whereby the MYC‐induced miRNA, miR‐18a 30, 31, targets GCLC and inhibits GSH synthesis, thereby contributing to GSH depletion in tumor cells (Fig 6). In vivo inhibition of miR‐18a results in upregulation of GCLC and a corresponding increase in GSH abundance in tumor tissues. Another MYC‐dependent miRNA, miR‐23a/b, has been implicated in the regulation of GLS, the enzyme that converts glutamine to glutamate 14. We propose that a MYC‐regulated miRNA network of miR‐23a/b and miR‐18a shapes the utilization of glutamine in MYC‐driven tumors.
Figure 6. Summary of miR18a‐dependent regulation of GSH metabolism in MYC‐driven murine liver tumors.
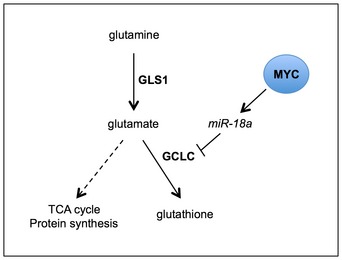
MYC suppresses GCLC via miR‐18a. Attenuated GCLC contributes to GSH depletion. Relative flux of glutamine‐derived carbons to other downstream pathways, such as the TCA cycle and protein synthesis, remains to be determined (dotted line).
GSH and GSSG were two of the most depleted metabolites in our global profiling. In addition to downregulation of GCLC, we found increased expression of glutaredoxin GLRX5 and a very small, but significant, increase in GGT1 expression in tumors compared to non‐tumor tissues (Fig 2B and D). Thus, it is possible that GSH depletion in MYC‐driven liver tumors results from multiple mechanisms, including decreased synthesis, elevated S‐glutathionylation, and elevated gamma‐glutamyl cycling, although we did not find strong metabolic support for the latter two mechanisms. Whether MYC regulates glutaredoxin activity or gamma‐glutamyl cycling is unknown. However, a recent report on the same LT2‐MYC model used in this study 45 found that Ggt1 transcript exhibits MYC‐dependent upregulation that is independent of transcriptional activation (Appendix Table S4). It is thus possible that MYC regulates other genes involved in GSH metabolism, perhaps also through post‐transcriptional means, such as miRNAs.
In this study, [13C]‐glutamine tracing experiments identified several metabolic differences between tumor and non‐tumor tissues. We observed increased incorporation of [13C]‐glutamine carbons into multiple metabolites, including the TCA cycle intermediates alpha‐ketoglutarate and malate, as well as pyruvate, in tumors relative to adjacent non‐tumor tissues (Fig EV3C). The TCA cycle is an important biosynthetic hub for proliferating cancer cells 46. Accordingly, prior studies of MYC‐overexpressing cell lines have characterized the flux of glutamine carbons away from the TCA cycle and into biosynthetic pathways, a process known as cataplerosis 10, 12. Thus, the differences in [13C]‐glutamine tracing we observed between tumor and non‐tumor tissue may reflect metabolic reprogramming that enhances glutamine utilization for biosynthesis in MYC‐driven liver tumor cells. Further studies are needed to define which pathways downstream of glutamine uptake or synthesis are critical for the proliferation and survival of MYC‐driven liver tumors (Fig 6).
In addition to the post‐transcriptional regulation by miR‐18a described in this study, GCLC is also transcriptionally regulated. The GCLC promoter contains several regulatory elements, including an E‐box, which may be bound by MYC or NRF2. Using cultured cells, different groups have shown opposing transcriptional effects of MYC binding to the GCLC promoter 47, 48. Thus, it is likely that GCLC expression is regulated both directly via MYC and indirectly via a MYC‐regulated miRNA, miR‐18a, in a context‐specific manner as shown in this study. However, a recent study of MYC‐dependent gene expression in the LT2‐MYC model 45 classified GCLC as having “secondary” (i.e., non‐transcriptional) MYC‐dependent repression (Appendix Table S4), lending further support to the miRNA‐dependent regulation described here.
MYC indirectly regulates GLS via suppression of specific miRNAs 14. Similarly, MYC indirectly regulates splicing of the key glycolytic enzyme PKM2 via RNA processing enzyme expression 7. Interestingly, we find that HNRNPA1, an RNA processing protein that regulates both PKM2 splicing 7 and miR‐18a processing 33, is upregulated in LT2‐MYC tumors in a MYC‐dependent manner (Fig EV4C). In addition to its capacity for direct transcriptional regulation, MYC may orchestrate tumor‐specific metabolic reprogramming through indirect, RNA‐dependent means (i.e., through miRNAs and the regulation of RNA processing enzymes such as HNRNPA1). These multiple levels of regulation may enable MYC to fine‐tune metabolic pathways to coordinate anabolic processes; future studies should investigate this possibility.
We found that miR‐18a expression is elevated in human HCC and correlates with altered GSH pathway gene expression 36 (Fig 4A–C). Further, we found that HCC patients with high serum and tissue AFP, a marker of aggressive disease, exhibit lower GSH abundance in their tumor tissues than patients with low AFP (Fig 4E and F). MiR‐18a and AFP are strongly correlated in human HCC (Fig 4D). Taken together, high tumor miR‐18a expression and/or high serum or tissue AFP levels may indicate tumor GSH synthesis suppression due to MYC activation. Future studies should elucidate the utility of stratification of HCCs by their miR‐18a or GSH status for exploring novel treatment strategies.
GSH is a major scavenger of reactive oxygen species (ROS) 26 and plays a critical role in detoxification of potential alkylating agents in the liver 49. A corollary of depleted GSH in MYC‐driven liver tumors may thus be increased sensitivity to ROS or alkylating agents, which can no longer be efficiently cleared from the tissue. Previous studies have shown that tumor cells may be able to survive loss of certain antioxidants by upregulating compensatory antioxidant systems such as thioredoxin 50 or NRF2 51, 52. We found that MYC‐driven liver tumors are sensitive to acute oxidative stress, as evidenced by cell loss, increased cell death, and a reduction in MYC expression in tumors following treatment with diquat (Fig 5). Whether GSH depletion in MYC‐driven tumors will render them more sensitive to chemotherapeutic drugs that rely on GSH for their detoxification is an important area for further investigation.
In summary, our work describes a novel example of metabolic reprogramming by MYC in vivo. We show that MYC attenuates GCLC via miR‐18a in a mouse model of liver cancer, contributing to GSH depletion and sensitivity to oxidative stress. Future studies on the role of GSH depletion or GCLC attenuation for tumor cell survival may identify novel treatment strategies for MYC‐expressing tumors.
Materials and Methods
Ethics statement
All protocols regarding animal studies were approved by the UCSF Institutional Animal Care and Use Committee (IACUC). Husbandry and housing were maintained by the UCSF Laboratory Animal Resource Center (LARC). Animals were handled in accordance with standards set forth by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC). ARRIVE guidelines 53 were consulted to accurately report the animal experiments described below.
LT2‐MYC tumor generation and regression
LT2‐MYC (LAP‐tTA × TetO‐MYC, FVB/n background from Taconic Biosciences) double‐transgenic mice have been described 22. Male LT2‐MYC mice were bred and maintained on doxycycline (200 mg/kg doxy chow, Bio‐Serv, ad libitum feeding) to suppress oncogene expression. At 8 weeks of age, doxycycline was removed from the diet (mice were placed on normal chow, ad libitum feeding) to induce MYC expression and tumorigenesis. Mice were monitored weekly for tumor development by inspection and palpating the abdomen. Average time to tumor detection was 10 weeks. To induce tumor regression, mice were placed back on a diet containing 200 mg/kg doxycycline, ad libitum feeding. Mice were euthanized as per ethical guidelines (i.e., reaching a body condition score, BCS, of 2 or less), and tumors or adjacent non‐tumor tissues were flash‐frozen in liquid nitrogen or prepared for histology as described below.
For histology, mouse livers were fixed in 4% paraformaldehyde in PBS at 4°C for 24 h and then switched to 70% ethanol. Paraffin‐embedded blocks and hematoxylin and eosin‐stained slides were prepared at the Gladstone Histology and Light Microscopy Core Facility.
mRNA microarray
Total RNA from four samples per genotype (LT2 Control, LT2‐MYC) was extracted as per manufacturer's instructions (mirVana™ miRNA isolation kit, Ambion). RNA quality was assessed using a Pico Chip on an Agilent 2100 Bioanalyzer (Agilent Technologies). Three LT2 Control and four LT2‐MYC samples were selected for Agilent stock mouse 44K (014868) array analysis—one LT2 Control RNA sample did not meet quality selection criteria and was thus removed from downstream applications. Sample preparation, labeling, and array hybridizations were performed according to standard protocols from UCSF Shared Microarray Core Facilities and Agilent Technologies (http://www.arrays.ucsf.edu; http://www.agilent.com). RNA was amplified and labeled with Cy3‐CTP using the Agilent Low RNA Input Fluorescent Linear Amplification kits following the manufacturer's protocol (Agilent). Labeled cRNA was assessed using Nandrop ND‐100 (Nanodrop Technologies Inc.). Cy3‐labeled target was hybridized to Agilent whole mouse genome 4x44K Ink‐jet arrays (Agilent). Hybridization samples were randomized on the 4x44K format to correct any batch bias. Hybridizations were performed for 14 h, according to the manufacturer's protocol (Agilent). Arrays were scanned using the Agilent microarray scanner (Agilent), and raw signal intensities were extracted with Feature Extraction v9.5 software (Agilent). Primary normalization and data extraction were performed by the Microarray Core Facility. Briefly, single channel data were normalized using quantile normalization method. No background subtraction was performed, and the median feature pixel intensity was used as the raw signal before normalization.
Metabolic analyses
Mass spectrometry analysis was performed to obtain global biochemical profiles of LT2 control liver tissue and LT2‐MYC liver tumor tissue (Metabolon Inc., Durham, NC, USA). Flash‐frozen tissue samples from seven mice were provided for each group. Samples were extracted and prepared for analysis using Metabolon's standard solvent extraction method. The extracted samples were split into equal parts for analysis on the GC/MS and LC/MS/MS platforms. Technical replicate samples were created from sample homogenates. The mView product specification includes all detectable compounds of known identity (named biochemicals). The Metabolon‐generated dataset (Dataset EV1) used in this study comprises a total of 334 named biochemicals. Initial statistical analysis was carried out by Metabolon. Briefly, following log transformation and imputation with minimum observed values for each compound (described previously in 54, 55, 56), Welch's two‐sample t‐test was used to identify biochemicals that differed significantly between control liver tissue and tumor tissue.
For the [U‐13C]‐glutamine isotopic tracing analyses (Fig 2E), MYC liver tumors were generated through hydrodynamic transfection of 8‐week‐old FVB/n male mice (Taconic Biosciences) (Fig EV3), as described previously 28. Five weeks following hydrodynamic transfection, 14 total mice were randomized into two groups (seven mice per group; labeled versus unlabeled glutamine). The labeled glutamine (Cambridge Isotope Laboratories Inc., CLM‐1822) or unlabeled glutamine (Sigma) was administered via i.v. injection, as described previously 11. Fifteen minutes after the final dose, the animals were euthanized and liver tumor and adjacent non‐tumor tissues were flash‐frozen and analyzed using a slight modification of previously described procedures 57. Briefly, 100 mg of frozen tissue were extracted in 300 μl of 40:40:20 acetonitrile:methanol:water with 1 nM final concentration of d3‐N15 serine (Cambridge Isotope Labs). Manual disruption of tissue was performed via TissueLyser using a 5‐mm stainless steel bead for 30 s (Qiagen). Metabolite‐containing supernatant was separated from insoluble tissue debris by refrigerated centrifugation at 20,000 g for 10 min. An aliquot of the supernatant was then injected into an Agilent 6460 QQQ LC‐MS/MS for targeted single‐reaction monitoring (SRM)‐based quantitation of metabolites. For separation of polar metabolites a Luna 5‐mm NH2 column (Phenomenex, 50 × 4.6 mm) was used for normal‐phase chromatography. The mobile phase was as follows: Buffer A, acetonitrile; Buffer B, 95:5 water/acetonitrile with either 0.1% formic acid or 0.2% ammonium hydroxide plus 50 mM ammonium acetate for positive and negative ionization mode, respectively. The flow rate for each run started at 0.2 ml/min for 5 min, followed by a gradient starting at 0% B and increasing linearly to 100% B over the course of 45 min with a flow rate of 0.7 ml/min, followed by an isocratic gradient of 100% B for 17 min at 0.7 ml/min before equilibrating for 8 min at 0% B with a flow rate of 0.7 ml/min. MS analysis was performed with an electrospray ionization (ESI) source on an Agilent 6430 QQQ LC‐MS/MS. The capillary voltage was set to 3.0 kV, and the fragmentor voltage was set to 100 V. The drying gas temperature was 350°C, the drying gas flow rate was 10 l/min, and the nebulizer pressure was 35 psi. Representative metabolites were quantified by integrating the area under the curve for the SRM of the transition from precursor to product ions at associated collision energies and normalized to internal standards and external standard curves. Expected expression of MYC and GCLC were confirmed in samples taken from the same tumors (Fig EV3A).
Microarray and metabolomics statistical analyses
Method for processing raw data into normalized expression values: Differential gene expression and metabolite abundance between LT2‐MYC tumors and LT2 control tissue was performed using the limma R package 58. Genes or metabolites that were significantly different between these groups at a false discovery rate of 0.05 were extracted for downstream analyses. Pathway enrichment within this set of genes or metabolites was quantified using the Fisher's exact test based on annotations from the Kyoto Encyclopedia of Genes and Genomes (KEGG) 25. Significantly enriched pathways were identified at a P‐value cutoff of 0.05.
Gene expression values for human liver tumor samples 36, 40 were sourced from the Gene Expression Omnibus (GEO) site (accession codes GSE14520 and GSE22058, respectively). For GSE14520, data from the two array platforms were combined, and batch corrections performed with ComBat from the sva R package 59. Differential gene expression between the non‐tumor (NT) and tumor (T) groups (GSE22058, Fig 4A) was quantified using the limma R package 60. Correlation plots of gene expression in tumors were generated using the gplots R package (GSE22058, Fig 4B and D) or Excel (GSE14520, Fig EV5A and B). Correlation P‐values were generated using unpaired, two‐sided t‐tests.
Human orthologs of dysregulated glutathione pathway genes in mice were identified using homology group definitions compiled and published by the Mouse Genome Database Group 61. Heatmaps and clustering analyses were performed using the gplots and cluster R packages, respectively.
Raw metabolite abundance values were obtained for the tumor samples from 39. Missing values were imputed with the minimum abundance across all samples for the respective metabolites. The resulting metabolite levels were then normalized to those from matched distal non‐tumor samples from the same patients, and log‐transformed. The tumor samples were dichotomized based on AFP expression using a cut‐point at the 50th percentile mark and statistical significance of the differences in metabolite abundance between the groups was determined using a Mann–Whitney U‐test in the limma R package 60.
Raw metabolite abundance values were obtained for the tumor samples from 40. Raw metabolite abundance values were log‐transformed, and missing values imputed using the K‐nearest neighbors algorithm within the impute R package 62; R package version 1.44.0. Statistical significance of the differences in metabolite abundance between the MH and HpSC groups were quantified using a Mann–Whitney U‐test in the limma R package.
Glutathione assay
The GSH‐Glo Glutathione Assay kit (Promega) was used as per the manufacturer's instructions. Briefly, flash‐frozen tissue samples were homogenized in ice‐cold PBS containing 2 mM EDTA (1 ml PBS/EDTA per 10 mg tissue) using a Dounce homogenizer. The extracts were centrifuged at 4°C (10,000 g, 10 min), and the supernatant was collected and used immediately for the assay at a dilution of 1:10 in PBS/EDTA. Luminescence was read on a Tecan Safire II plate reader.
Protein preparation and Western blot analysis
Cultured cells or flash‐frozen tissues were homogenized in ice‐cold radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris–HCl pH 7.6, 150 mM NaCl, 0.5% sodium deoxycholate, 1% NP‐40, 0.1% SDS, 2 mM EDTA) containing COMPLETE protease inhibitor cocktail (Roche) and phosphatase inhibitors (Santa Cruz Biotechnology). Protein concentrations were determined by performing DC Protein Assay (Bio‐Rad) using BSA as standard. Protein extracts were resolved using 4–12% Bis‐Tris SDS–PAGE gels (Invitrogen) in a Bolt apparatus with 1× MOPS buffer (Invitrogen). Transfer to nitrocellulose membranes (Life Technologies) was performed on an iBlot apparatus (Invitrogen). Membranes were probed with primary antibodies overnight on a 4°C shaker and then incubated with horseradish peroxidase (HRP)‐conjugated secondary antibodies, and signals were visualized with ECL (Bio‐Rad). The following primary antibodies were purchased and used as indicated by the manufacturer: AFP (Thermo Scientific, 710486); c‐MYC (MYC) (Abcam, ab32072); GCLC (Santa Cruz, sc‐22755); GSS (Sigma, HPA054508); GLRX5 (Sigma, HPA042465); GGT1 (Sigma, HPA045635); GSR (Invitrogen, PA5‐29945); G6PD (GeneTex, GTX101212); HNRNPA1 (Sigma, HPA001609); β‐actin (actin) (Santa Cruz, sc‐47778, HRP). MYC and AFP antibody use for Western blot with samples from the LT2‐MYC model was published previously 23. Actin antibody use for Western blot with samples from murine liver cancer cell lines was published previously 24. All Sigma antibodies (GSS, GLRX5, GGT1, HNRNPA1) were selected based on Western blot validation (human tissue only) in the Human Protein Atlas (www.proteinatlas.org). The antibodies for GCLC, GSS, GLRX5, GGT1, G6PD, and HNRNPA1 are supported for Western blot application (reactivity: human) by Antibodypedia (www.antibodypedia.com). The antibody for GSR is supported for Western blot application (reactivity: human and mouse) by Antibodypedia (www.antibodypedia.com). Because metabolic enzymes are highly conserved, we assumed that antibodies that were validated for Western blot application in human tissue would also be appropriate for application in murine tissue.
Murine liver tumor cell lines
The EC4 conditional line used in Fig 3A and C was a gift of D. Felsher at Stanford University. Similar lines were described previously 29. EC4 cells were grown in high glucose DMEM supplemented with 10% fetal bovine serum and 1× each of glutamine, non‐essential amino acids, and sodium pyruvate. To turn off transgene expression in EC4 cells, 8 ng/ml doxycycline (Sigma) in fresh media was added to cells. The LT2M cell line used in the in vitro LNA experiment (Fig 3E) was isolated and established from an LT2‐MYC mouse liver tumor by Dr. Andrei Goga at UCSF. After establishing this line, it was grown and expanded further in RPMI 1640 media supplemented with 10% fetal bovine serum. To make the immortalized LT2MR cell line used in the luciferase experiments (Fig 3D), LT2M cells were engineered to stably express RAS by retroviral infection with pMSCV‐HRAS V12 virus. No cell line used in this article was authenticated prior to use. All lines were tested monthly for mycoplasma contamination and found to be negative.
Real‐time quantitative PCR
Total RNA from liver samples or cultured cells was extracted using mirVana™ miRNA isolation kit (Ambion) and DNase treated with Turbo DNA‐free DNase Treatment kit (Ambion) as per manufacturer's protocol. For analysis of mRNA transcripts, cDNA was synthesized from one microgram of DNase I‐treated total RNA using iScript™ cDNA synthesis Kit (Bio‐rad). Real‐time PCR was performed using TaqMan probes (Applied Biosystems) for Gclc (Mm00802655_m1), and Gapdh (Mm99999915_g1) as endogenous control. For analysis of miRNAs, cDNA was synthesized from 10 ng of DNase I‐treated total RNA using the TaqMan microRNA reverse transcription kit (Applied Biosystems PN4366596) with RT primers from TaqMan sets for miR18a (Cat #002422), and snoRNA202 (Cat #4427975) as endogenous control. The reaction mix was prepared according to the protocol. Real‐time PCR was performed using TaqMan Universal Master Mix II, no UNG (Applied Biosystems PN 4440041) according to the protocol, using 1.33 μl of undiluted cDNA per reaction and RNA‐specific Taqman hydrolysis probes. Samples were run in triplicate on a Real‐Time Thermal Cycler (Bio‐Rad Laboratories), and variation was calculated using the ΔΔC t method with respective endogenous controls. Significance of differences in gene expression was determined by performing unpaired, two‐sided t‐tests on the replicate values for each gene in control and experimental groups.
Luciferase assays
A 467‐bp fragment of the Gclc 3′ UTR containing the putative miR‐18a binding site was PCR amplified from genomic DNA of LT2MR cells 24. The following primers were used to amplify the Gclc 3′ UTR fragment: GCLC 3′ UTR‐Short_For: caccGGCATTCCAGAGTTTCAAATGT and GCLC 3′ UTR‐Short_Rev: CAGCCTGTCAATCTGCTCCT. To make the mutated binding site construct, four bases of the putative miR‐18a binding site on Gclc 3′ UTR were mutated using site‐directed mutagenesis, as per manufacturer's instructions (QuikChange Lightning Site‐Directed Mutagenesis Kit, Agilent). The following primers were designed (miR‐18a seed sequence is underlined and the mutated bases are in bold): Forward: TGCCCTCCGTGGGTGAGGTAGCAGACCTGTGATATTTC; Reverse: GAAATATCACAGGTCTGCTACCTCACCCACGGAGGGCA. The PCR products (Gclc 3′ UTR WT and mutant) were then cloned by Topo cloning into pMSCV‐Luciferase reporter vectors.
LT2MR cells were plated in each well of a 12‐well dish, 75,000 cells/well. Co‐transfection of the pMSCV‐Luciferase reporter vector containing the Gclc 3′ UTR (WT or mutant) (1 μg/well), a Renilla‐Luciferase (Renilla‐Luc) reporter construct (100 ng/well), and either mir‐18a mimic (50 nM) or control mimic (50 nM) was carried out. Dharmafect Duo (Dharmacon) was used as a transfection reagent. The Dual Luciferase Reporter Assay System (Promega) was used as per product instructions. 250 μl of Passive Lysis buffer (Promega) was added to each well of the 12‐well plate, 48 h post‐transfection. The plate was covered with aluminum foil and placed in −20°C overnight. Luciferase assays were performed on a luminometer. Firefly luciferase activity was normalized to Renilla relative luminescence units (RLUs) for each sample. Six wells were treated per condition, and the experiment was repeated three times overall.
Locked nucleic acid experiments
LT2M cells were plated in each well of a 6‐well dish, 75,000 cells/well. Cells were transfected with 50 nM of either control or 18a LNA (Exiqon) using RNAiMax transfection reagent (Invitrogen) as per manufacturer's instruction. Cells were trypsinized and pelleted 48 h post‐transfection. Pellets were lysed and protein extracts made for subsequent Western blot analysis. Three wells were treated per condition, and each well was prepared independently for Western blot analysis. The experiment was repeated twice overall.
LT2‐MYC mice from three separate litters were induced at 8 weeks of age, by taking mice off doxy chow. After 8 weeks of tumor induction, mice were randomized into experimental groups. The mice received either miR‐18a (n = 4) or control (scramble) (n = 4) LNA, 25 mg/kg in saline, delivered in six i.p. injections over 3 weeks (two injections per week). Mice were euthanized and tissues collected 2 days after the final injection, at ~11 weeks induction total. Tumor and adjacent non‐tumor tissue were flash‐frozen for subsequent analyses. One mouse that received the 18a LNA treatment did not have appreciable tumor burden at euthanization (i.e., not enough tissue for downstream analysis) and so was not included in further analyses.
Diquat experiments
LT2‐MYC mice from multiple litters were induced at 8 weeks of age, by taking mice off of doxy chow. After 10–12 weeks of tumor induction, mice were randomized into experimental groups. The mice received 50 mg/kg diquat in saline, or saline alone, delivered once by i.p. injection. Mice were euthanized and tissues collected at 6 h (two experimental cohorts, saline n = 4, diquat n = 4) and 24 h (four experimental cohorts, saline n = 8, diquat n = 14) following treatment. Tumor and adjacent non‐tumor tissue were flash‐frozen for subsequent analyses. Additional tissue was collected for histology, as described below. Mice had variable levels of tumor burden (from none to majority of liver tissue) when euthanized; thus, not all samples were available for all downstream analyses.
Histological analyses of murine liver tumor samples
Mouse liver tissue samples were fixed in 4% paraformaldehyde in PBS at 4°C for 24 h and then switched to 70% ethanol. Paraffin‐embedded blocks and hematoxylin and eosin‐stained slides were prepared at the Gladstone Histology and Light Microscopy Core Facility. TUNEL staining was performed using the ApopTag Peroxidase In Situ Apoptosis Detection Kit (EMD Millipore/Calbiochem) according to the manufacturer's instructions. Immunohistochemical staining of MYC (Abcam ab32072; dilution as indicated by the manufacturer) was performed following xylene deparaffinization, rehydration, heat‐induced epitope retrieval with 10 mM sodium citrate buffer (0.05% Tween, pH 6.0), quenching of endogenous peroxidase activity by hydrogen peroxide incubation, and blocking in 5% normal goat serum in PBS. Goat anti‐rabbit IgG‐biotin secondary antibody was used as indicated by the manufacturer (Santa Cruz, sc‐2040). Samples were detected with VECTASTAIN Elite ABC Reagent (Vector Labs) and Vector DAB substrate kit (Vector Labs) and counterstained with hematoxylin.
To quantify cell loss, slides were blinded and a pathologist (K.J.E.) assessed the approximate total acellular tissue area (including abnormalities such as stroma, fat, and necrosis) in 10–30 high‐power fields for each sample. To quantify cell death, slides were blinded and the first author (B.A.) counted the number of TUNEL‐positive cells in 10 high‐power fields for each sample. For MYC staining, the following scale was established by K.J.E. to quantify MYC‐positive cells in tumor tissues: 1+ (1–50% of tumor cells staining positive for MYC); 2+ (51–74%); 3+ (75–84%); 4+ (85–100%). K.J.E. performed the MYC quantification on blinded slides.
Statistical analysis
Statistical analyses of human and mouse gene expression and metabolite profiling are described above. Unless otherwise noted in the appropriate figure legend, for all other comparisons, unpaired, two‐sided t‐tests were used. We generally used two‐sided t‐tests because we assumed a normal distribution in which an effect could be measured in either direction. We used one‐sided t‐tests when we assumed a normal distribution in which an effect could be measured in one direction. We used Mann–Whitney U‐tests when we assumed that the data were not normally distributed (Fig 4A, E and F). For the large (profiling) data sets, the distribution and variance of the data meet the assumptions of the statistical test used. No statistical method was used to predetermine sample size. The investigators were not blinded to allocation for the in vivo diquat and isotopic tracing experiments. The investigators were blinded to allocation for the in vivo 18a LNA experiments. For all in vivo studies, mice were randomized to treatment groups when tumors were detected by visual inspection or abdomen palpation in one or more representative animals. For LT2‐MYC studies, mice were randomized within cages, to minimize skewing effects from litters. We did not use statistical methods to assess whether our data met the assumptions of the tests used. We did not calculate an estimate of variation within each group of data, nor did we assess whether the variance was similar between groups that were statistically compared.
The sample size for all experiments (in vitro and in vivo) was not chosen with consideration of adequate power to detect a prespecified effect size. For in vitro studies, all completed experiments are reported. For in vivo studies, the number of indicated mice represents the total number of mice processed for each experiment. Mice with excessive tumor burden at the start of an in vivo study, as indicated by swollen abdomen or low BCS, were removed from the study and euthanized as per ethical guidelines. For the 18a LNA and diquat treatment studies, mice were euthanized at the study endpoint as indicated, unless they failed to meet the predetermined UCSF IACUC quality‐of‐life guidelines (i.e., BCS < 2). No mice that completed the studies' predetermined duration were excluded from analyses. No samples were fully processed for metabolomic, qPCR, Western blot, or immunohistochemical analysis and then excluded.
Data availability
Primary data
Gene expression values for human liver tumor samples 36, 40 are available from the GEO site (accession codes GSE14520 and GSE22058, respectively). Metabolite abundance values for human liver tumor samples were shared upon request to the respective authors 39, 40. miRNA expression values for murine liver tumors 24 are available at GEO accession code GSE44570. mRNA expression values for LT2‐MYC tumor formation and regression 23 are available at GEO accession code GSE28198. Metabolomic profiling data for LT2‐MYC tumors are available as Dataset EV1 with this article.
Referenced data
Summary data of MYC‐dependent regulation of glutathione pathway genes (Appendix Table S3) were extracted from Supplemental Data published previously 45.
Code availability
All codes used for this project have been deposited to Github (https://github.com/snjvb/liver_glut_met).
Author contributions
BA designed and conducted all of the experiments (except the initial transcriptomic and metabolomic profiling, the in vitro luciferase and LNA experiments, and the analysis of human HCC datasets) and wrote the manuscript. RC performed Western blot analysis and GSH quantification for the in vivo miR‐18a LNA experiment and provided intellectual input and valuable discussion. SB performed bioinformatic analyses of the data from the gene expression and metabolomic profiling of mice and human samples and provided valuable discussion. AB performed the initial gene expression and metabolomic profiling of the murine tumor samples, quantification of Gclc transcript expression in tumors, and the in vitro luciferase and LNA experiments and provided valuable discussion. RAK performed the mass spectrometry and metabolomic analysis for the isotopic tracing experiment. LL designed reporter constructs for and assisted with the in vitro luciferase experiment. KJE performed the quantification of cell loss and MYC staining for the in vivo diquat experiment. OM performed the quantification of miR‐18a expression for the in vivo LNA experiment. KK provided tumor‐bearing LT2‐MYC mice for diquat experiments. QH and GX provided the human HCC GSH abundance and AFP expression data. DKN supervised the mass spectrometry and metabolomics analyses and provided valuable discussion. AG supervised all of the studies and provided valuable discussion and intellectual input. All authors have approved the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 5
Acknowledgements
We thank Alicia Zhou and Dai Horiuchi for helpful input on the manuscript. We also thank J. Michael Bishop and Bishop laboratory members for thoughtful discussion, reagents, and technical assistance. We thank Florian Muller for helpful discussions and advice. Finally, we thank Mercedes Joaquin for her care of the animal colony. This material is based upon work supported by the National Science Foundation under Grant No. 1144247 (to B.A.), NIH R01‐CA136717, NIH R01‐CA170447, and NIH U19 CA179512 (to A.G.), UCSF Liver Center grant P30DK026743, a V‐Foundation Scholar Award, a Leukemia and Lymphoma Scholar Award (to A.G.), NIH NCI R01CA172667, NIH NCI 5K08CA172288 (to K.J.E.), the NIH Diabetes, Endocrinology and Metabolism training grant T32DK007418 (to R.C.), and NIH F99CA212488 (to R.C.), American Cancer Society Research Scholar Award (RSG‐14‐242‐01‐TBE), and the State Key Science & Technology Project for Infectious Diseases of China (2012ZX10002‐011) and the key foundations (21435006) from the National Natural Science Foundation of China.
EMBO Reports (2017) 18: 569–585
References
- 1. Dang CV (2012) MYC on the path to cancer. Cell 149: 22–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Horiuchi D, Anderton B, Goga A (2014) Taking on challenging targets: making MYC druggable. Am Soc Clin Oncol Educ Book 34: e497–e502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dang CV (2013) MYC, metabolism, cell growth, and tumorigenesis. Cold Spring Harb Perspect Med 3: a014217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Li B, Simon MC (2013) Molecular pathways: targeting MYC‐induced metabolic reprogramming and oncogenic stress in cancer. Clin Cancer Res 19: 5835–5841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Osthus RC, Shim H, Kim S, Li Q, Reddy R, Mukherjee M, Xu Y, Wonsey D, Lee LA, Dang CV (2000) Deregulation of glucose transporter 1 and glycolytic gene expression by c‐Myc. J Biol Chem 275: 21797–21800 [DOI] [PubMed] [Google Scholar]
- 6. Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, Royer RE, Vander Jagt DL, Semenza GL, Dang CV (2010) Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci USA 107: 2037–2042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. David CJ, Chen M, Assanah M, Canoll P, Manley JL (2010) HnRNP proteins controlled by c‐Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature 463: 364–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Heiden Vander MG, Lunt SY, Dayton TL, Fiske BP, Israelsen WJ, Mattaini KR, Vokes NI, Stephanopoulos G, Cantley LC, Metallo CM et al (2011) Metabolic pathway alterations that support: cell proliferation. Cold Spring Harb Symp Quant Biol 76: 325–334 [DOI] [PubMed] [Google Scholar]
- 9. Yuneva M, Zamboni N, Oefner P, Sachidanandam R, Lazebnik Y (2007) Deficiency in glutamine but not glucose induces MYC‐dependent apoptosis in human cells. J Cell Biol 178: 93–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang X‐Y, Pfeiffer HK, Nissim I, Daikhin E, Yudkoff M, McMahon SB et al (2008) Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci USA 105: 18782–18787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yuneva MO, Fan TWM, Allen TD, Higashi RM, Ferraris DV, Tsukamoto T, Matés JM, Alonso FJ, Wang C, Seo Y et al (2012) The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab 15: 157–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J, Tsukamoto T, Rojas CJ, Slusher BS, Zhang H et al (2012) Glucose‐independent glutamine metabolism via TCA cycling for proliferation and survival in b cells. Cell Metab 15: 110–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bott AJ, Peng IC, Fan Y, Faubert B, Zhao L, Li J, Neidler S, Sun Y, Jaber N, Krokowski D et al (2015) Oncogenic MYC induces expression of glutamine synthetase through promoter demethylation. Cell Metab 22: 1068–1077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gao P, Tchernyshyov I, Chang T‐C, Lee Y‐S, Kita K, Ochi T, Zeller KI, De Marzo AM, Van Eyk JE, Mendell JT et al (2009) c‐Myc suppression of miR‐23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 458: 762–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Xiang Y, Stine ZE, Xia J, Lu Y, O'Connor RS, Altman BJ, Hsieh AL, Gouw AM, Thomas AG, Gao P et al (2015) Targeted inhibition of tumor‐specific glutaminase diminishes cell‐autonomous tumorigenesis. J Clin Invest 125: 2293–2306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kaposi‐Novak P, Libbrecht L, Woo HG, Lee YH, Sears NC, Conner EA, Factor VM, Roskams T, Thorgeirsson SS (2009) Central role of c‐Myc during malignant conversion in human hepatocarcinogenesis. Cancer Res 69: 2775–2782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sanyal AJ, Yoon SK, Lencioni R (2010) The etiology of hepatocellular carcinoma and consequences for treatment. Oncologist 15(Suppl 4): 14–22 [DOI] [PubMed] [Google Scholar]
- 18. Schlaeger C, Longerich T, Schiller C, Bewerunge P, Mehrabi A, Toedt G, Kleeff J, Ehemann V, Eils R, Lichter P et al (2008) Etiology‐dependent molecular mechanisms in human hepatocarcinogenesis. Hepatology 47: 511–520 [DOI] [PubMed] [Google Scholar]
- 19. Lin CP, Liu CR, Lee CN, Chan TS, Liu HE (2010) Targeting c‐Myc as a novel approach for hepatocellular carcinoma. World J Hepatol 2: 16–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kawate S, Fukusato T, Ohwada S, Watanuki A, Morishita Y (1999) Amplification of c‐myc in hepatocellular carcinoma: correlation with clinicopathologic features, proliferative activity and p53 overexpression. Oncology 57: 157–163 [DOI] [PubMed] [Google Scholar]
- 21. Peng SY, Lai PL, Hsu HC (1993) Amplification of the c‐myc gene in human hepatocellular carcinoma: biologic significance. J Formos Med Assoc 92: 866–870 [PubMed] [Google Scholar]
- 22. Shachaf CM, Kopelman AM, Arvanitis C, Karlsson A, Beer S, Mandl S, Bachmann MH, Borowsky AD, Ruebner B, Cardiff RD et al (2004) MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature 431: 1112–1117 [DOI] [PubMed] [Google Scholar]
- 23. Hu S, Balakrishnan A, Bok RA, Anderton B, Larson PEZ, Nelson SJ, Kurhanewicz J, Vigneron DB, Goga A (2011) 13C‐pyruvate imaging reveals alterations in glycolysis that precede c‐Myc‐induced tumor formation and regression. Cell Metab 14: 131–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lim L, Balakrishnan A, Huskey N, Jones KD, Jodari M, Ng R, Song G, Riordan J, Anderton B, Cheung ST et al (2014) MicroRNA‐494 within an oncogenic microRNA megacluster regulates G1/S transition in liver tumorigenesis through suppression of mutated in colorectal cancer. Hepatology 59: 202–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tanabe M, Kanehisa M (2012) Using the KEGG database resource. Curr Protoc Bioinformatics 38: 1.12.1–1.12.43 [DOI] [PubMed] [Google Scholar]
- 26. Wu G, Fang Y‐Z, Yang S, Lupton JR, Turner ND (2004) Glutathione metabolism and its implications for health. J Nutr 134: 489–492 [DOI] [PubMed] [Google Scholar]
- 27. Lu SC (2013) Glutathione synthesis. Biochim Biophys Acta 1830: 3143–3153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tward AD, Jones KD, Yant S, Kay MA, Wang R, Bishop JM (2005) Genomic progression in mouse models for liver tumors. Cold Spring Harb Symp Quant Biol 70: 217–224 [DOI] [PubMed] [Google Scholar]
- 29. Cao Z, Fan‐Minogue H, Bellovin DI, Yevtodiyenko A, Arzeno J, Yang Q, Gambhir SS, Felsher DW (2011) MYC phosphorylation, activation, and tumorigenic potential in hepatocellular carcinoma are regulated by HMG‐CoA reductase. Cancer Res 71: 2286–2297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. He L, Thomson JM, Hemann MT, Hernando‐Monge E, Mu D, Goodson S, Powers S, Cordon‐Cardo C, Lowe SW, Hannon GJ et al (2005) A microRNA polycistron as a potential human oncogene. Nature 435: 828–833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dews M, Homayouni A, Yu D, Murphy D, Sevignani C, Wentzel E, Furth EE, Lee WM, Enders GH, Mendell JT et al (2006) Augmentation of tumor angiogenesis by a Myc‐activated microRNA cluster. Nat Genet 38: 1060–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. O'Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT (2005) c‐Myc‐regulated microRNAs modulate E2F1 expression. Nature 435: 839–843 [DOI] [PubMed] [Google Scholar]
- 33. Guil S, Cáceres JF (2007) The multifunctional RNA‐binding protein hnRNP A1 is required for processing of miR‐18a. Nat Struct Mol Biol 14: 591–596 [DOI] [PubMed] [Google Scholar]
- 34. Liu WH, Yeh SH, Lu CC, Yu SL, Chen HY, Lin CY, Chen DS, Chen PJ (2009) MicroRNA‐18a prevents estrogen receptor‐α expression, promoting proliferation of hepatocellular carcinoma cells. Gastroenterology 136: 683–693 [DOI] [PubMed] [Google Scholar]
- 35. Li L, Guo Z, Wang J, Mao Y, Gao Q (2012) Serum miR‐18a: a potential marker for hepatitis B virus‐related hepatocellular carcinoma screening. Dig Dis Sci 57: 2910–2916 [DOI] [PubMed] [Google Scholar]
- 36. Burchard J, Zhang C, Liu AM, Poon RTP, Lee NPY, Wong K‐F, Sham PC, Lam BY, Ferguson MD, Tokiwa G et al (2010) microRNA‐122 as a regulator of mitochondrial metabolic gene network in hepatocellular carcinoma. Mol Syst Biol 6: 402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ben‐Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A, Weinberg RA (2008) An embryonic stem cell‐like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet 40: 499–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pedica F, Ruzzenente A, Bagante F, Capelli P, Cataldo I, Pedron S, Iacono C, Chilosi M, Scarpa A, Brunelli M et al (2013) A re‐emerging marker for prognosis in hepatocellular carcinoma: the add‐value of FISHing c‐myc gene for early relapse. PLoS One 8: e68206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Huang Q, Tan Y, Yin P, Ye G, Gao P, Lu X, Wang H, Xu G (2013) Metabolic characterization of hepatocellular carcinoma using nontargeted tissue metabolomics. Cancer Res 73: 4992–5002 [DOI] [PubMed] [Google Scholar]
- 40. Budhu A, Roessler S, Zhao X, Yu Z, Forgues M, Ji J, Karoly E, Qin LX, Ye QH, Jia HL et al (2013) Integrated metabolite and gene expression profiles identify lipid biomarkers associated with progression of hepatocellular carcinoma and patient outcomes. Gastroenterology 144: 1066–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fussell KC, Udasin RG, Gray JP, Mishin V, Smith PJS, Heck DE, Laskin JD (2011) Redox cycling and increased oxygen utilization contribute to diquat‐induced oxidative stress and cytotoxicity in Chinese hamster ovary cells overexpressing NADPH‐cytochrome P450 reductase. Free Radic Biol Med 50: 874–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Han E‐S, Muller FL, Pérez VI, Qi W, Liang H, Xi L, Fu C, Doyle E, Hickey M, Cornell J et al (2008) The in vivo gene expression signature of oxidative stress. Physiol Genomics 34: 112–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Porro A, Iraci N, Soverini S, Diolaiti D, Gherardi S, Terragna C, Durante S, Valli E, Kalebic T, Bernardoni R et al (2011) c‐MYC oncoprotein dictates transcriptional profiles of ATP‐binding cassette transporter genes in chronic myelogenous leukemia CD34+ hematopoietic progenitor cells. Mol Cancer Res 9: 1054–1066 [DOI] [PubMed] [Google Scholar]
- 44. Nikiforov MA, Chandriani S, O'Connell B, Petrenko O, Kotenko I, Beavis A, Sedivy JM, Cole MD (2002) A functional screen for Myc‐responsive genes reveals serine hydroxymethyltransferase, a major source of the one‐carbon unit for cell metabolism. Mol Cell Biol 22: 5793–5800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kress TR, Pellanda P, Pellegrinet L, Bianchi V, Nicoli P, Doni M, Recordati C, Bianchi S, Rotta L, Capra T et al (2016) Identification of Myc‐dependent transcriptional programs in oncogene‐addicted liver tumors. Cancer Res 76: 3463–3472 [DOI] [PubMed] [Google Scholar]
- 46. DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, Thompson CB (2007) Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci USA 104: 19345–19350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Levy S, Forman HJ (2010) c‐Myc is a Nrf2‐interacting protein that negatively regulates phase II genes through their electrophile responsive elements. IUBMB Life 62: 237–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Benassi B, Fanciulli M, Fiorentino F, Porrello A, Chiorino G, Loda M, Zupi G, Biroccio A (2006) c‐Myc phosphorylation is required for cellular response to oxidative stress. Mol Cell 21: 509–519 [DOI] [PubMed] [Google Scholar]
- 49. Hinchman CA, Ballatori N (1994) Glutathione conjugation and conversion to mercapturic acids can occur as an intrahepatic process. J Toxicol Environ Health 41: 387–409 [DOI] [PubMed] [Google Scholar]
- 50. Arnér ESJ, Holmgren A (2006) The thioredoxin system in cancer. Semin Cancer Biol 16: 420–426 [DOI] [PubMed] [Google Scholar]
- 51. Singh A, Bodas M, Wakabayashi N, Bunz F, Biswal S (2010) Gain of Nrf2 function in non‐small‐cell lung cancer cells confers radioresistance. Antioxid Redox Signal 13: 1627–1637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shibata T, Ohta T, Tong KI, Kokubu A, Odogawa R, Tsuta K, Asamura H, Yamamoto M, Hirohashi S (2008) Cancer related mutations in NRF2 impair its recognition by Keap1‐Cul3 E3 ligase and promote malignancy. Proc Natl Acad Sci USA 105: 13568–13573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010) Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Adams SB, Setton LA, Kensicki E, Bolognesi MP, Toth AP, Nettles DL (2012) Global metabolic profiling of human osteoarthritic synovium. Osteoarthr Cartil 20: 64–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chen X, Xie C, Sun L, Ding J, Cai H (2015) Longitudinal metabolomics profiling of Parkinson's disease‐related α‐synuclein A53T transgenic mice. PLoS One 10: e0136612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chen Y, Tang Y, Zhang YC, Huang XH, Xie YQ, Xiang Y (2015) A metabolomic study of rats with doxorubicin‐induced cardiomyopathy and Shengmai injection treatment. PLoS One 10: e0125209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Benjamin DI, Louie SM, Mulvihill MM, Kohnz RA, Li DS, Chan LG, Sorrentino A, Bandyopadhyay S, Cozzo A, Ohiri A et al (2014) Inositol phosphate recycling regulates glycolytic and lipid metabolism that drives cancer aggressiveness. ACS Chem Biol 9: 1340–1350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Smyth G (2005) limma: Linear models for microarray data In Bioinformatics and computational biology solutions using R and bioconductor, Gentleman R, Carey VJ, Huber W, Irizarry RA, Dudoit S. (eds), pp 397–420. New York: Springer. [Google Scholar]
- 59. Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD (2012) The SVA package for removing batch effects and other unwanted variation in high‐throughput experiments. Bioinformatics 28: 882–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK (2015) limma powers differential expression analyses for RNA‐sequencing and microarray studies. Nucleic Acids Res 43: e47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Blake JA, Bult CJ, Eppig JT, Kadin JA, Richardson JE (2014) The Mouse Genome Database: integration of and access to knowledge about the laboratory mouse. Nucleic Acids Res 42: D810–D817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hastie T, Tibshirani R, Sherlock G (1999) Imputing missing data for gene expression arrays. http://ww.web.stanford.edu/~hastie/Papers/missing.pdf
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Dataset EV1
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 5
Data Availability Statement
Primary data
Gene expression values for human liver tumor samples 36, 40 are available from the GEO site (accession codes GSE14520 and GSE22058, respectively). Metabolite abundance values for human liver tumor samples were shared upon request to the respective authors 39, 40. miRNA expression values for murine liver tumors 24 are available at GEO accession code GSE44570. mRNA expression values for LT2‐MYC tumor formation and regression 23 are available at GEO accession code GSE28198. Metabolomic profiling data for LT2‐MYC tumors are available as Dataset EV1 with this article.
Referenced data
Summary data of MYC‐dependent regulation of glutathione pathway genes (Appendix Table S3) were extracted from Supplemental Data published previously 45.