

Abstract
Background
Activation of type IIB activin receptor (ActRIIB) in skeletal muscle leads to muscle atrophy because of increased muscle protein degradation. However, the intracellular signalling mechanism that mediates ActRIIB‐activated muscle catabolism is poorly defined.
Methods
We investigated the role of p38β mitogen‐activated protein kinases (MAPK) in mediating ActRIIB ligand activin A‐activated muscle catabolic pathways in C2C12 myotubes and in mice with perturbation of this kinase pharmacologically and genetically.
Results
Treatment of C2C12 myotubes with activin A or myostatin rapidly activated p38 MAPK and its effector C/EBPβ within 1 h. Paradoxically, Akt was activated at the same time through a p38 MAPK‐independent mechanism. These events were followed by up‐regulation of ubiquitin ligases atrogin1 (MAFbx) and UBR2 (E3α‐II), as well as increase in LC3‐II, a marker of autophagosome formation, leading to myofibrillar protein loss and myotube atrophy. The catabolic effects of activin A were abolished by p38α/β MAPK inhibitor SB202190. Using small interfering RNA‐mediated gene knockdown, we found that the catabolic activity of activin A was dependent on p38β MAPK specifically. Importantly, systemic administration of activin A to mice similarly activated the catabolic pathways in vivo, and this effect was blocked by SB202190. Further, activin A failed to activate the catabolic pathways in mice with muscle‐specific knockout of p38β MAPK. Interestingly, activin A up‐regulated MuRF1 in a p38 MAPK‐independent manner, and MuRF1 did not appear responsible for activin A‐induced myosin heavy chain loss and muscle atrophy.
Conclusions
ActRIIB‐mediated activation of muscle catabolism is dependent on p38β MAPK‐activated signalling.
Keywords: ActRIIB, Activin A, Myostatin, Muscle wasting, Cachexia, p38β MAPK
Introduction
Cachexia is a complex wasting syndrome characterized by systemic inflammation and progressive loss of skeletal muscle mass (muscle wasting). Many diseases can cause cachexia including cancer, AIDS, sepsis, diabetes, chronic renal failure, congestive heart failure, and chronic obstructive pulmonary disease. Cachexia contributes significantly to the morbidity and mortality of patients with these diseases.1 Although it has become clear that cachectic muscle wasting is primarily caused by accelerated muscle protein degradation, the precise mechanism through which the catabolic pathways in muscle are remotely activated by diseases in disparate locations is still undefined.2
In various diseases, abnormal increase of certain humoral factors can activate the cellular pathways for protein degradation in muscle, including inflammatory cytokines such as tumour necrosis factor alpha (TNFα), interleukin (IL)‐6, IL‐1β, and TNF‐like weak inducer of apoptosis, glucocorticoids, lipopolysaccharide (LPS), and cancer‐released factors such as proteolysis‐inducing factor.3 The breakdown of myofibrillar proteins is thought mediated mainly by the ubiquitin–proteasome pathway (UPP), which is capable of targeting specific proteins via E3 ubiquitin ligases.4 At least three E3s, atrogin1 (MAFbx),5, 6 MuRF1,6 and UBR2 (E3α‐II),7, 8 are up‐regulated in cachectic muscle in various disease models, which are rate limiting for UPP‐mediated muscle protein degradation and considered as markers of muscle catabolism. In addition, the autophagy–lysosome pathway (ALP) is activated in cachectic muscle,9, 10, 11 which may mediate the loss of organelles including mitochondria and cytosolic proteins.12 These two cellular proteolytic systems are often coordinately activated by diverse catabolic stimuli in a stimulus‐dependent manner. For example, fasting, denervation, or disuse inactivates Akt, resulting in activation of FoxO transcription factors to increase the activity of both the UPP and ALP.13, 14 On the other hand, inflammatory conditions and oxidative stress activate the UPP and ALP through the activation of p38 mitogen‐activated protein kinases (MAPK).15, 16, 17 At least three isoforms of the p38 MAPK family are expressed in skeletal muscle. The α‐isoform is well established as an epigenetic regulator essential for the initiation of myogenesis,18, 19 whereas the γ isoform regulates the expansion of myogenic precursor cells,20 endurance exercise‐induced mitochondrial biogenesis and angiogenesis,21 and glucose uptake.22 On the contrary, the β‐isoform appears responsible for the catabolic effect of p38 MAPK because of its activation of transcription factor C/EBPβ23 that up‐regulates atrogin124 and UBR2.8 In addition, Smad2/3 transcription factors that are activated by the TGFβ family of cytokines including activins and myostatin promote muscle protein breakdown.25 Thus, identifying the signalling pathway that mediates muscle catabolism by a particular stimulus is important for both mechanistic and therapeutic considerations.
Recent data indicate that activation of ActRIIB by its agonists, activins or myostatin, may cause muscle wasting associated with various diseases.25 Activin A is a major form of the activins. Normally, activin A serves as an endocrine factor to stimulate FSH biosynthesis and secretion in the pituitary gland.26 In many catabolic disease states, circulating activin A levels rise as a paracrine/autocrine factor generated by activated macrophages27 or certain types of cancer cells.28 Myostatin is predominantly expressed in skeletal muscle and secreted from muscle cells as a paracrine/autocrine factor.29 Similar to activin A, myostatin may also be synthesized and released by certain cancer cells and thus contributes to cancer‐induced muscle wasting.30, 31 Therefore, several drugs that antagonize ActRIIB activity have been developed and are in clinical trials for the treatment of various forms of muscle atrophy. However, the signalling mechanism through which ActRIIB mediates muscle catabolism is poorly understood. ActRIIB belongs to the TGFβ receptor superfamily and activates Smad2/3 signalling that promotes muscle catabolism.32 This led to a popular belief that the Smad2/3 signalling pathway is responsible for ActRIIB‐mediated muscle catabolism.25 However, recent data revealed that Smad3 knockout in mice does not spare the mice from muscle atrophy induced by the ActRIIB agonist myostatin,33 suggesting that Smad2/3 signalling is non‐essential for ActRIIB‐mediated muscle catabolism. As a member of the TGFβ receptor superfamily, ActRIIB activation in muscle cells leads to activation of TAK‐1,34 a kinase that activates a cascade of kinases resulting in the activation of p38 MAPK.35 Given that the activation of p38β MAPK mediates up‐regulation of E3 ligases atrogin1 and UBR2 through the transcription factor C/EBPβ,8, 23, 24 we hypothesized that ActRIIB mediates muscle catabolism through the p38β MAPK‐signalling pathway. In the current study, we tested this hypothesis and found evidence that ActRIIB‐mediated activation of muscle catabolic pathways is dependent on p38β MAPK.
Materials and methods
Myogenic cell culture
Murine C2C12 myoblasts (American Type Culture Collection) were cultured in growth medium (Dulbecco's modified eagle's medium supplemented with 10% foetal bovine serum) at 37°C with 5% CO2. At 85–90% confluence, myoblast differentiation was induced by incubation for 96 h in differentiation medium (Dulbecco's modified eagle's medium supplemented with 4% heat‐inactivated horse serum) to form myotubes. Myotubes were treated with activin A or myostatin (3 ng/mL) for indicated time and replaced every 24 h. Pretreatment of SB202190 or SP600125 (10 μM, dissolved in DMSO with 0.1% final concentration, Sigma‐Aldrich, St. Louis, MO) for 30 min was carried out when indicated. All cell culture experiments were independently replicated three times as indicated (N = 3).
Animal use
Experimental protocols were approved in advance by the institutional Animal Welfare Committee at the University of Texas Health Science Center at Houston. Mice with muscle‐specific knockout of p38β (p38β MKO) were created by crossbreeding floxed‐p38β mice (p38βf/f) in C57BL/6 background21 with muscle creatine kinase‐Cre (MCK‐Cre, The Jackson Laboratory, Bar Harbor, ME). Loss of the p38β gene was previously shown not affecting muscle phenotype.21 Activin A dissolved in phosphate‐buffered saline (PBS) was intraperitoneally (i.p.) injected into 7‐week‐old male mice (0.1 mg/kg) with PBS as control. SB202190 was i.p. injected (5 mg/kg) 30 min prior to activin A as needed. Tibialis anterior (TA) was collected in 8 h after activin A injection for analyses from six mice per group.
Transfection of small interfering RNA
Predesigned small interfering RNAs (siRNAs) specific for p38α and p38β were purchased from Sigma‐Aldrich. The IDs of p38α and p38β were SASI_Mm01_00020743 and SASI_Mm01_00044863, respectively. Control siRNA was purchased from Ambion (Austin, TX). These siRNAs were introduced into C2C12 myoblasts using the jetPRIME reagent (Polyplus‐transfection Inc., Illkirch, France) according to the manufacturer's protocol. In 24 h, myoblasts were differentiated, and experiments were started in another 96 h when myotubes were formed. Because of the role of p38α MAPK in promoting myogenic differentiation, we observed a delay in differentiation in p38α MAPK‐knockdown cells during the early stage (first 48 h). However, differentiation in these cells caught up later, and at 96 h, there was no significant difference in myotube formation between control and p38α knockdown cells. Knockdown of p38β MAPK did not alter differentiation.
Real‐time PCR
Total RNA was isolated from myotubes or muscle by using TRIzol reagent (Invitrogen, Carlsbad, CA). Real‐time PCR was performed as described previously.24 Sequences of specific primers are atrogin1 (sense: 5′‐CACATTCTCT‐CCTGGAAGGGC‐3′, antisense: 5′‐TTGATAAAGTCTTGAGGGGAAAGTG‐3′); UBR2 (sense: 5′‐TATTCTCCTCCTTACCTTG‐3′, antisense: 5′‐CGAAACCGCTCTTGGCATA‐3′); LC3b (sense: 5′‐CGTCCTGGACAAGACCAAGT‐3′, antisense: 5′‐ATTGCTGTCCCGAATGTCTC‐3′); and glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) (sense: 5′‐CATGGCCTTCCGTGTTCCTA‐3′, antisense: 5′‐GCGGCACGTCAGATCCA‐3′). Data were normalized to GAPDH.
Western blot analysis
Western blot analysis was carried out as described previously.16 Antibodies to total and/or phosphorylated p38MAPK (T181/Y182), p‐C/EBPβ (Thr‐188), Akt (S‐473), and total p38α and p38β were from Cell Signaling Technology (Beverly, MA). Antibody to total C/EBPβ was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Antibody to atrogin1/MAFbx was from ECM Biosciences (Versailles, KY). Antibodies to UBR2 and LC3‐II were obtained from Novus Biologicals (Littleton, CO). Anti‐myosin heavy chain (MHC) antibody (MF‐20) was from R&D Systems (Minneapolis, MN). Data were normalized to α‐tubulin (antibody was from Development Studies Hybridoma Bank at the University of Iowa, Iowa City, IA).
Fluorescence microscopy
C2C12 myotubes were stained with anti‐MHC antibody (MF‐20) and fluorescein isothiocyanate‐conjugated secondary antibody and examined using a Zeiss Axioskop 40 microscope and a Zeiss Axiocam MRM camera system controlled by Axiovision Release 4.6 imaging software. Acquired images were analysed for myotube diameter using the method of Menconi et al. 36 with modifications. Briefly, myotube diameters were measured in a total of 100 myotubes from ≥10 random fields using computerized image analysis (Scion Image, Frederick, MD, USA) at three points along their length. The average of diameters at the three points was used for each myotube. The measurements were conducted in a masked fashion.
Statistical analysis
Data were analysed with one‐way analysis of variance using the SigmaStat software (Systat Software, San Jose, CA) as indicated. When applicable, control samples from independent experiments were normalized to a value of 1 without showing variations (actual variations were within a normal range). A P‐value < 0.05 was considered to be statistically significant. Data are presented as the mean ± standard error.
Results
Activin A activates the p38β MAPK‐C/EBPβ catabolic signalling and the Akt anabolic signalling in C2C12 myotubes
To investigate the catabolic signalling pathways activated by ActRIIB in muscle cells, we treated C2C12 myotubes with endotoxin‐free recombinant activin A, the major agonist of ActRIIB, and monitored its downstream signalling events. Through a preliminary study involving multiple doses, we observed that 3 ng/mL of activin A provoked a robust yet non‐plateaued catabolic response in myotubes (data not shown). Using this concentration, we observed that activin A activated p38 MAPK and its effector C/EBPβ (phosphorylation of Thr‐188) at 1 h (Figure 1A). For comparison, we also monitored Akt activity by measuring the phosphorylation state of its S‐473 residue, which inversely regulates muscle catabolism through FoxO1/3.37, 38 Levels of pS473‐Akt were increased by activin A, similar to previous observations in myotubes treated with tumour cell conditioned medium24 or TNFα.39 To determine whether activin A induces C/EBPβ and Akt activation through p38 MAPK, myotubes were pre‐treated with p38α/β MAPK inhibitor SB202190, which prevented activation of C/EBPβ but not Akt (Figure 1A). To verify the specificity of SB202190 inhibition of C/EBPβ activation, additional experiment was performed by pre‐treating myotubes with c‐Jun N‐terminal kinase (JNK) inhibitor SP600125, which did not alter activin A‐induced C/EBPβ activation (Figure 1B). Then, we treated myotubes with another ActRIIB agonist myostatin and observed similar activation of p38 MAPK, C/EBPβ, and Akt (Figure 1C). Thus, the activin A effects observed are attributable to ActRIIB activation. Further, by knocking down the expression of p38α MAPK or p38β MAPK utilizing specific siRNAs, we confirmed that p38β MAPK, not p38α MAPK, mediates C/EBPβ activation induced by activin A (Figure 1D). These data indicate that ActRIIB activation by activin A or myostatin rapidly activates both p38 MAPK and Akt in myotubes in a parallel fashion. Given that Akt activation inhibits muscle catabolism, Akt does not appear to be part of activin A‐activated catabolic signalling. On the contrary, the p38β MAPK‐C/EBPβ signalling pathway appears involved in activin A‐induced catabolic signalling.
Figure 1.

Activin A activates the p38β mitogen‐activated protein kinases (MAPK)‐C/EBPβ catabolic signalling and the Akt anabolic signalling in C2C12 myotubes. (A) Activin A activates p38 MAPK‐C/EBPβ and Akt signalling. C2C12 myotubes were treated with activin A (Act A, 3 ng/mL) and/or SB202190 (SB, 10 μM) for 1 h as indicated, and cell lysate was analysed by Western blotting. (B) JNK inhibitor SP600125 does not block activin A activation of C/EBPβ. SP600125 (SP, 10 μM) was used as in the place of SB in (A), and its effect on C/EBPβ activation was analysed. (C) Myostatin activates p38 MAPK‐C/EBPβ and Akt signalling. C2C12 myotubes were treated with myostatin (3 ng/mL) and/or SB202190 (SB, 10 μM) for 1 h as indicated, and cell lysate was analysed by Western blotting. (D) p38β MAPK specifically mediates C/EBPβ activation by activin A. C2C12 myoblasts transfected with siRNA as indicated were differentiated for 96 h to form myotubes and then treated with activin A for 1 h. Cell lysate was analysed by Western blotting. Data were analysed with analysis of variance. Asterisk (*) denotes a difference (P < 0.05).
Activin A up‐regulates the expression of E3 ubiquitin ligases and activates autophagy through the activation of p38β MAPK in C2C12 myotubes
Next, we investigated the signalling mechanism through which activin A up‐regulates E3 ubiquitin ligases that are implicated in muscle catabolism, that is, atrogin1, MuRF1, and UBR2. We found that activin A treatment of myotubes for 8 h increased the levels of all three E3s. In addition, the up‐regulation of atrogin1 and UBR2 was blocked by SB202190. However, the up‐regulation of MuRF1 was not affected by the p38 MAPK inhibitor (Figure 2A). To verify the specificity of the SB202190 effect on atrogin1 and UBR2 levels, additional experiment was performed by pre‐treating myotubes with JNK inhibitor SP600125, which did not alter activin A‐induced up‐regulation of the two ligases (Figure 2B). Utilizing siRNA‐mediated gene knocked down, we confirmed that the p38β MAPK isoform mediated activin A up‐regulation of atrogin1 and UBR2, but not MuRF1 (Figure 2C). To determine whether p38β MAPK mediates activin A‐induced up‐regulation of atrogin1 and UBR2 through a transcriptional mechanism, we analysed the mRNA levels and found that activin A up‐regulated the atrogin1 and UBR2 mRNA, which was blocked by either SB202190 (Figure 2D) or p38β MAPK knockdown (Figure 2E). Therefore, activin A up‐regulates atrogin1 and UBR2 gene expression via the activation of p38β MAPK and up‐regulates MuRF1 via a third signalling pathway other than p38β MAPK‐ or Akt‐mediated signalling.
Figure 2.

Activin A up‐regulates the expression of E3 ubiquitin ligases UBR2 and atrogin1 through the activation of p38β MAPK in C2C12 myotubes. (A) Activin A up‐regulates UBR2 and atrogin1 via p38 MAPK. C2C12 myotubes were treated with activin A with or without the presence of SB202190 for 8 h. Cell lysate was analysed by Western blotting. (B) JNK inhibition does not affect activin A up‐regulation of UBR2 and atrogin1. (C) Activin A up‐regulates UBR2 and atrogin1 via p38β MAPK. C2C12 myoblasts transfected with small interfering RNA (siRNA) as indicated were differentiated for 96 h to form myotubes and then treated with activin A for 8 h. E3 levels in cell lysate were determined by Western blotting. (D) Activin A up‐regulates UBR2 and atrogin1 gene expression via p38 MAPK. C2C12 myotubes were treated as described in (A) for 4 h. Real‐time PCR was performed to determine mRNA levels of the two genes. (E) Activin A up‐regulation of the UBR2 and atrogin1 gene requires p38β MAPK. C2C12 myoblasts transfected with siRNA as indicated were differentiated for 96 h to form myotubes and then treated with activin A for 4 h. Real‐time PCR was performed to determine mRNA levels of the two genes. Data were analysed with analysis of variance. Asterisk (*) denotes a difference (P < 0.05).
In addition to activating the UPP, ActRIIB activation also leads to autophagy activation.40 However, the signalling pathway through which ActRIIB activates autophagy is undefined. Activation of p38 MAPK15 or inactivation of Akt41 activates autophagy in muscle cells. However, the fact that Akt is activated by activin A precludes its involvement in mediating autophagy activation by ActRIIB. Therefore, we examined whether activin A activates autophagy via p38 MAPK. We monitored autophagy activity by analysing levels of LC3‐II, the marker of autophagosome formation,42 and observed that activin A treatment of myotubes for 8 h increased LC3‐II levels, which was blocked by SB202190 (Figure 3A), but not by SP600125 (Figure 3B). Further, activin A effect on LC3 levels was blocked by p38β MAPK knockdown specifically (Figure 3C). Therefore, ActRIIB activation in myotubes stimulates autophagy activity via p38β MAPK.
Figure 3.

Activin A induces autophagy activation in C2C12 myotubes via the activation of p38β MAPK. Cell lysates derived from Figure 2A–C were further analysed for LC3 levels by Western blotting. LC3‐II levels were normalized to α‐tubulin. Asterisk (*) denotes a difference (P < 0.05) between the bracketed columns as determined by analysis of variance.
Activin A induces myotube atrophy by activating p38β MAPK
To assess whether p38β MAPK is critical to activin A‐induced myotube atrophy, we treated myotubes with activin A for 72 h and observed that activin A induced MHC loss, which was blocked by SB202190 (Figure 4A). As expected, SP600125 failed to prevent the reduction of MHC induced by activin A (Figure 4B). Knockdown of p38β MAPK spared MHC loss (Figure 4C). Further, by measuring the diameter of myotubes, we observed that activin A induced loss of myotube mass, which was blocked by SB202190 (Figure 4D), but not SP600125 (Figure 4E), and by knockdown of p38β MAPK (Figure 4F). Therefore, activin A‐induced activation of p38β MAPK is critical to the development of myofibrillar protein loss and myotube atrophy.
Figure 4.
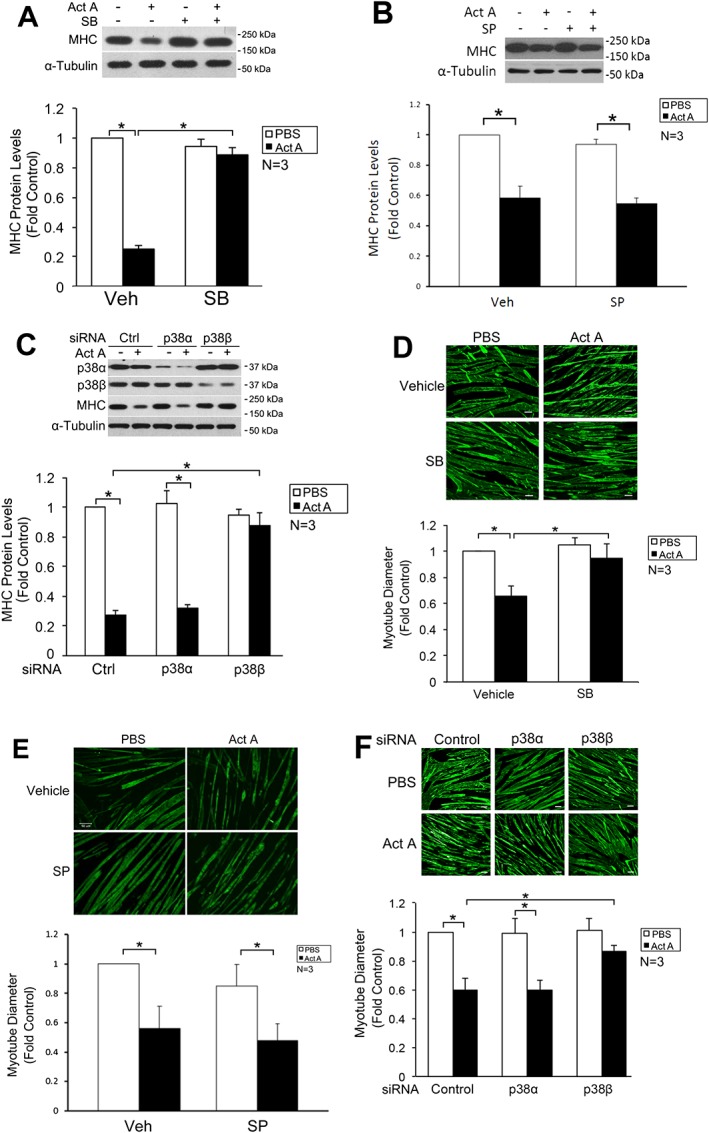
Activin A induces myotube atrophy by activating p38β MAPK. (A) Activin A induces myosin heavy chain (MHC) loss through the activation of p38 MAPK. C2C12 myotubes were treated with activin A for 72 h with or without the presence of SB202190. MHC in cell lysate was analysed by Western blotting. (B) Activin A‐induced MHC loss is not blocked by JNK inhibition. C2C12 myotubes were treated with activin A for 72 h with or without the presence of SP600125. MHC in cell lysate was analysed by Western blotting. (C) Activin A induces MHC loss through the activation of p38β MAPK. Myoblasts were transfected with siRNA as indicated and differentiated into myotubes for 96 h. Then, myotubes were treated with activin A for 72 h. MHC in cell lysate was analysed by Western blotting. (D–F) Activin A induces myotube atrophy through the activation of p38β MAPK. Myotubes derived from (A–C) were subjected to immunofluorescence staining of MHC. Diameter of myotubes was then determined as described in Methods section. Bar = 50 µm. Asterisk (*) denotes a difference (P < 0.05) between the bracketed columns as determined by analysis of variance.
Activin A‐induced muscle catabolic response in mice is dependent on p38β MAPK
To verify whether p38β MAPK signalling in muscle mediates the catabolic response to activin A in vivo, we administered activin A systemically to mice (i.p. 100 µg/kg, extrapolated from the in vitro dose used) and examined markers of the catabolic pathways in TA in 8 h. Similar to myotubes, activin A administration resulted in activation of p38 MAPK (Figure 5A) as well as C/EBPβ and Akt (Figure 5B) in control mice (p38βf/f) in vivo. In addition, we observed up‐regulation of atrogin1, UBR2, and MuRF1 and increase in LC3‐II in control mice. Pre‐treating mice with SB202190 blocked C/EBPβ activation, atrogin1, and UBR2 up‐regulation as well as LC3‐II increase, but not Akt activation and MuRF1 up‐regulation. Further, we administered activin A to mice with muscle‐specific knockout of p38β MAPK (p38β MKO) to study its in vivo effects. In the absence of p38β MAPK, activin A failed to induce C/EBPβ activation, atrogin1, and UBR2 up‐regulation as well as LC3‐II increase. On the other hand, Akt activation and MuRF1 up‐regulation were not affected by p38β MAPK knockout (Figure 5B). To determine whether activin A promotes the gene expression of atrogin1, UBR2, and LC3b in vivo, we further analysed their mRNA levels in TA. We observed increase in the three mRNAs by activin A, which was abolished by SB202190 or p38β MAPK knockout (Figure 5C). Therefore, we conclude that activin A activates muscle catabolic pathways in vivo primarily through the activation of p38β MAPK.
Figure 5.

Activin A‐induced muscle catabolic response in mice is dependent on p38β MAPK. (A) Systemic administration of activin A activates p38 MAPK in mouse muscle. Activin A (or vehicle phosphate‐buffered saline (PBS)) was injected i.p. (100 µg/kg) to 7‐week‐old male control mice (p38βf/f). In 8 h, the tibialis anterior (TA) muscle was collected for analysis by Western blotting. (B) Activin A‐induced catabolic response in mouse muscle is abolished by inhibition or knockout of p38β MAPK. Activin A (or vehicle PBS) was injected i.p. (100 µg/kg) to 7‐week‐old male control mice (p38βf/f) and mice with muscle‐specific knockout of p38β MAPK (p38β MKO). When indicated, SB202190 was i.p. injected (5 mg/kg) 30 min prior to activin A injection. In 8 h, the TA muscle was collected for analyses by Western blotting. (C) Activin A acts through p38β MAPK to up‐regulate the gene transcription of UBR2, atrogin1, and LC3b in mouse muscle. TA samples obtained were further analysed with real‐time PCR for mRNA levels of the three genes. Asterisk (*) denotes a difference (P < 0.05) between the bracketed columns as determined by analysis of variance.
Discussion
The current study reveals that ActRIIB activation by activin A induces muscle catabolism primarily through the activation of p38β MAPK‐mediated catabolic signalling that activates the UPP and the ALP. This finding is important because of the current confusion on the signalling mechanism of ActRIIB‐mediated muscle catabolism, given that ActRIIB is widely considered as a therapeutic target for various forms of muscle atrophy. Previous studies demonstrated the catabolic effect of activin A on muscle by systemically overexpressing activin A in mice43 where levels of activin A that muscles were exposed to was uncontrolled. By administrating activin A, we show here that activin A provokes a catabolic response in a dose comparable with that of other catabolic cytokines such as TNFα or IL‐6 in vitro and in vivo,16, 44 which puts activin A in the same pathological context as other known mediators of muscle wasting.
The signalling mechanism through which ActRIIB mediates accelerated muscle protein degradation is more complex than originally thought. Previously, the Smad family of transcription factors and Akt‐mediated signalling were considered as the major mediators of the catabolic action of ActRIIB in skeletal muscle.25 However, recent data refuted a key role of Smads in ActRIIB‐mediated muscle atrophy,33 and our present data suggest a non‐essential role of Akt in this process. Instead, we obtained in vitro and in vivo evidence that p38β MAPK is required for ActRIIB‐mediated activation of catabolic pathways in muscle.
Although we showed previously that p38β MAPK up‐regulates atrogin1 and UBR2,8, 23 this is the first time we demonstrate that p38β MAPK also mediates the activation of autophagy. It is known for several years that p38 MAPK mediates autophagy activation in response to pathogen challenges as part of the innate immune response.45 Similarly, such pathogen as LPS activates autophagy in skeletal muscle cells via the activation of p38 MAPK.15 However, the specific isoform of p38 MAPK that mediates autophagy activation was unknown. We present in vitro and in vivo evidence in the current study that activin A induces autophagy activation in muscle cells via p38β MAPK. This finding identifies an ActRIIB‐activated signalling pathway in muscle that coordinately activates the two major cellular protein degradation pathways, the UPP and the ALP, and allows intervention of ActRIIB‐mediated activation of both pathways by targeting abnormal activation of a single kinase—p38β MAPK.
While we confirmed that atrogin1 and UBR2 up‐regulation by activin A was mediated by p38β MAPK but not Akt, we also showed that MuRF1 up‐regulation by activin A was dependent on neither p38 MAPK nor Akt. Sriram et al. showed previously that MuRF1 is up‐regulated by ActRIIB agonist myostatin independent of Smad3 or NF‐kB.33 Thus, up‐regulation of MuRF1 by activin A may be mediated by an unknown mechanism. MuRF1 up‐regulation has been considered critical to muscle atrophy induced by diverse stimuli because of its direct role in targeting myofibrillar proteins including MHC for degradation.46, 47 However, in the present study, we observed that perturbation of p38β MAPK blocked activin A‐induced MHC loss and myotube atrophy without affecting the up‐regulation of MuRF1. Therefore, MuRF1 appears not essential for the degradation of MHC and myotube atrophy induced by activin A. Thus, activin A‐induced degradation of MHC is likely mediated by unknown mechanisms. In fact, MuRF1 is not up‐regulated in cachectic muscle of certain tumour‐bearing mice with severe MHC loss,24 which supports the presence of unknown mechanisms that mediate MHC degradation associated with cancer cachexia.
As a stress‐activated kinase, muscle p38 MAPK is activated by many catabolic stimuli in addition to ActRIIB agonists, including cancer,24 LPS,15, 17 pro‐inflammatory cytokines,16 oxidative stress, and dexamethasone.17 Thus, p38β MAPK appears to be a central mediator of muscle wasting associated with various disease conditions and therefore a common therapeutic target for muscle wasting associated with various diseases. On the other hand, Akt, FoxOs, and Smads appear to be critical to muscle atrophy associated with physiological stress, such as fasting, disuse, and denervation as shown by other investigators.13, 14 Considering that p38α MAPK is responsible for many of the functions of p38 MAPK ranging from responding to inflammatory stimuli48 and mediating ischaemic preconditioning in the heart49 to regulating myogenesis in striated muscles,18, 50 developing p38β MAPK‐specific inhibitors is a promising strategy for combating cachectic muscle wasting associated with various diseases.
Conflict of interest
The authors certify that they have no conflict of interest.
Acknowledgement
This study was supported by an R01 grant from the National Institute of Arthritis and Musculoskeletal and Skin Diseases to Y.‐P.L. (AR063786). We thank Boehringer Ingelheim Pharmaceuticals, Inc. for providing floxed‐p38 mice. The authors certify that they comply with the ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2015.51
Ding, H. , Zhang, G. , Sin, K. W. T. , Liu, Z. , Lin, R.‐K. , Li, M. , and Li, Y.‐P. (2017) Activin A induces skeletal muscle catabolism via p38β mitogen‐activated protein kinase. Journal of Cachexia, Sarcopenia and Muscle, 8: 202–212. doi: 10.1002/jcsm.12145.
References
- 1. Evans WJ, Morley JE, Argiles J, Bales C, Baracos V, Guttridge D, et al Clin Nutr 2008;27:793–799. [DOI] [PubMed] [Google Scholar]
- 2. Cohen S, Nathan JA, Goldberg AL. Muscle wasting in disease: molecular mechanisms and promising therapies. Nat Rev Drug Discov 2015;14:58–74. [DOI] [PubMed] [Google Scholar]
- 3. Fearon KC, Glass DJ, Guttridge DC. Cancer cachexia: mediators, signaling, and metabolic pathways. Cell Metab 2012;16:153–166. [DOI] [PubMed] [Google Scholar]
- 4. Lecker SH, Goldberg AL, Mitch WE. Protein degradation by the ubiquitin–proteasome pathway in normal and disease states. J Am Soc Nephrol 2006;17:1807–1819. [DOI] [PubMed] [Google Scholar]
- 5. Gomes MD, Lecker SH, Jagoe RT, Navon A, Goldberg AL. Atrogin‐1, a muscle‐specific F‐box protein highly expressed during muscle atrophy. Proc Natl Acad Sci U S A 2001;98:14440–14445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, et al Science 2001;294:1704–1708. [DOI] [PubMed] [Google Scholar]
- 7. Kwak KS, Zhou X, Solomon V, Baracos VE, Davis J, Bannon AW, et al Regulation of protein catabolism by muscle‐specific and cytokine‐inducible ubiquitin ligase E3alpha‐II during cancer cachexia. Cancer Res 2004;64:8193–8198. [DOI] [PubMed] [Google Scholar]
- 8. Zhang G, Lin RK, Kwon YT, Li YP. Signaling mechanism of tumor cell‐induced up‐regulation of E3 ubiquitin ligase UBR2. FASEB J Official Publication of the Federation of American Societies for Experimental Biology 2013;27:2893–2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Penna F, Costamagna D, Pin F, Camperi A, Fanzani A, Chiarpotto EM, et al Autophagic degradation contributes to muscle wasting in cancer cachexia. Am J Pathol 2013;182:1367–1378. [DOI] [PubMed] [Google Scholar]
- 10. Paul PK, Gupta SK, Bhatnagar S, Panguluri SK, Darnay BG, Choi Y, et al J Cell Biol 2010;191:1395–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Op den Kamp CM, Langen RC, Snepvangers FJ, de Theije CC, Schellekens JM, Laugs F, et al Nuclear transcription factor kappa B activation and protein turnover adaptations in skeletal muscle of patients with progressive stages of lung cancer cachexia. Am J Clin Nutr 2013;98:738–748. [DOI] [PubMed] [Google Scholar]
- 12. Bechet D, Tassa A, Taillandier D, Combaret L, Attaix D. Lysosomal proteolysis in skeletal muscle. Int J Biochem Cell Biol 2005;37:2098–2114. [DOI] [PubMed] [Google Scholar]
- 13. Zhao J, Brault JJ, Schild A, Goldberg AL. Coordinate activation of autophagy and the proteasome pathway by FoxO transcription factor. Autophagy 2008;4:378–380. [DOI] [PubMed] [Google Scholar]
- 14. Milan G, Romanello V, Pescatore F, Armani A, Paik JH, Frasson L, et al Regulation of autophagy and the ubiquitin–proteasome system by the FoxO transcriptional network during muscle atrophy. Nat Commun 2015;6:6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Doyle A, Zhang G, Abdel Fattah EA, Eissa NT, Li YP. Toll‐like receptor 4 mediates lipopolysaccharide‐induced muscle catabolism via coordinate activation of ubiquitin–proteasome and autophagy–lysosome pathways. FASEB J 2011;25:99–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li YP, Chen Y, John J, Moylan J, Jin B, Mann DL, et al TNF‐alpha acts via p38 MAPK to stimulate expression of the ubiquitin ligase atrogin1/MAFbx in skeletal muscle. FASEB J 2005;19:362–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. McClung JM, Judge AR, Powers SK, Yan Z. p38 MAPK links oxidative stress to autophagy‐related gene expression in cachectic muscle wasting. Am J Physiol Cell Physiol 2010;298:C542–C549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Palacios D, Mozzetta C, Consalvi S, Caretti G, Saccone V, Proserpio V, et al TNF/p38alpha/polycomb signaling to Pax7 locus in satellite cells links inflammation to the epigenetic control of muscle regeneration. Cell Stem Cell 2010;7:455–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Forcales SV, Albini S, Giordani L, Malecova B, Cignolo L, Chernov A, et al Signal‐dependent incorporation of MyoD‐BAF60c into Brg1‐based SWI/SNF chromatin‐remodelling complex. EMBO J 2012;31:301–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gillespie MA, Le Grand F, Scime A, Kuang S, von Maltzahn J, Seale V, et al p38‐{gamma}‐dependent gene silencing restricts entry into the myogenic differentiation program. J Cell Biol 2009;187:991–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pogozelski AR, Geng T, Li P, Yin X, Lira VA, Zhang M, et al p38gamma mitogen‐activated protein kinase is a key regulator in skeletal muscle metabolic adaptation in mice. PLoS One 2009;4:e7934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ho RC, Alcazar O, Fujii N, Hirshman MF, Goodyear LJ. p38gamma MAPK regulation of glucose transporter expression and glucose uptake in L6 myotubes and mouse skeletal muscle. Am J Physiol Regul Integr Comp Physiol 2004;286:R342–R349. [DOI] [PubMed] [Google Scholar]
- 23. Zhang G, Li YP. p38beta MAPK upregulates atrogin1/MAFbx by specific phosphorylation of C/EBPbeta. Skelet Muscle 2012;2:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang G, Jin B, Li YP. C/EBPbeta mediates tumour‐induced ubiquitin ligase atrogin1/MAFbx upregulation and muscle wasting. EMBO J 2011;30:4323–4335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Han HQ, Zhou X, Mitch WE, Goldberg AL. Myostatin/activin pathway antagonism: molecular basis and therapeutic potential. Int J Biochem Cell Biol 2013;45:2333–2347. [DOI] [PubMed] [Google Scholar]
- 26. Woodruff TK, Krummen LA, Lyon RJ, Stocks DL, Mather JP. Recombinant human inhibin A and recombinant human activin A regulate pituitary and ovarian function in the adult female rat. Endocrinology 1993;132:2332–2341. [DOI] [PubMed] [Google Scholar]
- 27. Jones KL, Mansell A, Patella S, Scott BJ, Hedger MP, de Kretser DM, et al Activin A is a critical component of the inflammatory response, and its binding protein, follistatin, reduces mortality in endotoxemia. Proc Natl Acad Sci U S A 2007;104:16239–16244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhou X, Wang JL, Lu J, Song Y, Kwak KS, Jiao Q, et al Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell 2010;142:531–543. [DOI] [PubMed] [Google Scholar]
- 29. Lee SJ, McPherron AC. Regulation of myostatin activity and muscle growth. Proc Natl Acad Sci U S A 2001;98:9306–9311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lokireddy S, Wijesoma IW, Bonala S, Wei M, Sze SK, McFarlane C, et al Myostatin is a novel tumoral factor that induces cancer cachexia. Biochem J 2012;446:23–36. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 31. Zimmers TA, Davies MV, Koniaris LG, Haynes P, Esquela AF, Tomkinson KN, et al Induction of cachexia in mice by systemically administered myostatin. Science 2002;296:1486–1488. [DOI] [PubMed] [Google Scholar]
- 32. Sartori R, Milan G, Patron M, Mammucari C, Blaauw B, Abraham R, et al Smad2 and 3 transcription factors control muscle mass in adulthood. Am J Physiol Cell Physiol 2009;296:C1248–C1257. [DOI] [PubMed] [Google Scholar]
- 33. Sriram S, Subramanian S, Juvvuna PK, Ge X, Lokireddy S, McFarlane CD, et al Myostatin augments muscle‐specific ring finger protein‐1 expression through an NF‐kB independent mechanism in SMAD3 null muscle. Mol Endocrinol 2014;28:317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Huang Z, Chen D, Zhang K, Yu B, Chen X, Meng J. Regulation of myostatin signaling by c‐Jun N‐terminal kinase in C2C12 cells. Cell Signal 2007;19:2286–2295. [DOI] [PubMed] [Google Scholar]
- 35. Terada Y, Nakashima O, Inoshita S, Kuwahara M, Sasaki S, Marumo F. Mitogen‐activated protein kinase cascade and transcription factors: the opposite role of MKK3/6‐p38K and MKK1‐MAPK. Nephrol Dial Transplant 1999;14:45–47. [DOI] [PubMed] [Google Scholar]
- 36. Menconi M, Gonnella P, Petkova V, Lecker S, Hasselgren PO. Dexamethasone and corticosterone induce similar, but not identical, muscle wasting responses in cultured L6 and C2C12 myotubes. J Cell Biochem 2008;105:353–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, et al Foxo transcription factors induce the atrophy‐related ubiquitin ligase atrogin‐1 and cause skeletal muscle atrophy. Cell 2004;117:399–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Stitt TN, Drujan D, Clarke BA, Panaro F, Timofeyva Y, Kline WO, et al The IGF‐1/PI3K/Akt pathway prevents expression of muscle atrophy‐induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol Cell 2004;14:395–403. [DOI] [PubMed] [Google Scholar]
- 39. Moylan JS, Smith JD, Chambers MA, McLoughlin TJ, Reid MB. TNF induction of atrogin‐1/MAFbx mRNA depends on Foxo4 expression but not AKT‐Foxo1/3 signaling. Am J Physiol Cell Physiol 2008;295:C986–C993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lee JY, Hopkinson NS, Kemp PR. Myostatin induces autophagy in skeletal muscle in vitro. Biochem Biophys Res Commun 2011;415:632–636. [DOI] [PubMed] [Google Scholar]
- 41. Zhao J, Brault JJ, Schild A, Cao P, Sandri M, Schiaffino S, et al FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab 2007;6:472–483. [DOI] [PubMed] [Google Scholar]
- 42. Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, et al Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008;4:151–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chen JL, Walton KL, Winbanks CE, Murphy KT, Thomson RE, Makanji Y, et al Elevated expression of activins promotes muscle wasting and cachexia. FASEB J 2014;28:1711–1723. [DOI] [PubMed] [Google Scholar]
- 44. Bonetto A, Aydogdu T, Jin X, Zhang Z, Zhan R, Puzis L, et al JAK/STAT3 pathway inhibition blocks skeletal muscle wasting downstream of IL‐6 and in experimental cancer cachexia. Am J Physiol Endocrinol Metab 2012;303:E410–E421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Xu Y, Liu XD, Gong X, Eissa NT. Signaling pathway of autophagy associated with innate immunity. Autophagy 2008;4:110–112. [DOI] [PubMed] [Google Scholar]
- 46. Clarke BA, Drujan D, Willis MS, Murphy LO, Corpina RA, Burova E, et al The E3 Ligase MuRF1 degrades myosin heavy chain protein in dexamethasone‐treated skeletal muscle. Cell Metab 2007;6:376–385. [DOI] [PubMed] [Google Scholar]
- 47. Cohen S, Brault JJ, Gygi SP, Glass DJ, Valenzuela DM, Gartner C, et al During muscle atrophy, thick, but not thin, filament components are degraded by MuRF1‐dependent ubiquitylation. J Cell Biol 2009;185:1083–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. O'Keefe SJ, Mudgett JS, Cupo S, Parsons JN, Chartrain NA, Fitzgerald C, et al Chemical genetics define the roles of p38alpha and p38beta in acute and chronic inflammation. J Biol Chem 2007;282:34663–34671. [DOI] [PubMed] [Google Scholar]
- 49. Sicard P, Clark JE, Jacquet S, Mohammadi S, Arthur JS, O'Keefe SJ, et al The activation of p38 alpha, and not p38 beta, mitogen‐activated protein kinase is required for ischemic preconditioning. J Mol Cell Cardiol 2010;48:1324–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Brien P, Pugazhendhi D, Woodhouse S, Oxley D, Pell JM. p38alpha MAPK regulates adult muscle stem cell fate by restricting progenitor proliferation during postnatal growth and repair. Stem Cells 2013;31:1597–1610. [DOI] [PubMed] [Google Scholar]
- 51. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle. J Cachexia Sarcopenia Muscle 2015;6:315–316. [DOI] [PMC free article] [PubMed] [Google Scholar]