

Abstract
Interleukin 17 (IL‐17) is an important inducer of tissue inflammation and is involved in numerous autoimmune diseases. However, how its signal transduction is regulated is not well understood. Here, we report that nuclear Dbf2‐related kinase 1 (NDR1) functions as a positive regulator of IL‐17 signal transduction and IL‐17‐induced inflammation. NDR1 deficiency or knockdown inhibits the IL‐17‐induced phosphorylation of p38, ERK1/2, and p65 and the expression of chemokines and cytokines, whereas the overexpression of NDR1 promotes IL‐17‐induced signaling independent of its kinase activity. Mechanistically, NDR1 interacts with TRAF3 and prevents its binding to IL‐17R, which promotes the formation of an IL‐17R‐Act1‐TRAF6 complex and downstream signaling. Consistent with this, IL‐17‐induced inflammation is significantly reduced in NDR1‐deficient mice, and NDR1 deficiency significantly protects mice from MOG‐induced experimental autoimmune encephalomyelitis (EAE) and 2,4,6‐trinitrobenzenesulfonic acid (TNBS)‐induced colitis likely by its inhibition of IL‐17‐mediated signaling pathway. NDR1 expression is increased in the colons of ulcerative colitis (UC) patients. Taken together, these findings suggest that NDR1 is involved in the development of autoimmune diseases.
Keywords: IL‐17, inflammation, NDR1, TRAF3
Subject Categories: Immunology, Signal Transduction
Introduction
Interleukin 17 (IL‐17A or IL‐17) has been recognized as a pro‐inflammatory cytokine that is mainly produced by Th17 cells, a recently identified subset of CD4+ T helper cells 1. In addition to being produced by Th17 cells, IL‐17 is also produced by a variety of innate immune cells, including iNKT cells 2, δγ T cells 3, LTi‐like cells 4, and NK cells 5. IL‐17A belongs to the IL‐17 family, which consists of six molecules (IL‐17A to IL‐17F). Of all members of the IL‐17 family, IL‐17A and IL‐17F are the best studied. They share the highest amino acid sequence similarity and the same receptors and activate many common downstream signaling events 1. IL‐17 exerts its effects by promoting tissue‐resident cells to produce various matrix metalloproteinases and pro‐inflammatory molecules, including IL‐6, CXCL2, CXCL1, and CCL20, which act synergistically to induce the inflammatory response and recruit neutrophils to the inflamed tissues 1.
It has been shown that IL‐17 is responsible for the development of many autoimmune diseases, such as experimental autoimmune encephalomyelitis (EAE) 6, rheumatoid arthritis 7, and inflammatory bowel disease (IBD) 8. In addition, IL‐17 levels are increased in patients with multiple sclerosis (MS) and ulcerative colitis (UC) 9, 10. Furthermore, mice deficient in IL‐17R or IL‐17 show a reduced severity of EAE compared to that of wild‐type mice 11, 12, and IL‐17R‐deficient mice display more resistance to TNBS‐induced colitis than their wild‐type counterparts 8. These reports suggest that IL‐17 functions as a critical pro‐inflammatory factor in the pathogenesis of EAE and IBD, which indicates that IL‐17 may be a potential therapeutic target for treating autoimmune diseases.
The IL‐17 receptor family consists of five members (IL‐17RA to IL‐17RE). IL‐17RA and IL‐17RC are the receptors for IL‐17A and IL‐17F 1. IL‐17RA is ubiquitously expressed on various cell types, including epithelial cells, fibroblasts, and various myeloid cells, but the expression of IL‐17RC seems to be limited mostly to non‐hematopoietic cells 13. This may explain why IL‐17A and IL‐17F exert effects mainly on non‐hematopoietic cells and tissues. Upon binding with IL‐17A or IL‐17F, the IL‐17RA‐IL‐17RC heterodimeric receptor complex recruits tumor necrosis factor receptor‐associated factor 6 (TRAF6) via the adaptor protein nuclear factor‐κB activator 1 (Act1) to activate the MAPKs and NF‐κB pathways and induce the production of pro‐inflammatory cytokines and chemokines 1. This receptor complex also recruits tumor necrosis factor receptor‐associated factors 2 and 5 (TRAF2, TRAF5) to prolong the t1/2 of chemokines 14. Intriguingly, recent progress in studies of IL‐17 signaling has shown that TRAF3 interacts with IL‐17R to interfere with the recruitment of Act1 by IL‐17R and negatively regulates the IL‐17 signaling cascade 15. However, the details of how IL‐17 signaling is regulated remain elusive.
Nuclear Dbf2‐related kinase 1 (NDR1), also known as serine/threonine kinase 38 (STK38), belongs to the NDR/LATS kinase family, a subfamily of the AGC group of serine/threonine kinases 16. NDR1 positively regulates centrosome duplication in a kinase activity‐dependent manner 17. A recent study has identified NDR1 as a tumor suppressor in human colorectal cancer, where it acts by phosphorylating yes‐associated protein (YAP1) at the S127 site 18. NDR1 inhibits Toll‐like receptor 9 (TLR9)‐activated inflammatory responses by promoting MEKK2 ubiquitination in macrophages 19. However, the functions and pathway‐related activities of NDR1 in the inflammatory response are still poorly defined.
In this study, we identified NDR1 as an adaptor protein that positively modulates IL‐17‐mediated signaling and inflammation. NDR1 promotes the IL‐17‐triggered phosphorylation of p38, ERK1/2, and p65 in HeLa cervical carcinoma cells, mouse embryonic fibroblasts (MEFs), and astrocytes. NDR1 also enhances the IL‐17‐induced expression of inflammatory genes in vitro and in vivo. In addition, mice deficient in NDR1 are largely protected from MOG‐induced EAE and 2,4,6‐trinitrobenzenesulfonic acid (TNBS)‐induced IBD. Moreover, the expression of NDR1 in the colon mucosal epithelial cells of UC patients is increased. Further study showed that NDR1 directly interacts with TRAF3 and promotes the formation of the activation complex IL‐17R‐Act1‐TRAF6, which results in the positive regulation of IL‐17‐induced signal transduction and inflammatory factor production.
Results
NDR1 promotes IL‐17‐induced inflammation in vitro and in vivo
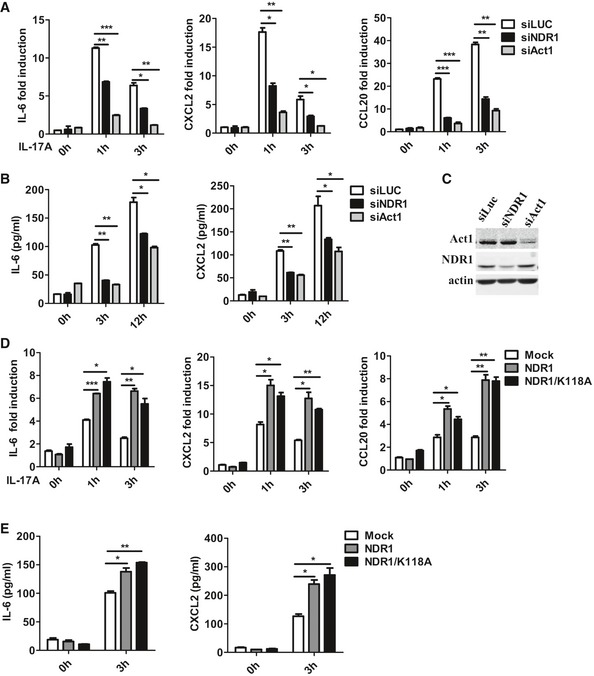
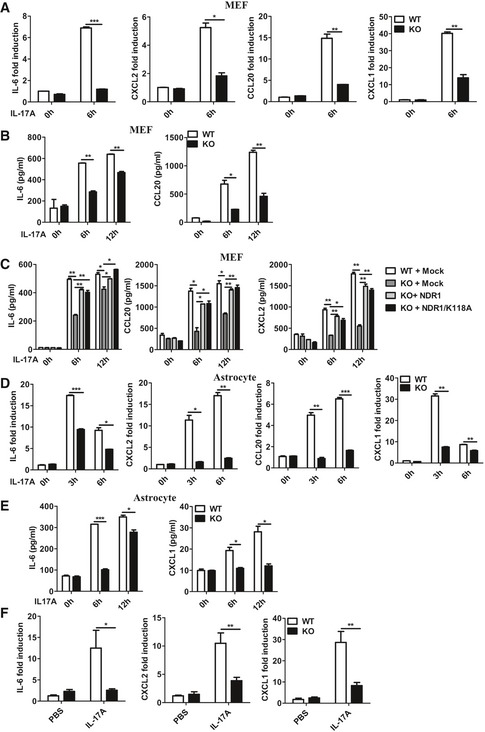
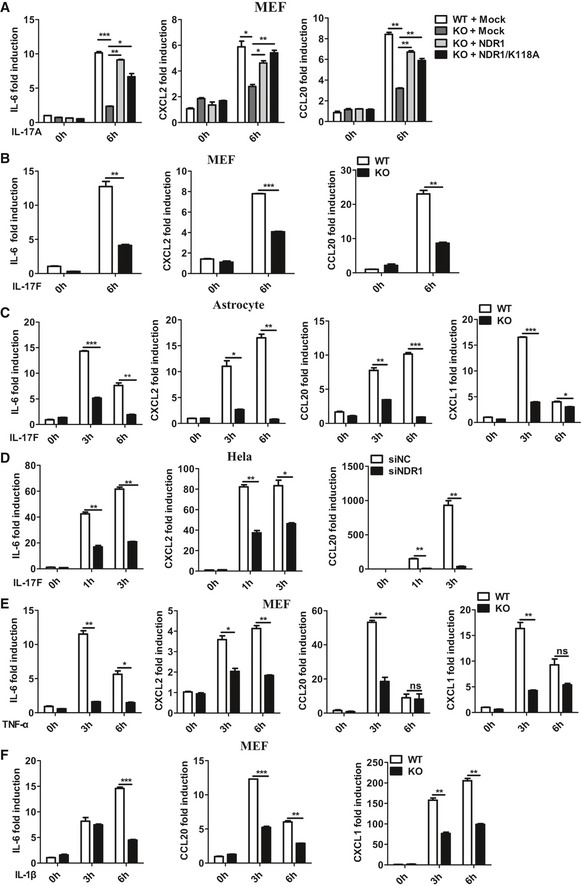
We transfected control small interfering RNA against luciferase genes (siLUC), Act1‐specific siRNA, or NDR1‐specific siRNA into HeLa cells to test whether NDR1 regulates IL‐17‐induced inflammatory cytokines and chemokines. Compared to the control siRNA, both NDR1‐specific siRNA and Act1‐specific siRNA significantly suppressed the IL‐17‐induced mRNA expression of IL‐6, CXCL2, and CCL20 in HeLa cells (Fig EV1A). The production of IL‐6 and CXCL2 was also decreased in NDR1‐silenced cells (Fig EV1B). Act1 plays an essential role in the positive regulation of IL‐17 signaling 20. Therefore, Act1‐specific siRNA, which efficiently inhibited Act1 expression (Fig EV1C), was used as the positive control. To further verify the role of NDR1 in IL‐17‐induced inflammation, plasmids expressing either the NDR1 protein or its kinase‐dead mutant K118A, which contains a single‐residue mutation at Lys118 (K118A) in the catalytic site of NDR1 16, were transfected into HeLa cells. The results in Fig EV1D and E show that both the overexpression of NDR1 and its kinase‐dead mutant K118A dramatically enhanced the IL‐17‐induced expression and production of IL‐6, CXCL2, and CCL20, which indicates that NDR1 kinase activation is not required for its function in IL‐17‐mediated signaling. We then examined the effect of NDR1 deficiency on the expression of IL‐17‐induced pro‐inflammatory molecules in mouse embryo fibroblasts (MEFs). Compared with wild‐type MEFs, NDR1‐deficient (Ndr1‐KO) MEFs showed much lower mRNA levels of IL‐6, CXCL2, CCL20, and CXCL1 (Fig 1A), and protein levels of IL‐6 and CCL20 (Fig 1B). Ndr1‐KO MEFs were transfected with plasmids expressing NDR1 or its kinase‐dead mutant K118A using a virus retroviral system. As shown in Figs 1C and EV2A, the overexpression of NDR1 or K118A in Ndr1‐KO MEFs restored IL‐17‐induced gene expression and production to a comparable level to that of the wild‐type MEFs. In addition, a deficiency of NDR1 in primary astrocytes resulted in much lower mRNA and protein level of pro‐inflammatory molecules induced by IL‐17 (Fig 1D and E). Furthermore, IL‐17F‐induced IL‐6, CXCL2, and CCL20 mRNA expression was inhibited in Ndr1‐KO MEFs (Fig EV2B), Ndr1‐KO primary astrocytes (Fig EV2C), and NDR1‐silenced HeLa cells (Fig EV2D). In addition, NDR1 deficiency inhibited the TNF‐α‐ and IL‐1β‐induced expression of pro‐inflammatory cytokines and chemokines, such as IL‐6, CXCL2, and CCL20 (Fig EV2E and F). Taken together, these results suggest that NDR1 functions as a positive regulator of IL‐17A‐ and IL‐17F‐induced inflammation in vitro. The efficiency of NDR1 knockdown, knockout, or overexpression was confirmed by immunoblot assay (see section “NDR1 promotes IL‐17‐mediated signaling independent of its kinase activity” below).
Figure EV1. NDR1 promotes the IL‐17‐induced expression of pro‐inflammatory cytokines in HeLa cells in a kinase activity‐independent manner.
-
A–CHeLa cells were transfected with NDR1 siRNA, Act1 siRNA (positive control) or luciferase siRNA (negative control) and then stimulated with IL‐17 (50 ng/ml) for 0, 1, 3, or 12 h. The mRNA levels and production of IL‐6, CXCL2, and CCL20 were analyzed by real‐time PCR (A) and ELISA (B), respectively. The efficiency of knockdown was detected by Western blot (C).
-
D, EHeLa cells were transfected with mock, NDR1, or NDR1/K118A plasmids and then stimulated with IL‐17 (50 ng/ml) for the indicated times. The induction of IL‐6, CXCL2, and CCL20 mRNA expression were analyzed by real‐time PCR (D) and ELISA (E), respectively.
Figure 1. NDR1 promotes IL‐17‐induced inflammation in vivo and in vitro .
-
A, BReal‐time PCR (A) and ELISA (B) analysis of IL‐6, CXCL2, CCL20, and CXCL1 mRNA expression and production in wild‐type (WT) and NDR1‐deficient homozygous (Ndr1‐KO) MEFs following stimulation with IL‐17 (100 ng/ml) for the indicated times.
-
CWT and Ndr1‐KO MEFs transfected with a retrovirus encoding mock, Flag‐NDR1, or Flag‐NDR1/K118A were treated with IL‐17 (100 ng/ml) for 0, 6, or 12 h. The protein levels of IL‐6, CCL20, and CXCL2 were analyzed by ELISA.
-
D, EReal‐time PCR (D) and ELISA (E) analysis IL‐6, CXCL2, CCL20, and CXCL1 mRNA expression and production in WT and Ndr1‐KO primary astrocytes following stimulation with IL‐17 (100 ng/ml) for the indicated times.
-
FNdr1‐KO mice (n = 5) and WT mice (n = 5) were treated by intraperitoneal injection of PBS or IL‐17 (0.5 μg in 200 μl PBS) for 24 h, and then, the peritoneal mesothelial cells were isolated to detect IL‐6, CXCL2, and CXCL1 mRNA expression.
Figure EV2. NDR1 promotes the IL‐17F‐induced expression of pro‐inflammatory cytokines.
-
AWT and Ndr1‐KO MEFs transfected with a retrovirus encoding mock, Flag‐NDR1, or Flag‐NDR1/K118A were treated with IL‐17 (100 ng/ml) for 0 or 6 h. The mRNA levels of IL‐6, CXCL2, and CCL20 were analyzed by real‐time PCR.
-
B, CWT and Ndr1‐KO MEFs (B) or primary astrocytes (C) were treated with IL‐17F (100 ng/ml) for the indicated times, and the induction of IL‐6, CXCL2, CCL20, and CXCL1 mRNA expression was analyzed by real‐time PCR.
-
DHeLa cells were transfected with NDR1 siRNA or control siRNA and then were treated with IL‐17F (50 ng/ml) for 0, 1, or 3 h, and the induction of IL‐6, CXCL2, and CCL20 mRNA expression was analyzed by real‐time PCR.
-
E, FWT and Ndr1‐KO MEFs were treated with TNF‐α (20 ng/ml) (D) or IL‐1β (10 ng/ml) (E) for the indicated times, and the induction of IL‐6, CXCL2, CCL20, and CXCL1 mRNA expression was analyzed by real‐time PCR.
To investigate the role of NDR1 in the IL‐17‐induced inflammatory response in vivo, NDR1 knockout (Ndr1‐KO) mice and control littermates (WT) were intraperitoneally injected with IL‐17. Compared with the expression in peritoneal mesothelial cells collected from WT mice, the expression of IL‐6, CXCL1, and CXCL2 mRNA was significantly decreased in those from Ndr1‐KO mice (Fig 1F). These results suggest that NDR1 promotes the IL‐17‐induced expression of pro‐inflammatory factors in vivo.
NDR1 promotes TNBS‐induced colitis
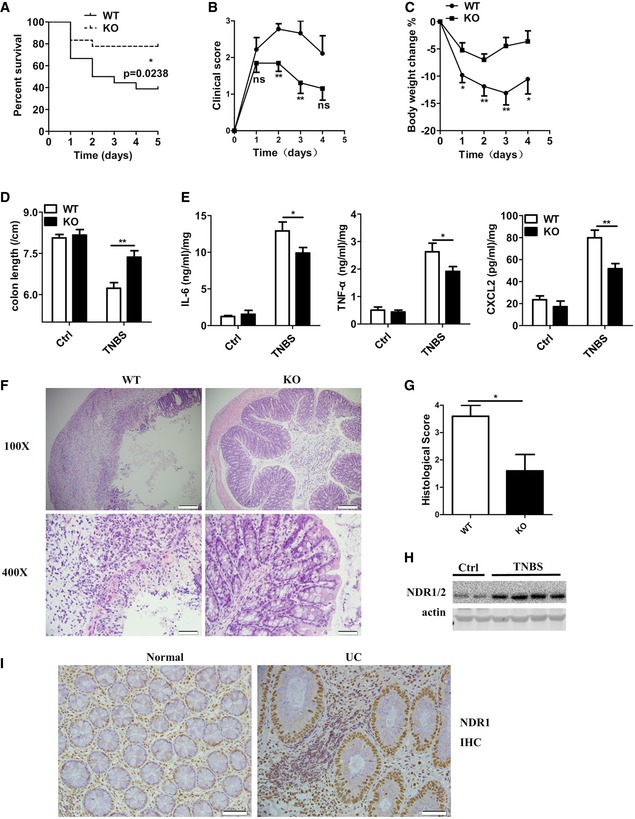
IL‐17R has been demonstrated to contribute to the development of 2,4,6‐trinitrobenzenesulfonic acid (TNBS)‐induced colitis, which is a model for autoimmune diseases that resembles human inflammatory bowel disease 8. To further investigate the role of NDR1 in IL‐17‐related autoimmune diseases, Ndr1‐KO mice and control littermates (WT) were challenged with TNBS as previously described 8. Notably, Ndr1‐KO mice exhibited a markedly prolonged survival (Fig 2A). Furthermore, Ndr1‐KO mice showed more resistance to TNBS‐induced colitis than WT mice, which was demonstrated by lower clinical scores (Fig 2B), less weight loss (Fig 2C), and reduced colon shortening (Fig 2D) in Ndr1‐KO mice. Consistent with these results, the amount of IL‐6, TNF‐α, and CXCL2 produced in the cultured whole colons of TNBS‐treated Ndr1‐KO mice was less than that of WT mice (Fig 2E). Histological analysis revealed a decreased infiltration of mononuclear cells, and less severe destruction of goblet cells and mucosal erosion in the colons of TNBS‐treated Ndr1‐KO mice compared to TNBS‐treated WT mice (Fig 2F and G). Notably, NDR1 expression was higher in the colons of TNBS‐treated WT mice than control mice (Fig 2H). These results indicate that NDR1 contributes to TNBS‐induced colitis and colonic damage.
Figure 2. NDR1 deficiency restricts TNBS‐induced colitis.
-
AWT (n = 18) and Ndr1‐KO (n = 18) mice were challenged intrarectally with 2,4,6‐trinitrobenzenesulfonic acid (TNBS) (200 mg/kg) to induce acute colitis. Mouse death was monitored until day 5. *P < 0.05 versus WT TNBS (Mantel–Cox test).
-
B–DIn a separate experiment, WT (n = 9) and Ndr1‐KO (n = 13) mice were treated with TNBS (150 mg/kg). Clinical scores (B), changes in body weight (C), and colon length (D) were measured in WT and Ndr1‐KO mice treated with TNBS for a total of 4 days. Mice were euthanized on day 4.
-
EELISA analysis of IL‐6, TNF‐α, and CXCL2 production by cultured whole‐colon tissue from the mice shown in (D), which were euthanized on day 4.
-
FHistology of colonic cross sections from mice treated as in (D). Scale bar of the upper panel, 200 μm; scale bar of the lower panel, 50 μm.
-
GSemiquantitative histological score was assessed as described in the Materials and Methods section. *P < 0.05 versus WT TNBS. WT (n = 5) and Ndr1‐KO (n = 5).
-
HWestern blotting analysis of NDR1 expression in TNBS‐induced colonic proteins.
-
IRepresentative NDR1‐antibody staining of human colon sections from non‐IBD normal controls and from UC patients. Scale bar, 50 μm.
We next examined NDR1 protein expression in colon samples of 34 UC patients and 29 control non‐IBD subjects by immunohistochemistry. The epithelial NDR1 level was enhanced in UC patients compared to that of normal control subjects (Fig 2I). Biopsies from all UC patients (34 of 34) exhibited strong NDR1 staining, whereas only 66% (19 of 29) of the normal control subjects showed strong NDR1 staining (Table 1, P = 0.004). Taken together, these results indicate that NDR1 might be involved in the development of human colitis.
Table 1.
NDR1 expression in UC patients from Xinhua Hospital
| Group types | Total no. studied | NDR1 expression | |||
|---|---|---|---|---|---|
| − (%) | + (%) | ++ (%) | +++ (%) | ||
| Normal | 29 | 0 (0.0%) | 0 (0.0%) | 10 (34%) | 19 (66%) |
| Ulcerative colitisa | 34 | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 34 (100.0%) |
Correlations were analyzed using Pearson's chi‐squared test.
P = 0.004 compared with normal colon tissues.
IL‐17A is a pivotal pro‐inflammatory cytokine secreted by Th17, a particular subset of T lymphocytes 1, and IL‐17A has been reported to be involved in the development of IBD 10. NDR1 was shown clearly to promote TNBS‐induced colitis and IL‐17‐mediated inflammation. We next explored whether NDR1 has effect on the production of Th17 cells in TNBS‐induced IBD. The mice were presensitized, and colitis was induced by rectal injection with TNBS. Consistent with our aforementioned results, Ndr1‐KO mice showed much lower disease severity, which was characterized by less weight loss (Fig EV3A), and less shrinkage in the colon length (Fig EV3B). To determine whether NDR1 deficiency affects the induction of Th17 cells in TNBS‐induced colitis, fluorescence‐activated cell sorter (FACS) analysis of intestinal lamina propria cells and mesenteric lymph node cells in WT and Ndr1‐KO mice was conducted on day 4 after induction of colitis. The frequency and the absolute number of Th17 cells (Fig EV3C–H) and regulatory T cells (Treg) (Appendix Fig S1A–D) in colons and mesenteric lymph nodes were comparable between TNBS‐treated WT and TNBS‐treated Ndr1‐KO mice. These results suggest that NDR1 has no effect on the production of Th17 or Treg cells in TNBS‐induced colitis. To further explore the role of NDR1 in the differentiation of Th17 and Treg cells, we performed an in vitro naive CD4+ T‐cell activation assay. Ablation of NDR1 had no effect on the production of Th17 effector cells (Fig EV3I and J) or Treg cells (Appendix Fig S1E and F). Taken together, these results suggest NDR1 contributes to TNBS‐induced colitis likely by its promotion of IL‐17‐mediated signaling rather than the source of IL‐17.
Figure EV3. NDR1 deficiency does not affect Th17 cell production in vivo and vitro.
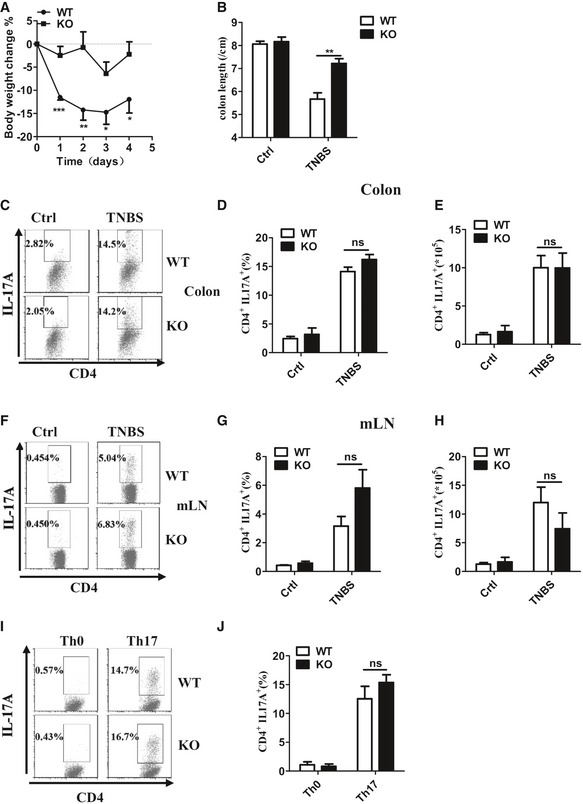
-
A, BWT (n = 6) and Ndr1‐KO (n = 5) mice were presensitized with 1% TNBS and then were rectally injected with TNBS at a dose of 150 mg/kg body weight on day 7. Changes in body weight (A) and colon length (B) were measured in WT and Ndr1‐KO mice with TNBS for a total of 4 days. Mice were euthanized on day 4.
-
C–EColons were collected on day 4; then, colonic propria cells were separated and stained with anti‐mouse markers CD4 and IL‐17A and then analyzed by flow cytometry. A representative plot of Th17 cell frequency (C) and a summary graph of Th17 cell frequency (D) and number (E) were showed.
-
F–HThe mesenteric lymph node cells were treated as in Fig 3C. A representative plot of Th17 cell frequency (F) and a summary graph of Th17 cell frequency (G) and number (H) were showed.
-
I, JNaive CD4+ T cells (CD44loCD62Lhi) isolated from Ndr1‐KO mice and control littermates (WT) were stimulated for 7 days with anti‐CD3 and anti‐CD28 under Th0 or Th17 conditions as described in the Materials and Methods section. Flow cytometry was used to measure the frequency of IL‐17‐producing cells, which is shown a representative plot (I) and a summary graph (J).
NDR1 contributes to EAE by its promotion of IL‐17‐mediated signaling
IL‐17 binds to IL‐17R expressed on epithelial cells, endothelial cells, and fibroblasts 1 and plays a critical role in the development of EAE 12. To further determine whether NDR1 promotes IL‐17‐induced inflammation in vivo, Ndr1‐KO and WT mice were irradiated to destroy bone marrow cells and then given a transplant of bone marrow cells from WT or Ndr1‐KO mice. Eight weeks after bone marrow transplantation, EAE was induced in the mice by immunizing them with myelin oligodendrocyte glycoprotein (MOG; MOG [35‐55]) 15. Whereas EAE was strongly induced in WT mice chimera (WT→WT) with a peak clinical score of 4.1 and WT mice transplanted with Ndr1‐KO bone marrow cells (KO→WT) with a peak clinical score of 3.9, the disease severity was significantly inhibited in Ndr1‐KO mice transplanted with WT bone marrow cells (WT→KO) with a peak clinical score of 2.5 (Fig 3A). Consistently, levels of myelin oligodendrocyte glycoprotein (MOG)‐induced pro‐inflammatory factors such as IL‐6, CXCL1, CXCL2, and TNF‐α in the spinal cords (Fig 3B) and brains (Fig 3C) of WT→KO chimeras were significantly lower than those in WT→WT chimeras and KO→WT chimeras, and the levels of these pro‐inflammatory factors were similar between WT→WT chimeras and KO→WT chimeras. Histological analysis by hematoxylin and eosin staining revealed a substantially lower level of perivascular infiltration of inflammatory cells in the spinal cords of WT→KO chimeras, and less demyelination was also observed by Luxol fast blue staining in WT→KO chimeras. However, perivascular infiltration of inflammatory cells and demyelination were comparable between the spinal cords of WT→WT chimeras and those of KO→WT (Fig 3D). These results collectively suggest that NDR1 deficiency in a non‐hematopoietic system restricts EAE development.
Figure 3. NDR1 deficiency in a non‐hematopoietic system restricts MOG‐induced EAE.
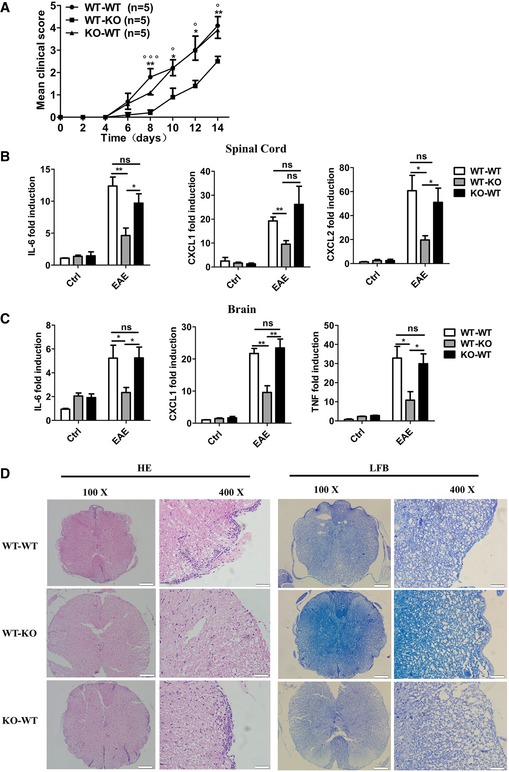
-
AMice with reconstituted bone marrow were immunized with MOG (35‐55) to induce EAE. Mean clinical scores were calculated every other day according to the standards described in the Materials and Methods section (*WT‐WT versus WT‐KO; °KO‐WT versus WT‐KO).
-
B, CIL‐6, TNF‐α, CXCL1, and CXCL2 mRNA in the spinal cords (B) or in the brains (C) were measured by real‐time PCR on day 14 after the second MOG immunization.
-
DHistology of the spinal cord was analyzed by hematoxylin and eosin (HE) or Luxol fast blue (LFB) staining on day 14 after the second MOG immunization. Scale bars for the left panel, 200 μm; scale bars for the right panel, 50 μm.
NDR1 was clearly shown to promote IL‐17‐mediated signaling and downstream gene induction of inflammatory molecules both in vitro and in vivo (Figs 1, EV2 and EV3). We next investigated whether NDR1‐mediated promotion of IL‐17‐mediated signaling was really responsible for the observed inhibitory effect on EAE in the WT→KO chimera mice. The blocking antibody of IL‐17A was used during the induction of EAE. Consistent with a previous report 6, treatment of the IL‐17‐blocking antibody greatly ameliorated EAE severity and delayed onset of disease in WT chimeras (Fig 4A). The WT→KO chimera mice exhibited much reduced EAE severity compared to WT chimeras, which was obliterated after the injection of IL‐17‐blocking antibody (Fig 4A). Parallel gene expression analyses revealed the induction of several known IL‐17 target genes, IL‐6, CXCL1, and CXCL2 in spinal cord (Fig 4B) and IL‐6, CXCL1, and TNF‐α in brain (Fig 4C) was substantially attenuated in the WT→KO chimeras compared to WT chimeras, but the expression of these genes in WT chimeras decreased to similar levels in WT→KO chimeras after treatment with IL‐17‐blocking antibody (Fig 4B and C). Consistently, histological analysis by hematoxylin and eosin staining revealed a markedly reduced perivascular infiltration of inflammatory cells in the spinal cords of WT→KO chimeras, and attenuated demyelination was also observed by Luxol fast blue staining in WT→KO chimeras. The IL‐17‐blocking antibody suppressed inflammatory infiltration and demyelination in WT chimeras to comparable levels of those in WT→KO chimeras (Fig 4D). These results collectively suggest that NDR1 deficiency in a non‐hematopoietic system restricts EAE development likely by its promotion of IL‐17‐mediated signaling.
Figure 4. NDR1 deficiency restricts MOG‐induced EAE by its promotion of IL‐17 signaling.
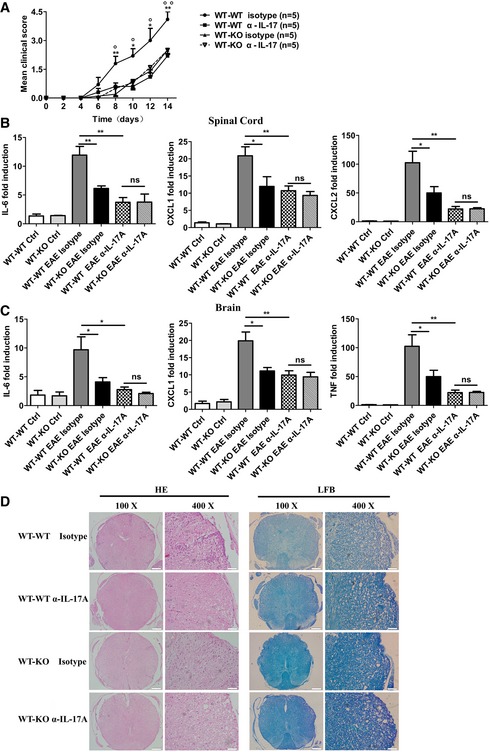
-
AMice with reconstituted bone marrow were immunized with MOG (35‐55) to induce EAE. The mice (n = 5/group) were treated with intraperitoneal injection of an anti‐IL‐17A antibody (100 μg/mouse each time) or appropriate isotype controls on days 7, 9, 11, and 13 after the second MOG immunization. Mean clinical scores were calculated every other day according to the standards described in the Materials and Methods section (*WT‐WT isotype versus WT‐KO isotype; °WT‐WT isotype versus WT‐WT α‐IL‐17).
-
B, CIL‐6, TNF‐α, CXCL1, and CXCL2 mRNA in the spinal cords (B) or in the brains (C) were measured by real‐time PCR on day 14 after the second MOG immunization.
-
DHistology of the spinal cord was analyzed by hematoxylin and eosin (HE) or Luxol fast blue (LFB) staining on day 14 after the second MOG immunization. Scale bars for the left panel, 200 μm; scale bars for the right panel, 50 μm.
To further prove that NDR1 contributes to EAE development by its promotion of IL‐17‐mediated signaling, Th17 cell transfer was conducted to induce EAE, which is normally used for assessing the IL‐17‐mediated effect on EAE induction. EAE induced by Th17 cell transfer was dramatically suppressed in the Ndr1‐KO mice versus WT mice (Appendix Fig S2A). Consistent with the clinical scores, histological analysis by hematoxylin and eosin staining showed markedly reduced inflammatory infiltration and demyelination in the spinal cords of Ndr1‐KO mice (Appendix Fig S2B). Furthermore, the induction of IL‐6, CXCL1, CXCL2, and CCL20 in spinal cord (Appendix Fig S2C) and brain (Appendix Fig S2D) was substantially attenuated in the Ndr1‐KO mice compared to the WT mice, indicating that NDR1 deficiency controls EAE development through its effects on the CNS‐resident cells.
NDR1 promotes IL‐17‐mediated signaling independent of its kinase activity
The underlying signaling mechanism by which NDR1 promotes IL‐17‐triggered inflammation was explored next. The IL‐17‐induced MAPKs and NF‐κB pathway are essential for expression of pro‐inflammatory factors 13. As shown in Fig 5A, the silencing of NDR1 in HeLa cells downregulated the IL‐17‐induced phosphorylation of p38, ERK1/2, and NF‐κB p65. Consistent with this, NDR1 deficiency in MEFs (Fig 5B) or astrocytes (Fig 5C) inhibited MAPKs and NF‐κB p65 activation induced by IL‐17. Furthermore, overexpression of NDR1 and its kinase mutant in Ndr1‐KO MEFs restored the IL‐17‐induced phosphorylation of the p38, ERK1/2, and NF‐κB p65 subunits (Fig 5D). Similarly, the reduction in IL‐17‐induced p38, ERK1/2, and NF‐κB p65 activation in NDR1‐silenced HeLa cells was prevented by the overexpression of NDR1 and its K118A mutants (Fig EV4A). Conversely, overexpression of NDR1 or NDR1 K118A in HeLa cells increased the IL‐17‐induced activation of p38, ERK1/2, and NF‐κB p65 (Fig EV4B). The IL‐17F‐induced activation of p38 and NF‐κB p65 was also noticeably decreased in NDR1‐silenced HeLa cells, Ndr1‐KO MEFs, and primary astrocytes (Fig EV4C–E). NDR1 deficiency also inhibited the TNF‐α‐ and IL‐1β‐induced activation of p38 and ERK1/2 (Fig EV4F and G). Taken together, these results suggest that NDR1 promotes the IL‐17‐mediated signaling pathway in a kinase activity‐independent manner.
Figure 5. NDR1 promotes IL‐17 signaling.
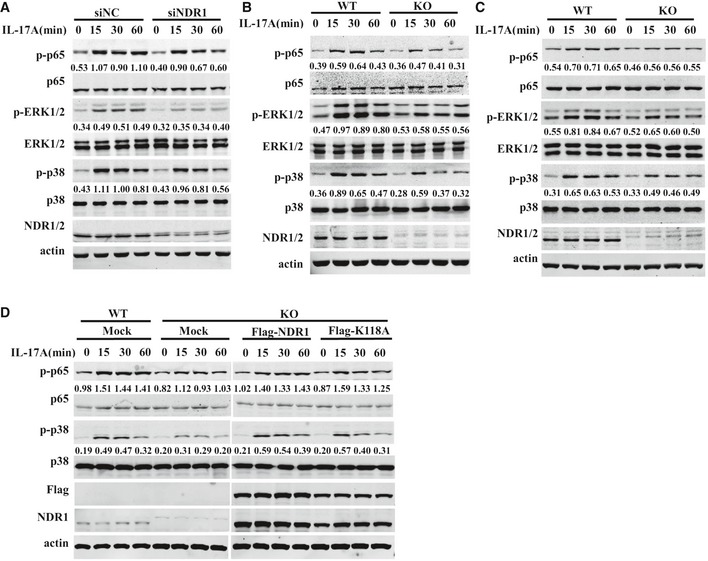
-
AHeLa cells were transfected with NDR1 siRNA or control siRNA and then stimulated with 50 ng/ml IL‐17 for 0, 15, 30, or 60 min. Whole cell lysates were immunoblotted with the indicated antibodies.
-
B, CWT and Ndr1‐KO MEFs (B) or primary astrocytes (C) were treated with 100 ng/ml IL‐17 for 0, 15, 30, or 60 min. Whole cell lysates were immunoblotted with the indicated antibodies.
-
DWT and Ndr1‐KO MEFs transfected with a retrovirus encoding mock, Flag‐NDR1, or Flag‐NDR1/K118A were treated with IL‐17 (100 ng/ml), and the whole cell lysates were immunoblotted with the indicated antibodies.
Figure EV4. NDR1 promotes IL‐17A and IL‐17F signaling.
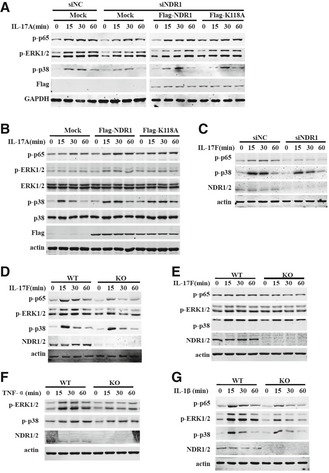
-
AHeLa cells were transfected with NDR1 siRNA or control siRNA for 24 h and then were transfected with empty vectors or plasmids encoding Flag‐NDR1 or Flag‐NDR1/K118A for 48 h. The cells were stimulated with 50 ng/ml IL‐17 for 0, 15, 30, or 60 min, and the whole cell lysates were immunoblotted with the indicated antibodies.
-
BHeLa cells were transfected with mock or plasmids encoding Flag‐NDR1 or Flag‐NDR1/K118A and then stimulated with IL‐17 (50 ng/ml) for 0, 15, 30, or 60 min. Whole cell lysates were immunoblotted with the indicated antibodies.
-
CHeLa cells were transfected with NDR1 siRNA or control siRNA and then were treated with IL‐17F (50 ng/ml) for 0, 15, 30, or 60 min. Whole cell lysates were immunoblotted with the indicated antibodies.
-
D, EWT and Ndr1‐KO MEFs (D) or primary astrocytes (E) were treated with IL‐17F (100 ng/ml) for 0, 15, 30, or 60 min. Whole cell lysates were immunoblotted with the indicated antibodies.
-
F, GWT and Ndr1‐KO MEFs were treated with TNF‐α (20 ng/ml) (F) or IL‐1β (10 ng/ml) (G) for 0, 15, 30, or 60 min. Whole cell lysates were immunoblotted with the indicated antibodies.
NDR1 interacts with TRAF3
To investigate the underlying mechanisms involved in the positive regulation of IL‐17‐mediated signaling by NDR1, we examined the effects of NDR1 on the expression of IL‐17R, Act1 20, 21, TRAF6 21, TRAF3 15, TRAF2, and TRAF5 14, which have been demonstrated to modulate IL‐17 signaling in HeLa cells and MEFs. NDR1 had no effect on the expression of these proteins (data not shown). Therefore, we immunoprecipitated NDR1 from lysates of HeLa cell stably overexpressing Flag‐NDR1 to determine which proteins might interact with NDR1 in IL‐17‐mediated signaling. The results showed that NDR1 physically associates with TRAF3 (Fig 6A) but not with TRAF2, TRAF6, TRAF4, TAK1, or Act1 (Appendix Fig S3A). When a TRAF3‐specific antibody was used in the immunoprecipitation assay, the association between TRAF3 and NDR1 was also detectable in HeLa cells (Fig 6B) and MEFs (Appendix Fig S3B). IL‐17 treatment promoted this association in a time‐dependent manner, and the interaction between these proteins peaked at 5 min after IL‐17 treatment (Fig 6B). When transiently overexpressed, NDR1 also interacted with TRAF3 (Fig 6C) but not with TRAF2, TRAF6, TAK1, or Act1 in HEK293T cells (data not shown). NDR1 directly interacted with TRAF3, as shown by the fact that Flag‐TRAF3 was able to pull down Myc‐NDR1 and vice versa (Fig 6D and E). NDR1 consists of a central kinase catalytic domain and a hydrophobic motif‐containing C‐terminal and N‐terminal 19. The different deletion constructs of NDR1 (Appendix Fig S3C) were constructed to detect their interactions with TRAF3. Domain‐mapping experiments showed that TRAF3 interacts with the central domain of NDR1 (NDR1 dS‐TKc) (Fig 6F). In addition, we constructed different deletion mutants of TRAF3 (Appendix Fig S3D) and examined which domain is required for TRAF3 to interact with NDR1. The results (Fig 6G) showed that only the full‐length and the TRAF deletion mutant interacted with NDR1, indicating that the RING‐finger, zinc‐finger, and coiled‐coil domains might be required for the association of TRAF3 with NDR1.
Figure 6. NDR1 interacts with TRAF3.
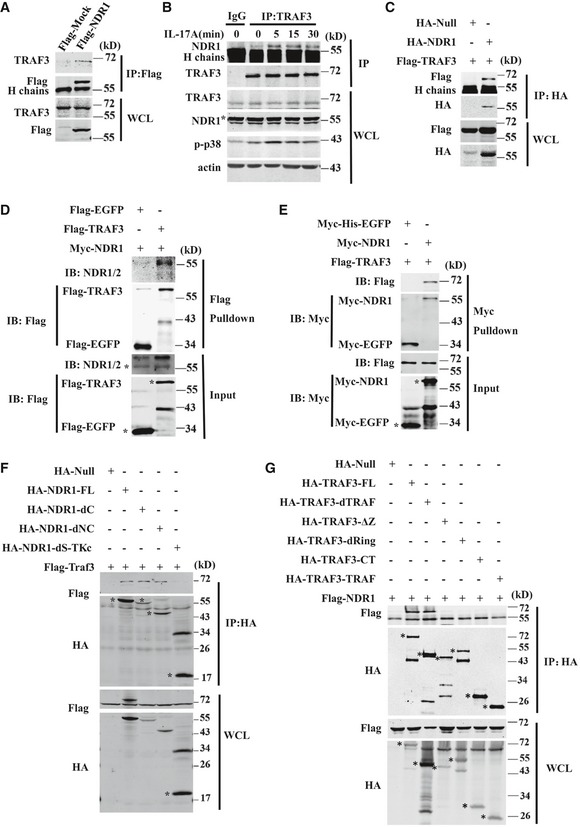
- Whole cell lysates of HeLa cells stably expressing Flag‐NDR1 or Flag‐mock were immunoprecipitated (IP) with anti‐Flag beads, which was followed by immunoblotting (IB) with anti‐Flag or anti‐TRAF3.
- Whole cell lysates of IL‐17‐treated HeLa cells were immunoprecipitated with anti‐TRAF3 or control IgG, which was followed by immunoblotting (IB) with anti‐TRAF3, anti‐NDR1, anti‐p‐p38, or anti‐actin.
- HEK293T cells were transfected with the combined plasmids as indicated. Whole cell lysates were immunoprecipitated (IP) with anti‐HA beads, which was followed by immunoblotting (IB) with anti‐Flag or anti‐HA.
- Recombinant Myc‐tagged NDR1 proteins were mixed with Flag‐tagged TRAF3 or Flag‐tagged EGFP‐purified proteins, and then, the proteins were pulled down with anti‐Flag beads.
- Recombinant Flag‐tagged TRAF3 proteins were mixed with Myc‐tagged NDR1 or Myc‐His‐tagged EGFP‐purified proteins, and then, the proteins were pulled down with protein A/G beads plus anti‐Myc.
- HA‐tagged full‐length NDR1, dC (1–440), dNC (90–440), or dS‐TKc truncation mutants were co‐expressed with Flag‐TRAF3 in HEK293T cells and then immunoprecipitated using anti‐HA beads.
- HA‐tagged full‐length TRAF3, dTRAF, dRing, ΔZinc, CT, or TRAF truncation mutants were co‐expressed with Flag‐NDR1 in HEK293T cells and then immunoprecipitated by anti‐HA beads.
NDR1 enhances IL‐17‐mediated signaling by targeting TRAF3
To further investigate whether NDR1 positively regulates IL‐17‐mediated signaling by targeting TRAF3, we transfected control siRNA or NDR1‐specific siRNA into Traf3 +/+ MEFs and Traf3 −/− MEFs and then detected IL‐17‐mediated signaling events by Western blot analysis. In line with previous findings 15, a TRAF3 deficiency greatly enhanced the IL‐17‐induced activation of p38, ERK1/2, and NF‐κB p65 and expression of cytokines (Fig 7A and B). The silencing of NDR1 impaired the phosphorylation of p38, ERK1/2, and NF‐κB p65 subunits that was induced by IL‐17 in Traf3 +/+ MEFs but did not affect the phosphorylation of p38, ERK1/2, and NF‐κB p65 subunits induced by IL‐17 in Traf3 −/− MEFs (Fig 7A). In addition, the silencing of NDR1 diminished the IL‐17‐induced IL‐6, CXCL2, CXCL1, and CCL20 expression in Traf3 +/+ MEFs but not in Traf3 −/− MEFs (Fig 7B). The production of IL‐6 and CCL20, as detected by ELISA, verified the results from the mRNA assay experiments (Fig 7C).
Figure 7. NDR1 promotes IL‐17 signaling by targeting TRAF3.
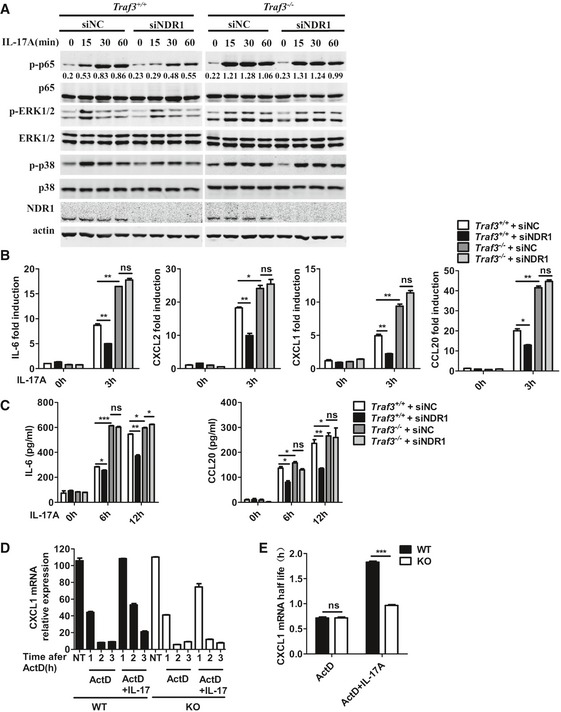
- Wild‐type (Traf3 +/+) and TRAF3‐deficient (Traf3 −/−) MEFs were transfected with control siRNA or NDR1 siRNA and stimulated with IL‐17 (100 ng/ml) for the indicated times. Whole lysates were subjected to Western blot. Numbers between two blots indicate densitometry of phospho‐p65 relative to that of total p65.
- Real‐time PCR analysis of IL‐6, CXCL2, CXCL1, and CCL20 mRNA expression in MEFs treated as described in (A).
- ELISA assay of IL‐6 and CCL20 production in MEFs from (B).
- WT and Ndr1‐KO MEFs were treated with TNF‐α (20 ng/ml) for 1 h. Fresh medium containing ActD alone or with IL‐17 was then added. Total RNA was collected at the indicated times, the levels of CXCL1 and actin mRNA were detected by real‐time PCR, and the percentage of remaining CXCL1 mRNA relative to actin is shown for each experimental condition.
- The mRNA level for CXCL1 was normalized to the Actin mRNA level and plotted as log of the percentage of remaining mRNA versus time. The best fit to liner decay was determined, and the t1/2 was calculate from the intersection at the point corresponding to 50% residual RNA.
TRAF3 has been shown to suppress the stabilization of the chemokine CXCL1 mRNA induced by IL‐17 15. We therefore examined whether NDR1 modulates the IL‐17‐mediated stabilization of CXCL1 mRNA. WT and Ndr1‐KO MEFs were pretreated with TNF‐α for 1 h and then treated with actinomycin D (to block transcription) alone or with IL‐17 for 1–3 h. IL‐17 prolonged the mRNA t1/2 of CXCL1 in both WT and Ndr1‐KO MEFs. The degradation of KC mRNA was more rapid in Ndr1‐KO MEFs than in WT MEFs (Fig 7D); thus, the half‐life of the CXCL1 chemokine induced by TNF‐α was extended in Ndr1‐KO MEFs (Fig 7E), which indicates NDR1 promotes the IL‐17‐mediated stabilization of KC mRNA. All together, these results suggest that NDR1 promotes IL‐17‐mediated signaling by targeting TRAF3.
It has been reported that TRAF3 interacts with the TNF receptor 2 (TNFR2) or TRAF2 and inhibits TNFR2 signaling 22. NDR1 deficiency inhibited the TNF‐α‐induced expression of pro‐inflammatory cytokines and activation of p38 and ERK1/2 (Figs EV2E and EV4F). Therefore, we next explored whether NDR1 regulates TNF‐α‐mediated signaling via TRAF3 using Traf3‐knockout MEFs. Silencing of NDR1 inhibited the phosphorylation of p38 and ERK1/2 and the induction of IL‐6, CXCL1, and CCL20 triggered by TNF‐α in WT MEFs. These processes were also abolished in Traf3 −/− MEFs (Fig EV5A and B). Taken together, these results demonstrate that NDR1 promotes TNF‐α‐mediated signaling via targeting TRAF3.
Figure EV5. NDR1 promotes TNF‐α signaling via targeting TRAF3.
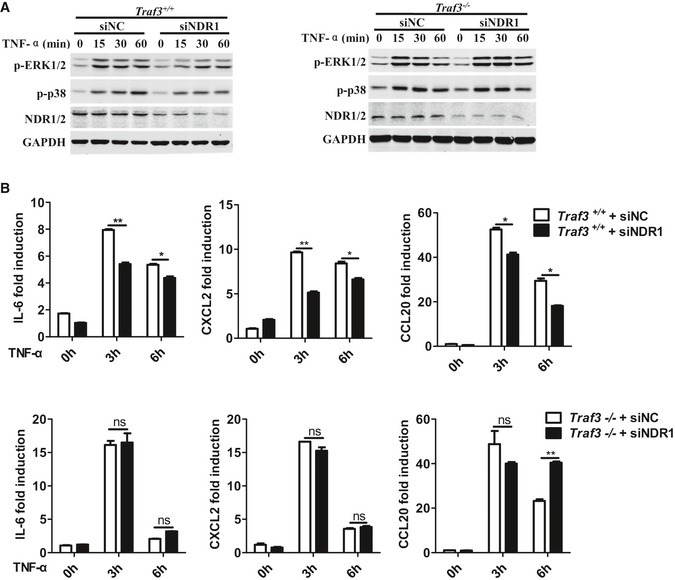
- Wild‐type (Traf3 +/+) and TRAF3‐deficient (Traf3 −/−) MEFs were transfected with NDR1 siRNA or control siRNA and then stimulated with TNF‐α (20 ng/ml) for the indicated times. Whole lysates were subjected to Western blot.
- Real‐time PCR analysis of IL‐6, CXCL2, and CCL20 mRNA expression in MEFs treated as described in (A). *P < 0.05 and **P < 0.01 (unpaired, two‐tailed Student's t‐test). Similar results were obtained in two independent experiments. Error bars are mean ± SEM values.
Source data are available online for this figure.
NDR1 competes with IL‐17R to interact with TRAF3
TRAF3 has been shown to negatively regulate IL‐17‐induced signaling by competing with Act1 to bind IL‐17R and interfere with the formation of the IL‐17R‐Act1‐TRAF6 complex 15. Because the binding of NDR1 with TRAF3 was promoted by IL‐17 treatment, we next explored whether NDR1 interferes with the interaction between IL‐17R and TRAF3. Because the commercially available anti‐IL‐17R antibody was not efficient in immunoprecipitating endogenous IL‐17R, HeLa cell lines stably expressing HA‐tagged IL‐17R were transfected with NDR1‐specific RNA or NDR1‐expressing plasmids, and co‐immunoprecipitation assays were conducted. The overexpression of NDR1 impaired the IL‐17‐induced interaction between IL‐17R and TRAF3 but promoted the interaction between IL‐17R and Act1 (Fig 8A). In contrast, silencing of NDR1 promoted the interaction of IL‐17R with TRAF3 but inhibited the interaction of IL‐17R with Act1 in IL‐17‐treated HeLa cells (Fig 8B). In addition, HeLa cells transfected with empty or NDR1 plasmids were treated with IL‐17 for different times and then immunoprecipitated with TRAF3‐ or Act1‐specific antibodies. As shown in Fig 8C and D, the overexpression of NDR1 impaired the IL‐17‐induced association of IL‐17R with TRAF3 (Fig 8C) but promoted the interaction of IL‐17R with Act1 (Fig 8D). To confirm these results, Ndr1‐KO MEFs were used to perform immunoprecipitation assay with TRAF3 or Act1 antibodies. As shown in Fig 8E and F, the IL‐17‐induced association of IL‐17R and TRAF3 was enhanced (Fig 8E), and the interaction of IL‐17R with Act1 was inhibited (Fig 8F), in Ndr1‐KO MEFs compared to WT MEFs. Collectively, these results suggest that NDR1 interferes with the association of IL‐17R and TRAF3 and promotes the formation of the IL‐17R‐Act1 complex, which is required for downstream signaling and cytokine induction.
Figure 8. NDR1 competes with IL‐17R to interact with TRAF3 and promote the formation of the IL‐17R‐ACT1 complex.
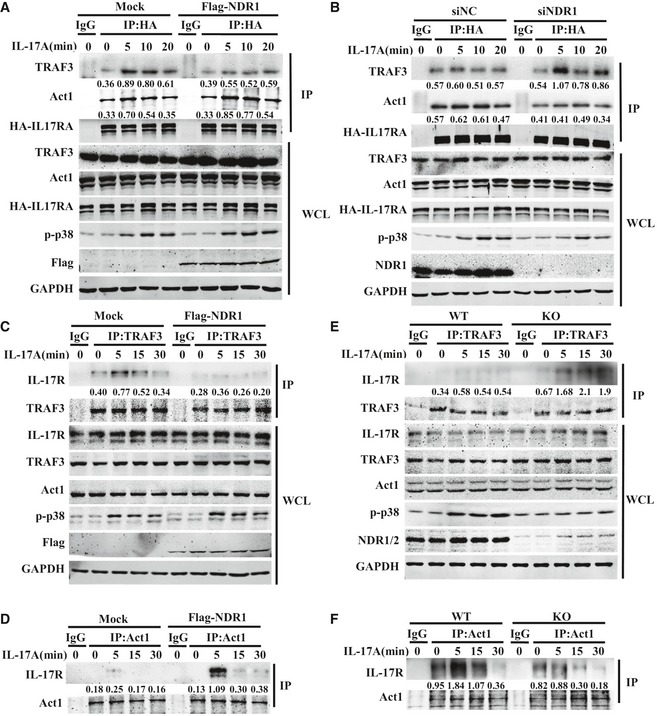
-
A, BHeLa cells stably expressing HA‐IL‐17RA were transfected with NDR1 siRNA or control siRNA (A) or plasmids encoding NDR1 (B) and then were treated with IL‐17 (50 ng/ml) for 0, 5, 10, or 20 min. Whole cell lysates were immunoprecipitated (IP) with anti‐HA or control IgG, which was followed by immunoblotting (IB) with the indicated antibodies. Numbers between two blots indicate densitometry of TRAF3 or ACT1 relative to HA‐IL‐17R in immunoprecipitates.
-
C, DHeLa cells were transfected with plasmids encoding NDR1 or with empty vectors and then stimulated with IL‐17 (50 ng/ml) for 0, 5, 15, or 30 min. Whole cell lysates were immunoprecipitated (IP) with anti‐TRAF3 (C) or anti‐Act1 (D), which was followed by immunoblotting with the indicated antibodies. Numbers between two blots indicate densitometry of IL‐17R relative to that of TRAF3 (C) or Act1 (D) in immunoprecipitates.
-
E, FWT and Ndr1‐KO MEFs were treated with 100 ng/ml IL‐17 for the indicated times. Whole cell lysates were immunoprecipitated (IP) with anti‐TRAF3 (E) or anti‐Act1 (F), which was followed by immunoblotting with the indicated antibodies. Numbers between two blots indicate densitometry of IL‐17R relative to that of TRAF3 (E) or Act1 (F) in immunoprecipitates.
Discussion
IL‐17 binds to the IL‐17 receptor and triggers a signaling pathway leading to the expression of cytokines and chemokines. The exploration of the signaling pathway linking this receptor activation to a specific physiological function and the details of the molecular mechanism are important for the development of new therapeutic strategies for IL‐17‐dependent inflammatory and autoimmune diseases. Here, we showed that NDR1 protein kinase promoted the formation of the IL‐17R and Act1 complex by targeting TRAF3 and enhanced the IL‐17‐induced production of pro‐inflammatory cytokines and chemokines. NDR1 deficiency reduced IL‐17‐induced peritonitis and EAE in mice.
NDR1 is involved in the regulation of BCR signaling via the modulation of MYC and p21 protein stability during the progression of the cell cycle 23. It is also involved in mediating both egress of thymocytes from the thymus and lymphocyte motility 24. The role of NDR1 in specific signaling pathways and inflammation is still poorly characterized. To date, only one published study has shown that NDR1 functions as a negative regulator in TLR9‐mediated signaling and inflammation in macrophages 19. In the current study, we identified NDR1 as positive regulator of IL‐17‐induced inflammation in vitro and in vivo. The IL‐17‐mediated expression of inflammatory genes was significantly decreased by NDR1 deficiency or knockdown. Interestingly, both the overexpression of NDR1 and its kinase‐dead mutant K118A robustly increased the IL‐17‐induced expression of pro‐inflammatory molecules. In addition to suppressing the response to IL‐17 in vitro, NDR1 deficiency also resulted in impaired inflammatory responses in vivo. The peritoneal mesothelial cells of Ndr1‐KO mice expressed lower levels of IL‐17‐induced genes than wild‐type (WT) control mice.
It has been well studied that IL‐17 cytokine plays critical roles in pathogenesis of IBD 20, 25. Here, we demonstrated Ndr1‐KO mice were also more resistant to TNBS‐induced colitis. However, NDR1‐KO mice did not show any defect in the production of Th17 cells which are reported to secrete IL‐17 cytokine during the phase of TNBS‐induced colitis 26. Consistently, NDR1 deficiency does not affect Th17 cell differentiation in vitro. Simultaneously, NDR1 deficiency in the non‐hematopoietic system of mice but not in the hematopoietic system correlated with a dramatically lower severity of EAE compared to WT mice. Furthermore, IL‐17‐blocking antibody treatment during the induction of EAE decreased EAE severity including clinical scores, pro‐inflammatory cytokine production, pro‐inflammatory infiltration, and demyelination in WT chimeras to paralleled levels of WT→KO chimeras. Taken together, NDR1 positively regulates the pathogenesis of IBD and EAE in vivo by the promotion of IL‐17‐mediated signaling rather than the induction of IL‐17 cells. Further study would be worthwhile to explore the importance of NDR1 in the control of other autoimmune diseases, such as rheumatoid arthritis and psoriasis.
It has been well documented that Act1 20, 21, TRAF6 21, TRAF4 27, TRAF3 15, TRAF2, and TRAF5 14 are involved in the activation of NF‐κB and MAPKs and in the stabilization of chemokine CXCL1 mRNA triggered by IL‐17. Here, we found that NDR1 greatly promoted the activation of NF‐κB and MAPKs and the stabilization of CXCL1 mRNA in response to stimulation with IL‐17, suggesting that NDR1 plays a general role in IL‐17 signaling. Co‐immunoprecipitation experiments showed that NDR1 specifically interacted with TRAF3, which has been reported to be a negative regulator of IL‐17R‐mediated signaling 15. We further demonstrated that NDR1 positively regulates IL‐17‐mediated signaling by targeting TRAF3. NDR1 inhibited the binding of TRAF3 to IL‐17R, which released IL‐17R and activated IL‐17R‐Act1‐TRAF6 complex. Consistent with these results, Ndr1‐KO MEFs or NDR1‐silenced HeLa cells showed impaired NF‐κB and MAPKs signaling and a decrease in the cytokine and chemokine production induced by IL‐17F, which is also signaled via the IL‐17R‐Act1‐TRAF6 complex 28.
TNF‐α signals through two distinct receptors, TNFR1 and TNFR2 29, and both of them are expressed on MEFs. We here showed that NDR1 promoted TNF‐α‐mediated signaling by targeting TRAF3. TRAF3 has been reported to form heterotrimers with TRAF2 to inhibit TNF signaling 30. It also has been reported to interact with TNFR2 and negatively regulate TNFR2 signaling 22. Therefore, NDR1 may directly interact with TRAF3 and inhibit the binding of TRAF3 to TRAF2 or TNFR2, leading to the enhanced TNF‐α‐mediated signaling. Upon binding with IL‐1β, the receptor IL‐1R recruits myeloid differentiation primary response gene 88 (MYD88) and interleukin‐1 receptor‐activated protein kinase (IRAK) 4 to form activated trimeric complex, followed by phosphorylation of IRAK1 and IRAK2, and then recruits TRAF6 to activate the IL‐1β pathway 31. In this study, NDR1 deficiency also suppressed the IL‐1β‐mediated signaling in MEFs. However, the role of TRAF3 in IL‐1β signaling remains unknown. NDR1 has been reported to phosphorylate YAP and negatively regulate YAP activity 18, and it also has been shown to suppress TLR9 signaling by the regulation of MEKK2 expression 19. Whether NDR1 positively modulates IL‐1β‐mediated signaling via targeting TRAF3 or via modulating expression or activity of some signaling events involved in IL‐1β pathway needs to be further explored.
In summary, our study identified a novel role of NDR1 in regulating IL‐17‐induced signaling and inflammation that is independent of its kinase activity. NDR1 interacts with TRAF3 and interferes with the association of TRAF3 and IL‐17R, resulting in increased formation of the activation complex IL‐17R‐Act1, which is required for the downstream signaling and production of pro‐inflammatory factors (Appendix Fig S4). NDR1‐deficient mice are resistant to MOG‐induced EAE and TNBS‐induced colitis, and NDR1 expression was increased in the colon tissue of UC patients. These results demonstrate that NDR1 positively modulates IL‐17‐mediated inflammation and may be a potential therapeutic target for the treatment of IBD and MS.
Materials and Methods
Reagents and antibodies
Recombinant IL‐17A (mouse and human), human TGF‐β, and mouse IL‐1β were purchased from Pepro Tech; human IL‐17F (11855‐HNAE) and TNF‐α were purchased from Sino Biological Inc; actinomycin D (ActD), antibody for NDR1 (12201‐5s), and anti‐Flag (M2) beads were purchased from Sigma‐Aldrich. Primary antibodies against p‐p65 (3033s), p‐p38 (4511S), p‐ERK1/2 (4370S), p65 (8242), p38 (8690), and ERK1/2 (4695) were purchased from Cell Signaling Technology. Primary antibodies for NDR1/2 (sc‐66998), TRAF3 (sc‐6933, sc‐1828), TRAF2 (sc‐877), TRAF6 (sc‐8409), TRAF4 (sc‐136107), IL‐17R (sc‐376600), Act1 (sc‐11444), and mouse control IgG were purchased from Santa Cruz Biotechnology. Antibodies for Flag, HA, and anti‐HA beads were purchased from Abmart. Antibody for GADPH (M130718) was purchased from Abcam. JetPEI was purchased from Polysciences; INTERFERin was purchased from Polyplus; and Lipofectamine RNAiMAX was purchased from Invitrogen. Recombinant mouse IL‐6 (575702), anti‐CD3 (B202621), anti‐CD28 (B209363), anti‐IL‐4 (B213843), anti‐IFN‐γ (B193599), and anti‐IL‐12/IL‐23P40 (B174522) were purchased from BioLegend. Phycoerythrin‐conjugated antibody to mouse IL‐17A, allophycocyanin‐conjugated antibody to mouse Foxp3, and fluorescein isothiocyanate‐conjugated antibody to mouse CD4 were purchased from BioLegend. IL‐17A neutralization antibody (16‐7173‐95) and relative isotype IgG were purchased from eBioscience. Mouse Naïve CD4+ T Cell Isolation Kit (15L66873) was purchased from STEMCELL Technologies.
Mice
NDR1 knockout (Ndr1‐KO) mice were kindly provided by Professor Hemmings Brain of Friedrich Miescher Institute for Biomedical Research, and 6‐ to 8‐week‐old Ndr1‐KO mice and their littermates with a C57BL/6 background were used in this study. C57BL/6J mice were purchased from Joint Ventures Sipper BK Experimental Animals (Shanghai, China). The progeny of Ndr1 +/− intercrosses were genotyped by PCR analysis of DNA isolated from the tail using the following three primers: 5′‐GTACATTAGGTAAGACTTGAGG‐3′, 5′‐CTAGCTCATCCAGCCATGTG‐3′, and 5′‐GCAGCGCATCGCCTTCTATC‐3′. All the mice were maintained under specific pathogen‐free conditions. All animal experiments were performed in accordance with protocols approved by the Scientific Investigation Board of Zhejiang University.
Human samples
Human paraffin‐embedded colon sections from IBD patients or normal control colon sections were obtained from Xinhua Hospital. The basic information from all the patients, including age, sex, and colitis location, is summarized in Appendix Table S1.
Plasmids
TRAF3 and NDR1 were amplified from HeLa cells by PCR and ligated into pcDNA3.1‐Flag/HA to construct Flag‐ or HA‐tagged expression plasmids, and truncated TRAF3 was amplified from vectors expressing full‐length TRAF3 and cloned into pcDNA3.1‐HA expression vectors. NDR1 and NDR1/K118A were ligated to pMXs‐IRES‐GFP vector for retrovirus‐mediated restoration of gene expression in MEFs.
Cell culture, plasmid transfection, and siRNA silencing
HeLa cells, human HEK293T cells, Plate E cells, HeLa cells stably expressing HA‐tagged IL‐17R (gifts from Prof. Y. Qian, Chinese Academy of Science), and Traf3 +/+ and Traf3 −/− MEFs (gifts from Prof. G. Cheng, University of California) were cultured in DMEM supplemented with 10% FBS. Primary MEFs were isolated from embryos on embryonic days 12.5–14.5 and were maintained in DMEM supplemented with 15% FBS, 100 μg/ml penicillin G, 100 μg/ml streptomycin. HEK293T cells and HeLa cells were transfected with plasmids, as indicated below, using PEI according to the manufacturer's protocol. HeLa cells were transfected with NDR1‐targeted siRNA (GUAUUAGCCAUAGACUCUAUUdTdT) using INTERFERin, and MEFs were transfected with NDR1‐targeted siRNA (AGACCAGCUGCGAUAUCUAUUdTdT) using Lipofectamine RNAiMAX according to the manufacturer's instructions.
Retrovirus‐delivered plasmid gene overexpression
Plate E cells were transfected with pMXs‐IRES‐GFP‐Flag‐NDR1, NDR1/K118A, or pMXs‐IRES‐GFP using the calcium phosphate transfection method for viral packing, and the supernatant suspension was harvested to obtain the retrovirus. WT and Ndr1‐KO MEFs were infected with the retrovirus supernatant suspension. After 2 days, the cells were stimulated with IL‐17A for the appropriate time before real‐time PCR and immunoblot analysis.
Quantitative real‐time PCR and ELISA
Total RNA was extracted using TRIzol reagent (Invitrogen) following the manufacturer's protocols. The cDNAs were synthesized using a cDNA synthesis kit (Takara) following the manufacturer's instructions. Real‐time PCR was conducted using SYBR Green (Takara). The primer sequences are shown in Appendix Table S2. The production of pro‐inflammatory molecules was detected using an ELISA kit (eBioscience) following the manufacturer's instructions.
Co‐immunoprecipitation and immunoblotting
Co‐immunoprecipitation and immunoblot analysis were performed as previously reported 32. Transfected HEK293T cells were lysed in a lysis buffer (20 mM Tris (pH 7.4), 150 mM NaCl, 1 mM EDTA, 10% glycerol, 1% NP‐40, and a cocktail of proteinase inhibitors). The supernatant of cell lysates was incubated with HA‐conjugated beads (Abmart) or anti‐Flag beads (Sigma‐Aldrich) for 3–4 h. Endogenous TRAF3 was immunoprecipitated using anti‐TRAF3 plus protein A/G beads. The beads were boiled to separate the proteins, and then, the samples were analyzed by Western blot.
Pull‐down assay
The fusion proteins of Flag‐TRAF3, Myc‐His‐EGFP, and Flag‐EGFP were expressed in HEK293T cells and purified according to standard protocols. Myc‐NDR1 (TP304133) was obtained from OriGene Technologies. For the Flag pull‐down assay, ~3 μg of the Myc‐NDR1 fusion protein bound to anti‐Flag beads was mixed with Flag‐EGFP or Flag‐TRAF3 incubated at 4°C with gentle mixing. For the Myc pull‐down assay, ~3 μg of the Flag‐TRAF3 fusion protein bound to anti‐Myc‐coupled beads was mixed with Myc‐His‐EGFP or Myc‐NDR1 and incubated at 4°C with gentle mixing. After an overnight incubation, the beads were washed three times with a cell lysis buffer and separated with an SDS sample buffer and analyzed with Western blot.
IL‐17‐induced peritoneal inflammatory responses
Male, age‐matched WT and Ndr1‐KO littermates were injected intraperitoneally with IL‐17 (0.5 μg per mouse) or PBS. Twenty‐four hours later, peritoneal mesothelial cells were isolated as previously described 33. To remove intraperitoneal leukocytes, the peritoneal cavity was washed with 5 ml PBS, and then, the peritoneal cavity was injected with 5 ml 0.25% trypsin. After 10 min, the trypsin solution was collected, and the peritoneal cavity was washed with 5 ml DMEM containing 10% FBS. The expression of pro‐inflammatory cytokines and chemokines in the cells was measured using real‐time PCR analysis.
Bone marrow chimeras
Recipient Ndr1‐KO mice and control littermates were irradiated by a sublethal dose of γ‐rays (9.0 Gy) to kill the bone marrow cells. At 6 h post‐irradiation, the recipients were injected with 1 × 107 bone marrow cells from donor wild‐type or Ndr1‐KO mice in the tail vein. Three experimental donation groups were established: WT→WT, WT→KO, and KO→WT. Eight weeks after bone marrow transplantation, blood was collected, and NDR1 genotyping analysis was performed to exclude mice in which the transplantation had failed.
Induction of EAE
EAE was induced by a subcutaneous immunization with 200 μg of MOG peptide (35‐55) (Sangon Biotech Co.) and 1 mg of a heat‐killed H37Ra strain of Mycobacterium tuberculosis (Difo) emulsified in complete Freund's adjuvant injected into the back and head region. This was followed by an injection of pertussis toxin (List Biological Laboratories) at a dose of 200 μg/mouse in the tail vein. Forty‐eight hours later, pertussis toxin was administered again. Disease severity was scored as previously described 15.
Adoptive transfer
WT mice were first immunized with MOG (35‐55) plus complete Freund's adjuvant to induce EAE. LNs were collected 10 d later, and cells were restimulated in vitro for 5 days with 20 μg/ml MOG (35‐55) in the presence of 20 ng/ml IL‐23 for Th17 cell differentiation. The MOG‐specific Th17 cells (3 × 107 cells/mouse) were adoptively transferred into sublethally irradiated (500 rads) WT and Ndr1‐KO mice, and disease severity was scored as described previously 34.
Isolation of astrocytes
Primary astrocytes were prepared as previously described 34, with minor modifications. In brief, the cerebral cortices were collected with smooth, fine forceps from 2‐day‐old mice and were trypsinized for 12 min at 37°C, plated in PDL‐coated flasks, and cultured with DMEM containing 10% FBS (Gibco). Astrocytes that had grown to confluence 10–14 days after plating were divided and used for the experiments.
CNS mRNA analysis
Fourteen days after the second immunization, brains and spinal cords of mice with EAE were collected, and mRNA was obtained from homogenized tissue.
TNBS‐induced colitis
The mice were rectally injected with 2,4,6‐trinitrobenzenesulfonic acid (TNBS) (Sigma) at a dose of 150–200 mg/kg body weight in 50% ethanol or with 50% ethanol alone through a catheter inserted into the colon 4 cm proximal to the anus, as previously described 8. Mice were sacrificed after treatment with TNBS for 4 days, and colon tissue was washed with cold PBS containing gentamycin, penicillin, and streptomycin. The colon tissue was cut into 1‐cm2 pieces. RNA was extracted from one piece with TRIzol, and the expression of pro‐inflammatory molecules was quantified using real‐time PCR. The other piece of colon tissue was washed three times with cold PBS containing 20 μg/ml gentamycin, 200 μg/ml penicillin G, and 200 μg/ml streptomycin to remove residual intestinal bacteria and then was incubated in supplemented culture medium containing 200 μg/ml penicillin G and 200 μg/ml streptomycin 35. The medium was collected after incubation for 24 h at 37°C, and the production of pro‐inflammatory molecules was determined with ELISA. For the detection of Th17 cells and Treg cells, the mice were presensitized with the solution 1% (wt/vol) TNBS diluted in acetone and olive oil which was applied to the backs of mice. Seven days later, the mice were rectally injected with TNBS at a dose of 150 mg/kg body weight as described above. Mice were sacrificed after treatment with TNBS for 4 days, and cells isolated from mesenteric lymph nodes and intestinal lamina propria were analyzed by FACS.
Isolation of intestinal lamina propria cells
The murine intestinal lamina propria cells were isolated as previously described 36. The isolated cells were incubated for 6 h in the presence of PMA, ionomycin, and brefeldin A, followed by staining with anti‐mouse markers: CD4, IL‐17A, Foxp3.
In vitro CD4+ T‐cell differentiation
Splenocytes, cells from mesenteric lymph nodes of age‐ and sex‐matched WT and Ndr1‐KO mice, were purified with Mouse Naïve CD4+ T Cell Isolation Kit. Purified naive CD4+ T cells were activated with 1 μg/ml of plate‐bound anti‐CD3 and 3 μg/ml anti‐CD28 under Th17 (10 μg/ml anti‐IL‐4, 10 μg/ml anti‐IFN‐γ, 10 μg/ml anti‐IL‐12/IL‐23p40, 20 ng/ml IL‐6, and 5 ng/ml TGF‐β), or Treg cells (10 μg/ml anti‐IL‐4, 10 μg/ml anti‐IFN‐γ, 10 μg/ml anti‐IL‐12/IL‐23p40, and 5 ng/ml TGF‐β) conditions. After 7 days of activation, the differentiated T cells were stained with Foxp3 to quantify the frequency of Treg cells or restimulated for 4 h with PMA and ionomycin in the presence of the protein transport inhibitor brefeldin A, followed by intracellular staining with IL‐17 to quantify the frequency of Th17 cells.
Histological analysis
Colon tissue and spinal cords were fixed in 4% neutral formalin. Paraffin‐embedded tissues were cut into sections and stained with hematoxylin and eosin. Histological scores were determined blindly based on the previously described criteria 36: 0 = normal; 1 = moderate mucosal inflammation without erosion or ulcer; 2 = severe mucosal inflammation with erosion; 3 = severe mucosal inflammation with ulcer (< 1 mm); and 4 = severe mucosal inflammation with ulcer (> 1 mm).
Statistical analysis
All data are expressed as the mean ± SEM. Results presented as fold induction were relative to that of the unstimulated cells or control group, and the baseline values were set as “1” in each panel. Statistical significance between two experimental groups was assessed using Student's t‐test. The clinical specimen analysis was evaluated using Pearson's chi‐squared tests with a 95% confidence interval (CI). A value of P < 0.05 was considered statistically significant.
Author contributions
XW designed the research; CM, WL, ZL, WT, and HL performed the research; RG improved the writing; YQ and HH contributed the reagents; XW and CM analyzed the data and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 2
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Source Data for Figure 8
Acknowledgements
This work was supported by grants from the National Basic Research Program of China (973) (2014CB542101), the National Natural Science Foundation of China (81230014, 31500705), and the Natural Science Foundation of Zhejiang Province (Y15H160029).
EMBO Reports (2017) 18: 586–602
References
- 1. Iwakura Y, Ishigame H, Saijo S, Nakae S (2011) Functional specialization of interleukin‐17 family members. Immunity 34: 149–162 [DOI] [PubMed] [Google Scholar]
- 2. Cella M, Fuchs A, Vermi W, Facchetti F, Otero K, Lennerz JK, Doherty JM, Mills JC, Colonna M (2009) A human natural killer cell subset provides an innate source of IL‐22 for mucosal immunity. Nature 457: 722–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH (2009) Interleukin‐1 and IL‐23 induce innate IL‐17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity 31: 331–341 [DOI] [PubMed] [Google Scholar]
- 4. Takatori H, Kanno Y, Watford WT, Tato CM, Weiss G, Ivanov II, Littman DR, O'Shea JJ (2009) Lymphoid tissue inducer‐like cells are an innate source of IL‐17 and IL‐22. J Exp Med 206: 35–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Passos ST, Silver JS, O'Hara AC, Sehy D, Stumhofer JS, Hunter CA (2010) IL‐6 promotes NK cell production of IL‐17 during toxoplasmosis. J Immunol 184: 1776–1783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q et al (2005) A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol 6: 1133–1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li N, Wang JC, Liang TH, Zhu MH, Wang JY, Fu XL, Zhou JR, Zheng SG, Chan P, Han J (2013) Pathologic finding of increased expression of interleukin‐17 in the synovial tissue of rheumatoid arthritis patients. Int J Clin Exp Pathol 6: 1375–1379 [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang Z, Zheng M, Bindas J, Schwarzenberger P, Kolls JK (2006) Critical role of IL‐17 receptor signaling in acute TNBS‐induced colitis. Inflamm Bowel Dis 12: 382–388 [DOI] [PubMed] [Google Scholar]
- 9. Venken K, Hellings N, Hensen K, Rummens JL, Stinissen P (2010) Memory CD4+ CD127 high T cells from patients with multiple sclerosis produce IL‐17 in response to myelin antigens. J Neuroimmunol 226: 185–191 [DOI] [PubMed] [Google Scholar]
- 10. Fonseca‐Camarillo G, Mendivil‐Rangel E, Furuzawa‐Carballeda J, Yamamoto‐Furusho JK (2011) Interleukin 17 gene and protein expression are increased in patients with ulcerative colitis. Inflamm Bowel Dis 17: E135–E136 [DOI] [PubMed] [Google Scholar]
- 11. Gonzalez‐Garcia I, Zhao Y, Ju S, Gu Q, Liu L, Kolls JK, Lu B (2009) IL‐17 signaling‐independent central nervous system autoimmunity is negatively regulated by TGF‐beta. J Immunol 182: 2665–2671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, Sudo K, Iwakura Y (2006) IL‐17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol 177: 566–573 [DOI] [PubMed] [Google Scholar]
- 13. Chang SH, Dong C (2011) Signaling of interleukin‐17 family cytokines in immunity and inflammation. Cell Signal 23: 1069–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sun D, Novotny M, Bulek K, Liu C, Li X, Hamilton T (2011) Treatment with IL‐17 prolongs the half‐life of chemokine CXCL1 mRNA via the adaptor TRAF5 and the splicing‐regulatory factor SF2 (ASF). Nat Immunol 12: 853–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhu S, Pan W, Shi P, Gao H, Zhao F, Song X, Liu Y, Zhao L, Li X, Shi Y et al (2010) Modulation of experimental autoimmune encephalomyelitis through TRAF3‐mediated suppression of interleukin 17 receptor signaling. J Exp Med 207: 2647–2662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vichalkovski A, Gresko E, Cornils H, Hergovich A, Schmitz D, Hemmings BA (2008) NDR kinase is activated by RASSF1A/MST1 in response to Fas receptor stimulation and promotes apoptosis. Curr Biol 18: 1889–1895 [DOI] [PubMed] [Google Scholar]
- 17. Hergovich A, Lamla S, Nigg EA, Hemmings BA (2007) Centrosome‐associated NDR kinase regulates centrosome duplication. Mol Cell 25: 625–634 [DOI] [PubMed] [Google Scholar]
- 18. Zhang L, Tang F, Terracciano L, Hynx D, Kohler R, Bichet S, Hess D, Cron P, Hemmings BA, Hergovich A et al (2015) NDR functions as a physiological YAP1 kinase in the intestinal epithelium. Curr Biol 25: 296–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wen M, Ma X, Cheng H, Jiang W, Xu X, Zhang Y, Zhang Y, Guo Z, Yu Y, Xu H et al (2015) Stk38 protein kinase preferentially inhibits TLR9‐activated inflammatory responses by promoting MEKK2 ubiquitination in macrophages. Nat Commun 6: 7167 [DOI] [PubMed] [Google Scholar]
- 20. Qian Y, Liu C, Hartupee J, Altuntas CZ, Gulen MF, Jane‐Wit D, Xiao J, Lu Y, Giltiay N, Liu J et al (2007) The adaptor Act1 is required for interleukin 17‐dependent signaling associated with autoimmune and inflammatory disease. Nat Immunol 8: 247–256 [DOI] [PubMed] [Google Scholar]
- 21. Liu C, Qian W, Qian Y, Giltiay NV, Lu Y, Swaidani S, Misra S, Deng L, Chen ZJ, Li X (2009) Act1, a U‐box E3 ubiquitin ligase for IL‐17 signaling. Sci Signal 2: ra63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cabal‐Hierro L, Rodriguez M, Artime N, Iglesias J, Ugarte L, Prado MA, Lazo PS (2014) TRAF‐mediated modulation of NF‐kB AND JNK activation by TNFR2. Cell Signal 26: 2658–2666 [DOI] [PubMed] [Google Scholar]
- 23. Bisikirska BC, Adam SJ, Alvarez MJ, Rajbhandari P, Cox R, Lefebvre C, Wang K, Rieckhof GE, Felsher DW, Califano A (2013) STK38 is a critical upstream regulator of MYC's oncogenic activity in human B‐cell lymphoma. Oncogene 32: 5283–5291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tang F, Gill J, Ficht X, Barthlott T, Cornils H, Schmitz‐Rohmer D, Hynx D, Zhou D, Zhang L, Xue G et al (2015) The kinases NDR1/2 act downstream of the Hippo homolog MST1 to mediate both egress of thymocytes from the thymus and lymphocyte motility. Sci Signal 8: ra100 [DOI] [PubMed] [Google Scholar]
- 25. Fujino S, Andoh A, Bamba S, Ogawa A, Hata K, Araki Y, Bamba T, Fujiyama Y (2003) Increased expression of interleukin 17 in inflammatory bowel disease. Gut 52: 65–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liu J, Han C, Xie B, Wu Y, Liu S, Chen K, Xia M, Zhang Y, Song L, Li Z et al (2014) Rhbdd3 controls autoimmunity by suppressing the production of IL‐6 by dendritic cells via K27‐linked ubiquitination of the regulator NEMO. Nat Immunol 15: 612–622 [DOI] [PubMed] [Google Scholar]
- 27. Wu L, Chen X, Zhao J, Martin B, Zepp JA, Ko JS, Gu C, Cai G, Ouyang W, Sen G et al (2015) A novel IL‐17 signaling pathway controlling keratinocyte proliferation and tumorigenesis via the TRAF4‐ERK5 axis. J Exp Med 212: 1571–1587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yang XO, Chang SH, Park H, Nurieva R, Shah B, Acero L, Wang YH, Schluns KS, Broaddus RR, Zhu Z et al (2008) Regulation of inflammatory responses by IL‐17F. J Exp Med 205: 1063–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cabal‐Hierro L, Lazo PS (2012) Signal transduction by tumor necrosis factor receptors. C Cell Signal 24: 1297–1305 [DOI] [PubMed] [Google Scholar]
- 30. He L, Grammer AC, Wu X, Lipsky PE (2004) TRAF3 forms heterotrimers with TRAF2 and modulates its ability to mediate NF‐{kappa}B activation. J Biol Chem 279: 55855–55865 [DOI] [PubMed] [Google Scholar]
- 31. Weber A, Wasiliew P, Kracht M (2010) Interleukin‐1 (IL‐1) pathway. Sci Signal 3: cm1 [DOI] [PubMed] [Google Scholar]
- 32. Li H, Huang F, Fan L, Jiang Y, Wang X, Li J, Wang Q, Pan H, Sun J, Cao X et al (2014) Phosphatidylethanolamine‐binding protein 4 is associated with breast cancer metastasis through Src‐mediated Akt tyrosine phosphorylation. Oncogene 33: 4589–4598 [DOI] [PubMed] [Google Scholar]
- 33. Zhong B, Liu X, Wang X, Chang SH, Liu X, Wang A, Reynolds JM, Dong C (2012) Negative regulation of IL‐17‐mediated signaling and inflammation by the ubiquitin‐specific protease USP25. Nat Immunol 13: 1110–1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang JM, Wang HK, Ye CQ, Ge W, Chen Y, Jiang ZL, Wu CP, Poo MM, Duan S (2003) ATP released by astrocytes mediates glutamatergic activity‐dependent heterosynaptic suppression. Neuron 40: 971–982 [DOI] [PubMed] [Google Scholar]
- 35. Wirtz S, Neufert C, Weigmann B, Neurath MF (2007) Chemically induced mouse models of intestinal inflammation. Nat Protoc 2: 541–546 [DOI] [PubMed] [Google Scholar]
- 36. Lin W, Ma C, Su F, Jiang Y, Lai R, Zhang T, Sun K, Fan L, Cai Z, Li Z et al (2016) Raf kinase inhibitor protein mediates intestinal epithelial cell apoptosis and promotes IBDs in humans and mice. Gut 66: 597–610 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 2
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Source Data for Figure 8