

Abstract
Transdermal delivery of hydrophilic drugs is challenging. This study presents a novel sustained epidermal powder delivery technology (sEPD) for safe, efficient, and sustained delivery of hydrophilic drugs across the skin. SEPD is based on coating powder drugs into high-aspect-ratio, micro-coating channels (MCCs) followed by topical application of powder drug-coated array patches onto ablative fractional laser-generated skin MCs to deliver drugs into the skin. We found sEPD could efficiently deliver chemical drugs without excipients and biologics drugs in the presence of sugar excipients into the skin with a duration of ~12 hours. Interestingly the sEPD significantly improved zidovudine bioavailability by ~100% as compared to oral gavage delivery. SEPD of insulin was found to maintain blood glucose levels in normal range for at least 6 hours in chemical-induced diabetes mice, while subcutaneous injection failed to maintain blood glucose levels in normal range. SEPD of anti-programmed death-1 antibody showed more potent anti-tumor efficacy than intraperitoneal injection in B16F10 melanoma models. Tiny skin MCs and ‘bulk’ drug powder inside relatively deep MCCs are crucial to induce the sustained drug release. The improved bioavailability and functionality warrants further development of the novel sEPD for clinical use.
Keywords: transdermal drug delivery, laser, micropore, microporation, microchannel, microneedle
Graphical abstract
Sustained epidermal powder delivery (sEPD) efficiently and sustainably delivers powder drugs coated inside micro-coating channels (MCCs) of a thick patch into the skin via tiny skin microchannels (MCs) owing to slow water evaporation from skin MCs to gradually dissolve topical drug powder, while control delivery induces more instant drug release.
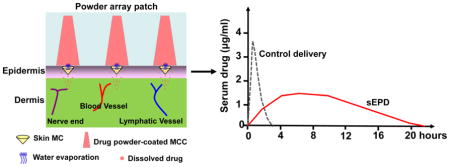
Introduction
Oral administration remains the most popular route for drug delivery due to its convenience and non-invasiveness [1]. Yet, drugs delivered orally can be extensively degraded in the gastrointestinal tract (GI) and metabolized in the liver before reaching systemic circulation. The GI tract degradation and first-pass metabolism present significant challenges for oral delivery of certain chemical drugs and most of the biologics drugs. Transdermal delivery has been actively pursued for alternative drug delivery with following advantages [2]. Firstly, transdermal delivery bypasses first-pass metabolism and avoids the harsh environment of the GI tract, potentially improving drug bioavailability. Most biologics drugs are compatible for transdermal drug delivery. Secondly, skin has a large surface area and is readily accessible. Thirdly, transdermal delivery can sustain drug release and potentially reduce dosing frequency. Sustained release is also likely to reduce peaking plasma drug levels and drug toxicity. Lastly, transdermal delivery can be needle-free, painless, and self-applicable with good patient compliance.
Despite these advantages, only a limited number of small hydrophobic drugs, like nicotine, fentanyl, and lidocaine, are approved for transdermal delivery [2, 3]. This is mainly because the superficial Stratum Corneum (SC) layer of the skin is impermeable to most of the hydrophilic molecules due to its highly compacted lipid structure [4]. While the SC layer is essential to protect from environmental pathogen invasion, it also presents as a formidable barrier for transdermal drug delivery. Different methods, like hydration, chemical enhancers, tape stripping, electric current, and ultrasound, have been explored to disrupt SC layer to facilitate transdermal drug delivery [2, 3, 5–9]. Despite years of research and development, little success has been achieved in this field either due to low efficient SC ablation or induction of skin irritation or other adverse reactions [2, 3].
Lasers have been explored to facilitate transdermal drug delivery [10, 11], but face similar challenges as mentioned above. In this regard, a low-fluence laser induces quick skin recovery, but it is unable to efficiently ablate SC layer [10, 12]. On the other hand, a high-fluence laser can efficiently ablate SC layer, but has a high risk of skin damage and infection [10, 12, 13]. This dilemma is efficiently addressed by the advent of an ablative fractional laser (AFL) technology. AFL is based on Fractional Photothermolysis [14], an innovative concept in skin resurfacing field. Instead of illuminating big laser beams with a few millimeters in diameter for full-surface SC ablation, AFL emits an array of focused laser beams with ten to hundred micrometers in diameter to vaporize tiny skin tissues and generate microchannels (MCs) in the skin surface [12, 15, 16]. These MCs can span from skin surface to deep epidermal or dermal tissue, depending on laser conditions. The micro-fractional laser ablation spares the majority of the skin and causes minimal skin reactions, leading to complete and fast skin recovery in 2~3 days [12, 15, 16]. The efficient SC ablation and quick skin recovery make AFL an attractive technology for transdermal drug delivery.
In the last decade, AFL has been found to efficiently enhance transdermal delivery of a variety of hydrophilic molecules, including small chemicals, macromolecules, and nanoparticles [12, 15–17]. Different dosage forms, like liquids, gels, lotions, and creams, show good delivery across AFL-treated skin [12, 15–17]. In pursuit of a more convenient and controlled delivery platform, we explored powder drug delivery through AFL-generated skin MCs [18, 19]. In that study powder drugs were coated onto adhesive patch surface in the same pattern as AFL-generated skin MCs [18, 19]. Powder drug-coated array patches were then topically applied onto AFL-generated skin MCs to deliver drugs into the skin via these MCs [18, 19]. We found the micro-fractional epidermal powder delivery (EPD) is capable of delivering both small chemicals and macromolecules into the skin with >80% drug doses delivered within 1 hour [19]. However, due to the limited surface coating, the amount of drugs that can be delivered per patch is rather low and may not be practical for high-dose drug delivery in humans. To increase the delivery capacity, this study explores the coating of powder drugs into high-aspect-ratio, micro-coating channels (MCCs) and then investigates drug delivery efficiency, pharmacokinetics, and bioavailability in preclinical animal models. We found the volumetric coating significantly improved drug coating capacity and prolonged drug release to ~12 hours. Small chemicals can be directly coated for high efficient delivery, while sugar excipients are required to induce high efficient delivery for biologics. Remarkably, the sustained epidermal powder delivery (sEPD) was found to significantly improve drug bioavailability and functionality in vivo when compared to oral or injection delivery.
Materials and Methods
Reagents
Sulforhodamine B (SRB, 230161), ovalbumin (OVA, A5503), zidovudine (AZT, A2169), mannitol (M4125), sucrose (S9378), trehalose (T5251) were purchased from Sigma (St. Louis, MO). AZT internal standard 3’-Azido-3’-deoxythymidine (AZT-IS, MG103) was purchased from Moravek Biochemicals (Brea, CA). Anti-mouse programmed death (PD)-1 (CD279) antibody (clone RMP1-14) and rat IgG2a isotope control were obtained from Bio X Cell (West Lebanon, NH).
Animals
BALB/c and C57BL/6 mice (male, 6–8 weeks old) were purchased from Charles River Laboratories (Wilmington, MA). Animals were housed in animal facilities of University of Rhode Island (URI) and anesthetized for hair removal, laser treatment, and patch application. All animal procedures were approved by Institutional Animal Care and Use Committees of URI.
Laser device
An UltraPulse Fractional CO2 Laser (Lumenis Inc.) was used in this study to generate patch MCCs and skin MCs.
Patch preparation, coating, and extraction
A 750μm-thick polycarbonate patch laminated with an adhesive layer was exposed to 5 pulses of AFL laser at 40mJ energy and 5% coverage to generate 9×9 array of MCCs in 6×6 mm2 area. Drug powder was repeatedly pushed into these MCCs with a spatula until full. Powder-coated 9×9 array patches were directly applied or cut into four 4×4 array patches and then applied onto AFL-treated skin. Powder array patches were immersed into phosphate buffer saline (PBS) with agitation to extract coated or remaining drugs.
Patch application
Dorsal mouse skin was exposed to AFL at 5mJ energy and 5% coverage to generate 4×4 MCs in 2×2 mm2 skin area unless otherwise specified. Powder array patches were then topically applied on the laser-treated skin with patch MCCs and skin MCs aligned. Patches were then firmly pressed on the skin to ensure a tight patch/skin contact. A narrow bandage was used to keep patches in position before removal at indicated times.
Gelatin skin model
Gelatin powder from porcine skin (60 bloom, type A, Electron Microscopy Sciences) was dissolved in warm water and then poured into 35mm petri dishes to form 5% gel with ~1 cm in thickness.
In vitro Franz Cell system
Franz Cell system with orifice diameter of 5mm and recipient chamber volume of 1.5ml were custom-made by PermeGear. Patch-applied skin was excised and mounted onto the surface of the recipient chamber. Donor chamber was laid atop and assembled with the help of a clamp. PBS (1.5ml) was added into the recipient chamber and bubbles were removed to ensure full skin contact with PBS. PBS in the recipient chamber was continuously stirred. At different times, 100μl solution was removed from the recipient chamber for quantification of drug concentrations. Equal volume of fresh PBS was added back to maintain an equal volume during the entire study.
Serum SRB quantification
Blood was collected into heparin-containing tubes and quickly centrifuged to separate serum from blood cells. Fluorescence intensity of SRB was measured at 565/585nm after 1:20 dilution of serum samples into PBS.
Oral gavage
Oral gavage was performed following a published protocol [20]. In brief, mice were restrained and a sterile plastic mouse-specific feeding tube (Cadence Science, Inc.) was inserted and advanced into the stomach. Solutions were slowly injected and feeding tube was pulled out afterwards.
LC-MS/MS quantification of AZT
Liquid chromatography-tandem mass spectrometry (LC-MS/MS) was used to quantify AZT levels as reported [21]. Patch extracts and AZT standards (5, 20, 50, 100, 200, 400ng/ml) were mixed with 100ng/ml AZT-IS. Serum samples were diluted by 20 times, mixed with 100ng/ml AZT-IS, filtered through 10kDa cutoff Amicon filter. Samples were loaded into an AB Sciex 4500 QTRAP LC-MS/MS equipped with Shimadzu LC-20AD pumps and a QTRAP 4500 System. A Synergi Hydro-RP 80A, 2.0×150 mm, 4μm particle size analytical column (Phenomonex, Torrance, CA) was used for sample separation. Acquisition was performed in multiple reaction monitoring (MRM) mode using m/z 268/127 for AZT and 271/130 for AZT-IS detection. A standard curve was generated by plotting peak area ratios of AZT to AZT-IS against AZT concentrations and used to quantify AZT levels in unknown samples.
Insulin biotinylation
Insulin (Humalog, insulin lispro injection, Eli Lilly & Co, Indianapolis, IN) was biotinylated using EZ-link Sulfo-NHS-biotinylation kit (Thermo Scientific, Rockford, IL) by following manufacturer's instructions. Briefly, insulin (400μl) was mixed with 10mM biotin reagent (482μl) and then incubated at room temperature for 0.5 hour followed by dialysis against PBS to remove biotin reagent. Biotin-insulin concentration was determined by BCA Protein Assay (Pierce, Rockford, IL). To prepare insulin powder, biotin-insulin was mixed with mannitol at 1:25 ratio (w/w) and then lyophilized.
Serum insulin detection
To detect biotin-insulin levels, ELISA plates were coated with 20μg/ml anti-insulin antibody (Clone: 3A6, Novus Biologicals) at 4 C overnight. After blocking in PBS supplemented with 2% fetal bovine serum (FBS), diluted serum samples were added and incubated at room temperature for 2 hours. After washing, streptavidin-HRP conjugates (Thermo Fisher) were added (1:4000) and plates were kept at room temperature for 1 hour. After washing, TMB substrates were added and reactions were stopped by 2M H2SO4. Absorbance at 450nm was read in a microplate reader (Molecular Device).
Diabetes model and blood glucose level (BGL) detection
Streptozotocin (STZ, Sigma, S0130) was used to induce diabetes in mice. In brief, STZ was dissolved in citric acid buffer (pH4.5), filtered through a 0.22μm membrane, and i.p. injected to BALB/c mice (6–8 weeks) at 160mg/kg within 15 minutes of preparation. BGLs were measured by AlphaTRAK 2 Blood Glucose Meter (Abott). Delivery was initiated when BGLs reached 400–500 mg/dl.
Tumor model
B16F10 melanoma cells (ATCC, CRL-6475) were cultured in DMEM media supplemented with 10% FBS, 2 mM L-glutamine, and 1% penicillin-streptomycin. Cells were harvested at ~80% confluent, washed in PBS, and 5×105 cells were s.c. injected to right flank back of C57BL/6 mice. Tumor growth was monitored by a digital caliper and tumor volume was calculated by the equation v=1/2×ab2, where a and b are long and short diameter of the tumor, respectively [22, 23].
Statistical analysis
Values were expressed as Mean ± SEM (standard error of mean). Student’s t-test was used to analyze the difference between groups. P value was calculated by PRISM software (GraphPad, San Diego, CA) and considered significant if it was less than 0.05.
Results
Sustained SRB delivery
Model drug SRB was first used to explore sEPD. Five pulses of AFL at 40mJ energy and 5% coverage were found to generate high-aspect-ratio, cone-shaped MCCs in a 750μm-thick patch with each MCC ~200μm in base diameter and ~600μm in depth (upper panels, figure 1A). Commercial SRB powder was directly coated into these MCCs (lower panels, figure 1A). We found ~8μg SRB powder could be coated into each MCC, corresponding to a 16-fold increase as compared to fractional surface coating.[19] Next laser at 5mJ energy and 5% coverage was used to generate skin MCs with an average diameter of 66μm in BALB/c mice (figure 1B).
Figure 1. Patch MCCs and skin MCs.
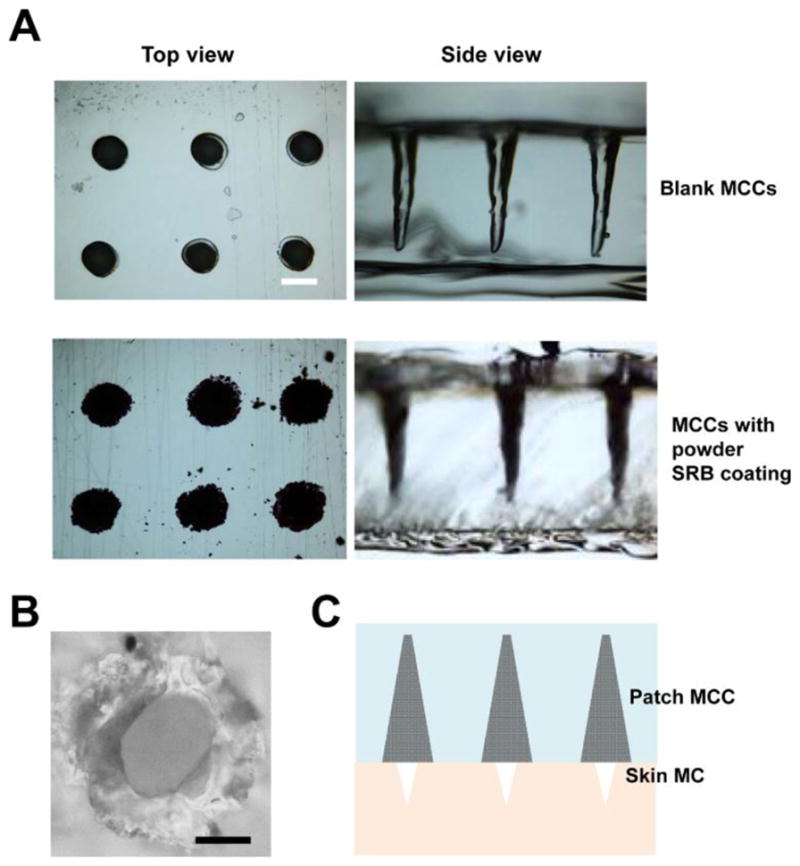
A. Top and side view of representative blank (upper panels) and powder SRB-coated MCCs (lower panels) under Nikon Eclipse E600 microscope. Scale: 200μm. B. Image of a representative skin MC under Nikon Eclipse E600 microscope. Scale: 40μm. C. Illustration of relative size of patch MCCs and skin MCs used to explore powder drug delivery in this study.
Powder SRB-coated array patches were then topically applied with patch MCC/skin MC aligned to explore powder SRB delivery via skin MCs. The relative size of patch MCCs and skin MCs in this study was shown in figure 1C. We first explored powder SRB delivery in in vitro Franz Cell systems (figure 2A). For comparison, powder SRB-coated array patches were also topically applied on tape-stripped or intact skin as our previous report.[15] As shown in figure 2B, powder SRB showed the highest delivery efficiency across AFL-treated skin. SRB levels in recipient chambers quickly rose to 62.9μg/ml in AFL group 2 days after delivery, while those in tape stripping group reached only 21.7μg/ml (figure 2B). SRB levels in recipient chambers of AFL group reached 75.5μg/ml on day 7, while those in tape stripping group only reached 54.1μg/ml (figure 2B). No significant delivery was observed in non-treated intact skin group and SRB levels was only 2.5μg/ml on day 7 (figure 2B). Early-phase delivery showed the same trend with SRB levels significantly higher in AFL group than those in tape stripping or intact skin group (figure 2C). Overall 85% and 60% SRB was delivered within 7 days across AFL-treated and tape-stripped skin, respectively, while less than 3% SRB was delivered across intact skin (figure 2D). These data indicated that powder SRB coated into MCCs of the thick patch could be efficiently and sustainably delivered through AFL-generated skin MCs.
Figure 2. Sustained powder SRB delivery in Franz cell systems.

A. Assembly of the Franz cell system. Skin of BALB/c mice was exposed to AFL at 5mJ energy and 5% coverage, or tape stripped, or left untreated followed by topical application of powder SRB-coated 4×4 array patches. Patch-applied skin was mounted onto the recipient chamber and then assembled with donor chamber. Recipient chamber was filled with 1.5mL PBS with continuous stirring for 7 days. B. Cumulative SRB concentrations in recipient chambers of different groups. n=4. C. Cumulative SRB levels in early-phase delivery of different groups. Student’s t-test was used to compare difference between AFL and tape stripping or non-treated control group at different time points. *, p<0.05; **, p<0.01. D. SRB delivery efficiency on day 7 in different groups.
Sustained powder SRB delivery was also explored in live BALB/c mice. Powder SRB-coated array patches were topically applied onto AFL-treated skin of BALB/c mice and patches were removed at different times. Pictures of the patches and skins were taken. Spot size of powder SRB coating gradually reduced on AFL-treated skin with significant reduction occurred between 3–9 hours (upper panels, figure 3A). Spots of SRB coating completely disappeared within 15 hours on AFL-treated skin, but remained with little change on intact skin even 24 hours later (upper panels, figure 3A). In parallel, pink-colored SRB emerged on AFL-treated skin right after patch application and remained visible in entire 24 hours (lower panels, figure 3A). SRB color in AFL-treated skin had no significant change in the first 3 hours, gradually increased after 5 hours and reduced after 12 hours, and remained visible at 24 hours (lower panels, figure 3A). Interestingly, the dark brown color of SRB, indicative of highly concentrated SRB solution, was evident in 11 out of 16 skin MCs between 5–9 hours, hinting significant SRB delivery during this period (lower panels, figure 3A). No significant pink color was found on intact skin even 24 hours later (lower panels, figure 3A). We further quantified SRB amount in patches before and 24 hours after delivery and found >90% SRB disappeared from patches applied onto AFL-treated skin, while >95% SRB remained on patches applied onto intact skin (figure 3B). Considering <0.5% SRB remained on skin surface in each group (data not shown), we concluded that powder SRB reached more than 90% delivery efficiency on AFL-treated skin and <5% delivery efficiency on intact skin (figure 3B). Serum SRB levels were detectable right after patch application and maintained at relatively low levels in the first 2 hours (figure 3C). Serum SRB levels increased and peaked between 6–8 hours and then declined to baseline after 15 hours (figure 3C). The timeline to reach peak serum levels correlated well with the above local delivery data. Dose-dependent SRB delivery was also evident with higher serum SRB levels in four-patch group as compared to one-patch group (figure 3C).
Figure 3. Sustained powder SRB delivery in vivo.

A. Pictures of patch (upper panels) and skin (lower panels) at different time points after topical application of powder SRB-coated 4×4 array patches onto AFL-treated or intact skin of BALB/c mice. Scale: 667μm. B. Delivery efficiency of powder SRB across laser-treated and non-treated control skin. n=4. C. Serum SRB levels at different times after topical application of four or one SRB-coated 4×4 array patches onto AFL-treated skin of BALB/c mice. n=4.
Gelatin skin to explore underlying mechanisms
To more closely observe powder delivery kinetics within MCCs, 5% gelatin gel that mimics toughness and elasticity of human skin was prepared and further layered with a 3M Tegaderm film to prevent water loss during the following studies [24]. Gelatin gel-based skin model (abbreviated as gelatin skin) was exposed to laser generating the same pattern of MCs followed by topical application of powder SRB-coated array patches as above. SRB was found to gradually release into AFL-treated gelatin skin. Pink colored SRB was visible right after patch application and then gradually expanded to surrounding areas (figure 4A). Patch MCC imaging revealed channel color change from initial black to dark pink at ~5 hours, then to light pink at ~9 hours, and finally to background color at ~19 hours (figure 4B). Enlarged pictures showed clear signs of water infiltration at 0.5 hour, significant water infiltration at 5 hours, significant SRB delivery at 9 hours, and almost complete SRB delivery at 19 hours. This pattern of channel color change reflected active water infiltration, drug dissolution and diffusion inside MCCs, which is consistent with the observed in vivo delivery (figure 3A).
Figure 4. Sustained powder SRB delivery in gelatin skin.

Powder SRB-coated 4×4 array patches were topically applied onto AFL-treated or intact gelatin skin. At different times, topical view of the delivery (A) and images of patch MCCs (B) were taken by a digital camera. Enlarged pictures of patch MCCs were shown in lower panels of B. Blue arrow points to signs of water infiltration at 0.5 hour or MCCs with clear color change at later time points.
Sustained AZT delivery with improved bioavailability
Next antiretroviral drug AZT (3'-azido-3'-deoxythymidine) was used to explore the sEPD in BALB/c mice. AZT belongs to nucleoside analog reverse-transcriptase inhibitor (NRTI) family and is taken twice daily orally together with other antiretroviral drugs to control human immunodeficiency virus (HIV) status [25]. Sustained delivery is promising to reduce dosing frequency and improve adherence of antiretroviral therapy. Commercial AZT powder was directly coated and about 7.5μg AZT powder could be coated into each MCC, similar to powder SRB coating. AZT powder-coated array patches were topically applied onto AFL-treated skin as above. Oral gavage of patch extracts served as control. As shown in figure 5A, off-white AZT powder almost completely disappeared from patches applied onto AFL-treated skin within 15 hours and MCCs after delivery were indistinguishable from those in blank patches. We further quantified AZT amount in patches before and after delivery and found >95% AZT was delivered in AFL-treated skin, while <5% AZT was delivered in intact skin (figure 5B). Serum AZT levels were detected by LC-MS/MS and peak area ratios were used to calculate serum AZT levels based on the standard curve (figure 5C). Serum AZT levels peaked at 2.7μg/ml at 30 minutes and declined to baseline at 3 hours in oral delivery, while serum AZT levels peaked at 1.0μg/ml at 4 hours and declined to baseline at ~15 hours in sEPD (figure 5D). We further calculated area under the concentration-to-time curve (AUC) and found AUC of oral delivery and sEPD was 2.8 and 5.5μg·hr/ml, respectively. The ~2-fold increase of AUC indicated improved bioavailability with sEPD. Considering water evaporation might play a crucial role in sEPD, laser parameters were modified. As shown in figure 5E, reduction of laser energy from 5 to 2.5mJ significantly reduced water evaporation measured by transepidermal water loss (TEWL) using Tewameter TM300 (Courage-Khazaka, Köln, Germany), while increase of laser pulses from 1 to 2 pulses significantly increased water evaporation. We found water evaporation was positively correlated with early-phase drug release. As shown in figure 5F, 2 pulses of AFL at 5mJ induced the quickest AZT release followed by 1 pulse of AFL at 5mJ and then 1 pulse of AFL at 2.5mJ (1 pulse was omitted hereafter for simplicity). Serum AZT levels in 5mJ/2pulse group peaked at a comparable level at 4 hours and then declined at a similar rate to that of 5mJ group (figure 5F). Serum AZT levels in 2.5mJ group peaked at 0.7μg/ml at 4 hours, which was significantly lower than that in 5mJ groups (figure 5F). Serum AZT levels in 2.5mJ group also declined with a slower rate than 5mJ groups and were still detectable at 15 hours (figure 5F).
Figure 5. Sustained AZT delivery.

A. Pictures of powder AZT-coated patches before (left) and 24 hours after delivery (middle) and also picture of blank patches (right). Scale: 667μm. B. Delivery efficiency of powder AZT-coated patches on AFL-treated or non-treated control skin at 24 hours. n=4. C. Standard curve used to quantify serum AZT levels. AZT levels were measured by LC-MS/MS method. Peak area ratios of AZT to AZT-IS of standard samples were plotted against AZT concentrations. D. Serum AZT levels in sEPD and oral gavage delivery. n=5–9. E. TEWL value of different AFL-treated skin of BALB/c mice. Student’s t-test was used to compare TEWL values between groups. n=3–5. F. Serum AZT levels at different times after topical application of powder AZT-coated array patches onto different AFL-treated skin of BALB/c mice. n=6–9.
Sustained OVA delivery in the presence of sugar excipients
Besides small chemicals, we also explored sEPD for biologics delivery. Model macromolecule OVA was explored first. Lyophilized OVA was similarly coated and topically applied onto laser-treated skin of BALB/c mice. Patches were removed 24 hours later and we found a significant amount of OVA powder remained inside MCCs of the thick patch (No excipient, figure 6A). The inefficient OVA delivery could be due to the formation of high viscosity of hydrogels and low diffusion capacity of macromolecules in hydrogels. Considering small sugar excipients, like mannitol, sucrose, trehalose, are less likely to form hydrogels due to their high water solubility and diffusion capacity, we explored whether incorporation of these excipients could increase powder OVA delivery. To this end, mannitol, sucrose, and trehalose were mixed with OVA at 25:1 weight ratio, lyophilized, coated and delivered as above. As shown in figure 6A, inclusion of mannitol, sucrose, or trehalose significantly increased OVA delivery as evidenced by the disappearance of the majority of OVA powder from MCCs within 24 hours. Due to the relatively non-hygroscopic feature of mannitol as compared to sucrose and trehalose [26], mannitol was selected to assist the delivery of other biologics in the following studies.
Figure 6. Increased powder OVA delivery in the presence of small sugar excipients.

Powder OVA-coated array patches in the presence or absence of mannitol, sucrose, or trehalose at a 1:25 mass ratio were topically applied onto AFL-generated skin MCs. Patches were removed 24 hours later and patch pictures before and after delivery were taken. Experiments were repeated 3 times and representative patch pictures were shown. Scale: 667μm.
Sustained insulin delivery with improved BGL control
Biologics drug insulin (Humalog) was then used to explore pharmacokinetics and bioavailability of sEPD. Currently most of the type I diabetes patients require multiple insulin injections per day to control their BGLs [27, 28]. Transdermal delivery eliminates needle injection and associated pain and is highly attractive for alternative insulin therapy. In this study, Humalog was conjugated to biotin to allow us to develop an ELISA assay to specifically detect human insulin in mouse serum (see Methods). We found biotin conjugation didn’t significantly affect blood glucose reduction of human insulin in STZ-induced diabetic mice (data not shown). Biotin-insulin (abbreviated as insulin) was mixed with mannitol, lyophilized, and coated as above. Powder insulin-coated array patches were topically applied onto AFL-treated skin of BALB/c mice. We found insulin powder completely disappeared from patches applied onto AFL-treated skin within 24 hours and MCCs after delivery were indistinguishable from those of blank patches (figure 7A). We further compared kinetics and bioactivity of insulin after sEPD and subcutaneous (s.c.) delivery in STZ-induced diabetic mice. As shown in figure 7B, serum insulin levels peaked 369ng/ml at ~1 hour in s.c. delivery and then declined to baseline at 4 hours. Insulin delivery was initiated right after patch application in sEPD and serum insulin levels were maintained between 40–100ng/ml from 0.5 to 8 hours (figure 7B). AUC was slightly increased in sEPD as compared to s.c. delivery (676 vs. 580ng·hr/ml). Remarkably, sEPD maintained a normal BGL (100–130mg/dl) for at least 6 hours despite a slower rate of BGL reduction at the beginning of the delivery (figure 7C). In contrast, BGLs in s.c. group quickly rose after reaching the normal value at 2 hours and were indistinguishable from those in non-treated group after 6 hours (figure 7C).
Figure 7. Sustained insulin delivery.

A. Pictures of patches before (left) and 24 hours after powder insulin delivery (middle) as well as picture of blank patches (right). Experiments were repeated 3 times and representative pictures were shown. Scale: 667μm. B–C. Powder insulin-coated patches were topically applied onto AFL-treated skin of STZ-induced diabetic mice or extracted for s.c. injection. At different time points, serum insulin levels (B) and BGLs (C) were measured. Non-treated mice served as control. n=7–9.
Improved anti-tumor immunotherapy of anti-PD-1 antibody
PD-1 is mainly expressed on activated T cell surface and is an important immune checkpoint with the major function to suppress immune responses [29]. Antibodies against PD-1 have been developed and two of these antibodies (Nivolumab, Pembrolizumab) were recently approved by FDA to treat metastatic melanoma, non-small cell lung cancer, and also other types of tumor at advanced stages [30–32]. A recent report indicated sustained local release of anti-PD-1 could significantly enhance anti-tumor immunotherapy [33]. Here we compared sEPD with systemic delivery of relatively low doses of anti-PD-1 antibody (0.72mg/kg) in immunotherapy against murine B16F10 melanoma. Mouse-origin anti-PD-1 antibodies were dialyzed, mixed with mannitol, lyophilized, and coated as in insulin delivery. Intraperitoneal (i.p.) injection of anti-PD-1 antibody, isotope control, and PBS served as controls. Treatment was initiated on day 5 and repeated twice on day 8 and 11. As shown in figure 8A, sEPD of anti-PD-1 antibody most significantly inhibited tumor growth when compared to that attained by i.p. injection of anti-PD-1 antibody or isotope control. SEPD and i.p. injection of anti-PD-1 antibody significantly reduced tumor size starting from day 12 and 14, respectively, while i.p. injection of isotope control significantly reduced tumor size from day 16 (figure 8B). No significant difference in tumor size was observed between i.p. injection of anti-PD-1 antibody and isotope control in entire period (figure 8B). Tumor size began to show significant difference between isotope control and sEPD (anti-PD-1) on day 14 and between i.p. (anti-PD-1) and sEPD (anti-PD-1) on day 16 (figure 8B). Tumor was dissected on day 16. As shown in figure 8C, tumor weight in sEPD and i.p. injection of anti-PD-1 antibody groups was significantly smaller than that in PBS group. No significant difference in tumor weight was found between PBS and isotope control group (figure 8C). Tumor weight was significantly smaller in sEPD (anti-PD-1) group as compared to i.p. (anti-PD-1) group (figure 8C). This study indicated that the novel sEPD could improve therapeutic efficacy of anti-PD-1 antibody in murine B16F10 melanoma models.
Figure 8. Improved efficacy of sEPD of anti-PD-1 antibody.

C57BL/6 mice were s.c. transplanted with B16F10 melanoma cells and then treated with sEPD of anti-PD-1 antibody, or i.p. injection of anti-PD-1 antibody or isotope control at the same antibody dose, or i.p. injection of PBS on day 5, 8, and 11. Patches were removed 24 hours later. A. Tumor size was monitored every other day from day 8. B. Days to reach significant difference of tumor sizes between groups and the significant levels. Two-way ANOVA and Bonferroni post-test were used to compare tumor sizes between groups. C–D. Mice were euthanized on day 16. Tumor was dissected and tumor images were shown in C (scale: 1 cm) and tumor weight was shown in D. Student’s t-test was used to compare tumor weight difference between groups. *, p<0.05; **, p<0.01; ***, p<0.001. n=5.
Discussion
This study explores a novel AFL-based transdermal delivery technology, called sEPD, for efficient and sustained delivery of powder hydrophilic drugs across the skin. SEPD is based on coating powder drugs into MCCs of a thick patch followed by topical application of powder drug-coated array patches onto AFL-generated skin MCs to deliver drugs into the skin. AFL has been actively explored to facilitate transdermal drug delivery in the last decade. In these studies, drugs were often prepared as liquids, gels, or creams and then topically applied onto AFL-treated skin [12, 15–17]. Most of these studies focused on local drug delivery to treat skin diseases [12, 15–17]. To our knowledge, this was the first study to explore systemic drug delivery in a powder form via AFL-generated skin MCs. We found water-soluble drugs SRB and AZT can be directly coated for high efficient delivery (figure 2–5), while biologics drugs OVA, insulin, and anti-PD-1 antibody require pre-mixing with small sugar excipients to induce high efficient delivery (figure 6–8). The low efficient biologics delivery in the absence of sugar excipients may be caused by formation of high viscosity of hydrogels and a low diffusion ability of macromolecules in hydrogels. Based on rough calculation, drug concentration will reach over 1, 000 mg/ml if 8μg drug powder is completely dissolved inside the cone-shaped MCC in figure 1A. Although this is not the case in sEPD, this calculation indicates that drugs are most likely delivered at saturated conditions at early stages of delivery considering rare drugs can have such a high water solubility. We expect drugs with high water solubility, low molecular weight, and low viscosity to have efficient sEPD. Yet whether these parameters can be established as quantitative criteria to predict efficiency of sEPD remains to be explored. In this study we found disaccharides, like mannitol, sucrose, and trehalose, were effective to enhance biologics delivery. Other water-soluble, small-molecular-weight chemicals, like amino acids, may also be able to enhance biologics delivery. The optimal excipients to enhance biologics delivery and maintain biologics activity in sEPD need to be explored in the future. In this study we found the relatively high mannitol to biologics ratio (25:1) rendered biologics delivery very similar to chemical drug delivery (figure 3, 5, 7).
SEPD induced a biphasic drug release with 3–15% drugs first released in 1–2 hours and the majority of the remaining drugs then released in the next 10–12 hours (figure 3C, 5D, 7B). Biphasic drug release is likely caused by differential powder dissolution in early and late phases. It was reported that interstitial fluid can quickly fill skin MCs after AFL treatment [34]. Patch application and firm pressing could bring lower portion of patch-coated drug powder in close contact with skin MC-filled interstitial fluid. The wetting and dissolution of this part of powder likely contribute to the early-phase drug release. The dissolution and delivery of remaining drug powder likely depend on continuous water evaporation from skin MCs. This pattern of in vivo delivery is recapitulated in powder SRB delivery in in vitro gelatin skin. We observed clear signs of water infiltration into lower portion of patch MCCs within 30 minutes and water infiltration into entire patch MCCs at 5 hours as evidenced by change of MCC color (figure 4B). In second phase of delivery, we observed uniform SRB color along long axis of each MCC, supporting relatively quick dissolution of entire remaining powder and continuous release rather than partial dissolution and pulsed release. SEPD takes advantage of interstitial water filled in and evaporated from skin MCs to gradually dissolve topical drug powder for sustained drug release. We found sEPD could be accelerated by increase of water evaporation and decelerated by reduction of water evaporation as shown in figure 5F. This finding supports generation of skin MCs that differ in water evaporation rate to modify drug release. Similar to traditional patches, daily activities, like hot baths, sunbaths, that increase skin temperature and water evaporation rate, should be avoided during sEPD. High-aspect-ratio MCCs are also key to induce sustained drug release as we found coating the same amount of powder on full patch surface induced much faster drug release (data not shown). Although the underlying reasons are unknown, we believe the overall distances that water molecules travel to dissolve the deepest drug powder in the coating or the lastly dissolved drug molecules travel to reach skin MCs could significantly impact the duration of drug release. Only one type of patch MCCs was explored for sustained drug release in this study. The impact of patch MCC shape and dimension on sustained drug release needs to be explored to better understand the potential of the novel delivery platform for clinical use.
SEPD was found to significantly improve drug bioavailability and functionality in vivo. For example, sEPD increased bioavailability of AZT by ~100% as compared to oral gavage delivery (figure 5D); sEPD of insulin maintained blood glucose levels in normal range much longer than s.c. injection delivery in STZ-induced diabetic mice (figure 7C); sEPD of anti-PD-1 antibody more significantly inhibited B16F10 tumor growth in murine models as compared to i.p. injection delivery (figure 8). The improved bioavailability and functionality are expected to be caused by sustained release considering the same doses were delivered between sEPD and control oral or injection delivery. The advantages of sEPD for different types of drug delivery warrants further development of this technology for clinical use. In this study we only explored powder drug delivery via AFL-generated skin MCs. Considering other technologies, like microneedles and radiofrequencies, can also generate similar skin MCs [35, 36], we are expecting similar deliveries after topical application of powder array patches on skin MCs generated by these technologies. Powder array patches in conjunction with these evolving skin MC-generating technologies will open a new avenue for safe and efficient transdermal delivery of hydrophilic drugs regardless of chemical structure and molecular weight.
Powder array patches have relatively high coating capacity considering no excipients are required for chemical drug delivery and only small sugar excipients are required for biologics drug delivery. In addition, drug coating amount can be conveniently expanded by enlarging patch MCC volume. SEPD is highly attractive to biologics drug delivery due to its needle-free, painless delivery, and sustained drug release with potentially improved bioavailability and functionality. The incorporation of sugar excipients, especially those with protein stabilization activities, is also promising to improve biologics stability and eliminate cold-chain storage. Furthermore, biologics powder array patches can be sealed in a small package to reduce shipping and storage cost. Direct powder delivery also eliminates human errors that may occur during reconstitution and injection of biologics drugs. SEPD is also very attractive to hydrophilic chemical drug delivery by bypassing the first-pass metabolism, avoiding the degradation of drugs in the GI tract, and sustaining drug release over time. SEPD generates lower peak serum drug levels and is likely to reduce toxicity for drugs with narrow therapeutic index. SEPD with multi-day sustained release is likely to reduce dosing frequency and improve adherence to treatment. SEPD significantly expand chemical drugs that can be delivered via the skin. A handheld laser ablation and patch application device can also be fabricated to facilitate clinical translation of this technology.
Conclusion
In summary, we developed a novel transdermal delivery platform, called sEPD, for safe, efficient, and sustained delivery of hydrophilic drugs across the skin. SEPD takes advantage of slow water evaporation from skin MCs to gradually dissolve topical ‘bulk’ drug powder inside deep MCCs to induce sustained drug release. Both chemical and biologics drugs can be efficiently and sustainably delivered by sEPD. Remarkably, we found sEPD could increase AZT bioavailability by ~100% as compared to oral gavage delivery, maintain BGLs in normal range much longer than s.c. injection delivery, and significantly improve immunotherapy efficacy of anti-PD-1 antibody against B16F10 melanoma. The improved bioavailability and functionality in vivo and the advantages of direct powder delivery warrant further investigation of sEPD in conjunction with skin MC-generating technologies, like AFL, microneedle, and radiofrequency, for hydrophilic drug delivery.
Acknowledgments
This work is partly supported by the National Institutes of Health grants DA033371 and AI107678 (to X.Y.C.). Microplate reader used in this work is supported by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health grant P20GM103430. The LC-MS/MS machine at a Rhode Island NSF EPSCoR research facility is supported in part by the National Science Foundation EPSCoR Cooperative Agreement #EPS-1004057.
Footnotes
The authors have no conflict of interest to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Gupta H, Bhandari D, Sharma A. Recent trends in oral drug delivery: a review. Recent Pat Drug Deliv Formul. 2009;3:162–173. doi: 10.2174/187221109788452267. [DOI] [PubMed] [Google Scholar]
- 2.Prausnitz MR, Langer R. Transdermal drug delivery. Nat Biotechnol. 2008;26:1261–1268. doi: 10.1038/nbt.1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Paudel KS, Milewski M, Swadley CL, Brogden NK, Ghosh P, Stinchcomb AL. Challenges and opportunities in dermal/transdermal delivery. Ther Deliv. 2010;1:109–131. doi: 10.4155/tde.10.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Elias PM. Structure and function of the stratum corneum permeability barrier. Drug Development Research. 1988;13:97–105. [Google Scholar]
- 5.Naik A, Kalia YN, Guy RH. Transdermal drug delivery: overcoming the skin's barrier function. Pharm Sci Technolo Today. 2000;3:318–326. doi: 10.1016/s1461-5347(00)00295-9. [DOI] [PubMed] [Google Scholar]
- 6.Benson HA. Transdermal drug delivery: penetration enhancement techniques. Curr Drug Deliv. 2005;2:23–33. doi: 10.2174/1567201052772915. [DOI] [PubMed] [Google Scholar]
- 7.Williams AC, Barry BW. Penetration enhancers. Adv Drug Deliv Rev. 2004;56:603–618. doi: 10.1016/j.addr.2003.10.025. [DOI] [PubMed] [Google Scholar]
- 8.Schoellhammer CM, Blankschtein D, Langer R. Skin permeabilization for transdermal drug delivery: recent advances and future prospects. Expert Opin Drug Deliv. 2014;11:393–407. doi: 10.1517/17425247.2014.875528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wong TW. Electrical, magnetic, photomechanical and cavitational waves to overcome skin barrier for transdermal drug delivery. J Control Release. 2014;193:257–269. doi: 10.1016/j.jconrel.2014.04.045. [DOI] [PubMed] [Google Scholar]
- 10.Lin CH, Aljuffali IA, Fang JY. Lasers as an approach for promoting drug delivery via skin. Expert Opin Drug Deliv. 2014;11:599–614. doi: 10.1517/17425247.2014.885501. [DOI] [PubMed] [Google Scholar]
- 11.Ali FR, Al-Niaimi F. Laser-assisted drug delivery in dermatology: from animal models to clinical practice. Lasers Med Sci. 2016;31:373–381. doi: 10.1007/s10103-015-1853-z. [DOI] [PubMed] [Google Scholar]
- 12.Lee WR, Shen SC, Al-Suwayeh SA, Yang HH, Yuan CY, Fang JY. Laser-assisted topical drug delivery by using a low-fluence fractional laser: imiquimod and macromolecules. J Control Release. 2011;153:240–248. doi: 10.1016/j.jconrel.2011.03.015. [DOI] [PubMed] [Google Scholar]
- 13.Scheiblhofer S, Thalhamer J, Weiss R. Laser microporation of the skin: prospects for painless application of protective and therapeutic vaccines. Expert Opin Drug Deliv. 2013;10:761–773. doi: 10.1517/17425247.2013.773970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Manstein D, Herron GS, Sink RK, Tanner H, Anderson RR. Fractional photothermolysis: a new concept for cutaneous remodeling using microscopic patterns of thermal injury. Lasers Surg Med. 2004;34:426–438. doi: 10.1002/lsm.20048. [DOI] [PubMed] [Google Scholar]
- 15.Chen X, Shah D, Kositratna G, Manstein D, Anderson RR, Wu MX. Facilitation of transcutaneous drug delivery and vaccine immunization by a safe laser technology. J Control Release. 2012;159:43–51. doi: 10.1016/j.jconrel.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haedersdal M, Sakamoto FH, Farinelli WA, Doukas AG, Tam J, Anderson RR. Fractional CO(2) laser-assisted drug delivery. Lasers Surg Med. 2010;42:113–122. doi: 10.1002/lsm.20860. [DOI] [PubMed] [Google Scholar]
- 17.Lee WR, Shen SC, Al-Suwayeh SA, Yang HH, Li YC, Fang JY. Skin permeation of small-molecule drugs, macromolecules, and nanoparticles mediated by a fractional carbon dioxide laser: the role of hair follicles. Pharm Res. 2013;30:792–802. doi: 10.1007/s11095-012-0920-4. [DOI] [PubMed] [Google Scholar]
- 18.Jia F, Liu S, Wu MX, Chen X. Micro-fractional Epidermal Powder Delivery for Skin Vaccination. Methods Mol Biol. 2016;1404:715–723. doi: 10.1007/978-1-4939-3389-1_46. [DOI] [PubMed] [Google Scholar]
- 19.Chen X, Kositratna G, Zhou C, Manstein D, Wu MX. Micro-fractional epidermal powder delivery for improved skin vaccination. J Control Release. 2014;192:310–316. doi: 10.1016/j.jconrel.2014.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Machholz E, Mulder G, Ruiz C, Corning BF, Pritchett-Corning KR. Manual restraint and common compound administration routes in mice and rats. J Vis Exp. 2012 doi: 10.3791/2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rower JE, Klein B, Bushman LR, Anderson PL. Validation of a sensitive LC/MS/MS method for the determination of zidovudine and lamivudine in human plasma. Biomed Chromatogr. 2012;26:12–20. doi: 10.1002/bmc.1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen X, Zeng Q, Wu MX. Improved efficacy of dendritic cell-based immunotherapy by cutaneous laser illumination. Clin Cancer Res. 2012;18:2240–2249. doi: 10.1158/1078-0432.CCR-11-2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen XY, Zhang W, Zhang W, Wu S, Bi F, Su YJ, Tan XY, Liu JN, Zhang J. Vaccination with viable human umbilical vein endothelial cells prevents metastatic tumors by attack on tumor vasculature with both cellular and humoral immunity. Clin Cancer Res. 2006;12:5834–5840. doi: 10.1158/1078-0432.CCR-06-1105. [DOI] [PubMed] [Google Scholar]
- 24.Park MA, Jang HJ, Sirotkin FV, Yoh JJ. Er:YAG laser pulse for small-dose splashback-free microjet transdermal drug delivery. Opt Lett. 2012;37:3894–3896. doi: 10.1364/ol.37.003894. [DOI] [PubMed] [Google Scholar]
- 25.Eron JJ, Yetzer ES, Ruane PJ, Becker S, Sawyer GA, Fisher RL, Tolson JM, Shaefer MS. Efficacy, safety, and adherence with a twice-daily combination lamivudine/zidovudine tablet formulation, plus a protease inhibitor, in HIV infection. AIDS. 2000;14:671–681. doi: 10.1097/00002030-200004140-00006. [DOI] [PubMed] [Google Scholar]
- 26.Fakes MG, Dali MV, Haby TA, Morris KR, Varia SA, Serajuddin AT. Moisture sorption behavior of selected bulking agents used in lyophilized products. PDA J Pharm Sci Technol. 2000;54:144–149. [PubMed] [Google Scholar]
- 27.Fry A. Insulin delivery device technology 2012: where are we after 90 years? J Diabetes Sci Technol. 2012;6:947–953. doi: 10.1177/193229681200600428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shah RB, Patel M, Maahs DM, Shah VN. Insulin delivery methods: Past, present and future. Int J Pharm Investig. 2016;6:1–9. doi: 10.4103/2230-973X.176456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leventakos K, Mansfield AS. Advances in the Treatment of Non-small Cell Lung Cancer: Focus on Nivolumab, Pembrolizumab, and Atezolizumab. BioDrugs. 2016;30:397–405. doi: 10.1007/s40259-016-0187-0. [DOI] [PubMed] [Google Scholar]
- 31.Marquez-Rodas I, Cerezuela P, Soria A, Berrocal A, Riso A, Gonzalez-Cao M, Martin-Algarra S. Immune checkpoint inhibitors: therapeutic advances in melanoma. Ann Transl Med. 2015;3:267. doi: 10.3978/j.issn.2305-5839.2015.10.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hamanishi J, Mandai M, Konishi I. Immune checkpoint inhibition in ovarian cancer. Int Immunol. 2016;28:339–348. doi: 10.1093/intimm/dxw020. [DOI] [PubMed] [Google Scholar]
- 33.Wang C, Ye Y, Hochu GM, Sadeghifar H, Gu Z. Enhanced Cancer Immunotherapy by Microneedle Patch-Assisted Delivery of Anti-PD1 Antibody. Nano Lett. 2016;16:2334–2340. doi: 10.1021/acs.nanolett.5b05030. [DOI] [PubMed] [Google Scholar]
- 34.Erlendsson AM, Doukas AG, Farinelli WA, Bhayana B, Anderson RR, Haedersdal M. Fractional laser-assisted drug delivery: Active filling of laser channels with pressure and vacuum alteration. Lasers Surg Med. 2016;48:116–124. doi: 10.1002/lsm.22374. [DOI] [PubMed] [Google Scholar]
- 35.Banga AK. Microporation applications for enhancing drug delivery. Expert Opin Drug Deliv. 2009;6:343–354. doi: 10.1517/17425240902841935. [DOI] [PubMed] [Google Scholar]
- 36.Stevenson CL, Santini JT, Jr, Langer R. Reservoir-based drug delivery systems utilizing microtechnology. Adv Drug Deliv Rev. 2012;64:1590–1602. doi: 10.1016/j.addr.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]